ZusammensetzungWirkstoffe
Trametinib als Trametinibdimethylsulfoxid
Hilfsstoffe
Tablettenkern:
Mannitol, Mikrokristalline Cellulose, Hypromellose, Croscarmellose-Natrium, Magnesiumstearat, Natriumlaurylsulfat, Kolloidales Siliciumdioxid,
Filmüberzug:
Titandioxid (E 171), Polyethylenglykol, gelbes Eisenoxid (E 172, bei 0.5 mg Tabletten), Polysorbat 80 und rotes Eisenoxid (E 172, bei 2 mg Tabletten).
Gesamtnatriumgehalt ist 0.199 mg bei den 0.5 mg Tabletten resp. 0.232 mg bei den 2 mg Tabletten.
Pulver zur Herstellung einer Lösung zum Einnehmen:
Sulfobutylbetadex-Natrium (9'400 mg), Sucralose (E955), Zitronensäure-Monohydrat (E330), Natriummonohydrogenphosphat, Kaliumsorbat (E202), Methylparahydroxybenzoat (E218, 75.20 mg), Erdbeeraroma.
1 Flasche enthält maximal 198 mg Natrium und 58 mg Kalium.
Jeder ml der rekonstituierten Lösung enthält 100 mg Sulfobutylbetadex-Natrium, 0.58 mg Kalium, 1,98 mg Natrium und 0,8 mg Methylparahydroxybenzoat (E218).
Indikationen/AnwendungsmöglichkeitenBefristet zugelassene Indikation
Niedriggradiges Gliom (Low Grade Gliom, LGG)
Mekinist in Kombination mit Dabrafenib ist angezeigt zur Behandlung von pädiatrischen Patienten ab 1 Jahr mit niedriggradigem Gliom (Low Grade Gliom, LGG) mit einer BRAF-V600E-Mutation, die eine systemische Therapie benötigen.
Aufgrund einer zum Zeitpunkt der Begutachtung des Gesuches unvollständigen klinischen Datenlage, wird/werden diese Indikation/en befristet zugelassen (Art. 9a Heilmittelgesetz). Die befristete Zulassung ist zwingend an die zeitgerechte Erfüllung von Auflagen gebunden. Nach deren Erfüllung kann die befristete Zulassung in eine ordentliche Zulassung überführt werden.
Ordentlich zugelassene Indiaktionen
Nicht resezierbares oder metastasiertes Melanom
Mekinist in Kombination mit Dabrafenib ist angezeigt zur Behandlung von erwachsenen Patienten mit nicht resezierbarem oder metastasiertem Melanom mit einer BRAF-V600-Mutation (V600E/K).
Adjuvante Behandlung des Melanoms
Mekinist in Kombination mit Dabrafenib ist angezeigt zur adjuvanten Behandlung von Patienten mit Melanom im Stadium III mit einer BRAF-V600-Mutation nach vollständiger Resektion.
Fortgeschrittenes oder metastasiertes, nicht-kleinzelliges Lungenkarzinom
Mekinist in Kombination mit Dabrafenib kann angewendet werden zur Behandlung von erwachsenen Patienten mit metastasiertem, nicht-kleinzelligem Lungenkarzinom (NSCLC; non-small cell lung cancer) mit einer BRAF-V600E Mutation.
Dosierung/AnwendungDie Behandlung mit Mekinist sollte von einem in der Anwendung onkologischer Arzneimittel erfahrenen Arzt eingeleitet und überwacht werden.
Mekinist ist in zwei Darreichungsformen erhältlich: als Tabletten und als Pulver zum Herstellen einer Lösung zum Einnehmen.
Vor der Einnahme von Mekinist in Kombination mit Dabrafenib soll das Vorliegen einer BRAF-V600-Mutation gemäss der zugelassenen Indikation anhand eines validierten Tests bestätigt sein. Mekinist in Kombination mit Dabrafenib soll nicht in Patienten angewandt werden, welche BRAF Wild-Typ Tumore aufweisen (siehe «Klinische Wirksamkeit»).
Übliche Dosierung
Filmtabletten
Bei erwachsenen Patienten, die Mekinist in Kombination mit Dabrafenib anwenden, beträgt die empfohlene Dosis für Mekinist Filmtabletten 2 mg oral einmal täglich und die empfohlene Dosis Dabrafenib 150 mg oral zweimal täglich unabhängig vom Körpergewicht (siehe «Klinische Wirksamkeit»).
Dosisanpassung aufgrund unerwünschter Wirkungen/Interaktionen
Bei unerwünschten Reaktionen kann eine Unterbrechung der Behandlung, eine Dosisreduktion oder der Behandlungsabbruch erforderlich werden (siehe «Tabelle 1 und 2»).
Tabelle 1: Empfohlene Dosisreduktion von Mekinist in Kombination mit Dabrafenib bei erwachsenen Patienten
|
Dosisreduktionen
|
Dosis von Mekinist
|
Dosis von Dabrafenib
| |
Startdosis
|
2 mg oral einmal täglich
|
150 mg oral zweimal täglich
| |
Erste Dosisreduktion
|
1.5 mg oral einmal täglich
|
100 mg oral zweimal täglich
| |
Zweite Dosisreduktion
|
1 mg oral einmal täglich
|
75 mg oral zweimal täglich
| |
Dritte Dosisreduktion
|
keine weitere Dosisreduktion
|
50 mg oral zweimal täglich
|
Die Empfehlungen zur Dosisanpassungen von Dabrafenib sind der Fachinformation von Dabrafenib zu entnehmen (siehe «Dosierung/Anwendung»). Mekinist 1 mg als Filmtabletten oral einmal täglich muss dauerhaft abgesetzt werden, wenn es nicht verträglich ist.
Pulver zur Herstellung einer Lösung zum Einnehmen
Die empfohlenen Dosierungen und Dosisreduktionen für Mekinist Pulver zur Herstellung einer Lösung zum Einnehmen basieren auf dem Körpergewicht (Tabelle 2)
Tabelle 2: Empfohlene körpergewichtsabhängige Dosierung und Dosisreduktion für Mekinist Pulver zur Herstellung einer Lösung zum Einnehmen
|
Körpergewicht (Kilogramm)
|
Empfohlene Dosis
Gesamtmenge der Lösung zum Einnehmen einmal täglich
(Gehalt an Trametinib)
|
Dosisreduktionen
| |
Erste Dosisreduktion (einmal täglich)
|
Zweite Dosisreduktion (einmal täglich)
| |
8 kg
|
6 ml (0.3 mg)
|
5 ml
|
3 ml
| |
9 kg
|
7 ml (0.35 mg)
|
5 ml
|
4 ml
| |
10 kg
|
7 ml (0.35 mg)
|
5 ml
|
4 ml
| |
11 kg
|
8 ml (0.4 mg)
|
6 ml
|
4 ml
| |
12 bis 13 kg
|
9 ml (0.45 mg)
|
7 ml
|
5 ml
| |
14 bis 17 kg
|
11 ml (0.55 mg)
|
8 ml
|
6 ml
| |
18 bis 21 kg
|
14 ml (0.7 mg)
|
11 ml
|
7 ml
| |
22 bis 25 kg
|
17 ml (0.85 mg)
|
13 ml
|
9 ml
| |
26 bis 29 kg
|
18 ml (0.9 mg)
|
14 ml
|
9 ml
| |
30 bis 33 kg
|
20 ml (1 mg)
|
15 ml
|
10 ml
| |
34 bis 37 kg
|
23 ml (1.15 mg)
|
17 ml
|
12 ml
| |
38 bis 41 kg
|
25 ml (1.25 mg)
|
19 ml
|
13 ml
| |
42 bis 45 kg
|
28 ml (1.4 mg)
|
21 ml
|
14 ml
| |
46 bis 50 kg
|
32 ml (1.6 mg)
|
24 ml
|
16 ml
| |
≥51 kg
|
40 ml (2 mg)
|
30 ml
|
20 ml
| |
Dauerhaft absetzen, wenn maximal zwei Dosisreduktionen nicht vertragen werden.
|
Mekinist Lösung zum Einnehmen ist dauerhaft abzusetzen, wenn es nach der zweiten Dosisreduktion nicht verträglich ist.
Tabelle 3: Empfohlene Dosierungsänderungen für Mekinist bei Nebenwirkungen
|
Schweregrad der Nebenwirkung [siehe Warnhinweise und Vorsichtsmassnahmen] a
|
Dosierungsänderung für Mekinistb
| |
Blutung
| |
·Schweregrad 3
|
Mekinist unterbrechen.
·Bei Besserung setzen Sie Mekinist mit einer niedrigeren Dosis fort.
·Falls keine Besserung eintritt, Mekinist dauerhaft absetzen.
| |
·Schweregrad 4
|
Mekinist dauerhaft absetzen.
| |
Venöse Thromboembolie
| |
·Unkomplizierte tiefe Venenthrombose (TVT) oder Lungenembolie (LE)
|
Unterbrechen Sie Mekinist für bis zu 3 Wochen.
·Bei Besserung auf Schweregrad 0-1 Mekinist mit niedrigerer Dosis wieder aufnehmen.
·Wenn keine Besserung eintritt, setzen Sie Mekinist dauerhaft ab.
| |
·Lebensbedrohliche PE
|
Mekinist dauerhaft absetzen.
| |
Kardiomyopathie
| |
·Asymptomatische, absolute Abnahme der linksventrikulären Ejektionsfraktion (LVEF) von 10 % oder mehr gegenüber dem Ausgangswert und unterhalb der institutionellen Untergrenze des Normalwerts (LLN)
|
Unterbrechen Sie Mekinist für bis zu 4 Wochen.
·Wenn sich der LVEF-Wert auf einen normalen Wert verbessert hat, setzen Sie die Behandlung mit Mekinist mit einer niedrigeren Dosis fort.
·Wenn sich der LVEF-Wert nicht auf den normalen Wert verbessert, muss Mekinist dauerhaft abgesetzt werden.
| |
·Symptomatische Kardiomyopathie
·Absoluter Rückgang der LVEF von mehr als 20 % gegenüber dem Ausgangswert, der unter LLN liegt
|
Mekinist dauerhaft absetzen.
| |
Okulare Toxizitäten
| |
·Abhebungen des retinalen Pigmentepithels (RPED)
|
Unterbrechen Sie Mekinist für bis zu 3 Wochen.
·Bei Besserung Mekinist mit gleicher oder niedrigerer Dosis wieder aufnehmen.
·Wenn keine Besserung eintritt, setzen Sie Mekinist dauerhaft ab oder nehmen Sie Mekinist mit einer niedrigeren Dosis wieder auf.
| |
·Netzhautvenenverschluss (RVO)
|
Mekinist dauerhaft absetzen.
| |
Lungen
| |
·Interstitielle Lungenerkrankung (ILD)/Pneumonitis
|
Mekinist dauerhaft absetzen.
| |
Fieberreaktionen
| |
·Fieber von 38°C bis 40°C (oder erste Symptome bei Rezidiv)
|
Unterbrechen Sie Mekinist, bis das Fieber abgeklungen ist, und setzen Sie dann Mekinist mit derselben oder einer niedrigeren Dosis fort.
| |
·Fieber über 40°C
·Fieber, kompliziert durch Schüttelfrost, Hypotonie, Dehydration oder Nierenversagen
|
·Unterbrechen Sie Mekinist bis die fieberhaften Reaktionen für mindestens 24 Stunden abgeklungen sind, und setzen Sie dann Mekinist mit einer niedrigeren Dosis fort.
Oder
·Mekinist dauerhaft absetzen.
| |
Hauttoxizitäten
| |
·Schweregrad 2, wenn nicht tolerierbar
·Schweregrad 3 oder 4
|
Unterbrechen Sie Mekinist für bis zu 3 Wochen.
·Bei Besserung setzen Sie Mekinist mit einer niedrigeren Dosis fort.
·Wenn keine Besserung eintritt, dauerhaft absetzen.
| |
·Schwere kutane Nebenwirkungen (SCARs)
|
Mekinist dauerhaft absetzen.
| |
Andere Nebenwirkungen
| |
·Schweregrad 2, wenn nicht tolerierbar
·Schweregrad 3
|
Mekinist unterbrechen.
·Bei Besserung auf Schweregrad 0-1 mit niedrigerer Dosis fortfahren.
·Wenn keine Besserung eintritt, dauerhaft absetzen.
| |
·Erstes Auftreten von Schweregrad 4
|
·Unterbrechen Sie Mekinist, bis sich die Behandlung auf Schweregrad 0-1 verbessert hat, und setzen Sie die Behandlung dann mit einer niedrigeren Dosis fort.
Oder
·Mekinist dauerhaft absetzen.
| |
·Rezidiv Schweregrad 4
|
Mekinist dauerhaft absetzen.
|
a National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) Version 4.0.
b Siehe Tabellen 1 und 2 für empfohlene Dosisreduktionen von Mekinist.
c Für die folgenden Nebenwirkungen werden keine Dosisanpassungen für Mekinist empfohlen, wenn es zusammen mit Dabrafenib verabreicht wird: nicht-kutane maligne Erkrankungen und Uveitis. Eine Dosisanpassung von Mekinist ist bei neuen primären Malignomen der Haut nicht erforderlich.
Beachten Sie die Fachinformation von Dabrafenib für Dosisanpassungen bei Nebenwirkungen im Zusammenhang mit Dabrafenib.
Sobald die unerwünschten Wirkungen bei einem Patienten wirksam unter Kontrolle gebracht wurden, kann eine Reeskalation der Dosis nach den gleichen Dosierungsschritten wie im Fall einer Deeskalation der Dosis erwogen werden. Die Mekinist-Dosis sollte 2 mg einmal täglich nicht überschreiten.
Verabreichungsschema
Mekinist und Dabrafenib sind ohne Nahrungsmittel, d.h. mindestens eine Stunde vor bzw. frühestens zwei Stunden nach einer Mahlzeit einzunehmen (siehe «Pharmakokinetik»).
Wenn Mekinist und Dabrafenib in Kombination eingenommen werden, ist die einmal tägliche Dosis Mekinist jeden Tag zur selben Uhrzeit entweder mit der morgendlichen oder der abendlichen Dabrafenib-Dosis einzunehmen. Die Pharmakokinetik der abendlichen Einnahme von Mekinist ist nicht untersucht worden, so dass die morgendliche Einnahme vorzuziehen ist.
Pulver zur Herstellung einer Lösung zum Einnehmen
Bei Mekinist Pulver zur Herstellung einer Lösung zum Einnehmen handelt es sich um eine Flasche mit Pulver, das der Patient zur individuellen Zubereitung oder als gebrauchsfertige Lösung verwenden kann. Nachdem die Lösung zubereitet wurde, muss sie innerhalb von 35 Tagen verbraucht werden. Nicht verbrauchte Lösung muss 35 Tage nach der Rekonstitution verworfen werden.
Bei der Anwendung von Mekinist als Lösung zum Einnehmen muss der Arzt bzw. die Ärztin die Patienteninformation und die Anweisung zur Zubereitung und Einnahme von Mekinist durchlesen und mit dem Patienten oder der/den Pflegeperson(en) besprechen. Der Arzt sollte sich vergewissern, dass der Patient oder die Pflegeperson(en) verstehen, wie Mekinist Pulver zur Herstellung einer Lösung zum Einnehmen mit Wasser zu mischen und die richtige Tagesdosis zu verabreichen ist.
Eine vollständige und bebilderte Gebrauchsanweisung für die Herstellung der Lösung zum Einnehmen finden Sie in der Rubrik «Hinweise für die Anwendung und Handhabung».
Therapiedauer
Bei Patienten mit inoperablem oder metastasiertem Melanom oder metastasiertem nicht-kleinzelligem Lungenkrebs wird die Fortführung der Therapie bis zum Fortschreiten der Krankheit oder bis zum Auftreten inakzeptabler Nebenwirkungen empfohlen.
Bei der adjuvanten Therapie des Melanoms ist die Therapiedauer auf maximal 1 Jahr begrenzt.
Bei pädiatrischen Patienten mit LGG wird eine Fortführung der Therapie bis zur Krankheitsprogression oder bis zu inakzeptabler Toxizität empfohlen. Dabei ist zu beachten, dass in den klinischen Studien bei pädiatrischen Patienten mit LGG deutliche Unterschiede in der Beurteilung der Wirksamkeit zwischen der unabhängigen Überprüfung und den Prüfzentren beobachtet wurden, die auch die Feststellung einer Krankheitsprogression umfassten (siehe auch «Klinische Wirksamkeit»). Darüber hinaus gibt es nur begrenzte Daten zur Langzeit-Anwendung und optimaler Therapiedauer der Behandlung mit Mekinist in Kombination mit Dabrafenib in der pädiatrischen Population (siehe «Warnhinweise und Vorsichtsmassnahmen»). Die mediane Therapiedauer in den relevanten klinischen Studien betrug bisher ca. 24 Monate (siehe «Klinische Wirksamkeit»). Weiterhin liegen nur begrenzte Daten über Patienten mit LGG im Alter von über 18 Jahren vor, die eine erste systemische Therapie benötigen. Daher sollte die Fortsetzung der Behandlung bis ins Erwachsenenalter auf der Grundlage der vom Arzt bzw. der Ärztin vorgenommenen Nutzen-Risiko-Abwägung erfolgen.
Behandlung von Pyrexie: Die Therapie sollte unterbrochen werden (Mekinist, wenn es als Monotherapie angewendet wird, bzw. sowohl Mekinist als auch Dabrafenib, wenn sie in Kombination angewendet werden), wenn die Körpertemperatur eines Patienten ≥38 °C (≥100,4 °F) beträgt. Im Falle eines Rezidivs kann die Therapie auch beim ersten Symptom einer Pyrexie unterbrochen werden. Eine Behandlung mit Antipyretika wie Ibuprofen oder Paracetamol sollte eingeleitet werden. Die Patienten sollten auf Anzeichen und Symptome einer Infektion untersucht werden (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»). Die Behandlung mit Mekinist bzw. sowohl mit Mekinist als auch mit Dabrafenib, wenn diese in Kombination angewendet werden, sollte wieder aufgenommen werden, wenn der Patient seit mindestens 24 Stunden symptomfrei ist: Entweder (1) in der gleichen Dosisstufe oder (2) um eine Dosisstufe reduziert, falls die Pyrexie rezidivierend auftritt und/oder von anderen schweren Symptomen wie Dehydratation, Hypotonie oder Nierenversagen begleitet war. Die Anwendung von oralen Kortikosteroiden sollte in den Fällen in Betracht gezogen werden, in denen Antipyretika nicht ausreichend wirken.
Wenn unter gleichzeitiger Behandlung mit Mekinist und Dabrafenib behandlungsbedingte Toxizitätserscheinungen auftreten, sollten die Dosen beider Arzneimittel gleichzeitig reduziert oder beide Behandlungen unterbrochen bzw. ganz abgebrochen werden, wobei die unten stehenden Ausnahmen gelten.
Ausnahmen, bei denen nur bei Mekinist eine Dosisänderung erforderlich ist:
·Reduzierte linksventrikuläre Ejektionsfraktion (LVEF)
·Netzhautvenenverschluss (RVO [retinal vein occlusion]), Ablösung des Netzhautpigmentepithels/Netzhautablösung (RPED [retinal pigment epithelial detachment]) und Chorioretinopathie
·Pneumonitis und interstitielle Lungenerkrankung (Interstitial Lung Disease, ILD)
Behandlung von LVEF-Reduktion/Linksventrikuläre Dysfunktion
LVEF soll vor Einleitung der Behandlung mit Mekinist in Kombination mit Dabrafenib sowie einen Monat nach Einleitung der Therapie und danach alle 3 Monate während der Behandlung mittels Ultraschall des Herzens oder mittels MUGA (multi-gated acquisition) Scan beurteilt werden.
Bei Patienten, die mit Mekinist in Kombination mit Dabrafenib behandelt werden, ist die Mekinist-Behandlung zu unterbrechen bei Patienten mit asymptomatischer, absoluter Abnahme der LVEF um >10% gegenüber Behandlungsbeginn und einer Ejektionsfraktion unterhalb der unteren Normbereichsgrenze (LLN) für die jeweilige Einrichtung (siehe «Warnhinweise und Vorsichtsmassnahmen»). Mit Dabrafenib kann in unveränderter Dosierung weiterbehandelt werden. Wenn die LVEF wiederhergestellt ist, kann erneut mit der Mekinist-Behandlung begonnen werden, allerdings in einer um eine Dosisstufe reduzierten Dosis bei sorgfältiger Überwachung. Bei linksventrikulärer kardialer Dysfunktion vom Grad 3 oder 4 oder wenn die LVEF nicht wiederhergestellt werden kann, ist Mekinist dauerhaft abzusetzen.
Behandlung von Netzhautvenenverschluss (RVO [retinal vein occlusion]), Ablösung des Netzhautpigmentepithels/Netzhautablösung (RPED [retinal pigment epithelial detachment]) und Chorioretinopathie
