ZusammensetzungWirkstoffe
Sparsentan
Hilfstoffe
Tablettenkern: silizifizierte mikrokristalline Cellulose, Laktose, Carboxymethylstärke-Natrium (Typ A), hochdisperses Siliciumdioxid, Magnesiumstearat
Filmüberzug: Poly(vinylalkohol), Macrogol 3350, Talk, Titandioxid (E171)
Filmtabletten zu 200 mg: 1 Filmtablette enthält 42 mg Laktose und max. 0,9 mg Natrium.
Filmtabletten zu 400 mg: 1 Filmtablette enthält 84 mg Laktose und max. 1,8 mg Natrium.
Indikationen/AnwendungsmöglichkeitenFilspari ist zur Behandlung von Erwachsenen mit primärer Immunglobulin-A-Nephropathie (IgAN) mit einer Protein-Ausscheidung im Urin von ≥ 1,0 g/Tag (oder einem Protein/Kreatinin-Quotient im Urin von ≥ 0,75 g/g) indiziert (siehe Abschnitt «Klinische Wirksamkeit»).
Aufgrund einer zum Zeitpunkt der Begutachtung des Gesuches unvollständigen klinischen Datenlage, wird diese Indikation befristet zugelassen (Art. 9a Heilmittelgesetz). Die befristete Zulassung ist zwingend an die zeitgerechte Erfüllung von Auflagen gebunden. Nach deren Erfüllung kann die befristete Zulassung in eine ordentliche Zulassung überführt werden.
Dosierung/AnwendungÜbliche Dosierung
Die Behandlung mit Filspari sollte mit einer Dosis von 200 mg einmal täglich über einen Zeitraum von 14 Tagen eingeleitet werden und dann je nach Verträglichkeit auf eine Erhaltungsdosis von 400 mg einmal täglich erhöht werden.
Für die Titration der anfänglichen Dosis von 200 mg einmal täglich auf die Erhaltungsdosis von 400 mg einmal täglich sind zum Erreichen der Erhaltungsdosis Filmtabletten von 200 mg und 400 mg erhältlich.
Bei Patienten mit Verträglichkeitsproblemen (systolischer Blutdruck [SBP] ≤ 100 mmHg, diastolischer Blutdruck ≤ 60 mmHg, sich verschlechterndem Ödem oder Hyperkaliämie) wird eine Anpassung der Begleitmedikation gefolgt von einer vorläufigen Dosisreduktion oder dem Absetzen von Filspari empfohlen (siehe "Warnhinweise und Vorsichtsmassnahmen" und "Klinische Wirksamkeit").
Wenn die Behandlung mit Filspari nach einer Unterbrechung wieder aufgenommen wird, sollte die Wiederholung des ursprünglichen Dosierungsschemas in Betracht gezogen werden. Bei anhaltender Hypotonie oder Veränderungen der Leberfunktion kann eine Unterbrechung der Behandlung mit oder ohne vorheriger Dosisreduktion von Filspari in Betracht gezogen werden (siehe "Warnhinweise und Vorsichtsmassnahmen").
Ausgelassene Dosen
Wird eine Dosis ausgelassen, sollte diese Dosis weggelassen und die nächste Dosis zum vorgesehenen Zeitpunkt eingenommen werden. Es sollten keine doppelten oder zusätzlichen Dosen eingenommen werden.
Ältere Patienten
Bei älteren Patienten wird keine Dosisanpassung empfohlen (siehe "Pharmakokinetik"). Bei älteren Patienten sollte die Behandlung mit Filspari mit einer Dosis von 200 mg einmal täglich über einen Zeitraum von 14 Tagen eingeleitet werden. Die Steigerung auf eine Erhaltungsdosis von 400 mg täglich sollte je nach Verträglichkeit mit Vorsicht vorgenommen werden (siehe "Warnhinweise und Vorsichtsmassnahmen").
Patienten mit Leberfunktionsstörung
Basierend auf Daten zur Pharmakokinetik ist bei Patienten mit leichter oder mittelschwerer Leberfunktionsstörung (Child-Pugh-Klassifikation A oder Child-Pugh B) keine Dosisanpassung von Filspari erforderlich (siehe "Pharmakokinetik").
Es gibt nur begrenzte klinische Erfahrungen mit mittelschwerer Leberfunktionsstörung. Daher sollte Filspari bei diesen Patienten nur mit Vorsicht angewendet werden (siehe "Warnhinweise und Vorsichtsmassnahmen").
Filspari wurde bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh-C-Klassifikation) nicht untersucht und wird daher für die Anwendung bei diesen Patienten nicht empfohlen.
Es gibt nur begrenzte klinische Erfahrungen mit Aspartat-Aminotransferase(AST)/Alanin-Aminotransferase(ALT)-Werten, die mehr als das 2fache der Obergrenze des Normalbereichs (ULN, upper limit of normal) betragen. Daher sollte von einem Beginn mit Filspari bei Patienten mit AST/ALT > 2 × ULN abgesehen werden (siehe "Warnhinweise und Vorsichtsmassnahmen").
Patienten mit Nierenfunktionsstörung
Bei Patienten mit leichter (chronische Nierenerkrankung [CKD] Stadium 2; geschätzte glomeruläre Filtrationsrate [eGFR] 60 bis 89 ml/min/1,73 m2) oder mittelschwerer (CKD-Stadien 3a und 3b; eGFR 30 bis 59 ml/min/1,73 m2) Nierenerkrankung ist keine Dosisanpassung erforderlich. Basierend auf pharmakokinetischen Daten kann keine Dosisanpassung für Patienten mit schwerer Nierenerkrankung (CKD-Stadium 4; eGFR < 30 ml/min/1,73 m2) empfohlen werden (siehe "Pharmakokinetik"). Da es nur begrenzte klinische Erfahrungen bei Patienten mit schweren Nierenerkrankungen gibt, wird Filspari für die Anwendung bei diesen Patienten nicht empfohlen (siehe "Warnhinweise und Vorsichtsmassnahmen").
Filspari wurde bei Patienten, die eine Nierentransplantation erhalten haben, nicht untersucht. Daher sollte Filspari bei diesen Patienten nur mit Vorsicht angewendet werden.
Bei Dialysepatienten wurde Filspari nicht untersucht. Bei diesen Patienten wird die Einleitung einer Behandlung mit Filspari nicht empfohlen.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Filspari bei Kindern unter 18 Jahren ist bisher noch nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Zum Einnehmen.
Es wird empfohlen, die Tabletten als Ganzes mit Wasser zu schlucken, um einen bitteren Geschmack zu vermeiden. Filspari kann mit oder ohne Nahrung eingenommen werden.
