ZusammensetzungWirkstoffe
Ocrelizumabum (gentechnologisch hergestellt unter Verwendung von CHO [Chinese Hamster Ovary]-Zellen).
Hilfsstoffe
Intravenöse Darreichungsform (i.v.)
Natrii acetas trihydricus 21,4 mg (entspricht pro Durchstechflasche 3,6 mg Natrium), acidum aceticum glaciale, α,α-trehalosum dihydricum, polysorbatum 20 (aus gentechnisch verändertem Mais hergestellt), aqua ad iniectabile.
1 ml Konzentrat enthält 0,36 mg Natrium.
Eine Durchstechflasche (10 ml) enthält 3,6 mg Natrium.
Subkutane Darreichungsform (s.c.)
Hyaluronidasum humanum (rHuPH20), natrii acetas trihydricus 50,1 mg (entspricht pro Durchstechflasche 8,4 mg Natrium), acidum aceticum glaciale, α,α-trehalosum dihydricum, polysorbatum 20 (aus gentechnisch verändertem Mais hergestellt), L-methioninum, aqua ad iniectabile.
1 ml Lösung enthält 0,37 mg Natrium.
Eine Durchstechflasche (23 ml) enthält 8,4 mg Natrium.
Indikationen/AnwendungsmöglichkeitenOcrevus ist für die Behandlung von erwachsenen Patienten mit aktiven schubförmigen Verlaufsformen der Multiplen Sklerose (MS) indiziert.
Ocrevus ist für die Behandlung von erwachsenen Patienten mit primär progredienter Multipler Sklerose (PPMS) zur Verlangsamung der Krankheitsprogression und zur Reduzierung der Verschlechterung der Gehgeschwindigkeit indiziert.
Dosierung/AnwendungDie Behandlung mit Ocrevus i.v. oder Ocrevus s.c. muss von einem in der Behandlung von MS-Patienten erfahrenen Neurologen begonnen und überwacht werden.
Es ist wichtig, die Produktkennzeichnung zu überprüfen, um sicherzustellen, dass der Patient die richtige Darreichungsform (Ocrevus i.v. oder Ocrevus s.c.) verordnungsgemäss und auf korrektem Weg erhält.
Patienten können die Behandlung mit Ocrevus i.v. oder s.c. beginnen. Patienten, die aktuell Ocrevus i.v. erhalten, können die Behandlung mit Ocrevus i.v. oder Ocrevus s.c. fortsetzen.
Ocrevus i.v.
Ocrevus i.v. ist nicht für die subkutane Anwendung bestimmt.
Ocrevus i.v.-Infusionen dürfen nur unter der unmittelbaren und engmaschigen Aufsicht von erfahrenem medizinischem Fachpersonal verabreicht werden.
Eine angemessene medizinische Versorgung einschliesslich einer vollständigen Ausrüstung zur Reanimation sowie Arzneimittel wie unter anderem Epinephrin (Adrenalin), Antihistaminika und Glucokortikoide sollten zum sofortigen Gebrauch verfügbar sein für den Fall schwerer unerwünschter Wirkungen, wie zum Beispiel schwerer infusionsbedingter Reaktionen oder Überempfindlichkeitsreaktionen.
Bei Patienten, die schwere pulmonale Symptome wie Bronchospasmus oder Asthma-Exazerbation entwickeln, muss die Infusion sofort und dauerhaft abgebrochen werden. Nach Durchführung einer symptomatischen Behandlung muss der Patient bis zum vollständigen Abklingen der pulmonalen Symptome überwacht werden, weil nach einer anfänglichen Besserung der klinischen Symptome eine Verschlechterung eintreten kann.
Während Ocrevus i.v.-Infusionen kann Hypotonie als Symptom einer infusionsbedingten Reaktion auftreten. Daher sollte die Unterbrechung einer antihypertensiven Behandlung 12 Stunden vor und während jeder Ocrevus i.v.-Infusion in Betracht gezogen werden. Ocrevus i.v. wird als intravenöse (i.v.) Infusion über eine separate Infusionsleitung verabreicht. Ocrevus i.v. darf nicht als schnelle i.v. Injektion oder Bolus injiziert und auch nicht unverdünnt infundiert werden.
Man verwende eine isotonische (0,9 %ige) Natriumchloridlösung als Infusionsmedium. Falls die i.v. Infusion nicht am gleichen Tag abgeschlossen werden kann, muss die übrig gebliebene Flüssigkeit des Infusionsbeutels entsorgt werden (siehe «Besondere Lagerungshinweise» und «Hinweise für die Handhabung und Entsorgung»).
Beobachten Sie alle Patienten bis mindestens eine Stunde nach dem Ende der Infusion (siehe «Warnhinweise und Vorsichtsmassnahmen, Infusionsbedingte Reaktionen und Injektionsreaktionen»).
Prämedikation zur Verringerung möglicher infusionsbedingter Reaktionen
Vor jeder Ocrevus i.v.-Infusion müssen die beiden folgenden Prämedikationen verabreicht werden, um die Häufigkeit und Schwere von infusionsbedingten Reaktionen zu verringern (siehe «Warnhinweise und Vorsichtsmassnahmen»):
·100 mg Methylprednisolon i.v. (oder Äquivalent) etwa 30 Minuten vor jeder Ocrevus i.v.-Infusion;
·ein Antihistaminikum etwa 30 bis 60 Minuten vor jeder Ocrevus i.v.-Infusion;
Zusätzlich kann auch eine Prämedikation mit einem Antipyretikum (z.B. Paracetamol) etwa 30 bis 60 Minuten vor jeder Ocrevus i.v.-Infusion erwogen werden.
Anwendung von Ocrevus i.v.
Therapieeinleitung
Die Anfangsdosis (Dosis 1) von 600 mg wird auf zwei separate i.v.-Infusionen verteilt verabreicht, d.h. in Form von zwei Infusionen zu jeweils 300 mg im Abstand von zwei Wochen.
Erhaltungstherapie
Nachfolgende Ocrevus i.v. Dosen werden alle 6 Monate als Einzeldosis von 600 mg durch eine i.v.- Infusion verabreicht (siehe Tabelle 1).
Wenn die Patienten bei vorgängigen Ocrevus i.v.-Infusionen keine schwerwiegende infusionsbedingte Reaktion (infusion-related reaction, IRR) entwickelten, kann die Infusionsdauer bei nachfolgenden Dosen (auf 2 Stunden) verkürzt werden (siehe Tabelle 1, Option 2).
Zwischen den einzelnen Ocrevus Dosen sollte ein Mindestabstand von 5 Monaten eingehalten werden.
Tabelle 1: Dosis und Therapieschema für Ocrevus i.v.
|
|
Zu verabreichende Dosis von Ocrevus i.v.*
|
Hinweis zur Infusion
| |
Therapieeinleitung
(600 mg)
auf 2 Infusionen aufgeteilt
|
Infusion 1
|
300 mg
in 250 ml
|
·Infusion mit 30 ml/Std. starten.
·Danach kann alle 30 Minuten um jeweils 30 ml/Std. bis auf maximal 180 ml/Std. erhöht werden.
·Jede Infusion sollte über einen Zeitraum von ungefähr 2,5 Std. hinweg verabreicht werden.
| |
Infusion 2
(2 Wochen später)
|
300 mg
in 250 ml
| |
Erhaltungstherapie**
(600 mg)
Einzelinfusion
einmal alle 6 Monate
|
Option 1
Infusion mit einer Dauer von ungefähr 3,5 Stunden
|
600 mg
in 500 ml
|
·Infusion mit 40 ml/Std. starten.
·Danach kann alle 30 Minuten um jeweils 40 ml/Std. bis auf maximal 200 ml/Std. erhöht werden.
·Jede Infusion sollte über einen Zeitraum von ungefähr 3,5 Std. hinweg verabreicht werden.
| |
ODER
| |
Option 2
Infusion mit einer Dauer von ungefähr 2 Stunden
|
600 mg
in 500 ml
|
·Infusion mit 100 ml/Std. in den ersten 15 Minuten starten.
·Infusion für die nächsten 15 Minuten auf 200 ml/Std. erhöhen.
·Infusion für die nächsten 30 Minuten auf 250 ml/Std. erhöhen.
·Infusion für die verbleibenden 60 Minuten auf 300 ml/Std. erhöhen.
·Jede Infusion sollte über ungefähr 2 Std. gegeben werden.
|
* Lösungen von Ocrevus für die i.v.-Infusion werden durch eine Verdünnung des Arzneimittels in einem Infusionsbeutel mit 0,9 %igem Natriumchlorid auf eine Arzneimittel-Endkonzentration von ca. 1,2 mg/ml hergestellt.
** Die erste Anschluss-Einzelinfusion sollte 6 Monate nach Infusion 1 der Anfangsdosis verabreicht werden.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Dosisanpassung während der Behandlung
Eine Dosisanpassung von Ocrevus i.v. wurde nicht untersucht und wird bei unauffälliger Verträglichkeit nicht empfohlen.
Dosisanpassung aufgrund unerwünschter Wirkungen/Interaktionen
Infusionsbedingte Reaktionen
Die Behandlung mit Ocrevus i.v. ist mit infusionsbedingten Reaktionen verbunden, die mit der Freisetzung von Zytokinen und/oder anderen chemischen Mediatoren zusammenhängen können. Generell sollten bei Auftreten von infusionsbedingten Reaktionen die folgenden Anpassungsrichtlinien beachtet werden. Weitere Informationen über infusionsbedingte Reaktionen finden Sie unter «Warnhinweise und Vorsichtsmassnahmen, Infusionsbedingte Reaktionen und Injektionsreaktionen».
Lebensbedrohende infusionsbedingte Reaktionen
Falls Anzeichen für lebensbedrohende oder zu Behinderungen führende infusionsbedingte Reaktionen auftreten, z.B. eine akute Überempfindlichkeit oder ein akutes Atemnotsyndrom, muss die Ocrevus i.v.-Infusion sofort gestoppt werden. Der Patient muss eine geeignete unterstützende Behandlung erhalten. Die Ocrevus i.v. Behandlung muss bei diesen Patienten dauerhaft beendet und darf nicht wieder aufgenommen werden.
Schwere infusionsbedingte Reaktionen
Die Infusion muss sofort gestoppt werden und der Patient muss eine symptomatische Behandlung erhalten, falls eine schwere infusionsbedingte Reaktion oder ein Symptomkomplex von Hautrötung, Fieber und Halsschmerzen auftreten sollten. Die Infusion darf erst nach dem Verschwinden aller Symptome wieder aufgenommen werden. Die Infusion sollte mit der Hälfte jener Infusionsrate wieder begonnen werden, die zu Beginn der infusionsbedingten Reaktion eingestellt war.
Leichte bis mittelschwere infusionsbedingte Reaktionen
Die Infusionsrate, die anfangs der infusionsbedingten Reaktion eingestellt war, sollte auf die Hälfte der Rate reduziert werden, wenn ein Patient eine leichte bis mittelschwere infusionsbedingte Reaktion erleidet (z.B. Kopfschmerzen). Diese reduzierte Rate sollte für mindestens 30 Minuten beibehalten werden. Sie kann dann wieder auf die für den Patienten anfänglich geplante Infusionsrate erhöht werden, wenn sie toleriert wird.
Ocrevus s.c.
Die erste Anwendung darf nur unter klinischer Beobachtung mit geeigneter medizinischer Betreuung erfolgen, um schwere Reaktionen wie schwere Injektionsreaktionen, Überempfindlichkeitsreaktionen und/oder anaphylaktische Reaktionen zu beherrschen (siehe «Warnhinweise und Vorsichtsmassnahmen, Infusionsbedingte Reaktionen und Injektionsreaktionen»).
Prämedikation gegen Injektionsreaktionen
Kurz vor jeder Ocrevus s.c.-Injektion müssen die beiden folgenden Prämedikationen verabreicht werden, um das Risiko lokaler und systemischer Injektionsreaktionen zu verringern (siehe «Warnhinweise und Vorsichtsmassnahmen, Infusionsbedingte Reaktionen und Injektionsreaktionen»):
·oral 20 mg Dexamethason (oder Äquivalent)
·orales Antihistaminikum (z.B. Desloratadin oder Äquivalent).
Zusätzlich kann auch die Gabe eines Antipyretikums (z.B. Paracetamol) kurz vor jeder Anwendung von Ocrevus s.c. in Betracht gezogen werden.
Anwendung von Ocrevus s.c.
Ocrevus s.c. ist nicht für die intravenöse Anwendung bestimmt und muss immer als subkutane Injektion durch eine erfahrene medizinische Fachperson angewendet werden (siehe «Hinweise für die Handhabung» und «Entsorgung»). Ocrevus s.c. vor der Anwendung aus dem Kühlschrank nehmen und die Lösung auf Raumtemperatur kommen lassen.
Bei der Anfangsdosis wird nach der Injektion eine mindestens einstündige Überwachung mit Zugang zu geeigneter medizinischer Unterstützung zur Behandlung schwerer Injektionsreaktionen empfohlen. Bei nachfolgenden Dosen liegen die Notwendigkeit und Dauer einer Überwachung nach der Injektion im Ermessen des behandelnden Arztes (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Es werden 23 ml (920 mg) Ocrevus s.c.-Lösung innerhalb von etwa 10 Minuten subkutan in das Abdomen gegeben, nach Möglichkeit unter Verwendung eines s.c.-Infusionssets (z.B. geflügelt/Butterfly). Ein etwaiges verbleibendes Restvolumen im s.c.-Infusionsset darf dem Patienten NICHT gegeben werden.