Falls Patienten unter der Behandlung mit Mekinist über neu aufgetretene Sehstörungen berichten, wie zentrale Visusstörung, Verschwommensehen oder Verlust des Sehvermögens, ist eine dringende ophthalmologischen Abklärung notwendig. Bei Patienten, bei denen eine RVO diagnostiziert wird, sollte die Behandlung mit Mekinist endgültig abgesetzt werden. Die Therapie mit Dabrafenib kann in gleicher Dosierung weitergeführt werden. Falls eine RPED oder eine Chorioretinopathie diagnostiziert wird, folgen Sie dem Dosisanpassungsplan für Mekinist in Tabelle 3 – die Behandlung mit Dabrafenib wird in derselben Dosis fortgesetzt (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Behandlung Pneumonitis und interstitielle Lungenerkrankung (ILD)
Bei Pneumonitis- und ILD-Ereignissen folgen Sie nur dem Dosisanpassungsplan für Mekinist in Tabelle 3; es ist keine Änderung der Dabrafenib-Dosis erforderlich.
Kombinationstherapie
Angaben zur Dosisänderung von Dabrafenib finden Sie in der vollständigen Arzneimittelinformation von Dabrafenib.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit leichter Leberfunktionsbeeinträchtigung ist keine Dosisanpassung erforderlich. In einer populationspharmakokinetischen Analyse unterschieden sich die orale Clearance von Trametinib, und damit auch die Exposition, nicht signifikant zwischen Patienten mit leichter Leberfunktionsbeeinträchtigung und lebergesunden Patienten. Verfügbare Daten von Personen mit mässiger bis hochgradiger Leberfunktionsbeeinträchtigung weisen auf eine begrenzte Auswirkung auf die Trametinib-Exposition hin (siehe «Pharmakokinetik»). Mekinist soll bei Personen mit mässiger bis hochgradiger Leberfunktionsbeeinträchtigung mit Vorsicht angewendet werden.
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter bis mässiger Beeinträchtigung der Nierenfunktion ist keine Dosisanpassung erforderlich. Eine leichte bis mässige Nierenfunktionsbeeinträchtigung hatte keine relevanten Auswirkungen auf die Populationspharmakokinetik von Trametinib (siehe «Pharmakokinetik»).
Es liegen keine klinischen Daten zu Mekinist bei Patienten mit schwerer Beeinträchtigung der Nierenfunktion vor, daher lassen sich bezüglich der Notwendigkeit einer Dosisanpassung keine Angaben machen.
Ältere Patienten
Bei Patienten über 65 Jahren ist keine Dosisanpassung erforderlich (siehe «Pharmakokinetik»).
Pädiatrische Population
Die Anwendung von Mekinist bei pädiatrischen Patienten im Alter von weniger als 1 Jahr ist nicht zugelassen. Aufgrund der renalen Toxizität von Dabrafenib in Untersuchungen zur juvenilen Toxizität bei Ratten wurden diese Patienten von klinischen Studien mit Mekinist in Kombination mit Dabrafenib ausgeschlossen. Für Kinder zwischen 1 und 2 Jahren liegen nur begrenzte Daten zur Sicherheit und Wirksamkeit vor (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Verspätete Dosisgabe
Wenn eine Dosis Mekinist vergessen wurde, sollte die Dosis nicht nachgeholt werden, wenn bis zur nächsten planmässigen Dosis weniger als 12 Stunden verbleiben. Wenn eine Dosis Dabrafenib vergessen wurde, sollte die Dosis nicht nachgeholt werden, wenn bis zur nächsten planmässigen Dosis weniger als 6 Stunden verbleiben.
KontraindikationenÜberempfindlichkeit gegen die Wirkstoffe oder einen der sonstigen Bestandteile.
Warnhinweise und VorsichtsmassnahmenDie Ansprechrate auf die Kombination war bei Patienten, die unter Vorbehandlung mit einem BRAF-Inhibitor eine Krankheitsprogression gezeigt hatten, deutlich niedriger als bei Patienten ohne eine solche Vorbehandlung. Deshalb sollten bei Patienten, die unter vorausgehender Behandlung mit einem BRAF-Inhibitor Krankheitsprogression gezeigt hatten, andere Behandlungsmöglichkeiten in Betracht gezogen werden, bevor die Kombination verabreicht wird.
Zusätzliche Informationen bezüglich Warnhinweisen und Vorsichtsmassnahmen in Zusammenhang mit einer Behandlung mit Dabrafenib finden Sie in der Dabrafenib Fachinformation.
Neue Malignitäten
Bei der Kombination von Mekinist und Dabrafenib können neue Malignitäten, sowohl kutane als auch nicht-kutane, auftreten.
Hyperglykämie
In klinischen Studien mit Mekinist, die mit Dabrafenib verabreicht wurden, benötigten 15% der Patienten mit Diabetes in der Vorgeschichte, die Mekinist mit Dabrafenib erhalten hatten, eine intensivere hypoglykämische Therapie. Hyperglykämie 3. und 4. Grades trat bei 2% der Patienten auf. Überwachen Sie den Serumglucosespiegel zu Beginn und falls klinisch angemessen, wenn Mekinist bei Patienten mit bereits bestehendem Diabetes oder Hyperglykämie mit Dabrafenib verabreicht wird.
Interstitielle Lungenerkrankung (ILD)
In verschiedenen klinischen Studien mit Mekinist traten bei 2% der Patienten ILD oder Pneumonitis auf. Mekinist muss bei Patienten mit neuen oder fortschreitenden pulmonalen Symptomen und Befunden, einschliesslich Husten, Atemnot, Hypoxie, Pleuraerguss oder Infiltraten, bis zur klinischen Abklärung ausgesetzt werden. Bei Patienten, bei denen eine behandlungsbedingte ILD oder Pneumonitis diagnostiziert wird, muss Mekinist dauerhaft abgesetzt werden.
Kutanes Plattenepithelkarzinom (cutaneous Squamous Cell Carcinoma, cuSCC)
Fälle von cuSCC (darunter auch diejenigen, die als Subtyp Keratoakanthom oder gemischtes Keratoakanthom klassifiziert werden) wurden bei Patienten, die mit Mekinist in Kombination mit Dabrafenib behandelt wurden, berichtet.
In einer Phase-3-Studie trat cuSCC bei 2 % der Patienten auf, die Mekinist in Kombination mit Dabrafenib erhielten, und bei 9 % der Patienten, die Dabrafenib als Monotherapie erhielten. Die mittlere Zeit bis zur Diagnose des ersten Auftretens von cuSCC im Kombinationstherapie-Arm betrug 222 Tage (bei einer Zeitspanne von 56 bis 328 Tagen) und 57 Tage (bei einer Zeitspanne von 9 bis 169 Tagen) im Dabrafenib-Monotherapie-Arm.
Vor dem Behandlungsbeginn mit Dabrafenib sollte eine Hautuntersuchung durchgeführt werden, ebenso während der Behandlung mit Dabrafenib alle 2 Monate während der gesamten Therapie. Die Überwachung sollte jeden 2. bis 3. Monat für 6 Monate nach Beendigung der Behandlung mit Dabrafenib weitergeführt werden oder so lange, bis eine neue antineoplastische Therapie begonnen wird.
Fälle von cuSCC sollten durch dermatologische Exzision behandelt werden, eine Änderung der Behandlung ist nicht erforderlich. Die Patienten sollten angewiesen werden, ihren Arzt sofort zu informieren, wenn sich neue Läsionen bilden.
Blutungen
Blutungsereignisse, darunter schwere Blutungsereignisse in kritische Körperbereiche und Organe, sind bei Patienten aufgetreten, die Mekinist in Kombination mit Dabrafenib angewendet haben (siehe «Unerwünschte Wirkungen»). In klinischen Studien mit dieser Kombination wurden in 17% der Patienten Blutungen berichtet, 3% der Patienten hatten gastrointestinale Blutungen. Sechs von 559 Patienten (1%) mit nicht resezierbarem oder metastasiertem Melanom, die Mekinist in Kombination mit Dabrafenib erhielten, hatten tödliche intrakranielle Blutungsereignisse. Drei Fälle wurden in einer Studie MEK115306 (COMBI-d) und drei in einer Studie MEK116513 (COMBI-v) beobachtet. Bei der adjuvanten Behandlung des Melanoms traten in der Phase-III-Studie keine tödlich verlaufenden Blutungsereignisse auf.
Zwei von 93 Teilnehmern (2 %), die Mekinist in Kombination mit Dabrafenib in einer Phase-II-Studie zu nicht-kleinzelligem Lungenkarzinom (NSCLC; non-small cell lung cancer) erhielten, hatten tödliche Blutungsereignisse (einen Fall von Subarachnoidalblutung und einen Fall von retroperitonealer Blutung).
Wenn Patienten Symptome einer intrakraniellen Blutung entwickeln, ist eine umgehende medizinische Versorgung erforderlich.
Das Risiko von Blutungsereignissen kann durch die begleitende Anwendung einer Antiplatelet- oder Antikoagulans-Therapie erhöht werden. Bei Blutungen sollten Patienten gemäss klinischer Indikation behandelt werden.
Bei Blutungen vom Grad 4 ist Mekinist dauerhaft abzusetzen, ebenso bei Grad 3 Blutungen, die sich nicht bessern.
Neues Primärmelanom
In einem Fall wurden in einer Phase-3-Studie bei einem Patienten, der Mekinist in Kombination mit Dabrafenib erhielt, neue Primärmelanome gemeldet, die 110 Tage nach Studienbeginn auftraten (< 1 %). Fälle von neuen Primärmelanomen können durch Exzision behandelt werden, eine Änderung der Behandlung ist nicht erforderlich. Die Überwachung von Hautläsionen sollte in gleicher Weise wie bei cuSCC erfolgen.
Nicht-kutane sekundäre/rezidivierende Malignität
In-Vitro-Experimente haben eine paradoxe Aktivierung des MAP-Kinase Signalwegs in BRAF-Wildtyp-Zellen mit RAS-Mutationen bei einer Exposition gegenüber BRAF-Inhibitoren gezeigt, was zu einem erhöhten Risiko nicht-kutaner Malignitäten bei mit Dabrafenib behandelten Patienten führen kann. Bei BRAF-Inhibitoren wurden Fälle von RAS-getriebenen Malignomen festgestellt. Patienten sollten gemäss klinischer Notwendigkeit überwacht werden. Vor der Fortführung der Behandlung mit Dabrafenib bei Patienten mit nicht-kutaner Malignität mit RAS-Mutation sollten die Vorteile und Risiken in Betracht gezogen werden. Unter der Kombinationstherapie von Mekinist mit Dabrafenib sind keine Dosisanpassungen für Mekinist erforderlich.
Nach dem Absetzen von Dabrafenib sollte die Überwachung nicht-kutaner sekundärer/rezidivierender Malignome mindestens 6 Monate weitergeführt werden, oder so lange, bis eine neue antineoplastische Therapie begonnen wird.
Pyrexie
In den klinischen Studien mit Mekinist in Kombination mit Dabrafenib wurde über das Auftreten von Pyrexie berichtet. Die Mehrheit der Pyrexie-Ereignisse trat innerhalb des ersten Therapiemonats auf. Etwa ein Drittel der Patienten unter der Kombinationstherapie, bei denen Pyrexie auftrat, zeigte drei oder mehr Ereignisse. Pyrexie kann von schwerem Schüttelfrost, Dehydration und Hypotonie begleitet werden, was in manchen Fällen zu akuter Niereninsuffizienz führen kann. Während und nach schweren Pyrexie-Ereignissen sollte das Serum-Kreatinin und die Nierenfunktion überwacht werden. Es wurden schwerwiegende, nicht infektiöse febrile Ereignisse beobachtet. Diese Ereignisse haben in klinischen Studien gut auf Dosisunterbrechungen und/oder Dosisreduzierungen sowie auf unterstützende Versorgung angesprochen (inklusive der Verabreichung von nicht-steroidalen und steroidalen Antipyretika). Ein studienübergreifender Vergleich bei 1810 Patienten, die mit der Kombinationstherapie behandelt wurden, zeigte eine Verringerung der Inzidenz von hochgradiger Pyrexie und anderen pyrexiebezogenen unerwünschten Ereignissen, wenn die Behandlung sowohl mit Mekinist als auch mit Dabrafenib unterbrochen wurde, im Vergleich zu einer Unterbrechung nur von Dabrafenib. Daher wird eine Unterbrechung sowohl von Mekinist als auch von Dabrafenib empfohlen, wenn die Körpertemperatur des Patienten ≥38 °C (≥100,4 °F) beträgt. Im Falle eines Rezidivs kann die Therapie auch beim ersten Symptom einer Pyrexie unterbrochen werden (siehe Abschnitte «Dosierung/Anwendung» und «Klinische Wirksamkeit»).
Siehe die Fachinformation von Dabrafenib für die weiteren Informationen zur Behandlung von Pyrexie (siehe «Dosierung/Anwendung»).
Kolitis und Perforation des Magens oder der Darmwand (Magen-/Darmdurchbruch)
Kolitis und eine Perforation des Magens oder der Darmwand (Magen-/Darmdurchbruch), einschliesslich Fälle mit tödlichem Ausgang, sind bei Patienten aufgetreten, die Mekinist als Monotherapie und in Kombination mit Dabrafenib einnahmen (siehe Rubrik «Unerwünschte Wirkungen»). Die Behandlung mit Mekinist als Monotherapie oder in Kombination mit Dabrafenib muss bei Patienten mit Risikofaktoren für einen Magen-/Darmdurchbruch (Perforation), einschliesslich einer Vorgeschichte von Divertikulitis, Metastasen des Gastrointestinaltrakts mit Vorsicht angewendet werden und die gleichzeitige Anwendung von Arzneimitteln, die bekanntermassen das Risiko einer gastrointestinalen Perforation mit sich bringen (z.B. Bevacizumab, Aflibercept und andere Angiogenese-Inhibitoren), ist zu vermeiden.
Wenn bei Patienten Symptome einer Kolitis oder eines Magen- oder Darmdurchbruchs auftreten, ist eine umgehende medizinische Versorgung notwendig.
LVEF-Reduktion/Linksventrikuläre Dysfunktion
Unter Mekinist, in Kombination mit Dabrafenib, wurde eine Senkung der LVEF gemeldet (siehe «Unerwünschte Wirkungen»). In klinischen Studien lag die mediane Zeit bis zum Beginn des ersten Auftretens von linksventrikulärer Dysfunktion, Herzinsuffizienz und LVEF-Abfall bei Patienten, die mit Mekinist in Kombination mit Dabrafenib behandelt wurden, zwischen zwei und fünf Monaten. Mekinist sollte bei Patienten mit Erkrankungen, die die linksventrikuläre Funktion einschränken könnten, mit Vorsicht angewendet werden. Die LVEF sollte bei allen Patienten vor Einleitungder Mekinist-Behandlung, einen Monat nach Beginn der Therapie und danach in ungefähr 3-monatigen Abständen während der Behandlung beurteilt werden (siehe «Dosierung/Anwendung»).
EKG
Mekinist als Monotherapie beeinflusst nicht oder nur geringgradig das QTc-Intervall. Dagegen wird unter Mekinist eine Senkung der Herzfrequenz sowie eine Verlängerung des PR Intervalls beobachtet.
Die Kombination von Mekinist mit Dabrafenib ist nicht in einer gezielten Studie bezüglich QTc Intervall untersucht worden.
Sehstörungen
Mit Mekinist wurden Erkrankungen im Zusammenhang mit Sehstörungen, insbesondere Ablösung des Netzhautpigmentepithels/Netzhautablösung (RPED), Chorioretinopathie und Netzhautvenenverschluss (RVO), beobachtet. In den klinischen Studien mit Mekinist wurde über Symptome wie Verschwommensehen, verminderte Sehschärfe und sonstige Sehstörungen berichtet. Mekinist wird bei Patienten mit anamnestisch bekannter RVO nicht empfohlen.
Zu Beginn und während der Behandlung mit Mekinist sollte, falls klinisch erforderlich, eine umfassende ophthalmologische Untersuchung durchgeführt werden. Falls Patienten unter der Behandlung mit Mekinist über neu aufgetretene Sehstörungen berichten, wird zu einer prompten ophthalmologischen Abklärung geraten. Wird eine retinale Anomalie festgestellt, sollte die Behandlung mit Mekinist umgehend unterbrochen werden und eine Überweisung an einen Netzhautspezialisten ist in Betracht zu ziehen. Falls eine RPED oder Chorioretinopathie diagnostiziert wird, sollte nach dem Dosismodifikationsschema in Tabelle 1 und 2 des Abschnitts «Dosierung/Anwendung» vorgegangen werden. Bei Patienten, bei denen eine RVO diagnostiziert wird, sollte die Behandlung mit Mekinist endgültig abgesetzt werden.
Hautausschlag
In klinischen Studien mit Mekinist in Kombination mit Dabrafenib kam es bei ca. 20 - 30 % der Patienten zu einem Hautausschlag (siehe «Unerwünschte Wirkungen»). Mehrheitlich waren diese Fälle vom Schweregrad 1 oder 2 und erforderten weder eine Unterbrechung der Behandlung noch eine Dosisreduktion.
Schwere unerwünschte Hautreaktionen
Es wurde über Fälle von schweren unerwünschten Hautreaktionen (SCARS, severe adverse skin reactions) unter der Therapie mit Mekinist in Kombination mit Dabrafenib berichtet. Zu diesen gehören u.a. das Stevens-JohnsonSyndrom und die Arzneimittelreaktion mit systemischen Symptomen und Eosinophilie (DRESS, drug reaction with systemic symptoms and eosinophilia), welche lebensbedrohlich oder tödlich verlaufen können. Daher müssen die Patienten vor Beginn der Behandlung über die jeweiligen Anzeichen und Symptome aufgeklärt werden. Während der Therapie sollten die Patienten engmaschig auf Hautreaktionen überwacht werden. Falls Anzeichen und Symptome einer schweren unerwünschten Hautreaktion auftreten, müssen Mekinist und Dabrafenib abgesetzt werden
Tiefenvenenthrombose (TVT)/Lungenembolie (LE)
TVT und LE können bei Anwendung von Mekinist in Kombination mit Dabrafenib auftreten. Wenn Patienten Symptome einer Lungenembolie oder Tiefenvenenthrombose entwickeln, ist eine umgehende medizinische Versorgung erforderlich.
Hemophagocytic lymphohistiocytosis (HLH)