Kontraindikationen·Überempfindlichkeit gegenüber dem Wirkstoff oder einem der anderen Inhaltsstoffe (siehe „Zusammensetzung“).
·Schwangerschaft (siehe "Warnhinweise und Vorsichtsmassnahmen" und "Schwangerschaft, Stillzeit")
·Gleichzeitige Verabreichung von Angiotensin-Rezeptorblockern (ARB), Endothelin-Rezeptorantagonisten (ERA) oder Reninhemmern (siehe "Warnhinweise und Vorsichtsmassnahmen" und "Interaktionen")
Warnhinweise und VorsichtsmassnahmenFrauen im gebärfähigen Alter
Die Behandlung mit Filspari darf bei Frauen im gebärfähigen Alter nur begonnen werden, wenn sichergestellt ist, dass keine Schwangerschaft vorliegt und eine wirksame Empfängnisverhütung eingesetzt wird (siehe "Kontraindikation" und "Schwangerschaft, Stillzeit").
Hypotonie
Hypotonie wurde mit der Anwendung von Hemmern des Renin-Angiotensin-Aldosteron-Systems (RAAS), einschliesslich Filspari, in Verbindung gebracht. Während der Behandlung mit Filspari kann es zu einer Hypotonie kommen, wovon am häufigsten bei älteren Patienten berichtet wurde (siehe "Unerwünschte Wirkungen").
Bei Patienten mit Hypotonie-Risiko sollte erwogen werden, ob andere Antihypertensiva weggelassen oder angepasst werden sollen und ein angemessener Volumenstatus aufrechterhalten werden soll. Wenn sich trotz Weglassen oder Reduzierung anderer Antihypertensiva eine Hypotonie entwickelt, sollte eine Dosisreduzierung oder -unterbrechung von Filspari in Betracht gezogen werden. Eine vorübergehende hypotensive Reaktion ist keine Kontraindikation für die weitere Verabreichung von Filspari. Die Behandlung kann wieder aufgenommen werden, sobald sich der Blutdruck stabilisiert hat.
Wenn die Hypotonie trotz Weglassen oder Reduzierung von Antihypertensiva anhält, sollte die Dosierung von Filspari auf die ursprüngliche Anfangsdosis reduziert werden, bis sich der Blutdruck stabilisiert. Eine Dosisunterbrechung bei der Behandlung mit Filspari sollte in Betracht gezogen werden, wenn die Symptome der Hypotonie nach einer 2-wöchigen Dosisreduzierung fortbestehen. Filspari sollte bei Patienten mit systolischen Blutdruckwerten ≤ 100 mmHg mit Vorsicht angewendet werden (siehe "Dosierung/Anwendung"). Bei Patienten mit einem systolischen Blutdruck mit Werten ≤ 100 mmHg sollte die Dosis von Filspari nicht erhöht werden (siehe "Dosierung/Anwendung").
Eingeschränkte Nierenfunktion
Ein vorübergehender Anstieg des Serumkreatinins wurde mit RAAS-Hemmern, einschliesslich Filspari, in Verbindung gebracht. Es kann zu einem vorübergehenden Anstieg des Serumkreatinins kommen, insbesondere zu Beginn der Behandlung mit Filspari (siehe "Unerwünschte Wirkungen"). Bei Risikopatienten sollten die Serumkreatinin- und Serumkaliumwerte regelmässig überwacht werden. Bei Patienten mit beidseitiger Stenose der Nierenarterie sollte Filspari mit Vorsicht angewendet werden.
Aufgrund der begrenzten klinischen Erfahrung bei Patienten mit einer eGFR < 30 ml/min/1,73 m2 wird Filspari für die Anwendung bei diesen Patienten nicht empfohlen (siehe "Dosierung/Anwendung").
Flüssigkeitsretention
Flüssigkeitsretention wurde mit Arzneimitteln in Verbindung gebracht, die den Endothelin-Typ-A-Rezeptor (ETAR) antagonisieren, einschliesslich Sparsentan. Während der Behandlung mit Filspari kann es zu einer Flüssigkeitsretention kommen (siehe "Unerwünschte Wirkungen").
Wenn sich während der Behandlung mit Filspari eine Flüssigkeitsretention entwickelt, wird eine Behandlung mit Diuretika empfohlen, oder es sollte die Dosis der vorhandenen Diuretika erhöht werden, bevor die Dosis von Filspari geändert wird. Bei Patienten, bei denen vor Behandlungsbeginn mit Filspari eine Flüssigkeitsretention festgestellt wurde, kann eine Behandlung mit Diuretika in Betracht gezogen werden.
Bei Patienten mit Herzinsuffizienz wurde Filspari nicht untersucht. Daher sollte Filspari bei Patienten mit Herzinsuffizienz nur mit Vorsicht angewendet werden.
Leberfunktion
Erhöhungen der ALT- oder AST-Werte von mindestens 3 × ULN wurden bei einer Behandlung mit Filspari beobachtet (siehe "Unerwünschte Wirkungen").
Bei mit Filspari behandelten Patienten wurden keine gleichzeitigen Erhöhungen des Bilirubins > 2 × ULN oder Fälle von Leberversagen beobachtet. Um das Risiko einer potenziellen schweren Lebertoxizität zu verringern, sollten daher die Serum-Aminotransferase-Werte und das Gesamtbilirubin vor Behandlungsbeginn überwacht werden, und dann sollte die Überwachung alle drei Monate fortgesetzt werden.
Die Patienten sollten auf Anzeichen einer Leberschädigung überwacht werden. Wenn Patienten eine anhaltende, ungeklärte, klinisch signifikante ALT- und/oder AST-Erhöhung entwickeln, oder wenn die Erhöhungen von einem Anstieg des Bilirubins > 2 × ULN begleitet werden, oder wenn die ALT- und/oder AST-Erhöhung von Anzeichen oder Symptomen einer Leberschädigung (z. B. Gelbsucht) begleitet wird, sollte die Filspari-Therapie abgebrochen werden.
Eine erneute Verabreichung von Filspari sollte nur in Erwägung gezogen werden, wenn die Leberenzymwerte und das Bilirubin auf die Ausgangswerte vor der Behandlung zurückgehen, sowie nur bei Patienten ohne klinische Symptome einer Lebertoxizität. Filspari sollte bei Patienten mit erhöhten Aminotransferasen (> 2 × ULN) vor Beginn der Behandlung nicht angewendet werden (siehe "Dosierung/Anwendung").
Es gibt nur begrenzte klinische Erfahrungen bei mittelschwerer Leberfunktionsstörung. Daher sollte Filspari bei diesen Patienten nur mit Vorsicht angewendet werden (siehe "Dosierung/Anwendung").
