ZusammensetzungWirkstoffe
Ranibizumabum*
*Ranibizumabum ist das Fragment eines humanisierten monoklonalen Antikörpers, das mit Hilfe rekombinanter DNA-Technologie in Escherichia coli hergestellt wurde.
Hilfsstoffe
α,α-trehalosum dihydricum
Histidinum
Histidini hydrochloridum monohydricum
Polysorbatum 20
Aqua ad iniectabile
Indikationen/AnwendungsmöglichkeitenRanivisio ist indiziert bei Erwachsenen für die Behandlung:
·der exsudativen (feuchten) altersbezogenen Makuladegeneration (feuchte AMD).
·eines Visusverlustes durch ein Diabetisches Makulaödem (DME).
·der mässig schweren bis schweren nicht-proliferativen diabetischen Retinopathie (NPDR) bzw. proliferativen diabetischen Retinopathie (PDR),
·eines Visusverlustes durch ein Makulaödem infolge eines retinalen Venenverschlusses (retinaler Venenastverschluss BRVO und retinaler Zentralvenenverschluss CRVO).
·einer aktiven, den Visus beeinträchtigenden choroidalen Neovaskularisation (CNV).
·eines Visusverlustes durch choroidale Neovaskularisation (CNV) infolge einer pathologischen Myopie (PM).
Ranivisio ist indiziert bei Frühgeborenen für die Behandlung der Frühgeborenen Retinopathie (RPM):
·RPM in Zone I (Krankheitsstadium 1+, 2+, 3 oder 3+), Zone II (Krankheitsstadium 3+) oder AP-RPM (aggressive posteriore RPM).
Dosierung/AnwendungRanivisio darf nur durch einen qualifizierten Ophthalmologen angewendet werden, der über eine adäquate Infrastruktur verfügt. Ranivisio wird in den Glaskörper (intravitreal) injiziert. Eine Ranivisio Durchstechflasche (Erwachsene und Frühgeborene) enthält 0.23 ml, eine Fertigspritze (nur für Erwachsene) enthält 0.165 ml: Beide sind zum einmaligen Gebrauch für einen Patienten bestimmt.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Übliche Dosierung
Dosierung für Erwachsene
Die empfohlene Dosis von 0.5 mg wird als intravitreale Einzelinjektion appliziert. Dies entspricht einem Injektionsvolumen von 0.05 ml. Der Abstand zwischen zwei Injektionen in dasselbe Auge darf nicht kürzer als 1 Monat sein.
Die Behandlung wird mit einer Injektion monatlich begonnen, bis der maximale Visus erreicht ist und/oder keine Anzeichen von Krankheitsaktivität mehr vorhanden sind. Bei Patienten mit feuchter AMD, DME, mässig schwerer bis schwerer NPDR oder PDR, und BRVO oder CRVO können initial drei oder mehr aufeinanderfolgende monatliche Injektionen notwendig sein.
Danach sollten die Abstände zwischen den Kontrolluntersuchungen und die Behandlungsintervalle vom Arzt festgelegt werden. Bei den Kontrolluntersuchungen wird die Krankheitsaktivität durch klinische Untersuchung, durch Kontrolle des Visus und/oder durch bildgebende Verfahren (z.B. optische Kohärenztomographie und/oder Fluoreszein-Angiographie) beurteilt.
Die Kontroll- und Behandlungsintervalle können gemäss der vorliegenden Krankheitsaktivität und des beobachteten Therapieverlaufs schrittweise verlängert oder verkürzt werden.
Falls aufgrund der ärztlichen Beurteilung angezeigt, kann Ranivisio auch monatlich an einem bestimmten Auge angewendet werden.
Die Behandlung eines Visusverlustes durch CNV muss individuell für jeden Patienten auf Grundlage der Krankheitsaktivität festgelegt werden. Für CNV infolge von einer pathologischen Myopie (PM) oder für andere, nicht im Rahmen einer AMD auftretende CNV, liegen keine Erfahrungen über einen Behandlungszeitraum von mehr als einem Jahr vor.
Dosierung für Frühgeborene
Die empfohlene Dosis für Ranivisio bei Frühgeborenen beträgt 0.1 mg, die als einmalige intravitreale Injektion appliziert wird. Dies entspricht einem Injektionsvolumen von 0.01 ml. Bei Frühgeborenen wird die Behandlung der Frühgeborenen-Retinopathie (RPM) mit einer Einzeldosis eingeleitet, die an beiden Augen am selben Tag verabreicht werden kann. Insgesamt können bei Anzeichen von Krankheitsaktivität bis zu drei Injektionen pro Auge innerhalb von sechs Monaten verabreicht werden. Wenn mehr als eine Injektion erforderlich ist, sollte der zeitliche Abstand zwischen zwei Injektionen mindestens einen Monat betragen.
Patienten mit Leberfunktionsstörungen
Es liegen keine Untersuchungen vor. Da die systemisch verfügbare Menge vernachlässigbar ist, sind keine speziellen Vorsichtsmassnahmen vorgesehen.
Patienten mit Nierenfunktionsstörungen
Für Patienten mit eingeschränkter Nierenfunktion ist keine Dosisanpassung notwendig (s. «Pharmakokinetik»).
Ältere Patienten (ab 65 Jahren)
Es ist keine Dosisanpassung notwendig.
Kinder und Jugendliche
Ranivisio wird für den Gebrauch bei Kindern und Jugendlichen nicht empfohlen, da nicht ausreichend Daten zur Sicherheit und Wirksamkeit in dieser Patientengruppe vorliegen. Zu jugendlichen Patienten im Alter von 12 bis 17 Jahren mit Visusverlust durch CNV liegen begrenzte Daten vor. (s. «Eigenschaften/Wirkungen»).
Art der Anwendung
Vor der intravitrealen Verabreichung sollte eine gründliche Anamnese hinsichtlich möglicher Überempfindlichkeitsreaktionen erhoben werden (s. «Warnhinweise und Vorsichtsmassnahmen»).
Die Verabreichung von Ranivisio muss unter aseptischen Bedingungen (geeignete Räumlichkeiten, keimfreie Abdeckung, Handschuhe, Utensilien) erfolgen. Geeignete Anästhesie und ein topisches Breitband-Mikrobizid zur Desinfektion der periokulären Haut, des Lids und der okulären Oberfläche sollten vor Durchführung der Injektion angewendet werden.
Der gesamte Inhalt der Durchstechflasche wird mit einer Spritze und einer 5 µm Filternadel entnommen. Vor der intravitrealen Injektion wird die Filternadel entfernt und die Injektionsnadel (13 mm, 30 Gauge) auf die Spritze aufgesetzt. Der Inhalt soll soweit ausgestossen werden, dass die Kolbenspitze der Spritze auf der Markierung für die 0.05 ml (50 µl) liegt. Der Inhalt einer Durchstechflasche ist zur Verabreichung einer Einzeldosis bestimmt. Die nicht gebrauchte Lösung ist zu verwerfen.
Bei Erwachsenen sollte die Injektionsnadel 3.5-4.0 mm hinter dem Limbus vollständig eingestochen werden. Dabei ist die horizontale Mittellinie zu vermeiden und die Nadel zum Zentrum des Augapfels zu richten.
Das Injektionsvolumen wird langsam abgegeben, und bei Folgeinjektionen ist auf einen Wechsel der Injektionsstelle auf der Sklera zu achten.
Nach der Injektion muss der Augeninnendruck des Patienten überwacht werden. Die Überwachung sollte die Kontrolle der Durchblutung der Sehnervenpapille unmittelbar nach der Injektion, Augeninnendruckmessung innert 30 Minuten, Ophthalmoskopie, Spaltlampenuntersuchung sowie Funduskontrolle nach 2-7 Tagen umfassen. Der Patient muss instruiert werden, allfällige Zeichen einer Endophthalmitis sofort dem Arzt zu melden (s. «Warnhinweise und Vorsichtsmassnahmen»).
Bei Frühgeborenen muss die Injektionsnadel 1.0 bis 2.0 mm hinter dem Limbus eingeführt werden, wobei die Nadel in Richtung des Sehnervs zeigt. Dann wird das Injektionsvolumen von 0.01 ml verabreicht.
KontraindikationenÜberempfindlichkeit gegenüber Ranibizumab oder einem der Hilfsstoffe.
Ranivisio ist kontraindiziert bei Patienten mit Infektionen im oder um das Auge, sowie bei Patienten mit aktiver intraokulärer Entzündung.
Warnhinweise und VorsichtsmassnahmenReaktionen durch intravitreale Injektionen
Bei einer intravitrealen Injektion kann es zu infektiösen Endophthalmitiden und Netzhautablösungen kommen. Bei der Injektion von Ranivisio sind aseptische Injektionstechniken anzuwenden. Zudem sollten die Patienten während der auf die Injektion folgenden Tage beobachtet werden, um eine Infektion frühzeitig zu erkennen und zu behandeln (s. «Unerwünschte Wirkungen»).
Bei Erwachsenen wurde ein vorübergehender Anstieg des Augeninnendrucks innerhalb von 60 Minuten nach der Injektion von Ranivisio beobachtet. Es wurde auch über langanhaltenden erhöhten intraokulären Druck berichtet. Sowohl der intraokuläre Druck als auch die Perfusion der zentralen Retinalarterie müssen überwacht und entsprechend behandelt werden (s. «Unerwünschte Wirkungen»).
Beidseitige Behandlung
Die Sicherheit und Wirksamkeit einer gleichzeitigen Behandlung mit Ranivisio an beiden Augen wurde nicht in dafür angelegten Studien geprüft.
Die begrenzt vorliegenden Daten zur bilateralen Anwendung von Ranivisio (einschliesslich einer Verabreichung am selben Tag) weisen, verglichen mit einer unilateralen Behandlung, nicht auf ein erhöhtes Risiko für systemische unerwünschte Ereignisse hin.
Arterielle thromboembolische Ereignisse
Es besteht ein potentielles Risiko für arterielle thromboembolische Ereignisse bei der intravitrealen Applikation von VEGF (vascular endothelial growth factor)-Inhibitoren. Bei Patienten mit einem bekannten Risiko für Schlaganfälle (die z.B. bereits einen Schlaganfall oder eine transiente ischämische Attacke erlitten hatten) ist das Risiko möglicherweise erhöht.
Immunogenität
In allen Behandlungsgruppen zeigten 0-3% der noch nicht behandelten Patienten eine Immunreaktion gegenüber Ranibizumab. Bei monatlicher Applikation wurden nach 12-24 Monaten schwache Antikörper-Titer in 1-6% der Patienten beobachtet. Diese Immunogenitäts-Daten spiegeln den Prozentsatz der Patienten, bei denen ein Elektrochemilumineszenz-Test positive Resultate ergab. Die Daten waren stark abhängig von der Sensitivität und der Spezifität des Tests. Die klinische Signifikanz der Immunreaktion auf Ranibizumab ist zur Zeit unklar. Bei einigen Patienten mit den höchsten Immunoreaktivitätstitern wurden Iritis und Vitritis beobachtet.
Patientengruppen mit eingeschränkt verfügbaren Daten
Die Anwendung von Ranibizumab ist bisher nicht bei Patienten mit aktiver systemischer Infektion oder mit weiteren Augenerkrankungen wie Netzhautablösung oder Makulaloch untersucht worden.