In der Erfahrung nach der Markteinführung von Mekinist in Kombination mit Tafinlar (dabrafenib) wurde HLH beobachtet. HLH ist ein potentiell lebensbedrohliches Syndrom mit pathologischer Aktivierung der Immunabwehr. Falls die HLH nicht frühzeitig erkannt und behandelt wird, verläuft sie häufig letal. Die Erkrankung ist gekennzeichnet durch klinische Anzeichen und Symptome einer schweren systemischen Entzündung wie Fieber, Hautausschlag, Hepatosplenomegalie, Zytopenien, Lymphadenopathie, neurologische Symptome, hohes Serum-Ferritin, Hypertriglyceridämie sowie Störungen der Leberfunktion und der Koagulation. Die Symptome treten in der Regel innerhalb von 2 Monaten nach Beginn der Behandlung auf, ein späteres Auftreten ist aber möglich. Bei Verdacht auf HLH sollte die Behandlung unterbrochen werden. Nach Bestätigung von HLH sollte die Behandlung abgebrochen und eine geeignete HLH-Behandlung eingeleitet werden.
Fertilität
Zu den Auswirkungen von Trametinib auf die Fertilität beim Menschen liegen keine Daten vor. Fertilitätsstudien an Tieren wurden zwar nicht durchgeführt, es konnten jedoch unerwünschte Wirkungen auf die weiblichen Fortpflanzungsorgane beobachtet werden (siehe «Präklinische Daten»). Trametinib beeinträchtigt möglicherweise die Fertilität beim Menschen.
Pädiatrische Population
Die sich in Entwicklung befindlichen pädiatrische Patienten unterliegen einer potentiellen Langzeittherapie mit Mekinist in Kombination mit Dabrafenib. Gleichzeitig wirken beide Arzneimittel auf eine Signaltransduktionskette, die eine wichtige Rolle bei der Regulierung von Zell- und Gewebeentwicklung spielt (siehe «Wirkungsmechanismus»). Vor diesem Hintergrund weisen die in der pädiatrischen Population vorliegenden Daten Limitationen auf (siehe auch «Dosierung/Anwendung»).
So liegen keine ausreichenden Daten zu Sicherheit und Wirksamkeit der Langzeit-Anwendung und optimaler Therapiedauer der Behandlung mit Mekinist in Kombination mit Dabrafenib in der pädiatrischen Population vor. Die mediane Therapie- und Nachbeobachtungsdauer betrug in den relevanten klinischen Studien bisher ca. 24 respektive 26 Monate (siehe «Klinische Wirksamkeit»). Die Langzeitfolgen der unter der kombinierten sehr häufig beobachteten unerwünschten Gewichtszunahmen sind derzeit unklar. Obwohl bisher keine Zunahme der Häufigkeiten sekundärer maligner Neoplasien in der pädiatrischen Population berichtet wurde, ist eine abschliessende Bewertung zum jetzigen Zeitpunkt nicht möglich (siehe «Unerwünschte Wirkungen»).
Für Kinder zwischen 1 und 2 Jahren liegen nur begrenzte Daten zur Sicherheit und Wirksamkeit vor.
Der Beitrag von Mekinist und Dabrafenib zur Wirksamkeit der Kombination bei pädiatrische Patienten mit LGG ist unklar, da ein direkter Vergleich zwischen Kombination und Monotherapien in einer ausreichend grossen Patientenpopulation fehlt.
Von klinischen Studien ausgeschlossene Patienten
Die gemeinsamen Ausschlusskriterien für alle Studien zu Mekinist waren: Wildtyp-BRAF oder eine Nicht-V600-Mutation; Vorgeschichte oder Hinweise auf ein kardiovaskuläres Risiko; Vorgeschichte von Pneumonitis oder interstitieller Lungenerkrankung; Vorgeschichte oder Hinweise auf ein Retinopathierisiko. Studienspezifische Ausschlusskriterien finden Sie im Abschnitt «Klinische Wirksamkeit».
Hilfsstoffe
Sulfobutylbetadex-Natrium
Dieses Arzneimittel enthält 100 mg Cyclodextrin pro mL. Wenden Sie dieses Arzneimittel nicht bei Kindern unter 2 Jahren an, es sei denn, Ihr Arzt bzw. Ihre Ärztin hat es empfohlen.
Methylparahydroxybenzoat (E218)
Kann allergische Reaktionen, auch Spätreaktionen, hervorrufen.
Kalium
Dieses Arzneimittel enthält Kalium, jedoch weniger als 1 mmol (39 mg) Kalium pro 2 mg Trametinib (Maximaldosis), d.h. es ist nahezu «kaliumfrei».
Natrium
Filmtabletten
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Filmtablette, d.h. es ist nahezu «natriumfrei».
Pulver zur Herstellung einer Lösung zum Einnehmen
Dieses Arzneimittel enthält 79.2 mg Natrium (Hauptbestandteil von Kochsalz/Speisesalz) pro 2 mg Trametinib (Maximaldosis). Dies entspricht 4% der für eine oder einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung.
InteraktionenBitte lesen Sie die Fachinformation zu Dabrafenib, um mehr über die Wechselwirkungen im Zusammenhang mit Dabrafenib zu erfahren.
Pharmakokinetische Interaktionen
Da Trametinib vorwiegend über die durch hydrolytische Enzyme (z.B. Carboxylesterasen) vermittelte Deacetylierung metaboliert wird, ist es unwahrscheinlich, dass dessen Pharmakokinetik von anderen Wirkstoffen durch metabolische Wechselwirkungen beeinflusst wird. Die Exposition mit Trametinib-Wiederholungsdosen wurde durch die gleichzeitige Gabe eines CYP3A4-Induktors nicht beeinflusst (siehe «Pharmakokinetik»).
Pharmakodynamische Interaktionen
Entsprechend in-vitro- und in-vivo-Daten ist es unwahrscheinlich, dass Trametinib die Pharmakokinetik anderer Arzneimittel durch Wechselwirkung mit CYP-Enzymen oder -Transportern signifikant beeinflusst (siehe «Pharmakokinetik»).
Die wiederholte Verabreichung einer einmaligen täglichen Dosis von 2 mg Trametinib hatte keinen Einfluss auf Cmax und AUC einer Einzeldosis des CYP2C8/CYP3A4-Substrats Dabrafenib.
Andere Interaktionen
Kombination mit Dabrafenib
Die gleichzeitige Verabreichung wiederholter Dosen Trametinib 2 mg einmal täglich und Dabrafenib 150 mg zweimal täglich führte nicht zu klinisch relevanten Cmax- und AUC-Änderungen bei Trametinib oder Dabrafenib, mit Anstiegen der Cmax und der AUC von Dabrafenib um 23 bzw. 16 %. Anhand einer populationspharmakokinetischen Analyse wurde für die gleichzeitige Verabreichung von Trametinib mit Dabrafenib ein geringfügiger Rückgang der Bioverfügbarkeit von Trametinib, entsprechend einer 12 %igen Abnahme der AUC, geschätzt. Zu Empfehlungen hinsichtlich der Wechselwirkungen von Dabrafenib mit anderen Arzneimitteln, siehe Dabrafenib Fachinformation.
Wirkung von MEKINIST auf andere Arzneimittel
Daten aus In-vitro- und In-vivo-Untersuchungen legen nahe, dass Trametinib die Pharmakokinetik anderer Arzneimittel wahrscheinlich nicht beeinflusst. Nach den Ergebnissen aus In-vitro-Studien ist Trametinib kein Inhibitor von CYP1A2, CYP2A6, CYP2B6, CYP2D6 und CYP3A4. In vitro hat sich Trametinib als Inhibitor von CYP2C8, CYP2C9 und CYP2C19, als Induktor von CYP3A4 sowie als Inhibitor der Transporter OAT1, OAT3, OCT2, MATE1, OATP1B1, OATP1B3, Pgp und BCRP erwiesen. Aufgrund der geringen Dosis und der geringen klinischen systemischen Exposition im Verhältnis zur in-vitro-Potenz von Inhibition bzw. Induktion gilt Trametinib nicht als in-vivo-Inhibitor bzw. -Induktor dieser Enzyme bzw. Transporter.
Wirkung anderer Arzneimittel auf MEKINIST
Daten aus In-vivo- und In-vitro-Untersuchungen legen nahe, dass die Pharmakokinetik von Trametinib wahrscheinlich nicht durch andere Arzneimittel beeinflusst wird. Trametinib ist kein Substrat von CYP-Enzymen oder der Transporter BCRP, OATP1B1, OATP1B3, OATP2B1, OCT1, MRP2 und MATE1. Trametinib wird über Carboxylesterasen deacetyliert; Arzneimittelwechselwirkungen, bei denen um Esterasen konkurriert wird, sind in der Literatur nicht beschrieben. Trametinib ist ein in-vitro-Substrat des Efflux-Transporters Pgp, eine Beeinflussung durch Inhibition dieses Transporters ist jedoch angesichts der hohen passiven Durchlässigkeit und der hohen Bioverfügbarkeit unwahrscheinlich. Nach gleichzeitiger Verabreichung von Trametinib mit dem CYP3A4-Induktor Dabrafenib entsprachen Cmax und AUC von Trametinib nach wiederholter Verabreichung der unter Monotherapie beobachteten Exposition; dies deutet darauf hin, dass ein CYP3A4-Induktor keinen Einfluss auf die Trametinib-Exposition besitzt.
Schwangerschaft, StillzeitSchwangerschaft
Mekinist kann dem Fötus schaden, wenn das Arzneimittel einer schwangeren Frau verabreicht wird. Es liegen keine Daten mit Mekinist bei Schwangeren vor. In tierexperimentellen Studien wurde eine Reproduktionstoxizität festgestellt (siehe «Präklinische Daten»).
Daher sollte Mekinist in Kombination mit Dabrafenib während der Schwangerschaft nicht angewendet werden, es sei denn, es ist klar notwendig. Vor Beginn einer Therapie soll ein Schwangerschaftstest durchgeführt werden.
Weibliche und männliche Patienten müssen wirksame Empfängnisverhütungsmethoden anwenden:
Frauen: Potenziell gebärfähige Frauen müssen während der Behandlung und in den ersten 4 Monaten nach Behandlungsende wirksame Methoden der Empfängnisverhütung anwenden. Wird Mekinist in Kombination mit Dabrafenib in der Schwangerschaft verabreicht oder wird eine Patientin während der Behandlung schwanger, so sollte sie über das potenzielle Risiko für das Ungeborene unterrichtet werden.
Frauen im gebärfähigen Alter, die Mekinist in Kombination mit Dabrafenib erhalten, sollten darüber aufgeklärt werden, dass der Kombinationspartner Dabrafenib die Wirksamkeit von Hormon-Verhütungsmitteln senken kann und dass alternative Empfängnisverhütungsmethoden, angewendet werden sollten (s. «Interaktionen»).
Männer: Männliche Patienten (auch solche, bei denen eine Vasektomie durchgeführt wurde) mit Sexualpartnerinnen, die schwanger sind, möglicherweise schwanger sind oder schwanger werden könnten, müssen während der Monotherapie mit Mekinist bzw. der Kombinationstherapie mit Mekinist und Dabrafenib sowie für mindestens 16 Wochen nach Beendigung der Behandlung mit Mekinist während des Geschlechtsverkehrs Kondome verwenden.
Stillzeit
Es liegen keine Daten über die Wirkung von Mekinist auf das gestillte Kind oder die Wirkung von Mekinist auf die Milchproduktion vor. Zur Ausscheidung von Trametinib in der Muttermilch ist nichts bekannt. Da viele Arzneimittel in der Muttermilch ausgeschieden werden, kann ein Risiko für das gestillte Kind nicht ausgeschlossen werden. Bei stillenden Müttern sollte Mekinist nicht angewendet werden. Unter Abwägung der Wichtigkeit des Stillens für das Kind gegen den Nutzen der Behandlung für die Mutter sollte entweder abgestillt oder die Behandlung mit Mekinist beendet werden.
Fertilität
Daten zur Fertilität beim Menschen mit Mekinist liegen nicht vor. Am Tier wurden unerwünschte Wirkungen auf die männlichen und weiblichen Fortpflanzungsorgane beobachtet (siehe «präklinische Daten»).
Männliche Patienten sollten auf das potenzielle Risiko einer möglicherweise irreversiblen Beeinträchtigung der Spermatogenese hingewiesen werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zu den Auswirkungen von Mekinist auf die Verkehrstüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen durchgeführt. Angesichts der Pharmakologie von Trametinib dürfte es zu keiner ungünstigen Beeinflussung derartiger Tätigkeiten kommen. Ob ein Patient in der Lage ist, Aufgaben zu bewältigen, die Urteilsvermögen und uneingeschränkte motorische bzw. kognitive Leistungsfähigkeit erfordern, sollte unter Berücksichtigung des klinischen Status des Patienten und des Profils unerwünschter Wirkungen von Mekinist beurteilt werden.
Unerwünschte WirkungenDie im Folgenden beschriebenen unerwünschten Arzneimittelwirkungen (UAW) berücksichtigen verschiedene Quellen von Sicherheitsinformationen, einschliesslich klinischer Studien, Berichte nach der Markteinführung und Literaturberichte.
Die Häufigkeit der in Tabelle 4 unten beschriebenen UAW basiert auf der integrierten Sicherheitspopulation von 1087 Patienten mit BRAF V600-mutiertem inoperablem oder metastasiertem Melanom Stadium III, BRAF V600-mutiertes Melanom nach vollständiger Resektion mit adjuvanter Behandlung und fortgeschrittenes NSCLC.
Alle Patienten wurden mit 2 mg Mekinist einmal täglich und 150 mg Dabrafenib zweimal täglich behandelt. Von diesen Patienten wurden 559 in zwei randomisierten Phase-III-Studien, MEK115306 (COMBI-d) und MEK116513 (COMBI-v), mit der Kombination für BRAF V600-mutiertes-Melanom behandelt. 435 wurden mit der Kombination in der adjuvanten Behandlung von BRAF V600-mutiertem Melanom im Stadium III nach vollständiger Resektion in einer randomisierten Phase-III-Studie BRF115532 (COMBI-AD) behandelt, und 93 wurden mit der Kombination für BRAF-V600-mutiertes NSCLC in einer nicht-randomisierten Phase-II-Multi-Kohortenstudie BRF113928 behandelt.
Die häufigsten Nebenwirkungen (Inzidenz >20 %) für Mekinist in Kombination mit Dabrafenib waren: Pyrexie, Müdigkeit, Übelkeit, Schüttelfrost, Kopfschmerzen, Durchfall, Erbrechen, Arthralgie und Hautausschlag.
Die unerwünschten Arzneimittelwirkungen sind nach Systemorganklasse und dann entsprechend ihrer Häufigkeit – die am häufigsten auftretenden zuerst – gemäss der folgenden Konvention aufgeführt: Sehr häufig (≥1/10); häufig (≥1/100, < 1/10); gelegentlich (≥1/1.000, < 1/100); selten (≥1/10.000, < 1/1.000), sehr selten (< 1/10.000), einschliesslich Einzelberichten. Innerhalb jeder Häufigkeitsgruppe sind die unerwünschten Wirkungen in absteigender Reihenfolge des Schweregrads angegeben.
Tabelle 4: Nebenwirkungen, die in der integrierten Sicherheitspopulation von Mekinist in Kombination mit Dabrafenib in den Studien MEK115306, MEK116513a, BRF113928 und BRF115532 (n = 1076) berichtet wurden
|
Infektionen und parasitäre Erkrankungen
| |
Sehr häufig
|
Nasopharyngitis (11 %)
| |
Häufig
|
Harnwegsinfektion, Cellulitis, Follikulitis, Paronychie, pustulärer Hautausschlag
| |
Gutartige, bösartige und unspezifische Neubildungen (einschl. Zysten und Polypen)
| |
Häufig
|
Kutanes Plattenepithelkarzinom (squamous cell carcinoma, SCC)b,Papillomec, seborrhoische Keratose
| |
Gelegentlich
|
Neues Primärmelanomd, Acrochordon (Fibrome)
| |
Erkrankungendes Blutes- und des Lymphsystems
| |
Häufig
|
Neutropenie, Anämie, Thrombozytopenie, Leukopenie
| |
Erkrankungen des Immunsystems
| |
Gelegentlich
|
Überempfindlichkeite ,Sarkoidose
| |
Nicht bekannt*
|
Hämophagozytische Lymphohistiozytose
| |
Stoffwechsel- und Ernährungsstörungen
| |
Sehr häufig
|
Verminderter Appetit (14 %)
| |
Häufig
|
Dehydration, Hyponaträmie, Hypophosphatämie, Hyperglykämie
| |
Erkrankungen des Nervensystems
| |
Sehr häufig
|
Kopfschmerzen (33%), Schwindelgefühl (11%)
| |
Augenerkrankungen
| |
Häufig
|
Verschwommene Sicht, Sehverschlechterung, Uveitis
| |
Gelegentlich
|
Chorioretinopathie, Ablösung des Netzhautpigmentepithels/ Netzhautablösung (RPED), periorbitales Ödem
| |
Herzerkrankungen
| |
Häufig
|
Verminderte Ejektionsfraktion
| |
Gelegentlich
|
Bradykardie
| |
Selten
|
Myokarditis*
| |
Gefässerkrankungen
| |
Sehr häufig
|
Hypertonie (19 %), Hämorrhagief (19%)
| |
Häufig
|
Hypotonie, Lymphödem
| |
Erkrankungen der Atemwege, des Brustraums und Mediastinums
| |
Sehr häufig
|
Husten (20 %)
| |
Häufig
|
Dyspnoe
| |
Gelegentlich
|
Pneumonitis
| |
Erkrankungen des Gastrointestinaltrakts
| |
Sehr häufig
|
Übelkeit (38 %), Diarrhö (32 %), Erbrechen (29 %), Bauchschmerzen (17%)g, Verstopfung (13 %)
| |
Häufig
|
Mundtrockenheit, Stomatitis
| |
Gelegentlich
|
Pankreatitis, Kolitis
| |
Selten
|
gastrointestinale Perforation
| |
Leber und Gallenerkrankungen
| |
Sehr häufig
|
Erhöhte Alaninaminotransferase (14 %), Erhöhte Aspartataminotransferase (13 %)
| |
Häufig
|
Erhöhte alkalische Phosphatase im Blut, Erhöhte Konzentrationen der Gamma-Glutamyltransferase
| |
Erkrankungen der Haut und des Unterhautgewebes
| |
Sehr häufig
|
Ausschlag (24 %), Trockene Haut (13 %), Pruritus (10 %), Erythemh (10%)
| |
Häufig
|
akneiforme Dermatitis, aktinische Keratose, Nachtschweiss, Hyperkeratose, Alopezie, Erythrodysästhesie-Syndrom der Handflächen und Fusssohlen, Hautläsion, Hyperhidrose, Hautfissuren, Pannikulitis, Lichtempfindlichkeiti
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
| |
Sehr häufig
|
Arthralgie (26%), Myalgie (15 %), Schmerzen in den Extremitäten (12 %), Muskelkrämpfek (10%)
| |
Häufig
|
Erhöhte Kreatinphosphokinase im Blut
| |
Erkrankungen der Nieren und Harnwege
| |
Häufig
|
Nierenversagen
| |
Gelegentlich
|
Nephritis
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
| |
Sehr häufig
|
Pyrexie (58 %), Müdigkeit (38 %), Schüttelfrost (33 %), peripheres Ödem (16 %), Asthenie (15 %), grippaler Infekt (11%)
| |
Häufig
|
Schleimhautentzündung, Gesichtsödem
|
a Das Sicherheitsprofil von MEK116513 ähnelt im Allgemeinen dem von MEK115306, mit folgenden Ausnahmen: 1) Die folgenden Nebenwirkungen haben eine höhere Häufigkeitskategorie im Vergleich zu MEK115306: Muskelkrämpfe (sehr häufig); Nierenversagen und Lymphödem (häufig); akutes Nierenversagen (gelegentlich); 2) Die folgenden Nebenwirkungen traten in MEK116513 auf, aber nicht in MEK115306: Herzinsuffizienz, linksventrikuläre Dysfunktion, interstitielle Lungenerkrankung (gelegentlich). 3) Die folgende Nebenwirkung ist in MEK116513 und BRF115532 aufgetreten, aber nicht in MEK115306 und BRF113928: Rhabdomyolyse (gelegentlich)
