Eigenschaften/WirkungenATC-Code
ATC-Code: S01LA04
Ranivisio ist ein Biosimilar.
Wirkungsmechanismus
Der Wirkstoff von Ranivisio (Ranibizumab) ist ein humanisiertes, rekombinantes monoklonales Antikörperfragment (Fab) gegen den humanen vaskulären endothelialen Wachstumsfaktor A (VEGF-A). Es bindet mit hoher Affinität an VEGF-A und an dessen Isoformen. Die Isoformen, wie z.B. VEGF121 und VEGF165 entstehen durch alternatives mRNA-Spleissen, die Isoform VEGF110 durch Proteolyse. Die Bindung von Ranibizumab an VEGF-A und dessen Isoformen inhibiert die Aktivierung der Rezeptoren VEGFR-1 und VEGFR-2 auf der Oberfläche der Endothelzellen.
Pharmakodynamik
Die Aktivierung der Rezeptoren VEGFR-1 und -2 führt zur Proliferation von Endothelzellen, zur Neovaskularisation und zum Flüssigkeitsaustritt aus den Gefässen. Es wird angenommen, dass alle diese Faktoren zur Progression der neovaskulären Form der altersbedingten Makuladegeneration (AMD), der Entwicklung von CNV, inklusive CNV infolge einer pathologischen Myopie (PM), von diabetischen Makulaödemen (DME) und von retinalen Venenverschlüssen, die zu Visusverlust führen (RVO), beitragen.
Klinische Wirksamkeit
Behandlung der feuchten AMD
Die klinische Wirksamkeit und Sicherheit von Ranibizumab zur Behandlung der feuchten AMD wurde in drei randomisierten, doppelt-maskierten Studien bei insgesamt 1'323 Patienten (Ranibizumab: N=879, Kontrollgruppen N=444) mit neovaskulärer AMD untersucht. Als Kontrollarm diente in der Studie MARINA Scheinbehandlungen und in der Studie ANCHOR eine aktive Kontrolle mittels PDT mit Visudyne. Eingeschlossen wurden Patienten mit Läsionen in der Grösse von bis zu 12 Papillenflächen und einer Sehschärfe von 20/40 bis 20/320 (nach Snellen) im Studienauge. Das Durchschnittsalter der Patienten lag bei 77 Jahren. In den klinischen Studien wurden die Patienten angewiesen, sich selbstständig antimikrobielle Augentropfen zu applizieren (viermal täglich, jeweils 3 Tage vor und nach jeder Injektion).
In die 24monatige Studie MARINA wurden 716 Patienten mit minimaler klassischer CNV oder okkulter CNV eingeschlossen. Sie erhielten monatliche intravitreale Injektionen von Ranibizumab 0.3 mg (N=238), Ranibizumab 0.5 mg (N=240) oder Scheininjektionen (N=238). Während der 24-monatigen Behandlungsphase erhielten die Patienten durchschnittlich 22 von 24 möglichen Behandlungen. Die Resultate der MARINA Studie nach 12 Monaten Behandlung wurden bei 90% der Patienten auch nach 24 Monaten Behandlung (1x/Monat) im Wesentlichen bestätigt.
In die 24monatige Studie ANCHOR wurden 423 Patienten mit vorwiegend klassischen CNV-Läsionen eingeschlossen. Sie erhielten monatliche intravitreale Injektionen von Ranibizumab 0.3 mg und Schein-PDT (N=143), intravitreale Injektionen von Ranibizumab 0.5 mg und Schein-PDT (N=140) oder intravitreale Scheininjektionen und aktive Verteporfin-PDT (N=143). Die erste Schein- bzw. aktive Verteporfin-PDT wurde gemeinsam mit der anfänglichen Ranibizumab-Injektion appliziert. Danach erfolgte die Behandlung im Abstand von 3 Monaten, wenn am Studienauge weiterhin oder erneut Flüssigkeit aus den Gefässen austrat (Fluoreszein-Angiographie).
Die Resultate sind in den nachfolgenden Tabellen zusammengefasst:
Tabelle 0-1 Studie MARINA: Resultate nach 12 und 24 Monaten
|
Veränderung der Sehschärfe (in Buchstaben, ETDRS)
|
Monat
|
Scheinbehandlung
(n=238)
|
Ranibizumab 0.5 mg
(n=240)
|
Differenz
(95% CI)a
| |
Sehschärfe-Verlust <15 Buchstaben (%)b
|
Monat 12
|
62%
|
95%
|
32%
(26%, 39%)
| |
Monat 24
|
53%
|
90%
|
37%
(29%, 44%)
| |
Sehschärfe-Gewinn ≥15 Buchstaben (%)b
|
Monat 12
|
5%
|
34%
|
29%
(22%, 35%)
| |
Monat 24
|
4%
|
33%
|
29%
(23%, 35%)
| |
Durchschnittliche Veränderung der Sehschärfeb(Buchstaben)
|
Monat 12
|
-10.5 (16.6)
|
+7.2 (14.4)
|
17.5
(14.8, 20.2)
| |
Monat 24
|
-14.9 (18.7)
|
+6.6 (16.5)
|
21.1
(18.1, 24.2)
| |
a
nach Stratifizierung
b p <0.01
|
Tabelle 0-2 Studie ANCHOR: Resultate nach 12 und 24 Monaten
|
Veränderung der Sehschärfe
|
|
Verteporfin PDT
(n=143)
|
Ranibizumab 0.5 mg
(n=140)
|
Differenz
(95% CI) a
| |
Sehschärfe-Verlust <15 Buchstaben (%)b
|
Monat 12
|
64%
|
96%
|
33%
(25%, 41%)
| |
Monat 24
|
66%
|
90%
|
25%
(16%, 34%)
| |
Sehschärfe-Gewinn ≥15 Buchstaben (%)b
|
Monat 12
|
6%
|
40%
|
35%
(26%, 44%)
| |
Monat 24
|
6%
|
41%
|
35%
(26%, 44%)
| |
Durchschnittliche Veränderung der Sehschärfeb (Buchstaben)
|
Monat 12
|
-9.5 (16.4)
|
+11.3 (14.6)
|
21.1
(17.5, 24.6)
| |
Monat 24
|
-9.8 (16.4)
|
+10.7 (16.5)
|
20.7
(16.8, 24.7)
| |
a
nach Stratifizierung
b p <0.01
|
In den beiden Studien MARINA und ANCHOR resultierte die unter der Behandlung mit 0.5 mg Ranibizumab nach Monat 12 beobachtete Verbesserung des Visus in einem Nutzen für den Patienten, gemessen anhand der drei Subskalen des National Eye Institute Visual Function Questionnaire (VFQ-25), die zuvor als sekundäre Endpunkte für die Wirksamkeit festgelegt worden waren (Tätigkeiten mit Bezug auf Nah- und Weitsehen sowie weitere, vom Sehen abhängige Tätigkeiten). Alle Unterschiede zwischen Ranibizumab 0.5 mg und den zwei Kontrollgruppen waren statistisch signifikant und klinisch relevant, mit p-Werten zwischen 0.009 bis <0.0001.