Ranivisio und Laserphotokoagulation
Sowohl die Anwendung von Ranibizumab bei Patienten, welche sich in der Vorgeschichte einer Laserphotokoagulation unterzogen haben als auch eine gleichzeitige Anwendung von Ranibizumab und Laserphotokoagulation wurden untersucht. Falls Ranivisio am selben Tag wie eine Laserphotokoagulation gegeben werden sollte, darf die Injektion frühestens 30 Minuten nach der Laserphotokoagulation erfolgen.
Begleitbehandlung mit anderen anti-VEGF(Vascular Endothelial Growth Factor)-Arzneimitteln:
Ranivisio sollte nicht gleichzeitig mit anderen Anti-VEGF-Arzneimitteln (systemisch oder okular) verabreicht werden.
Aussetzen der Ranivisio-Behandlung bei Erwachsenen
In folgenden Fällen sollte die Dosis ausgesetzt und die Behandlung nicht vor der nächsten planmässigen Behandlung wieder aufgenommen werden:
·Abnahme der bestkorrigierten Sehschärfe (best corrected visual acuityBCVA) von ≥30 Buchstaben im Vergleich zur letzten Bestimmung der Sehschärfe;
·Augeninnendruck von ≥30 mmHg;
·Netzhautriss;
·subretinale Blutung, bei der das Zentrum der Fovea betroffen ist oder die Grösse der Blutung ≥50 % der gesamten betroffenen Läsion beträgt;
·innerhalb der vorangegangenen oder nächsten 28 Tage durchgeführte oder geplante intraokulare Operationen.
Dauer der Behandlung
Für CNV infolge einer pathologischen Myopie (PM) oder für andere, nicht im Rahmen einer AMD auftretende CNV, liegen keine Erfahrungen über einen Behandlungszeitraum von mehr als einem Jahr vor.
InteraktionenEs wurden keine speziellen Interaktionsstudien durchgeführt.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter/Kontrazeption bei Frauen
Gebärfähige Frauen sollten während und bis 30 Tage nach Behandlung wirksame Methoden zur Schwangerschaftsverhütung anwenden.
Schwangerschaft
Es gibt keine Daten zur Anwendung von Ranibizumab bei schwangeren Frauen.
Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädigende Wirkungen in Bezug auf Schwangerschaft oder embryonale/fötale Entwicklung (s. «Präklinische Daten»). Ranibizumab hemmt den VEGF-A, einen wichtigen angiogenen Faktor für die Bildung von neuen Blutgefässen während der embryonalen und fötalen Entwicklung und Plazentabildung. Die systemische Exposition von Ranibizumab ist nach der okulären Anwendung gering. Wegen des Wirkungsmechanismus muss Ranibizumab als potentiell teratogen und embryo-/fetotoxisch angesehen werden und darf während der Schwangerschaft nicht angewendet werden, es sei denn es ist klar notwendig. Bei Patientinnen, die schwanger werden möchten, sollte Ranibizumab 3 Monate vor der Empfängnis abgesetzt werden.
Stillzeit
Begrenzte Daten deuten darauf hin, dass Ranibizumab in der Muttermilch vorhanden ist und die VEGF-Produktion unterdrücken kann. Die Wirkung von Ranibizumab auf den gestillten Säugling und die Wirkung von Ranibizumab auf die Milchproduktion/Ausscheidung sind nicht bekannt. Da zahlreiche Substanzen in die Muttermilch ausgeschieden werden, und die Möglichkeit einer Resorption und Beeinträchtigung des Wachstums und der Entwicklung des Kindes besteht, wird als Vorsichtsmassnahme das Stillen während der Behandlung mit Ranivisio nicht empfohlen. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
Der Einfluss von Ranibizumab auf die männliche und weibliche Fertilität wurde nicht untersucht.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDie Behandlung mit Ranivisio kann vorübergehende Sehstörungen verursachen, wodurch die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen beeinflusst werden können (s. «Unerwünschte Wirkungen»). Patienten, die solche Anzeichen verspüren, dürfen nicht Auto fahren oder Maschinen bedienen, bis diese vorübergehenden Sehstörungen abgeklungen sind.
Unerwünschte WirkungenAus den drei klinischen Studien der Phase III zur Behandlung der feuchten AMD bilden insgesamt 1'315 Patienten die Basis für die Sicherheitspopulation. Alle Patienten wurden mindestens 24 Monate mit Ranibizumab behandelt, mit der empfohlenen Dosis von 0.5 mg wurden 440 Patienten therapiert.
Schwerwiegende unerwünschte Ereignisse im Zusammenhang mit dem Injektionsvorgang waren: Endophthalmitis, rhegmatogene Netzhautablösung, Einriss der Retina und iatrogener traumatischer Katarakt (s. «Warnhinweise und Vorsichtsmassnahmen»).
Bei mit Ranibizumab behandelten Patienten wurden auch intraokuläre Entzündung und erhöhter Augeninnendruck beobachtet (s. «Warnhinweise und Vorsichtsmassnahmen»).
Bei Patienten mit feuchter AMD, die in den kontrollierten Phase-III-Studien zur feuchten AMD FVF2598g (MARINA), FVF2587g (ANCHOR) und FVF3192g (PIER) mit 0.5 mg Ranibizumab behandelt wurden, traten die unten aufgelisteten unerwünschten Ereignisse häufiger auf (mindestens 2 Prozentpunkte) als bei Patienten der Kontrollgruppen (Scheininjektion (s. «Eigenschaften/Wirkungen») oder PDT mit Verteporfin). Deshalb wurden sie als potenzielle unerwünschte Arzneimittelwirkungen (UAWs) beschrieben. Die unten aufgeführten Daten zur Sicherheit beinhalten darüber hinaus alle unerwünschten Ereignisse von denen angenommen wird, dass sie zumindest potenziell durch die Injektion als solche oder durch das Arzneimittel verursacht werden und bei den 440 Patienten auftraten, die mit 0.5 mg Ranibizumab gegen feuchte AMD in der Kombinationstherapie behandelt wurden.
Patienten Kollektiv mit DR
Die Sicherheit von Ranibizumab wurde in einer 24-monatigen klinischen Studie in der Protocol-S-Studie untersucht, die u.a. 191 mit Ranibizumab behandelte Patienten mit diabetischer Retinopathie (DR) einschloss. Die beobachteten okulären und nicht-okulären Ereignisse standen im Einklang mit dem, was bei einer diabetischen Patientenpopulation mit DR zu erwarten wäre, oder sie ähnelten im Hinblick auf ihre Häufigkeit und Schwere den Ereignissen, die in früheren klinischen Studien zu Ranibizumab beobachtet wurden.
Patienten Kollektiv CNV
Die Sicherheit von Ranibizumab wurde in einer 12-monatigen klinischen Studie (MINERVA) untersucht, an der 171 mit Ranibizumab behandelte Patienten mit Visusverlust durch CNV teilnahmen (s. «Eigenschaften/Wirkungen»). Das Sicherheitsprofil bei diesen Patienten war konsistent mit jenem früherer klinischer Studien mit Ranibizumab.
Patienten Kollektiv CNV infolge von PM
Die Sicherheit von Ranibizumab wurde in der 12-monatigen klinischen Studie RADIANCE untersucht. An dieser Studie nahmen 224 Patienten mit CNV infolge von PM teil, die mit Ranibizumab behandelt wurden (s. «Eigenschaften/Wirkungen»). Häufigkeit und Schwere der in dieser Studie aufgetretenen okulären und nicht-okulären Ereignisse waren mit denen in den Studien bei feuchter AMD vergleichbar.
Population mit Frühgeborenen Retinopathie (RPM)
·Die Sicherheit von Ranibizumab 0.1 mg wurde in einer sechsmonatigen klinischen Studie (RAINBOW) untersucht, die 76 mit Ranibizumab behandelte Frühgeborene mit RPM umfasste (s. «Eigenschaften/Wirkungen»). Die okulären Nebenwirkungen, die in der RAINBOW-Studie beobachtet wurden, waren konsistent mit jenen, die auch bei Erwachsenen unter der Behandlung mit Ranibizumab 0.5 mg auftraten oder die ein Ereignis im Rahmen der RPM darstellen. Die nicht-okulären Nebenwirkungen in dieser klinischen Studie entsprachen generell denen, die bei diesen Patienten mit multiplen Komorbiditäten aufgrund der Frühgeburt zu erwarten wären. Aus theoretischen Gründen muss in Betracht gezogen werden, dass die Reifung anderer Organe nach einer Behandlung mit anti-VEGFs verzögert werden könnte, und darum Komplikationen auftreten könnten.
·Die Langzeitsicherheit bei Frühgeborenen mit RPM wurde in der RAINBOW-Erweiterungsstudie bis zum Alter von 5 Jahren nachgewiesen und zeigte keine neuen Sicherheitssignale. Das in der RAINBOW-Erweiterungsstudie beobachtete Sicherheitsprofil für Ranibizumab 0,1 mg stimmte mit dem in der 24-wöchigen RAINBOW-Hauptstudie für 0,1 mg beobachteten Profil überein.
Liste der unerwünschten Wirkungen
Häufigkeiten: «Sehr häufig» (≥1/10), «häufig» (≥1/100 <1/10), «gelegentlich» (≥1/1'000 <1/100), «selten» (≥1/10'000 <1/1'000), «sehr selten» (<1/10'000), «nicht bekannt» (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Infektionen und parasitäre Erkrankungen
Sehr häufig: Nasopharyngitis (12.5-16.4%).
Häufig: Influenza, Harnwegsinfektionen.
Erkrankungen des Blutes und des Lymphsystems
Häufig: Anämie.
Erkrankungen des Immunsystems
Häufig: Hypersensitivitätsreaktionen.
Psychiatrische Erkrankungen
Häufig: Angstzustände.
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (8-15%).
Häufig: Schlaganfall.
Augenerkrankungen
Sehr häufig: Intraokuläre Entzündungen (10-18%), Glaskörperentzündung (2.3-10%), Glaskörperabhebung (18-19%), Netzhautblutungen (25%), Sehstörungen (6.6-10.5%), Augenschmerzen (27-32%), Mouches volantes (7-25%), Bindehautblutung (55-72%), Augenirritation (12-15%), Fremdkörpergefühl im Auge (12-15%), verstärkter Tränenfluss (10-14%), Blepharitis (8-11%), Pruritus (8.5-10%).
Häufig: Retina-Degeneration, Störungen der Retina, Retina-Abhebung, Risse der Retina, Abhebung des retinalen Pigmentepithels, Risse im retinalen Pigmentepithel, Seh-Verschlechterung, Glaskörperblutungen und –Störungen, Uveitis, Iritis, Iridocyclitis, (subkapsuläre) Katarakt, posteriore Kapselsack-Trübung, Keratitis punctata, Kornea-Abrasionen, Trübungen des Kammerwassers, verschwommenes Sehen, Blutungen an der Injektionsstelle, Augenblutungen, (allergische) Konjunktivitis, Ausscheidungen am Auge, Photopsie, Photophobie, Augenbeschwerden, Schmerzen und Ödeme des Augenlids, Hyperämie der Konjunktiva.
