ZusammensetzungWirkstoffe
Bimekizumab, aus gentechnisch veränderten CHO (Chinese Hamster Ovary)-Zellen hergestellt.
Hilfsstoffe
Glycin, Natriumacetat-Trihydrat (E 262), Eisessig, Polysorbat 80, Wasser für Injektionszwecke benötigt für ein Volumen von 1 ml.
Jeder Fertigpen (1 ml) enthält 0,45 mg Natrium.
Jede Fertigspritze (1 ml) enthält 0,45 mg Natrium.
Indikationen/AnwendungsmöglichkeitenPlaque-Psoriasis
Bimzelx wird angewendet zur Behandlung erwachsener Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine systemische Therapie infrage kommen.
Psoriasis-Arthritis
Bimzelx wird allein oder in Kombination mit Methotrexat zur Behandlung erwachsener Patienten mit aktiver Psoriasis-Arthritis angewendet, die auf ein oder mehrere krankheitsmodifizierende Antirheumatika (disease-modifying antirheumatic drugs, DMARDs) unzureichend angesprochen oder diese nicht vertragen haben.
Axiale Spondyloarthritis
Nicht-röntgenologische axiale Spondyloarthritis (nr-axSpA)
Bimzelx wird zur Behandlung erwachsener Patienten mit schwerer aktiver nicht-röntgenologischer axialer Spondyloarthritis mit objektiven Anzeichen einer Entzündung, welche auf nicht-steroidale Antirheumatika (NSARs oder non-steroidal anti-inflammatory drugs, NSAIDs) unzureichend angesprochen oder diese nicht vertragen haben, angewendet. Objektive Anzeichen einer Entzündung können mittels erhöhtes C-reaktives Protein (CRP) und Magnetresonanztomographie (MRT) nachgewiesen werden.
Ankylosierende Spondylitis (AS, röntgenologische axiale Spondyloarthritis)
Bimzelx wird zur Behandlung erwachsener Patienten mit schwerer aktiver ankylosierender Spondylitis (AS), welche auf eine konventionelle Therapie unzureichend angesprochen oder diese nicht vertragen haben, angewendet.
Dosierung/AnwendungBimzelx ist zur Anwendung unter Anleitung und Überwachung durch einen Arzt vorgesehen, der in der Diagnose und Behandlung der Erkrankungen erfahren ist, für die Bimzelx indiziert ist.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Plaque-Psoriasis
Die empfohlene Dosis Bimzelx für erwachsene Patienten mit Plaque-Psoriasis beträgt 320 mg (verabreicht als 2 subkutane Injektionen zu jeweils 160 mg) in Woche 0, 4, 8, 12, 16 und danach alle 8 Wochen.
Psoriasis-Arthritis
Die empfohlene Dosis für erwachsene Patienten mit aktiver Psoriasis-Arthritis beträgt 160 mg (verabreicht als 1 subkutane Injektion von 160 mg) alle 4 Wochen.
Für Psoriasis-Arthritis-Patienten mit gleichzeitig bestehender mittelschwerer bis schwerer Plaque-Psoriasis ist die empfohlene Dosis die gleiche wie für Plaque-Psoriasis [320 mg (verabreicht als 2 subkutane Injektionen zu jeweils 160 mg) in Woche 0, 4, 8, 12, 16 und danach alle 8 Wochen]. Nach 16 Wochen wird eine regelmässige Bewertung der Wirksamkeit empfohlen und wenn ein ausreichendes klinisches Ansprechen in den Gelenken nicht aufrechterhalten werden kann, kann eine Umstellung auf 160 mg alle 4 Wochen in Betracht gezogen werden.
Axiale Spondyloarthritis (nr-axSpA und AS)
Die empfohlene Dosis für erwachsene Patienten mit axialer Spondyloarthritis beträgt 160 mg (verabreicht als 1 subkutane Injektion) alle 4 Wochen.
Bei Patienten in den zuvor genannten Indikationen, bei denen sich nach 16 Behandlungswochen keine Besserung zeigt, ist ein Absetzen der Behandlung in Erwägung zu ziehen.
Spezielle Dosierungsanweisungen
Übergewichtige Patienten mit Plaque-Psoriasis
Bei einigen Patienten mit Plaque-Psoriasis (einschliesslich Psoriasis-Arthritis mit gleichzeitig bestehender mittelschwerer bis schwerer Psoriasis) und mit einem Körpergewicht ≥120 kg, die bis Woche 16 keine vollständige Symptomfreiheit der Haut erreicht haben, könnten 320 mg alle 4 Wochen nach Woche 16 das Ansprechen auf die Behandlung weiter verbessern (siehe Klinische Wirksamkeit).
Patienten mit Leberfunktionsstörungen
Bimzelx wurde in dieser Patientengruppe nicht untersucht. Auf Grundlage der Pharmakokinetik werden Dosisanpassungen nicht als notwendig erachtet (siehe Pharmakokinetik).
Patienten mit Nierenfunktionsstörungen
Bimzelx wurde in dieser Patientengruppe nicht untersucht. Auf Grundlage der Pharmakokinetik werden Dosisanpassungen nicht als notwendig erachtet (siehe Pharmakokinetik).
Ältere Patienten
Eine Dosisanpassung ist nicht erforderlich (siehe Pharmakokinetik).
Kinder und Jugendliche
Bimzelx ist für die Anwendung in der pädiatrischen Population nicht zugelassen.
Art der Anwendung
Bimzelx wird als subkutane Injektion verabreicht.
Geeignete Injektionsstellen sind Oberschenkel, Abdomen und Oberarm. Es ist auf einen Wechsel der Injektionsstellen zu achten. Injektionen sollten nicht in Psoriasisplaques oder Bereiche erfolgen, in denen die Haut empfindlich, geschädigt, erythematös oder verhärtet ist.
Nach einer entsprechenden Einweisung in die subkutane Injektionstechnik können sich Patienten Bimzelx selbst injizieren, falls ihr Arzt dies für angemessen hält, und mit ärztlicher Nachsorge nach Bedarf. Die Patienten sind anzuweisen, die gesamte Menge Bimzelx gemäss den Hinweisen für die Handhabung in der Packungsbeilage zu injizieren (siehe Patienteninformation).
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Zusammensetzung.
Klinisch relevante aktive Infektionen (z.B. aktive Tuberkulose)
Warnhinweise und VorsichtsmassnahmenInfektionen
Bimzelx kann das Risiko von Infektionen, wie Infektionen der oberen Atemwege und oraler Candidose, erhöhen (siehe Unerwünschte Wirkungen).
Bei Patienten mit einer chronischen Infektion oder einer rezidivierenden Infektion in der Anamnese sollte Bimzelx mit Vorsicht angewendet werden. Bei Patienten mit einer klinisch relevanten aktiven Infektion (insbesondere HIV-, HBV- oder HCV-Infektionen) sollte die Behandlung mit Bimekizumab nicht eingeleitet werden, bis die Infektion abgeklungen ist oder angemessen behandelt wird. Mit Bimzelx behandelte Patienten sind anzuweisen, ärztlichen Rat einzuholen, wenn Anzeichen oder Symptome einer klinisch bedeutsamen chronischen oder akuten Infektion auftreten. Bei Entwicklung einer Infektion, ist der Patient sorgfältig zu überwachen. Wenn sich daraus eine schwere Infektion entwickelt oder eine Infektion nicht auf die Standardtherapie anspricht, soll die Behandlung abgebrochen werden, bis die Infektion abgeklungen ist.
Untersuchung auf Tuberkulose (TB) vor Behandlungsbeginn
In den klinischen Studien entwickelten Patienten mit latenter TB, die Bimzelx und eine Anti-TB-Therapie erhielten, keine aktive TB. Vor Beginn der Behandlung mit Bimzelx sind die Patienten auf eine TB-Infektion zu untersuchen. Bimzelx darf nicht bei Patienten mit aktiver TB angewendet werden. Patienten, die Bimzelx erhalten, müssen auf Anzeichen und Symptome einer aktiven TB überwacht werden. Bei Patienten mit latenter oder aktiver TB in der Vorgeschichte, bei denen nicht bestätigt werden kann, dass sie eine adäquate Behandlung erhalten haben, sollte vor Beginn der Behandlung mit Bimzelx eine Anti-TB-Therapie in Erwägung gezogen werden.
Maligne Erkrankungen
In klinischen Studien bis zu einem Jahr wurde bei der Behandlung mit Bimzelx kein erhöhtes Risiko für maligne Erkrankungen beobachtet. Resultate von Untersuchungen zur Langzeitsicherheit sind noch nicht verfügbar.
Da es sich bei Psoriasis-Patienten um eine Risikopopulation handelt, sollten Patienten vor und während der Behandlung mit Bimzelx auf das Vorliegen von Hauttumoren untersucht werden.
Entzündliche Darmerkrankungen
Fälle von neu aufgetretenen oder sich verschlechternden bestehenden entzündlichen Darmerkrankungen wurden unter Bimzelx berichtet. Bimzelx wird bei Patienten mit entzündlicher Darmerkrankung nicht empfohlen. Wenn ein Patient Anzeichen und Symptome einer entzündlichen Darmerkrankung entwickelt oder eine vorbestehende entzündliche Darmerkrankung sich verschlechtert, sollte Bimzelx abgesetzt werden und eine geeignete medizinische Behandlung eingeleitet werden.
Hypersensitivitätsreaktionen
Im Falle des Auftretens einer schwerwiegenden Überempfindlichkeitsreaktion muss die Anwendung von Bimzelx unverzüglich abgebrochen und eine geeignete Behandlung eingeleitet werden.
Impfungen
Vor Einleitung einer Therapie mit Bimzelx wird die Durchführung aller altersgemässen Impfungen in Übereinstimmung mit den aktuellen Impfempfehlungen empfohlen. Patienten, die mit Bimzelx behandelt werden, dürfen keine Lebendimpfstoffe erhalten. Patienten, die mit Bimzelx behandelt werden, können inaktivierte oder Totimpfstoffe erhalten. Gesunde Personen, die zwei Wochen vor der Impfung mit einem inaktivierten Impfstoff gegen saisonale Influenza eine Einzeldosis von 320 mg Bimzelx erhalten hatten, zeigten eine vergleichbare Antikörperantwort wie Personen, die vor der Impfung kein Bimzelx erhalten hatten.
Hilfsstoffe
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Fertigpen, d.h. es ist nahezu «natriumfrei».
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Fertigspritze, d.h. es ist nahezu «natriumfrei».
InteraktionenEs wurden keine Studien zur Erfassung von CYP450-Wechselwirkungen bei Menschen durchgeführt. Es gibt keine direkte Evidenz für die Bedeutung von IL-17A oder IL-17F bei der Expression von CYP450-Enzymen. Die Bildung einiger CYP450-Enzyme wird durch erhöhte Zytokinspiegel im Zuge chronischer Entzündungen unterdrückt. Daher können entzündungshemmende Behandlungen, etwa mit dem IL-17A- und IL-17F-Inhibitor Bimzelx, zu einer Normalisierung der CYP450-Spiegel und zu einer damit einhergehenden geringeren Exposition von CYP450-metabolisierten Begleitmedikationen führen. Daher kann eine klinisch relevante Wirkung auf CYP450-Substrate mit einer geringen therapeutischen Breite, bei denen die Dosis individuell angepasst wird (z.B. Warfarin), nicht ausgeschlossen werden. Bei Patienten, die mit dieser Art von Arzneimitteln behandelt werden, sollte zu Beginn einer Bimzelx-Therapie eine therapeutische Überwachung in Betracht gezogen werden.
Analysen der populationspharmakokinetischen (PK) Daten zeigten, dass die Clearance von Bimekizumab nicht durch die gleichzeitige Verabreichung von konventionellen krankheitsmodifizierenden Antirheumatika (conventional disease modifying antirheumatic drugs, cDMARDs), einschliesslich Methotrexat, oder durch eine vorherige Exposition gegenüber Biologika beeinflusst wurde.
Schwangerschaft, StillzeitSchwangerschaft
Bisher liegen nur begrenzte Erfahrungen mit der Anwendung von Bimzelx bei Schwangeren vor. Frauen im gebärfähigen Alter müssen während der Behandlung und für mindestens 17 Wochen nach der Behandlung eine zuverlässige Verhütungsmethode anwenden. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf Schwangerschaft, embryonale/fötale Entwicklung, Geburt oder postnatale Entwicklung (siehe Präklinische Daten). Während der Schwangerschaft sollte Bimzelx nur angewendet werden, wenn der Nutzen für die Mutter klar das potentielle Risiko für den Fötus überwiegt.