In die Studie PIER wurden 184 Patienten mit CNV-Läsionen (mit und ohne klassische Anteile) eingeschlossen. Sie erhielten während der ersten 3 Monate je eine monatliche intravitreale Injektion von Ranibizumab 0.3 mg bzw. von Ranibizumab 0.5 mg oder intravitreale Scheininjektion. Weitere Injektionen von Ranibizumab erfolgten im Abstand von 3 Monaten. Nach Monat 14 konnten diejenigen Patienten, welche eine Scheininjektion erhielten, ebenfalls mit Ranibizumab behandelt werden und ab dem Monat 19 waren häufigere Injektionen von Ranibizumab möglich. Die mit Ranibizumab behandelten Patienten in PIER erhielten durchschnittlich 10 Behandlungen innert 24 Monaten.
Der primäre Wirksamkeits-Endpunkt war die mittlere Veränderung der Sehschärfe während der 12 Monate. Nach einem anfänglichen Anstieg in der Zeit der monatlichen Injektionen, verloren die Patienten in der Phase der 3-monatlichen Injektionen an Sehschärfe, die nach 12 Monaten zum Ausgangspunkt zurückkehrte und dieser Effekt wurde in den meisten mit Ranibizumab behandelnden Patienten (82%) bei Monat 24 aufrechterhalten. Die Daten von einer begrenzten Anzahl derjenigen Patienten, welche nach über einem Jahr Scheinbehandlung zu einer Behandlung mit Ranibizumab gewechselt haben, deuten darauf hin, dass ein früher Therapiebeginn mit besserer Erhaltung der Sehschärfe assoziiert ist.
Tabelle 0-3 Studie PIER: Resultate nach 12 Monaten
|
Veränderung der Sehschärfe
|
Scheinbehandlung (n=63)
|
Ranibizumab 0.5 mg
(n=61)
|
Differenz
(95% CI)a
| |
Sehschärfe-Verlust <15 Buchstaben (%)b
|
49
|
90
|
37
(23, 52)
| |
Sehschärfe-Gewinn ≥15 Buchstaben (%)b
|
10
|
13
|
2
(-8, 12)
| |
Durchschnittliche Veränderung der Sehschärfeb
|
-16.3 (22.3)
|
-0.2 (13.1)
|
14.7
(8.2, 21.2)
| |
a
nach Stratifizierung
b p <0.0001
|
Die Phase-IIIb Studie SAILOR wurde bei Behandlungsnaïven, wie auch vorbehandelten Patienten mit CNV infolge von AMD durchgeführt. SAILOR war eine 1-Jahres Multizentrische Studie. Das primäre Ziel der Studie war die Abschätzung der Inzidenz von okulären und nicht-okulären unerwünschten Wirkungen während der 12 Behandlungsmonate. 2378 Patienten wurden in 2 Gruppen randomisiert und erhielten während 3 Monaten 0.3 mg bzw. 0.5 mg Ranibizumab jeden Monat appliziert; anschliessend wurde je nach Befund weiterbehandelt, mit Abständen von mindestens 1 Monat.
Insgesamt bestand kein Ungleichgewicht zwischen den beiden Gruppen in Bezug auf okuläre und nicht-okuläre unerwünschte Wirkungen. Es gab insbesondere auch kein Ungleichgewicht zwischen den beiden Gruppen in Bezug auf die Zahl der Schlaganfälle. Unter 0.3 mg waren es 8/1'169 Patienten (0.7%, 95%CI: 0.3% bis 1.3%). Unter 0.5 mg waren es 15/1'209 Patienten (1.2%, 95% CI: 0.7% bis 2%). Patienten mit bekannten Risiko-Faktoren wie z.B. ein vorangegangener Schlaganfall oder eine vorangegangene transiente ischämische Attacke, haben vermutlich ein erhöhtes Risiko für einen Schlaganfall während der Behandlung mit Ranibizumab.
Behandlung des Visusverlustes durch DME
Die klinische Wirksamkeit und Sicherheit von Ranibizumab bei Patienten mit Visusverlust durch ein Diabetisches Makulaödem (DME) wurden in der Studie RESTORE bei insgesamt 345 Patienten mit Visusverlust durch DME untersucht. Die Studie hatte 3 Arme: In Arm 1 wurde Patienten (N=116) initial Ranibizumab 0.5 mg intravitreal als Monotherapie injiziert und eine Schein-Laserphotokoagulation durchgeführt. In Arm 2 (N=118) wurde initial Ranibizumab 0.5 mg intravitreal injiziert und eine Laserphotokoagulation durchgeführt. In Arm 3 (N=111) wurde eine initiale Monotherapie mit Laserphotokoagulation und eine Scheininjektion durchgeführt.
Die Behandlung mit Ranibizumab wurde mit monatlichen intravitrealen Injektionen fortgesetzt und unterbrochen, wenn sich der Visus des Patienten unter Ranibizumab an drei aufeinanderfolgenden Untersuchungsterminen stabilisiert hatte. Nach einer Unterbrechung wurde die Behandlung wieder aufgenommen, wenn sich der Visus des Patienten durch Fortschreiten des DME verschlechtert hatte. Wiederbehandlungen mit Laserphotokoagulation wurden am gleichen Tag, mindestens 30 Minuten vor der Ranibizumab Injektion, gemäss der ETDRS-Kriterien durchgeführt.