Gelegentlich: Endophthalmitis, Hypopyon, Hyphaema, Keratopathie, Verklebung der Iris, Kornea-Einschmelzungen und –Ödeme, Streifen (Striae) der Kornea, Schmerzen und Irritationen an der Einstichstelle, Erblindung, Irritationen des Augenlids.
Herzerkrankungen, Gefässerkrankungen
Arterielle thromboembolische Ereignisse nach Definition der Antiplatelet Trialists' Collaboration (1994) wie gefässbedingte Todesfälle, nicht-fatale Myokardinfarkte, nicht-fatale ischämische Schlaganfälle und nicht-fatale hämorrhagische Schlaganfälle wurden mit der systemischen Verfügbarkeit von hochpotenten VEGF-Inhibitoren in Zusammenhang gebracht. Im ersten Jahr betrug der Anteil der thromboembolischen Ereignisse in den beiden mit Ranibizumab behandelten Patientengruppen (0.3 und 0.5 mg) 2.3%; im Vergleich dazu waren es in der Kontrollgruppe nur 1.3%. Im 2. Jahr der Studie MARINA waren es in beiden Behandlungsgruppen 3.0%, während dem es in der Kontrollgruppe 3.2% waren.
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Häufig: Husten.
Erkrankungen des Gastrointestinaltrakts
Häufig: Nausea.
Erkrankungen der Haut und des Unterhautgewebes
Gelegentlich: Allergische Reaktionen (Ausschlag, Urticaria, Pruritus, Erythema).
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Gelenkschmerzen (8-12%).
Untersuchungen
Erhöhter Augeninnendruck.
Immunogenität
Wie bei allen therapeutischen Proteinen besteht die Möglichkeit einer Immunantwort bei Patienten, die mit Ranibizumab behandelt wurden. Die Immunogenitätsdaten spiegeln den Prozentsatz von Patienten wider, die in Immunoassays positiv auf Antikörper gegen Ranibizumab getestet wurden. Die Daten waren stark abhängig von der Sensitivität und der Spezifität der Assays.
In den AMD Studien zeigten in allen Behandlungsgruppen 0% bis 3% der noch nicht behandelten Patienten eine Immunreaktion gegenüber Ranibizumab. Nach der monatlichen Verabreichung von Ranibizumab über 12 bis 24 Monate wurden die Antikörper gegen Ranibizumab bei ungefähr 1% bis 8% der Patienten mit neovaskulärer AMD nachgewiesen.
In den DME Studien zeigten in allen Behandlungsgruppen 0% bis 2% der noch nicht behandelten Patienten eine Immunreaktion gegenüber Ranibizumab. Nach der monatlichen Verabreichung von Ranibizumab über 12 Monate wurden die Antikörper gegen Ranibizumab bei ungefähr 2% bis 4% der Patienten mit DME detektiert.
In den RVO Studien zeigten in allen Behandlungsgruppen 2% bis 3% der noch nicht behandelten Patienten eine Immunreaktion gegenüber Ranibizumab. Nach der monatlichen Verabreichung von Ranibizumab über 12 Monate wurden die Antikörper gegen Ranibizumab bei ungefähr 4% bis 5% der Patienten mit RVO detektiert.
Die klinische Signifikanz der Immunoreaktivität auf Ranibizumab ist zur Zeit unklar.
In den gepoolten Daten der abgeschlossenen, randomisierten, doppelblinden, klinischen Studien traten bei DME Patienten nicht-schwerwiegende, nicht- okuläre Wundinfektionen/-entzündungen unter 0.5 mg Ranibizumab häufiger auf als unter den Kontrollbehandlungen (1.85/100 Patientenjahre vs. 0.27/100 Patientenjahre). Ein Zusammenhang mit Ranibizumab ist bislang nicht bekannt. Es besteht jedoch das theoretische Risiko eines Zusammenhangs dieser Ereignisse mit der VEGF-Inhibition.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungAus den klinischen Studien zur feuchten AMD und aus Post-Marketing-Beobachtungen wurden Fälle von unbeabsichtigter Überdosierung (Injektion von grösseren Volumina als den empfohlenen 0.05 ml Ranibizumab) berichtet.
Anzeichen und Symptome
Die dabei am häufigsten auftretenden unerwünschten Wirkungen sind erhöhter Augeninnendruck und Augenschmerzen.
Behandlung
Falls eine zu hohe Dosis verabreicht wurde, sollte der Augeninnendruck überwacht und je nach Einschätzung durch den behandelnden Arzt gegebenenfalls behandelt werden.
In klinischen Studien erhielten Patienten mit feuchter AMD und DME Dosierungen von bis zu 2 mg Ranibizumab in einem Injektionsvolumen von 0.05 ml bis 0.10 ml. Art und Häufigkeit der unerwünschten okulären und systemischen Ereignisse entsprachen den für die Dosis 0.5 mg (in 0.05 ml) Ranibizumab berichteten.
Eigenschaften/WirkungenATC-Code
ATC-Code: S01LA04
Ranivisio ist ein Biosimilar.
Wirkungsmechanismus
Der Wirkstoff von Ranivisio (Ranibizumab) ist ein humanisiertes, rekombinantes monoklonales Antikörperfragment (Fab) gegen den humanen vaskulären endothelialen Wachstumsfaktor A (VEGF-A). Es bindet mit hoher Affinität an VEGF-A und an dessen Isoformen. Die Isoformen, wie z.B. VEGF121 und VEGF165 entstehen durch alternatives mRNA-Spleissen, die Isoform VEGF110 durch Proteolyse. Die Bindung von Ranibizumab an VEGF-A und dessen Isoformen inhibiert die Aktivierung der Rezeptoren VEGFR-1 und VEGFR-2 auf der Oberfläche der Endothelzellen.
Pharmakodynamik
Die Aktivierung der Rezeptoren VEGFR-1 und -2 führt zur Proliferation von Endothelzellen, zur Neovaskularisation und zum Flüssigkeitsaustritt aus den Gefässen. Es wird angenommen, dass alle diese Faktoren zur Progression der neovaskulären Form der altersbedingten Makuladegeneration (AMD), der Entwicklung von CNV, inklusive CNV infolge einer pathologischen Myopie (PM), von diabetischen Makulaödemen (DME) und von retinalen Venenverschlüssen, die zu Visusverlust führen (RVO), beitragen.
Klinische Wirksamkeit
Behandlung der feuchten AMD
Die klinische Wirksamkeit und Sicherheit von Ranibizumab zur Behandlung der feuchten AMD wurde in drei randomisierten, doppelt-maskierten Studien bei insgesamt 1'323 Patienten (Ranibizumab: N=879, Kontrollgruppen N=444) mit neovaskulärer AMD untersucht. Als Kontrollarm diente in der Studie MARINA Scheinbehandlungen und in der Studie ANCHOR eine aktive Kontrolle mittels PDT mit Visudyne. Eingeschlossen wurden Patienten mit Läsionen in der Grösse von bis zu 12 Papillenflächen und einer Sehschärfe von 20/40 bis 20/320 (nach Snellen) im Studienauge. Das Durchschnittsalter der Patienten lag bei 77 Jahren. In den klinischen Studien wurden die Patienten angewiesen, sich selbstständig antimikrobielle Augentropfen zu applizieren (viermal täglich, jeweils 3 Tage vor und nach jeder Injektion).
In die 24monatige Studie MARINA wurden 716 Patienten mit minimaler klassischer CNV oder okkulter CNV eingeschlossen. Sie erhielten monatliche intravitreale Injektionen von Ranibizumab 0.3 mg (N=238), Ranibizumab 0.5 mg (N=240) oder Scheininjektionen (N=238). Während der 24-monatigen Behandlungsphase erhielten die Patienten durchschnittlich 22 von 24 möglichen Behandlungen. Die Resultate der MARINA Studie nach 12 Monaten Behandlung wurden bei 90% der Patienten auch nach 24 Monaten Behandlung (1x/Monat) im Wesentlichen bestätigt.
In die 24monatige Studie ANCHOR wurden 423 Patienten mit vorwiegend klassischen CNV-Läsionen eingeschlossen. Sie erhielten monatliche intravitreale Injektionen von Ranibizumab 0.3 mg und Schein-PDT (N=143), intravitreale Injektionen von Ranibizumab 0.5 mg und Schein-PDT (N=140) oder intravitreale Scheininjektionen und aktive Verteporfin-PDT (N=143). Die erste Schein- bzw. aktive Verteporfin-PDT wurde gemeinsam mit der anfänglichen Ranibizumab-Injektion appliziert. Danach erfolgte die Behandlung im Abstand von 3 Monaten, wenn am Studienauge weiterhin oder erneut Flüssigkeit aus den Gefässen austrat (Fluoreszein-Angiographie).
Die Resultate sind in den nachfolgenden Tabellen zusammengefasst:
Tabelle 0-1 Studie MARINA: Resultate nach 12 und 24 Monaten
|
Veränderung der Sehschärfe (in Buchstaben, ETDRS)
|
Monat
|
Scheinbehandlung
(n=238)
|
Ranibizumab 0.5 mg
(n=240)
|
Differenz
(95% CI)a
| |
Sehschärfe-Verlust <15 Buchstaben (%)b
|
Monat 12
|
62%
|
95%
|
32%
(26%, 39%)
| |
Monat 24
|
53%
|
90%
|
37%
(29%, 44%)
| |
Sehschärfe-Gewinn ≥15 Buchstaben (%)b
|
Monat 12
|
5%
|
34%
|
29%
(22%, 35%)
| |
Monat 24
|
4%
|
33%
|
29%
(23%, 35%)
| |
Durchschnittliche Veränderung der Sehschärfeb(Buchstaben)
|
Monat 12
|
-10.5 (16.6)
|
+7.2 (14.4)
|
17.5
(14.8, 20.2)
| |
Monat 24
|
-14.9 (18.7)
|
+6.6 (16.5)
|
21.1
(18.1, 24.2)
| |
a
nach Stratifizierung
b p <0.01
|
Tabelle 0-2 Studie ANCHOR: Resultate nach 12 und 24 Monaten
|
Veränderung der Sehschärfe
|
|
Verteporfin PDT
(n=143)
|
Ranibizumab 0.5 mg
(n=140)
|
Differenz
(95% CI) a
| |
Sehschärfe-Verlust <15 Buchstaben (%)b
|
Monat 12
|
64%
|
96%
|
33%
(25%, 41%)
| |
Monat 24
|
66%
|
90%
|
25%
(16%, 34%)
| |
Sehschärfe-Gewinn ≥15 Buchstaben (%)b
|
Monat 12
|
6%
|
40%
|
35%
(26%, 44%)
| |
Monat 24
|
6%
|
41%
|
35%
(26%, 44%)
| |
Durchschnittliche Veränderung der Sehschärfeb (Buchstaben)
|
Monat 12
|
-9.5 (16.4)
|
+11.3 (14.6)
|
21.1
(17.5, 24.6)
| |
Monat 24
|
-9.8 (16.4)
|
+10.7 (16.5)
|
20.7
(16.8, 24.7)
| |
a
nach Stratifizierung
b p <0.01
|
In den beiden Studien MARINA und ANCHOR resultierte die unter der Behandlung mit 0.5 mg Ranibizumab nach Monat 12 beobachtete Verbesserung des Visus in einem Nutzen für den Patienten, gemessen anhand der drei Subskalen des National Eye Institute Visual Function Questionnaire (VFQ-25), die zuvor als sekundäre Endpunkte für die Wirksamkeit festgelegt worden waren (Tätigkeiten mit Bezug auf Nah- und Weitsehen sowie weitere, vom Sehen abhängige Tätigkeiten). Alle Unterschiede zwischen Ranibizumab 0.5 mg und den zwei Kontrollgruppen waren statistisch signifikant und klinisch relevant, mit p-Werten zwischen 0.009 bis <0.0001.
