ZusammensetzungWirkstoffe
Omalizumabum
Hilfsstoffe
Injektionslösung in Fertigpen:
Arginini hydrochloridum, Histidini hydrochloridum monohydricum, Histidinum, Polysorbatum 20, Aqua ad iniectabile q.s. ad solutionem
Indikationen/AnwendungsmöglichkeitenAllergisches Asthma
Xolair wird in Kombination mit anderen Asthmatherapien zur verbesserten Asthmakontrolle bei Erwachsenen und Kindern (ab 6 Jahren) mit schwerem persistierendem allergischem Asthma (positiver Hauttest oder in vitro–Reaktivität gegen ein ganzjährig auftretendes Aeroallergen) angewendet, falls diese trotz täglicher Therapie mit hoch dosierten inhalativen Kortikosteroiden und einem langwirksamen Beta2-Agonisten sowohl eine reduzierte Lungenfunktion (FEV1<80%) haben als auch unter häufigen Symptomen während des Tages oder nächtlichem Erwachen leiden und Asthma-Exazerbationen hatten.
Nasenpolypen
Xolair (Omalizumab) ist bei Erwachsenen (ab 18 Jahren) zur Behandlung von Nasenpolypen indiziert, die auf intranasale Kortikosteroide nicht ausreichend ansprechen.
Chronische spontane Urtikaria (CSU)
Zusatztherapie für Erwachsene und Jugendliche (ab 12 Jahren) mit langwieriger* chronischer spontaner Urtikaria (CSU), welche mit einer H1 Antihistaminika-Therapie nicht kontrolliert werden kann und für welche in einer Abklärung durch einen mit diesem Krankheitsbild vertrauten Arzt keine andere zugrunde liegenden Krankheit gefunden wurde.
(*In den Zulassungsstudien wurden Patienten mit einer CSU-Krankheitsdauer von 6 Monaten bis 66 Jahren, durchschnittlich 6 Jahren, untersucht)
Dosierung/AnwendungAllergisches Asthma
Übliche Dosierung
Erwachsene und Kinder ab 6 Jahren
Die Xolair Fertigpen sind nicht für die Anwendung bei Patienten unter 12 Jahren bestimmt. Die Xolair 75 mg Fertigspritze und die Xolair 150 mg Fertigspritze oder das Xolair Pulver und Lösungsmittel für die Injektionslösung können bei Kindern im Alter von 6 bis 11 Jahren mit allergischem Asthma angewendet werden.
Die geeignete Dosis und Verabreichungsfrequenz von Xolair wird anhand des vor Behandlungsbeginn gemessenen Immunglobulin E (IgE)-Basiswertes (I.E./ml) im Serum und des Körpergewichtes (kg) ermittelt. Vor der ersten Anwendung ist es erforderlich den IgE-Wert des Patienten zur Dosisfestlegung von Xolair mit einem kommerziell erhältlichen Gesamt-Serum-IgE-Test zu bestimmen. Basierend auf diesen Messungen werden 75-600 mg Xolair in 1 bis 2 Injektionen pro Verabreichung benötigt.
Für Patienten mit einem IgE-Wert unter 76 I.E./ml war ein Nutzen weniger wahrscheinlich (s. «Pharmakodynamik»). Verschreibende Ärzte sollten vor Beginn der Therapie sicherstellen, dass Patienten mit einem IgE-Wert unter 76 I.E./ml eine eindeutige in vitro Reaktivität (RAST) gegenüber einem ganzjährig auftretenden Allergen zeigen.
Siehe Tabelle 1 als Umrechnungsschema und Tabellen 2 und 3 zur Dosisfindung.
Patienten, deren Basis-IgE-Spiegel oder Körpergewicht in Kilogramm ausserhalb der Grenzen der Dosierungstabellen liegen (≤20 oder >150 kg KG), sollten nicht mit Xolair behandelt werden.
|
|
Tabelle 1: Umrechnung der Dosierung auf die Anzahl der Fertigpen, Anzahl der Injektionen und injiziertes Gesamtvolumen pro Verabreichung
| |
Dosis (mg)
|
Anzahl der Fertigpen
|
Anzahl der Injektionen
|
Injiziertes
Gesamtvolumen (ml)
| |
75 mga
|
150 mgb
|
300 mgc,*
| |
75
|
1
|
0
|
0
|
1
|
0.5
| |
150
|
0
|
1
|
0
|
1
|
1.0
| |
225
|
1
|
1
|
0
|
2
|
1.5
| |
300
|
0
|
0
|
1
|
1
|
2.0
| |
375
|
1
|
0
|
1
|
2
|
2.5
| |
450
|
0
|
1
|
1
|
2
|
3.0
| |
600
|
0
|
0
|
2
|
2
|
4.0
| |
|
a
0.5 ml = Maximalvolumen pro Fertigpen (Xolair 75 mg).
b 1.0 ml = Maximalvolumen pro Fertigpen (Xolair 150 mg).
c 2.0 ml = Maximalvolumen pro Fertigpen (Xolair 300 mg).
|
*Die Xolair 300 mg Fertigspritze und alle Dosisstärken des Xolair Fertigpens sind nicht für die Anwendung bei Patienten unter 12 Jahren bestimmt.