Die Resultate sind in den folgenden Tabellen zusammengefasst:
Tabelle 0-4 Studie RESTORE: Resultate nach 12 Monaten
|
Veränderung der bestmöglich korrigierten Sehschärfe
|
Ranibizumab
0.5 mg
(n=115)
|
Ranibizumab
0.5 mg + Laser
(n=118)
|
Laser
(n=110)
| |
Durchschnittliche Veränderung des best korrigierten Visus vom 1. zum 12. Monat in Buchstaben, verglichen mit dem Visus zu Beginn der Studie (Standardabweichung)a
|
6.1 (6.4)
|
5.9 (7.9)
|
0.8 (8.6)
| |
Durchschnittliche Veränderung des best korrigierten Visus im 12. Monat in Buchstaben, verglichen mit dem Visus zu Beginn der Studie (Standardabweichung)
|
6.8 (8.3)a
|
6.4 (11.8)b
|
0.9 (11.4)
| |
Zunahme des best korrigierten Visus ≥10 Buchstaben (% der Patienten)
|
37.4c
|
43.2
|
15.5
| |
Zunahme des best korrigierten Visus ≥15 Buchstaben (% der Patienten)
|
22.6d
|
22.9e
|
8.2
| |
a
p <0.0001, b p=0.0004, c p=0.0001, d p=0.0032, e p=0.0021
|
Bei der RESTORE-Erweiterungsstudie handelte es sich um eine offene, multizentrische, 24-monatige Erweiterungsstudie. 240 Patienten, die die 12-monatige Kernstudie abgeschlossen hatten, traten in die Erweiterungsstudie ein und wurden nach Bedarf (PRN: pro re nata) im selben Auge, das in der Kernstudie als Studienauge definiert worden war, mit 0.5 mg Ranibizumab behandelt. Die Behandlung wurde nach einem Abfall der BCVA aufgrund von DME monatlich bis zum Erreichen einer stabilen BCVA verabreicht. Ausserdem erfolgte eine Laserbehandlung gemäss den ETDRS-Leitlinien, falls dies vom Prüfarzt als notwendig erachtet wurde.
Die durchschnittliche Anzahl von Ranibizumab-Injektionen bei Patienten, die in der Kernstudie mit Ranibizumab behandelt worden waren, betrug in der 24-monatigen Erweiterungsphase 6.4. Von den 74 Patienten aus dem Laserbehandlungsarm der Kernstudie erhielten 59 (79%) Patienten zu irgendeinem Zeitpunkt während der Erweiterungsphase Ranibizumab. Die durchschnittliche Anzahl von Ranibizumab-Injektionen während der 24-monatigen Erweiterungsphase betrug bei diesen 59 Patienten 8.1 Injektionen. Die Anteile der Patienten, die während der Erweiterungsphase keine Behandlung mit Ranibizumab benötigten, betrugen in den Gruppen, die zuvor eine Behandlung mit Ranibizumab, Ranibizumab + Laser bzw. Laser allein erhalten hatten, 19%, 25% bzw. 20%.
Die Ergebnisse sind in der folgenden Tabelle zusammengefasst:
Tabelle 0-5 Resultate nach 36 Monaten in der RESTORE-Erweiterungsstudie
|
Ergebnisparameter gegenüber Baseline in der Kernstudie
|
Vorher Ranibizumab
0.5 mg
n = 83
|
Vorher Ranibizumab
0.5 mg + Laser
n = 83
|
Vorher Laser
n = 74*
| |
Mittlere Veränderung der BCVA gegenüber Baseline in der Kernstudie nach 36 Monaten (SD)
|
8.0 (10.09)
|
6.7 ( 9.59)
|
6.0 ( 9.35)
| |
Gewinn von ≥10 Buchstaben gegenüber Baseline in der Kernstudie bzw. BCVA ≥84 (%) nach 36 Monaten
|
39 (47.0)
|
37 (44.6)
|
31 (41.9)
| |
Gewinn von ≥15 Buchstaben gegenüber Baseline in der Kernstudie bzw. BCVA ≥84 (%) nach 36 Monaten
|
23 (27.7)
|
25 (30.1)
|
16 (21.6)
| |
n = Anzahl der Patienten, für die Werte sowohl bei Baseline (Kernstudie, Monat 0) als auch beim Besuch in Monat 36 verfügbar waren.
* 59 (79%) der 74 Patienten mit vorheriger Laserbehandlung erhielten in der Erweiterungsstudie Ranibizumab.
|
Das in dieser 24-monatigen Erweiterungsstudie beobachtete Langzeitsicherheitsprofil von Ranibizumab deckt sich mit dem bekannten Sicherheitsprofil von Ranibizumab.
In der Phase-IIIb-Studie RETAIN wurden 372 Patienten mit Visusverlust durch DME randomisiert folgenden intravitrealen Injektionen zugeteilt:
·Ranibizumab 0.5 mg mit gleichzeitiger Laserphotokoagulation im Rahmen eines «Treat and Extend»(TE)-Schemas (n=121),
·Ranibizumab 0.5 mg als Monotherapie im Rahmen eines TE-Schemas (n = 128) oder
·Ranibizumab 0.5 mg als Monotherapie im Rahmen eines PRN-Schemas (n = 123).
·In allen Gruppen wurde die Ranibizumab-Behandlung mit monatlichen intravitrealen Injektionen eingeleitet und bis zur Erreichung einer stabilen BCVA bei mindestens drei aufeinanderfolgenden monatlichen Untersuchungen fortgeführt. Die Laserphotokoagulation erfolgte bei Baseline am selben Tag wie die erste Ranibizumab-Injektion und anschliessend nach Bedarf gemäss den ETDRS-Kriterien. Im Rahmen des TE-Schemas wurde Ranibizumab im weiteren Verlauf in Abständen von 2- oder maximal 3 Monaten verabreicht. Beim PRN-Schema wurde die BCVA monatlich beurteilt, und Ranibizumab wurde dann, falls erforderlich, beim selben Besuch verabreicht. In allen Gruppen wurde die monatliche Behandlung nach einem Abfall der BCVA aufgrund einer DME-Progression wieder aufgenommen und bis zum erneuten Erreichen einer stabilen BCVA fortgeführt. Die Studiendauer betrug 24 Monate.
·In der RETAIN-Studie betrug die Zahl der Injektionen im Durchschnitt (Median) 12.4 (12.0) bei der TE-Ranibizumab + Laser-Behandlungsgruppe, 12.8 (12.0) bei der TE-Ranibizumab-Monotherapie- Behandlungsgruppe und 10.7 (10.0) bei der PRN-Ranibizumab-Behandlungsgruppe. Die Anzahl der benötigten, planmässigen Behandlungstermine war nach 3 initialen monatlichen Behandlungsterminen 13 unter dem TE-Schema, verglichen mit 20 monatlichen Terminen, die unter dem PRN-Schema benötigt wurden. Unter beiden Behandlungsschemata hielten mehr als 70% der Patienten ihre BCVA bei einer Visitenfrequenz von ≥2 Monaten aufrecht. Zusätzliche Laserbehandlungen waren unter dem entsprechenden TE-Schema nicht mit einer geringeren durchschnittlichen Zahl von Ranibizumab-Injektionen assoziiert.