In die Studie PIER wurden 184 Patienten mit CNV-Läsionen (mit und ohne klassische Anteile) eingeschlossen. Sie erhielten während der ersten 3 Monate je eine monatliche intravitreale Injektion von Ranibizumab 0.3 mg bzw. von Ranibizumab 0.5 mg oder intravitreale Scheininjektion. Weitere Injektionen von Ranibizumab erfolgten im Abstand von 3 Monaten. Nach Monat 14 konnten diejenigen Patienten, welche eine Scheininjektion erhielten, ebenfalls mit Ranibizumab behandelt werden und ab dem Monat 19 waren häufigere Injektionen von Ranibizumab möglich. Die mit Ranibizumab behandelten Patienten in PIER erhielten durchschnittlich 10 Behandlungen innert 24 Monaten.
Der primäre Wirksamkeits-Endpunkt war die mittlere Veränderung der Sehschärfe während der 12 Monate. Nach einem anfänglichen Anstieg in der Zeit der monatlichen Injektionen, verloren die Patienten in der Phase der 3-monatlichen Injektionen an Sehschärfe, die nach 12 Monaten zum Ausgangspunkt zurückkehrte und dieser Effekt wurde in den meisten mit Ranibizumab behandelnden Patienten (82%) bei Monat 24 aufrechterhalten. Die Daten von einer begrenzten Anzahl derjenigen Patienten, welche nach über einem Jahr Scheinbehandlung zu einer Behandlung mit Ranibizumab gewechselt haben, deuten darauf hin, dass ein früher Therapiebeginn mit besserer Erhaltung der Sehschärfe assoziiert ist.
Tabelle 0-3 Studie PIER: Resultate nach 12 Monaten
|
Veränderung der Sehschärfe
|
Scheinbehandlung (n=63)
|
Ranibizumab 0.5 mg
(n=61)
|
Differenz
(95% CI)a
| |
Sehschärfe-Verlust <15 Buchstaben (%)b
|
49
|
90
|
37
(23, 52)
| |
Sehschärfe-Gewinn ≥15 Buchstaben (%)b
|
10
|
13
|
2
(-8, 12)
| |
Durchschnittliche Veränderung der Sehschärfeb
|
-16.3 (22.3)
|
-0.2 (13.1)
|
14.7
(8.2, 21.2)
| |
a
nach Stratifizierung
b p <0.0001
|
Die Phase-IIIb Studie SAILOR wurde bei Behandlungsnaïven, wie auch vorbehandelten Patienten mit CNV infolge von AMD durchgeführt. SAILOR war eine 1-Jahres Multizentrische Studie. Das primäre Ziel der Studie war die Abschätzung der Inzidenz von okulären und nicht-okulären unerwünschten Wirkungen während der 12 Behandlungsmonate. 2378 Patienten wurden in 2 Gruppen randomisiert und erhielten während 3 Monaten 0.3 mg bzw. 0.5 mg Ranibizumab jeden Monat appliziert; anschliessend wurde je nach Befund weiterbehandelt, mit Abständen von mindestens 1 Monat.
Insgesamt bestand kein Ungleichgewicht zwischen den beiden Gruppen in Bezug auf okuläre und nicht-okuläre unerwünschte Wirkungen. Es gab insbesondere auch kein Ungleichgewicht zwischen den beiden Gruppen in Bezug auf die Zahl der Schlaganfälle. Unter 0.3 mg waren es 8/1'169 Patienten (0.7%, 95%CI: 0.3% bis 1.3%). Unter 0.5 mg waren es 15/1'209 Patienten (1.2%, 95% CI: 0.7% bis 2%). Patienten mit bekannten Risiko-Faktoren wie z.B. ein vorangegangener Schlaganfall oder eine vorangegangene transiente ischämische Attacke, haben vermutlich ein erhöhtes Risiko für einen Schlaganfall während der Behandlung mit Ranibizumab.
Behandlung des Visusverlustes durch DME
Die klinische Wirksamkeit und Sicherheit von Ranibizumab bei Patienten mit Visusverlust durch ein Diabetisches Makulaödem (DME) wurden in der Studie RESTORE bei insgesamt 345 Patienten mit Visusverlust durch DME untersucht. Die Studie hatte 3 Arme: In Arm 1 wurde Patienten (N=116) initial Ranibizumab 0.5 mg intravitreal als Monotherapie injiziert und eine Schein-Laserphotokoagulation durchgeführt. In Arm 2 (N=118) wurde initial Ranibizumab 0.5 mg intravitreal injiziert und eine Laserphotokoagulation durchgeführt. In Arm 3 (N=111) wurde eine initiale Monotherapie mit Laserphotokoagulation und eine Scheininjektion durchgeführt.
Die Behandlung mit Ranibizumab wurde mit monatlichen intravitrealen Injektionen fortgesetzt und unterbrochen, wenn sich der Visus des Patienten unter Ranibizumab an drei aufeinanderfolgenden Untersuchungsterminen stabilisiert hatte. Nach einer Unterbrechung wurde die Behandlung wieder aufgenommen, wenn sich der Visus des Patienten durch Fortschreiten des DME verschlechtert hatte. Wiederbehandlungen mit Laserphotokoagulation wurden am gleichen Tag, mindestens 30 Minuten vor der Ranibizumab Injektion, gemäss der ETDRS-Kriterien durchgeführt.
Die Resultate sind in den folgenden Tabellen zusammengefasst:
Tabelle 0-4 Studie RESTORE: Resultate nach 12 Monaten
|
Veränderung der bestmöglich korrigierten Sehschärfe
|
Ranibizumab
0.5 mg
(n=115)
|
Ranibizumab
0.5 mg + Laser
(n=118)
|
Laser
(n=110)
| |
Durchschnittliche Veränderung des best korrigierten Visus vom 1. zum 12. Monat in Buchstaben, verglichen mit dem Visus zu Beginn der Studie (Standardabweichung)a
|
6.1 (6.4)
|
5.9 (7.9)
|
0.8 (8.6)
| |
Durchschnittliche Veränderung des best korrigierten Visus im 12. Monat in Buchstaben, verglichen mit dem Visus zu Beginn der Studie (Standardabweichung)
|
6.8 (8.3)a
|
6.4 (11.8)b
|
0.9 (11.4)
| |
Zunahme des best korrigierten Visus ≥10 Buchstaben (% der Patienten)
|
37.4c
|
43.2
|
15.5
| |
Zunahme des best korrigierten Visus ≥15 Buchstaben (% der Patienten)
|
22.6d
|
22.9e
|
8.2
| |
a
p <0.0001, b p=0.0004, c p=0.0001, d p=0.0032, e p=0.0021
|
Bei der RESTORE-Erweiterungsstudie handelte es sich um eine offene, multizentrische, 24-monatige Erweiterungsstudie. 240 Patienten, die die 12-monatige Kernstudie abgeschlossen hatten, traten in die Erweiterungsstudie ein und wurden nach Bedarf (PRN: pro re nata) im selben Auge, das in der Kernstudie als Studienauge definiert worden war, mit 0.5 mg Ranibizumab behandelt. Die Behandlung wurde nach einem Abfall der BCVA aufgrund von DME monatlich bis zum Erreichen einer stabilen BCVA verabreicht. Ausserdem erfolgte eine Laserbehandlung gemäss den ETDRS-Leitlinien, falls dies vom Prüfarzt als notwendig erachtet wurde.
Die durchschnittliche Anzahl von Ranibizumab-Injektionen bei Patienten, die in der Kernstudie mit Ranibizumab behandelt worden waren, betrug in der 24-monatigen Erweiterungsphase 6.4. Von den 74 Patienten aus dem Laserbehandlungsarm der Kernstudie erhielten 59 (79%) Patienten zu irgendeinem Zeitpunkt während der Erweiterungsphase Ranibizumab. Die durchschnittliche Anzahl von Ranibizumab-Injektionen während der 24-monatigen Erweiterungsphase betrug bei diesen 59 Patienten 8.1 Injektionen. Die Anteile der Patienten, die während der Erweiterungsphase keine Behandlung mit Ranibizumab benötigten, betrugen in den Gruppen, die zuvor eine Behandlung mit Ranibizumab, Ranibizumab + Laser bzw. Laser allein erhalten hatten, 19%, 25% bzw. 20%.
Die Ergebnisse sind in der folgenden Tabelle zusammengefasst:
Tabelle 0-5 Resultate nach 36 Monaten in der RESTORE-Erweiterungsstudie
|
Ergebnisparameter gegenüber Baseline in der Kernstudie
|
Vorher Ranibizumab
0.5 mg
n = 83
|
Vorher Ranibizumab
0.5 mg + Laser
n = 83
|
Vorher Laser
n = 74*
| |
Mittlere Veränderung der BCVA gegenüber Baseline in der Kernstudie nach 36 Monaten (SD)
|
8.0 (10.09)
|
6.7 ( 9.59)
|
6.0 ( 9.35)
| |
Gewinn von ≥10 Buchstaben gegenüber Baseline in der Kernstudie bzw. BCVA ≥84 (%) nach 36 Monaten
|
39 (47.0)
|
37 (44.6)
|
31 (41.9)
| |
Gewinn von ≥15 Buchstaben gegenüber Baseline in der Kernstudie bzw. BCVA ≥84 (%) nach 36 Monaten
|
23 (27.7)
|
25 (30.1)
|
16 (21.6)
| |
n = Anzahl der Patienten, für die Werte sowohl bei Baseline (Kernstudie, Monat 0) als auch beim Besuch in Monat 36 verfügbar waren.
* 59 (79%) der 74 Patienten mit vorheriger Laserbehandlung erhielten in der Erweiterungsstudie Ranibizumab.
|
Das in dieser 24-monatigen Erweiterungsstudie beobachtete Langzeitsicherheitsprofil von Ranibizumab deckt sich mit dem bekannten Sicherheitsprofil von Ranibizumab.
In der Phase-IIIb-Studie RETAIN wurden 372 Patienten mit Visusverlust durch DME randomisiert folgenden intravitrealen Injektionen zugeteilt:
·Ranibizumab 0.5 mg mit gleichzeitiger Laserphotokoagulation im Rahmen eines «Treat and Extend»(TE)-Schemas (n=121),
·Ranibizumab 0.5 mg als Monotherapie im Rahmen eines TE-Schemas (n = 128) oder
·Ranibizumab 0.5 mg als Monotherapie im Rahmen eines PRN-Schemas (n = 123).