Die Injektion sollte am Abdomen durchgeführt werden, ausgenommen der 5 cm um den Bauchnabel herum. Ocrevus s.c.-Injektionen sollten nicht an Stellen durchgeführt werden, in denen die Haut gerötet, empfindlich oder verhärtet ist oder ein Bluterguss vorliegt oder an denen sich Muttermale oder Narben befinden.
Dosis und Behandlungsplan
Ocrevus s.c. wird alle 6 Monate als subkutane Injektion von 920 mg gegeben.
Es ist keine Aufteilung der Anfangsdosis oder nachfolgender Dosen in separate Injektionen erforderlich.
Zwischen jeder Ocrevus-Dosis sollte ein zeitlicher Mindestabstand von 5 Monaten eingehalten werden.
Ocrevus i.v. und s.c.
Patienten mit Leberfunktionsstörungen
Die Sicherheit und Wirksamkeit von Ocrevus bei Patienten mit Leberfunktionsstörungen sind nicht formal untersucht worden. Patienten mit leichter Leberfunktionsstörung wurden in die klinischen Studien eingeschlossen. Es gibt keine Erfahrung in Patienten mit mässiger und schwerer Leberinsuffizienz. Ocrevus ist ein monoklonaler Antikörper und wird über eine Katabolisierung eliminiert (eher als über hepatische Metabolisierung). Deshalb wird nicht erwartet, dass bei Patienten mit Leberfunktionsstörungen eine Dosisanpassung erforderlich ist (siehe «Pharmakokinetik, Kinetik spezieller Patientengruppen, Leberfunktionsstörungen»).
Patienten mit Nierenfunktionsstörungen
Die Sicherheit und Wirksamkeit von Ocrevus bei Patienten mit Nierenfunktionsstörungen sind nicht formal untersucht worden. Patienten mit leichter Niereninsuffizienz wurden in die klinischen Studien eingeschlossen. Es gibt keine Erfahrung in Patienten mit mässiger und schwerer Niereninsuffizienz. Ocrevus ist ein monoklonaler Antikörper und wird über eine Katabolisierung eliminiert (eher als über renale Ausscheidung). Deshalb wird nicht erwartet, dass bei Patienten mit Nierenfunktionsstörungen eine Dosisanpassung erforderlich ist (siehe «Pharmakokinetik, Kinetik spezieller Patientengruppen, Nierenfunktionsstörungen»).
Ältere Patienten
Die Sicherheit und Wirksamkeit von Ocrevus bei Patienten > 55 Jahren wurde nicht belegt.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Ocrevus bei Kindern und Jugendlichen (< 18 Jahren) sind nicht untersucht worden.
Verspätete Dosisgabe
Falls eine geplante Infusion mit Ocrevus ausgelassen wird, sollte diese so schnell wie möglich nachgeholt werden; es sollte nicht bis zur nächsten geplanten Gabe gewartet werden. Zwischen den Dosisgaben von Ocrevus sollte das 6-monatige Behandlungsintervall (mindestens 5 Monate) eingehalten werden.
KontraindikationenÜberempfindlichkeit gegen Ocrelizumab oder einen der Hilfsstoffe
Patienten mit schwerer Herzinsuffizienz (NYHA-Stadium IV)
Patienten mit schwerer Immunsuppression, einschliesslich solcher Patienten, die aktuell eine immunsuppressive Behandlung erhalten (ausgenommen sind symptomatische Behandlungen mit Kortikosteroiden gegen Rezidive) oder deren Immunsystem durch vorausgehende Therapien geschwächt ist (siehe «Warnhinweise und Vorsichtsmassnahmen, Behandlung mit Immunsuppressiva vor, während oder nach der Anwendung von Ocrevus»)
Vorliegen einer aktiven Infektion (siehe «Warnhinweise und Vorsichtsmassnahmen»)
Bestehende aktive maligne Erkrankungen, mit Ausnahme von Patienten mit kutanem Basalzellkarzinom
Therapiebeginn während der Schwangerschaft.
Warnhinweise und VorsichtsmassnahmenVor jeder Infusion muss das medizinische Fachpersonal sicherstellen, dass der Patient die Sicherheitsinformation gelesen und verstanden hat.
Infusionsbedingte Reaktionen (IRRs) und Injektionsreaktionen (IRs)
IRRs sind mit der Anwendung von Ocrevus i.v. und IRs mit der Anwendung von Ocrevus s.c. verbunden. IRRs und IRs können mit der Freisetzung von Zytokinen und/oder anderen chemischen Mediatoren zusammenhängen. Ärzte sollten Patienten umfassend informieren, dass IRRs und IRs während oder innerhalb von 24 Stunden nach der Gabe der Behandlung auftreten können.
Auch eine Überempfindlichkeitsreaktion kann auftreten (eine akute allergische Reaktion auf das Arzneimittel). IRRs und IRs sind klinisch von den akuten Überempfindlichkeitsreaktionen Typ 1 (IgE-vermittelt) nicht zu unterscheiden (siehe «Warnhinweise und Vorsichtsmassnahmen, Überempfindlichkeitsreaktionen»).
Bezüglich der Prämedikation zur Reduzierung der Häufigkeit und Schwere von IRRs und des Risikos von IRs siehe «Dosierung/Anwendung».
Infusionsbedingte Reaktionen bei Anwendung von Ocrevus i.v.
Die Symptome der infusionsbedingten Reaktionen können während jeder Infusion auftreten, am häufigsten werden diese aber während der ersten Infusion verzeichnet (siehe «Unerwünschte Wirkungen»). Diese Reaktionen können sich als Juckreiz, Hautausschlag, Urtikaria, Erythem, Rachenreizung, oropharyngeale Schmerzen, Atemnot, Rachen- oder Larynxödem, Wallungen, erniedrigter Blutdruck, Fieber, Müdigkeit, Kopfschmerzen, Schwindel, Übelkeit, Tachykardie und Anaphylaxie manifestieren. Bei der Anwendung von intravenös verabreichtem Ocrelizumab wurden schwerwiegende Reaktionen im Zusammenhang mit IRRs berichtet, von denen einige einen Krankenhausaufenthalt erforderlich machten. Patienten mit einer Ocrevus Therapie sollten bis mindestens eine Stunde nach Beendigung der Infusion im Hinblick auf jedes einzelne infusionsbedingte Reaktions-Symptom beobachtet werden.
Massnahmen bei infusionsbedingten Reaktionen bei Anwendung von Ocrevus i.v.
Für Massnahmen bei Patienten mit lebensbedrohlichen schweren, oder leichten bis mittelstarken infusionsbedingten Reaktions-Symptomen siehe «Dosierung/Anwendung, Dosierungsanpassungen».
Bei Patienten mit schweren pulmonalen Symptomen, wie Bronchospasmus oder Asthma-Exazerbation, muss die Infusion sofort und dauerhaft abgebrochen werden. Nach Durchführung der symptomatischen Behandlung muss der Patient, bis die pulmonalen Symptome ganz abgeklungen sind, überwacht werden, weil nach einer anfänglichen Besserung eine Verschlechterung auftreten könnte.
Ein erniedrigter Blutdruck als infusionsbedingtes Reaktions-Symptom kann während jeder Ocrevus-Infusion auftreten. Daher sollte die Unterbrechung einer antihypertensiven Behandlung 12 Stunden vor und während jeder Ocrevus-Infusion in Betracht gezogen werden. Patienten mit einer anamnestischen kongestiven Herzinsuffizienz (New York Heart Association III & IV) wurden nicht untersucht (siehe «Kontraindikationen»).
Injektionsreaktionen bei Anwendung von Ocrevus s.c.
IR-Symptome können während oder innerhalb von 24 Stunden nach einer Injektion auftreten. Bei der ersten Injektion wurde häufiger über IR-Symptome berichtet. Bei den IRs kann es sich um lokale IRs oder systemische IRs handeln.
Häufige Symptome lokaler IRs an der Injektionsstelle sind Erythem, Schmerzen, Schwellung und Juckreiz. Zu den häufigen Symptomen systemischer IRs zählen Kopfschmerzen und Übelkeit (siehe «Unerwünschte Wirkungen»). Kurz vor der Injektion müssen die Patienten eine Prämedikation erhalten, um das Risiko von IRs zu reduzieren (siehe «Dosierung/Anwendung»). Patienten, die mit der Anfangsdosis von Ocrevus s.c. behandelt werden, sollen nach Abschluss der Injektion mindestens eine Stunde lang auf Anzeichen einer schweren IR beobachtet werden. Bei der Initialdosis des Arzneimittels müssen geeignete Mittel zur Behandlung von schweren IRs, Überempfindlichkeitsreaktionen und/oder anaphylaktischen Reaktionen bereitstehen.
Für die Folgedosen liegt die Notwendigkeit einer Überwachung nach der Injektion im Ermessen des behandelnden Arztes. Beim Auftreten von IRs wird eine symptomatische Behandlung empfohlen.
Bei Anzeichen einer lebensbedrohlichen IR ist die Gabe von Ocrevus s.c. sofort zu beenden, und es sollen unterstützende Behandlungsmassnahmen eingeleitet werden. Bei diesen Patienten muss Ocrevus dauerhaft abgesetzt werden.
Wenn bei einem Patienten eine schwere IR auftritt, ist die Injektion sofort zu unterbrechen und der Patient symptomatisch zu behandeln. Die Injektion soll erst dann zu Ende geführt werden, wenn alle Symptome abgeklungen sind.
Überempfindlichkeitsreaktionen
Überempfindlichkeitsreaktionen können auftreten (akute IgE vermittelte allergische Reaktion auf das Arzneimittel). Hinsichtlich der Symptome kann eine Überempfindlichkeitsreaktion schwer von infusionsbedingten Reaktionen oder Injektionsreaktionen zu unterscheiden sein. Eine Überempfindlichkeitsreaktion kann während jeder Anwendung auftreten, in der Regel aber nicht während der ersten. Bei nachfolgenden Anwendungen, die schwerere Symptome als bisher oder neue schwere Symptome auslösen, sollte sofort an eine mögliche Überempfindlichkeitsreaktion gedacht werden. Die Anwendung und Behandlung muss sofort und dauerhaft gestoppt werden, wenn eine Überempfindlichkeitsreaktion während der Anwendung vermutet wird. Patienten mit bekannter IgE-vermittelter Überempfindlichkeit auf Ocrelizumab oder einen der Hilfsstoffe dürfen nicht behandelt werden (siehe «Kontraindikationen»).
Infektionen
Ocrevus darf bei Patienten mit einer aktiven, schweren Infektion (wie z.B. Tuberkulose, Sepsis und opportunistische Infektionen) oder einer stark eingeschränkten Immunabwehr (z.B. bei stark reduzierter CD4 oder CD8 Zellzahl) nicht verabreicht werden. Bei Patienten mit einer aktiven Infektion muss mit der Gabe von Ocrevus zugewartet werden, bis die Infektion abgeheilt ist (siehe «Kontraindikationen»).
Weitere Informationen zu den Risikofaktoren für schwerwiegende Infektionen, die mit anderen Erkrankungen als MS in Zusammenhang stehen, finden Sie im Abschnitt «Unerwünschte Wirkungen» (schwere Infektionen aus klinischen Studien zu anderen Autoimmunerkrankungen als MS).
Während der Behandlung mit Ocrevus können schwerwiegende Infektionen, einschliesslich Todesfälle (v.a. im Rahmen von Pneumonien) auftreten (siehe «Unerwünschte Wirkungen»). Die Häufigkeit von tödlich verlaufenden Infektionen, die unter der Behandlung mit Ocrevus berichtet wurden, liegt im Rahmen der Häufigkeit von tödlich verlaufenden Infektionen, die bei mit Placebo behandelten Patienten in anderen MS-Studien berichtet wurden.
Bei Patienten, die Anzeichen oder Symptome einer Infektion im Anschluss an eine Behandlung mit Ocrevus berichten, sollten diese rasch abgeklärt und die Patienten entsprechend behandelt werden. Vor einer weiteren Behandlung sind die Patienten erneut auf ein potenzielles Infektionsrisiko zu untersuchen.
Progressive multifokale Leukoenzephalopathie (PML)
Infektionen mit dem JC-Virus, die zu PML führten, wurden bei Patienten beobachtet, die mit anti-CD20-Antikörpern einschliesslich Ocrevus behandelt wurden und vorwiegend mit Risikofaktoren (z.B. Patientenpopulation, Mehrfachtherapie mit Immunsuppressiva, andere vorgängige DMTs (krankheitsmodifizierende Therapeutika) oder niedrige Lymphozytenzahlen) belastet waren.
Da das Risiko einer PML unter Ocrevus nicht ausgeschlossen werden kann, sollten Ärzte bzgl. Frühzeichen und Symptome einer PML wachsam sein, die jede Art von neu auftretenden oder sich verschlechternden neurologischen Zeichen oder Symptomen beinhalten und den Symptomen eines MS-Schubs gleichen können.