Die Ergebnisse sind in der folgenden Tabelle zusammengefasst:
Tabelle 0-6 Resultate in der RETAIN-Studie
|
Ergebnisparameter gegenüber Baseline
|
TE Ranibizumab
0.5 mg + Laser
n = 117
|
TE Ranibizumab
0.5 mg allein
n = 125
|
PRN Ranibizumab
0.5 mg
n = 117
| |
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 12 (SD)
|
5.9 (5.5)b
|
6.1 (5.7)b
|
6.2 (6.0)
| |
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 24 (SD)
|
6.8 (6.0)
|
6.6 (7.1)
|
7.0 (6.4)
| |
Mittlere Veränderung der BCVA nach 24 Monaten (SD)
|
8.3 (8.1)
|
6.5 (10.9)
|
8.1 (8.5)
| |
Gewinn von ≥10 Buchstaben bzw. BCVA ≥84 (%) nach 24 Monaten
|
43.6
|
40.8
|
45.3
| |
Gewinn von ≥15 Buchstaben bzw. BCVA ≥84 (%) nach 24 Monaten
|
25.6
|
28.0
|
30.8
| |
Verlust von ≥10 Buchstaben nach 24 Monaten
|
2.6
|
7.2
|
3.4
| |
Verlust von ≥15 Buchstaben nach 24 Monaten
|
0.9
|
4.0
|
2.6
| |
b
p <0.0001
|
In den DME-Studien ging die BCVA-Verbesserung in allen Behandlungsgruppen mit einer allmählichen Verringerung der mittleren CRT einher.
Die Behandlung der mässig schweren bis schweren nicht-proliferativen diabetischen Retinopathie (NPDR) bzw. proliferativen diabetischen Retinopathie (PDR)
Die klinische Sicherheit und Wirksamkeit von Ranibizumab bei Patienten mit proliferativer diabetischer Retinopathie (PDR) wurden in eine multizentrische, randomisierte, aktiv kontrollierte Nichtunterlegenheitsstudie der Phase III im Parallel-Design (Protocol S), an der 305 Patienten (394 Studienaugen) mit PDR mit oder ohne DME (diabetisches Makulaödem) bei Baseline teilnahmen und in der 0.5 mg intravitreal injiziertes Ranibizumab mit der Standardbehandlung mit panretinale Photokoagulation (PRP) verglichen wurde, beurteilt. Insgesamt wurden 191 Augen (48.5%) randomisiert und der Behandlung mit 0.5 mg Ranibizumab und 203 Augen (51.5%) wurden randomisiert und der Behandlung mit PRP zugewiesen. Insgesamt 88 Augen (22.3%) wiesen bei Baseline ein DME auf: 42 (22.0%) und 46 (22.7%) der Augen in der Ranibizumab- bzw. der PRP-Gruppe. Insgesamt 306 Augen (77.7%) wiesen bei Baseline kein DME auf: 149 (78.0%) und 157 (77.3%) der Augen in der Ranibizumab- bzw. der PRP-Gruppe.
Nach 2 Jahren Behandlung hatte sich der BCVA (best corrected visual acuity) Score in der Ranibizumab-Gruppe von der Baseline um +2.7 Buchstaben und in der PRP Gruppe von der Baseline um -0.7 Buchstaben verändert. Die Differenz von 3.5 Buchstaben lag innerhalb der non-inferiority margin, sodass die Non-Inferiorität von Ranibizumab versus PRP bestätigt wurde.
Die Änderung des Schweregrads der diabetischen Retinopathie wurde anhand von Fundusfotos unter Verwendung des Schweregrad-Scores für diabetische Retinopathie (DRSS) aus der Studie zur frühzeitigen Behandlung der diabetischen Retinopathie (ETDRS) beurteilt. In dieser Studie war bei 41.8% der Augen unter der Behandlung mit Ranibizumab (n=189) in Monat 12 eine Verbesserung beim DRSS um mindestens 2 Stufen eingetreten, verglichen mit 14.6% bei den mit PRP behandelten Augen (n=199).
In einer Metaanalyse von 3 randomisierten, doppelblinden, aktiv kontrollierten Phase-III-Studien [D2301 (RESTORE), D2303 (REVEAL) und D2305 (REFINE)], durchgeführt in insgesamt 875 Patienten mit DME, zeigten 48,4% der 315 mit Ranibizumab behandelten Patienten mit mittelschwerer bis schwerer NPDR oder PDR (n = 192) eine Verbesserung der DRSS um mindestens 2 Stufen im 12. Monat, verglichen mit 14,6% der Laserbehandelte Patienten (n = 123).
Behandlung des Visusverlustes durch ein Makulaödem infolge RVO
Die klinische Sicherheit und Wirksamkeit von Ranibizumab bei Patienten mit Visusverlust durch ein Makulaödem infolge eines retinalen Venenverschlusses (RVO) wurden in zwei randomisierten, doppelt-maskierten, kontrollierten Studien BRAVO (N=397) und CRUISE (N=392) untersucht. In beiden Studien erhielten die Patienten entweder 0.3 mg Ranibizumab oder 0.5 mg Ranibizumab oder eine Scheinbehandlung. In BRAVO, war eine Laserphotokoagulation als Rettungsbehandlung zu jedem Zeitpunkt während der Studie ab dem 3. Monat in allen Studienarmen erlaubt. Die Resultate aus BRAVO und CRUISE sind in folgenden Tabellen zusammengestellt.
Tabelle 0-7 Studie BRAVO: Resultate bei Monat 6 und 12
|
Veränderung der Sehschärfe
|
Scheinbehandlung
(n=132)
|
Ranibizumab 0.5 mg
(n=131)
| |
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 6 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studiea
|
+7.3
|
+18.3
| |
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 12 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studie
|
+12.1
|
+18.3
| |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 6
|
28.8%
|
61.1%
| |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 12
|
43.9%
|
60.3%
| |
Anteil der Patienten mit Laserrescue über 12 Monaten
|
61.4%
|
34.4%
| |
a
p<0.0001
|
Tabelle 0-8 Studie CRUISE: Resultate bei Monat 6 und 12
|
Veränderung der Sehschärfe
|
Scheinbehandlung
(n=132)
|
Ranibizumab 0.5 mg
(n=131)
| |
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 6 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studiea
|
+0.8
|
+14.9
| |
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 12 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studie
|
+7.3
|
+13.9
| |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 6
|
16.9%
|
47.7%
| |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 12
|
33.1%
|
50.8%
| |
a
p<0.0001
|
Mit Ranibizumab behandelte Patienten zeigten in beiden Studien (BRAVO und CRUISE) eine kontinuierliche Verringerung der zentralen Netzhautdicke.