·In allen Gruppen wurde die Ranibizumab-Behandlung mit monatlichen intravitrealen Injektionen eingeleitet und bis zur Erreichung einer stabilen BCVA bei mindestens drei aufeinanderfolgenden monatlichen Untersuchungen fortgeführt. Die Laserphotokoagulation erfolgte bei Baseline am selben Tag wie die erste Ranibizumab-Injektion und anschliessend nach Bedarf gemäss den ETDRS-Kriterien. Im Rahmen des TE-Schemas wurde Ranibizumab im weiteren Verlauf in Abständen von 2- oder maximal 3 Monaten verabreicht. Beim PRN-Schema wurde die BCVA monatlich beurteilt, und Ranibizumab wurde dann, falls erforderlich, beim selben Besuch verabreicht. In allen Gruppen wurde die monatliche Behandlung nach einem Abfall der BCVA aufgrund einer DME-Progression wieder aufgenommen und bis zum erneuten Erreichen einer stabilen BCVA fortgeführt. Die Studiendauer betrug 24 Monate.
·In der RETAIN-Studie betrug die Zahl der Injektionen im Durchschnitt (Median) 12.4 (12.0) bei der TE-Ranibizumab + Laser-Behandlungsgruppe, 12.8 (12.0) bei der TE-Ranibizumab-Monotherapie- Behandlungsgruppe und 10.7 (10.0) bei der PRN-Ranibizumab-Behandlungsgruppe. Die Anzahl der benötigten, planmässigen Behandlungstermine war nach 3 initialen monatlichen Behandlungsterminen 13 unter dem TE-Schema, verglichen mit 20 monatlichen Terminen, die unter dem PRN-Schema benötigt wurden. Unter beiden Behandlungsschemata hielten mehr als 70% der Patienten ihre BCVA bei einer Visitenfrequenz von ≥2 Monaten aufrecht. Zusätzliche Laserbehandlungen waren unter dem entsprechenden TE-Schema nicht mit einer geringeren durchschnittlichen Zahl von Ranibizumab-Injektionen assoziiert.
Die Ergebnisse sind in der folgenden Tabelle zusammengefasst:
Tabelle 0-6 Resultate in der RETAIN-Studie
|
Ergebnisparameter gegenüber Baseline
|
TE Ranibizumab
0.5 mg + Laser
n = 117
|
TE Ranibizumab
0.5 mg allein
n = 125
|
PRN Ranibizumab
0.5 mg
n = 117
| |
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 12 (SD)
|
5.9 (5.5)b
|
6.1 (5.7)b
|
6.2 (6.0)
| |
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 24 (SD)
|
6.8 (6.0)
|
6.6 (7.1)
|
7.0 (6.4)
| |
Mittlere Veränderung der BCVA nach 24 Monaten (SD)
|
8.3 (8.1)
|
6.5 (10.9)
|
8.1 (8.5)
| |
Gewinn von ≥10 Buchstaben bzw. BCVA ≥84 (%) nach 24 Monaten
|
43.6
|
40.8
|
45.3
| |
Gewinn von ≥15 Buchstaben bzw. BCVA ≥84 (%) nach 24 Monaten
|
25.6
|
28.0
|
30.8
| |
Verlust von ≥10 Buchstaben nach 24 Monaten
|
2.6
|
7.2
|
3.4
| |
Verlust von ≥15 Buchstaben nach 24 Monaten
|
0.9
|
4.0
|
2.6
| |
b
p <0.0001
|
In den DME-Studien ging die BCVA-Verbesserung in allen Behandlungsgruppen mit einer allmählichen Verringerung der mittleren CRT einher.
Die Behandlung der mässig schweren bis schweren nicht-proliferativen diabetischen Retinopathie (NPDR) bzw. proliferativen diabetischen Retinopathie (PDR)
Die klinische Sicherheit und Wirksamkeit von Ranibizumab bei Patienten mit proliferativer diabetischer Retinopathie (PDR) wurden in eine multizentrische, randomisierte, aktiv kontrollierte Nichtunterlegenheitsstudie der Phase III im Parallel-Design (Protocol S), an der 305 Patienten (394 Studienaugen) mit PDR mit oder ohne DME (diabetisches Makulaödem) bei Baseline teilnahmen und in der 0.5 mg intravitreal injiziertes Ranibizumab mit der Standardbehandlung mit panretinale Photokoagulation (PRP) verglichen wurde, beurteilt. Insgesamt wurden 191 Augen (48.5%) randomisiert und der Behandlung mit 0.5 mg Ranibizumab und 203 Augen (51.5%) wurden randomisiert und der Behandlung mit PRP zugewiesen. Insgesamt 88 Augen (22.3%) wiesen bei Baseline ein DME auf: 42 (22.0%) und 46 (22.7%) der Augen in der Ranibizumab- bzw. der PRP-Gruppe. Insgesamt 306 Augen (77.7%) wiesen bei Baseline kein DME auf: 149 (78.0%) und 157 (77.3%) der Augen in der Ranibizumab- bzw. der PRP-Gruppe.
Nach 2 Jahren Behandlung hatte sich der BCVA (best corrected visual acuity) Score in der Ranibizumab-Gruppe von der Baseline um +2.7 Buchstaben und in der PRP Gruppe von der Baseline um -0.7 Buchstaben verändert. Die Differenz von 3.5 Buchstaben lag innerhalb der non-inferiority margin, sodass die Non-Inferiorität von Ranibizumab versus PRP bestätigt wurde.
Die Änderung des Schweregrads der diabetischen Retinopathie wurde anhand von Fundusfotos unter Verwendung des Schweregrad-Scores für diabetische Retinopathie (DRSS) aus der Studie zur frühzeitigen Behandlung der diabetischen Retinopathie (ETDRS) beurteilt. In dieser Studie war bei 41.8% der Augen unter der Behandlung mit Ranibizumab (n=189) in Monat 12 eine Verbesserung beim DRSS um mindestens 2 Stufen eingetreten, verglichen mit 14.6% bei den mit PRP behandelten Augen (n=199).
In einer Metaanalyse von 3 randomisierten, doppelblinden, aktiv kontrollierten Phase-III-Studien [D2301 (RESTORE), D2303 (REVEAL) und D2305 (REFINE)], durchgeführt in insgesamt 875 Patienten mit DME, zeigten 48,4% der 315 mit Ranibizumab behandelten Patienten mit mittelschwerer bis schwerer NPDR oder PDR (n = 192) eine Verbesserung der DRSS um mindestens 2 Stufen im 12. Monat, verglichen mit 14,6% der Laserbehandelte Patienten (n = 123).
Behandlung des Visusverlustes durch ein Makulaödem infolge RVO
Die klinische Sicherheit und Wirksamkeit von Ranibizumab bei Patienten mit Visusverlust durch ein Makulaödem infolge eines retinalen Venenverschlusses (RVO) wurden in zwei randomisierten, doppelt-maskierten, kontrollierten Studien BRAVO (N=397) und CRUISE (N=392) untersucht. In beiden Studien erhielten die Patienten entweder 0.3 mg Ranibizumab oder 0.5 mg Ranibizumab oder eine Scheinbehandlung. In BRAVO, war eine Laserphotokoagulation als Rettungsbehandlung zu jedem Zeitpunkt während der Studie ab dem 3. Monat in allen Studienarmen erlaubt. Die Resultate aus BRAVO und CRUISE sind in folgenden Tabellen zusammengestellt.
Tabelle 0-7 Studie BRAVO: Resultate bei Monat 6 und 12
|
Veränderung der Sehschärfe
|
Scheinbehandlung
(n=132)
|
Ranibizumab 0.5 mg
(n=131)
| |
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 6 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studiea
|
+7.3
|
+18.3
| |
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 12 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studie
|
+12.1
|
+18.3
| |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 6
|
28.8%
|
61.1%
| |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 12
|
43.9%
|
60.3%
| |
Anteil der Patienten mit Laserrescue über 12 Monaten
|
61.4%
|
34.4%
| |
a
p<0.0001
|
Tabelle 0-8 Studie CRUISE: Resultate bei Monat 6 und 12
|
Veränderung der Sehschärfe
|
Scheinbehandlung
(n=132)
|
Ranibizumab 0.5 mg
(n=131)
| |
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 6 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studiea
|
+0.8
|
+14.9
| |
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 12 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studie
|
+7.3
|
+13.9
| |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 6
|
16.9%
|
47.7%
| |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 12
|
33.1%
|
50.8%
| |
a
p<0.0001
|
Mit Ranibizumab behandelte Patienten zeigten in beiden Studien (BRAVO und CRUISE) eine kontinuierliche Verringerung der zentralen Netzhautdicke.
Neben der Visusverbesserung nach Behandlung mit Ranibizumab bei Monat 6 und 12 wurde auch die Lebensqualität der Patienten anhand des National Eye Institute Visual Function Questionnaire (VFQ-25) erhoben. Die Unterschiede zwischen der Ranibizumab 0.5 mg Gruppe und der Kontrollgruppe wurden bei Monat 6 mit p-Werten zwischen 0.02 und 0.0002 ermittelt.
Die Resultate der HORIZON Verlängerungsstudie zu BRAVO und CRUISE zeigten nach 12 Monaten folgendes:
Die reduzierte Behandlungsfrequenz in der Studie HORIZON hatte wenig Auswirkung auf BRVO-Patienten, welche ihre initiale Visusverbesserung, wie sie in der Studie BRAVO beobachtet wurde (+17.5 Buchstaben nach 24 Monaten mit einer Dosis von 0.5 mg und durchschnittlich 2.4 Injektionen im zweiten Jahr) behielten.
Hingegen zeigte sich die reduzierte Behandlungsfrequenz bei CRVO-Patienten mit einer Verringerung der in der Studie CRUISE gewonnenen Sehschärfe (+12 Buchstaben nach 24 Monaten mit einer Dosis von 0.5 mg und durchschnittlich 3.8 Injektionen im zweiten Jahr).
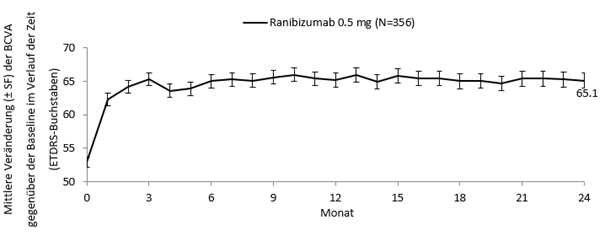
Studie E2401 (CRYSTAL) und Studie E2402 (BRIGHTER)
In den Studien BRIGHTER und CRYSTAL wurden die klinische Sicherheit und Wirksamkeit von Ranibizumab über 24 Monate bei Patienten mit Sehstörungen wegen eines RVO-bedingten Makulaödems bewertet. In die Studien wurden Patienten mit BRVO (n = 455) bzw. CRVO (n = 357) rekrutiert. In beiden Studien erhielten die Patienten 0.5 mg Ranibizumab nach Bedarf. BRIGHTER war eine 3-armige, randomisierte, aktiv kontrollierte Studie, in der 0.5 mg Ranibizumab als Monotherapie mit Ranibizumab in Kombination mit Laserphotokoagulation und mit Laserphotokoagulation alleine verglichen wurden. Nach 6 Monaten konnten die Teilnehmer im Arm mit der Laser-Monotherapie 0.5 mg Ranibizumab erhalten. CRYSTAL war eine einarmige Studie mit einer Monotherapie mit 0.5 mg Ranibizumab.