PML ist eine opportunistische Infektion, die durch das JC-Virus verursacht wird und tödlich verlaufen oder zu schweren Behinderungen führen kann. PML kann nur bei einer vorliegenden JCV-Infektion auftreten. Es sollte darauf hingewiesen werden, dass ein negativer anti-JCV-Antikörper-Test die Möglichkeit einer anschliessenden JCV-Infektion nicht ausschliesst. Eine PML verläuft oftmals tödlich und resistent gegenüber sämtlichen Therapien. Die Symptome der PML sind vielfältig, schreiten über Tage bis Wochen fort und können zunehmende Schwäche einer Körperseite oder Ungeschicklichkeit der Gliedmassen, Gleichgewichtsstörungen, Sehstörungen sowie Veränderungen des Denkens, des Gedächtnisses und der Orientierung umfassen, die zu Verwirrung und Persönlichkeitsveränderungen führen.
Falls eine PML vermutet wird, muss die Ocrevus Gabe unterbrochen werden. Bei Verdacht auf eine PML sollte eine Evaluation anhand eines MRTs (vorzugsweise mit Kontrastmittel) im Vergleich zu einem vor der Behandlung angefertigten MRT (vorzugsweise nicht älter als 3 Monate) und eines bestätigenden Liquortests mit Bestimmung der viralen JC-DNA sowie wiederholter neurologischer Untersuchungen erfolgen.
Falls die PML bestätigt ist, muss die Behandlung dauerhaft abgebrochen werden.
Immunvermittelte Kolitis
Bei Patienten, die Ocrevus nach der Markteinführung erhielten, wurde über eine immunvermittelte Kolitis berichtet, die sich als schwere und akut auftretende Form der Kolitis darstellen kann. Einige Fälle von Kolitis waren schwerwiegend und erforderten einen Krankenhausaufenthalt, und bei einigen wenigen Patienten war ein chirurgischer Eingriff erforderlich. Bei vielen dieser Patienten waren systemische Kortikosteroide erforderlich. Die Zeitspanne zwischen dem Beginn der Behandlung und dem Auftreten der Symptome reichte in diesen Fällen von einigen Wochen bis zu Jahren. Überwachen Sie die Patienten während der Behandlung mit Ocrevus auf immunvermittelte Kolitis und untersuchen Sie sie umgehend, wenn Anzeichen und Symptome auftreten, die auf eine immunvermittelte Kolitis hindeuten könnten, wie z.B. neuer oder anhaltender Durchfall oder andere gastrointestinale Anzeichen und Symptome.
Hepatitis-B-Reaktivierung
Im Rahmen der Anwendungsbeobachtung nach der Markteinführung kam es unter Behandlung mit Ocrevus zu einer Reaktivierung des Hepatitis-B-Virus (HBV). Bei Patienten unter Behandlung mit Anti-CD20-Antikörpern traten fulminante Hepatitis, Leberversagen und Tod infolge einer Reaktivierung von HBV auf.
Ein HBV-Screening gemäss den örtlichen Richtlinien sollte bei allen Patienten vor Behandlungsbeginn mit Ocrevus durchgeführt werden. Patienten mit einer aktiven HBV-Infektion (d.h. eine aktive Infektion bestätigt durch positive Befunde bei Tests auf HBsAg und anti HB) dürfen nicht mit Ocrevus behandelt werden (siehe «Kontraindikationen»). Patienten mit positiver Serologie (d.h. negativ für HBsAg und positiv für HB Core-Antikörper [HBcAb+] und HBV-Träger [positiv für Oberflächenantigen, HBsAg+]) sollten vor Beginn der Behandlung einen Spezialisten für Lebererkrankungen konsultieren und nach lokalen medizinischen Standards überwacht und betreut werden, um eine Hepatitis-B-Reaktivierung zu verhindern.
Späte Neutropenie
Es wurden Fälle von spät auftretenden Neutropenien berichtet. Obwohl einige Fälle dem Schweregrad 3 oder 4 entsprachen, handelte es sich bei der Mehrzahl der Fälle um Grad 1 oder 2. Fälle von spät auftretender Neutropenie wurden mindestens 4 Wochen nach der letzten Infusion von Ocrevus gemeldet. Bei Patienten mit Anzeichen und Symptomen einer Infektion wird die Bestimmung der neutrophilen Granulozyten im Blut empfohlen (siehe «Unerwünschte Wirkungen»).
Behandlung mit Immunsuppressiva vor, während oder nach der Behandlung mit Ocrevus
Bei anderen Autoimmunerkrankungen führte die gleichzeitige Anwendung von Ocrevus und immunsuppressiven Arzneimitteln (z.B. chronische Kortikosteroide, nicht-biologische und biologische krankheitsmodifizierende Antirheumatika [DMARDs], Mycophenolat-Mofetil, Cyclophosphamid, Azathioprin) zu einer Zunahme von schwerwiegenden Infektionen, einschliesslich opportunistischer Infektionen. Die Infektionen umfassten unter anderem atypische Pneumonie und Pneumocystis jirovecii-Pneumonie, Varizellen-Pneumonie, Tuberkulose und Histoplasmose. Einige dieser Infektionen verliefen in seltenen Fällen tödlich. Eine explorative Analyse identifizierte die folgenden mit einem Risiko für schwerwiegende Infektionen einhergehenden Faktoren: höhere Ocrevus-Dosen als bei MS empfohlen, andere Begleiterkrankungen, chronische Anwendung von Immunsuppressiva bzw. Kortikosteroiden sowie asiatische Patienten. Die gleichzeitige Anwendung von anderen Immunsuppressiva und Ocrevus mit Ausnahme von Kortikosteroiden zur symptomatischen Behandlung von Schüben wird nicht empfohlen.
Bei Einleitung einer Behandlung mit Ocrevus nach einer immunsuppressiven Therapie bzw. bei Einleitung einer immunsuppressiven Therapie nach einer Behandlung mit Ocrevus muss das Potenzial für überlappende pharmakodynamische Wirkungen berücksichtigt werden (siehe «Wirkungsmechanismus/Pharmakodynamik»). Bei der Verschreibung von Ocrevus ist Vorsicht unter Berücksichtigung der Pharmakodynamik anderer krankheitsmodifizierender MS-Therapeutika geboten. Ocrevus wurde nicht in Kombination mit anderen krankheitsmodifizierenden MS-Therapeutika untersucht.
Impfungen
Der Arzt sollte den Impfstatus der Patienten überprüfen, die regional gültigen Impfempfehlungen für Schutzimpfungen beachten und wichtige Schutzimpfungen auffrischen, bevor eine Behandlung mit Ocrevus eingeleitet wird. Die Impfungen sollten mindestens 6 Wochen vor der ersten Anwendung mit Ocrevus abgeschlossen sein.
Die Sicherheit einer Immunisierung mit Lebendimpfstoffen oder attenuierten Lebendimpfstoffen nach einer Ocrevus Therapie wurde nicht untersucht. Eine solche Impfung ist während der Behandlung und bis zur Repletion der B-Zellen nicht empfohlen (die Zeit bis zur B-Zell Repletion betrug im Median 72 Wochen, siehe «Wirkungsmechanismus/Pharmakodynamik»).
Nach einer zweijährigen Behandlung mit Ocrevus i.v. war der Anteil an Patienten mit positiven Antikörpertitern gegen S. pneumoniae, Mumps, Röteln und Varizellen ähnlich wie die Anteile vor Behandlungsbeginn.
In einer randomisierten unverblindeten Studie (Parallelgruppen: Ocrevus i.v. versus keine bzw. andere immunmodulatorische Therapie) entwickelten Patienten mit RMS, die mit Ocrevus i.v. behandelt wurden, eine zum Teil deutlich abgeschwächte humorale Immunantwort gegen Tetanustoxoid (positive IgG-Antwort bei 23,9 % versus 54,5 %), 23-valentes Pneumokokkenpolysaccharid (Reduktion einer positiven Immunantwort um bis zu 2/3, eine weitere Booster-Auffrischimpfung führte zu keiner relevanten Erhöhung), Neoantigen von Schlitzschnecken-Hämocyanin und saisonale Influenzaimpfstoffe (seroprotektive Titer variierten für die saisonalen Influenzaimpfstoffe zwischen 55,6-80 % versus 75-97 %).
Es wird empfohlen, bei allen Impfungen, ausser mit Lebendimpfstoffen oder lebend-attenuierten Impfstoffen, die lokalen Impfempfehlungen (einschliesslich inaktivierter saisonaler Influenza Impfung) einzuhalten. Es sollte in Betracht gezogen werden, die impfstoffinduzierten Immuntiter zu messen, um zu überprüfen, ob die geimpften Personen eine schützende Immunantwort ausbilden können, weil die Wirksamkeit der Impfung unter Umständen vermindert ist.
Exposition gegenüber Ocrelizumab in utero und Impfung von Neugeborenen und Säuglingen mit Lebendimpfstoffen oder lebend-attenuierten Impfstoffen
Aufgrund einer möglichen B-Zell-Depletion bei Neugeborenen und Säuglingen von Müttern, die während der Schwangerschaft Ocrevus erhalten haben, müssen Säuglinge hinsichtlich einer B-Zell-Depletion überwacht werden. Vor einer Impfung ist die Anzahl der CD19 positiven B-Zellen bei Neugeborenen und Säuglingen zu bestimmen. Eine Impfung mit Lebendimpfstoffen oder lebend-attenuierten Impfstoffen soll erst nach vollständiger Normalisierung der Anzahl der B-Zellen erfolgen. Sicherheit und Zeitpunkt der Immunisierung sind mit dem zuständigen Pädiater zu besprechen.
Malignome
In klinischen Studien wurden Fälle von malignen Erkrankungen (darunter 6 Fälle von Mammakarzinomen unter Ocrevus, keine Fälle in den Kontrollarmen (Rebif® oder Placebo) der kontrollierten Studien) berichtet. Die Inzidenz lag im Rahmen der bei MS-Patienten zu erwartenden Hintergrundrate.
Mit Ausnahme von Patienten mit kutanem Basalzellkarzinom dürfen Patienten mit bestehenden aktiven malignen Erkrankungen (einschliesslich Patienten, die hinsichtlich der Rezidivierung einer malignen Erkrankung aktiv überwacht werden) nicht mit Ocrevus behandelt werden (siehe «Kontraindikationen»). Bei Patienten mit bekannten Risikofaktoren für Malignitäten sollte das Nutzen-Risiko-Verhältnis von Ocrevus sorgfältig abgewogen werden und vor sowie während der Behandlung eine entsprechende Tumorüberwachung durchgeführt werden.
Hautreaktionen
Unter Behandlung mit Ocrevus wurden Fälle von Pyoderma gangraenosum beschrieben. Bei anderen anti-CD20-Antikörpern wurden auch andere schwere Hautreaktionen wie toxische epidermale Nekrolyse (Lyell-Syndrom) und Stevens-Johnson-Syndrom beobachtet. Zur Differenzierung verschiedener Hautreaktionen und Festlegung der anschliessenden Behandlung ist eine Hautbiopsie hilfreich. Für den Fall, dass ein derartiges Ereignis eintritt, ist ein Abbruch der Behandlung in Erwägung zu ziehen.
Weitere Hinweise
Ocrevus, Konzentrat zur Herstellung einer Infusionslösung enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d.h. es ist nahezu «natriumfrei».
Ocrevus, Injektionslösung enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d.h. es ist nahezu «natriumfrei».
Roche stellt, im Rahmen der risikominimierenden Massnahmen, Schulungsmaterial und Informationsbroschüren für Fachpersonen und Patienten zur Verfügung.
InteraktionenEs wurden keine formalen Arzneimittel-Interaktionsstudien durchgeführt. Ein Risiko für Interaktionen mit gleichzeitig angewendeten Arzneimitteln kann nicht ausgeschlossen werden.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Ein Therapiebeginn während der Schwangerschaft darf nicht erfolgen (siehe «Kontraindikationen»). Frauen im gebärfähigen Alter sollten während der Ocrevus Behandlung und bis 6 Monate nach der letzten Ocrevus-Dosis eine zuverlässige Empfängnisverhütung anwenden (siehe «Pharmakokinetik, Elimination»).
Schwangerschaft
Ocrevus ist ein humanisierter monoklonaler Antikörper vom Immunglobulin-G1-Subtyp; Immunglobuline passieren bekanntermassen die Plazentaschranke. Tierexperimentelle Studien haben keine teratogenen Wirkungen gezeigt, es wurde jedoch Reproduktionstoxizität beobachtet (siehe «Präklinische Daten»).
Es liegen keine ausreichenden und gut kontrollierten Daten aus Studien bei schwangeren Frauen vor; jedoch sind eine vorübergehende periphere B-Zell-Verminderung und eine Lymphopenie bei Säuglingen bekannt, die von Müttern geboren wurden, die andere anti-CD20-Antikörper während der Schwangerschaft erhalten hatten. B-Zell-Verminderung in utero wurde auch in tierexperimentellen Studien beobachtet.