Duale Blockade des Renin-Angiotensin-Aldosteron-Systems (RAAS)
Es gibt Anhaltspunkte dafür, dass durch die gleichzeitige Anwendung von Angiotensinkonversionsenzym(ACE)-Hemmern, Angiotensin-II-Rezeptorblockern oder Aliskiren das Risiko für Hypotonie, Hyperkaliämie und eingeschränkte Nierenfunktion (einschliesslich akutem Nierenversagen) erhöht wird. Eine duale Blockade des RAAS durch eine kombinierte Anwendung von ACE-Hemmern und Angiotensin-II-Rezeptorblockern (teilweise ein Mechanismus von Sparsentan) oder Reninhemmern wird daher nicht empfohlen (siehe "Interaktionen" und "Klinische Wirksamkeit”). Wird die Therapie einer dualen Blockade als unabdingbar erachtet, sollte diese nur unter Aufsicht eines Spezialisten und unter häufiger engmaschiger Überwachung der Nierenfunktion, der Elektrolyte und des Blutdrucks durchgeführt werden.
Hyperkaliämie
Die Behandlung sollte nicht begonnen werden bei Patienten mit einem Kaliumspiegel im Serum > 5,5 mmol/l. Wie bei anderen Medizinprodukten, die das Renin-Angiotensin-Aldosteron-System beeinflussen, kann es während der Behandlung mit Filspari zu einer Hyperkaliämie kommen, besonders bei Vorhandensein einer Nierenfunktionsbeeinträchtigung und/oder Herzinsuffizienz. Es wird bei Risikopatienten eine genaue Überwachung des Kaliums im Serum empfohlen. Wenn bei Patienten eine klinisch signifikante Hyperkaliämie auftritt, wird eine Anpassung der Begleitmedikation, eine vorläufige Dosisreduktion oder das Absetzen empfohlen. Ist das Kalium im Serum > 5,5 mmol/l sollte das Absetzen in Betracht gezogen werden.
Patienten mit der seltenen hereditären Galaktoseunverträglichkeit, völligem Laktasemangel oder Glukose-Galaktose-Malabsorption sollten dieses Arzneimittel nicht einnehmen.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Tablette, d. h. es ist nahezu „natriumfrei“.
InteraktionenGleichzeitige Anwendung mit ARB, ERA und Reninhemmern
Die gleichzeitige Anwendung von Sparsentan mit ERA wie Bosentan, Ambrisentan, Macitentan, Sitaxentan, ARB wie Irbesartan, Losartan, Varsartan, Candesartan, Telmisartan oder Reninhemmern wie Aliskiren ist kontraindiziert (siehe "Kontraindikationen").
Gleichzeitige Anwendung mit ACE- und Mineralkortikoid (Aldosteron)-Rezeptorhemmern
Die gleichzeitige Verabreichung von Sparsentan mit Mineralokortikoid (Aldosteron)-Rezeptorhemmern wie Spironolacton und Finerenon ist voraussichtlich mit einem erhöhten Risiko einer Hyperkaliämie verbunden.
Es liegen keine Daten zur Kombination von Sparsentan mit ACE-Hemmern wie Enalapril oder Lisinopril vor. Daten aus klinischen Studien haben gezeigt, dass die duale Blockade des Renin-Angiotensin-Aldosteron-Systems (RAAS) durch die kombinierte Einnahme von ACE-Hemmern und Angiotensin-II-Rezeptorblockern oder Aliskiren im Vergleich zu der Anwendung eines einfachen am RAAS ansetzenden Wirkstoffs mit häufigerem Auftreten unerwünschter Ereignisse wie Hypotonie, Hyperkaliämie und eingeschränkter Nierenfunktion (einschliesslich akutem Nierenversagen) verbunden ist (siehe "Pharmakokinetik").
Die Anwendung von Sparsentan in Kombination mit ACE-Hemmern wie Enalapril oder Lisinopril sollte mit Vorsicht erfolgen, und es sollten der Blutdruck, der Kaliumspiegel und die Nierenfunktion überwacht werden (siehe "Warnhinweise und Vorsichtsmassnahmen").
Gleichzeitige Anwendung mit Kaliumpräparaten und kaliumsparenden Diuretika
Da bei Patienten, die mit Arzneimitteln behandelt werden, die den Angiotensin-II-Rezeptor Typ 1 (AT1R) antagonisieren, eine Hyperkaliämie auftreten kann (siehe "Unerwünschte Wirkungen"), kann die gleichzeitige Anwendung von Kaliumpräparaten, kaliumsparenden Diuretika wie Spironolacton, Eplerenon, Triamteren oder Amilorid oder kaliumhaltigen Salzersatzstoffen das Risiko einer Hyperkaliämie erhöhen und wird nicht empfohlen.
Auswirkungen anderer Arzneimittel auf Sparsentan
Sparsentan wird hauptsächlich durch Cytochrom P450 (CYP)3A metabolisiert.
Starke und mässige CYP3A-Inhibitoren
Die gleichzeitige Verabreichung von Sparsentan mit Itraconazol (starker CYP3A-Inhibitor) erhöhte die Cmax von Sparsentan um das 1,3-Fache und die AUC0-inf um das 2,7-Fache%. Die gleichzeitige Verabreichung mit einem starken CYP3A-Inhibitor wie Boceprevir, Telaprevir, Clarithromycin, Indinavir, Lopinavir/Ritonavir, Itraconazol, Nefazodon, Ritonavir, Grapefruit und Grapefruitsaft, wird nicht empfohlen.
Die gleichzeitige Verabreichung von Sparsentan mit Cyclosporin (mässiger CYP3A-Inhibitor) erhöhte die Cmax von Sparsentan um das 1,4-Fache und die AUC0inf um das 1,7-Fache. Die gleichzeitige Verabreichung mit einem mässigen CYP3A-Inhibitor wie Conivaptan, Fluconazol und Nelfinavir sollte mit Vorsicht erfolgen.
CYP3A-Induktoren
Sparsentan ist ein CYP3A-Substrat. Durch die gleichzeitige Verabreichung eines mässigen oder starken CYP3A-Induktors wie Rifampicin, Efavirenz, Dexamethason, Carbamazepin, Phenytoin und Phenobarbital wird die Exposition von Sparsentan verringert, wodurch die Wirksamkeit von Sparsentan beeinträchtigt werden könnte. Die gleichzeitige Verabreichung mit einem mässigen oder starken CYP3A-Induktor wird daher nicht empfohlen.