b schliesst die, SCC der Haut, SCC in-situ (Morbus Bowen) und Keratoakanthom ein
c schliesst Papillome, Hautpapillome ein
d schliesst bösartiges Melanom; metastasiertes, bösartiges Melanom und oberflächlich spreitendes Melanom im Stadium III ein
e schliesst Überempfindlichkeit gegenüber Arzneimittel ein
f Blutungen von verschiedenen Stellen, einschliesslich intrakranieller Blutungen und tödlicher Blutungen
g schliesst Abdominalschmerz, Oberbauchschmerzen und Unterbauchschmerzen ein
h schliesst Erythem, generalisiertes Erythem ein
i Fälle von Lichtempfindlichkeit wurden auch während der Post-Marketing-Erfahrung beobachtet. Alle in den klinischen Studien COMBI-d und COMBI-v berichteten Fälle waren vom Grad 1 und es war keine Dosisänderung erforderlich.
k schliesst Muskelkrämpfe und muskuloskelettale Steifigkeit ein
*Häufigkeit basiert auf dem Bericht nach der Markteinführung
Spezialpopulationen
Pädiatrische Patienten
Mekinist in Kombination mit Dabrafenib
Die Sicherheit von Mekinist in Kombination mit Dabrafenib wurde bei 171 pädiatrischen Patienten in zwei Studien untersucht (Studie DRB436G2201; n=123 und Studie TMT212 X2101, n = 48), davon 159 mit BRAF V600E-Mutation-positivem Gliom.
Das allgemeine Sicherheitsprofil in der pädiatrischen Population war ähnlich wie das bei Erwachsenen beobachtete Sicherheitsprofil. Die am häufigsten gemeldeten unerwünschten Arzneimittelwirkungen (≥20 %) waren Fieber (Pyrexie) (65%), Hautausschlag (47%), Kopfschmerzen (39%), Erbrechen (38%), trockene Haut (34%), Müdigkeit (Fatigue) (33%), Diarrhoe (30%), Blutungen (29%), Neutropenie (25%), Übelkeit (25%), akneiforme Dermatitis (25%), Bauchschmerzen (23%) und Husten (21%).Im pädiatrischen Sicherheitspool wurde eine unerwünschte Arzneimittelwirkung in Form einer Gewichtszunahme mit einer Häufigkeit von 15,2 % (sehr häufig) festgestellt. Bei 51 von 171 Patienten (29.8 %) war der BMI im Vergleich zu Baseline um ≥2 BMI-Perzentilkategorien gestiegen.
In Studie DRB436G2201 wurde bei 20,5% (15 von 73) der pädiatrischen Patienten, die mit Mekinist in Kombination mit Dabrafenib behandelt wurden, eine COVID-19 Infektion berichtet, in einem Fall vom Schweregrad ≥3.
Im pädiatrischen Sicherheitspool wurde bei 8,7% (14 von 161) der pädiatrischen Patienten unter kombinierter Behandlung mit Mekinist und Dabrafenib eine Abnahme der linksventrikulären Ejektionsfraktion (LVEF) von 10 % oder mehr gegenüber dem Ausgangswert und unterhalb der institutionellen Untergrenze des Normalwerts (LLN) beobachtet.
Weitere unerwünschte Arzneimittelwirkungen, die bei pädiatrischen Patienten im Vergleich zu erwachsenen Patienten häufiger auftraten, waren Neutropenie, akneiforme Dermatitis, Paronychie, Anämie, Leukopenie (sehr häufig); Bradykardie, generalisierte exfoliative Dermatitis, Überempfindlichkeit und Pankreatitis (häufig). Darüber hinaus war in der LGG-Kohorte der Studie G2201 die relative Inzidenz von Lymphozytenerhöhungen, Magnesiumerhöhungen und niedrigem systolischem Blutdruck bei zielgerichteter Therapie höher als bei Chemotherapie.
Tabelle 5: Häufigste unerwünschte Arzneimittelwirkungen vom Grad 3/4 (≥2%) bei Mekinist in Kombination mit Dabrafenib bei pädiatrischen Patienten
|
Unerwünschte Arzneimittelwirkungen
|
Mekinist in Kombination mit Dabrafenib
N=171
| |
Grade 3 und 4 (%)
| |
Neutropenie
|
25 (15)
| |
Fieber (Pyrexie)1
|
15 (9)
| |
Alanin-Aminotransferase erhöht²
|
10 (6)
| |
Aspartat-Aminotransferase erhöht3
|
6 (4)
| |
Gewichtszunahme (Gewicht erhöht)
|
7 (4)
| |
Kopfschmerzen
|
5 (3)
| |
Erbrechen
|
5 (3)
| |
Hypotension
|
4 (2)
| |
Hautausschlag4
|
4 (2)
| |
Alkalische Phosphatase im Blut erhöht
|
4 (2)
| |
1
Neutropenie umfasst Neutropenie, Neutrophilenzahl erniedrigt und febrile Neutropenie.
2 ALT umfasst Alanin-Aminotransferase erhöht und Transaminasen erhöht.
3 AST umfasst Aspartat-Aminotransferase erhöht und Transaminasen erhöht.
4 Ausschlag umfasst Ausschlag, Ausschlag makulo-papulös, Ausschlag papulös, erythematöser Hautausschlag, Ausschlag papulös und Ausschlag makulös.
|
Bei pädiatrischen Patienten kleiner 6 Jahre wurden unter kombinierter Behandlung mit Mekinist und Dabrafenib mehr schwerwiegende Ereignisse berichtet als bei Patienten im Alter zwischen 6 und 12 Jahren. Fieber trat bei Kindern kleiner 6 Jahre häufiger auf als bei älteren pädiatrischen Patienten.
Der pädiatrische Sicherheitspool enthält nur vier Kinder im Alter zwischen 1 und 2 Jahren, so dass das Sicherheitsprofil in dieser Alterkategorie unvollständig charakterisiert ist.
QT-Verlängerung
Für Informationen zu QT-Verlängerung siehe «Eigenschaften/Wirkungen».
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungFälle einer Überdosierung von Trametinib von mehr als 4 mg täglich wurden aus den klinischen Studien nicht gemeldet. Im Rahmen klinischer Studien wurden Dosen von bis zu 4 mg oral einmal täglich und Stossdosen von 10 mg oral einmal täglich über zwei aufeinanderfolgende Tage hinweg untersucht.
Behandlung
Das Vorgehen zur Behandlung einer Überdosierung richtet sich nach den klinischen Erfordernissen bzw., sofern verfügbar, nach den Empfehlungen des jeweiligen toxikologischen Informationszentrums. Für den Fall einer Überdosierung von Trametinib ist keine spezifische Behandlung verfügbar. Im Falle einer Überdosierung sollte der Patient die jeweils angemessene unterstützende Behandlung erhalten und entsprechend überwacht werden. Eine Hämodialyse dürfte aufgrund der hohen Plasmaproteinbindung von Trametinib keine Steigerung der Elimination bewirken.
Eigenschaften/WirkungenATC-Code
L01EE01
Wirkungsmechanismus
Trametinib ist ein reversibler, hoch selektiver, allosterischer Hemmer der Aktivierung und Kinaseaktivität von MEK 1 und MEK 2 (MEK= Mitogen-aktivierte, durch extrazelluäre Signale regulierte Kinasen). MEK-Proteine sind kritische Komponenten des durch extrazelluläre Signale regulierten Kinase (ERK)-Wegs. Bei Melanomen und anderen Krebserkrankungen wird dieser Weg häufig durch mutierte Formen von BRAF aktiviert, was nachfolgend die MEK aktiviert und das Tumorwachstum anregt. Trametinib hemmt diese Aktivierung der MEK durch BRAF sowie die MEK-Kinaseaktivität. Das Wachstum von BRAF-V600-mutierten Melanomzelllinien wird durch Trametinib gehemmt und in Tiermodellen mit BRAF-V600-mutiertem Melanom konnte eine Antitumor Wirkung gezeigt werden.
Dabrafenib ist ein potenter, selektiver, ATP-kompetitiver Hemmer von BRAF-V600-mutierten Kinasen und Wildtyp-BRAF- und CRAF-Kinasen. Onkogene Mutationen des BRAF-Gens führen zu einer konstitutiven Aktivierung des RAS/RAF/MEK/ERK-Wegs und zu einer Stimulierung des Tumorzellenwachstums.
Trametinib und Dabrafenib hemmen innerhalb dieser Signaltransduktionskette die beiden Kinasen MEK und BRAF; die Kombination beider Wirkstoffe führt zu einer dualen, wirksamen Hemmung der Signaltransduktionskette. Die Kombination aus Trametinib und Dabrafenib hat sich bei BRAF-V600-mutierten Melanom-Zelllinien in vitro als synergistisch erwiesen und führt zu einer Verzögerung der Resistenzentwicklung in vivo bei BRAF-V600-mutierten Melanom-Heterotransplantaten.
Pharmakodynamik
Trametinib supprimierte die Konzentrationen der phosphorylierten ERK bei BRAF-mutierten Melanom-Zelllinien und in Melanom-Heterotransplantat-Modellen.
Bei Personen mit BRAF- und NRAS-mutiertem Melanom führte die Verabreichung von Trametinib zu dosisabhängigen Veränderungen der Tumor-Biomarker, insbesondere zur Hemmung der phosphorylierten ERK, der Hemmung des Ki67 (einem Marker für die Zellproliferation) sowie zu einem Anstieg des p27 (einem Apoptose-Marker). Die durchschnittlichen Trametinib-Konzentrationen nach wiederholter Verabreichung von 2 mg einmal täglich übersteigen die präklinische Zielkonzentration im 24-Stunden-Dosierungsintervall und sorgen daher für eine anhaltende Inhibierung des MEK-Signalweges.
Kardiale Elektrophysiologie
Studie MEK111054
Das QT-verlängernde Potenzial von Trametinib wurde in einer speziell darauf abgestimmten Stand-alone-Studie der Phase I mit 35 Teilnehmern mit soliden Tumoren (wovon 32 Teilnehmer die Studie abgeschlossen hatten) untersucht.
Diese erhielten 3 mg entsprechendes Placebo am Studientag 1 mit darauffolgender, einmal täglicher 2 mg Trametinib-Dosis sowie 2 Tabletten zu je 0.5 mg entsprechendes Placebo an den Studientagen 2 bis 14, gefolgt von einer einzelnen 3 mg Trametinib Dosis am Studientag 15 (supratherapeutic dose).
Diese Studie zeigte keine QTcF-Intervall-verlängernde Wirkung von Trametinib nach wiederholter Gabe von 2 mg Trametinib, inklusive der einmaligen Gabe der supratherapeutic dose von 3 mg am Studientag 15. Die Analyse der Beziehung zwischen der Veränderung des QTcF-Intervalls seit Baseline und den Plasmakonzentrationen von Trametinib und der vorhergesagten Änderung der QTcF-Strecke zeigt, dass kein eindeutiger Bezug des QTcF-Intervalls zu Trametinib-Plasmakonzentrationen vorlag, bei einer geringen Neigung der modellierten Regressionsgerade; Tendenz von 0.0577 msec/ng/ml (95%-KI -0.124, 0.239).
Die Kombination von Trametinib mit Dabrafenib ist nicht in einer gezielten Studie bezüglich QTc Intervall untersucht worden.
Bei einer 1,5-fachen der empfohlenen Höchstdosis verlängert Mekinist den QT-Intervall nicht klinisch signifikant.
Klinische Wirksamkeit
Nicht resezierbares oder metastasiertes Melanom
Wirksamkeit und Sicherheit der empfohlenen Mekinist-Dosis (einmal täglich 2 mg) in Kombination mit Dabrafenib (zweimal täglich 150 mg) zur Behandlung von erwachsenen Patienten mit nicht resezierbarem oder metastasiertem Melanom mit einer BRAF-V600-Mutation wurden in zwei pivotalen Studien der Phase III untersucht.
Die Kombination von Trametinib 2 mg QD und Dabrafenib 150 mg BID zeigte in einer frühen Studie begrenzte klinische Aktivität bei einer geringen Anzahl von 26 Patienten, bei denen es nach einer Therapie mit einem BRAF-Inhibitor zur Krankheitsprogression kam. Die vom Prüfarzt bewertete, bestätigte Ansprechrate lag bei 15 % (95-%-KI: 4.4, 34.9) und mittlere PFS lag bei 3.6 Monaten (95-%-KI: 1.9, 5.2). Ähnlich waren die Resultate bei den 45 Patienten, die von der Dabrafenib-Monotherapie in die Kombination von Trametinib 2 mg QD und Dabrafenib 150 mg BID in Teil C der Studie wechselten. Bei diesen Patienten wurde eine bestätigte Ansprechrate von 13 % (95-%-KI: 5.0, 27.0) mit einem mittleren PFS von 3.6 Monaten beobachtet (95-%-KI: 2, 4). Diese Ansprechrate ist deutlich tiefer als bei Patienten, die keine Krankheitsprogression unter Vorbehandlung mit einem BRAF-Inhibitor gezeigt hatten, so dass eine Anwendung von Trametinib in Kombination mit Dabrafenib bei diesen Patienten erst nach Abwägen weiterer Therapieoptionen in Betracht gezogen werden sollte.
MEK115306 (COMBI-d)
MEK115306 (COMBI-d) war eine randomisierte, doppelblinde Phase-III-Studie zum Vergleich der Kombination von Trametinib und Dabrafenib gegenüber Dabrafenib und Placebo in der Erstlinientherapie bei Patienten mit nicht resezierbarem (Stadium IIIC) oder metastasiertem (Stadium IV) BRAF-V600E/K-Mutations-positivem kutanem Melanom. Der primäre Endpunkt der Studie war das vom Prüfarzt bewertete progressionsfreie Überleben (PFS). Sekundäre Endpunkte waren das mediane Gesamtüberleben (overall survival, OS), die Gesamtansprechrate (overall response rate, ORR) und die Ansprechdauer (duration of response, DOR). Die Studienteilnehmer wurden nach Laktatdehydrogenase (LDH)-Aktivität (> obere Normgrenze (ONG) versus ≤ ONG) und BRAF-Mutation (V600E versus V600K) stratifiziert.
Es wurden insgesamt 423 Studienteilnehmer im Verhältnis von 1:1 randomisiert und dem Kombinationstherapie-Arm (einmal täglich Trametinib 2 mg und zweimal täglich Dabrafenib 150 mg) (N = 211) bzw. dem Dabrafenib-Monotherapie-Arm (zweimal täglich 150 mg) (N = 212) zugeordnet. Die Merkmale bei Baseline waren in den beiden Behandlungsgruppen ausgeglichen. Bei den meisten Patienten lag eine BRAF-V600E-Mutation vor (85 %); bei den übrigen 15 % der Patienten lag eine BRAF-V600K-Mutation vor.
Zum Zeitpunkt der Primäranalyse des PFS wurden unter der Kombinationstherapie ein medianes PFS von 9.3 Monaten unter der Kombination von Trametinib und Dabrafenib beobachtet und von 8.8 Monaten unter der Dabrafenib-Monotherapie (HR = 0.75, 95%-KI: 0.57, 0.99, p = 0.035). ORR war 67 % vs. 51 % (p = 0.0014) und DOR betrug 9.2 vs. 10.2 Monate bei der Kombination gegenüber der Dabrafenib-Monotherapie. In einer späteren Analyse, welche zusammenfiel mit der Hauptanalyse des OS (siehe hier weiter unten) wurde ein neu deutlicherer Unterschied zugunsten der Kombination von Trametinib und Dabrafenib als in der vorausgegangenen Hauptanalyse dieses Parameters beobachtet, mit 11.0 Monaten im PFS unter der Kombination (95%-KI: 8.0, 13.9) und weiter von 8.8 Monaten (95%-KI: 5.9, 9.3) unter der Dabrafenib-Monotherapie (HR = 0.67, 95%-KI: 0.53, 0.84, p<0.001). In dieser Analyse betrug die ORR 69 % gegenüber 53 % (p = 0.0014) und die DOR betrug 12.9 gegenüber 10.6 Monaten bei der Kombinationstherapie respektive bei Behandlung mit der Dabrafenib-Monotherapie.
Zum Zeitpunkt der Hauptanalyse des OS-Analyse wurden 222 Todesfälle (52.5 %) aus der randomisierten (oder ITT-) Population berichtet [Kombination 99 Todesfälle (47 %) und Dabrafenib 123 Todesfälle (58 %)]. Die mediane Nachbeobachtungszeit für die Studienbehandlung betrug 20 Monate im Kombinationstherapie-Arm und 16 Monate im Dabrafenib-Monotherapie-Arm. Die Studie MEK115306 zeigte eine statistisch signifikante Senkung des Todesrisikos von 29 % im Kombinationstherapie-Arm im Vergleich zum Dabrafenib-Monotherapie-Arm (HR = 0.71, 95%-KI: 0.55, 0.92; p = 0.011). Das mediane OS betrug 25.1 Monate im Kombinationstherapie-Arm und 18.7 Monate im Dabrafenib-Monotherapie-Arm. Die ermittelten OS-Werte für 12 Monate (74 %) und 24 Monate (51.4 %) waren zudem im Kombinationsarm höher als die im Dabrafenib-Monotherapie-Arm (67.6 bzw. 42.1 %).
Eine Analyse des Gesamtüberlebens (OS) nach 5 Jahren ergab, dass das mediane Gesamtüberleben (OS) unter der Kombinationstherapie etwa 7 Monate länger war als das mediane Gesamtüberleben (OS) unter der Dabrafenib-Monotherapie (25,8 Monate (95%-KI: 19.2, 38.2) gegenüber 18.7 Monaten (95%-KI: 15.2, 23.1)) mit einer Hazard Ratio von 0,80 (95%-KI: 0.63, 1.01).. Die 5-Jahres-Gesamtüberlebensrate betrug 32% (95%-KI: 25.1, 38.3) unter der Kombinationstherapie gegenüber 27% (95%-KI: 20.7, 33.0) unter der Dabrafenib-Monotherapie. Nach 5 Jahren war das mediane progressionsfreie Überleben (PFS) für die Kombinationstherapie 10.2 Monate (95%-Kl: 8.1, 12.8) gegenüber 8.8 Monate (95%-Kl: 5.9, 9.3) für Dabrafenib-Monotherapie mit einer Hazard Ratio von 0,73 (95%-KI: 0.59, 0.91).
MEK116513 (COMBI-v)
Die Studie MEK116513 war eine 2-armige, randomisierte, offene Phase-III-Studie zum Vergleich von Trametinib und Dabrafenib als Kombinationstherapie mit Vemurafenib als Monotherapie bei BRAF-V600-Mutations-positivem metastasiertem Melanom. Der primäre Endpunkt der Studie war das Gesamtüberleben (OS). Die Studienteilnehmer wurden nach Laktatdehydrogenase (LDH)-Aktivität (> obere Normgrenze (ONG) versus ≤ ONG) und BRAF-Mutation (V600E versus V600K) stratifiziert.
Insgesamt wurden 704 Studienteilnehmer im Verhältnis 1:1 randomisiert und entweder dem Kombinationstherapie-Arm (einmal täglich Trametinib 2 mg und zweimal täglich Dabrafenib 150 mg) oder dem Vemurafenib-Monotherapie-Arm (zweimal täglich 960 mg) zugewiesen. Bei der Mehrzahl der Studienteilnehmer lag eine BRAF-V600E-Mutation vor (89 %). 10 % der Patienten hatten eine BRAF-V600K Mutation und 1 Patient (<1 %) zeigte beide Mutationen (BRAF V600E/K).
Die OS-Analyse wurde durchgeführt, als insgesamt 222 Todesfälle aufgetreten waren (77 % der erforderlichen Ereignisse für die finale Analyse). Die unabhängige Monitorengruppe (Independent Data Monitoring Committee, IDMC) empfahl den Abbruch der Studie, da die OS-Ergebnisse die zuvor festgelegte statistische Wirksamkeitsgrenze überschritten hatten. In der Folge galt die OS-Zwischenanalyse als finale OS-Vergleichsanalyse.