Bei Neugeborenen und Säuglingen, die im Mutterleib Ocrevus ausgesetzt waren, wird empfohlen, Impfungen mit Lebendimpfstoffen oder lebend-attenuierten Impfstoffen auf einen späteren Zeitpunkt zu verschieben. Die B-Zell-Zahlen bei menschlichen Neugeborenen und Säuglingen, die im Mutterleib Ocrevus ausgesetzt waren, wurden in klinischen Studien nicht untersucht. Die mögliche Dauer der B-Zell-Depletion bei Neugeborenen und Säuglingen ist nicht bekannt (siehe «Warnhinweise und Vorsichtsmassnahmen, Impfungen»).
Bei intrauterin exponierten Neugeborenen, deren B-Zell-Zahlen nicht im Normalbereich liegen, sollte das Aufschieben von Impfungen mit Lebendimpfstoffen bzw. lebend-attenuierten Impfstoffen erwogen werden.
Ocrevus sollte während einer Schwangerschaft nicht angewendet werden, es sei denn, der mögliche Nutzen für die Mutter überwiegt gegenüber dem möglichen Risiko für den Fötus.
Wehen und Entbindung
Die sichere Anwendung von Ocrevus während Wehen und Entbindung wurde nicht untersucht.
Stillzeit
Es ist nicht bekannt, ob Ocrevus in die menschliche Muttermilch ausgeschieden wird, oder ob es Auswirkungen auf das gestillte Kind und auf die Milchproduktion hat. In Studien an Tieren wurde die Ausscheidung von Ocrelizumab in die Muttermilch nachgewiesen (siehe «Präklinische Daten»). Frauen sollten angehalten werden, das Stillen während der Ocrelizumab Therapie einzustellen, weil das menschliche IgG in die Muttermilch ausgeschieden wird und nicht bekannt ist, wie hoch das Potential für eine Verminderung der B-Zellen bei einer Ocrevus Aufnahme ist.
Fertilität
Präklinische Daten basierend auf Studien zur männlichen und weiblichen Fertilität bei Cynomolgus-Affen lassen keine besonderen Gefahren für den Menschen erkennen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenOcrevus selbst hat keinen bzw. einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen. Der Einfluss der Prämedikation mit Antihistaminika ist jedoch zu beachten. Nach Infusionsreaktionen sollte die Stabilisierung des Zustandes des Patienten abgewartet werden, bevor Fahrzeuge geführt oder Maschinen bedient werden.
Unerwünschte WirkungenKlinische Studien
Das Sicherheitsprofil von Ocrelizumab basiert auf Daten von Patienten mit RMS und PPMS, die Ocrelizumab intravenös oder subkutan erhalten haben.
Die Sicherheit von Ocrevus wurde auf der Grundlage von pivotalen klinischen MS-Studien an 1'311 Patienten unter Verwendung von Ocrevus i.v. beurteilt, davon 825 Patienten mit schubförmiger Multipler Sklerose (Relapsing Multiple Sclerosis, RMS) in zwei identischen aktiv-kontrollierten klinischen Studien und 486 Patienten in einer Placebo-kontrollierten Studie mit Patienten mit primär progredienter Multipler Sklerose (PPMS) (siehe «Eigenschaften/Wirkungen, klinische Wirksamkeit»). Die am häufigsten berichteten unerwünschten Arzneimittelwirkungen (UAW) waren infusionsbedingte Reaktionen und Atemwegsinfektionen.
Die Häufigkeitskategorien werden definiert als sehr häufig (≥1/10), häufig (≥1/100 bis < 1/10), gelegentlich (≥1/1000 bis < 1/100), selten (≥1/10'000 bis < 1/1000), sehr selten (< 1/10'000) und nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden). Die unerwünschten Wirkungen sind in abnehmender Häufigkeit angegeben.
Zusammenfassung der unter Ocrevus i.v. aufgetretenen UAW bei RMS oder PPMS
Infektionen und parasitäre Erkrankungen
Sehr häufig: Infektion der oberen Atemwege (RMS: 15,2 %; PPMS: 12,1 %), Nasopharyngitis (PPMS: 24,1 %; RMS: 14,9 %), Influenza (PPMS: 11,7 %; RMS: 4,6 %).
Häufig: Bronchitis, Sinusitis, Gastroenteritis, virale Infektion, oraler Herpes, Infektion der Atemwege, Zellulitis, Herpes Zoster, Konjunktivitis.
Nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden): PML* und immunvermittelte Kolitis* (siehe «Warnhinweise und Vorsichtsmassnahmen»).
* Nach der Markteinführung beobachtet.
Erkrankungen des Blutes und des Lymphsystems
Häufig: Neutropenie.
Nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden): Spätes Auftreten einer Neutropenie*.
* Nach der Markteinführung beobachtet.
Erkrankungen des Immunsystems
Nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden): Überempfindlichkeit* (siehe «Warnhinweise und Vorsichtsmassnahmen»).
* Nach der Markteinführung beobachtet.
Erkrankungen der Atemwege, des Brustraums und des Mediastinums
Häufig: Husten, Katarrh.
Erkrankungen der Haut und des Unterhautgewebes
Nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden): Pyoderma gangraenosum* (siehe «Warnhinweise und Vorsichtsmassnahmen»).
* Nach der Markteinführung beobachtet.
Untersuchungen
Sehr häufig: Verminderte IgM-Serumspiegel (RMS: 16,5 %, PPMS: 15,5 %).
Häufig: Verminderte IgG-Serumspiegel.
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Sehr häufig: Infusionsbedingte Reaktionen (PPMS: 40,1 %; RMS: 34,3 %) (Symptome, die bis 24 Std. nach einer Infusion als infusionsbedingte Reaktionen gemeldet wurden, sind unten als «Infusionsbedingte Reaktionen» beschrieben).
Ocrevus s.c.
Die Sicherheit von Ocrevus s.c. wurde auf der Grundlage von klinischen MS-Studien an 312 Patienten unter Anwendung von Ocrevus s.c. untersucht, darunter Patienten aus der pivotalen Studie OCARINA II und Patienten aus der Studie OCARINA I. Von diesen 312 Patienten erhielten 181 Patienten aus der Studie OCARINA II und 118 Patienten aus der Studie OCARINA I mindestens eine 920-mg-Dosis Ocrevus s.c.
Die Sicherheitsdaten zu Ocrevus s.c. stimmten mit dem vorstehend beschriebenen bekannten Sicherheitsprofil von Ocrevus i.v. überein, mit Ausnahme der sehr häufigen IRs als UAW bei subkutaner Gabe.
Beschreibung ausgewählter Nebenwirkungen aus klinischen Studien
Infusionsbedingte Reaktionen (IRRs) bei Anwendung von Ocrevus i.v.
Die in den Studien zu RMS und PPMS im Rahmen von infusionsbedingten Reaktionen aufgetretenen Symptome umfassten unter anderem: Juckreiz, Hautausschlag, Urtikaria, Erythem, Wallungen, Hypotonie, Fieber, Müdigkeit, Kopfschmerzen, Schwindel, Halsreizungen, oropharyngeale Schmerzen, Dyspnoe, Rachen- oder Kehlkopf Ödeme, Übelkeit, Tachykardie. In den kontrollierten klinischen Studien gab es keine tödlichen infusionsbedingten Reaktionen.
In den aktiv-kontrollierten (RMS) klinischen Studien waren infusionsbedingte Reaktionen das häufigste unerwünschte Ereignis bei mit Ocrevus i.v. 600 mg behandelten Patienten, mit einer Gesamtinzidenz von 34,3 % verglichen mit einer Inzidenz von 9,9 % in der Interferon-beta-1a-Behandlungsgruppe (Placebo-Infusion). Die Inzidenz von infusionsbedingten Reaktionen war während der Anfangsdosis/Dosis 1 bei Infusion 1 am höchsten (27,5 %) und verringerte sich im Laufe der Zeit auf < 10 % bei Dosis 4. Die Mehrheit der infusionsbedingten Reaktionen in beiden Behandlungsgruppen waren leicht (21,7%) bis mittelschwer (10,1%); 2,4 % hatten schwere infusionsbedingte Reaktionen und 0,1 % lebensbedrohliche infusionsbedingte Reaktionen (siehe «Warnhinweise und Vorsichtsmassnahmen, Infusionsbedingte Reaktionen und Injektionsreaktionen»).
In der Placebo-kontrollierten (PPMS) klinischen Studie waren infusionsbedingte Reaktionen die häufigsten UAW mit einer Inzidenz von 40,1 % verglichen mit 25,5 % in der Placebo-Gruppe. Die Inzidenz von infusionsbedingten Reaktionen war am höchsten während der Anfangsdosis/ersten Dosis bei Infusion 1 (27,4 %) und nahm mit den weiteren Dosen auf < 10 % bei Dosis 4 ab. Ein grösserer Anteil an Patienten in jeder Gruppe erlitt infusionsbedingte Reaktionen bei der ersten Infusion jeder Dosis als bei der zweiten Infusion dieser Dosis. Die Mehrheit der infusionsbedingten Reaktionen unter Ocrevus war leicht (26,7 %) bis mittelschwer (11,9 %); 1,4 % hatten schwer und niemand lebensbedrohliche infusionsbedingte Reaktionen (siehe «Warnhinweise und Vorsichtsmassnahmen, Infusionsbedingte Reaktionen und Injektionsreaktionen»).
Alternative kürzere Infusion der Erhaltungstherapie
In einer Studie (MA30143 Teilstudie zur Verkürzung der Infusionsdauer) zur Charakterisierung des Sicherheitsprofils kürzerer (2-stündiger) Ocrevus-Infusionen bei Patienten mit schubförmig remittierenden Verlaufsformen der Multiplen Sklerose waren Inzidenz, Intensität und Art der IRR-Symptome vergleichbar mit denjenigen die bei ungefähr 3,5-stündigen Infusionen auftraten (siehe «Klinische Wirksamkeit»). In beiden Infusionsgruppen war die Gesamtzahl der erforderlichen Interventionen gering, allerdings waren in der Gruppe mit den kürzeren (2-stündigen) Infusionen mehr Interventionen (Reduzierung der Geschwindigkeit oder zeitweise Unterbrechungen der Infusion) zum Management der IRR-Symptome erforderlich als in der Gruppe mit über 3,5 Stunden gegebenen Infusionen (8,7 % vs. 4,8 %).
Injektionsreaktionen (IRs) bei Anwendung von Ocrevus s.c.
Basierend auf den beobachteten Symptomen werden IRs in systemische IRs und lokale IRs unterteilt.
In der Studie OCARINA II (vorgängig nicht mit Ocrelizumab behandelte Patienten) waren die häufigsten Symptome, die bei systemischen IRs und lokalen IRs berichtet wurden: Kopfschmerzen (2.5%), Übelkeit (1.7%), Erythem an der Injektionsstelle (29.7%), Schmerzen an der Injektionsstelle (14.4%), Schwellung an der Injektionsstelle (8.5%) und Juckreiz an der Injektionsstelle (6.8%). 118 Patienten erhielten die erste Injektion. IRs traten bei 48,3 % dieser Patienten nach der ersten Injektion auf. Bei 11,0 % bzw. 45,8 % dieser 118 Patienten trat mindestens ein Ereignis einer systemischen IR bzw. einer lokalen IR auf. Unter den Patienten mit IRs traten diese bei der Mehrzahl der Patienten (82,5 %) innerhalb von 24 Stunden nach dem Ende der Injektion auf und nicht während der Injektion. Alle IRs waren nicht schwerwiegend und leicht (71,9 %) oder mittelschwer (28,1 %). Die mediane Dauer betrug bei systemischen IRs 3 Tage und bei lokalen IRs 4 Tage. Bei allen Patienten klangen die IRs vollständig ab, 26,3 % benötigten eine symptomatische Behandlung.
In der Studie OCARINA I erhielten 125 Patienten eine oder mehrere Injektionen von 1'200 mg Ocrevus s.c.. Von den 125 Patienten, die die erste Injektion erhielten, trat bei 16,0 % mindestens ein systemisches IR-Ereignis und bei 64,0 % mindestens ein lokales IR-Ereignis auf. Bei den 104 Patienten, die die zweite Injektion erhielten, reduzierte sich die Inzidenz systemischer IRs und lokaler IRs auf 7,7 % bzw. 37,5 %. Alle IRs waren nicht schwerwiegend, wobei alle bis auf eine IR bei der ersten Injektion leicht bis mittelschwer waren. Alle IRs bei der zweiten Injektion waren nicht schwerwiegend und leicht bis mittelschwer. Von den Patienten mit IRs benötigten 21,2 % nach der ersten Injektion und 17,9 % nach der zweiten Injektion eine symptomatische Behandlung.
Infektionen
Die Ocrevus Behandlung war nicht mit einer Zunahme von schweren Infektionen assoziiert (bei RMS-Patienten war die Rate an schweren Infektionen niedriger (Ocrevus 1,3 %) als unter Interferon beta-1a (2,9 %), und bei PPMS-Patienten war die Rate ähnlich wie unter Placebo (6,2 % versus 6,7 %)).
In den aktiv-kontrollierten klinischen Studien (RMS) mit Ocrevus i.v. und der Placebo-kontrollierten klinischen Studie (PPMS) wurden Atemwegs- und Herpes-Infektionen (beide überwiegend leicht bis mittelschwer) häufiger im Ocrevus-Therapiearm beobachtet.