Stillzeit
Es ist nicht bekannt, ob Bimzelx in die Muttermilch übergeht oder vom Säugling systemisch resorbiert wird. Die Vorteile des Stillens für die Entwicklung und Gesundheit, die klinische Notwendigkeit einer Behandlung mit Bimzelx für die Mutter, sowie mögliche negative Auswirkungen auf das gestillte Kind durch Bimzelx oder die Grunderkrankung der Mutter sind bei der Entscheidung zu berücksichtigen.
Fertilität
Die Wirkung von Bimzelx auf die Fertilität des Menschen wurde nicht untersucht. Tierexperimentelle Studien weisen nicht auf direkte oder indirekte schädliche Wirkungen auf die Fertilität hin (siehe Präklinische Daten).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDer Einfluss von Bimzelx auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen wurde nicht spezifisch untersucht.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Klinische Studien
Übersicht
Insgesamt wurden 4821 Patienten in verblindeten und unverblindeten klinischen Studien zu Plaque-Psoriasis, Psoriasis-Arthritis und axialer Spondyloarthritis (nr-axSpA und AS) mit Bimekizumab behandelt; dies entspricht einer Exposition von 8733,0 Patientenjahren. Davon waren mehr als 3900 Patienten mindestens ein Jahr lang Bimekizumab exponiert. Insgesamt ist das Sicherheitsprofil von Bimekizumab über alle Indikationen hinweg konsistent.
Die am häufigsten berichteten unerwünschten Wirkungen waren Infektionen der oberen Atemwege (14.5%, 14.6% bzw. 16.3% bei Plaque Psoriasis [PSO], Psoriasis-Arthritis [PsA] bzw. axSpA) und orale Candidose (7,3 %, 2,3 % bzw. 3,7 % bei PSO, PsA bzw. axSpA).
Liste der unerwünschten Wirkungen
Die unerwünschten Wirkungen von Bimekizumab sind nach MedDRA-Systemorganklassen und Häufigkeit gemäss folgender Konvention geordnet:
«sehr häufig» (≥1/10)
«häufig» (≥1/100, <1/10),
«gelegentlich» (≥/1'000, <1/100)
«selten» (≥1/10'000, <1/1'000)
«sehr selten» (<1/10'000)
«nicht bekannt» (kann aus den verfügbaren Daten nicht abgeschätzt werden)
Tabelle 1: Liste der unerwünschten Wirkungen in klinischen Studien
|
Systemorganklasse
|
Häufigkeit
|
Nebenwirkung
| |
Infektionen und parasitäre Erkrankungen
|
Sehr häufig
|
Infektionen der oberen Atemwege
| |
Häufig
|
Orale Candidose, Tinea-Infektionen, Infektionen des Ohrs, Herpes-simplex-Infektionen, oropharyngeale Candidose, Gastroenteritis, Follikulitis, Vulvovaginale Candidose
| |
Gelegentlich
|
Konjunktivitis, Schleimhaut- und kutane Candidose (einschliesslich ösophageale Candidose)
| |
Erkrankungen des Blutes und des Lymphsystems
|
Gelegentlich
|
Neutropenie
| |
Erkrankungen des Nervensystems
|
Häufig
|
Kopfschmerzen
| |
Erkrankungen des Gastrointestinaltrakts
|
Gelegentlich
|
Entzündliche Darmerkrankung
| |
Erkrankungen der Haut und des Unterhautgewebes
|
Häufig
|
Dermatitis und Ekzem, Akne, Ausschlag
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Häufig
|
Reaktionen an der Injektionsstellea, Müdigkeit
| |
a)
Umfasst: Erythem, Reaktionen, Ödeme, Schmerzen und Schwellungen an der Injektionsstelle.
|
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Infektionen
Im placebokontrollierten Zeitraum der klinischen Phase-III-Studien zur Plaque-Psoriasis wurden bei 36,0 % der Patienten, die bis zu 16 Wochen mit Bimekizumab behandelt worden waren, im Vergleich zu 22,5 % der mit Placebo behandelten Patienten Infektionen berichtet. Bei den meisten Infektionen handelte es sich um nicht schwerwiegende, leicht bis mittelschwer verlaufende Infektionen der oberen Atemwege wie Nasopharyngitis. Schwerwiegende Infektionen traten bei 0,3 % der mit Bimekizumab und 0 % der mit Placebo behandelten Patienten auf. Die mit Bimekizumab behandelten Patienten wiesen entsprechend dem Wirkmechanismus höhere Raten von oralen und oropharyngealen Candidosen auf (7,3 % bzw. 1,2 % im Vergleich zu 0 % bei den mit Placebo behandelten Patienten). Die meisten Fälle waren nicht schwerwiegend, verliefen leicht oder mittelschwer und erforderten kein Absetzen der Behandlung.
Über den gesamten Behandlungszeitraum der Phase-III-Studien zur Plaque-Psoriasis wurden bei 63,2 % der mit Bimekizumab behandelten Patienten Infektionen berichtet (120,4 pro 100 Patientenjahre). Schwerwiegende Infektionen wurden bei 1,5 % der mit Bimekizumab behandelten Patienten gemeldet (1,6 pro 100 Patientenjahre) (siehe Warnhinweise und Vorsichtsmassnahmen).
Die in den klinischen Phase-III-Studien bei PsA und axSpA (nr-axSpA und AS) beobachteten Infektionsraten waren ähnlich wie die bei Plaque-Psoriasis beobachteten Infektionsraten, abgesehen von den Raten orale und oropharyngeale Candidose bei mit Bimekizumab behandelten Patienten, die mit 2,3 % bzw. 0 % bei PsA und 3,7 % bzw. 0,3 % bei axSpA im Vergleich zu 0 % unter Placebo niedriger waren.
Neutropenie
In klinischen Phase-III-Studien an Patienten mit Plaque-Psoriasis wurden unter Bimekizumab Neutropenien beobachtet. In dem 16-wöchigen placebokontrollierten Zeitraum wurden Neutropenien Grad 3/4 sowohl unter Bimekizumab als auch unter Placebo bei je 0,6 % der Patienten beobachtet. Über den gesamten Behandlungszeitraum der Phase-III-Studien wurden bei 1 % der mit Bimekizumab behandelten Patienten Neutropenien Grad 3/4 beobachtet.
Die Häufigkeit von Neutropenien in klinischen Studien zu PsA und axSpA (nr-axSpA und AS) war vergleichbar mit der Häufigkeit, die in Studien zu Plaque-Psoriasis beobachtet wurde.
Die meisten Fälle waren vorübergehend und erforderten keinen Behandlungsabbruch. Die Neutropenien gingen nicht mit schwerwiegenden Infektionen einher.
Immunogenität
Wie alle therapeutischen Proteine ist Bimekizumab potenziell immunogen. Der Nachweis einer Bildung von Anti-Wirkstoff-Antikörpern hängt in hohem Masse von der Sensitivität und Spezifität des verwendeten Assays ab. Zudem wird die beobachtete Inzidenz positiver Nachweise von Anti-Wirkstoff-Antikörpern (einschliesslich neutralisierender Antikörper) möglicherweise durch weitere Faktoren wie das Testverfahren, das Probenhandling, den Zeitpunkt der Probennahme, die Begleitmedikation oder die Grunderkrankung beeinflusst. Aus diesen Gründen kann ein Vergleich der Inzidenz von Antikörpern gegen Bimekizumab mit der Inzidenz von Antikörpern gegen andere Präparate irreführend sein.
Plaque-Psoriasis
Ungefähr 45 % der Plaque-Psoriasis-Patienten, die bis zu 56 Wochen mit Bimekizumab in der empfohlenen Dosierung (320 mg alle 4 Wochen bis Woche 16 und danach 320 mg alle 8 Wochen) behandelt wurden, entwickelten Antikörper gegen den Wirkstoff. Von den Patienten, die Anti-Wirkstoff-Antikörper entwickelten, hatten etwa 34 % (16 % aller mit Bimekizumab behandelten Patienten) Antikörper, die als neutralisierend klassifiziert wurden.
Psoriasis-Arthritis
Ungefähr 31 % der Psoriasis-Arthritis-Patienten, die mit Bimekizumab in der empfohlenen Dosierung (160 mg alle 4 Wochen) bis Woche 16 behandelt wurden, hatten Antikörper gegen den Wirkstoff. Von den Patienten mit Anti-Wirkstoff-Antikörpern hatten etwa 33 % (10 % aller mit Bimekizumab behandelten Patienten) Antikörper, die als neutralisierend klassifiziert wurden. In Woche 52 hatten ungefähr 47 % der nicht mit einem biologischen krankheitsmodifizierenden Antirheumatikum (biologic disease-modifying anti-rheumatic drug, bDMARD) behandelten Psoriasis-Arthritis-Patienten, die in der BE OPTIMAL-Studie mit Bimekizumab in der empfohlenen Dosierung (160 mg alle 4 Wochen) behandelt wurden, Antikörper gegen den Wirkstoff. Von den Patienten mit Anti-Wirkstoff-Antikörpern hatten etwa 38 % (18 % aller in der BE OPTIMAL-Studie mit Bimekizumab behandelten Patienten) Antikörper, die als neutralisierend klassifiziert wurden.
Axiale Spondyloarthritis (nr-axSpA und AS)
Ungefähr 57 % der Patienten mit nr-axSpA, die bis zu 52 Wochen mit Bimekizumab in der empfohlenen Dosierung (160 mg alle 4 Wochen) behandelt wurden, entwickelten Antikörper gegen den Wirkstoff. Von den Patienten mit Anti-Wirkstoff-Antikörpern hatten etwa 44 % (25 % aller mit Bimekizumab behandelten Patienten) Antikörper, die als neutralisierend klassifiziert wurden.
Ungefähr 44 % der Patienten mit AS, die bis zu 52 Wochen mit Bimekizumab in der empfohlenen Dosierung (160 mg alle 4 Wochen) behandelt wurden, entwickelten Antikörper gegen den Wirkstoff. Von den Patienten mit Anti-Wirkstoff-Antikörpern hatten etwa 44 % (20 % aller mit Bimekizumab behandelten Patienten) Antikörper, die als neutralisierend klassifiziert wurden.
Indikationsübergreifend hatte die Entwicklung von Anti-Bimekizumab-Antikörpern keine klinisch bedeutsamen Auswirkungen auf das klinische Ansprechen und ein Zusammenhang zwischen Immunogenität und behandlungsbedingten unerwünschten Ereignissen wurde nicht eindeutig nachgewiesen.
Hypersensitivitätsreaktionen:
Unter Behandlung mit IL-17-Inhibitoren wurden schwerwiegende Hypersensitivitätsreaktionen einschliesslich anaphylaktischer Reaktionen beobachtet.
Ältere Patienten (≥65 Jahre)
Für diese Altersgruppe liegen nur begrenzte Daten vor. Im placebokontrollierten Zeitraum der klinischen Phase-III-Studien an Patienten mit Plaque-Psoriasis wurde orale Candidose bei 18,2 % der Patienten ≥65 Jahre gegenüber 6,3 % bei Patienten < 65 Jahre beobachtet. Von Dermatitis und Ekzem waren 7,3 % der Patienten ≥65 Jahre gegenüber 2,8 % der Patienten < 65 Jahre betroffen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEinzeldosen von 640 mg intravenös oder 640 mg subkutan, gefolgt von 320 mg subkutan alle zwei Wochen für fünf Dosen, wurden in klinischen Studien ohne dosislimitierende Toxizität verabreicht. Im Falle einer Überdosierung wird empfohlen, den Patienten auf Anzeichen und Symptome von Nebenwirkungen zu überwachen und umgehend eine geeignete symptomatische Behandlung einzuleiten.