Die wichtigsten funktionellen und anatomischen Ergebnisse der BRIGHTER- und der CRYSTAL-Studie sind in Tabelle 0-9 und in den Abbildungen 1-0 und 2-0 dargestellt.
Tabelle 0-9 Ergebnisse in Monat 6 (BRIGHTER) und Monat 24 (BRIGHTER und CRYSTAL)
|
|
BRIGHTER
|
CRYSTAL
| |
|
Ranibizumab 0.5 mg
n = 180
|
Ranibizumab 0.5 mg
+ Laser
n = 178
|
Laser*
n = 90
|
Ranibizumab 0.5 mg
(n = 356)
| |
Mittlere Veränderung der BCVA in Monat 6b(Buchstaben) (SD)
|
+14.8
(10.7)
|
+14.8
(11.13)
|
+6.0
(14.27)
|
+12.0
(13.95)
| |
Mittlere Veränderung der BCVA in Monat 24b(Buchstaben) (SD)
|
+15.5
(13.91)
|
+17.3
(12.61)
|
+11.6
(16.09)
|
+12.1
(18.60)
| |
Anteil der Patienten, die bei der BCVA im Monat 24 ≥15 Buchstaben erreichten
|
52.8%
|
59.6%
|
43.3%
|
49.2%
| |
Mittlere Anzahl an Injektionen (SD) (Monate 0-23)
|
11.4
(5.81)
|
11.3
(6.02)
|
N/A
|
13.1
(6.39)
| |
*
Ab Monat 6 war eine Behandlung mit 0.5 mg Ranibizumab zulässig (24 Patienten wurden nur mit Laser behandelt).
b p <0.0001 für beide Vergleiche in der BRIGHTER-Studie in Monat 6: Ranibizumab 0.5 mg gegenüber Laser und Ranibizumab 0.5 mg + Laser gegenüber Laser.
|
Abbildung 1-0 BRIGHTER: Mittlere Veränderung der BCVA gegenüber der Baseline über einen Zeitraum von 24 Monaten
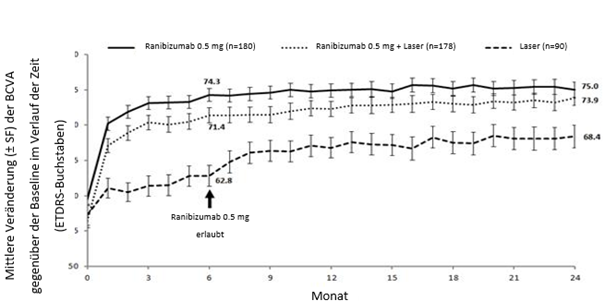
Abbildung 2-0 CRYSTAL: Mittlere Veränderung der BCVA gegenüber der Baseline über einen Zeitraum von 24 Monaten
Die Verbesserung des Visus fiel bei Patienten mit oder ohne Netzhautischämie ähnlich aus: In der BRIGHTER-Studie wiesen Patienten mit Netzhautischämie (n = 87) oder ohne Netzhautischämie (n = 35), die mit einer Ranibizumab-Monotherapie behandelt wurden, in Monat 24 eine mittlere Veränderung gegenüber der Baseline von +15.4 bzw. +12.9 Buchstaben auf. In der CRYSTAL-Studie wiesen Patienten mit Netzhautischämie (n = 107) oder ohne Netzhautischämie (n = 109) eine mittlere Veränderung gegenüber der Baseline von +11.1 bzw. +12.9 Buchstaben auf.
Bei Patienten mit einer Krankheitsdauer von <3 Monaten war in der BRIGHTER- bzw. CRYSTAL-Studie in Monat 1 eine Verbesserung der Sehschärfe von 13.3 bzw. 10.0 Buchstaben und in Monat 24 von 17.7 bzw. 13.2 Buchstaben zu beobachten. Bei einer Krankheitsdauer von ≥12 Monaten waren es in Monat 1 4.8 bzw. 7.9 Buchstaben, in Monat 24 waren es 8.4 bzw. 8.6 Buchstaben. Eine Initiierung der Behandlung bei Diagnosestellung ist zu erwägen.
Das Sicherheitsprofil von Ranibizumab, das in diesen 24-monatigen Studien beobachtet wurde, entspricht dem aus früheren Studien bekannten Sicherheitsprofil von Ranibizumab.
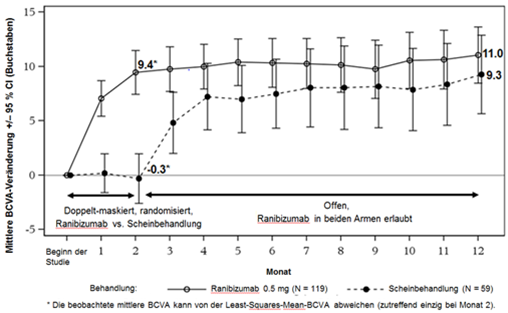
Behandlung eines Visusverlustes durch CNV – Studie G2301 (MINERVA)
Die klinische Sicherheit und Wirksamkeit von Ranibizumab bei Patienten mit Visusverlust durch CNV infolge von anderen Ätiologien als neovaskulärer AMD und PM wurden auf Grundlage der 12-Monats-Daten der randomisierten, doppelt-maskierten, Scheinbehandlungs-kontrollierten Studie G2301 (MINERVA) beurteilt. Für die Analyse wurden fünf Untergruppen nach Ätiologie vordefiniert (angioide Streifen, postentzündliche Retinochoroidopathie, zentrale seröse Chorioretinopathie, idiopathische Chorioretinopathie und andere Ätiologien). 178 Patienten wurden im Verhältnis 2:1 in einen der folgenden Arme randomisiert:
Ranibizumab 0.5 mg zu Beginn der Studie, gefolgt von einem individuellen Dosierungsschema je nach Krankheitsaktivität.
Scheininjektion zu Beginn der Studie, gefolgt von einem individuellen Dosierungsschema je nach Krankheitsaktivität.
Ab Monat 2 erhielten alle Patienten eine Behandlung mit Ranibizumab nach Bedarf. Primärendpunkt war die Veränderung der besten korrigierten Sehschärfe (BCVA) vom Beginn der Studie bis Monat 2.
Die wichtigsten Ergebnisse von MINERVA sind in Tabellen 0-10 und 0-11 sowie in Abbildung 3-0 zusammengestellt.
Tabelle 0-10 Resultate nach 2 Monaten (MINERVA)
|
|
Ranibizumab 0.5 mg
(n = 119)
|
Scheinbehandlung
(n = 59)
| |
Mittlere BCVA-Veränderung vom Beginn der Studie bis Monat 2 (Buchstaben) (Least-Squares-Mittelwert) a
|
+9.5
|
-0.4
| |
Anteil Patienten mit einem Gewinn von ≥15 Buchstaben seit Beginn der Studie oder einem Wert von 84 Buchstaben in Monat 2
|
31.4%
|
12.3%
| |
Anteil Patienten, die vom Beginn der Studie bis Monat 2 nicht mehr als 15 Buchstaben verloren
|
99.2%
|
94.7%
| |
Verringerung der mittleren Foveadicke vom Beginn der Studie bis Monat 2 (Least-Squares-Mittelwert) a
|
77 µm
|
-9.8 µm
| |
a
Einseitiger Vergleich (p <0.001) mit Scheinbehandlungskontrolle
|
Abbildung 3-0: Mittlere Veränderung der BCVA vom Beginn der Studie bis Monat 12 im Zeitverlauf (MINERVA)
Beim Vergleich von Ranibizumab mit den Scheininjektionen in Monat 2 wurde eine konsistente Wirkung beobachtet, dies sowohl in der gesamten Studienpopulation, als auch in den einzelnen, gemäss der Ätiologie definierten Untergruppen.
Tabelle 0-11 Gesamtwirkung der Behandlung und Wirkung der Behandlung in den zu Beginn der Studie gemäss der Ätiologie definierten Untergruppen für die primäre Variable in Monat 2 (MINERVA)
|
Insgesamt und gegliedert nach Ätiologie bei Studienbeginn
|
Behandlungswirkung im Vergleich zur Scheinbehandlung (Buchstaben)
|
Patientenzahlen
(n) (Behandlung + Scheinbehandlung)
| |
Gesamt
|
9.9
|
175*
| |
Angioide Streifen
|
14.6
|
27
| |
Postentzündliche Retinochoroidopathie
|
6.5
|
27
| |
Zentrale seröse Chorioretinopathie
|
5.0
|
23
| |
Idiopathische Chorioretinopathie
|
11.4
|
62
| |
Sonstige Ätiologiena
|
10.6
|
36
| |
a
CNV-Ätiologien, die nicht unter die anderen Untergruppen fallen
* Anzahl Patienten mit für die Analyse verfügbaren Daten
|
Die Verbesserung des Visus wurde von einer Verringerung der mittleren Foveadicke über einen Zeitraum von 12 Monaten begleitet.
Die mittlere Anzahl der Ranibizumab-Injektionen über einen Zeitraum von 12 Monaten in das Studienauge betrug im Ranibizumab-Arm 5.8 und in der Gruppe mit Scheininjektionen 5.4.
Kinder und Jugendliche
Fünf jugendliche Patienten im Alter von 12 bis 17 Jahren mit Visusverlust infolge CNV (1x subfoveale CNV bei einer Drusenpapille; je 1x juxtafoveale CNV und subfoveale CNV bei idiopathischer CNV; 2x subfoveale CNV bei Best disease) erhielten eine initiale Behandlung mit Ranibizumab 0.5 mg gefolgt von einem individualisierten Behandlungsschema auf Grundlage von Anzeichen der Krankheitsaktivität (z.B. Beeinträchtigung der Sehschärfe, intra-/subretinale Flüssigkeit, Blutungen oder Leckagen). Die BCVA-Veränderung vom Beginn der Studie bis Monat 12 verbesserte sich bei allen fünf Patienten und reichte von +5 bis +38 Buchstaben. Die Verbesserung des Visus wurde von einer Stabilisierung oder Verringerung der mittleren Foveadicke über einen Zeitraum von 12 Monaten begleitet (ΔCSFT0-12 Mte.: -286 μm bis +10 μm). Während der 12 Monate wurden 2 bis 5 Injektionen ins Studienauge verabreicht (s. «Dosierung/Anwendung»).