Der Gesamtanteil der Patienten mit einer schwerwiegenden Infektion war unter Ocrevus mit den entsprechenden Häufigkeiten in den Kontrollarmen (Placebo und IFN) vergleichbar. Lebensbedrohliche (Grad 4) Infektionen waren unter der Behandlung mit Ocrevus gering, aber häufiger als in den Kontrollarmen (0,2 % unter OCR im Vergleich zu 0 % unter IFN bei RMS bzw. 1,6 % unter OCR im Vergleich zu 0,4 % unter Placebo bei PPMS). Diese Infektionen führten zu keiner Behandlungseinschränkung. Bei PPMS-Patienten mit Schluckstörungen besteht ein erhöhtes Risiko für eine Aspirationspneumonie. Eine Behandlung mit Ocrevus kann bei diesen Patienten das Risiko für eine schwere Pneumonie weiter erhöhen. Ärzte sollen bei Patienten, die eine Pneumonie aufweisen, umgehend entsprechende Massnahmen einleiten.
Infektionen der Atemwege
Der Anteil an Atemwegs-Infektionen war höher bei den mit Ocrevus behandelten Patienten als bei denjenigen, die mit Interferon beta-1a oder Placebo behandelt wurden. Die Infektionen waren überwiegend leicht bis mittelschwer und waren hauptsächlich Infektionen der oberen Atemwege (einschliesslich Nasopharyngitiden) und Bronchitiden. Es kam zu letal verlaufenden Pneumonien unter Ocrevus. Die Häufigkeit von letal verlaufenden Pneumonien, die unter der Behandlung mit Ocrevus berichtet wurden, liegt im Rahmen der Häufigkeit von letal verlaufenden Pneumonien, die bei mit Placebo behandelten Patienten in anderen MS-Studien berichtet wurden.
Herpes
In den aktiv-kontrollierten (RMS) klinischen Studien mit Ocrevus i.v. wurden Herpes-Infektionen häufiger bei mit Ocrevus behandelten Patienten festgestellt als bei denjenigen mit Interferon beta-1a; diese umfassen Herpes zoster (2,1 % vs. 1,0 %), Herpes simplex (0,7 % vs. 0,1 %) und oralen Herpes (3,0 % vs. 2,2 %), genitalen Herpes (0,1 % vs. 0 %) und generalisierte Herpes-Virus-Infektionen (0,1 % vs. 0 %). Die Infektionen waren überwiegend leicht bis mittelschwer und die Patienten erholten sich unter Standard-Behandlungen. Es gab keine Berichte von disseminiertem Herpes.
In der Placebo-kontrollierten (PPMS) klinischen Studie mit Ocrevus i.v. wurde im Ocrevus-Therapiearm ein höherer Anteil an Patienten mit Herpes simplex (2,7 % vs. 0,8 %) beobachtet.
Schwere Infektionen aus klinischen Studien zu anderen Autoimmunerkrankungen als MS
Ocrevus gleichzeitig in Kombination mit immunsuppressiven Medikamenten (z.B. chronische Steroide, nicht-biologische und biologische krankheitsmodifizierende Antirheumatika [DMARD's], Mycophenolatmofetil, Cyclophosphamid, Azathioprin) wurde bei anderen Autoimmunerkrankungen untersucht.
Die Mehrzahl der zur Verfügung stehenden Daten stammt aus Studien bei Patienten mit einer rheumatoiden Arthritis (RA), wo ein Ungleichgewicht an schwerwiegenden Infektionen beobachtet wurde, einschliesslich, aber nicht ausschliesslich beschränkt auf atypische Pneumonien, Pneumocystis jirovecii-Pneumonie, Varizellen-Pneumonie, Tuberkulose und Histoplasmose in der Ocrevus-Immunsuppressivum-Gruppe. In seltenen Fällen waren einige dieser Infektionen tödlich. Schwerwiegende Infektionen wurden häufiger in der 1000 mg-Dosis-Gruppe berichtet als in der 400 mg-Dosis-Gruppe oder in der Immunsuppressivum-Placebo-Gruppe.
Die Risikofaktoren für schwere Infektionen in diesen Studien beinhalteten: andere Komorbiditäten, chronische Verwendung von Immunsuppressiva/Steroiden und ethnische Herkunft aus Asien.
Laborwertanomalien
Immunglobuline
Die Behandlung mit Ocrevus führte über den kontrollierten Untersuchungszeitraum der Studien mit Ocrevus i.v. zu einer Abnahme der Gesamt-Immunglobuline, vor allem bedingt durch eine Reduktion der IgM.
Bei den aktiv-kontrollierten (RMS) Studien betrugen zu Beginn im Ocrevus i.v. Arm die Anteile an Patienten mit IgG, IgA bzw. IgM Werten unterhalb der Untergrenze des Normwertes (< LLN) 0,5 %, 1,5 % bzw. 0,1 %. Nach 96-wöchiger Behandlung betrugen die Anteile der mit Ocrevus i.v. behandelten Patienten mit IgG, IgA bzw. IgM Werten < LLN 1,5 %, 2,4 % bzw. 16,5 %.
Bei der Placebo-kontrollierten (PPMS) Studie betrugen zu Beginn im Ocrevus i.v. Arm die Anteile an Patienten mit IgG, IgA bzw. IgM Werten < LLN 0,0 %, 0,2 % bzw. 0,2 %. Nach 120-wöchiger Behandlung betrugen die Anteile der mit Ocrevus i.v. behandelten Patienten mit IgG, IgA bzw. IgM Werten < LLN 1,1 %, 0,5 % bzw.15,5 %.
Die gepoolten Daten der pivotalen klinischen Studien über Ocrevus i.v. (RMS und PPMS) und ihrer unverblindeten Verlängerungen (die Exposition dauerte bis zu etwa sieben Jahre an) scheinen einen Zusammenhang zwischen verringerten Immunglobulin-Werten und schwerwiegenden Infektionen (SI) zu zeigen, und zwar am deutlichsten im Hinblick auf IgG (bei 0,5 % der Patienten trat während einer Periode mit IgG-Werten < des unteren Normalwerts [LLN] eine SI auf). Die Art, der Schweregrad, die Latenzzeit, die Dauer und die Folgen der SI, die während der Episoden mit Immunglobulin-Werten unter dem LLN beobachtet wurden, standen mit den Daten im Einklang, die allgemein für SI bei mit Ocrevus behandelten Patienten beobachtet wurden.
Neutrophile
Insgesamt betrachtet war in den meisten Fällen die Verminderung der Neutrophilenzahl vorübergehend (nur einmal bei einem mit Ocrevus i.v. behandelten Patienten beobachtet) und vom Schweregrad 1 oder 2. Eine Neutropenie kann mehrere Monate nach der Anwendung von Ocrevus i.v. auftreten (siehe «Warnhinweise und Vorsichtsmassnahmen»).
In der aktiv-kontrollierten (RMS) Behandlungsperiode nahm die Zahl der Neutrophilen bei den Ocrevus i.v. Patienten in 14,7 % ab im Vergleich zu 40,9 % bei den Patienten mit Interferon beta-1a Therapie. In der Placebo-kontrollierten (PPMS) klinischen Studie war der Anteil mit verringerten Neutrophilen bei den Ocrevus Patienten etwas höher (12,9 %) als bei den Placebo-Patienten (10,0 %); von diesen hatten ungefähr 1 % der Patienten in der Ocrevus-Gruppe eine Neutropenie vom Grad 4, verglichen mit 0 % in der Placebo-Gruppe.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungAnzeichen und Symptome
Es gibt nur begrenzte Erfahrungen aus klinischen Studien mit höheren Dosen als die zugelassene Dosis von Ocrevus. Die bisher höchste getestete Dosis bei MS-Patienten betrug 2000 mg, als zwei je 1000 mg i.v. Infusionen im Abstand von 2 Wochen (Phase-II-Dosisfindungsstudie in RRMS), und 1200 mg als s.c.-Injektion (Phase-Ib-Dosisfindungsstudie). Die unerwünschten Arzneimittelwirkungen entsprachen dem Ocrevus Sicherheitsprofil in den entscheidenden klinischen Studien.
Behandlung
Es gibt kein spezifisches Antidot im Falle einer Überdosierung. Die Infusion bzw. Injektion muss sofort gestoppt und der Patient auf Infusionsreaktionen und Injektionsreaktionen hin beobachtet werden (siehe «Warnhinweise und Vorsichtsmassnahmen, Infusionsbedingte Reaktionen und Injektionsreaktionen»).
Eigenschaften/WirkungenATC-Code
L04AG08
Wirkungsmechanismus
Ocrelizumab ist ein rekombinanter humanisierter monoklonaler Antikörper, der selektiv die CD20 exprimierenden B-Zellen zum Ziel hat.
CD20 ist ein Oberflächen-Antigen, das auf Pro-B-Zellen, reifen und Gedächtnis-B-Zellen gefunden wird, jedoch durch lymphoide Stammzellen und Plasmazellen nicht exprimiert wird.
Die genauen Mechanismen, durch welche Ocrelizumab bei MS seine therapeutischen klinischen Wirkungen ausübt, sind nicht vollständig aufgeklärt, aber es wird eine Immunmodulation durch die Verringerung der Anzahl und Funktion der CD20-exprimierenden B-Zellen angenommen. Nach der Bindung an die Zelloberfläche, vermindert Ocrelizumab selektiv die CD20-exprimierenden B-Zellen durch eine Antikörper-abhängige zelluläre Phagozytose (ADCP), eine Antikörper-abhängige zellvermittelte Zytotoxizität (ADCC), eine Komplement-abhängige Zytotoxizität (CDC) und durch Apoptose. Die Fähigkeit der B-Zell-Wiederherstellung und die vorbestehende humorale Immunität bleiben erhalten. Darüber hinaus sind die angeborene Immunität und die Gesamt-T-Zellzahlen nicht betroffen.
Pharmakodynamik
Die Behandlung mit Ocrevus führt zu einem schnellen Abfall der CD19-exprimierenden B-Zellen im Blut innert 14 Tagen nach der Behandlung (erster Zeitpunkt der Beurteilung), was einen erwarteten pharmakologischen Effekt darstellt. Dieser hielt während des Behandlungszeitraumes mit Ocrevus i.v. an. Für die Zählung der B-Zellen wird CD19 verwendet, da das Vorhandensein von Ocrevus die Erkennung von CD20 durch den Test stört.
Die Phase-III-Studien zeigten, dass zwischen jeder Dosis von Ocrevus i.v. bei bis zu 5 % der Patienten eine Erholung der B-Zellzahl (> Untergrenze des Normwertes (LLN) oder Ausgangswert) stattfand, zumindest zu einem Zeitpunkt. Das Ausmass und die Dauer der B-Zell-Verminderung war in den PPMS und RMS Studien beständig.
Die längste Nachbeobachtungszeit nach der letzten Ocrevus i.v. Infusion (Phase II WA21493, n = 51) zeigt auf, dass die mediane Zeit bis zur Wiederherstellung der B-Zellen (Rückkehr zum Ausgangswert/LLN je nachdem, was zuerst eintrat) 72 Wochen betrug (Bereich 27 – 175 Wochen). Neunzig Prozent aller Patienten hatten ihre B-Zell-Population innert etwa zweieinhalb Jahre nach der letzten i.v. Infusion wieder auf das Niveau des LLN oder des Ausgangwertes aufgefüllt.
Klinische Wirksamkeit
Ocrevus i.v.
Schubförmige Verlaufsformen der MS
Die Wirksamkeit und Sicherheit von Ocrevus wurden in zwei randomisierten, doppelblinden, Double-Dummy, aktiv vergleichend-kontrollierten klinischen Studien (WA21092, WA21093) mit identischem Design bei Patienten mit schubförmigen Verlaufsformen der MS (gemäss McDonald-Kriterien 2010) ausgewertet. Studiendesign und die Ausgangs-Charakteristika der Studienpopulation sind in Tabelle 2 zusammengefasst.
Die demographischen Merkmale und Ausgangs-Charakteristika waren in beiden Behandlungsgruppen ausgewogen. Die Patienten der Gruppe A erhielten Ocrevus 600 mg alle 6 Monate (Dosis 1 als 2 x je 300 mg i.v. Infusionen im Abstand von 2 Wochen), die anschliessenden Dosen wurden als eine einzelne 600 mg i.v. Infusion verabreicht. Die Patienten der Gruppe B erhielten Interferon beta-1a (Rebif®) 44 μg s.c. 3 mal wöchentlich.
Die Ergebnisse dieser Studien zeigen, dass Ocrevus, im Vergleich zu Interferon beta-1a 44 µg subkutan, signifikant Schübe, die mittels MRT gemessene subklinische Krankheitsaktivität sowie die Krankheitsprogression vermindert.
Die klinischen Schlüsselbefunde und die Auswirkungen im MRT sind in der Tabelle 3 und Abbildung 1 aufgeführt.
Tabelle 2: Studiendesign und demographische Charakteristika
|
|
Studie 1
|
Studie 2
| |
Name der Studie
|
WA21092 (OPERA I) (n = 821)
|
WA21093 (OPERA II) (n = 835)
| |
Studiendesign
| |
Studienpopulation
|
Patienten mit schubförmigen Verlaufsformen der MS
| |
Anamnese beim Screening
|
Mindestens zwei Schübe in den beiden vorangegangenen Jahren oder ein Schub im vergangenen Jahr; EDSS zwischen 0 und 5,5, inklusive
| |
Studiendauer
|
2 Jahre (96 Wochen)
| |
Behandlungsgruppen
|
Gruppe A: Ocrevus 600 mg alle 24 Wochen i.v.