Eigenschaften/WirkungenATC-Code
L04AC21
Wirkungsmechanismus
Bimekizumab ist ein humanisierter monoklonaler IgG1/κ-Antikörper der selektiv und mit hoher Affinität an die IL-17A-, IL-17F- und IL-17AF-Zytokine bindet und so deren Interaktion mit dem IL-17RA/IL-17RC-Rezeptorkomplex blockiert. Erhöhte Konzentrationen von IL-17A und IL-17F wurden mit der Pathogenese von verschiedenen immunvermittelten entzündlichen Erkrankungen, einschliesslich Plaque Psoriasis, Psoriasis-Arthritis und axialer Spondyloarthritis, in Verbindung verbracht. IL-17A und IL-17F kooperieren und/oder synergieren mit anderen entzündlichen Zytokinen bei der Auslösung einer Entzündung. IL-17F wird in erheblichen Mengen von Zellen des angeborenen Immunsystemszellen produziert. Diese Produktion kann unabhängig von IL-23 erfolgen. Bimekizumab hemmt die proinflammatorischen Zytokine, was zu einer Normalisierung der entzündeten Haut und einer deutlichen Abnahme der lokalen und systemischen Entzündung sowie in der Folge zu einer Besserung der klinischen Anzeichen und Symptome der Psoriasis, Psoriasis-Arthritis und axialen Spondyloarthritis führt. In In-vitro-Modellen zeigte Bimekizumab eine stärkere hemmende Wirkung auf die mit Psoriasis verbundene Genexpression, Zytokinproduktion, Migration der Entzündungszellen und pathologische Osteogenese im Vergleich zu einer alleinigen IL-17A vermittelten Hemmung.
Pharmakodynamik
Es wurden keine formalen Studien zur Pharmakodynamik von Bimekizumab durchgeführt.
Klinische Wirksamkeit
Plaque Psoriasis
Die Sicherheit und Wirksamkeit von Bimekizumab wurde bei 1'480 Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis in drei multizentrischen, randomisierten, Placebo- und/oder aktiv kontrollierten Phase-III-Studien beurteilt. Die Patienten waren mindestens 18 Jahre alt, hatten einen PASI (Psoriasis Area and Severity Index)-Score ≥12 und einen IGA (Investigators Global Assessment)-Score ≥3 auf einer 5-Punkte-Skala, mindestens 10 % ihrer Körperoberfläche war von Psoriasis betroffen (BSA [Body Surface Area]) ≥10 %) und sie waren Kandidaten für eine systemische Psoriasis-Therapie und/oder Phototherapie. Die Wirksamkeit und Sicherheit von Bimekizumab wurden gegenüber Placebo und Ustekinumab (BE VIVID – PS0009), gegenüber Placebo (BE READY – PS0013) und gegenüber Adalimumab (BE SURE – PS0008) beurteilt.
In der Studie BE VIVID wurden 567 Patienten über 52 Wochen untersucht. Die Patienten wurden auf Bimekizumab (320 mg alle 4 Wochen), Ustekinumab (je nach Gewicht des Patienten 45 mg oder 90 mg zu Baseline, in Woche 4 und dann alle 12 Wochen) oder Placebo für die ersten 16 Wochen, gefolgt von Bimekizumab (320 mg alle 4 Wochen) randomisiert.
In der Studie BE READY wurden 435 Patienten über 56 Wochen untersucht. Die Patienten wurden auf Bimekizumab 320 mg alle 4 Wochen oder Placebo randomisiert. In Woche 16 traten Patienten, die ein PASI-90-Ansprechen erreicht hatten, in die 40-wöchige randomisierte Absetz-Phase ein. Patienten, die anfänglich auf Bimekizumab 320 mg alle 4 Wochen randomisiert worden waren, wurden erneut randomisiert und erhielten dann Bimekizumab 320 mg alle 4 Wochen, Bimekizumab 320 mg alle 8 Wochen oder Placebo (d.h. Absetzen von Bimekizumab). Patienten, die anfänglich auf Placebo randomisiert worden waren, erhielten weiterhin Placebo, wenn sie PASI-90-Responder waren. Patienten, die in Woche 16 kein PASI-90-Ansprechen erreicht hatten, gingen in einen unverblindeten Escape-Arm über und erhielten Bimekizumab 320 mg alle 4 Wochen für 12 Wochen. Patienten mit Rezidiv (kein PASI-75-Ansprechen) während der randomisierten Absetz-Phase gingen ebenfalls in den 12-wöchigen Escape-Arm über.
Die Studie BE SURE untersuchte 478 Patienten über 56 Wochen. Die Patienten wurden auf Bimekizumab 320 mg alle 4 Wochen bis Woche 56, Bimekizumab 320 mg alle 4 Wochen bis Woche 16, gefolgt von Bimekizumab 320 mg alle 8 Wochen bis Woche 56, oder Adalimumab gemäss Zulassungsempfehlung bis Woche 24, gefolgt von Bimekizumab 320 mg alle 4 Wochen bis Woche 56, randomisiert.
Die Baseline-Charakteristika waren in allen 3 Studien ähnlich. Im Median waren 20 % der Körperoberfläche der Patienten von Psoriasis betroffen (BSA 20 %), der mediane PASI-Score lag bei 18 und anhand des IGA-Scores war die Erkrankung bei 33 % der Patienten als schwerwiegend einzustufen. Die medianen Baseline-Scores für Schmerzen, Juckreiz und Schuppung lagen gemäss Patienten-Symptom-Tagebuch (Patient Symptoms Diary, PSD) zwischen 6 und 7 auf einer Punkteskala von 0–10, und der mediane Gesamtscore des Dermatology Life Quality Index (DLQI) betrug bei Baseline 9.
In allen 3 Studien zusammen hatten 38 % der Patienten eine vorherige biologische Therapie erhalten, 23 % mindestens einen Anti-IL17-Wirkstoff und 13 % mindestens einen TNF-Antagonisten. 22 % hatten keinerlei systemische Therapie (nicht-biologische und biologische Wirkstoffe eingeschlossen) erhalten und 39 % der Patienten waren zuvor mit Phototherapie oder Photochemotherapie behandelt worden.
Die Wirksamkeit von Bimekizumab wurde bezüglich der Auswirkung auf die Hauterkrankung insgesamt, auf spezifische Körperstellen (Kopfhaut, Nägel, Handflächen und Fusssohlen), auf die von den Patienten berichteten Symptome und bezüglich der Auswirkungen auf die Lebensqualität bewertet. Die beiden co-primären Endpunkte in allen 3 Studien waren der Anteil der Patienten, die 1) ein PASI-90-Ansprechen und 2) ein IGA-Ansprechen «erscheinungsfrei oder fast erscheinungsfrei» (IGA 0/1 mit Verbesserung um mindestens zwei Skalenpunkte im Vergleich zu Baseline) in Woche 16 erreichten. Das PASI-100- und IGA-0-Ansprechen zu Woche 16 und das PASI-75-Ansprechen zu Woche 4 waren die wichtigsten sekundären Endpunkte in allen 3 Studien.
Hauterkrankung insgesamt
Die Behandlung mit Bimekizumab führte zu einer signifikanten Verbesserung aller Krankheitsaktivitätsparameter im Vergleich zu Placebo, Ustekinumab oder Adalimumab in Woche 16. Die wichtigsten Ergebnisse zur Wirksamkeit sind in Tabelle 2 zusammengefasst.
Tabelle 2: Zusammenfassung des klinischen Ansprechens in BE VIVID, BE READY und BE SURE
|
|
BE VIVID
|
BE READY
|
BE SURE
| |
|
Placebo
(n = 83)
n (%)
|
BKZ 320 mg Q4W
(n = 321)
n (%)
|
Ustekinumab
(n = 163)
n (%)
|
Placebo
(n = 86)
n (%)
|
BKZ 320 mg Q4W
(n = 349)
n (%)
|
BKZ 320 mg Q4W
(n = 319)
n (%)
|
Adalimumab
(n = 159)
n (%)
| |
PASI 100
Woche 16
|
0 (0,0)
|
188 (58,6)a
|
34 (20,9)
|
1 (1,2)
|
238 (68,2)a
|
194 (60,8)a
|
38 (23,9)
| |
PASI 90
Woche 16
|
4 (4,8)
|
273 (85,0)a, b
|
81 (49,7)
|
1 (1,2)
|
317 (90,8)a
|
275 (86,2)a
|
75 (47,2)
| |
PASI 75
Woche 4
Woche 16
|
2 (2,4)
6 (7,2)
|
247 (76,9)a, b
296 (92,2)
|
25 (15,3)
119 (73,0)
|
1 (1,2)
2 (2,3)
|
265 (75,9)a
333 (95,4)
|
244 (76,5)a
295 (92,5)
|
50 (31,4)
110 (69,2)
| |
IGA 0
Woche 16
|
0 (0,0)
|
188 (58,6)a
|
36 (22,1)
|
1 (1,2)
|
243 (69,6)a
|
-
|
-
| |
IGA 0/1
Woche 16
|
4 (4,8)
|
270 (84,1)a, b
|
87 (53,4)
|
1 (1,2)
|
323 (92,6)a
|
272 (85,3)a
|
91 (57,2)
| |
Absoluter PASI ≤2
Woche 16
|
3 (3,6)
|
273 (85,0)
|
84 (51,5)
|
1 (1,2)
|
315 (90,3)
|
280 (87,8)
|
86 (54,1)
| |
Schmerzen PSD (n)
Woche 16
|
(n = 54)
9 (16,7)
|
(n = 229)
177 (77,3)a
|
(n = 107)
73 (68,2)
|
(n = 67)
6 (9,0)
|
(n = 255)
201 (78,8)a
|
-
|
-
| |
Juckreiz PSD (n)
Woche 16
|
(n = 61)
8 (13,1)
|
(n = 244)
187 (76,6)a
|
(n = 117)
77 (65,8)
|
(n = 72)
4 (5,6)
|
(n = 278)
210 (75,5)a
|
-
|
-
| |
Schuppung PSD (n)
Woche 16
|
(n = 63)
8 (12,7)
|
(n = 246)
193 (78,5)a
|
(n = 116)
69 (59,5)
|
(n = 70)
4 (5,7)
|
(n = 286)
223 (78,0)a
|
-
|
-
|
BKZ 320 mg Q4W = Bimekizumab alle 4 Wochen. Non-Responder-Imputation (NRI) wurde verwendet.
Ansprechen IGA 0/1 war definiert als erscheinungsfrei (0) oder fast erscheinungsfrei (1) mit einer Besserung um mindestens 2 Kategorien in Woche 16 gegenüber Baseline. Ansprechen IGA 0 war definiert als erscheinungsfrei (0) mit einer Besserung um mindestens 2 Kategorien in Woche 16 gegenüber Baseline.
PSD ist das Patienten-Symptom-Tagebuch (Patient Symptoms Diary). PSD-Ansprechen ist definiert als Veränderung von Baseline bis Woche 16 ≥ einen definierten Schwellenwert (1,98, 2,39 bzw. 2,86 für Schmerzen, Juckreiz und Schuppung).
a) p < 0,001 gegenüber Placebo (BE VIVID und BE READY), gegenüber Adalimumab (BE SURE), auf Multiplizität adjustiert.
b) p < 0,001 gegenüber Ustekinumab (BE VIVID), auf Multiplizität adjustiert.
Die Behandlung mit Bimekizumab war mit einem schnellen Wirkeintritt assoziiert. In der Studie BE VIVID waren die PASI-90-Ansprechraten in Woche 2 bzw. Woche 4 unter Bimekizumab (12,1 % bzw. 43,6 %) höher als unter Placebo (1,2 % bzw. 2,4 %) und Ustekinumab (1,2 % bzw. 3,1 %).
Abbildung 1: PASI-90-Ansprechraten im Zeitverlauf der Studie BE VIVID
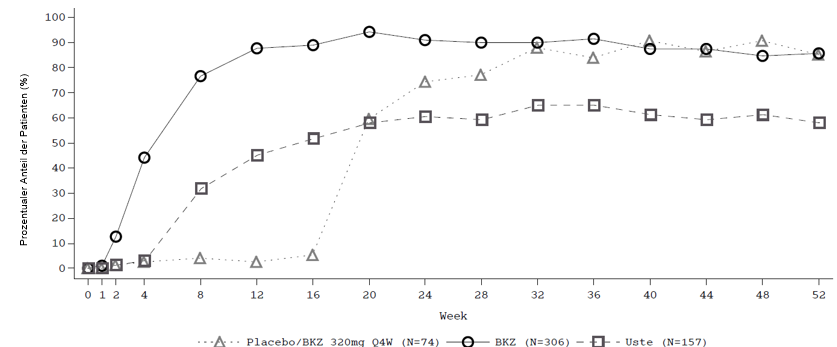
BKZ = Bimekizumab, Uste = Ustekinumab; NRI wird verwendet.