Arzneimittel zur Reduzierung der Magensäure
Gemäss einer populationspharmakokinetischen (PK) Analyse hätte die gleichzeitige Verabreichung eines Arzneimittels zur Reduzierung der Säure während der Behandlung mit Sparsentan keinen signifikanten Einfluss auf die Variabilität von Sparsentan PK. Arzneimittel, die den pH-Wert im Magen verändern, wie Antazida, Protonenpumpenhemmer und Histamin-2-Rezeptoragonisten, können gleichzeitig mit Sparsentan angewendet werden.
Auswirkungen von Sparsentan auf andere Arzneimittel
In vitro wurde CYP3A durch Sparsentan gehemmt und induziert, während CYP2B6, CYP2C9 sowie CYP2C19 induziert wurden.
Die gleichzeitige Verabreichung von Sparsentan im Steady State mit dem CYP3A4-Substrat Midazolam hatte keine Auswirkungen auf die systemische Exposition von Midazolam. Die gleichzeitige Verabreichung von Sparsentan im Steady State mit dem CYP2B6-Substrat Bupropion verringerte sowohl die Cmax von Bupropion als auch die AUC0-inf um das 1,5-Fache.
Eine Dosisanpassung ist nicht erforderlich, wenn Sparsentan im Steady State mit einem CYP3A4- oder CYP2B6-Substrat kombiniert wird.
Die Signifikanz der CYP2C9- und CYP2C19-Induktion durch Sparsentan wurde nicht in einer klinischen Studie untersucht. Die gleichzeitige Verabreichung von Sparsentan mit einem CYP2C9-Substrat wie S-Warfarin, Phenytoin und Ibuprofen oder CYP2C19-Substrate wie Omeprazol und Phenytoin sollte nur mit Vorsicht erfolgen.
Die Signifikanz der CYP3A4-Induktion nach einer Einzeldosis Sparsentan wurde nicht in einer klinischen Studie untersucht. Sparsentan ist ein Inhibitor von CYP3A4 und kann bei der Einleitung einer Behandlung mit Sparsentan Auswirkungen auf die PK von Arzneimitteln haben, die Substrate von CYP3A4 sind. Daher sollte die Einleitung einer Behandlung mit Sparsentan als Begleitmedikation mit einem CYP3A4-Substrat wie Alfentanil, Conivaptan, Indinavir, Cyclosporin und Tacrolimus nur mit Vorsicht erfolgen.
In vitro ist Sparsentan ein Inhibitor von Pgp, BCRP, OATP1B3 und OAT3 in relevanten Konzentrationen.
Die Signifikanz der Pgp-Hemmung durch Sparsentan wurde nicht in einer klinischen Studie untersucht.
Die gleichzeitige Verabreichung von Sparsentan mit Pgp-hemmenden Substraten sollte nur mit Vorsicht erfolgen, wenn bekannt ist, dass die Pgp-Hemmung eine signifikante Auswirkung auf die Resorption hat.
Die gleichzeitige Verabreichung von Sparsentan mit Pitavastatin (einem Substrat von OATP1B1, OATP1B3 und BCRP) verringerte die Cmax von Pitavastatin um um das 1,2-Fache und die AUC0-inf um das 1,4-Fache. Eine Dosisanpassung ist nicht erforderlich, wenn Sparsentan mit einem OATP1B1-, OATP1B3- oder BCRP-Substrat kombiniert wird.
Es wurde keine klinische Studie zur Untersuchung der Auswirkung von Sparsentan auf ein empfindliches OAT3-Substrat durchgeführt. Bei einer Dosis von 800 mg scheint Sparsentan jedoch keine Auswirkungen auf den Biomarker 6β-Hydroxycortisol (Substrat von OAT3) zu haben, was darauf hindeutet, dass die klinische Wirkung höchstwahrscheinlich begrenzt ist.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Die Behandlung mit Filspari darf bei Frauen im gebärfähigen Alter nur begonnen werden, wenn sichergestellt ist, dass keine Schwangerschaft vorliegt. Frauen im gebärfähigen Alter müssen während der Behandlung und bis zu einen Monat nach Behandlungsende eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Bisher liegen keine oder nur sehr begrenzte Erfahrungen zur Anwendung von Filspari bei Schwangeren vor.
Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe "Präklinische Daten").
Filspari ist während der Schwangerschaft kontraindiziert (siehe "Kontraindikationen").
Stillzeit
Physikalisch-chemische Daten deuten auf die Ausscheidung von Sparsentan in die Muttermilch beim Menschen hin. Ein Risiko für Neugeborene/Säuglinge kann nicht ausgeschlossen werden. Filspari sollte in der Stillzeit nicht angewendet werden.
Fertilität
Es liegen keine Daten zu den Auswirkungen von Sparsentan auf die Fertilität bei Menschen vor. Tierexperimentelle Daten ergaben keine Hinweise auf eine Beeinträchtigung der männlichen oder weiblichen Fertilität (siehe "Präklinische Daten").
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenFilspari kann einen geringen Einfluss auf die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen haben.
Es wurden keine Studien zu den Auswirkungen von Filspari auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt. Es sollte jedoch beachtet werden, dass bei der Einnahme von Filspari Schwindel auftreten kann (siehe "Unerwünschte Wirkungen"). Patienten mit Schwindel sollten darauf hingewiesen werden, das Führen von Fahrzeugen oder Bedienen von Maschinen solange zu unterlassen, bis die Symptome abgeklungen sind.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die am häufigsten gemeldeten unerwünschten Arzneimittelwirkungen (UAW) waren Hypotonie (9 %), Hyperkaliämie (7 %), Schwindel (7 %) und periphere Ödeme (5 %). Die am häufigsten gemeldete schwerwiegende Nebenwirkung war eine akute Nierenschädigung (1 %).
Liste der unerwünschten Wirkungen
Unterstützende Sicherheitsdaten wurden aus 27 klinischen Studien gewonnen, an denen mehr als 500 Patienten teilnahmen, die Filspari bei chronischen Nierenerkrankungen, einschliesslich IgAN, erhielten (siehe "Klinische Wirksamkeit").
Die unerwünschten Wirkungen sind nach MedDRA-Systemorganklassen und Häufigkeit gemäss folgender Konvention geordnet:
„sehr häufig“ (≥1/10)
„häufig“ (≥1/100, <1/10),
„gelegentlich“ (≥1/1‘000, <1/100)
„selten“ (≥1/10‘000, <1/1‘000)
„sehr selten“ (<1/10‘000)
Tabelle 1: In klinischen Studien beobachtete unerwünschte Arzneimittelwirkungen
|
Systemorganklasse
|
Häufig
|
Gelegentlich
| |
Erkrankungen des Blutes und des Lymphsystems
|
-
|
Anämie
| |
Stoffwechsel- und Ernährungsstörungen
|
Hyperkaliämie
|
-
| |
Erkrankungen des Nervensystems
|
Schwindel
Kopfschmerzen
|
-
| |
Gefässerkrankungen
|
Hypotonie
Orthostasesyndrom
|
-
| |
Erkrankungen der Nieren und Harnwege
|
Niereninsuffizienz
akute Nierenschädigung
|
-
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
peripheres Ödem
Ermüdung
|
-
| |
Untersuchungen
|
Kreatinin im Blut erhöht
Transaminasen erhöhta
|
-
|
a Erhöhte Transaminasen umfasst bevorzugte Begriffe wie erhöhte Alanin-Aminotransferasen, erhöhte Aspartat-Aminotransferasen, erhöhte Gamma-Glutamyltransferasen, erhöhte Leberenzyme und erhöhte Transaminasen.