Gruppe B: Interferon beta-1a (Rebif®), 44 μg 3x / Woche s.c (IFN)
| |
Ausgangs-Charakteristika
|
Ocrevus 600 mg
(n = 410)
|
IFN 44 µg
(n = 411)
|
Ocrevus 600 mg
(n = 417)
|
IFN 44 µg
(n = 418)
| |
Mittleres Alter (Jahre)
|
37,1
|
36,9
|
37,2
|
37,4
| |
Altersspanne (Jahre) bei Einschluss in die Studie
|
18 - 56
|
18 - 55
|
18 - 55
|
18 - 55
| |
Geschlechterverteilung (% Männer / % Frauen)
|
34,1 / 65,9
|
33,8 / 66,2
|
35,0 / 65,0
|
33,0 / 67,0
| |
Durchschnittliche/mediane Dauer seit Beginn der MS Symptome (Jahre)
|
6,74 / 4,88
|
6,25 / 4,62
|
6,72 / 5,16
|
6,68 / 5,07
| |
Durchschnittliche/mediane Dauer seit der Diagnose (Jahre)
|
3,82 / 1,53
|
3,71 / 1,57
|
4,15 / 2,10
|
4,13 / 1,84
| |
Durchschnittliche Anzahl der Schübe im letzten Jahr
|
1,31
|
1,33
|
1,32
|
1,34
| |
Durchschnitt des EDSS (Mittelwert)*
|
2,82
|
2,71
|
2,73
|
2,79
| |
Therapienaive Patienten auf vorangegangene krankheitsmodifizierende MS-Therapien (%)**
|
73,4
|
71,0
|
72,7
|
74,9
| |
Durchschnittliche Anzahl Gd- aufnehmender T1 Läsionen
|
1,69
|
1,87
|
1,82
|
1,95
| |
Durchschnittliche Anzahl hyperintenser T2 Läsionen
|
51,04
|
51,06
|
49,26
|
51,01
|
* Expanded Disability Status Scale.
** Patienten, welche in den 2 Jahren vor Randomisierung keine spezifische MS Medikation hatten.
Tabelle 3: Klinische Schlüsselbefunde und Endpunkte im MRT in den Studien WA21092 und WA21093
|
Endpunkte
|
Studie 1: WA21092
(OPERA I)
|
Studie 2: WA21093
(OPERA II)
| |
Ocrevus 600 mg
(n = 410)
|
IFN 44 μg
(n = 411)
|
Ocrevus 600 mg
(n = 417)
|
IFN 44 μg
(n = 418)
| |
Klinische Endpunkte
|
| |
Annualisierte Schubrate (primärer Endpunkt)
|
0,156
|
0,292
|
0,155
|
0,290
| |
Relative Verminderung
|
46 % (p < 0,0001)
|
47 % (p < 0,0001)
| |
Verhältnis der Patienten mit einer 12-wöchigen bestätigten Behinderungs-Progression3
|
9,8 % Ocrevus vs.15,2 % IFN
| |
Risiko Verminderung (gepoolte Analyse1)
|
40 %
(p = 0,0006)
| |
Risiko Verminderung (Individuelle Studien2)
|
43 % (p = 0,0139)
|
37 % (p = 0,0169)
| |
Verhältnis der Patienten mit einer 24-wöchigen bestätigten Progression der Behinderung3
|
7,6 % Ocrevus vs. 12,0 % IFN
| |
Risiko Verminderung (gepoolte Analyse1)
|
40 %
(p = 0,0025)
| |
Risiko Verminderung (Individuelle Studien2)
|
43 % (p = 0,0278)
|
37 % (p = 0,0370)
| |
Verhältnis der Patienten mit mindestens einer 12-wöchigen bestätigten Verbesserung der Behinderung4 (gepoolt)
|
20,7 % Ocrevus vs. 15,6 % IFN
| |
Relative Verbesserung (gepoolte Analyse1)
|
33 % (p = 0,0194)
| |
Relative Verbesserung (Individuelle Studien2)
|
61 % (p = 0,0106)
|
14 % (p = 0,4019)
| |
Durchschnittliche Veränderung vom Ausgangswert im Multiple Sclerosis Functional Composite (MSFC)
|
0,213
|
0,174
|
0,276
|
0,169
| |
Differenz
|
0,039 (p = 0,3261)
|
0,107 (p = 0,0040)
| |
Verhältnis der Patienten ohne messbare Krankheitsaktivität (NEDA)5
|
48 %
|
29 %
|
48 %
|
25 %
| |
Relative Verbesserung2
|
64 % (p < 0,0001)
|
89 % (p < 0,0001)
| |
MRT Endpunkte
| |
Durchschnittl. Anzahl Gd-markierter T1 Läsionen im MRT Scan
|
0,016
|
0,286
|
0,021
|
0,416
| |
Relative Verminderung
|
94 % (p < 0,0001)
|
95 % (p < 0,0001)
| |
Durchschnittl. Anzahl von neuen und/oder vergrösserten hyperintensen T2 Läsionen im MRT Scan
|
0,323
|
1,413
|
0,325
|
1,904
| |
Relative Verminderung
|
77 % (p < 0,0001)
|
83 % (p < 0,0001)
| |
Durchschnittl. Anzahl von neuen T1-hypointensen Läsionen (chronic black holes) im MRT Scan
|
0,420
|
0,982
|
0,449
|
1,255
| |
Relative Verminderung
|
57 % (p < 0,0001)
|
64 % (p < 0,0001)
| |
Prozentuale Änderung des Hirnvolumens zwischen Wochen 24 bis 96
|
-0,572
|
-0,741
|
-0,638
|
-0,750
| |
Relative Verminderung an Hirnvolumenverlust
|
22,8 % (p = 0,0042)2
|
14,9 % (p = 0,0900)
| |
Lebensqualität
| |
Durchschnittl. Änderung gegenüber Ausgangswert im SF-36 Physical Component Summary
|
0,036
|
-0,657
|
0,326
|
-0,833
| |
Differenz
|
0,693 (p = 0,2193)
|
1,159 (p = 0,0404)6
|
1 Daten prospektiv gepoolt aus Studien 1 & 2.
2 Nicht konfirmatorische p-Wertanalyse; nicht Teil der vorbestimmten Testhierarchie.
3 Definiert als eine Zunahme von ≥1,0 Punkte von der Baseline Expanded Disability Status Scale (EDSS) Score für Patienten mit einem Score von 5,5 oder weniger, oder ≥0,5, wenn der Baseline Score > 5,5 war, nach Kaplan-Meier-Schätzungen in Woche 96.
4 Definiert als Abnahme von ≥1,0 Punkte vom Baseline EDSS Score für Patienten mit einem Ausgangs-EDSS-Score ≥2 und ≤5,5 oder ≥0,5, wenn der Baseline Score > 5,5 war. Patienten mit Baseline-Score < 2 wurden nicht in die Analyse miteinbezogen.
5 NEDA definiert als Abwesenheit von im Protokoll definierten Schüben, bestätigte Progression der Behinderung (CDP) und fehlende MRT-Aktivität (weder Gd-anreichernde T1-Läsionen noch neue oder sich vergrössernde T2-Läsionen) während der gesamten 96-wöchigen Behandlung.
6 Explorative Ergebnisse auf Basis der gesamten ITT-Population.
Abbildung 1: Kaplan-Meier-Plot der Zeit bis zum Beginn einer mindestens 24 Wochen anhaltenden bestätigten Progression der Behinderung, bei welcher das initiale Ereignis der neurologischen Verschlechterung in der doppelblinden Behandlungsphase auftrat (gepoolte ITT Population)*
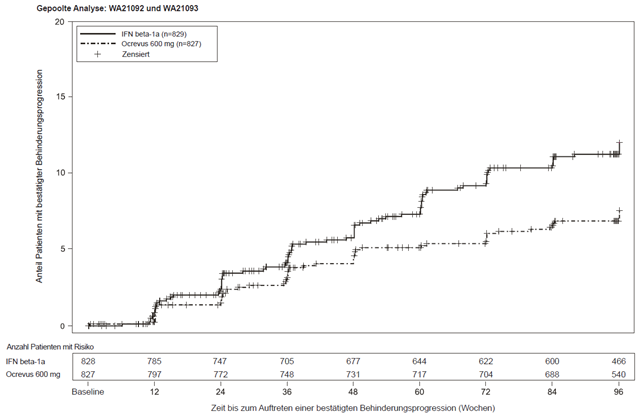
* Im Voraus spezifizierte gepoolte Analyse von OPERA I & II.
Die Ergebnisse der im Voraus spezifizierten gepoolten Analysen der Zeit bis zum Beginn einer mindestens 24 Wochen anhaltenden bestätigten Progression der Behinderung zeigten, dass eine Behandlung mit Ocrelizumab eine 40 %ige Risikoreduktion verglichen mit Interferon beta-1a bewirkte (p = 0,0025).
Teilstudie zur Verkürzung der Infusionsdauer
Die Sicherheit der kürzeren (2-stündigen) Ocrevus i.v.-Infusion wurde in einer prospektiven, multizentrischen, randomisierten, doppelblinden, kontrollierten Teilstudie der Studie MA30143 (Ensemble-Studie) mit Parallelarmen bei Patienten mit schubförmig remittierenden Verlaufsformen der Multiplen Sklerose ohne vorgängige krankheitsmodifizierende Behandlung beurteilt. Die erste Dosis von Ocrevus i.v. wurde als zwei 300 mg Infusionen (insgesamt 600 mg) gegeben, zwischen denen ein Zeitabstand von 14 Tagen lag. Ab der zweiten Dosis (Dosis 2 bis 6) wurden die Patienten im Verhältnis 1:1 entweder in die Gruppe mit konventioneller Infusion von Ocrevus i.v. (alle 24 Wochen über ungefähr 3,5 Stunden) oder in die Gruppe mit kürzerer Infusion von Ocrevus i.v. (alle 24 Wochen über ungefähr 2 Stunden) randomisiert. Die Randomisierung wurde nach Region und Dosis, bei der die Patienten zuerst randomisiert worden waren, stratifiziert.
Der primäre Endpunkt war der Anteil der Patienten mit IRRs während oder innerhalb von 24 Stunden nach der ersten randomisierten Infusion von Ocrevus. Die primäre Analyse wurde durchgeführt, als 580 Patienten randomisiert worden waren. Der Anteil der Patienten mit IRRs während oder innerhalb von 24 Stunden nach der ersten randomisierten Infusion betrug in der Gruppe mit kürzerer Infusion 24,6 % im Vergleich zu 23,1 % in der Gruppe mit konventioneller Infusion. Der Gruppenunterschied nach Stratifizierung war ähnlich. Insgesamt waren die IRRs bei allen randomisierten Dosen mehrheitlich leicht oder mittelschwer und es gab nur zwei schwere IRRs (eine IRR je Gruppe). Es traten keine lebensbedrohlichen, tödlichen oder schwerwiegenden IRRs auf.
Primär progrediente Form der MS
Die Wirksamkeit und Sicherheit von Ocrevus wurden auch in einer randomisierten, doppelblinden, Placebo-kontrollierten klinischen Studie bei Patienten mit primär progredienter MS (Studie WA25046) bewertet. Das Studiendesign und die Ausgangs-Charakteristika der Studienpopulation sind in Tabelle 4 dargestellt.
Die demographischen Merkmale und Ausgangs-Charakteristika waren in beiden Behandlungsgruppen ausgewogen.
Die Patienten mit Ocrevus (Gruppe A) erhielten alle 6 Monate 600 mg (als 2 x je 300 mg i.v. Infusion im Abstand von 2 Wochen). Die Patienten der Gruppe B hingegen erhielten ein Placebo. Die 600 mg Infusionen bei der RMS und die 2 x je 300 mg Infusionen bei der PPMS zeigten konsistente PK- und PD-Profile. Die Profile der infusionsbedingten Reaktionen pro Infusion waren auch ähnlich, unabhängig davon, ob die 600 mg Dosis als Einmaldosis mit einer 600 mg Infusion oder als getrennte Infusionen zu je 300 mg-Infusionen und im Abstand von 2 Wochen verabreicht wurden (siehe «Unerwünschte Wirkungen, Infusionsbedingte Reaktionen» und «Pharmakokinetik»). Da insgesamt mehr Infusionen zu je 300 mg verabreicht wurden, war in dieser Gruppe die Gesamtzahl der infusionsbedingten Reaktionen höher. Deshalb wird nach der Dosis 1 (Anfangsdosis) empfohlen, Ocrevus nachfolgend nur noch als Einmal-Infusion mit 600 mg zu verabreichen (siehe Tabelle 1), um so die Gesamtzahl an Infusionen und die damit verbundenen Infusionsreaktionen zu reduzieren (und die gleichzeitige Exposition gegenüber prophylaktisch verabreichtem Methylprednisolon).