Hinweis: Die Patienten aus der Gruppe Placebo/BKZ wechselten in der Erhaltungsbehandlungsphase ab Woche 16 von Placebo zu BKZ.
In der BE VIVID-Studie erreichten zu Woche 52, im Vergleich zu Ustekinumab, Patienten unter Bimekizumab höhere PASI-90- (81,6 % Bimekizumab gegenüber 55,8 % Ustekinumab, p < 0,001), IGA 0/1- (77,9 % Bimekizumab gegenüber 60,7 % Ustekinumab,p < 0,001) und PASI-100-Ansprechraten (64,2 % Bimekizumab gegenüber 38,0 % Ustekinumab).
In der BE SURE-Studie erreichte zu Woche 24 ein höherer Prozentsatz der mit Bimekizumab behandelten Patienten ein PASI-90-Ansprechen und IGA0/1-Ansprechen im Vergleich zu Adalimumab (85,6 % bzw. 86,5 % gegenüber 51,6 % bzw. 57,9 %,p < 0,001). Unter den 65 Adalimumab-Non-Respondern in Woche 24 (< PASI 90) erreichten 78,5 % nach 16 Behandlungswochen mit Bimekizumab ein PASI-90-Ansprechen. Bei Patienten, die von Adalimumab auf Bimekizumab umgestellt wurden, ergaben sich keine neuen Befunde bezüglich der Sicherheit. In Woche 56 erreichten 70,2 % der mit Bimekizumab behandelten Patienten ein PASI-100-Ansprechen.
Abbildung 2: PASI-90-Ansprechraten im Zeitverlauf der Studie BE SURE
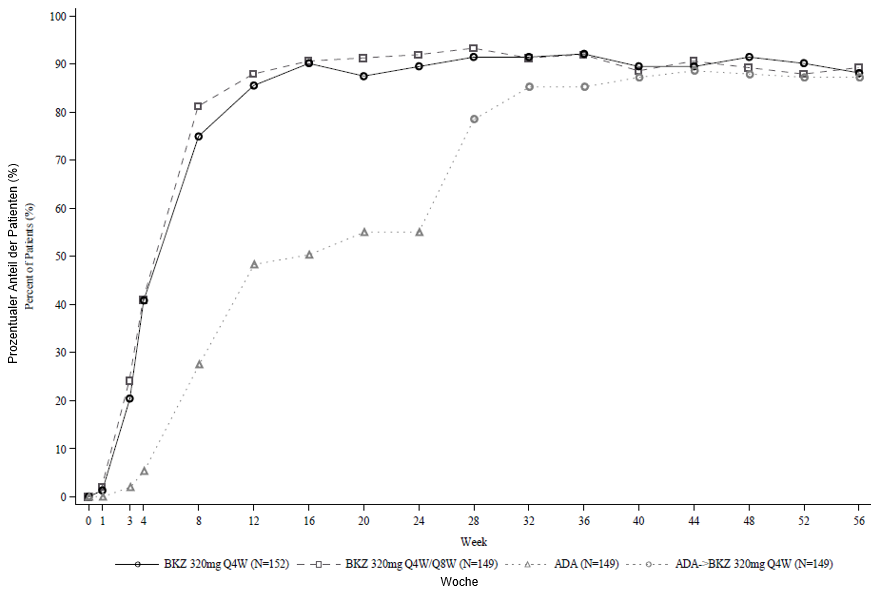
BKZ 320 mg Q4W = Bimekizumab alle 4 Wochen; BKZ 320 mg Q8W = Bimekizumab alle 8 Wochen; ADA = Adalimumab.
Hinweis: Enthalten sind nur Patienten, die in Woche 24 oder später Bimekizumab erhielten. Patienten aus der Gruppe BKZ Q4W/Q8W wechselten in Woche 16 von einer Q4W- zu einer Q8W-Dosierung. Patienten aus der Gruppe ADA/BKZ 320 mg Q4W wechselten in Woche 24 von ADA zu BKZ Q4W. NRI wird verwendet.
Die Wirksamkeit von Bimekizumab wurde unabhängig von Alter, Geschlecht, ethnischer Zugehörigkeit, Krankheitsdauer, Körpergewicht, PASI-Schweregrad bei Baseline und vorheriger Behandlung mit einem Biologikum nachgewiesen. Bimekizumab war wirksam bei Patienten mit vorheriger Biologikum-Exposition, einschliesslich Anti-TNF/Anti-IL-17, und bei Patienten ohne vorherige systemische Therapie. Basierend auf populationspharmakokinetischen/-pharmakodynamischen (PK/PD-)Analysen und gestützt durch klinische Daten, profitierten Patienten mit höherem Körpergewicht (≥120 kg), die in Woche 16 keine vollständige Symptomfreiheit der Haut erreicht hatten, von einer fortgesetzten Behandlung mit Bimekizumab 320 mg alle vier Wochen (Q4W) nach den ersten 16 Behandlungswochen.
In der BE SURE-Studie erhielten die Patienten Bimekizumab 320 mg Q4W bis Woche 16, gefolgt von einer Dosierung Q4W oder alle acht Wochen (Q8W) bis Woche 56, unabhängig vom Responder-Status in Woche 16. Bei Patienten in der Gruppe ≥120 kg (N=37) verbesserte sich im Arm mit Q4W-Erhaltungstherapie das PASI-100-Ansprechen zwischen Woche 16 (23,5 %) und Woche 56 (70,6 %) stärker als im Arm mit Q8W-Erhaltungstherapie (Woche 16: 45,0 % vs. Woche 56: 60,0 %).
Aufrechterhaltung des Ansprechens
Tabelle 3: Fortdauer des Ansprechens in Woche 52 bei Respondern in Woche 16*
|
PASI 100
|
PASI 90
|
IGA 0/1
|
Absoluter PASI ≤2
| |
BKZ 320 mg Q4W/Q4W
(N = 355)
n (%)
|
BKZ 320 mg Q4W/Q8W
(N = 182)
n (%)
|
BKZ 320 mg Q4W/Q4W
(N = 516)
n (%)
|
BKZ 320 mg Q4W/Q8W
(N = 237)
n (%)
|
BKZ 320 mg Q4W/Q4W
(N = 511)
n (%)
|
BKZ 320 mg Q4W/Q8W
(N = 234)
n (%)
|
BKZ 320 mg Q4W/Q4W
(N = 511)
n (%)
|
BKZ 320 mg Q4W/Q8W
(N = 238)
n (%)
| |
295 (83,1)
|
161 (88,5)
|
464 (89,9)
|
214 (90,3)
|
447 (87,5)
|
214 (91,5)
|
460 (90,0)
|
215 (90,3)
|
* Integrierte Analyse von BE VIVID, BE READY und BE SURE. NRI wird verwendet.
BKZ 320 mg Q4W: Bimekizumab 320 mg alle 4 Wochen, gefolgt von Bimekizumab 320 mg alle 4 Wochen ab Woche 16. BKZ 320 mg Q8W: Bimekizumab 320 mg alle 4 Wochen, gefolgt von Bimekizumab 320 mg alle 8 Wochen ab Woche 16.
Aufrechterhaltung des PASI-90-Ansprechens (nach Absetzen von Bimekizumab)
Abbildung 3: PASI-90-Responder-Raten im Zeitverlauf – randomisierte Auswasch-Phase in BE READY
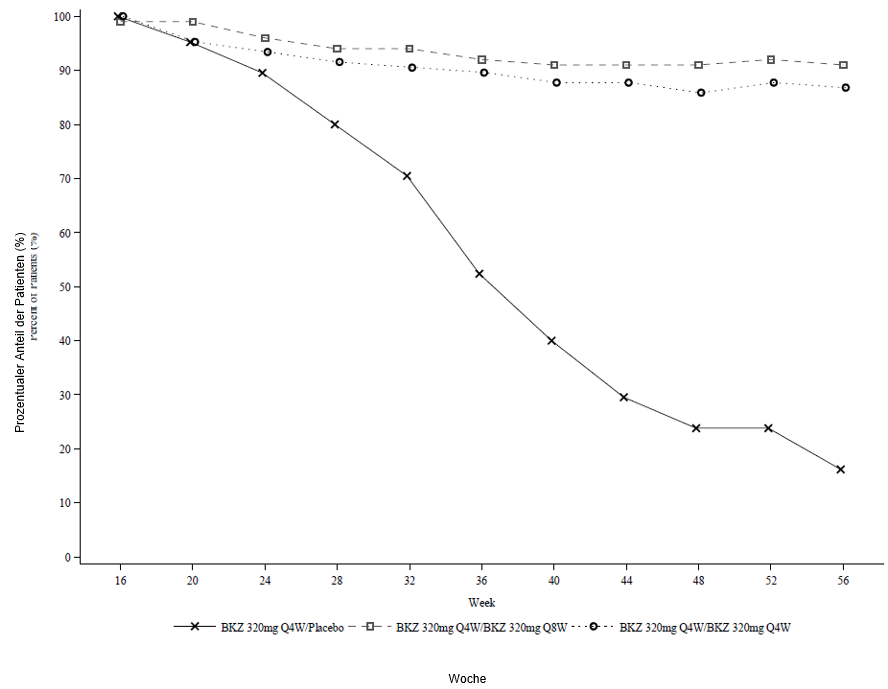
NRI wird verwendet.
In BE READY betrug für PASI-90-Responder aus Woche 16, die dann auf Placebo randomisiert wurden und nicht länger Bimekizumab erhielten, die mediane Zeit bis zum Rezidiv, definiert als Verlust des PASI-75-Ansprechens, ungefähr 28 Wochen (32 Wochen nach der letzten Bimekizumab-Dosis). Von diesen Patienten erreichten 88,1 % innerhalb von 12 Wochen nach Wiederaufnahme der Behandlung mit Bimekizumab 320 mg alle 4 Wochen wieder ein PASI-90-Ansprechen.
Einzelne Körperregionen
Bei Patienten, die mit Bimekizumab behandelt wurden, zeigte sich in Woche 16 in den Studien BE VIVID und BE READY gegenüber Placebo eine signifikante Verbesserung der Psoriasis an Kopfhaut, Nägeln, Handflächen und Fusssohlen (siehe Tabelle 4).
Tabelle 4: Ansprechen in einzelnen Körperregionen in BE VIVID und BE READY in Woche 16
|
|
BE VIVID
|
BE READY
| |
|
Placebo
|
BKZ 320 mg Q4W
|
Ustekinumab
|
Placebo
|
BKZ 320 mg
Q4W
| |
Kopfhaut IGA (n)a
Kopfhaut IGA 0/1, n (%)
|
(72)
11 (15,3)
|
(285)
240 (84,2)b
|
(146)
103 (70,5)
|
(74)
5 (6,8)
|
(310)
286 (92,3)b
| |
pp-IGA (n)a
pp-IGA 0/1, n (%)
|
(29)
7 (24,1)
|
(105)
85 (81,0)
|
(47)
39 (83,0)
|
(31)
10 (32,3)
|
(97)
91 (93,8)
| |
mNAPSI 100 (n)a
mNAPSI 100, n (%)
|
(51)
4 (7,8)
|
(194)
57 (29,4)
|
(109)
15 (13,8)
|
(50)
3 (6,0)
|
(210)
73 (34,8)
|
NRI wird verwendet.
a) Nur Patienten mit einem Investigator Global Assessment (IGA)-Score der Kopfhaut von 2 oder höher, einem palmoplantaren IGA von 2 oder höher und einem mNAPSI (modifizierten Nail Psoriasis and Severity Index)-Score > 0 bei Baseline. Kopfhaut-Ansprechen IGA 0/1 und pp-Ansprechen IGA 0/1 waren definiert als erscheinungsfrei (0) oder fast erscheinungsfrei (1) mit einer Besserung um ≥2 Kategorien gegenüber Baseline.
b) p < 0,001 gegenüber Placebo, für Multiplizität korrigiert.
Das IGA-Ansprechen an der Kopfhaut und der palmoplantaren Region blieb bis Woche 52/56 erhalten. Die Nagel-Psoriasis besserte sich über Woche 16 hinaus. In BE VIVID erreichte in Woche 52 ein höherer Anteil der mit Bimekizumab behandelten Patienten eine völlige Symptomfreiheit der Nägel (mNAPSI 100) im Vergleich zu mit Ustekinumab behandelten Patienten (60,3 % gegenüber 40,4 %). In BE READY erreichten in Woche 56 unter Bimekizumab 320 mg alle 8 Wochen bzw. Bimekizumab 320 mg alle 4 Wochen 67,7 % bzw. 69,8 % der PASI-90-Responder aus Woche 16 eine völlige Symptomfreiheit der Nägel.