Beschreibung ausgewählter Nebenwirkungen
Hämoglobin erniedrigt
Bei PROTECT wurde eine Anämie oder vermindertes Hämoglobin bei 2 (< 1 %) mit Sparsentan behandelten Personen im Vergleich zu 2 (< 1 %) mit Irbesartan behandelten Personen als UAW gemeldet. Insgesamt wurde zu einem beliebigen Zeitpunkt nach der Behandlung bei 5 (2,5 %) Personen in der Sparsentan-Behandlungsgruppe und bei 3 (1,5 %) Personen in der Irbesartan-Behandlungsgruppe ein Hämoglobinwert von ≤ 9 g/dl festgestellt. Man geht davon aus, dass dieser Rückgang zum Teil auf eine Hämodilution zurückzuführen ist. Es gab keine Behandlungsabbrüche aufgrund von Anämie.
Unerwünschte die Leber betreffende Ereignisse
Bei PROTECT hatten insgesamt 6 (3 %) Teilnehmer der Sparsentan-Gruppe und 4 (2 %) Teilnehmer der Irbesartan-Gruppe erhöhte Lebertransaminasewerte, die die obere Normalgrenze ohne Erhöhung des Gesamtbilirubins um mehr als das 3fache überschritten, nachdem sie das Studienmedikament für jeweils 168 bis 407 Tage erhalten haben. Bei allen Ereignissen handelte es sich um nicht schwerwiegende und asymptomatische Ereignisse, die meistens von leichter oder moderater Intensität. Alle Ereignisse waren reversibel und wurden als mögliche ursächliche Faktoren oder als eventuell zur Erhöhung der Transaminasen beitragend, oder als andere Gründe identifiziert. In der Sparsentan-Gruppe wurde das Studienmedikament bei 3 Teilnehmern nach positiver Rechallenge abgesetzt, während bei 2 Teilnehmern die Behandlung mit Sparsentan ohne erneut erhöhte Leberenzyme wieder aufgenommen wurde.
Akute Nierenschädigung (AKI)
Bei PROTECT wurden für 4 (2 %) Teilnehmer der Sparsentan-Gruppe und 2 (1 %) Teilnehmer der Irbesartan-Gruppe UAW in Form einer akuten Nierenschädigung gemeldet. Für vier Teilnehmer (2 %), die Sparsentan erhalten hatten, wurden schwerwiegende AKI gemeldet, die alle reversibel waren. Bei keiner der schwerwiegenden AKI war eine Dialyse erforderlich. In der Sparsentan-Gruppe wurde das Studienmedikament bei 3 Teilnehmern abgesetzt.
Hyperkaliämie
Bei PROTECT wurde für 18 (9 %) mit Sparsentan behandelte Teilnehmer eine Hyperkaliämie als UAW gemeldet, in der mit Irbesartan behandelten Gruppe waren es im Vergleich dazu 16 (8 %) Teilnehmer. Bei den mit Sparsentan behandelten Teilnehmern waren alle Ereignisse nicht schwerwiegend, es handelte sich bei den meisten um Ereignisse von leichter bis mässiger Intensität, die alle reversibel waren. Es gab keine Behandlungsabbrüche aufgrund von Hyperkaliämie. Das Risiko einer Hyperkaliämie steigt bei Patienten mit einer niedrigen eGFR.
Hypotonie
Bei PROTECT wurde für jeweils 10 % und 8 % der mit Sparsentan behandelten Patienten ein SBP < 100 mmHg bzw. eine Senkung des SBP um mehr als 30 mmHg gemeldet, dem gegenüber standen 9 % bzw. 6 % bei Irbesartan. Bei den mit Sparsentan behandelten Teilnehmern waren nur 15 Teilnehmer (7,4 %) über 65 Jahre alt. Eine Hypotonie wurde bei 17 (9 %) Teilnehmern im Alter < 65 Jahre und bei 5 (33 %) Teilnehmern im Alter von 65 bis 74 Jahre gemeldet.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungFilspari wurde gesunden Personen in Dosen von bis zu 1600 mg/Tag verabreicht, ohne dass es Hinweise auf dosislimitierende Toxizitäten gab. Patienten, bei denen es zu einer Überdosierung kommt (möglicherweise mit Anzeichen und Symptomen einer Hypotonie), sollten sorgfältig überwacht werden und eine angemessene symptomatische Behandlung erhalten.
Eigenschaften/WirkungenATC-Code
C09XX01
Wirkungsmechanismus
Sparsentan ist ein dualer Endothelin-Angiotensin-Rezeptorantagonist.
Es ist ein einzelnes Molekül, das als hochaffiner, doppelt wirkender Antagonist sowohl des ETAR als auch des AT1R fungiert. Endothelin-1 über ETAR und Angiotensin II über AT1R vermitteln Prozesse, die durch hämodynamische Wirkungen und Mesangialzellproliferation, die erhöhte Expression und Aktivität proinflammatorischer und profibrotischer Mediatoren, Podozytenschädigung und oxidativen Stress zur Progression von IgAN führen. Sparsentan hemmt die Aktivierung sowohl von ETAR als auch AT1R und verringert dadurch die Proteinurie und verlangsamt das Fortschreiten der Nierenerkrankung.
Pharmakodynamik
In einer randomisierten positiv- und placebokontrollierten Studie mit gesunden Teilnehmern bewirkte Sparsentan eine leichte QTcF-Verlängerung mit einem maximalen Effekt von 8,8 ms (90-%-KI: 5,9; 11,8) bei 800 mg und 8,1 ms (90-%-KI: 5,2; 11,0) bei 1600 mg. In einer zusätzlichen Studie mit gesunden Teilnehmern mit einer Sparsentan-Exposition von mehr als dem 2-Fachen der empfohlenen Höchstdosis für den Menschen trat keine relevante QTcF-Verlängerung auf; der maximale Effekt lag bei 8,3 (6,69; 9,90) ms. Daher ist es unwahrscheinlich, dass Sparsentan eine klinisch relevante Wirkung auf die QT-Verlängerung hat.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von Sparsentan wurden in PROTECT mit Patienten mit IgAN untersucht.