Behandlung eines Visusverlustes durch CNV infolge von PM
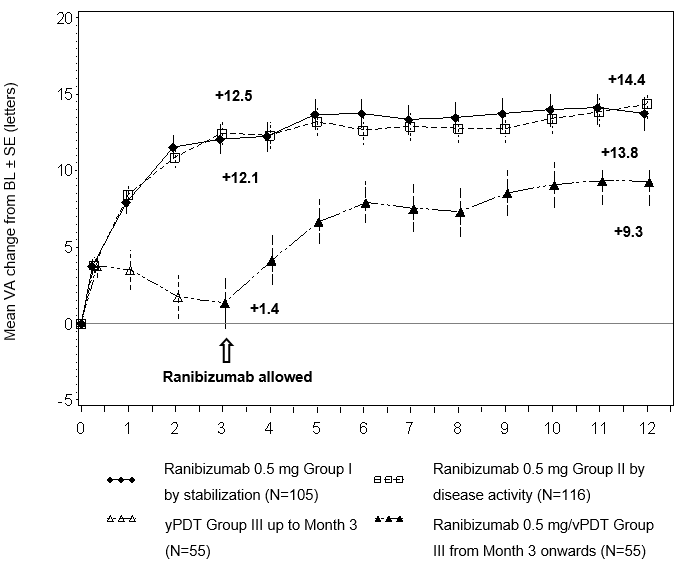
Die klinische Sicherheit und Wirksamkeit von Ranibizumab bei Patienten mit Visusverlust durch CNV infolge von PM wurden ausgehend von 12-Monats-Daten aus der randomisierten, doppelt maskierten, kontrollierten Pivotstudie RADIANCE beurteilt. Diese Studie war dazu ausgelegt, zwei verschiedene Dosierungsschemata von 0.5 mg Ranibizumab per intravitrealer Injektion im Vergleich zu Verteporfin PDT (vPDT, photodynamische Therapie mit Visudyne) zu evaluieren.
Die 277 Patienten wurden in einen der folgenden Arme randomisiert:
Gruppe I (0.5 mg Ranibizumab, Dosierungsplan beruhte auf Stabilitäts-Kriterien, definiert als keine Veränderung der bestkorrigierten Sehschärfe (BCVA) im Vergleich zu zwei vorhergehenden monatlichen Untersuchungen)
Gruppe II (0.5 mg Ranibizumab, Dosierungsplan beruhte auf Krankheitsaktivitäts-Kriterien, definiert als Visusverlust aufgrund von intra- oder subretinaler Flüssigkeit oder aktivem Flüssigkeitsaustritt infolge der CNV-Läsion mit Nachweis per OCT und/oder FA).
Gruppe III (vPDT - die Patienten konnten ab Monat 3 mit Ranibizumab behandelt werden)
Im Lauf der 12 Studienmonate erhielten die Patienten in Gruppe I durchschnittlich 4.6 Injektionen (zwischen 1 und 11 Injektionen) und in Gruppe II durchschnittlich 3.5 Injektionen (zwischen 1 und 12 Injektionen). In Gruppe II (in welcher Patienten die empfohlene Behandlung basierend auf Krankheitsaktivität erhielten, s. «Dosierung/Anwendung»), benötigten 50.9% der Patienten eine oder zwei Injektionen, 34.5% der Patienten drei bis fünf Injektionen und 14.7% der Patienten 6 bis 12 Injektionen innerhalb der 12-monatigen Studienperiode. 62.9% der Gruppe II-Patienten benötigten in den letzten sechs Monaten der Studie keine weiteren Injektionen.
Die wichtigsten Behandlungsergebnisse der Studie RADIANCE sind in Tabelle 0-12 und Abbildung 0-4 zusammengefasst.
Tabelle 0-12 Behandlungsergebnis bei Monat 3 und Monat 12 (RADIANCE)
|
|
Gruppe I
0.5 mg Ranibizumab
Stabilität der Sehschärfe
(n=105)
|
Gruppe II
0.5 mg Ranibizumab
Krankheitsaktivität
(n=116)
|
Gruppe III
vPDT*
(n=55)
| |
Monat 3
| |
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 3 gegenüber Baselinea(Buchstaben)
|
+10.5
|
+10.6
|
+2.2
| |
Anteil der Patienten mit Verbesserung der BCVA um
|
|
|
| |
≥10 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben
|
61.9%
|
65.5%
|
27.3%
| |
≥15 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben
|
38.1%
|
43.1%
|
14.5%
| |
Monat 12
| |
Anzahl der Injektionen bis Monat 12:
|
|
|
| |
Durchschnitt
|
4.6
|
3.5
|
N/A
| |
Medianwert
|
4.0
|
2.0
|
N/A
| |
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 12 gegenüber Baseline (Buchstaben)
|
+12.8
|
12.5
|
N/A
| |
Anteil der Patienten mit Verbesserung der BCVA um
|
|
|
| |
≥10 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben
|
69.5%
|
69.0%
|
N/A
| |
≥15 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben
|
53.3%
|
51.7
|
N/A
| |
* Vergleichende Kontrolle bis Monat 3. Die in die vPDT-Gruppe randomisierten Patienten konnten ab Monat 3 eine Behandlung mit Ranibizumab erhalten (in Gruppe III erhielten 38 Patienten Ranibizumab ab Monat 3): p<0.00001 für den Vergleich mit der vPDT-Kontrolle
|
Abbildung 0-4 Mittlere Veränderung der BCVA gegenüber Baseline BCVA im Zeitverlauf bis Monat 12 (RADIANCE)
BL = Baseline; SE = Standardfehler des Mittelwerts.
Abbildung:
Mittlere Verbesserung der Sehschärfe gegenüber BL ± SE (Buchstaben)
Gruppe I mit 0.5 mg Ranibizumab nach Stabilisierung (N=105)
Gruppe II mit 0.5 mg Ranibizumab nach Krankheitsaktivität (N=116)
Gruppe III mit Visudyne-PDT (N=55)
Die in die vPDT-Gruppe randomisierten Patienten konnten ab Monat 3 eine Behandlung mit Ranibizumab erhalten.
Die Visusverbesserung war von einer Reduzierung der zentralen Netzhautdicke begleitet.
In den Ranibizumab-Behandlungsarmen wurde gegenüber der vPDT-Gruppe (p-Wert <0.05) ein von den Patienten subjektiv angegebener Nutzen in Bezug auf die Verbesserung des kombinierten Ergebnisses wie auch des Ergebnisses in mehreren Subskalen (allgemeiner Visus, Nahsichtaktivitäten, psychische Verfassung und unabhängiger Funktionsstatus) des Fragebogens VFQ-25 festgestellt.
Behandlung der RPM bei Frühgeborenen: Studie H2301 (RAINBOW)
Die klinische Sicherheit und Wirksamkeit von Ranibizumab 0.1 mg bei der Behandlung der RPM bei Frühgeborenen wurden anhand der Sechsmonatsdaten aus der randomisierten, offenen, dreiarmigen Parallelgruppen-Überlegenheitsstudie H2301 (RAINBOW) untersucht, die darauf ausgelegt war, die Anwendung von Ranibizumab in Dosen von 0.1 mg und 0.2 mg, verabreicht als intravitreale Injektionen, im Vergleich zur Lasertherapie zu beurteilen. Geeignete Patienten mussten einen der folgenden Netzhautbefunde in beiden Augen aufweisen:
·Zone I, Krankheitsstadium 1+, 2+, 3 oder 3+ oder
·Zone II, Krankheitsstadium 3+ oder
·Aggressive posteriore RPM (AP-RPM)
Für diese Studie wurden 225 Patienten im Verhältnis 1:1:1 randomisiert, um Ranibizumab intravitreal 0.1 mg (n=77) bzw. 0.2 mg (n=74) oder eine Lasertherapie (n=74) zu erhalten.
Der Behandlungserfolg, gemessen anhand des Ausbleibens einer aktiven RPM und des Fehlens ungünstiger struktureller Auswirkungen in beiden Augen 24 Wochen nach der ersten Studienbehandlung, betrug 75 % in der Ranibizumab 0.1 mg-Gruppe und 66.2 % in der Lasertherapie-Gruppe. Die Mehrheit der mit Ranibizumab 0.1 mg behandelten Patienten (77.6 %) erhielt eine einzige Injektion pro Auge.
Aus der 0.1 mg Ranibizumab-Behandlungsgruppe wechselten weniger Patienten aufgrund fehlenden Ansprechens zu einem anderen Behandlungsmodus als in der Lasertherapie-Gruppe (16.9 % vs. 24.3 %). Ungünstige strukturelle Auswirkungen waren in der 0.1 mg Ranibizumab-Behandlungsgruppe (5 Patienten, 6.7 %) weniger häufig als in der Lasertherapie-Gruppe (7 Patienten, 10.1 %). Darüber hinaus erreichten 75 % der Patienten innerhalb von 8 Tagen ein Abklingen der Erkrankung unter Ranibizumab 0.1 mg im Vergleich zu 22.5 Tagen in der Lasertherapie-Gruppe.
Studie H2301E1 (RAINBOW Erweiterungsstudie)
Die Langzeitwirksamkeit und Sicherheit von Ranibizumab 0,1 mg zur Behandlung von RPM bei Frühgeborenen wurde in der Studie H2301E1 (RAINBOW-Erweiterungsstudie), einer Erweiterung der Studie H2301 (RAINBOW), an Patienten bis zum Erreichen des 5. Lebensjahrs untersucht.
Das Hauptziel bestand darin, die Beurteilung der Sehfunktion beim Termin der Patienten bei Erreichen ihres 5. Lebensjahres zu bewerten, indem die Sehschärfe anhand der Early Treatment Diabetic Retinopathy Study (ETDRS) mit Lea-Symbol-Optotypen am besser sehenden Auge (dem Auge mit dem höheren ETDRS-Score) beurteilt wurde.
Bei 78,2% (43/55) der Patienten in der Ranibizumab 0,1 mg-Gruppe und bei 76,6% (36/47) der Patienten in der Lasertherapie-Gruppe wurde beim Termin bei Erreichen des 5. Lebensjahres ein ETDRS-Score aufgezeichnet. Der Unterschied im Mittelwert (SE) der kleinsten Quadrate (LS) zwischen der Ranibizumab 0,1 mg-Gruppe und der Lasertherapie-Gruppe betrug 2,5 (95-%-KI: -3,4, 8,3). Die kategorisierten Ergebnisse der Sehschärfe des besseren Auges bei Erreichen des 5. Lebensjahres der Patienten sind in Tabelle 13 dargestellt. Insgesamt wurde das Fehlen aller okulären Strukturanomalien bis zum Erreichen des 5. Lebensjahrs bei einem höheren Anteil der Patienten in der Ranibizumab 0,1 mg-Gruppe [93,8% (61/65)] als in der Lasertherapie-Gruppe [88,7% (47/53)] beobachtet.
Tabelle 13 Auswirkungen auf die Sehschärfe im besser sehenden Auge¹ beim Termin bei Erreichen des 5. Lebensjahrs der Patienten
|
Kategorie der Sehschärfe
|
Ranibizumab 0.1 mg
N=65
n (%)
|
Laser
N=54
n (%)
| |
≥1 bis ≤34 Buchstaben
|
3 (4.6)
|
2 (3.7)
| |
≥35 bis ≤70 Buchstaben
|
25 (38.5)
|
23 (42.6)
| |
≥71 Buchstaben
|
15 (23.1)
|
11 (20.4)
| |
¹ Das besser sehende Auge ist das Auge mit dem höheren ETDRS-Buchstaben-Score bei Erreichen des 5.Lebensjahrs. Wenn beide Augen den gleichen ETDRS-Buchstaben-Score haben, wird das rechte Auge als das besser sehende Auge angegeben.
|
PharmakokinetikAbsorption
Die monatliche, intravitreale Verabreichung von Ranibizumab bei Patienten mit neovaskulärer AMD führt zu allgemein tiefen Serumkonzentrationen von Ranibizumab mit der maximalen Serumkonzentration (Cmax) deutlich unter der Konzentration, welche zu einer 50%igen Hemmung der VEGFs führte (11-27 ng/ml im Zellproliferations-Assay).