Gesundheitsbezogene Lebensqualität / von Patienten berichtete Ergebnisse
In allen 3 Studien erreichte in Woche 16, im Vergleich zu Placebo oder einem aktiven Vergleichsmedikament, ein grösserer Anteil der mit Bimekizumab behandelten Patienten eine nicht mehr durch die Psoriasis eingeschränkte Lebensqualität (gemessen anhand des Dermatology Life Quality Index, DLQI).
In BE READY lag der Prozentsatz der Patienten mit einem DLQI von 0/1 (keine Auswirkungen der Psoriasis auf die gesundheitsbezogene Lebensqualität) in Woche 16 bei 75,6 % in der Bimekizumab-Gruppe bzw. bei 5,8 % in der Placebo-Gruppe.
In BE VIVID betrug die DLQI 0/1 Ansprechrate in Woche 16 in der Bimekizumab-, Ustekinumab- und Placebogruppe 67,3 %, 42,3 % bzw. 12,0 %. Die DLQI 0/1-Ansprechraten stiegen über Woche 16 hinaus an und blieben bis Woche 52 erhalten (74,8 % bei mit Bimekizumab 320 mg Q4W behandelten Patienten).
In BE SURE betrug die DLQI 0/1-Ansprechrate in Woche 16 in der Bimekizumab- und Adalimumab-Gruppe 63,0 % bzw. 46,5 %. Die DLQI 0/1-Ansprechrate in Woche 56 betrug 78,9 % bei mit Bimekizumab 320 mg Q8W bzw. 74,1 % bei mit Bimekizumab 320 mg Q4W behandelten Patienten.
Psoriasis-Arthritis (PsA)
Die Sicherheit und Wirksamkeit von Bimekizumab wurden bei 1112 erwachsenen Patienten (mindestens 18 Jahre alt) mit aktiver Psoriasis-Arthritis (PsA) in zwei multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studien (PA0010 – BE OPTIMAL und PA0011 – BE COMPLETE) untersucht. Die BE OPTIMAL-Studie umfasste einen Arm mit aktiver Referenzbehandlung (Adalimumab) (N = 140).
In beiden Studien war bei den Patienten vor mindestens 6 Monaten eine aktive Psoriasis-Arthritis gemäss den CASPAR-Kriterien (Classification Criteria for Psoriatic Arthritis) diagnostiziert worden, und die Krankheit war aktiv mit einer Anzahl druckschmerzempfindlicher Gelenke (tender joint count, TJC) ≥3 und einer Anzahl geschwollener Gelenke (swollen joint count, SJC) ≥3. Die Patienten hatten die Diagnose PsA in BE OPTIMAL im Median seit 3,6 Jahren und in BE COMPLETE seit 6,8 Jahren. In diese Studien wurden Patienten mit allen Subtypen der PsA aufgenommen, darunter polyartikuläre symmetrische Arthritis, oligoartikuläre asymmetrische Arthritis, Arthritis der distalen Interphalangealgelenke, prädominante Spondylitis und Arthritis mutilans. Bei Baseline hatten 55,9 % der Patienten ≥3 % Körperoberfläche (Body Surface Area, BSA) mit aktiver Plaque-Psoriasis. 10,4 % der Patienten hatten eine mittelschwere bis schwere Plaque-Psoriasis, und 31,9 % bzw. 12,3 % hatten bei Baseline eine Enthesitis bzw. eine Daktylitis. Der primäre Wirksamkeitsendpunkt in beiden Studien war das ACR-(American College of Rheumatology-)-50-Ansprechen in Woche 16.
Die wichtigsten sekundären Endpunkte in Woche 16 waren in beiden Studien wie folgt: Veränderung gegenüber Baseline des Health Assessment Questionnaire - Disability Index (cfB HAQ-DI), Verringe-rung des Psoriasis Area and Severity Index um 90 % gegenüber Baseline (PASI90), Veränderung gegenüber Baseline des Scores im Short Form-36-Gesundheitsfragebogen (Short Form 36-item Health Survey (SF-36) Physical Component Summary (PCS)), Ansprechen auf die minimale Krankheitsaktivität (Minimal Disease Activity (MDA)), sowie Enthesitis- und Daktylitis-freier Zustand, welche auf gepoolten Daten aus beiden Studien beruhen. In der BE OPTIMAL-Studie war die Veränderung gegenüber Base-line des Van der Heijde modified Total Sharp Score (vdHmTSS) ebenfalls ein wichtiger sekundärer Endpunkt.
In der BE OPTIMAL-Studie wurden 852 Patienten untersucht, die zuvor kein biologisches krankheitsmodifizierendes Antirheumatikum (bDMARD) zur Behandlung von Psoriasis-Arthritis oder Psoriasis erhalten hatten. Die Patienten wurden randomisiert (3:2:1) und erhielten entweder Bimekizumab 160 mg alle 4 Wochen bis Woche 52 oder Placebo bis Woche 16, gefolgt von Bimekizumab 160 mg alle 4 Wochen bis Woche 52 oder einem aktiven Referenzbehandlungarm (Adalimumab 40 mg alle 2 Wochen) bis Woche 52. In dieser Studie hatten 78,3 % der Patienten eine Vorbehandlung mit ≥1 cDMARD erhalten, 21,7 % der Patienten hatten keine Vorbehandlung mit cDMARDs erhalten. Bei Baseline erhielten 58,2 % der Patienten gleichzeitig Methotrexat (MTX), 11,3 % erhielten gleichzeitig cDMARDs ausser MTX, und 30,5 % erhielten keine cDMARDs.
In der BE COMPLETE-Studie wurden 400 Patienten eingeschlossen, die auf eine Behandlung mit einem oder zwei Tumornekrosefaktor-alpha-Inhibitoren (Anti-TNFα-IR) zur Behandlung von Psoriasis-Arthritis oder Psoriasis unzureichend angesprochen (mangelnde Wirksamkeit) oder diese nicht vertragen hatten. Die Patienten wurden auf Bimekizumab 160 mg alle 4 Wochen oder Placebo bis Woche 16 randomisiert (2:1). Bei Baseline erhielten 42,5 % der Patienten gleichzeitig MTX, 8,0 % erhielten gleichzeitig cDMARDs ausser MTX, und 49,5 % erhielten keine cDMARDs. In dieser Studie sprachen 76,5 % der Teilnehmer unzureichend auf einen TNFα-Hemmer an, 11,3 % sprachen unzureichend auf zwei TNFα-Hemmer an und 12,3 % vertrugen TNFα-Hemmer nicht.
Anzeichen und Symptome
Bei bDMARDs-naiven Patienten (BE OPTIMAL) und anti-TNFα-IR-Patienten (BE COMPLETE) führte die Behandlung mit Bimekizumab im Vergleich zu Placebo in Woche 16 zu einer signifikanten Verbesserung der Symptome und der Krankheitsaktivität, wobei in beiden Patientenpopulationen ähnliche Ansprechraten beobachtet wurden (siehe Tabelle 5). In der Beurteilung nach ACR 50, MDA, PASI 90 blieb das klinische Ansprechen in BE OPTIMAL bis zur Woche 52 erhalten.
Tabelle 5: Klinisches Ansprechen in den Studien BE OPTIMAL und BE COMPLETE
|
|
BE OPTIMAL (bDMARD-naiv)
|
BE COMPLETE (anti-TNFα-IR)
| |
|
Placebo
(N = 281)
n (%)
|
BKZ 160 mg Q4W
(N = 431)
n (%)
|
Unterschied gegenüber Placebo (95%-KI)(d)
|
Referenz-behandlung(e) (Adalimumab)
(N = 140)
n (%)
|
Placebo
(N = 133)
n (%)
|
BKZ 160 mg Q4W
(N = 267)
n (%)
|
Unterschied gegenüber Placebo (95%-KI)(d)
| |
ACR 50
|
|
|
|
|
|
|
| |
Woche 16
|
28 (10,0)
|
189 (43,9)*
|
33,9 (27,4; 40,4)
|
64 (45,7)
|
9 (6,8)
|
116 (43,4)*
|
36,7 (27,7; 45,7)
| |
Woche 24
|
-
|
196 (45,5)
|
|
66 (47,1)
|
|
|
| |
Woche 52
|
|
235 (54,5)
|
|
70 (50,0)
|
|
|
| |
MDA(a)
|
|
|
|
|
|
|
| |
Woche 16
|
37 (13,2)
|
194 (45,0)*
|
31,8 (25,2; 38,5)
|
63 (45,0)
|
8 (6,0)
|
118 (44,2)*
|
38,2 (29,2; 47,2)
| |
Woche 24
|
-
|
209 (48,5)
|
|
67 (47,9)
|
|
|
| |
Woche 52
|
|
237 (55,0)
|
|
74 (52,9)
|
|
|
| |
Patienten mit ≥3 % BSA
|
(N = 140)
|
(N = 217)
|
|
(N = 68)
|
(N = 88)
|
(N = 176)
|
| |
PASI 90
|
|
|
|
|
|
|
| |
Woche 16
|
4 (2,9)
|
133 (61,3)*
|
58,4 (49,9; 66,9)
|
28 (41,2)
|
6 (6,8)
|
121 (68,8)*
|
61,9 (51,5; 72,4)
| |
Woche 24
|
-
|
158 (72,8)
|
|
32 (47,1)
|
|
|
| |
Woche 52
|
|
155 (71,4)
|
|
41 (60,3)
|
|
|
| |
Patienten mit LDI > 0 (b)
|
(N = 47)
|
(N = 90)
|
|
|
| |
Frei von Daktylitis (b)
Woche 16
|
24 (51,1)
|
68 (75,6)***
|
24,5 (8,4; 40,6)
|
| |
Patienten mit LEI > 0 (c)
|
(N = 106)
|
(N = 249)
|
|
| |
Frei von Enthesitis (c)
Woche 16
|
37 (34,9)
|
124 (49,8)**
|
14,9 (3,7; 26,1)
|
|
|
BKZ 160 mg Q4W = Bimekizumab 160 mg alle 4 Wochen. KI = Konfidenzintervall, NC = nicht berechenbar (not calculable).
(a) Ein Patient wurde mit minimaler Krankheitsaktivität (Minimal Disease Activity, MDA) eingestuft, wenn er 5 der 7 folgenden Kriterien erfüllte: Anzahl druckschmerzempfindlicher Gelenke < 1; Anzahl geschwollener Gelenke < 1; Psoriasis Activity and Severity Index < 1 oder Körperoberfläche < 3; vom Patienten angegebene Schmerzen auf Visueller Analogskala (VAS) < 15; globale Krankheitsaktivität des Patienten auf VAS < 20; Health Assessment Questionnaire Disability Index < 0,5; schmerzende entheseale Punkte < 1
(b) Auf der Grundlage gepoolter Daten aus den Studien BE OPTIMAL und BE COMPLETE für Patienten mit einem Leeds Dactylitis Index (LDI) > 0 zu Baseline. Frei von Daktylitis bedeutet LDI = 0
(c) Auf der Grundlage gepoolter Daten aus den Studien BE OPTIMAL und BE COMPLETE für Patienten mit einem Leeds Enthesitis Index (LEI) > 0 zu Baseline. Frei von Enthesitis bedeutet LEI = 0
(d) Unbereinigte Unterschiede werden angezeigt
(e) Kein statistischer Vergleich mit Bimekizumab oder Placebo durchgeführt
* p < 0,001 gegenüber Placebo, multiplizitätsbereinigt. ** p = 0,008 im Vergleich zu Placebo, multiplizitätsbereinigt. *** p = 0,002 im Vergleich zu Placebo, multiplizitätsbereinigt. NRI wird verwendet. Andere Endpunkte in Woche 16 und alle Endpunkte in Woche 24 und Woche 52 waren nicht Teil der sequenziellen Testhierarchie und alle Vergleiche sind nominal.
In BE OPTIMAL waren die Ergebnisse für mit cDMARD vorbehandelte Patienten ähnlich (ACR50 Woche 16: Bimekizumab 160mg Q4W: 43.5%, Placebo: 9.5%).