PROTECT ist eine randomisierte, doppelblinde (110 Wochen), aktiv kontrollierte, multizentrische, globale Phase-III-Studie mit Patienten mit IgAN. An der Studie nahmen Patienten im Alter von ≥ 18 Jahren teil, darunter 15 (8 %) mit Sparsentan behandelte Patienten im Alter von > 65 Jahren, mit einer eGFR ≥ 30 ml/min/1,73 m2 und einer Gesamtproteinausscheidung im Urin von ≥ 1,0 g/Tag. Vor der Studienteilnahme hatten die Patienten mindestens 3 Monate lang die maximal verträgliche Dosis eines ACE-Hemmers und/oder eines ARB eingenommen. Die ACE-Hemmer- und/oder ARB-Therapie wurde vor der Verabreichung von Sparsentan abgesetzt. Patienten mit einem Kalium-Ausgangswert von mehr als 5,5 mmol/l wurden ausgeschlossen.
Insgesamt wurden 404 Patienten randomisiert und erhielten Sparsentan (n = 202) oder Irbesartan (n = 202). Die Behandlung wurde mit 200 mg Sparsentan einmal täglich oder 150 mg Irbesartan einmal täglich begonnen. Nach 14 Tagen sollte die Dosis je nach Verträglichkeit auf die empfohlene Dosis von 400 mg Sparsentan einmal täglich oder 300 mg Irbesartan einmal täglich titriert werden. Die Dosisverträglichkeit wurde definiert als systolischer Blutdruck > 100 mmHg und diastolischer Blutdruck > 60 mmHg nach 2 Wochen sowie keine UE (z. B. Verschlechterung des Ödems) oder Laborbefunde (z. B. Serumkalium > 5,5 mEq/l [5,5 mmol/l]). Inhibitoren des RAAS- oder Endothelin-Systems waren während der Studie verboten. Andere Klassen von Antihypertensiva waren je nach Bedarf zugelassen, um den Zielblutdruck zu erreichen. Die Behandlung mit Immunsuppressiva war während der Studie nach dem Ermessen des Forschers zulässig.
Die Ausgangsdaten bzgl. eGFR und Proteinurie waren zwischen den Behandlungsgruppen vergleichbar. Die Gesamtpopulation hatte eine mittlere (SD) eGFR von 57 (24) ml/min/1,73 m2 und ein medianes Urinprotein/Kreatinin(UP/K)-Verhältnis von 1,24 g/g (Interquartilsabstand: 0,83; 1,77). Das Durchschnittsalter betrug 46 Jahre (Spanne 18 bis 76 Jahre); 70 % waren männlich, 67 % Weiss, 28 % Asiatisch, 1 % Schwarz oder Afroamerikanisch und 3 % gehörten einer anderen Ethnie an.
Die primäre (Interims-)Proteinurieanalyse wurde 36 Wochen nach Randomisierung von etwa 280 Teilnehmern durchgeführt, um zu bestimmen, ob der Behandlungseffekt des primären Wirksamkeitsendpunktes, die Veränderung des UP/K in Woche 36 gegenüber dem Ausgangswert, statistisch signifikant ist. Der primäre Endpunkt der Studie, die Veränderung des UP/K-Verhältnisses gegenüber dem Ausgangswert in Woche 36, wurde erreicht. Der geometrische Mittelwert des UP/K in Woche 36 betrug 0,62 g/g in der Sparsentan-Gruppe gegenüber 1,07 g/g in der Irbesartan-Gruppe. Der geometrische Mittelwert der kleinsten Quadrate (LS, von engl. least squares) für die prozentuale Veränderung von UP/K gegenüber dem Ausgangswert in Woche 36 betrug -49,8 % (95-%-Konfidenzintervall [KI]): -54,98, -43,95) in der Sparsentan-Gruppe gegenüber -15,1 % (95-%-KI: -23,72, -5,39) in der Irbesartan-Gruppe (p<0,0001; (Abbildung 1). Bei der endgültigen Analyse zeigte Sparsentan eine schnelle und nachhaltige Wirkung der antiproteinurischen Behandlung über einen Zeitraum von 2 Jahren mit einem einem geometrischen Mittelwert des UP/K in Woche 110 von 0,64 g/g in der Sparsentan-Gruppe gegenüber 1,09 g/g in der Irbesartan-Gruppe, was eine mittlere Reduzierung von 43 % gegenüber dem Ausgangswert (95 % KI: -49,75, -34,97) im Vergleich zu nur 4,4 % bei einer Behandlung mit Irbesartan (95 % KI: -15,84, 8,70) darstellt. Eine Verbesserung der Proteinurie-Reduktion wurde mit Sparsentan bereits nach 4 Wochen fortlaufend beobachtet und hielt bis Woche 110 an (Abbildung 1).
Abbildung 1: Prozentuale Veränderung gegenüber dem Ausgangswert des Verhältnisses Urinprotein/Kreatinin nach Visite (PROTECT)
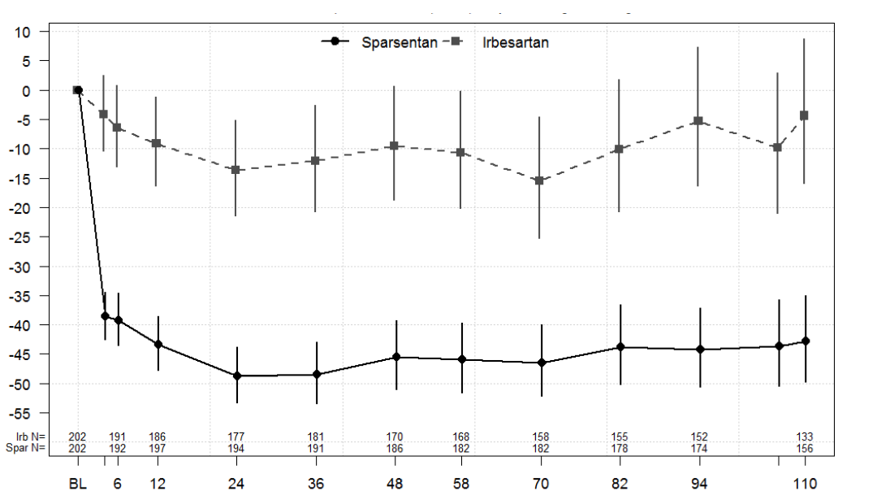
Hinweise: Der bereinigte geometrische Mittelwert der kleinsten Quadrate für das Verhältnis UP/K im Vergleich zum Ausgangswert basierte auf einem Längsschnittmodell mit wiederholten Messungen, das nach eGFR- und Proteinurie-Screening stratifiziert war, und wird als prozentuale Veränderung zusammen mit dem jeweiligen 95-%-KI angegeben. Die Analyse umfasst UP/K-Daten während der Doppelblindphase von allen Patienten, die randomisiert wurden und mindestens eine Dosis des Studienmedikaments erhielten. Der Ausgangswert wurde definiert als die letzte nicht fehlende Beobachtung vor der und einschliesslich der Verabreichung der ersten Dosis.