Neben der Visusverbesserung nach Behandlung mit Ranibizumab bei Monat 6 und 12 wurde auch die Lebensqualität der Patienten anhand des National Eye Institute Visual Function Questionnaire (VFQ-25) erhoben. Die Unterschiede zwischen der Ranibizumab 0.5 mg Gruppe und der Kontrollgruppe wurden bei Monat 6 mit p-Werten zwischen 0.02 und 0.0002 ermittelt.
Die Resultate der HORIZON Verlängerungsstudie zu BRAVO und CRUISE zeigten nach 12 Monaten folgendes:
Die reduzierte Behandlungsfrequenz in der Studie HORIZON hatte wenig Auswirkung auf BRVO-Patienten, welche ihre initiale Visusverbesserung, wie sie in der Studie BRAVO beobachtet wurde (+17.5 Buchstaben nach 24 Monaten mit einer Dosis von 0.5 mg und durchschnittlich 2.4 Injektionen im zweiten Jahr) behielten.
Hingegen zeigte sich die reduzierte Behandlungsfrequenz bei CRVO-Patienten mit einer Verringerung der in der Studie CRUISE gewonnenen Sehschärfe (+12 Buchstaben nach 24 Monaten mit einer Dosis von 0.5 mg und durchschnittlich 3.8 Injektionen im zweiten Jahr).
Studie E2401 (CRYSTAL) und Studie E2402 (BRIGHTER)
In den Studien BRIGHTER und CRYSTAL wurden die klinische Sicherheit und Wirksamkeit von Ranibizumab über 24 Monate bei Patienten mit Sehstörungen wegen eines RVO-bedingten Makulaödems bewertet. In die Studien wurden Patienten mit BRVO (n = 455) bzw. CRVO (n = 357) rekrutiert. In beiden Studien erhielten die Patienten 0.5 mg Ranibizumab nach Bedarf. BRIGHTER war eine 3-armige, randomisierte, aktiv kontrollierte Studie, in der 0.5 mg Ranibizumab als Monotherapie mit Ranibizumab in Kombination mit Laserphotokoagulation und mit Laserphotokoagulation alleine verglichen wurden. Nach 6 Monaten konnten die Teilnehmer im Arm mit der Laser-Monotherapie 0.5 mg Ranibizumab erhalten. CRYSTAL war eine einarmige Studie mit einer Monotherapie mit 0.5 mg Ranibizumab.
Die wichtigsten funktionellen und anatomischen Ergebnisse der BRIGHTER- und der CRYSTAL-Studie sind in Tabelle 0-9 und in den Abbildungen 1-0 und 2-0 dargestellt.
Tabelle 0-9 Ergebnisse in Monat 6 (BRIGHTER) und Monat 24 (BRIGHTER und CRYSTAL)
|
|
BRIGHTER
|
CRYSTAL
| |
|
Ranibizumab 0.5 mg
n = 180
|
Ranibizumab 0.5 mg
+ Laser
n = 178
|
Laser*
n = 90
|
Ranibizumab 0.5 mg
(n = 356)
| |
Mittlere Veränderung der BCVA in Monat 6b(Buchstaben) (SD)
|
+14.8
(10.7)
|
+14.8
(11.13)
|
+6.0
(14.27)
|
+12.0
(13.95)
| |
Mittlere Veränderung der BCVA in Monat 24b(Buchstaben) (SD)
|
+15.5
(13.91)
|
+17.3
(12.61)
|
+11.6
(16.09)
|
+12.1
(18.60)
| |
Anteil der Patienten, die bei der BCVA im Monat 24 ≥15 Buchstaben erreichten
|
52.8%
|
59.6%
|
43.3%
|
49.2%
| |
Mittlere Anzahl an Injektionen (SD) (Monate 0-23)
|
11.4
(5.81)
|
11.3
(6.02)
|
N/A
|
13.1
(6.39)
| |
*
Ab Monat 6 war eine Behandlung mit 0.5 mg Ranibizumab zulässig (24 Patienten wurden nur mit Laser behandelt).
b p <0.0001 für beide Vergleiche in der BRIGHTER-Studie in Monat 6: Ranibizumab 0.5 mg gegenüber Laser und Ranibizumab 0.5 mg + Laser gegenüber Laser.
|
Abbildung 1-0 BRIGHTER: Mittlere Veränderung der BCVA gegenüber der Baseline über einen Zeitraum von 24 Monaten
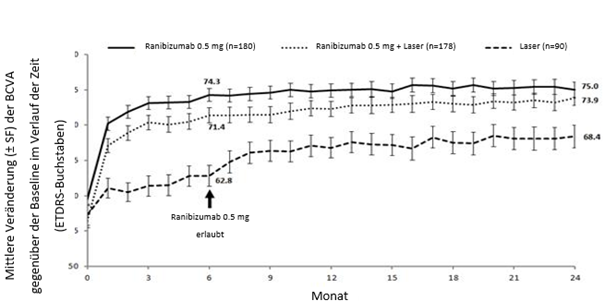
Abbildung 2-0 CRYSTAL: Mittlere Veränderung der BCVA gegenüber der Baseline über einen Zeitraum von 24 Monaten
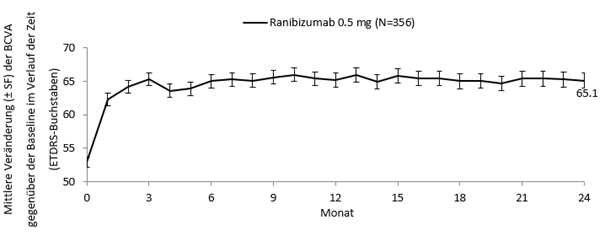
Die Verbesserung des Visus fiel bei Patienten mit oder ohne Netzhautischämie ähnlich aus: In der BRIGHTER-Studie wiesen Patienten mit Netzhautischämie (n = 87) oder ohne Netzhautischämie (n = 35), die mit einer Ranibizumab-Monotherapie behandelt wurden, in Monat 24 eine mittlere Veränderung gegenüber der Baseline von +15.4 bzw. +12.9 Buchstaben auf. In der CRYSTAL-Studie wiesen Patienten mit Netzhautischämie (n = 107) oder ohne Netzhautischämie (n = 109) eine mittlere Veränderung gegenüber der Baseline von +11.1 bzw. +12.9 Buchstaben auf.