Tabelle 4: Studiendesign und Ausgangs-Charakteristika der Studie WA25046
|
Studienname
|
Studie WA25046 ORATORIO (n = 732)
| |
Studiendesign
| |
Studienpopulation
|
Patienten mit primär progredienter Form der MS
| |
Studiendauer
|
Ereignisgesteuert (Minimum 120 Wochen und 253 bestätigte Ereignisse einer Progression der Behinderung)
Mediane Nachbeobachtungszeit: Ocrevus 3,0 Jahre, Placebo 2,8 Jahre
| |
Anamnese beim Screening
|
Alter 18 – 55 Jahre,
EDSS von 3,0 – 6,5,
hierin Score ≥ 2,0 im funktionellen System der Pyramidenbahn aufgrund der Befunde in den unteren Extremitäten.
| |
Behandlungsgruppen
|
Gruppe A: Ocrevus 600 mg
Gruppe B: Placebo, 2:1 randomisiert
| |
Ausgangs-Charakteristika
|
Ocrevus 600 mg (n = 488)
|
Placebo (n = 244)
| |
Mittleres Alter (Jahre)
|
44,7
|
44,4
| |
Altersspanne (Jahre) bei Einschluss in die Studie
|
20 - 56
|
18 - 56
| |
Geschlechterverteilung (% männlich / % weiblich)
|
51,4 / 48,6
|
49,2 / 50,8
| |
Durchschnittl./mediane Dauer seit Beginn der MS Symptome (in Jahren)
|
6,7 / 6,0
|
6,1 / 5,5
| |
Durchschnittl./mediane Krankheitsdauer seit der PPMS Diagnose (in Jahren)
|
2,9 / 1,6
|
2,8 / 1,3
| |
Durchschnitt des EDSS (Mittelwert)
|
4,7
|
4,7
|
Die klinischen Endpunkte und Wirkungen im MRT sind in Tabelle 5 und Abbildung 2 aufgeführt.
Tabelle 5: Die klinischen Endpunkte und Endpunkte im MRT in der Studie WA25046 (PPMS)
|
|
Studie 3
| |
Endpunkte
|
WA25046 (ORATORIO)
| |
Ocrevus 600 mg
(n = 488)
|
Placebo
(n = 244)
| |
Klinische Endpunkte
| |
Primärer Wirksamkeitsendpunkt
Anteil der Patienten mit einer innerhalb 12 Wochen bestätigten Progression der Behinderung1 (Primärer Endpunkt)
|
30,2 %
|
34,0 %
| |
Risiko Verminderung
|
24 %
(p = 0,0321)
| |
Anteil der Patienten mit einer innerhalb 24 Wochen bestätigten Progression der Behinderung1
|
28,3 %
|
32,7 %
| |
Risiko Verminderung
|
25 %
(p = 0,0365)
| |
Prozentuale Veränderung der gemessenen 25-Fuss Gehzeit ab Studienbeginn bis Woche 120
|
38,9
|
55,1
| |
Relative Reduktion der Progressionsrate der Gehzeit
|
29,4 %
(p = 0,0404)
| |
Endpunkte im MRT
| |
Prozentuale Veränderung des Volumens der hyperintensen T2 Läsionen ab Studienbeginn bis Woche 120
|
-3,4
|
7,4
| |
(p < 0,0001)
| |
Prozentuale Veränderung des Hirnvolumens von Woche 24 bis Woche 120
|
-0,902
|
-1,093
| |
Rate der relativen Reduktion des Hirnvolumenverlustes
|
17,5 %
(p = 0,0206)
| |
Lebensqualität
| |
Durchschn. Änderung gegenüber Ausgangswert im SF-36 Physical Component Summary
|
-0,731
|
-1,108
| |
Differenz
|
0,377
(p = 0,6034)
|
1 Definiert als eine Zunahme von ≥1,0 Punkten von der Baseline des EDSS Score für Patienten mit einem Baseline Score von 5,5 oder weniger, oder ≥0,5 falls der Baseline Score > 5,5 beträgt; Kaplan-Meier Schätzungen bei Woche 120.
Abbildung 2: Kaplan-Meier Plot der Zeit bis zum Beginn der bestätigten Progression der Behinderung, während mindestens 12 Wochen aufrechterhalten, wobei das initiale Ereignis der neurologischen Verschlechterung in die doppelblinde Behandlungsphase fällt (ITT-Population)*
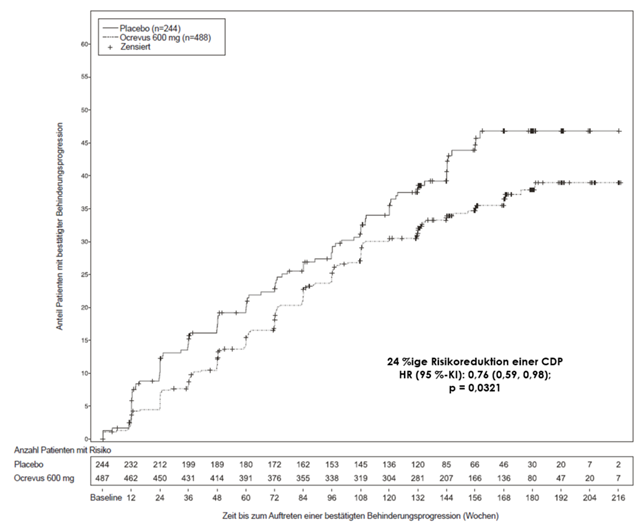
* Alle Patienten in dieser Analyse durchliefen eine mindestens 120 Wochen dauernde Nachbeobachtung. Die primäre Analyse basiert auf allen aufgetretenen Ereignissen.
In der verlängerten Kontrollperiode (Extended Controlled Period [ECP]), die eine doppelt verblindete Behandlung und ca. 9 zusätzliche Monate kontrollierte Nachbeobachtung vor der Fortsetzung in der Open-Label-Extension (OLE) oder bis zum Abbruch der Studienbehandlung beinhaltet, wurden Post-hoc-Analysen durchgeführt. Der Anteil der Patienten mit einer 24-wöchigen bestätigten Behinderungsprogression von EDSS ≥ 7,0 (24W-CDP von EDSS ≥ 7,0; Zeit bis zur Rollstuhlpflicht) betrug 9,1 % in der Placebo-Gruppe im Vergleich zu 4,8 % in der Ocrevus-Gruppe in Woche 144, was zu einer 47 %igen Risikoreduktion der Zeit bis zur Rollstuhlpflicht (HR: 0,53, [0,31; 0,92]) während der ECP führte. Da diese Ergebnisse explorativer Natur waren und Daten nach Entblindung einschlossen, sollten diese Ergebnisse mit Vorsicht interpretiert werden.
Ocrevus s.c.
OCARINA II
Die Studie CN42097 (OCARINA II) war eine multizentrische, randomisierte, Open-Label-Studie mit Parallelarmen zur Beurteilung der Pharmakokinetik, Pharmakodynamik, Sicherheit und Immunogenität sowie der radiologischen und klinischen Wirkungen von Ocrevus s.c. im Vergleich zu Ocrevus i.v. bei Patienten mit RMS oder PPMS. OCARINA II war dazu ausgelegt, die Nichtunterlegenheit der Behandlung mit Ocrevus s.c. gegenüber Ocrevus i.v. auf der Grundlage des primären PK-Endpunkts der Fläche unter der Konzentrations-Zeit-Kurve (AUC) bis Woche 12 nach der Injektion/Infusion (AUCW1-12) aufzuzeigen.
Insgesamt 236 Patienten mit RMS oder PPMS (213 Patienten mit RMS, 23 Patienten mit PPMS) wurden im Verhältnis 1:1 in den s.c. Arm oder i.v. Arm randomisiert. Während des kontrollierten Zeitraums (Tag 0 bis Woche 24) erhielten die Patienten entweder eine einzelne s.c.-Injektion von 920 mg an Studientag 1 oder zwei i.v.-Infusionen von 300 mg an Studientag 1 bzw. 14. Nach dem kontrollierten Zeitraum hatten alle Patienten die Möglichkeit, in den Wochen 24 und 48 (Dosis 2 und 3) weitere Injektionen von 920 mg s.c. zu erhalten. Patienten wurden ausgeschlossen, wenn sie innerhalb der vergangenen 24 Monate eine Behandlung mit anti-CD20-Antikörpern, einschliesslich Ocrelizumab, erhalten hatten.
Die Patienten waren 18–65 Jahre alt und hatten beim Screening einen EDSS zwischen 0 und 6,5. Die demografischen Merkmale waren ähnlich und die Baseline-Merkmale in den beiden Behandlungsgruppen waren gut ausgewogen. Das Durchschnittsalter betrug im s.c. Arm 39,9 Jahre und im i.v. Arm 40,0 Jahre. Im s.c. Arm waren 34,7 % der Patienten männlich und im i.v. Arm 40,7 % der Patienten. Die mittlere/mediane Dauer seit der MS-Diagnose betrug im s.c. Arm 5,70/3,10 Jahre und im i.v. Arm 4,78/2,35 Jahre.
Die Nichtunterlegenheit der Ocrelizumab-Exposition nach Gabe von 920 mg Ocrevus s.c. im Vergleich zu 600 mg Ocrevus i.v. wurde anhand des primären PK-Endpunkts, AUC bis Woche 12 (AUCW1-12) nach der Injektion, nachgewiesen (siehe «Pharmakokinetik»).
Immunogenität
Die Daten zur Immunogenität sind stark abhängig von der Empfindlichkeit und Spezifität der verwendeten Testverfahren. Zusätzlich kann die in einem Testverfahren beobachtete Häufigkeit eines positiven Ergebnisses durch verschiedene Faktoren beeinflusst werden, so auch durch die Probenhandhabung, den Zeitpunkt der Probenentnahme, eine Medikamenten Interferenz, die Begleitmedikation und die zu Grunde liegende Erkrankung. Daher kann der Vergleich der Inzidenz von Antikörpern gegen Ocrevus mit der Inzidenz von Antikörpern gegen andere Produkte je nachdem irreführend sein.
Ocrevus i.v.
Die Patienten in den MS-Studien (WA21092, WA21093 und WA25046) wurden zu mehreren Zeitpunkten (zu Beginn und dann alle 6 Monate bis nach Abschluss der Behandlung und der Studie) auf anti-Arzneimittel Antikörper (anti-drug antibodies, ADAs) getestet. Bei den mit Ocrelizumab behandelten 1311 Patienten wurden 12 (~ 1 %) als positiv getestet für in der Behandlung entstandene ADAs, davon 2 Patienten mit neutralisierenden Antikörpern. Die Auswirkungen der unter der Behandlung aufgetretenen ADAs auf die Sicherheit und Wirksamkeit können aufgrund der niedrigen Inzidenz von mit Ocrevus assoziierten ADAs nicht beurteilt werden.
Ocrevus s.c.
In den Studien OCARINA II und OCARINA I entwickelte keiner der Patienten behandlungsbedingte ADAs gegen Ocrelizumab.
Die Inzidenz behandlungsbedingter Anti-rHuPH20(Hyaluronidase)-Antikörper bei Patienten unter Behandlung mit Ocrevus s.c. in der Studie OCARINA I betrug 2,3 % (3/132). Keiner der Patienten in der Studie OCARINA II entwickelte behandlungsbedingte Anti-rHuPH20-Antikörper.
PharmakokinetikIn den MS-Studien wurde die Pharmakokinetik von Ocrevus in einer populationspharmakokinetischen Analyse untersucht. Bei niedrigen Dosen war die Ocrevus Clearance konzentrationsabhängig. Im therapeutischen Dosisbereich ist die Pharmakokinetik von Ocrevus linear.
Ocrevus i.v.
Die Gesamtexposition (die Fläche unter der Kurve (die AUC) über die 24 Wochen Dosierungsintervalle) war in der Studie mit 2 x je 300 mg bei PPMS und jener mit 1 x 600 mg bei RMS ähnlich, was erwartet wurde, da identische Dosen von 600 mg i.v. verabreicht wurden. Die Fläche unter der Kurve (die AUC) nach der vierten Dosis von 600 mg Ocrevus betrug 3510 µg/ml•Tag, die mittlere maximale Konzentration (Cmax) 212 µg/ml bei der RMS (600 mg Infusion) und 141 µg/ml bei der PPMS (300 mg / Infusion).
Ocrevus s.c.
Nach Gabe von 920 mg Ocrevus s.c. betrug die vorausgesagte mittlere Exposition (AUC über das 24-wöchige Dosierungsintervall) 3'730 µg/ml•Tag.
Der primäre PK-Endpunkt in der Studie OCARINA II, AUCW1-12, nach Gabe von 920 mg Ocrevus s.c. erwies sich gegenüber der Anwendung von Ocrevus i.v. als nicht unterlegen. Das Verhältnis der geometrischen Mittelwerte (Geometric Mean Ratios, GMRs) für AUCW1-12 betrug 1,29 (90 %-KI: 1,23–1,35).
Absorption
Ocrevus wird mittels einer i.v. Infusion oder einer s.c.-Injektion verabreicht. Die Bioverfügbarkeit nach s.c. Gabe von 920 mg Ocrevus s.c. wurde auf 81 % geschätzt. Die mittlere Cmax betrug 132 µg/ml und tmax wurde nach ungefähr 4 Tagen erreicht (Bereich 2–13 Tage).
Distribution
Die populationspharmakokinetische Schätzung des zentralen Verteilungsvolumens war 2,78 l. Das periphere Volumen wurde auf 2,68 l geschätzt und die interkompartimentelle Clearance auf 0,294 l/Tag.