Abkürzungen: KI = Konfidenzintervall; eGFR = geschätzte glomeruläre Filtrationsrate; LS = kleinste Quadrate (von engl. least squares); UP/K = Verhältnis Urinprotein/Kreatinin.
Geschätzte GFR
Zum Zeitpunkt der bestätigenden Analyse betrug die Verbesserung der dauerhaften eGFR-Senkung in zwei Jahren (ab 6 Wochen) bei Sparsentan 1,1 ml/min/1,73 m2 pro Jahr im Vergleich zu Irbesartan [95-%-KI -0,07, 2,12; p = 0,037), und die entsprechende Verbesserung der gesamten eGFR-Senkung (ab Baseline) betrug 1,0 ml/min/1,73 m2 in zwei Jahren (95-%-KI: -0,03, 1,94; p = 0,058). Die absolute Veränderung der eGFR gegenüber dem Ausgangswert nach 2 Jahren betrug -5,8 ml/min/1,73 m2 (95-%-KI: -7,38, -4,24) für Sparsentan im Vergleich zu -9,5 ml/min/1,73 m2 (95-%-KI: -11,17, -7,89) für Irbesartan.
Zusätzliche Informationen
In zwei grossen randomisierten, kontrollierten Studien (ONTARGET (ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial) und VA NEPHRON-D (The Veterans Affairs Nephropathy in Diabetes)) wurde die kombinierte Anwendung von ACE-Hemmern und Angiotensin-II-Rezeptorblockern untersucht. Die Studie ONTARGET wurde bei Patienten mit kardiovaskulären oder cerebrovaskulären Erkrankungen oder Diabetes mellitus Typ 2 in der Anamnese durchgeführt, bei denen Hinweise auf Endorganschäden vorlagen. VA NEPHRON-D war eine Studie für Patienten mit Diabetes mellitus Typ 2 und diabetischer Nephropathie. Diese Studien zeigten keine signifikante positive Wirkung auf renale und/oder kardiovaskuläre Ergebnisse und Mortalität, wobei im Vergleich zur Monotherapie ein erhöhtes Risiko für Hyperkaliämie, akute Nierenschädigung und/oder Hypotonie beobachtet wurde. Angesichts ihrer ähnlichen pharmakodynamischen Eigenschaften sind diese Ergebnisse auch für andere ACE-Hemmer und Angiotensin-II-Rezeptorblocker relevant. Daher sollte bei Patienten mit diabetischer Nephropathie keine Begleittherapie mit ACE-Hemmern und Angiotensin-II-Rezeptorblockern durchgeführt werden. Die Studie ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints) war darauf ausgelegt, den Nutzen einer Hinzunahme von Aliskiren zu einer Standardtherapie mit einem ACE-Hemmer oder einem Angiotensin-II-Rezeptorblocker bei Patienten mit Diabetes mellitus Typ 2und chronischer Nierenerkrankung, kardiovaskulärer Erkrankung oder beidem zu prüfen. Die Studie wurde aufgrund eines erhöhten Risikos unerwünschter Ergebnisse vorzeitig beendet. In der Aliskiren-Gruppe traten zahlenmässig häufiger Fälle von kardiovaskulärem Tod und Schlaganfall auf als in der Placebogruppe, und unerwünschte Ereignisse sowie schwerwiegende unerwünschte Ereignisse von Interesse (Hyperkaliämie, Hypotonie und Nierenfunktionsstörungen) wurden in der Aliskiren-Gruppe häufiger gemeldet als in der Placebogruppe.
PharmakokinetikAbsorption
Nach einer oralen Einzeldosis von 400 mg Sparsentan beträgt die mittlere Zeit bis zur Spitzenplasmakonzentration etwa 3 Stunden.
Nach einer oralen Einzeldosis von 400 mg Sparsentan beträgt der geometrische Mittelwert der Cmax 6,97 μg/ml und die AUC 83 μg × h/ml. Die Steady-State-Plasmaspiegel werden innerhalb von 7 Tagen erreicht, ohne dass es bei der empfohlenen Dosierung zu einer Akkumulation der Exposition kommt.
Nach einer Dosis von 400 mg Sparsentan täglich beträgt der geometrische Mittelwert der Cmax im Steady State 6,47 μg/ml und die AUC im Steady State 63,6 μg × h/ml.
Wechselwirkung mit Nahrungsmitteln
Bei Dosen von bis zu 400 mg war die Auswirkung einer fettreichen Mahlzeit auf die Sparsentan-Exposition nicht klinisch relevant. Sparsentan kann mit oder ohne Nahrung eingenommen werden.
Distribution
Gemäss einer populationspharmakokinetischen Analyse beträgt das ersichtliche Verteilungsvolumen im Steady State 61,4 l.
Sparsentan ist in hohem Masse (> 99 %) an menschliche Plasmaproteine gebunden, wobei es bevorzugt an Albumin und mässig an saures Alpha-1-Glykoprotein bindet.
Metabolismus
Sparsentan wird hauptsächlich durch CYP3A4 mit einem geringen Anteil von CYP2C8, 2C9 und 3A5 metabolisiert. Die Muttersubstanz hat den überwiegenden Anteil im menschlichen Plasma und macht etwa 90 % der gesamten Radioaktivität im Blutkreislauf aus. Ein hydroxylierter Metabolit mit geringerem Anteil war der einzige Metabolit im Plasma, der > 1 % der gesamten Radioaktivität ausmachte (etwa 3 %). Der Hauptstoffwechselweg von Sparsentan war die Oxidation und Dealkylierung, und es wurden 9 Metaboliten im menschlichen Stuhl, Plasma und Urin identifiziert.
Elimination
Die Clearance von Sparsentan ist zeitabhängig. Gemäss einer populationspharmakokinetischen Analyse beträgt die ersichtliche Clearance 3,88 l/h und steigt im Steady State auf 5,11 l/h an.
Die Halbwertszeit von Sparsentan im Steady State wird auf 9,6 Stunden geschätzt.
Nach einer Einzeldosis von 400 mg radioaktiv markiertem Sparsentan wurden 82 % der dosierten Radioaktivität innerhalb eines 10-tägigen Sammelzeitraums wiedergefunden: 80 % über den Stuhl, davon 9 % in unveränderter Form, und 2 % über den Urin, davon nur eine vernachlässigbare Menge in unveränderter Form.