Die OS-Analyse für die Studie MEK116513 basierte auf 222 Todesfällen (32 %) [Kombination; 100 Todesfälle (28 %) und Vemurafenib 122 Todesfälle (35 %)]. Die mediane Nachbeobachtungszeit für die Studienbehandlung betrug 11 Monate im Kombinationstherapie-Arm und 9 Monate im Vemurafenib-Arm. Die Studie MEK116513 zeigte eine statistisch signifikante Senkung des Todesrisikos von 31 % im Kombinationstherapie-Arm im Vergleich zu Vemurafenib (HR = 0.69, 95%-KI: 0.53, 0.89; p = 0.005). Das mediane OS wurde für den Kombinationstherapie-Arm noch nicht erreicht und betrug für den Vemurafenib-Monotherapie-Arm 17.2 Monate.
Das beobachtete mediane PFS betrug 11.4 Monate bei der Kombinationstherapie von Trametinib und Dabrafenib und 7.3 Monate unter Vemurafenib-Monotherapie (HR = 0.56, 95%-KI: 0.46, 0.69, p < 0.001). Die ORR betrug 64 % versus 51 % (p = 0.0005) und die DOR betrug 13.8 versus 7.5 Monate bei der Kombinationstherapie gegenüber der Vemurafenib-Monotherapie.
Eine Analyse des Gesamtüberlebens (OS) nach 5 Jahren ergab, dass das mediane Gesamtüberleben (OS) unter der Kombinationstherapie etwa 8 Monate länger war als das mediane Gesamtüberleben (OS) unter der Vemurafenib-Monotherapie (26.0 Monate (95%-KI: 22.1, 33.8) gegenüber 17.8 Monaten (95%-KI: 15.6, 20.7), mit einer Hazard ratio von 0.70 (95%-KI: 0.58, 0.84). Die 5-Jahres-Gesamtüberlebensrate betrug 36% (95%-KI: 30.5, 40.9) unter der Kombinationstherapie gegenüber 23% (95%-KI: 18.1, 27.4) unter der Vemurafenib-Monotherapie.
BRF117277 / DRB436B2204 (COMBI-MB) – Patienten mit Hirnmetastasen eines metastasierten Melanoms
Die Wirksamkeit und Sicherheit von Mekinist in Kombination mit Dabrafenib bei Patienten mit BRAF-Mutation-positivem Melanom mit Hirnmetastasen wurde in einer nicht-randomisierten, offenen, multizentrischen Phase-II-Studie (COMBI-MB-Studie) untersucht. Es wurden insgesamt 125 Patienten in vier Kohorten aufgenommen:
Kohorte A: Melanom-Patienten mit BRAFV600E-Mutation und asymptomatischen Hirnmetastasen ohne vorherige lokale zielgerichtete Therapie der Hirnmetastasen bei einem ECOG-Leistungsstatus von 0 oder 1.
Kohorte B: Melanom-Patienten mit BRAFV600E-Mutation und asymptomatischen Hirnmetastasen mit vorheriger lokaler zielgerichteter Therapie der Hirnmetastasen bei einem ECOG-Leistungsstatus von 0 oder 1.
Kohorte C: Melanom-Patienten mit BRAFV600D/K/R-Mutation und asymptomatischen Hirnmetastasen mit oder ohne vorherige lokale zielgerichtete Therapie der Hirnmetastasen bei einem ECOG-Leistungsstatus von 0 oder 1.
Kohorte D: Melanom-Patienten mit BRAFV600D/E/K/R-Mutation und symptomatischen Hirnmetastasen, mit oder ohne vorherige lokale zielgerichtete Therapie der Hirnmetastasen bei einem ECOG-Leistungsstatus von 0 oder 1 oder 2.
Insgesamt hatten 104/125 Patienten eine V600E Mutation, 18 Patienten eine V600K Mutation und nur 3 Patienten eine V600R Mutation. Kein Patient hatte eine V600D-Mutation.
Der primäre Endpunkt der Studie war das intrakranielle Ansprechen in Kohorte A, definiert als der Prozentsatz der Patienten mit einem bestätigten intrakraniellen Ansprechen, das vom vom Prüfarzt anhand der RECIST-Kriterien (Response Evaluation Criteria in Solid Tumors, Kriterien für die Bewertung des Ansprechens der Behandlung bei soliden Tumoren), Version 1.1, bewertete wurde. Die Wirksamkeitsergebnisse sind in Tabelle 6 zusammengefasst. Die sekundären Endpunkte waren die Dauer des intrakraniellen Ansprechens, die Gesamtansprechrate sowohl der intrakraniellen wie auch der extrakraniellen Tumormanifestationen, das PFS und das OS. Die Wirksamkeitsergebnisse sind in Tabelle 6 zusammengefasst.
Tabelle 6: COMBI-MB - Wirksamkeitsergebnisse nach Einschätzung des Prüfarztes
|
|
Alle behandelten Patienten
| |
Endpunkte/ Beurteilung
|
Kohorte A
N=76
|
Kohorte B
N=16
|
Kohorte C
N=16
|
Kohorte D
N=17
| |
Intrakranielle Ansprechrate, % (95 % KI)
|
| |
|
59 %
(47.3, 70.4)
|
56 %
(29.9, 80.2)
|
44 %
(19.8, 70.1)
|
59 %
(32.9, 81.6)
| |
Dauer des intrakraniellen Ansprechens, Medianwert, Monate (95%-KI)
| |
|
6.5
(4.9, 8.6)
|
7.3
(3.6, 12.6)
|
8.3
(1.3, 15.0)
|
4.5
(2.8, 5.9)
| |
Gesamtansprechen (intra- und extrakraniell) ORR, % (95 % KI)
| |
|
59 %
(47.3, 70.4)
|
56 %
(29.9, 80.2)
|
44 %
(19.8, 70.1)
|
65 %
(38.3, 85.8)
| |
Medianwert PFS, Monate (95 % KI)
| |
|
5.7
(5.3, 7.3)
|
7.2
(4.7, 14.6)
|
3.7
(1.7, 6.5)
|
5.5
(3.7, 11.6)
| |
Medianwert OS, Monate (95 % KI)
| |
Medianwert, Monate
|
10.8
(8.7, 17.9)
|
24.3
(7.9, NR)
|
10.1
(4.6, 17.6)
|
11.5
(6.8, 22.4)
| |
KI = Konfidenzintervall
NR = Ohne Angabe
|
Adjuvante Behandlung des Melanoms
Studie BRF115532/DRB436F2301 (COMBI-AD)
Die Wirksamkeit und Sicherheit von Mekinist in Kombination mit Dabrafenib wurde in einer multizentrischen, randomisierten, doppelblinden, Placebo-kontrollierten Phase-III-Studie bei Patienten mit Melanom im Stadium III mit einer BRAF-V600-Mutation nach vollständiger Resektion untersucht.
Die Patienten wurden im Verhältnis 1:1 randomisiert und erhielten entweder eine Kombinationstherapie mit Mekinist 2 mg einmal täglich und Dabrafenib150 mg zweimal täglich oder zwei Placebos über einen Zeitraum von 12 Monaten. Nur Patienten mit einer vollständigen Melanomresektion und vollständiger Lymphadenektomie innerhalb von 12 Wochen vor der Randomisierung wurden in die Studie aufgenommen. Eine vorherige systemische Krebsbehandlung, einschliesslich Strahlentherapie, war nicht erlaubt. Teilnahmeberechtigt waren Patienten mit einer Vorgeschichte früherer Malignität, wenn sie mindestens 5 Jahre krankheitsfrei waren. Patienten mit bösartigen Tumoren mit bestätigten aktivierenden RAS-Mutationen waren nicht teilnahmeberechtigt. Die Patienten wurden nach dem BRAF-Mutationsstatus (V600E oder V600K) und dem Erkrankungsstadium vor der Operation stratifiziert (nach Stadium-III-Substadium, d.h. unterschiedliche Lymphknotenbeteiligungen, primäre Tumorgrösse und Ulzeration). Der primäre Endpunkt war das vom Prüfer beurteilte rückfallfreie Überleben (relapse-free survival, RFS), definiert als der Zeitraum ab Randomisierung bis zum Wiederauftreten des Tumors oder Tod jeglicher Ursache. Eine radiologische Tumorbeurteilung wurde in den ersten zwei Jahren alle 3 Monate und danach alle 6 Monate durchgeführt, bis der erste Rückfall beobachtet wurde. Sekundäre Endpunkte waren das Gesamtüberleben (OS; wichtiger sekundärer Endpunkt) und das fernmetastasenfreie Überleben (distant metastasis-free survival, DMFS).
Insgesamt 870 Patienten wurden zur Kombinationstherapie (n = 438) und zu Placebo (n = 432) randomisiert. Die Studie nahm Patienten mit Erkrankung aller Stadium-III-Substadien vor der Resektion auf; 18 % dieser Patienten wiesen nur mikroskopisch nachweisbare Lymphknotenbeteiligung und keine primäre Tumorulzeration auf. Die Mehrheit der Patienten hatte eine BRAF-V600E-Mutation (91 %). Zum Zeitpunkt der Primäranalyse, die mediane Dauer der Nachbeobachtung (Zeit ab Randomisierung bis zum letzten Kontakt oder Tod) betrug 2,83 Jahre in der Behandlungsgruppe und 2,75 Jahre in der Placebogruppe.
Die Ergebnisse der Primäranalyse des RFS sind in Tabelle 7 dargestellt. Die Studie zeigte einen statistisch signifikanten Unterschied für den primären Endpunkt hinsichtlich des RFS zwischen den Behandlungsarmen. Bei der Behandlungsgruppe, die Mekinist und Dabrafenib als Kombinationstherapie erhielt, lag die Risikoreduktion bei 53 % im Vergleich zur Placebo-Gruppe.
Tabelle 7: COMBI-AD Primäranalyse – Ergebnisse des rückfallfreien Überlebens
|
|
Mekinist + Dabrafenib
|
Placebo
| |
RFS-Parameter
|
N = 438
|
N = 432
| |
Anzahl der Ereignisse - n (%)
Rezidiv
Rezidiviert mit Fernmetastasen
Tod
|
166 (38 %)
163 (37 %)
103 (24 %)
3 (< 1 %)
|
248 (57 %)
247 (57 %)
133 (31 %)
1 (< 1 %)
| |
Medianwert (Monate)
(95 % KI)
|
NE
(44,5, nicht schätzbar (NE))
|
16,6
(12,7; 22,1)
| |
Hazard Ratio[1]
(95 % KI)
P-Wert[2]
|
0,47
(0,39; 0,58)
1,53×10-14
| |
1-Jahres-Rate (95 % KI)
|
0,88 (0,85, 0,91)
|
0,56 (0,51, 0,61)
| |
2-Jahres-Rate (95 % KI)
|
0,67 (0,63, 0,72)
|
0,44 (0,40, 0,49)
| |
3-Jahres-Rate (95 % KI)
|
0,58 (0,54, 0,64)
|
0,39 (0,35, 0,44)
| |
[1] Die Hazard Ratio ergibt sich aus dem stratifizierten Pike-Modell.
[2] Der P-Wert wird aus dem zweiseitigen stratifizierten Logrank-Test ermittelt (Stratifikationsfaktoren waren Krankheitsstadium, IIIA vs. IIIB vs. IIIC, und BRAF- V600-Mutationstyp - V600E vs. V600K).
NE = nicht schätzbar
|
Basierend auf 153 Ereignissen (60 (14 %) im Kombinationstherapie-Arm und 93 (22 %) im Placebo-Arm), was einem Informationsanteil von 26 % des Gesamtziels von 597 OS-Ereignissen entspricht, betrug die geschätzte Hazard Ratio für OS 0,57 (95 % KI: 0,42, 0,79; p = 0,0006) zum Zeitpunkt der Primäranalyse. Diese Ergebnisse entsprachen nicht der präspezifizierten Grenze, um bei dieser ersten OS-Zwischenanalyse statistische Signifikanz zu erlangen (HR = 0,50; p = 0,000019). Schätzungen zum Überleben 1 und 2 Jahre nach Randomisierung betrugen 97 % und 91 % im Kombinationstherapie-Arm und 94 % und 83 % im Placebo-Arm.
Basierend auf aktualisierten Daten aus weiterer 29-monatigen Nachbeobachtungszeit im Vergleich zur Primäranalyse (Mindestnachbeobachtung von 59 Monaten) wurde der Vorteil des rückfallfreien Überleben mit einer geschätzte Hazard Ratio von 0.51 (51% KI: 0,42, 0,61) aufrechterhalten. Die 5-jährige rückfallfreie Überlebensrate betrug 52 % (95 % ki: 48, 58) im Kombinationstherapie-Arm im Vergleich zu 36 % (95 % KI: 32, 41) im Placebo Arm.
Fortgeschrittenes NSCLC
Studie BRF113928
Die Wirksamkeit und Sicherheit von Mekinist in Kombination mit Dabrafenib wurde in einer multizentrischen, nicht-randomisierten, offenen Phase-II-Studie bei 57 mit Chemotherapie vorbehandelten Patienten mit metastasiertem BRAF-V600E-Mutations-positivem NSCLC untersucht. Der primäre Endpunkt war die vom Prüfarzt beurteilte Gesamtansprechrate (overall response rate, ORR) unter Verwendung der RECIST-Kriterien (Response Evaluation Criteria in Solid Tumors, Kriterien für die Bewertung des Ansprechens der Behandlung bei soliden Tumoren). Die sekundären Endpunkte umfassten die Ansprechdauer (duration of response, DoR), das progressionsfreie Überleben (PFS) und das Gesamtüberleben (overall survival, OS). ORR, DoR und PFS wurden von einem unabhängigen Prüfungsausschuss (Independent Review Committee, IRC) in Form einer Empfindlichkeitsanalyse beurteilt.
Zum Zeitpunkt der Primäranalysis, der vom Prüfarzt beurteilten Gesamtansprechrate (overall response rate, ORR) betrug die ORR in der zuvor bereits behandelten Population 66.7% (95% CI, 52.9%, 78.6%). Damit wurde die statistische Signifikanz in Ablehnung der Nullhypothese erfüllt, dass die ORR von Dabrafenib in Kombination mit Mekinist für diese NSCLC-Population höchstens 30 % betrug. Die Ergebnisse für die von einem unabhängigen Prüfungsausschuss (Independent Review Committee, IRC) beurteilte Gesamtansprechrate (overall response rate, ORR) entsprachen der Prüfarztbewertung. Die endgültige Analyse der Wirksamkeit, die 5 Jahre nach der ersten Dosis des letzten Probanden durchgeführt wurde, wird in Tabelle 8.
Tabelle 8: ORR nach Einschätzung des Prüfarztes und durch unabhängige Röntgenuntersuchung
|
Gruppe, bei der Röntgenuntersuchung durchgeführt wurde
|
Prüfer
|
Unabhängig
| |
Kategorie
|
N = 57
|
N = 57
| |
Gesamtansprechen – n (%)
| |
ORR (CR + PR)
|
39 (68.4%)
|
36 (63.2%)
| |
(95%-KI)
|
(54.8, 80.1)
|
(49.3, 75.6)
| |
Dauer des Ansprechens
| |
Anzahl der Responder
|
39
|
36
| |
Anzahl der Patienten mit Krankheitsprogress oder Todesfälle - n (%)
|
27 (71)
|
20(56)
| |
Medianwert DoR, Monate
(95%-KI)
|
9.8
(6.9, 18.3)
|
12.6
(5.8, 26.2)
| |
Progressionsfreies Überleben
| |
Krankheitsprogress oder Tod - n (%)
|
41(72)
|
38 (67)
| |
Medianwert PFS, Monate
(95%-KI)
|
10.2
(6.9, 16.7)
|
8.6
(5.2, 16.8)
| |
Gesamtüberleben
| |
Anzahl der Todesfälle - n (%)
|
33 (58%)
| |
Medianwert, Monate
(95%-KI)
|
18.2
(14.3, 28.6)
| |
Konfidenzintervall (KI) errechnet mit der exakten Methode (das sogenannte Clopper-Pearson-Intervall).
CR=komplette Remission; PR=partielle Remission; SD=stabile Erkrankung; PD=Krankheitsprogress
|
In die Studie BRF113928 wurden auch 36 Patienten mit BRAF V600E-mutationspositivem NSCLC eingeschlossen, die Dabrafenib in Kombination mit Trametinib als Erstlinientherapie bei einer metastasierenden Erkrankung erhielten. Bei der Abschlussanalyse nach 5 Jahren für alle Studienteilnehmer, die mit der Erstlinientherapie behandelt wurden, lag der primäre Endpunkt der vom Prüfarzt beurteilten Gesamtansprechrate (overall response rate, ORR) bei 63.9 % (95%-KI, 46.2 %, 79.2 %). Damit wurde die statistische Signifikanz in Ablehnung der Nullhypothese erfüllt, dass die ORR von Dabrafenib in Kombination mit Trametinib für diese NSCLC-Population höchstens 30 % betrug. Die vom Prüfarzt beurteilte mediane Dauer der Remission (DoR) betrug 10.2 Monate (95%-KI: 8.3, 15.2), das vom Prüfarzt beurteilte mediane progressionsfreie Überleben betrug 10.8 Monate (95%-KI: 7.0, 14.5), das vom Prüfarzt beurteilte mediane Gesamtüberleben betrug 17.3 Monate (95%-KI: 12.3, 40.2), wobei 61 % der Todesfälle zum Zeitpunkt der Analyse vorlagen.
Niedriggradiges Gliom (LGG)
Studie DRB436G2201
Die klinische Wirksamkeit und Sicherheit der Kombinationstherapie von Mekinist plus Dabrafenib bei pädiatrischen Patienten im Alter von 1 bis < 18 Jahren mit BRAF V600E-Mutation-positivem Gliom wurde in der multizentrischen, offenen klinischen Phase-II-Studie CDRB436G2201 untersucht. Patienten mit niedriggradigem Gliom (WHO-Grad 1 und 2), die eine systemische Ersttherapie benötigten und zuvor keine Strahlentherapie erhalten hatten, wurden im Verhältnis 2:1 zu Trametinib plus Dabrafenib (D+T) oder Carboplatin plus Vincristin (C+V) randomisiert. Etwa 83% aller Patienten hatten sich einem vorgängigen chirurgischen Eingriff unterzogen; nur zwei Patienten (beide im D+T Arm) hatten nach dem Eingriff keine Resterkrankung.
Patienten mit einem Karnofsky/Lansky-Performance-Score von < 50 %, Patienten ohne ausreichende Knochenmark-, Nieren-, Leber- und Herzfunktion sowie Patienten mit unkontrollierten oder signifikanten Erkrankungen, einschliesslich Herzerkrankungen, Diabetes mellitus, Bluthochdruck, Lebererkrankungen oder Infektionen von der Teilnahme ausgeschlossen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Der BRAF-Mutationsstatus wurde prospektiv durch einen lokalen Test oder, wenn ein lokaler Test nicht verfügbar war, durch einen Echtzeit-PCR-Test des Zentrallabors ermittelt. Darüber hinaus wurden retrospektive Tests von verfügbaren Tumorproben durch das Zentrallabor durchgeführt, um die BRAF-V600E-Mutation zu bestätigen.
Die Dosierung von Tafinlar und Mekinist war alters- und gewichtsabhängig, wobei Tafinlar oral in einer Dosierung von 2.625 mg/kg zweimal täglich für die Altersgruppe < 12 Jahre und 2.5 mg/kg zweimal täglich für die Altersgruppe 12 Jahre und älter verabreicht wurde; Mekinist wurde oral in einer Dosierung von 0.032 mg/kg einmal täglich für die Altersgruppe < 6 Jahre und 0.025 mg/kg einmal täglich für die Altersgruppe 6 Jahre und älter verabreicht. Die Höchstdosis von Tafinlar wurde auf 150 mg zweimal täglich und die von Mekinist auf 2 mg einmal täglich begrenzt. Carboplatin und Vincristin wurden je nach Alter und Körperoberfläche in einer Dosierung von 175 mg/m2 bzw. 1.5 mg/m2 als 10-wöchige Induktionstherapie, gefolgt von acht 6-wöchigen Zyklen Erhaltungstherapie, verabreicht.
Der primäre Wirksamkeitsendpunkt in beiden Kohorten war die Gesamtansprechrate (ORR, Summe der Patienten mit bestätigter kompletter Remission/CR und partieller Remission/PR) durch unabhängige Überprüfung auf der Grundlage der RANO-Kriterien (2017). Die primäre Analyse wurde durchgeführt, wenn alle Patienten in beiden Kohorten mindestens 32 Wochen der Therapie abgeschlossen hatten.