Bei Patienten mit einer Krankheitsdauer von <3 Monaten war in der BRIGHTER- bzw. CRYSTAL-Studie in Monat 1 eine Verbesserung der Sehschärfe von 13.3 bzw. 10.0 Buchstaben und in Monat 24 von 17.7 bzw. 13.2 Buchstaben zu beobachten. Bei einer Krankheitsdauer von ≥12 Monaten waren es in Monat 1 4.8 bzw. 7.9 Buchstaben, in Monat 24 waren es 8.4 bzw. 8.6 Buchstaben. Eine Initiierung der Behandlung bei Diagnosestellung ist zu erwägen.
Das Sicherheitsprofil von Ranibizumab, das in diesen 24-monatigen Studien beobachtet wurde, entspricht dem aus früheren Studien bekannten Sicherheitsprofil von Ranibizumab.
Behandlung eines Visusverlustes durch CNV – Studie G2301 (MINERVA)
Die klinische Sicherheit und Wirksamkeit von Ranibizumab bei Patienten mit Visusverlust durch CNV infolge von anderen Ätiologien als neovaskulärer AMD und PM wurden auf Grundlage der 12-Monats-Daten der randomisierten, doppelt-maskierten, Scheinbehandlungs-kontrollierten Studie G2301 (MINERVA) beurteilt. Für die Analyse wurden fünf Untergruppen nach Ätiologie vordefiniert (angioide Streifen, postentzündliche Retinochoroidopathie, zentrale seröse Chorioretinopathie, idiopathische Chorioretinopathie und andere Ätiologien). 178 Patienten wurden im Verhältnis 2:1 in einen der folgenden Arme randomisiert:
Ranibizumab 0.5 mg zu Beginn der Studie, gefolgt von einem individuellen Dosierungsschema je nach Krankheitsaktivität.
Scheininjektion zu Beginn der Studie, gefolgt von einem individuellen Dosierungsschema je nach Krankheitsaktivität.
Ab Monat 2 erhielten alle Patienten eine Behandlung mit Ranibizumab nach Bedarf. Primärendpunkt war die Veränderung der besten korrigierten Sehschärfe (BCVA) vom Beginn der Studie bis Monat 2.
Die wichtigsten Ergebnisse von MINERVA sind in Tabellen 0-10 und 0-11 sowie in Abbildung 3-0 zusammengestellt.
Tabelle 0-10 Resultate nach 2 Monaten (MINERVA)
|
|
Ranibizumab 0.5 mg
(n = 119)
|
Scheinbehandlung
(n = 59)
| |
Mittlere BCVA-Veränderung vom Beginn der Studie bis Monat 2 (Buchstaben) (Least-Squares-Mittelwert) a
|
+9.5
|
-0.4
| |
Anteil Patienten mit einem Gewinn von ≥15 Buchstaben seit Beginn der Studie oder einem Wert von 84 Buchstaben in Monat 2
|
31.4%
|
12.3%
| |
Anteil Patienten, die vom Beginn der Studie bis Monat 2 nicht mehr als 15 Buchstaben verloren
|
99.2%
|
94.7%
| |
Verringerung der mittleren Foveadicke vom Beginn der Studie bis Monat 2 (Least-Squares-Mittelwert) a
|
77 µm
|
-9.8 µm
| |
a
Einseitiger Vergleich (p <0.001) mit Scheinbehandlungskontrolle
|
Abbildung 3-0: Mittlere Veränderung der BCVA vom Beginn der Studie bis Monat 12 im Zeitverlauf (MINERVA)
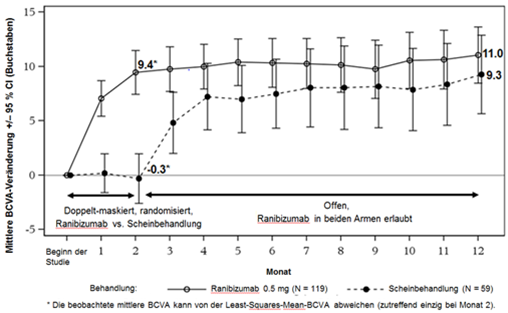
Beim Vergleich von Ranibizumab mit den Scheininjektionen in Monat 2 wurde eine konsistente Wirkung beobachtet, dies sowohl in der gesamten Studienpopulation, als auch in den einzelnen, gemäss der Ätiologie definierten Untergruppen.
Tabelle 0-11 Gesamtwirkung der Behandlung und Wirkung der Behandlung in den zu Beginn der Studie gemäss der Ätiologie definierten Untergruppen für die primäre Variable in Monat 2 (MINERVA)
|
Insgesamt und gegliedert nach Ätiologie bei Studienbeginn
|
Behandlungswirkung im Vergleich zur Scheinbehandlung (Buchstaben)
|
Patientenzahlen
(n) (Behandlung + Scheinbehandlung)
| |
Gesamt
|
9.9
|
175*
| |
Angioide Streifen
|
14.6
|
27
| |
Postentzündliche Retinochoroidopathie
|
6.5
|
27
| |
Zentrale seröse Chorioretinopathie
|
5.0
|
23
| |
Idiopathische Chorioretinopathie
|
11.4
|
62
| |
Sonstige Ätiologiena
|
10.6
|
36
| |
a
CNV-Ätiologien, die nicht unter die anderen Untergruppen fallen
* Anzahl Patienten mit für die Analyse verfügbaren Daten
|
Die Verbesserung des Visus wurde von einer Verringerung der mittleren Foveadicke über einen Zeitraum von 12 Monaten begleitet.
Die mittlere Anzahl der Ranibizumab-Injektionen über einen Zeitraum von 12 Monaten in das Studienauge betrug im Ranibizumab-Arm 5.8 und in der Gruppe mit Scheininjektionen 5.4.