Linearität/Nicht-Linearität
Die Cmax und die AUC von Sparsentan steigen nach der Verabreichung von Einzeldosen von 200 mg bis 1600 mg weniger als proportional an. Sparsentan zeigte eine zeitabhängige Pharmakokinetik mit keiner Cmax-Akkumulation und verringerter AUC im Steady State nach einer Dosis von 400 oder 800 mg täglich.
Kinetik spezieller Patientengruppen
Ältere Patienten
Die populationspharmakokinetische Analyse ergab keinen signifikanten Einfluss des Alters auf die Plasmaexposition von Sparsentan. Bei älteren Patienten ist keine Dosisanpassung erforderlich (siehe "Dosierung/Anwendung"). Bei Patienten im Alter von > 75 Jahren wurde Sparsentan nicht untersucht.
Leberfunktionsstörung
In einer einschlägigen Studie zu Leberfunktionsstörungen war die systemische Exposition nach einer Einzeldosis von 400 mg Sparsentan bei Patienten mit bestehender (Baseline) leichter oder mittelschwerer Leberfunktionsstörung (Child-Pugh-Klassifikation A oder ChildPugh B) ähnlich wie bei Patienten mit normaler Leberfunktion. Bei Patienten mit leichter oder mittelschwerer Leberfunktionsstörung ist keine Dosisanpassung erforderlich. Bei Patienten mit mittelschwerer Leberfunktionsstörung sollte Sparsentan mit Vorsicht angewendet werden (siehe "Dosierung/Anwendung" und "Warnhinweise und Vorsichtsmassnahmen").
Zu Patienten mit schwerer Leberfunktionsstörung liegen keine Daten vor, daher wird Sparsentan bei diesen Patienten (Child-Pugh-C-Klassifikation) nicht empfohlen (siehe "Dosierung/Anwendung").
Nierenfunktionsstörung
Laut einer populationspharmakokinetischen Analyse bei chronisch nierenkranken Patienten mit leichter (Kreatinin-Clearance 60-89 ml/min), mittelschwerer (Kreatinin-Clearance 30-59 ml/min) und schwerer (Kreatinin-Clearance 15-29 ml/min) Nierenerkrankung hat eine Niereninsuffizienz im Vergleich zu einer normalen Nierenfunktion keine klinisch bedeutsamen Auswirkungen auf die Pharmakokinetik (Kreatinin-Clearance ≥ 90 ml/min). Zu Patienten mit terminaler Niereninsuffizienz (Kreatinin-Clearance < 15 ml/min) liegen keine Daten vor.
Aufgrund der begrenzten verfügbaren Daten kann für Patienten mit schwerer Nierenerkrankung (eGFR < 30 ml/min/1,73 m2, siehe "Dosierung/Anwendung") keine Dosisanpassung empfohlen werden. Sparsentan wurde bei Dialysepatienten oder Patienten mit schwerer Nierenerkrankung nicht untersucht, daher wird die Behandlung mit Sparsentan bei diesen Patienten nicht empfohlen. Sparsentan wurde bei Patienten, die eine Nierentransplantation erhalten haben, nicht untersucht, daher sollte Sparsentan bei dieser Patientenpopulation mit Vorsicht angewendet werden (siehe "Dosierung/Anwendung").
Sonstige besondere Personengruppen
Populationspharmakokinetische Analysen deuten auf keinen klinisch bedeutsamen Einfluss von Alter, Geschlecht oder Ethnie auf die Pharmakokinetik von Sparsentan.
Präklinische DatenIn Studien zur embryofetalen Entwicklung bei Ratten und Kaninchen wurde bei beiden Arten eine Entwicklungstoxizität festgestellt. Bei Ratten wurden bei allen getesteten Sparsentan-Dosen dosisabhängige teratogene Wirkungen in Form von kraniofazialen Fehlbildungen, Skelettanomalien, erhöhter embryofetaler Letalität und einem reduzierten fetalen Gewicht beobachtet, und zwar bei Expositionen, die 8-Fach bzw. 13-Fach über der AUC für 800 mg/Tag bzw. 400 mg/Tag beim Menschen lagen. Bei Kaninchen traten keine fetalen Missbildungen oder Auswirkungen auf die Lebensfähigkeit des Embryos oder Fetus oder auf das Wachstum des Fetus auf, aber bei einer Exposition von ca. dem 0,10- und 0,2-Fachen der AUC beim Menschen bei 800 mg/Tag und 400 mg/Tag kam es zu einer Zunahme von Skelettveränderungen (überzählige Halsrippen).
In der prä- und postnatalen Entwicklungsstudie bei Ratten wurde beim ~8-Fachen und 13-Fachen der AUC des Menschen eine maternale Toxizität einschliesslich Tod und beim ~2-Fachen und 3-Fachen der AUC des Menschen bei 800 mg/Tag und 400 mg/Tag eine maternale Toxizität beobachtet. Beim ~8-Fachen und 13-Fachen der AUC des Menschen traten ein Anstieg der Sterblichkeit und ein vermindertes Wachstum der Jungtiere auf und beim ~2-Fachen und 3-Fachen der AUC des Menschen bei 800 mg/Tag und 400 mg/Tag ein vermindertes Wachstum.
Studien mit Jungtieren
Studien mit Jungtieren von Ratten haben gezeigt, dass bis zu einer Dosierung von 10 mg/kg/Tag keine allgemeinen toxikologischen Nebenwirkungen und bis zu einer Dosierung von 60 mg/kg/Tag keine reproduktive Toxizität bei männlichen oder weiblichen Tieren zu beobachten war, wenn mit der Verabreichung am postnatalen Tag (PND) 14 begonnen wurde (dies entspricht einjährigen Kindern). Vaskuläre Toxizität trat bei Dosen ≥ 3 mg/kg/Tag auf, wenn die Verabreichung am PND 7 (entspricht Neugeborenen) begann.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit „EXP“ bezeichneten Datum verwendet werden.»
Besondere Lagerungshinweise
Bei 15 - 30°C lagern.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Zulassungsnummer69241 (Swissmedic)
PackungenFilmtabletten zu 200 mg/400 mg: Packungen mit je 30 Filmtabletten in einer Flasche. (B)
Filmtabletten 400 mg: Packungen mit je 30 Filmtabletten in einer Flasche. (B)
Filmtabletten 400 mg: 3 Packungen mit je 30 Filmtabletten in einer Flasche. (B)
ZulassungsinhaberinVifor (International) Inc., St. Gallen
Stand der InformationMai 2024
|