In der Kohorte der niedriggradigen Gliome (LGG) der Studie G2201 wurden 110 Patienten nach dem Zufallsprinzip für D+T (n=73) oder C+V (n=37) ausgewählt. Das Durchschnittsalter betrug 9,5 Jahren, wobei 34 Patienten (30.9 %) 12 Monate bis < 6 Jahre, 36 Patienten (32.7 %) 6 bis < 12 Jahre und 40 Patienten (36.4 %) 12 bis < 18 Jahre alt waren; 60 % waren weiblich. Zum Zeitpunkt der Primäranalyse betrug die mediane Therapiedauer im D+T Arm 76 Wochen, die mediane Nachbeobachtungszeit in der LGG Kohorte 18,9 Monate. Die ORR im D+T-Arm (46.6 %) zeigte eine statistisch signifikante Verbesserung gegenüber dem C+V-Arm (10.8 %), mit einer Odds Ratio (95% KI) von 7,19 (2.3, 22.4) und einem einseitigen p-Wert < 0.001 (Tabelle 9). Die anschliessende hierarchische Prüfung ergab ebenfalls eine Verbesserung des progressionsfreien Überlebens (PFS) gegenüber der Chemotherapie, mit einer hazard Ratio (95% KI) von 0,31 (0.17,0.55); (einseitiger p-Wert des Log-Rang-Tests< 0.001). Es wurden Unterschiede in der Beurteilung der Wirksamkeit zwischen der unabhängigen Überprüfung und den Prüfzentren beobachtet, welche auch die Feststellung von kompletten und partiellen Remissionen sowie Krankheitsprogressionen umfassten. Die Rate der Übereinstimmung betrug im D+T Arm insgesamt 52%.
Als funktionelle Parameter wurden Veränderungen der Krampfanfallhäufigkeit («seizure activity») und des Visus («visual acuity») vor und nach Therapie untersucht. Für beide Parameter war keine Verbesserung unter Behandlung mit D+T im Vergleich zum Kontrollarm nachweisbar.
Tabelle 9: Ansprechen und progressionsfreies Überleben in der Studie G2201 (LGG-Kohorte)
|
|
Dabrafenib + Trametinib
N=73
|
Carboplatin plus Vincristin
N=37
| |
Bestes Gesamtansprechen
| |
Komplette Remission (CR), n (%)
|
2 (2.7)
|
1 (2.7)
| |
Partielle Remission (PR), n (%)
|
32 (43.8)
|
3 (8.1)
| |
Stabile Erkrankung (SD), n (%)
|
30 (41.1)
|
15 (40.5)
| |
Krankheitsprogress (PD), n (%)
|
8 (11.0)
|
12 (32.4)
| |
Unbekannt, n (%)
|
1 (1.4)
|
6 (16.2)
| |
Gesamtansprechenrate (ORR)
| |
ORR (CR+PR), 95%-KI, p-Wert
|
46.6 % (34.8 – 58.6 %), p<0.001
|
10.8 % (3.0 - 25.4 %)
| |
Odds Ratio
|
7.19 (2.3 - 22.4)
| |
Progressionsfreies Überleben
| |
Medianwert (Monate)
|
20.1 (12.8, NE)
|
7.4 (3.6, 11.8)
| |
Hazard Ratio (95%-KI), p-Wert
|
0.31 (0.17-0.55), p<0.001
|
PharmakokinetikAbsorption
Trametinib wird oral mit einer medianen Zeit von 1.5 Stunden von der Verabreichung bis zum Erreichen der Plasma-Spitzenkonzentration absorbiert. Die durchschnittliche absolute Bioverfügbarkeit einer Einzeldosis in Form einer 2-mg-Tablette beträgt 72 % derjenigen nach einer intravenösen (IV) Mikrodosis. Der Anstieg der Exposition (Cmax und AUC) verhielt sich nach wiederholter Verabreichung dosisproportional. Nach Verabreichung von 2 mg täglich betrugen die geometrischen Mittelwerte von Cmax, AUC(0τ) und der Konzentration vor Verabreichung 22.2 ng/ml, 370 ng*hr/ml bzw. 12.1 ng/ml, bei niedrigem Spitzen-Tal-Verhältnis (1.8). Die interindividuelle Variabilität war gering (< 28 %). Nach Verabreichung in Kombination mit Dabrafenib 150 mg zweimal täglich betrugen die geometrischen Mittelwerte von Cmax, AUC(0-τ) und der Trametinib Konzentration vor Verabreichung 22.6 ng/ml, 356 ng*hr/ml bzw. 10.9 ng/ml. Die Verabreichung einer Einzeldosis Trametinib Filmtabletten zusammen mit einer fettreichen, hochkalorigen Mahlzeit führte zu einer Abnahme von Cmax und AUC um 70 % bzw. 10 % gegenüber den Werten bei nüchterner Einnahme (siehe «Dosierung/Anwendung»). Trametinib Tabletten und Pulver zur Herstellung einer Lösung zum Einnehmen sind Darreichungsformen mit sofortiger Freisetzung, die ähnliche Auswirkungen auf die PK haben dürften.
Distribution
Trametinib bindet zu 97.4 % an humane Plasmaproteine. Trametinib weist nach Verabreichung einer intravenösen Mikrodosis von 5 µg ein Verteilungsvolumen von 1060 l auf.
Metabolismus
In-vitro- und in-vivo-Studien zeigten, dass Trametinib vorwiegend per Deacetylierung allein oder mit Monooxygenierung oder in Kombination mit Glucuronidierungs-Biotransformationswegen verstoffwechselt wird. Die Deacetylierung wird durch Carboxylesterasen (d.h. Carboxylesterase 1b/c und 2) vermittelt und kann auch durch andere hydrolytische Enzyme vermittelt werden.
Elimination
Trametinib akkumuliert bei wiederholter Verabreichung einer Dosis von 2 mg einmal täglich mit einem durchschnittlichen Akkumulationsverhältnis von 6.0. Die terminale Halbwertszeit liegt nach einer Einzeldosis im Durchschnitt bei 127 Stunden (5.3 Tage). Der Steady State wurde an Tag 15 erreicht. Die Plasma-Clearance nach intravenöser Verabreichung beträgt 3.21 l/h.
Die Gesamtwiederfindung einer oralen Einzeldosis von radioaktiv markiertem Trametinib in einer Lösung ist nach 10-tägiger Probenentnahmephase aufgrund der langen Halbwertszeit gering (< 50 %).
Das arzneimittel-bezogene Material wurde in erster Linie mit den Fäzes ausgeschieden (≥81 % der ausgeschiedenen Radioaktivität), lediglich ≤19 % befanden sich im Urin. Weniger als 0,1 % der ausgeschiedenen Dosis wurde in Form der Muttersubstanz im Urin wiedergefunden.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Eine Populationspharmakokinetische Analyse und Daten aus einer klinisch-pharmakologischen Studie an Patienten mit normaler Leberfunktion oder mit leichter, mässiger oder hochgradieger Billirubin- und/oder AST-Erhöhung (nach der Klassifizierung des National Cancer Institut [NCI]) weisen darauf hin, dass die Leberfunktion die orale Clearance von Trametinib nicht signifikant beeinflusst.
Nierenfunktionsstörungen
Eine eingeschränkte Nierenfunktion hat angesichts der geringen renalen Ausscheidung von Trametinib wahrscheinlich keinen klinisch relevanten Einfluss auf dessen Pharmakokinetik. Die Pharmakokinetik von Trametinib wurde anhand einer populationspharmakokinetischen Analyse an 223 in klinische Studien eingeschlossenen Patienten mit leicht eingeschränkter Nierenfunktion und 35 Patienten mit mässig eingeschränkter Nierenfunktion untersucht. Eine leicht bis mässig eingeschränkte Nierenfunktion hatte keinen Einfluss auf die Trametinib-Exposition (< 6 % in beiden Gruppen). Daten zu Patienten mit hochgradig eingeschränkter Nierenfunktion liegen nicht vor (siehe «Dosierung/Anwendung»).
Ältere Patienten
Gemäss der populationspharmakokinetischen Analyse hatte das Alter keinen signifikanten Einfluss auf die Pharmakokinetik von Trametinib.
Pädiatrische Population
Die Pharmakokinetik von Trametinib bei Gliomen und anderen soliden Tumoren wurde bei 244 pädiatrischen Patienten (1 bis < 18 Jahre alt) nach einmaliger oder wiederholter gewichtsangepasster Dosierung untersucht. Die pharmakokinetischen Eigenschaften (Absorptionsrate, Metabolitenverhältnisse, Arzneimittel- Clearance) von Trametinib bei pädiatrischen Patienten sind mit denen von erwachsenen Patienten vergleichbar. Es wurde festgestellt, dass das Gewicht die orale Clearance von Trametinib beeinflusst. Die pharmakokinetischen Expositionen von Trametinib bei der empfohlenen an das Körpergewicht angepassten Dosierung bei pädiatrischen Patienten lagen im Bereich der bei Erwachsenen beobachteten Expositionen.
Präklinische DatenTrametinib in Kombination mit Dabrafenib
Bei Hunden, denen vier Wochen lang Trametinib und Dabrafenib in Kombination verabreicht wurden, wurden ähnliche Toxizitätserscheinungen festgestellt wie in präklinischen Studien, die mit den einzelnen Substanzen allein durchgeführt wurden. In Hunden, denen die Kombination aus Trametinib und Dabrafenib verabreicht wurde, wurden dieselben testikulären Wirkungen, bestehend aus Degeneration und sekundärer Nebenhoden Oligospermie, beobachtet, wie in Hunden, denen entweder Trametinib oder Dabrafenib allein verabreicht wurde. Siehe dazu auch die Fachinformation für Dabrafenib.
Mutagenität / Karzinogenität
Trametinib hat sich in vitro an Bakterien und Säugerzellkulturen sowie in vivo im Mikronukleustest bei Nagetieren als nicht genotoxisch erwiesen. Karzinogenitätsstudien mit Trametinib wurden nicht durchgeführt.
Reproduktionstoxizität
Fertilität
Es liegen für Trametinib keine Daten am Menschen vor. Fertilitätsstudien mit Trametinib wurden nicht durchgeführt. In Studien mit wiederholter Verabreichung wurde bei erwachsenen und jungen Ratten wurden bei ≥0.016 mg/kg/Tag (ca. das 0.3-Fache der klinischen Exposition beim Menschen, basierend auf dem AUC) Veränderungen der Follikelreifung beobachtet, die sich in der Zunahme von zystischen Follikeln und der Abnahme von zystischen Gelbkörpern äusserte.
Ausserdem wurden bei jungen Ratten, die Trametinib erhielten, verminderte Ovarialgewichte, leichte Verzögerungen bei den Anzeichen für die weibliche sexuelle Reifung (Vaginaöffnung und vermehrte Inzidenz vorstehender terminaler Endknospen in der Milchdrüse) und leichte Hypertrophie des Oberflächenepithels des Uterus beobachtet. Alle diese Wirkungen waren nach einem behandlungsfreien Zeitraum reversibel und der Pharmakologie zuzuschreiben. In Toxizitätsstudien mit Ratten und Hunden mit einer Dauer von bis zu 13 Wochen wurden jedoch keine Behandlungseffekte auf das männliche Reproduktionsgewebe beobachtet.
Trächtigkeit
In reproduktionstoxikologischen Studien an Ratten wurden bei Dosen von ≥ 0.031 mg/kg/Tag (entspricht, bezogen auf die AUC, ca. dem 0.3-Fachen der klinischen Exposition beim Menschen) maternale Toxizität und eine Toxizität auf die Entwicklung der Föten beobachtet (reduzierte Fetalgewichte). Bei trächtigen Kaninchen wurde bei Dosen von ≥ 0.039 mg/kg/Tag (entspricht, bezogen auf die AUC, ca. dem 0.1-Fachen der klinischen Exposition beim Menschen) maternale Toxizität und eine Toxizität auf die Entwicklung der Föten (reduzierte Fetalgewichte und gehäuftes Auftreten von Ossifizierungsdefekten) beobachtet.
Weitere Daten (Lokale Toxizität, Phototoxizität, Immunotoxizität)
In Studien mit wiederholter Gabe wurden nach Exposition mit Trametinib Effekte vorwiegend an der Haut, im Magen-Darm-Trakt, im hämatologischen System, an Knochen und in der Leber gefunden. Die meisten der Befunde waren nach einem Arzneimittel-freien Erholungszeitraum reversibel. In Studien mit wiederholter Verabreichung an Ratten wurden nach 8 Wochen bei ≥0.062 mg/kg/Tag (entspricht, bezogen auf die AUC, ca. dem 0.8 -Fachen der klinischen Exposition beim Menschen) Leberzellnekrose und erhöhte Transaminasenwerte festgestellt.
Bei Mäusen wurden bei ≥0.25 mg/kg/Tag Trametinib (entspricht, bezogen auf die AUC, ca. dem 3 -Fachen der klinischen Exposition beim Menschen) über bis zu drei Wochen nach 3 Wochen eine verminderte Herzfrequenz, ein vermindertes Herzgewicht und eine reduzierte linksventrikuläre Funktion ohne kardiale histopathologische Anzeichen festgestellt. Bei Ratten wurden bei Dosen von ≥1 mg/kg/Tag (entspricht, bezogen auf die AUC, ca. dem 12 -Fachen der klinischen Exposition beim Menschen) Myokardkalzifizierung und nekrose, einhergehend mit erhöhten Serumphosphatkonzentrationen, beobachtet. Bei jungen Ratten wurden bei 0.35 mg/kg/Tag (ca. das 2-Fache der klinischen Exposition beim Menschen, basierend auf dem AUC) erhöhte Herzgewichte ohne histopathologische Befunde beobachtet.
Bei adulten Ratten war die Mineralisierung multipler Organe mit erhöhten Phosphor-Serumkonzentrationen assoziiert und eng mit Nekrosen im Herz, der Leber und der Niere sowie Lungenblutungen verbunden, bei einer Exposition vergleichbar der Humanexposition in den klinischen Studien. Bei Ratten wurde eine Hypertrophie der Wachstumsfuge sowie ein erhöhter Knochenstoffwechsel beobachtet, die Hypertrophie der Wachstumsfuge wird jedoch nicht als klinisch relevant für erwachsene Menschen eingeschätzt.
In Ratten und Hunden, denen Trametinib in klinischen oder subklinischen Dosen verabreicht wurde, wurden Knochenmarknekrosen, Lymphatrophien des Thymus und des mit dem Darm assoziierten Lymphgewebes (GALT) sowie lymphatische Nekrosen in den Lymphknoten, der Milz und im Thymus beobachtet, die das Potential für eine Beeinträchtigung der Immunfunktion haben.
Bei juvenilen Ratten wurden bei dem ungefähr zweifachen der klinischen Exposition beim Erwachsenen auf Basis der AUC erhöhte Herzgewichte ohne histopathologische Befunde beobachtet.
Trametinib war in einem in-vitro-Neutralrotaufnahme (Neutral Red Uptake, NRU)-Assay der Mäusefibroblasten 3T3 bei signifikant höheren Konzentrationen als den klinischen Expositionen (IC50 bei 2.92 µg/ml, ≥130-Faches der klinischen Exposition, basierend auf Cmax) phototoxisch, was darauf hinweist, dass ein geringes Phototoxizitätsrisiko für Patienten besteht, die Trametinib anwenden.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach dem ersten Öffnen der Flasche (Filmtabletten) bzw. nach Rekonstitution des Pulvers zur Herstellung einer Lösung zum Einnehmen:
Filmtabletten
Nach dem Öffnen kann die Flasche 30 Tage lang bei unter 30°C gelagert werden.
Pulver zur Herstellung einer Lösung zum Einnehmen:
Die rekonstituierte Lösung ist 35 Tage haltbar. Die Flasche mit der rekonstituierten Lösung ist im Umkarton unter 25°C zu lagern und darf nicht eingefroren werden.
Nicht verwendete Lösung ist 35 Tage nach der Rekonstitution zu entsorgen.
Besondere Lagerungshinweise
Filmtabletten
Nicht über 25°C lagern.
In der Originalverpackung aufbewahren.
Den Behälter fest verschlossen halten.
Im Originalbehälter fest verschlossen lagern, um den Inhalt vor Licht und Feuchtigkeit zu schützen. Das Trocknungsmittel nicht entfernen oder einnehmen.
Pulver zur Herstellung einer Lösung zum Einnehmen:
Bis zur Rekonstitution im Kühlschrank bei 2°C bis 8°C aufbewahren.
In der Originalverpackung aufbewahren, um das Pulver vor Licht und Feuchtigkeit zu schützen.
Die Flasche fest verschlossen halten.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer65883, 69134 (Swissmedic)
PackungenMekinist 0.5 mg: Packungen zu 7 und 30 Filmtabletten (A)
Mekinist 2 mg: Packungen zu 7 und 30 Filmtabletten (A)
Mekinist 4.7 mg: 1 Flasche mit Pulver zur Herstellung einer Lösung zum Einnehmen, 1 orale Dosierspritze und 1 Flaschenadaptor (A):
ZulassungsinhaberinNovartis Pharma Schweiz AG, Risch; Domizil: 6343 Rotkreuz
Stand der InformationMärz 2023
Sonstige Hinweise - Hinweise für die Anwendung und Handhabung
|
GEBRAUCHSANWEISUNG
MEKINIST 4.7 mg Pulver zur Herstellung einer Lösung zum Einnehmen
Trametinib
Zum Einnehmen
| |
Diese Gebrauchsanweisung enthält Informationen zur Einnahme von Mekinist
| |
Wichtige Informationen, die Sie vor der Einnahme von Mekinist wissen müssen
| |
·Bitte lesen Sie diese Gebrauchsanweisung sorgfältig durch, bevor Sie mit der Anwendung von Mekinist beginnen.
·Bitten Sie Ihren Arzt bzw. Ihre Ärztin, Ihnen zu zeigen, wie Sie Mekinist richtig einnehmen. Nehmen Sie Mekinist immer genau nach Anweisung Ihres Arztes bzw. Ihrer Ärztin ein.
·Wenn Sie Fragen zur Zubereitung oder Einnahme von Mekinist haben, wenden Sie sich an Ihren Arzt bzw. Ihre Ärztin.
·Sie erhalten eine Mekinist-Flasche mit Pulver (damit Sie die Lösung selbst zubereiten können) ODER als gebrauchsfertige Lösung. Prüfen Sie, ob Sie ein Pulver oder eine Lösung erhalten haben. Befolgen Sie dann die entsprechenden nachstehenden Anweisungen.
·Die zubereitete Lösung muss innerhalb von 35 Tagen verwendet werden.
·Entsorgen Sie Arzneimittel nicht im Abwasser oder Haushaltsabfall. Fragen Sie Ihren Apotheker bzw. Ihre Apothekerin, wie das Arzneimittel zu entsorgen ist, wenn Sie es nicht mehr verwenden. Sie tragen damit zum Schutz der Umwelt bei.
REINIGUNG VON VERSCHÜTTETEN FLÜSSIGKEITEN
| |
Wenn Mekinist auf die Haut gelangt, waschen Sie den Bereich gut mit Wasser und Seife ab. Wenn Mekinist in die Augen gelangt, spülen Sie Ihre Augen mit Wasser aus.
Befolgen Sie diese Schritte, wenn Sie Mekinist Lösung zum Einnehmen verschütten:
·Ziehen Sie Plastikhandschuhe an.
·Saugen Sie die Lösung mit einem saugfähigen Material, wie z.B. Papiertüchern, vollständig auf.
·Geben Sie das saugfähige Material in einen verschliessbaren Plastikbeutel.
·Wischen Sie alle Oberflächen, die mit der Lösung in Berührung gekommen sind, mit einem Alkoholtupfer ab.
·Geben Sie den Beutel, die Handschuhe und die Tücher in einen zweiten Plastikbeutel und verschliessen Sie ihn.
·Entsorgen Sie Arzneimittel nicht im Abwasser oder Haushaltsabfall. Fragen Sie Ihren Apotheker bzw. Ihre Apothekerin, wie das Arzneimittel zu entsorgen ist, wenn Sie es nicht mehr verwenden. Sie tragen damit zum Schutz der Umwelt bei.
| |
Pulver für die Zubereitung
Wenn Sie Mekinist als Pulver erhalten, sollte die Packung Folgendes enthalten:
1.1 Flasche mit Schutzkappe mit Pulver
2.1 Applikationsspritze für Zubereitungen zum Einnehmen
3.1 Flaschenadapter (an der Applikationsspritze für Zubereitungen zum Einnehmen befestigt)
Gebrauchsanweisung und Packungsbeilage (nicht abgebildet).
|
|