Kinder und Jugendliche
Fünf jugendliche Patienten im Alter von 12 bis 17 Jahren mit Visusverlust infolge CNV (1x subfoveale CNV bei einer Drusenpapille; je 1x juxtafoveale CNV und subfoveale CNV bei idiopathischer CNV; 2x subfoveale CNV bei Best disease) erhielten eine initiale Behandlung mit Ranibizumab 0.5 mg gefolgt von einem individualisierten Behandlungsschema auf Grundlage von Anzeichen der Krankheitsaktivität (z.B. Beeinträchtigung der Sehschärfe, intra-/subretinale Flüssigkeit, Blutungen oder Leckagen). Die BCVA-Veränderung vom Beginn der Studie bis Monat 12 verbesserte sich bei allen fünf Patienten und reichte von +5 bis +38 Buchstaben. Die Verbesserung des Visus wurde von einer Stabilisierung oder Verringerung der mittleren Foveadicke über einen Zeitraum von 12 Monaten begleitet (ΔCSFT0-12 Mte.: -286 μm bis +10 μm). Während der 12 Monate wurden 2 bis 5 Injektionen ins Studienauge verabreicht (s. «Dosierung/Anwendung»).
Behandlung eines Visusverlustes durch CNV infolge von PM
Die klinische Sicherheit und Wirksamkeit von Ranibizumab bei Patienten mit Visusverlust durch CNV infolge von PM wurden ausgehend von 12-Monats-Daten aus der randomisierten, doppelt maskierten, kontrollierten Pivotstudie RADIANCE beurteilt. Diese Studie war dazu ausgelegt, zwei verschiedene Dosierungsschemata von 0.5 mg Ranibizumab per intravitrealer Injektion im Vergleich zu Verteporfin PDT (vPDT, photodynamische Therapie mit Visudyne) zu evaluieren.
Die 277 Patienten wurden in einen der folgenden Arme randomisiert:
Gruppe I (0.5 mg Ranibizumab, Dosierungsplan beruhte auf Stabilitäts-Kriterien, definiert als keine Veränderung der bestkorrigierten Sehschärfe (BCVA) im Vergleich zu zwei vorhergehenden monatlichen Untersuchungen)
Gruppe II (0.5 mg Ranibizumab, Dosierungsplan beruhte auf Krankheitsaktivitäts-Kriterien, definiert als Visusverlust aufgrund von intra- oder subretinaler Flüssigkeit oder aktivem Flüssigkeitsaustritt infolge der CNV-Läsion mit Nachweis per OCT und/oder FA).
Gruppe III (vPDT - die Patienten konnten ab Monat 3 mit Ranibizumab behandelt werden)
Im Lauf der 12 Studienmonate erhielten die Patienten in Gruppe I durchschnittlich 4.6 Injektionen (zwischen 1 und 11 Injektionen) und in Gruppe II durchschnittlich 3.5 Injektionen (zwischen 1 und 12 Injektionen). In Gruppe II (in welcher Patienten die empfohlene Behandlung basierend auf Krankheitsaktivität erhielten, s. «Dosierung/Anwendung»), benötigten 50.9% der Patienten eine oder zwei Injektionen, 34.5% der Patienten drei bis fünf Injektionen und 14.7% der Patienten 6 bis 12 Injektionen innerhalb der 12-monatigen Studienperiode. 62.9% der Gruppe II-Patienten benötigten in den letzten sechs Monaten der Studie keine weiteren Injektionen.
Die wichtigsten Behandlungsergebnisse der Studie RADIANCE sind in Tabelle 0-12 und Abbildung 0-4 zusammengefasst.
Tabelle 0-12 Behandlungsergebnis bei Monat 3 und Monat 12 (RADIANCE)
|
|
Gruppe I
0.5 mg Ranibizumab
Stabilität der Sehschärfe
(n=105)
|
Gruppe II
0.5 mg Ranibizumab
Krankheitsaktivität
(n=116)
|
Gruppe III
vPDT*
(n=55)
| |
Monat 3
| |
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 3 gegenüber Baselinea(Buchstaben)
|
+10.5
|
+10.6
|
+2.2
| |
Anteil der Patienten mit Verbesserung der BCVA um
|
|
|
| |
≥10 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben
|
61.9%
|
65.5%
|
27.3%
| |
≥15 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben
|
38.1%
|
43.1%
|
14.5%
| |
Monat 12
| |
Anzahl der Injektionen bis Monat 12:
|
|
|
| |
Durchschnitt
|
4.6
|
3.5
|
N/A
| |
Medianwert
|
4.0
|
2.0
|
N/A
| |
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 12 gegenüber Baseline (Buchstaben)
|
+12.8
|
12.5
|
N/A
| |
Anteil der Patienten mit Verbesserung der BCVA um
|
|
|
| |
≥10 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben
|
69.5%
|
69.0%
|
N/A
| |
≥15 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben
|
53.3%
|
51.7
|
N/A
| |
* Vergleichende Kontrolle bis Monat 3. Die in die vPDT-Gruppe randomisierten Patienten konnten ab Monat 3 eine Behandlung mit Ranibizumab erhalten (in Gruppe III erhielten 38 Patienten Ranibizumab ab Monat 3): p<0.00001 für den Vergleich mit der vPDT-Kontrolle
|
Abbildung 0-4 Mittlere Veränderung der BCVA gegenüber Baseline BCVA im Zeitverlauf bis Monat 12 (RADIANCE)
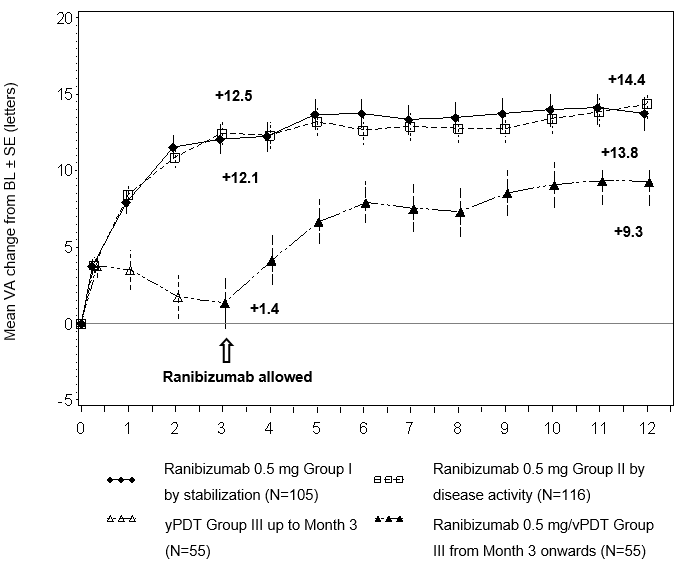
BL = Baseline; SE = Standardfehler des Mittelwerts.