In BE OPTIMAL wurden unter Bimekizumab in Woche 16 bei jeder der einzelnen ACR-Komponenten Verbesserungen gegenüber Baseline festgestellt, die bis zu Woche 52 anhielten.
Das Ansprechen auf die Behandlung mit Bimekizumab war in Woche 4 bei ACR 50 (BE OPTIMAL 17,6 % gegenüber 3,2 %, nominal p < 0,001 und BE COMPLETE 16,1 % gegenüber 1,5 %, nominal p < 0,001) signifikant höher als unter Placebo.
Bei 87,2 % der mit Bimekizumab behandelten Patienten, die in BE OPTIMAL in Woche 16 ein ACR-50-Ansprechen erreichten, blieb dieses Ansprechen bis Woche 52 erhalten.
Die Wirksamkeit und Sicherheit von Bimekizumab wurde unabhängig von Alter, Geschlecht, ethnischer Zugehörigkeit, Baseline-Körpergewicht, Psoriasis-Beteiligung zu Baseline, CRP zu Baseline, Krankheitsdauer und vorheriger Anwendung von cDMARDs nachgewiesen. In beiden Studien wurde ein vergleichbares Ansprechen auf Bimekizumab beobachtet, unabhängig davon, ob die Patienten gleichzeitig cDMARDs, einschliesslich MTX, erhielten oder nicht.
Radiologisches Ansprechen
In BE OPTIMAL wurde die Hemmung des Fortschreitens der strukturellen Schädigung radiologisch beurteilt und als Veränderung des Van der Heijde Modified Total Sharp Scores (vdHmTSS) und seiner Komponenten, Erosion Score (ES) und Joint Space Narrowing Score (JSN) in Woche 16 gegenüber Baseline ausgedrückt (siehe Tabelle 6).
Tabelle 6: Veränderung des vdHmTSS in BE OPTIMAL in Woche 16
|
|
Placebo
|
BKZ 160 mg Q4W
|
Unterschied gegenüber Placebo (95%-KI)a)
| |
Population mit erhöhtem hs-CRP und/oder mindestens 1 Knochenerosion zu Baseline
|
(N = 227)
|
(N = 361)
|
| |
Mittlere Veränderung gegenüber Baseline (SE)
|
0,36 (0,10)
|
0,04 (0,05)*
|
-0,32 ( -0,35; -0,30)
| |
Gesamtpopulation
|
(N = 269)
|
(N = 420)
|
| |
Mittlere Veränderung gegenüber Baseline (SE)
|
0,32 (0,09)
|
0,04 (0,04)*
|
-0,26 ( -0,29; -0,23)
|
*p = 0,001 im Vergleich zu Placebo. p-Werte basieren auf einer referenzbasierten Imputation unter Verwendung des Unterschieds im Kleinste-Quadrate-Mittelwert anhand eines ANCOVA-Modells mit der Behandlung, der Knochenerosion zu Baseline und der Region als feste Effekte und dem Baseline-Score als Kovariate.
Die zusammenfassenden Daten für Woche 16 basieren auf dem ersten Wertesatz für die Primäranalyse.
a) Unbereinigte Unterschiede werden angezeigt
Bimekizumab hemmte das Fortschreiten der Gelenkschädigung bis Woche 16 sowohl in der Population mit erhöhtem hs-CRP und/oder mindestens einer Knochenerosion zu Baseline als auch in der Gesamtpopulation im Vergleich zu Placebo signifikant. Während die referenzbasierte Imputation als Methode für den Umgang mit fehlenden Daten im statistischen Testverfahren zum Vergleich von Bimekizumab und Placebo festgelegt wurde, wurden die Veränderungen gegenüber Baseline sowohl in der Population mit erhöhtem hs-CRP und/oder mindestens einer Knochenerosion zu Baseline als auch in der Gesamtpopulation in Woche 16 im Bimekizumab-Arm (mittlere Veränderung gegenüber Baseline 0,01 bzw. 0,01) und im Adalimumab-Arm (mittlere Veränderung gegenüber Baseline -0,05 bzw. -0,03) ebenfalls mittels standardmässiger multipler Imputation berechnet. Die Hemmung des Fortschreitens der Gelenkschädigung wurde sowohl in der Population mit erhöhtem hs-CRP und/oder mindestens einer Knochenerosion zu Baseline als auch in der Gesamtpopulation bis Woche 52 sowohl im Bimekizumab-Arm (mittlere Veränderung gegenüber Baseline 0,10 bzw. 0,10) als auch im Adalimumab-Arm (mittlere Veränderung gegenüber Baseline -0,17 bzw. -0,12) aufrechterhalten.
Der beobachtete Anteil der Patienten, die von der Randomisierung bis Woche 52 kein radiologisch nachweisbares Fortschreiten der Gelenkschädigung (definiert als eine Veränderung des mTSS von ≤0,5 gegenüber Baseline) aufwiesen, betrug in der Population mit erhöhtem hs-CRP und/oder mindestens einer Knochenerosion 87,9 % (N = 276/314) für Bimekizumab und 84,8 % (N = 168/198) für Studienteilnehmer unter Placebo, die auf Bimekizumab umgestellt wurden, sowie 94,1 % (N = 96/102) für Adalimumab. Vergleichbare Raten wurden in der Gesamtpopulation beobachtet (89,3 % (N = 326/365) für Bimekizumab und 87,3 % (N = 207/237) für Studienteilnehmer unter Placebo, die auf Bimekizumab umgestellt wurden, sowie 94,1 % (N = 111/118) für Adalimumab).
Körperliche Funktionsfähigkeit und weitere gesundheitsbezogene Ergebnisse
Sowohl bDMARD-naive (BE OPTIMAL) als auch Anti-TNFα-IR-Patienten (BE COMPLETE), die Bimekizumab erhielten, zeigten in Woche 16 eine signifikante Verbesserung der körperlichen Funktionsfähigkeit gegenüber Baseline im Vergleich zu Placebo-Patienten (p < 0,001), bewertet anhand des HAQ-DI (Veränderung des Kleinste-Quadrate-Mittelwerts gegenüber Baseline: -0,3 versus -0,1 in BE OPTIMAL bzw. -0,3 versus 0 in BE COMPLETE). In beiden Studien erreichte ein grösserer Anteil der Patienten in der Bimekizumab-Gruppe in Woche 16 eine klinisch bedeutsame Verringerung des HAQ-DI-Wertes um mindestens 0,35 gegenüber Baseline als in der Placebo-Gruppe.
Mit Bimekizumab behandelte Patienten zeigten in Woche 16 im Vergleich zu Placebo eine signifikante Verbesserung des Scores im Short Form-36-Gesundheitsfragebogen (Short Form-36 item Health Survey Physical Component Summary, SF-36 PCS) (Veränderung des Kleinste-Quadrate-Mittelwerts gegenüber Baseline: 6,3 versus 1,9, p < 0,001 in BE OPTIMAL und 6,2 versus 0,1, p < 0,001 in BE COMPLETE).
In beiden Studien berichteten die mit Bimekizumab behandelten Patienten im Vergleich zur Placebogruppe in Woche 16 über eine deutliche Verringerung der Müdigkeit gegenüber Baseline, gemessen anhand des FACIT-(Functional Assessment of Chronic Illness Therapy-)Fatigue-Scores. In der mit Bimekizumab behandelten Gruppe wurde im Vergleich zur Placebogruppe in Woche 16 auch eine deutliche Verbesserung des Psoriasis-Arthritis-Impact-of-Disease-12-(PsAID-12-)Scores gegenüber Baseline beobachtet.
Bei Patienten mit axialer Beteiligung zu Baseline, etwa 74 % der Patienten (definiert als ein BASDAI-Wert [Bath Ankylosing Spondylitis Disease Activity Index] ≥4), hatte sich der BASDAI-Wert in Woche 16 gegenüber Baseline im Vergleich zu Placebo stärker verbessert.
Die bis Woche 16 erzielten Verbesserungen bei allen Messgrössen der körperlichen Funktionsfähigkeit und anderen oben erwähnten gesundheitsbezogenen Ergebnisse (HAQ-DI-, SF-36 PCS-, FACIT-Fatigue-, PsAID-12-Werte und BASDAI) blieben in BE OPTIMAL bis Woche 52 erhalten.
In der BE OPTIMAL-Studie erreichten in Woche 52 65,5 % der mit Bimekizumab behandelten Patienten eine völlige Symptomfreiheit der Nägel (mNAPSI-Abheilung bei Patienten mit einem mNAPSI-Wert von über 0 zu Baseline).
Axiale Spondyloarthritis (nr-axSpA und AS)
Die Sicherheit und Wirksamkeit von Bimekizumab wurde bei 586 erwachsenen Patienten (mindestens 18 Jahre alt) mit aktiver axialer Spondyloarthritis (axSpA) in zwei multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studien untersucht, eine bei nicht-röntgenologischer axialer Spondyloarthritis (nr-axSpA) und eine bei ankylosierender Spondylitis (AS), auch als röntgenologische axiale Spondyloarthritis (axSpA) bezeichnet. Der primäre Endpunkt war in beiden Studien der Anteil der Patienten, die in Woche 16 ein ASAS (Assessment of SpondyloArthritis International Society)-40-Ansprechen erreichten. Beide Patientengruppen hatten übereinstimmende Ergebnisse.
In der Studie BE MOBILE 1 (AS0010) wurden 254 Patienten mit aktiver nr-axSpA untersucht. Die Patienten hatten eine axSpA (Alter bei Beginn der Symptome < 45 Jahre), die den ASAS-Klassifizierungskriterien entsprach, und eine aktive Erkrankung, definiert als Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 und Wirbelsäulenschmerzen ≥4 auf einer numerischen Bewertungsskala von 0 bis 10 (nach BASDAI Frage 2), sowie keine Anzeichen für radiologisch nachweisbare Veränderungen in den Iliosakralgelenken, die den modifizierten New-York-Klassifikationskriterien der AS entsprechen würden. Die Patienten zeigten auch objektive Anzeichen einer Entzündung, festgestellt durch erhöhtes C-reaktives Protein (CRP) und/oder mittels Nachweis einer Sakroiliitis in der Magnetresonanztomographie (MRT) sowie ein unzureichendes Ansprechen auf zwei verschiedene nicht-steroidale Antirheumatika (NSARs oder non-steroidal anti-inflammatory drugs, NSAIDs) beziehungsweise eine Unverträglichkeit oder Gegenanzeige gegen NSARs in der Vorgeschichte. Die Patienten wurden randomisiert (1:1) und erhielten entweder Bimekizumab 160 mg alle 4 Wochen bis Woche 52 oder Placebo bis Woche 16, gefolgt von Bimekizumab 160 mg alle 4 Wochen bis Woche 52. Bei Studienbeginn betrug die mittlere Dauer der nr-axSpA-Symptome 9 Jahre (Median 5,5 Jahre). 10,6 % der Patienten wurden zuvor mit einem Anti-TNFα-Wirkstoff behandelt.
In der BE MOBILE 2-Studie (AS0011) wurden 332 Patienten mit aktiver AS mit dokumentiertem radiologischem Nachweis (Röntgen) untersucht, die die modifizierten New-York-Klassifikationskriterien der AS erfüllten. Die Patienten hatten eine aktive Erkrankung, definiert als BASDAI ≥4 und Wirbelsäulenschmerzen ≥4 auf einer numerischen Bewertungsskala von 0 bis 10 (nach BASDAI Frage 2). Die Patienten mussten in der Vergangenheit unzureichend auf 2 verschiedene NSARs angesprochen haben oder eine Unverträglichkeit oder eine Kontraindikation für NSARs aufweisen. Die Patienten wurden randomisiert (2:1) und erhielten entweder Bimekizumab 160 mg alle 4 Wochen bis Woche 52 oder Placebo bis Woche 16, gefolgt von Bimekizumab 160 mg alle 4 Wochen bis Woche 52. Bei Studienbeginn betrug die mittlere Dauer der AS-Symptome 13,5 Jahre (Median 11 Jahre). 16,3 % der Patienten wurden zuvor mit einem Anti-TNFα-Wirkstoff behandelt.