| |
Übersicht über wiederverwendbare Applikationsspritzen für Zubereitungen zum Einnehmen:
|

Setzen Sie den Flaschenadapter noch nicht ein.
Lesen Sie die wichtigen Informationen über Mekinist oben und gehen Sie dann zu den Anweisungen für die Zubereitung in Abschnitt A weiter.
| |
Weiter zu Abschnitt A
| |
ODER
| |
Gebrauchsfertige Lösung
Wenn Sie MEKINIST als gebrauchsfertige Lösung erhalten, sollte die Packung Folgendes enthalten:
1.1 Flasche mit Schutzkappe mit gebrauchsfertiger Lösung
2.1 Applikationsspritze für Zubereitungen zum Einnehmen
3.1 Flaschenadapter (bereits eingesetzt)
|
|

| |
Übersicht über wiederverwendbare Applikationsspritzen für Zubereitungen zum Einnehmen:
|

Entfernen Sie den Adapter nicht von der Flasche.
Lesen Sie die wichtigen Informationen über Mekinist oben und gehen Sie dann zu den Anweisungen zur Verabreichung in Abschnitt B.
| |
Weiter zu Abschnitt B
| |
ABSCHNITT A. ZUBEREITUNG
| |
Befolgen Sie die Anweisungen in Abschnitt A zur Zubereitung, wenn Sie Mekinist als Pulver erhalten haben.
Im Falle eines Verschüttens oder eines Kontakts von Mekinist-Lösung mit der Haut oder den Augen, befolgen Sie die Informationen im Abschnitt «Reinigung von verschütteten Flüssigkeiten».
Waschen und trocknen Sie Ihre Hände, bevor Sie Mekinist zubereiten.
Um Mekinist zuzubereiten, benötigen Sie:
1.Flasche mit Pulver und Schutzkappe
2.Applikationsspritze für Zubereitungen zum Einnehmen
3.Adapter (an der Applikationsspritze für Zubereitungen zum Einnehmen befestigt)
Sie benötigen ausserdem stilles Trinkwasser in Flaschen (nicht enthalten).
Wenden Sie sich an Ihren Apotheker bzw. Ihre Apothekerin, wenn das unter Punkt 1, 2 oder 3 genannte Material in Ihrer Packung fehlt.
|
|