Metabolismus
Der Ocrevus Metabolismus ist nicht speziell untersucht worden, da Antikörper hauptsächlich durch Katabolismus eliminiert werden.
Elimination
Die konstante Clearance wurde auf 0,17 l/Tag geschätzt. Die terminale Halbwertszeit betrug 26 Tage.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Es wurde diesbezüglich keine formale pharmakokinetische Studie durchgeführt. Die Patienten mit einer leichten Lebereinschränkung wurden in die klinischen Studien aufgenommen und es wurden keine Veränderungen der Ocrevus Pharmakokinetik bei diesen Patienten beobachtet.
Nierenfunktionsstörungen
Es wurde keine formale pharmakokinetische Studie durchgeführt. Die Patienten mit einer leichten Niereneinschränkung wurden in die klinischen Studien aufgenommen und es wurden keine Veränderungen der Ocrevus Pharmakokinetik bei diesen Patienten beobachtet.
Ältere Patienten
Es wurden keine Studien zur Untersuchung der Pharmakokinetik von Ocrevus bei älteren Patienten (≥58 Jahre) durchgeführt.
Kinder und Jugendliche
Es wurden keine Studien zur Untersuchung der Pharmakokinetik von Ocrevus bei Kindern und Jugendlichen (< 18 Jahren) durchgeführt.
Präklinische DatenSicherheitspharmakologie
Die präklinischen Daten weisen keine besonderen Gefahren für den Menschen basierend auf den konventionellen Studien zur pharmakologischen Sicherheit und der akuten und wiederholten Dosis-Toxizität nach.
Genotoxizität
Es wurden keine Studien zur Abklärung der Genotoxizität durchgeführt.
Kanzerogenität
Es wurden keine Studien zur Abklärung der Kanzerogenität durchgeführt.
Reproduktionstoxizität
Die präklinischen Daten, basierend auf Studien der männlichen und weiblichen Fruchtbarkeit bei Affen, weisen keine besonderen Gefahren für den Menschen nach.
In einer embryo-fötalen Entwicklungsstudie an Affen wurden nach Verabreichung von 75/100 mg/kg (Loading Dosis/Studiendosis) Ocrevus keine Hinweise auf eine maternale Toxizität, Teratogenität oder Embryotoxität gefunden. Da IgG-Moleküle die Plazentaschranke passieren, verursacht Ocrevus eine Verminderung der B-Zellzahlen in den Feten behandelter Affen.
In einer prä- und postnatalen Entwicklungsstudie bei Affen wurden Ocrevus Gaben (15/20 und 75/100 mg/kg Loading-Dosis/Studien-Dosis, entsprechend einem Dosis-Äquivalent beim Menschen von etwa 3'000 mg (ca. 5 x die klinische Dosis) bzw. 15'000 mg (ca. 25 x die klinische Dosis)) mit einer Glomerulopathie (7/24 Tiere), mit einer der Bildung von lymphoiden Follikeln im Knochenmark (9/24 Tiere) und einer lymphoplasmazytoiden Entzündung in der Niere (2/24 Tiere) assoziiert. Das Hodengewicht der Neugeborenen war in der 75/100 mg/kg Gruppe im Vergleich zur Kontrollgruppe signifikant reduziert. Es gab zwei Todesfälle (2/24 Tiere) in der Studie. Der eine Fall war auf Schwächlichkeit wegen Frühgeburt gekoppelt mit einer opportunistischen Infektion zurückzuführen, der andere auf eine infektiöse Meningoenzephalitis des Neugeborenen mit Beteiligung des Kleinhirns wegen einer aktiven Infektion (Mastitis) des Muttertieres. Der Verlauf beider Neugeborenen-Infektionen war möglicherweise durch die B-Zell-Depletion beeinflusst worden. Neugeborene Nachkömmlinge von Muttertieren, denen Ocrevus verabreicht wurde, zeigten in der postnatalen Phase aufgebrauchte B-Zell-Populationen.
Messbare Mengen (ca. 0,2 % des steady-state Serumtalspiegels) von Ocrevus wurden in der Muttermilch während der Stillzeit nachgewiesen.
Sonstige Angaben
Die subkutane Gabe von Ocrelizumab mit Hyaluronidase wurde in Studien zur lokalen Verträglichkeit bei Ratten und Minischweinen gut vertragen.
Es wurden keine Studien zu Karzinogenität, Genotoxizität oder Fertilität mit rekombinanter humaner Hyaluronidase durchgeführt. Reproduktionstoxikologische Studien bei Mäusen mit rHuPH20, in denen die NEL(No Effect Level)-Dosis > 1'100-mal höher als die empfohlene klinische Dosis war, ergaben embryofetale Verluste, und es gab keine Hinweise auf Teratogenität.
Sonstige HinweiseOcrevus i.v.
Inkompatibilitäten
Es wurden keine Inkompatibilitäten zwischen Ocrevus i.v. und Polyvinylchlorid (PVC), Polyolefin (PO) Beutel oder einem i.v.-Verabreichungsset beobachtet.
Es dürfen keine anderen als die unter Hinweise für die Handhabung erwähnten Verdünnungsmittel für Ocrevus i.v. eingesetzt werden, da deren Verwendung nicht untersucht wurde.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit der zubereiteten Infusionslösung
Das Präparat enthält keine Konservierungsmittel und ist nur für den einmaligen Gebrauch bestimmt.
Die hergestellte Infusionslösung sollte sofort verwendet werden. Wenn sie nicht sofort verwendet wird, kann sie bis zu 24 Stunden bei 2–8 °C und 8 Stunden bei Raumtemperatur gelagert werden.
Falls eine i.v. Infusionslösung nicht am gleichen Tag aufgebraucht wird, muss die übrig gebliebene Lösung entsorgt werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Im Kühlschrank (2-8 °C) lagern.
Nicht schütteln. Nicht einfrieren.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Hinweise für die Handhabung
Ocrevus i.v.-Infusionen müssen von einer medizinischen Fachperson unter aseptischen Bedingungen hergestellt werden. Zur Zubereitung der verdünnten Infusionslösung sind eine sterile Nadel und Spritze zu verwenden.
Ocrevus i.v. kann feine durchscheinende und/oder reflektierende Partikel mit einer verstärkten Opaleszenz enthalten. Die Lösung darf nicht verwendet werden, wenn sie verfärbt ist oder wenn sie frei bewegliche Fremdpartikel enthält.
Ocrevus i.v. muss vor der Verabreichung verdünnt werden. Lösungen von Ocrevus i.v. für die i.v. Verabreichung werden durch Verdünnung des Arzneimittels in einem Infusionsbeutel mit 0,9 %igem Natriumchlorid (300 mg / 250 ml oder 600 mg / 500 ml) auf eine Arzneimittel-Endkonzentration von ca. 1,2 mg/ml hergestellt.
Die verdünnte Infusionslösung muss als eine Infusion ausgestattet mit einem 0,2 oder 0,22 Mikrometer In-line-Filter verabreicht werden.
Der Inhalt des Infusionsbeutels muss vor Beginn der i.v. Applikation Raumtemperatur erreichen, um eine unerwünschte Infusionsreaktion aufgrund der niedrigen Lösungs-Temperatur zu vermeiden.
Ocrevus s.c.
Inkompatibilitäten
Es wurden keine Inkompatibilitäten zwischen Ocrevus s.c. und Polypropylen (PP), Polycarbonat (PC), Polyethylen (PE), Polyvinylchlorid (PVC), Polyurethan (PUR) und Edelstahl festgestellt.
Haltbarkeit
Dieses Arzneimittel nach Ablauf des auf dem Behälter angegebenen Verfalldatums («EXP») nicht mehr verwenden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Im Kühlschrank (2-8 °C) lagern.
Nicht schütteln. Nicht einfrieren.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ungeöffnet kann die Durchstechflasche gegebenenfalls bis zu 12 Stunden bei ≤25 °C bleiben.
Die ungeöffneten Durchstechflaschen können mehrmals in den Kühlschrank zurückgestellt werden, d.h. sie können insgesamt maximal 12 Stunden ohne Kühlung bei ≤25 °C gelagert werden.
Lagerung der Spritze
·Aus mikrobiologischer Sicht ist das Produkt unmittelbar nach dem Überführen aus der Durchstechflasche in die Spritze zu verwenden, da es kein antimikrobielles Konservierungsmittel enthält. Falls es nicht sofort verwendet wird, liegen die Aufbrauchsfristen und Lagerbedingungen vor dem Gebrauch in der Verantwortung des Anwenders und sollten normalerweise nicht länger als 24 Stunden bei 2 °C bis 8 °C betragen.
·Wenn die Zubereitung unter kontrollierten und validierten aseptischen Bedingungen erfolgt ist, kann die verschlossene Spritze bis zu 30 Tage bei 2 °C bis 8 °C im Kühlschrank und danach 8 Stunden bei diffusem Tageslicht bei Temperaturen ≤30 °C gelagert werden.
Hinweise für die Handhabung
Ocrevus s.c. ist von medizinischem Fachpersonal unter Einhaltung aseptischer Verfahren vorzubereiten.
Ocrevus s.c. ist eine gebrauchsfertige Lösung mit einer Einzeldosis nur für die subkutane Injektion und sollte nicht verdünnt oder mit anderen Arzneimitteln gemischt werden.
Die Ocrevus s.c. Lösung per Sichtprüfung auf Vorhandensein von Partikeln oder Verfärbungen prüfen. Die Lösung darf nicht verwendet werden, wenn Verfärbungen vorliegen oder Fremdpartikel vorhanden sind.
Vorbereitung der Spritze
Die Durchstechflasche vor der Verwendung aus dem Kühlschrank nehmen und die Lösung auf Raumtemperatur kommen lassen.
Den gesamten Inhalt der Ocrevus s.c.-Lösung mit einer Spritze und einer Transferkanüle (21 G wird empfohlen) aus der Durchstechflasche aufziehen.
Die Transferkanüle abnehmen und für die Injektion ein s.c.-Infusionsset (z.B. geflügelt/Butterfly) mit einer Kanüle mit 24–26 G anbringen. Es sollte ein s.c.-Infusionsset mit einem Restvolumen von NICHT mehr als 0,8 ml verwendet werden.
Den s.c.-Infusionsschlauch mit der Arzneimittellösung füllen, um die Luft im Infusionsschlauch zu entfernen. Die Befüllung stoppen, bevor die Flüssigkeit die Kanüle erreicht.
Es ist darauf zu achten, dass die Spritze nach dem Vorfüllen des Schlauchs und dem Entfernen der Überschussmenge genau 23 ml Arzneimittellösung enthält.
Das Arzneimittel sofort injizieren, um ein Verstopfen der Kanüle zu vermeiden.
Die sofortige Anwendung wird empfohlen, da Ocrevus s.c. kein antimikrobielles Konservierungsmittel enthält. Wenn die Dosis nicht sofort gegeben wird, siehe «Aufbewahrung der Spritze» weiter unten. Die Spritze NICHT aufbewahren, wenn sie bereits vorbereitet und an dem vorgefüllten s.c.-Infusionsset befestigt wurde.
Aufbewahrung der Spritze
·Wenn die Dosis nicht sofort gegeben werden soll, kann unter Anwendung einer aseptischen Technik der gesamte Inhalt von Ocrevus s.c. aus der Durchstechflasche in die Spritze aufgezogen werden, um das Dosisvolumen (23 ml) plus das Füllvolumen für das s.c.-Infusionsset zu berücksichtigen. Die Transferkanüle durch eine Spritzenverschlusskappe ersetzen. Zur Aufbewahrung KEIN s.c.-Infusionsset anbringen.
·Wenn die Spritze im Kühlschrank aufbewahrt wurde, muss sie vor der Anwendung Raumtemperatur annehmen können.
Ocrevus i.v. und s.c.
Entsorgung nicht gebrauchter/abgelaufener Arzneimittel
Die Freisetzung von Arzneimitteln in die Umwelt sollte minimiert werden. Das Arzneimittel darf nicht im Abwasser oder durch den Hausmüll entsorgt werden. Verwenden Sie - falls an Ihrem Standort möglich - etablierte «Sammelsysteme».
Die folgenden Punkte sollten strikt in Bezug auf die Verwendung und Entsorgung von Spritzen und anderen medizinischen scharfen Gegenstände eingehalten werden:
·Nadeln und Spritzen sollten niemals wiederverwendet werden.
·Legen Sie alle verwendeten Nadeln und Spritzen in einen Behälter für spitze Gegenstände (Stichverletzungssichere Einwegbehälter).
Nicht verwendetes Arzneimittel und/oder das Abfallmaterial sind entsprechend den nationalen Anforderungen zu entsorgen. Nadeln und Spritzen sollten niemals wiederverwendet werden.
Zulassungsnummer66185, 68988 (Swissmedic).
PackungenOcrevus, Durchstechflasche mit 10 ml Konzentrat zur Herstellung einer Infusionslösung (300 mg/10 ml) (i.v.): 1 [A]
Ocrevus subkutan, Durchstechflasche mit 23 ml Injektionslösung (920 mg/23 ml) (s.c.): 1 [A]
ZulassungsinhaberinRoche Pharma (Schweiz) AG, Basel.
Stand der InformationAugust 2024.
|