Klinisches Ansprechen
Die Behandlung mit Bimekizumab führte sowohl in der Patientengruppe mit nr-axSpA als auch in jener mit AS im Vergleich zu Placebo in Woche 16 zu einer signifikanten Besserung der Symptome und der Krankheitsaktivität (siehe Tabelle 7). Das klinische Ansprechen blieb in der Beurteilung gemäss allen in Tabelle 7 dargestellten Endpunkten in beiden Patientengruppen bis Woche 52 erhalten.
Tabelle 7: Klinisches Ansprechen in BE MOBILE 1 und BE MOBILE 2
|
|
BE MOBILE 1 (nr-axSpA)
|
BE MOBILE 2 (AS)
| |
|
Placebo
(N = 126)
n (%)
|
BKZ 160 mg Q4W
(N = 128)
n (%)
|
Unterschied gegenüber Placebo (95%-KI)a)
|
Placebo
(N = 111)
n (%)
|
BKZ 160 mg Q4W
(N = 221)
n (%)
|
Unterschied gegenüber Placebo (95%-KI)a)
| |
ASAS 40
|
|
|
|
|
|
| |
Woche 16
Woche 52
|
27 (21,4)
|
61 (47,7)*
78 (60,9)
|
26,2 (14,9; 37,5)
|
25 (22,5)
|
99 (44,8)*
129 (58,4)
|
22,3 (11,5; 33,0)
| |
ASAS-40 ohne anti-TNFα-Vorbehandlung
|
|
|
|
|
|
| |
Woche 16
Woche 52
|
(N = 109)
25 (22,9)
|
(N = 118)
55 (46,6)
73 (61,9)
|
24,8 (12,4; 37,1)
|
(N = 94)
22 (23,4)
|
(N = 184)
84 (45,7)*
108 (58,7)
|
22,3 (10,5; 34,0)
| |
ASAS 20
|
|
|
|
|
|
| |
Woche 16
Woche 52
|
48 (38,1)
|
88 (68,8)*
94 (73,4)
|
30,7 (19,0; 42,3)
|
48 (43,2)
|
146 (66,1)*
158 (71,5)
|
22,8 (11,8; 33,8)
| |
ASDAS – erhebliche Verbesserung
|
|
|
|
|
|
| |
Woche 16
Woche 52
|
9 (7,1)
|
35 (27,3)*
47 (36,7)
|
20,2 (11,2; 29,3)
|
6 (5,4)
|
57 (25,8)*
71 (32,1)
|
20,4 (11,7; 29,1)
| |
BASDAI-50
|
|
|
|
|
|
| |
Woche 16
Woche 52
|
27 (21,4)
|
60 (46,9)
69 (53,9)
|
25,3 (14,0; 36,6)
|
29 (26,1)
|
103 (46,6)
119 (53,8)
|
20,5 (9,6; 31,4)
|
BKZ 160 mg Q4W = Bimekizumab 160 mg alle 4 Wochen. ASDAS = Ankylosing Spondylitis Disease Activity Score.
NRI wird verwendet.
a) Unbereinigte Unterschiede werden dargestellt.
* p < 0,001 gegenüber Placebo, für Multiplizität korrigiert.
Der Anteil der Patienten in BE MOBILE 1, die in Woche 16 einen ASDAS < 2,1 (eine Kombination aus ASDAS-ID [inactive disease] und ASDAS-LD [low disease]) erreichten, betrug in der Bimekizumab-Gruppe 46,1 % und 21,1 % in der Placebogruppe (multiple Imputation). In Woche 52 erreichten 61,6 % der Patienten in der Bimekizumab-Gruppe einen ASDAS < 2,1, davon 25,2 % mit inaktiver Krankheit (ASDAS < 1,3).
Der Anteil der Patienten in BE MOBILE 2, die in Woche 16 einen ASDAS < 2,1 (eine Kombination aus ASDAS-ID und ASDAS-LD) erreichten, betrug in der Bimekizumab-Gruppe 44,8 % und 17,4 % in der Placebogruppe (multiple Imputation). In Woche 52 erreichten 57,1 % der Patienten in der Bimekizumab-Gruppe einen ASDAS < 2,1, davon 23,4 % mit inaktiver Krankheit (ASDAS < 1,3).
Alle vier ASAS-40-Komponenten (Gesamtwirbelsäulenschmerzen, Morgensteifigkeit, Bath Ankylosing Spondylitis Functional Index [BASFI] und Patient's Global Assessment of Disease Activity [PGADA]) verbesserten sich unter der Bimekizumab-Behandlung und trugen zum gesamten ASAS-40-Ansprechen in Woche 16 bei. Diese Verbesserungen blieben in beiden Patientengruppen bis Woche 52 erhalten.
Die Verbesserungen bei anderen Wirksamkeitsparametern sind in Tabelle 8 dargestellt.
Tabelle 8: Weitere Wirksamkeitsparameter in BE MOBILE 1 und BE MOBILE 2
|
|
BE MOBILE 1 (nr-axSpA)
|
BE MOBILE 2 (AS)
| |
|
Placebo
(N = 126)
|
BKZ 160 mg Q4W
(N = 128)
|
Placebo
(N = 111)
|
BKZ 160 mg Q4W
(N = 221)
| |
Nächtliche Wirbelsäulenschmerzen
|
|
|
|
| |
Baseline
Mittlere Veränderung in Woche 16 gegenüber Baseline
Mittlere Veränderung in Woche 52 gegenüber Baseline
|
6,7
-1,7
|
6,9
-3,6*
-4,3
|
6,8
-1,9
|
6,6
-3,3*
-4,1
| |
BASDAI
|
|
|
|
| |
Baseline
Mittlere Veränderung in Woche 16 gegenüber Baseline
Mittlere Veränderung in Woche 52 gegenüber Baseline
|
6,7
-1,5
|
6,9
-3,1*
-3,9
|
6,5
-1,9
|
6,5
-2,9*
-3,6
| |
BASMI
|
|
|
|
| |
Baseline
Mittlere Veränderung in Woche 16 gegenüber Baseline
Mittlere Veränderung in Woche 52 gegenüber Baseline
|
3,0
-0,1
|
2,9
-0,4
-0,6
|
3,8
-0,2
|
3,9
-0,5**
-0,7
| |
hs-CRP (mg/l)
|
|
|
|
| |
Baseline (geometrisches Mittel)
Verhältnis zu Baseline in Woche 16
Verhältnis zu Baseline in Woche 52
|
5,0
0,8
|
4,6
0,4
0,4
|
6,7
0,9
|
6,5
0,4
0,3
|
BASMI = Bath Ankylosing Spondylitis Metrology Index. Hs-CRP = hochsensitives C-reaktives Protein
MI wird verwendet.
*p < 0,001 referenzbasierte Imputation, gegenüber Placebo, für Multiplizität korrigiert. **p < 0,01 referenzbasierte Imputation, gegenüber Placebo, für Multiplizität korrigiert.
Die Behandlung mit Bimekizumab war sowohl in der nr-axSpA- als auch in der AS-Patientengruppe mit einem schnellen Wirkeintritt assoziiert.
Abbildung 4: ASAS-40-Ansprechen im Zeitverlauf bis Woche 52 in BE MOBILE 1 (NRI)
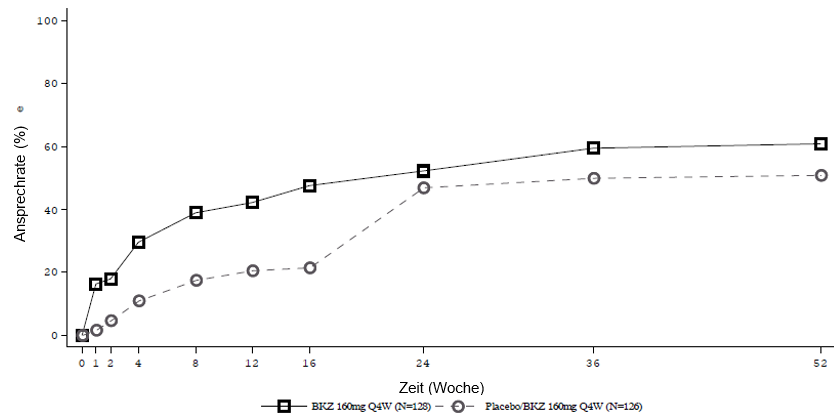
Patienten, die Placebo erhielten, wurden in Woche 16 auf Bimekizumab 160 mg Q4W umgestellt.
Abbildung 5: ASAS-40-Ansprechen im Zeitverlauf bis Woche 52 in BE MOBILE 2 (NRI)
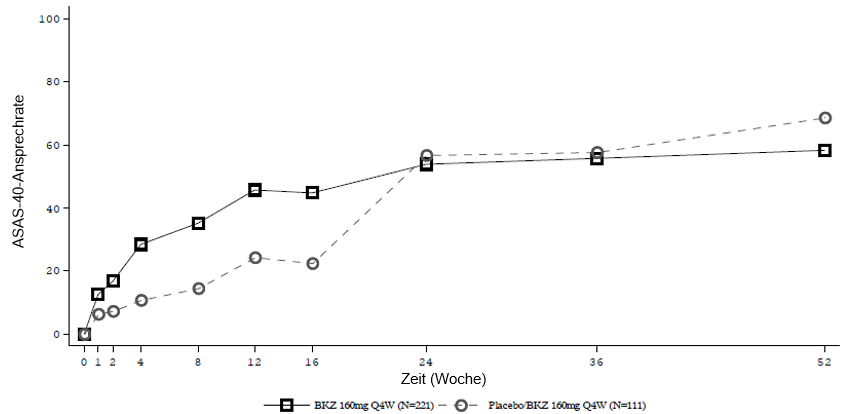
Patienten, die Placebo erhielten, wurden in Woche 16 auf Bimekizumab 160 mg Q4W umgestellt.
In einer integrierten Analyse zu BE MOBILE 1 und BE MOBILE 2 blieb bei den mit Bimekizumab behandelten Patienten, die in Woche 16 ein ASAS-40-Ansprechen erreichten, dieses Ansprechen bei 82,1 % bis Woche 52 erhalten.
Die Wirksamkeit von Bimekizumab wurde unabhängig von Alter, Geschlecht, ethnischer Zugehörigkeit, Krankheitsdauer, Entzündungsstatus zu Baseline, Baseline-ASDAS und begleitenden cDMARDs nachgewiesen.
In Woche 16 war bei Patienten mit Enthesitis bei Baseline der Anteil der Patienten (NRI) mit abklingender Enthesitis gemäss Maastricht Ankylosing Spondylitis Enthesitis Index (MASES) unter Bimekizumab grösser als unter Placebo (BE MOBILE 1: 51,1 % gegenüber 23,9 % und BE MOBILE 2: 51,5 % gegenüber 32,8 %). Das Abklingen der Enthesitis blieb unter Bimekizumab in beiden Studien bis zur Woche 52 erhalten (BE MOBILE 1: 54,3 % und BE MOBILE 2: 50,8 %).
Verringerung der Entzündung
Bimekizumab verringerte die Entzündungswerte gemäss hs-CRP (siehe Tabelle 9) und gemäss MRT in einer Bildgebungs-Substudie. Die Entzündungszeichen wurden zu Baseline und in Woche 16 mittels MRT beurteilt und als Veränderung im SPARCC (Spondyloarthritis Research Consortium of Canada)-Score für die Iliosakralgelenke und im ASspiMRI-a(Ankylosing Spondylitis spine Magnetic Resonance Imagine-activity)-Score in der Berlin-Modifikation für die Wirbelsäule gegenüber Baseline ausgedrückt. Eine Verringerung der Entzündungszeichen sowohl in den Iliosakralgelenken als auch in der Wirbelsäule wurde bei den mit Bimekizumab behandelten Patienten im Vergleich zu Placebo beobachtet (siehe Tabelle 9). Die Verringerung der Entzündung gemäss hs-CRP und gemäss MRT blieb bis Woche 52 erhalten.