| |
1. Prüfen Sie, ob sich in der Flasche Pulver oder eine flüssige Lösung befindet.
·Wenn sich in der Flasche Pulver befindet, fahren Sie mit Schritt 2 unten fort.
·Wenn sich in der Flasche eine Lösung befindet, fahren Sie mit Abschnitt B Schritt 2 fort.
·Wenn sich in der Flasche eine Lösung befindet, geben Sie kein weiteres Wasser in die Flasche.
|
|

| |
2. Schütteln Sie die Flasche nicht.
Klopfen Sie fest auf die Seiten der Flasche, um das Pulver zu lösen.
|
|

| |
3. Entfernen Sie die kindersichere Schutzkappe, indem Sie sie herunterdrücken und gegen den Uhrzeigersinn drehen.
|
|

| |
4. Nehmen Sie den Adapter von der Spritze ab.
|
|

| |
5. Füllen Sie ein Trinkglas mit stillem Trinkwasser.*
·Füllen Sie mit der Applikationsspritze für Zubereitungen zum Einnehmen Wasser in die Flasche bis zum blauen Pfeil auf dem Flaschenetikett. Dies entspricht einer Zugabe von 90 mL Wasser.
Hinweis: Wenn sich Schaum bildet, füllen Sie bis zur Oberkante des Wassers auf, nicht bis zur Oberkante des Schaums.
*Bei Zubereitung in der Apotheke kann auch steriles oder gereinigtes Wasser verwendet werden.
|
|

| |
6. Setzen Sie die Schutzkappe wieder auf die Flasche und drehen Sie sie zum Verschliessen im Uhrzeigersinn. Vergewissern Sie sich, dass die Schutzkappe sicher auf der Flasche aufsitzt, bevor Sie mit dem nächsten Schritt fortfahren.
|
|
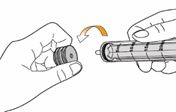
| |
7. Drehen Sie die Flasche wiederholt auf den Kopf und dann wieder in eine aufrechte Position, bis sich das Pulver vollständig aufgelöst hat. Sie können die Flasche auch leicht schütteln.
Hinweis: Das vollständige Auflösen des Pulvers kann 5 oder mehr Minuten dauern.
|
|

| |
8. Stellen Sie die Flasche für mindestens 5 Minuten aufrecht auf eine flache Oberfläche.
·Warten Sie, bis kein Schaum mehr zu sehen ist, bevor Sie mit dem nächsten Schritt fortfahren.
|
|

| |
9. Nachdem sich das Pulver vollständig aufgelöst hat, befindet sich das Wasser in der Flasche möglicherweise unterhalb des blauen Pfeils.*
·Nehmen Sie bei Bedarf die Schutzkappe ab und füllen Sie klares Wasser bis zum blauen Pfeil nach.
Hinweis: Sie haben jetzt insgesamt 90 ml Wasser hinzugefügt.
|
|

| |
10. Stellen Sie die Flasche auf den Tisch und halten Sie sie fest. Stecken Sie mit der anderen Hand den Adapter mit dem Daumen oder der Handfläche in die Flasche.
·Der Adapter muss bündig mit der Flasche abschliessen, es dürfen keine Rillen zu sehen sein. Drücken Sie kräftig, bis er vollständig eingeführt ist.
Hinweis: Das Einsetzen des Adapters kann einen hohen Kraftaufwand erfordern.
|
|

|

| |
11. Setzen Sie die Schutzkappe wieder auf die Flasche und drehen Sie sie zum Verschliessen im Uhrzeigersinn.
·Vergewissern Sie sich, dass die Schutzkappe sicher befestigt ist.
Hinweis: Wenn die Schutzkappe nicht fest sitzt, überprüfen Sie, ob der Adapter vollständig eingesetzt ist.
|
|

| |
12. Schreiben Sie das Verfalldatum auf den Umkarton. Das Verfalldatum beträgt 35 Tage nach Herstellung der Lösung.
·Wenn nach 35 Tagen noch Lösung übrig ist, fragen Sie Ihren Apotheker bzw. Ihre Apothekerin, wie Sie diese entsorgen können und nehmen Sie eine neue Flasche Mekinist zur Hand.
Entsorgen Sie Arzneimittel nicht im Abwasser oder Haushaltsabfall.
Hinweis: Füllen Sie nach Abschluss der Zubereitungsschritte kein weiteres Wasser mehr in diese Flasche.
|
|

| |
13. Um die Lösung zu verabreichen, folgen Sie den Anweisungen in Abschnitt B.
Um die Lösung über eine Ernährungssonde zu verabreichen, gehen Sie zu Abschnitt C.
| |
ABSCHNITT B. ANWENDUNG
| |
Zur Verabreichung von Mekinist als gebrauchsfertige Lösung benötigen Sie:
1.Lösung in der Flasche
2.Applikationsspritze für Zubereitungen zum Einnehmen
3.Adapter (bereits im Flaschenhals eingesetzt)
Wenden Sie sich an Ihren Apotheker bzw. Ihre Apothekerin, wenn Ihnen eines oder mehrere dieser Hilfsmittel fehlen.
Verabreichen Sie Mekinist nicht, wenn mehr als 35 Tage seit dem Datum vergangen sind, das auf dem Umkarton hinter «Lösung zubereitet am» steht.
Im Falle eines Verschüttens oder eines Kontakts von Mekinist-Lösung mit der Haut oder den Augen, befolgen Sie die Informationen im Abschnitt «Reinigung von verschütteten Flüssigkeiten».
Waschen und trocknen Sie Ihre Hände, bevor Sie Mekinest verabreichen.
|
|

| |
1. Prüfen Sie, ob sich in der Flasche Pulver oder eine flüssige Lösung befindet.
·Wenn sich in der Flasche Pulver befindet, fahren Sie mit Abschnitt A Schritt 1 unten fort.
·Wenn sich in der Flasche Lösung befindet, fahren Sie mit Schritt 2 unten fort.
·Wenn sich in der Flasche eine Lösung befindet, geben Sie kein weiteres Wasser in die Flasche
|
|

| |
2. Überprüfen Sie das Datum der Rekonstitution (Zubereitung) der Lösung auf dem Flaschenkarton. Verabreichen Sie Mekinist nicht, wenn das Verfalldatum überschritten ist oder kein Datum angegeben ist.
Hinweis: Das aufgedruckte Verfalldatum auf der rechten Seite des Flaschenetiketts gilt NICHT für die Lösung. Dieses aufgedruckte Verfalldatum gilt nur für das Pulver.
|
|

| |
3. Schwenken Sie die Flasche vorsichtig 30 Sekunden lang, um die Lösung zu mischen.
·Wenn Schaum entsteht, lassen Sie die Flasche stehen, bis kein Schaum mehr zu sehen ist.
|
|

| |
·Entfernen Sie die kindersichere Schutzkappe, indem Sie die Schutzkappe herunterdrücken und gegen den Uhrzeigersinn drehen.
|
|

| |
·Prüfen Sie, ob bereits ein Flaschenadapter in den Flaschenhals eingesetzt ist.
Wenn er nicht eingesetzt ist, nehmen Sie den Adapter von der Spritze ab und setzen Sie ihn ein. Für die weitere Anleitung siehe Schritt 4 und Schritt 10 in Abschnitt A.
Wenden Sie sich an Ihren Arzt bzw. Ihre Ärztin, wenn Sie unsicher sind oder der Adapter fehlt.
|
|

| |
·Drücken Sie den Kolben bis zum Anschlag in die Applikationsspritze für Zubereitungen zum Einnehmen, um die gesamte Luft darin zu entfernen.
|
|

| |
4. Stellen Sie die Flasche auf eine flache Oberfläche und halten Sie sie aufrecht.
·Stecken Sie die Spitze der Applikationsspritze für Zubereitungen zum Einnehmen in die Öffnung des Flaschenadapters.
·Vergewissern Sie sich, dass die Applikationsspritze sicher befestigt ist.
WICHTIGER HINWEIS: Aufgrund des Luftdrucks bewegt sich der Kolben möglicherweise, wenn Sie Ihre Dosis in Schritt 7 abmessen. Halten Sie den Kolben fest, damit er sich nicht bewegt.
|
|

| |
5. Drehen Sie die Flasche vorsichtig auf den Kopf und ziehen Sie am Kolben, um Ihre Dosis abzumessen. Mit der Spitze nach oben muss die Oberseite des schwarzen Stopfens mit Ihrer verschriebenen Dosis übereinstimmen.
·Wenn grosse Luftblasen in der Spritze erscheinen, drücken Sie das Arzneimittel zurück in die Flasche und ziehen Sie Ihre Dosis erneut auf. Machen Sie das so lange, bis keine Luftblasen mehr zu sehen sind. Falls noch kleine Luftblasen zu sehen sind, ist das in Ordnung.
|
|

|

| |
6. Halten Sie den Kolben weiterhin fest, drehen Sie die Flasche wieder um und stellen Sie sie auf eine flache Oberfläche. Während Sie den Kolben noch festhalten, ziehen Sie die Applikationsspritze für Zubereitungen zum Einnehmen vorsichtig nach oben aus der Flasche.
|
|

| |
7. Vergewissern Sie sich, dass die Oberseite des schwarzen Stopfens auf die vorgeschriebene Dosis eingestellt ist. Wenn nicht, wiederholen Sie die Schritte 5-6.
·Wird das Arzneimittel direkt über den Mund eingenommen (durch Schlucken), fahren Sie mit Schritt 8 fort.
·Wird das Arzneimittel über eine Ernährungssonde verabreicht, gehen Sie zu Abschnitt C weiter.
|
|

| |
8. WICHTIGER HINWEIS: Achten Sie bei der Verabreichung an ein Kind darauf, dass es aufrecht sitzt.
·Platzieren Sie das Ende der Applikationsspritze so im Mund, dass die Spitze die Innenseite einer der Wangen berührt.
·Drücken Sie den Kolben langsam ganz nach unten, um die volle Dosis Mekinist zu verabreichen.
ACHTUNG: Wenn Sie Mekinist in den Rachen verabreichen oder den Kolben zu schnell drücken, kann es zum Ersticken kommen.
|
|
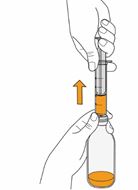
| |
9. Stellen Sie sicher, dass sich keine Lösung mehr in der Spritze befindet.
·Wenn sich noch Lösung in der Spritze befindet, verabreichen Sie diese.
Hinweis: Wenn Ihre Dosis grösser ist als das Fassungsvermögen der Spritze, wiederholen Sie die Verabreichung, bis das gesamte Volumen abgegeben wurde.
|
|

| |
10.Entfernen Sie den Adapter nicht.
·Setzen Sie die Schutzkappe wieder auf die Flasche und drehen Sie sie zum Verschliessen im Uhrzeigersinn.
·Vergewissern Sie sich, dass der Verschluss fest auf der Flasche sitzt.
|
|

| |
11.Anweisungen zur Reinigung und Lagerung finden Sie in Abschnitt D.
|
| |
ABSCHNITT C. VERABREICHUNG ÜBER DIE ERNÄHRUNGSSONDE
| |
Bitte befolgen Sie diesen Abschnitt nur, wenn Sie Mekinist über eine Ernährungssonde verabreichen wollen.
Zur Verabreichung über eine Ernährungssonde lesen Sie bitte die folgenden Informationen und fahren Sie dann mit Schritt 1 fort.
·Die Lösung ist für die Verabreichung über eine Ernährungssonde geeignet.
·Verwenden Sie eine nasogastrische (NG) oder gastrische (G) Ernährungssonde mit einer Mindestgrösse von 4 Ch.
·Verwenden Sie zur Verabreichung von Mekinist immer die in dieser Packung enthaltene 20-ml-Applikationsspritze für Zubereitungen zum Einnehmen.
·Möglicherweise benötigen Sie einen ENFIT-Adapter (nicht in der Packung enthalten), um die 20-ml-Applikationsspritze an die Ernährungssonde anzuschliessen.
| |
1.Spülen Sie die Ernährungssonde gemäss den Anweisungen des Herstellers unmittelbar vor der Verabreichung von Mekinist.
| |
2.Führen Sie die Schritte 1-7 in Abschnitt B aus und gehen Sie dann zu Schritt 3 in diesem Abschnitt über.
| |
3.Schliessen Sie die 20-ml-Spritze mit Mekinist an die Ernährungssonde an. Möglicherweise benötigen Sie einen ENFIT-Adapter, um die Spritze mit der Sonde zu verbinden.
| |
4.Üben Sie gleichmässigen Druck aus, um die Lösung in die Ernährungssonde zu geben.
| |
5.Stellen Sie sicher, dass sich kein Mekinist mehr in der Spritze befindet. Wenn sich noch Lösung in der Spritze befindet, verabreichen Sie diese.
| |
6.Spülen Sie die Sonde erneut gemäss den Anweisungen des Herstellers.
| |
7.Gehen Sie zu Abschnitt D für die Reinigung.
| |
ABSCHNITT D. REINIGUNG
| |
Im Falle eines Verschüttens oder eines Kontakts von Mekinist-Lösung mit der Haut oder den Augen, befolgen Sie die Informationen im Abschnitt «Reinigung von verschütteten Flüssigkeiten».
Bewahren Sie Ihre Applikationsspritze für Zubereitungen zum Einnehmen getrennt von Ihren Küchenutensilien auf.
| |
1. Füllen Sie ein Glas mit warmem Seifenwasser.
|
|

| |
2. Legen Sie die Applikationsspritze in das Glas mit dem warmen Wasser. Ziehen Sie vier- bis fünfmal Wasser in die Applikationsspritze hinein und wieder heraus.
|
|

| |
3. Entfernen Sie den Kolben aus dem Spritzenzylinder.
|
|

| |
4. Spülen Sie das Glas, den Kolben und den Spritzenzylinder unter warmem Leitungswasser ab.
|
|

| |
5. Lassen Sie den Kolben und den Spritzenzylinder vor dem nächsten Gebrauch auf einer trockenen Oberfläche an der Luft trocknen.
Bewahren Sie die Spritze immer ausserhalb der Reichweite von Kindern auf.
Hinweis: Verwenden Sie eine neue Applikationsspritze für jede neue Flasche Mekinist.
|
|

| |
AUFBEWAHRUNG
| |
Lagerung des Pulvers für die Herstellung der Lösung
Mekinist Pulver ausserhalb der Sicht und Reichweite von Kindern aufbewahren.
Bis zur Rekonstitution im Kühlschrank bei 2°C bis 8°C aufbewahren.
In der Originalverpackung aufrecht aufbewahren, um das Pulver vor Licht und Feuchtigkeit zu schützen. Die Flasche fest verschlossen halten.
Das Pulver nicht mehr verwenden, nachdem das auf der rechten Seite des Etiketts aufgedruckte Verfalldatum abgelaufen ist.
|
|

| |
Aufbewahrung der gebrauchsfertigen Lösung
Mekinist Lösung ausserhalb der Sicht und Reichweite von Kindern aufbewahren.
Die Flasche mit der Lösung ist im Umkarton unter 25°C zu lagern.
Die Flasche nicht einfrieren.
Die Lösung aufrecht, in der mitgelieferten Schachtel, vor direktem Licht geschützt und mit fest verschlossener Schutzkappe aufbewahren.
Die Lösung nicht mehr verwenden, wenn mehr als 35 Tage nach dem Datum auf der Packung unter «Lösung hergestellt am» vergangen sind.
|
|

| |
Aufbewahrung von Applikationsspritze für Zubereitungen zum Einnehmen
·Die Applikationsspritzen ausserhalb der Sicht und Reichweite von Kindern aufbewahren.
·Die Applikationsspritze in der mitgelieferten Schachtel zusammen mit Ihrem Pulver/ihrer Lösung aufbewahren.
|
|

| |
ENTSORGUNG
| |
·Fragen Sie Ihren Apotheker, wie Sie abgelaufene oder nicht mehr benötigte Arzneimittel entsorgen können.
·Verwenden Sie für jede neue Flasche Mekinist eine neue Spritze. Fragen Sie Ihren Apotheker, wie Sie die nicht mehr benötigten Spritzen entsorgen können.
·Entsorgen Sie Arzneimittel nicht im Abwasser oder Haushaltsabfall.
·Sie tragen damit zum Schutz der Umwelt bei.
|
|