Distribution
Die maximale Serumkonzentration (Cmax) liegt im Allgemeinen in einem Bereich von 0.46 bis 1.76 ng/ml, die minimale Serumkonzentration (Cmin) im Bereich von 0.04 bis 0.29 ng/l. Die Cmax im Serum war über einen Dosisbereich von 0.05 bis 1.0 mg/Auge proportional zur verabreichten Dosierung. Die Serumkonzentration in Patienten mit DME und RVO war ähnlich der in Patienten mit neovaskulärer AMD.
Metabolismus
Nicht zutreffend.
Elimination
Basierend auf Ranibizumab Serumkonzentrationen liegt die durchschnittliche Eliminationshalbwertzeit von Ranibizumab im Glaskörper bei Patienten mit neovaskulärer AMD bei etwa 9 Tagen. Es wird angenommen, dass die Serumkonzentrationen von Ranibizumab 90'000-mal tiefer sind als im Glaskörper.
Leberfunktionsstörungen
Es liegen keine spezifischen Untersuchungen vor.
Nierenfunktionsstörungen
Es wurden keine prospektiven Studien zur Pharmakokinetik von Ranibizumab bei Patienten mit eingeschränkter Nierenfunktion durchgeführt. In einer Populationsanalyse pharmakokinetischer Daten bei Patienten mit neovaskulärer AMD wiesen 68% (N=136/200) der Patienten eine eingeschränkte Nierenfunktion auf (46.5% leicht, 20% mässig und 1.5% schwer). Bei den untersuchten Patienten mit RVO hatten 48.2% (N=253/525) eine eingeschränkte Nierenfunktion (36.4% leicht, 9.5% mittel und 2.3% schwer). Die beobachtete Reduktion der systemischen Clearance von Ranibizumab war statistisch nicht signifikant.
Pädiatrische Population (Frühgeborene mit RPM)
Nach intravitrealer Verabreichung von Ranibizumab 0.1 mg an jedem Auge bei Frühgeborenen mit einer RPM lag die Serumkonzentration von Ranibizumab höher als bei erwachsenen Patienten mit neovaskulärer AMD unter der Behandlung mit 0.5 mg in jedem Auge. Gemäss einer pharmakokinetischen Analyse der Population waren die Unterschiede bei der Cmax und der AUCinf ca. 8- bzw. 5-fach höher. Die scheinbare systemische Halbwertszeit betrug ca. 6 Tage. Bei dieser Analyse wurde kein Zusammenhang zwischen der systemischen Konzentration von Ranibizumab und derjenigen des VEGF festgestellt.
Präklinische DatenToxizität bei wiederholter Gabe
Die bilaterale intravitreale Verabreichung von Ranibizumab an Cynomolgus-Affen in Dosierungen von 0.25 mg/Auge bis zu 2.0 mg/Auge alle 2 Wochen während bis zu 26 Wochen ergab dosisabhängige okuläre Effekte.
In der Vorderkammer wurde ein dosisabhängiges Ansteigen von «Flare» und Zellen beobachtet, mit einem Maximum 2 Tage nach der Injektion. Die Intensität der Entzündungsreaktion verminderte sich üblicherweise bei folgenden Injektionen oder in der Erholungsphase, war aber nicht in allen Fällen vollständig reversibel. In der hinteren Kammer kam es zu reversiblen fokalen perivenösen retinalen Blutungen, die hauptsächlich nach der ersten Verabreichung auftraten, und zu vitrealen Zellinfiltrationen und „mouches volantes“, die tendenziell dosisabhängig waren und bis zum Behandlungsende persistierten. In der Studie über 26 Wochen nahm die Intensität der Entzündung mit der Anzahl Injektionen zu und der Versuch musste frühzeitig abgebrochen werden. Eine Reversibilität der Befunde zeichnete sich in der Erholungsphase ab. Eine Kataraktbildung wurde bei einigen Tieren nach einer relativ langen Phase starker Entzündung beobachtet, was darauf hindeutet, dass die Linsenveränderungen eher sekundär auf die schwere Entzündung folgten. Ein transienter Anstieg des Augeninnendruckes nach Verabreichung wurde unabhängig von der Dosis nach intravitrealer Injektion beobachtet.
Die mikroskopischen Veränderungen in den Augengeweben wurden alle mit der Entzündung in Zusammenhang gebracht und deuteten nicht auf degenerative Prozesse in irgendwelchen Augenstrukturen hin. In einigen Fällen wurden granulomatöse entzündliche Veränderungen am Sehnervenkopf beobachtet. Diese Veränderungen des posterioren Segmentes nahmen während der Erholungsphase ab und verschwanden in einigen Fällen ganz.
Anschliessend an die intravitreale Injektion wurden bei Serumspiegeln, die mehr als 100 fach höher waren als die die bei normaler therapeutischer Anwendung beim Menschen auftreten, keine Zeichen einer systemischen Toxizität beobachtet. Antikörper im Serum, bzw. im Glaskörper gegen Ranibizumab wurden nicht bei allen der behandelten Tieren gefunden.
Die präklinischen Unterlagen decken lediglich die systemische Exposition nach intravitrealer Anwendung ab.
Genotoxizität, Kanzerogenität und Immunotoxizität
Es liegen keine Daten zur Mutagenität, Karzinogenität und Immunotoxizität von Ranibizumab bei Tieren vor.
Reproduktionstoxizität
Bei der intravitrealen Behandlung von trächtigen Affenweibchen mit Ranizumab mit einer maximalen systematischen Exposition, entsprechend dem 0.9 bis 7-Fachen der schlimmstenfalls anzunehmenden klinischen Exposition, wurden keine embryo/fetotoxischen oder teratogenen Effekte gefunden, und es kam zu keiner Auswirkung auf Gewicht und Struktur der Plazenta; trotzdem sollte Ranibizumab aufgrund seines pharmakologischen Effekts als potenziell teratogen und embryo-/fetotoxisch angesehen werden.
Das Fehlen von Ranibizumab/vermittelten Effekten auf die embryo-fötale Entwicklung ist wohl hauptsächlich darauf zurückzuführen, dass das Fab-Fragment die Plazentaschranke nicht passieren kann. Dennoch wurde ein Fall mit hohen maternalen Ranibizumab-Serumspiegeln und der Nachweisbarkeit von Ranibizumab in fötalem Serum beschrieben; dies lässt darauf schliessen, dass der Anti-Ranibizumab-Antikörper als Trägerprotein (mit Fc-Region) für Ranibizumab fungierte, auf diese Weise die Clearance im Serum der Mutter verminderte und das Passieren der Plazentaschranke ermöglichte. Da die Untersuchungen zur embryo-fötalen Entwicklung an gesunden trächtigen Tieren durchgeführt wurden und die Durchgängigkeit der Plazenta für ein Fab-Fragment durch eine Erkrankung modifiziert werden könnte, sollten die Studienergebnisse mit Vorsicht beurteilt werden.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatilibitätsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit „EXP“ bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern.
Nicht einfrieren.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Die ungeöffnete Durchstechflasche kann vor Gebrauch bis zu 24 Stunden bei Raumtemperatur (25 °C) aufbewahrt werden.
Hinweise für die Handhabung
Die Durchstechflasche ist nur für den einmaligen Gebrauch bestimmt. Nach der Injektion müssen nicht verwendete Produktreste verworfen werden. Jede Durchstechflasche, die Anzeichen einer Beschädigung oder Manipulation aufweist, darf nicht benutzt werden. Die Sterilität kann nicht garantiert werden, sollte die Versiegelung der Packung eine Beschädigung aufweisen.
Zur Vorbereitung und intravitrealen Injektion werden die folgenden nicht in der Packung enthaltenen Einzelteile zum einmaligen Gebrauch benötigt:
eine 5µm-Filterkanüle (18 G)
für erwachsene Patienten: eine sterile 1ml-Spritze (mit einer 0,05ml-Markierung) und eine Injektionskanüle (30 G x ½")
Zur Vorbereitung von Ranivisio für die intravitreale Anwendung folgen Sie bitte der Gebrauchsanweisung am Ende der Fachinformation.
Zulassungsnummer69068 (Swissmedic)
Packungen1 Durchstechflasche (Typ-I-Glas) mit Gummistopfen (Chlorbutyl) zu 0,23 ml. [B]
ZulassungsinhaberinBioeq AG
6300 Zug
Auslieferung
Medvisis Switzerland AG
4614 Hägendorf
Stand der InformationApril 2024
Hinweise zur Vorbereitung und Anwendung
Um Ranivisio für die intravitreale Anwendung bei Erwachsenen und Frühgeborenen vorzubereiten, bitte die folgenden Instruktionen beachten:
1. Vor dem Aufziehen den Deckel der Durchstechflasche entfernen und das Septum der Durchstechflasche reinigen (z. B. mit einem 70%igen Alkoholtupfer).
2. Eine 5µm-Filterkanüle (18 G x 1½", 1,2 mm x 40 mm) wird unter sterilen Bedingungen auf eine 1ml-Spritze gesteckt. Die stumpfe Filterkanüle in das Zentrum des Gummistopfens drücken, bis die Nadel die Unterkante der Durchstechflache berührt.
3. Den gesamten Inhalt aus der Durchstechflasche aufziehen, dabei die Durchstechflasche in senkrechter, leicht geneigter Position halten, um die vollständige Entnahme zu erleichtern.
4. Während des Entleerungsvorgangs der Durchstechflasche den Spritzenstempel ausreichend anziehen, um die Filterkanüle vollständig zu entleeren.
5. Die stumpfe Filterkanüle von der Spritze abnehmen, dabei die stumpfe Filterkanüle in der Durchstechflasche belassen. Nach dem Aufziehen wird diese Filterkanüle verworfen, sie darf nicht zur intravitrealen Injektion verwendet werden.
6. Eine Injektionskanüle (30 G x ½", 0,3 mm x 13 mm) unter sterilen Bedingungen fest auf die Spritze aufstecken.
7. Vorsichtig die Schutzkappe von der Injektionskanüle abziehen, ohne dabei die Injektionskanüle von der Spritze abzutrennen.
Zu beachten: Während des Abziehens der Schutzkappe sollte die Injektionskanüle an ihrer Aufsteckkappe gehalten werden.
8. Vorsichtig die Luft zusammen mit der überschüssigen Lösung herausdrücken und stellen Sie die Dosierung auf die 0,05ml-Markierung der Spritze für Erwachsene und auf die 0,01-ml-Markierung für Frühgeborene ein. Die Spritze ist nun fertig zur Injektion.
Zu beachten: Die Injektionskanüle nicht abwischen und den Spritzenkolben nicht zurückziehen.
Stecken Sie die Kappe nach der Injektion nicht wieder auf die Nadel auf oder entfernen Sie die Nadel von der Spritze. Entsorgen Sie die verwendete Spritze zusammen mit der Nadel in einem durchstichsicheren Behältnis entsprechend den nationalen Anforderungen.
|