Tabelle 9: Verringerung der Entzündung gemäss MRT in BE MOBILE 1 und BE MOBILE 2
|
|
BE MOBILE 1 (nr-axSpA)
|
BE MOBILE 2 (AS)
| |
|
Placebo
|
BKZ 160 mg Q4W
|
Placebo
|
BKZ 160 mg Q4W
| |
SPARCC-Score
|
|
|
|
| |
Mittlere Veränderung in Woche 16 gegenüber Baseline a)
|
-1,56
(N = 62)
|
-6,15
(N = 78)
|
0,59
(N = 46)
|
-4,51
(N = 81)
| |
Mittlere Veränderung in Woche 52 gegenüber Baseline a)
|
|
-7,57
(N = 67)
|
|
-4,67
(N = 78)
| |
ASspiMRI-a-Score (in der Berlin Modifikation)
|
|
|
|
| |
Mittlere Veränderung in Woche 16 gegenüber Baseline a)
|
0,03
(N = 60)
|
-0,36
(N = 74)
|
-0,34
(N = 46)
|
-2,23
(N = 81)
| |
Mittlere Veränderung in Woche 52 gegenüber Baseline a)
|
|
-0,70
(N = 65)
|
|
-2,38
(N = 77)
|
a)Die Werte für die Veränderung gegenüber Baseline basieren auf den beobachteten Fällen und wurden durch die zentrale Auswertung des Datensatzes für Woche 52 ermittelt.
Körperliche Funktionsfähigkeit und weitere gesundheitsbezogene Ergebnisse
Die mit Bimekizumab behandelten Patienten zeigten eine signifikante Verbesserung der körperlichen Funktion gemäss BASFI-Beurteilung gegenüber Baseline im Vergleich zu Placebo (Veränderung des Kleinste-Quadrate-Mittelwerts in Woche 16 gegenüber Baseline in BE MOBILE 1: -2,4 gegenüber - 0,9, p < 0,001 und in BE MOBILE 2: -2,0 gegenüber -1,0, p < 0,001). Die mit Bimekizumab behandelten Patienten zeigten eine signifikante Verbesserung im SF-36 PCS-Score gegenüber Baseline im Vergleich zu Placebo (Veränderung des Kleinste-Quadrate-Mittelwerts in Woche 16 gegenüber Baseline in BE MOBILE 1: 9,3 gegenüber 5,4, p < 0,001 und in BE MOBILE 2: 8,5 gegenüber 5,2, p < 0,001).
Die mit Bimekizumab behandelten Patienten zeigten eine signifikante Verbesserung der gesundheitsbezogenen Lebensqualität gemäss ASQoL (AS Quality of Life Questionnaire) gegenüber Baseline im Vergleich zu Placebo (Veränderung des Kleinste-Quadrate-Mittelwerts in Woche 16 gegenüber Baseline in BE MOBILE 1: -4,9 gegenüber -2,3, p < 0,001 und in BE MOBILE 2: -4,6 gegenüber -3,0, p < 0,001) sowie eine bedeutsame Verringerung der Ermüdung gemäss FACIT-Fatigue-Score (mittlere Veränderung in Woche 16 gegenüber Baseline in BE MOBILE 1: 8,5 für Bimekizumab gegenüber 3,9 für Placebo und in BE MOBILE 2: 8,4 für Bimekizumab gegenüber 5,0 für Placebo).
Die bis Woche 16 erzielten Verbesserungen bei allen Messgrössen der körperlichen Funktion und den anderen zuvor erwähnten gesundheitsbezogenen Ergebnissen (BASFI-, SF-36 PCS-, ASQoL- und FACIT-Fatigue-Score) blieben in beiden Studien bis Woche 52 erhalten.
Extraartikuläre Manifestation
In gepoolten Daten aus BE MOBILE 1 (nr-axSpA) und BE MOBILE 2 (AS) war in Woche 16 der Anteil der Patienten, die eine Uveitis entwickelten, unter Bimekizumab (0,6 %) geringer als unter Placebo (4,6 %). Die Uveitis-Inzidenz blieb unter Langzeitbehandlung mit Bimekizumab gering (1,2/100 Patientenjahre in den gepoolten Phase-II/III-Studien).
PharmakokinetikDie pharmakokinetischen (PK) Eigenschaften von Bimekizumab waren bei Patienten mit Plaque-Psoriasis, Psoriasis-Arthritis und axialer Spondyloarthritis (nr-axSpA und AS) vergleichbar.
Absorption
Auf Basis der populationspharmakokinetischen Analyse erreichte Bimekizumab nach einer subkutanen Einzeldosis von 320 mg bei Plaque-Psoriasis-Patienten eine mediane (2,5. und 97,5. Perzentil) maximale Plasmakonzentration von 25 (12–50) μg/ml zwischen 3 und 4 Tagen nach der Dosisgabe.
Die populationspharmakokinetische Analyse zeigte, dass Bimekizumab bei gesunden Freiwilligen mit einer durchschnittlichen absoluten Bioverfügbarkeit von 70,1 % resorbiert wurde.
Distribution
Auf Basis populationspharmakokinetischer Analysen betrug das mediane Verteilungsvolumen (V/F) (Variationskoeffizient %) bei Patienten mit Plaque-Psoriasis im Steady-State 11,2 l (30,5 %).
Metabolismus
Bimekizumab ist ein monoklonaler Antikörper und es wird erwartet, dass dieser, analog zu endogenen Immunglobulinen, über katabole Wege in kleine Peptide und Aminosäuren abgebaut wird.
Elimination
Auf Basis populationspharmakokinetischer Analysen betrug in klinischen Studien an Patienten mit Plaque-Psoriasis die mediane scheinbare Clearance (CL/F) (Variationskoeffizient %) von Bimekizumab 0,337 l/Tag (32,7 %) und die mittlere terminale Halbwertszeit 23 Tage.
Basierend auf simulierten Daten liegen die mediane (2,5. und 97,5. Perzentil) Spitzen- und Talkonzentration im Steady-State nach subkutaner Verabreichung von 320 mg alle 4 Wochen bei 43 (20–91) µg/ml bzw. 20 (7–50) µg/ml und der Steady-State wird bei vierwöchentlicher Dosisgabe nach etwa 16 Wochen erreicht. Im Vergleich zur Exposition nach einer Einzeldosis zeigte die populationspharmakokinetische Analyse, dass die Patienten nach wiederholter vierwöchentlicher Verabreichung einen 1,74-fachen Anstieg der maximalen Plasmakonzentrationen und der Fläche unter der Kurve (AUC) aufwiesen.
Nach der Umstellung von einer Dosierung 320 mg alle 4 Wochen auf 320 mg alle 8 Wochen in Woche 16 wurde der Steady-State etwa 16 Wochen nach der Umstellung erreicht. Die medianen (2,5. und 97,5. Perzentil) Spitzen- und Tal-Plasmakonzentrationen liegen bei 30 (14–60) μg/ml bzw. 5 (1–16) μg/ml.
Linearität/Nicht Linearität
Bimekizumab zeigte bei Patienten mit Plaque-Psoriasis nach mehrfacher subkutaner Verabreichung eine dosisproportionale Pharmakokinetik über einen Dosisbereich von 64 mg bis 480 mg, wobei die scheinbare Clearance dosisunabhängig war.
Pharmakokinetische/pharmakodynamische Zusammenhänge
Ein populationspharmakokinetisches/-pharmakodynamisches Modell wurde unter Verwendung aller verfügbaren Daten bei Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis entwickelt. Die Analyse zeigte, dass höhere Bimekizumab-Konzentrationen mit einem besseren PASI (Psoriasis Area and Severity Index)- und IGA (Investigators Global Assessment)-Ansprechen assoziiert sind und dass eine Dosis von 320 mg in Woche 0, 4, 8, 12, 16 und danach alle 8 Wochen bei der Mehrheit der Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis einen maximalen Nutzen bietet (siehe Kinetik spezieller Patientengruppen, Körpergewicht).
Kinetik spezieller Patientengruppen
Leber- und Nierenfunktionsstörungen
Es wurden keine spezifischen Studien zur Ermittlung der Auswirkungen einer eingeschränkten Nieren- oder Leberfunktion auf die Pharmakokinetik von Bimekizumab durchgeführt. Es wird erwartet, dass die renale Elimination von intaktem Bimekizumab, einem monoklonalen IgG-Antikörper, gering und von untergeordneter Bedeutung ist. IgG-Antikörper werden hauptsächlich über den intrazellulären Katabolismus eliminiert und es ist daher auch nicht zu erwarten, dass eine Leberfunktionsstörung die Clearance von Bimekizumab beeinflusst. Auf Basis populationspharmakokinetischer Analysen hatten Leberfunktionsmarker (ALT/Bilirubin) keine Auswirkung auf die Clearance von Bimekizumab bei Patienten mit Plaque-Psoriasis.
Ältere Patienten
Auf Basis der populationspharmakokinetischen Analyse mit einer begrenzten Anzahl von älteren Patienten (N = 337 für Alter ≥65 Jahre und N = 45 für Alter ≥75 Jahre) war die scheinbare Clearance (CL/F) bei älteren Patienten und Patienten unter 65 Jahren vergleichbar. Eine Dosisanpassung ist nicht erforderlich.
Körpergewicht
Im populationspharmakokinetischen Modell zeigte sich eine abnehmende Exposition mit steigendem Körpergewicht. Nach diesem Modell ist die durchschnittliche Plasmakonzentration bei erwachsenen Patienten mit einem Gewicht ≥120 kg nach subkutaner Injektion von 320 mg um mindestens 30 % niedriger als bei erwachsenen Patienten mit einem Körpergewicht von 90 kg. Eine Dosisanpassung kann bei manchen Patienten angezeigt sein (siehe Dosierung/Anwendung).
Ethnische Zugehörigkeit / Geschlecht
In einer klinischen Studie zur Pharmakokinetik wurden hinsichtlich der Bimekizumab-Exposition keine klinisch bedeutsamen Unterschiede zwischen japanischen oder chinesischen und kaukasischen Studienteilnehmern beobachtet. Eine Dosisanpassung ist nicht erforderlich. Eine Population pharmakokinetische Analyse deutet darauf hin, dass Frauen im Vergleich zu Männern eine um 10 % schnellere scheinbare Clearance (CL/F) haben, die klinisch nicht bedeutsam ist. Eine Dosisanpassung ist nicht erforderlich.
Präklinische DatenBasierend auf Gewebe-Kreuzreaktivitätstests, Studien zur Toxizität bei wiederholter Gabe (einschliesslich sicherheitspharmakologischer Endpunkte und Bewertung fertilitätsbezogener Endpunkte) sowie der Beurteilung der prä- und postnatalen Entwicklung lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Genotoxizität / Kanzerogenität
Zur Mutagenität und Kanzerogenität von Bimekizumab wurden keine Studien durchgeführt. Eine Schädigung der DNA oder Chromosomen ist bei monoklonalen Antikörpern jedoch nicht zu erwarten. In einer 26-wöchigen Studie zur chronischen Toxikologie an Cynomolgus-Affen wurden bei einer Dosis, die dem 109-fachen der humantherapeutischen Exposition bei 320 mg alle 4 Wochen entspricht, keine präneoplastischen oder neoplastischen Läsionen beobachtet.
Reproduktionstoxizität
In einer erweiterten Studie zur peri- und postnatalen Entwicklung an Cynomolgus-Affen zeigten sich unter Bimekizumab keine Auswirkungen auf Schwangerschaft, Geburt, Säuglingsüberleben, fetale und postnatale Entwicklung bei Verabreichung während der Organogenese und bis zur Geburt in einer Dosis, die basierend auf der AUC dem 27-Fachen der menschlichen Exposition bei 320 mg alle 4 Wochen entsprach. Bei der Geburt waren die Serumkonzentrationen von Bimekizumab bei den neugeborenen Affen vergleichbar mit jenen der Mütter.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern.
Nicht einfrieren.
Fertigpen im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Fertigspritze im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Der Bimzelx-Fertigpen und die Bimzelx-Fertigspritze können bei Raumtemperatur (bis zu 25 °C) für einen einmaligen Zeitraum von maximal 25 Tagen lichtgeschützt aufbewahrt werden. Wurde das Arzneimittel aus dem Kühlschrank genommen und bei Raumtemperatur aufbewahrt, verwerfen Sie es nach 25 Tagen oder nach Ablauf des auf dem Umkarton aufgedruckten Verfalldatums, je nachdem, was zuerst eintritt. In das Feld auf dem Umkarton kann das Datum der Entnahme aus dem Kühlschrank eingetragen werden.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer68548, 68612 (Swissmedic)
PackungenBimzelx 160 mg, Injektionslösung im Fertigpen
1 Fertigpen (B).
2 Fertigpens (B).
Bimzelx 160 mg, Injektionslösung in einer Fertigspritze
1 Fertigspritze (B).
2 Fertigspritzen (B).
ZulassungsinhaberinUCB-Pharma AG, Bulle
Stand der InformationJuli 2024
|