Abbildung:
Mittlere Verbesserung der Sehschärfe gegenüber BL ± SE (Buchstaben)
Gruppe I mit 0.5 mg Ranibizumab nach Stabilisierung (N=105)
Gruppe II mit 0.5 mg Ranibizumab nach Krankheitsaktivität (N=116)
Gruppe III mit Visudyne-PDT (N=55)
Die in die vPDT-Gruppe randomisierten Patienten konnten ab Monat 3 eine Behandlung mit Ranibizumab erhalten.
Die Visusverbesserung war von einer Reduzierung der zentralen Netzhautdicke begleitet.
In den Ranibizumab-Behandlungsarmen wurde gegenüber der vPDT-Gruppe (p-Wert <0.05) ein von den Patienten subjektiv angegebener Nutzen in Bezug auf die Verbesserung des kombinierten Ergebnisses wie auch des Ergebnisses in mehreren Subskalen (allgemeiner Visus, Nahsichtaktivitäten, psychische Verfassung und unabhängiger Funktionsstatus) des Fragebogens VFQ-25 festgestellt.
Behandlung der RPM bei Frühgeborenen: Studie H2301 (RAINBOW)
Die klinische Sicherheit und Wirksamkeit von Ranibizumab 0.1 mg bei der Behandlung der RPM bei Frühgeborenen wurden anhand der Sechsmonatsdaten aus der randomisierten, offenen, dreiarmigen Parallelgruppen-Überlegenheitsstudie H2301 (RAINBOW) untersucht, die darauf ausgelegt war, die Anwendung von Ranibizumab in Dosen von 0.1 mg und 0.2 mg, verabreicht als intravitreale Injektionen, im Vergleich zur Lasertherapie zu beurteilen. Geeignete Patienten mussten einen der folgenden Netzhautbefunde in beiden Augen aufweisen:
·Zone I, Krankheitsstadium 1+, 2+, 3 oder 3+ oder
·Zone II, Krankheitsstadium 3+ oder
·Aggressive posteriore RPM (AP-RPM)
Für diese Studie wurden 225 Patienten im Verhältnis 1:1:1 randomisiert, um Ranibizumab intravitreal 0.1 mg (n=77) bzw. 0.2 mg (n=74) oder eine Lasertherapie (n=74) zu erhalten.
Der Behandlungserfolg, gemessen anhand des Ausbleibens einer aktiven RPM und des Fehlens ungünstiger struktureller Auswirkungen in beiden Augen 24 Wochen nach der ersten Studienbehandlung, betrug 75 % in der Ranibizumab 0.1 mg-Gruppe und 66.2 % in der Lasertherapie-Gruppe. Die Mehrheit der mit Ranibizumab 0.1 mg behandelten Patienten (77.6 %) erhielt eine einzige Injektion pro Auge.
Aus der 0.1 mg Ranibizumab-Behandlungsgruppe wechselten weniger Patienten aufgrund fehlenden Ansprechens zu einem anderen Behandlungsmodus als in der Lasertherapie-Gruppe (16.9 % vs. 24.3 %). Ungünstige strukturelle Auswirkungen waren in der 0.1 mg Ranibizumab-Behandlungsgruppe (5 Patienten, 6.7 %) weniger häufig als in der Lasertherapie-Gruppe (7 Patienten, 10.1 %). Darüber hinaus erreichten 75 % der Patienten innerhalb von 8 Tagen ein Abklingen der Erkrankung unter Ranibizumab 0.1 mg im Vergleich zu 22.5 Tagen in der Lasertherapie-Gruppe.
Studie H2301E1 (RAINBOW Erweiterungsstudie)
Die Langzeitwirksamkeit und Sicherheit von Ranibizumab 0,1 mg zur Behandlung von RPM bei Frühgeborenen wurde in der Studie H2301E1 (RAINBOW-Erweiterungsstudie), einer Erweiterung der Studie H2301 (RAINBOW), an Patienten bis zum Erreichen des 5. Lebensjahrs untersucht.
Das Hauptziel bestand darin, die Beurteilung der Sehfunktion beim Termin der Patienten bei Erreichen ihres 5. Lebensjahres zu bewerten, indem die Sehschärfe anhand der Early Treatment Diabetic Retinopathy Study (ETDRS) mit Lea-Symbol-Optotypen am besser sehenden Auge (dem Auge mit dem höheren ETDRS-Score) beurteilt wurde.
Bei 78,2% (43/55) der Patienten in der Ranibizumab 0,1 mg-Gruppe und bei 76,6% (36/47) der Patienten in der Lasertherapie-Gruppe wurde beim Termin bei Erreichen des 5. Lebensjahres ein ETDRS-Score aufgezeichnet. Der Unterschied im Mittelwert (SE) der kleinsten Quadrate (LS) zwischen der Ranibizumab 0,1 mg-Gruppe und der Lasertherapie-Gruppe betrug 2,5 (95-%-KI: -3,4, 8,3). Die kategorisierten Ergebnisse der Sehschärfe des besseren Auges bei Erreichen des 5. Lebensjahres der Patienten sind in Tabelle 13 dargestellt. Insgesamt wurde das Fehlen aller okulären Strukturanomalien bis zum Erreichen des 5. Lebensjahrs bei einem höheren Anteil der Patienten in der Ranibizumab 0,1 mg-Gruppe [93,8% (61/65)] als in der Lasertherapie-Gruppe [88,7% (47/53)] beobachtet.
Tabelle 13 Auswirkungen auf die Sehschärfe im besser sehenden Auge¹ beim Termin bei Erreichen des 5. Lebensjahrs der Patienten
|
Kategorie der Sehschärfe
|
Ranibizumab 0.1 mg
N=65
n (%)
|
Laser
N=54
n (%)
| |
≥1 bis ≤34 Buchstaben
|
3 (4.6)
|
2 (3.7)
| |
≥35 bis ≤70 Buchstaben
|
25 (38.5)
|
23 (42.6)
| |
≥71 Buchstaben
|
15 (23.1)
|
11 (20.4)
| |
¹ Das besser sehende Auge ist das Auge mit dem höheren ETDRS-Buchstaben-Score bei Erreichen des 5.Lebensjahrs. Wenn beide Augen den gleichen ETDRS-Buchstaben-Score haben, wird das rechte Auge als das besser sehende Auge angegeben.
|
|