Diese Tabelle stellt die geringste Anzahl von Injektionen für die Patienten dar. Es sind jedoch auch andere Kombinationen aus Spritze und Pen möglich, um die gewünschte Dosis zu erreichen.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Therapiedauer
In klinischen Studien zu allergischem Asthma wurde eine Reduktion der Asthma-Exazerbationen und der Anwendung einer Bedarfsmedikation sowie eine Verbesserung der Symptombewertung während der ersten 16 Behandlungswochen beobachtet. 16 Wochen nach Beginn der Therapie mit Xolair sollte bei den Patienten die Wirksamkeit der Behandlung durch den Arzt überprüft werden, bevor weitere Injektionen verabreicht werden. Die Entscheidung zur Weiterbehandlung mit Xolair sollte auf einer merklichen Verbesserung der allgemeinen Asthma-Kontrolle basieren (s. «Pharmakodynamik»).
Ein Therapieabbruch ergibt im Allgemeinen eine Rückkehr zu einem erhöhten Spiegel an freiem IgE und den damit verbundenen Symptomen.
Dosisanpassung
Die Gesamt-IgE-Spiegel sind während der Behandlung erhöht und bleiben bis zu einem Jahr nach Beendigung der Therapie erhöht. Daher kann eine erneute Messung der IgE-Spiegel während der Behandlung unter Xolair nicht zu einer erneuten Dosisfindung für Xolair verwendet werden. Die Dosisfindung nach Unterbrechung von weniger als 1 Jahr muss daher anhand der Serum-IgE-Spiegel erfolgen, welche bei der ursprünglichen Dosisfindung ermittelt wurden (s. «Eigenschaften/Wirkungen»). Gesamt-IgE-Spiegel sollten nur erneut zur Dosisfindung getestet werden, falls die Behandlung mit Xolair für ein Jahr oder länger unterbrochen wurde.
Die Dosierung sollte bei signifikanter Änderung des Körpergewichtes angepasst werden (s. Tabellen 2 und 3).
|
Tabelle 2: VERABREICHUNG ALLE 4 WOCHEN Allergisches Asthma und Nasenpolypen. Xolair-Dosierung (mg pro Dosis) bei subkutaner Verabreichung alle 4 Wochen
| |
|
Körpergewicht (kg)
| |
IgE-Basiswert (I.E./ml)
|
>20-25
|
>25-30
|
>30-40
|
>40-50
|
>50-60
|
>60-70
|
>70-80
|
>80-90
|
>90-125
|
>125-150
| |
≥30–100
|
75
|
75
|
75
|
150
|
150
|
150
|
150
|
150
|
300
|
300
| |
>100–200
|
150
|
150
|
150
|
300
|
300
|
300
|
300
|
300
|
450
|
600
| |
>200–300
|
150
|
150
|
225
|
300
|
300
|
450
|
450
|
450
|
600
|
| |
>300–400
|
225
|
225
|
300
|
450
|
450
|
450
|
600
|
600
|
|
| |
>400–500
|
225
|
300
|
450
|
450
|
600
|
600
|
|
|
|
| |
>500–600
|
300
|
300
|
450
|
600
|
600
|
|
|
|
|
| |
>600–700
|
300
|
|
450
|
600
|
|
VERABREICHUNG ALLE
2 WOCHEN
SIEHE TABELLE 3
|
|
Tabelle 3: VERABREICHUNG ALLE 2 WOCHEN. Allergisches Asthma und Nasenpolypen. Xolair-Dosierung (mg pro Dosis) bei subkutaner Verabreichung alle 2 Wochen
| |
|
Körpergewicht (kg)
| |
IgE-Basiswert (I.E./ml)
|
>20-25
|
>25-30
|
>30-40
|
>40-50
|
>50-60
|
>60-70
|
>70-80
|
>80-90
|
>90-125
|
>125-150
| |
≥30–100
|
VERABREICHUNG ALLE 4 WOCHEN SIEHE TABELLE 2
|
|
|
|
|
| |
>100–200
|
|
|
|
|
| |
>200–300
|
|
|
|
|
|
|
|
|
|
375
| |
>300–400
|
|
|
|
|
|
|
|
|
|
| |
>400–500
|
|
|
|
|
|
|
375
|
375
|
|
| |
>500–600
|
|
|
|
|
|
375
|
–Ungenügende Daten für eine Dosierungsempfehlung
| |
>600–700
|
|
225
|
|
|
375
|
|
Die empfohlene Maximaldosis beträgt 375 mg Omalizumab alle 2 Wochen.
Chronische spontane Urtikaria (CSU)
Übliche Dosierung
Die empfohlene initiale Dosis beträgt 300 mg als subkutane Injektion alle 4 Wochen. Die meisten Patienten, welche auf die Therapie ansprechen, zeigen eine Besserung bereits innerhalb von 4 Wochen nach der ersten Dosis. Bei den Patienten, die nur teilweise auf die erste Behandlung ansprechen, ist eine weitere Verbesserung der Symptome unter Fortführung der Behandlung möglich.
Die Erfahrungen aus klinischen Studien in dieser Indikation über eine Behandlungsdauer von mehr als 6 Monaten sind begrenzt.
Der behandelnde Arzt sollte in regelmässigen Abständen die Notwendigkeit der Fortsetzung der Therapie überprüfen.
Die klinischen Daten zeigen, dass einige Patienten unter Xolair 150 mg alle 4 Wochen eine Kontrolle ihrer Symptome erreichen können. Daher kann bei Patienten, die unter Xolair 300 mg zusammen mit H1 Anthihistaminika frei von Juckreiz und Quaddeln sind, versucht werden, die Dosis auf 150 mg alle vier Wochen zu reduzieren.
Nasenpolypen (ab 18 Jahren)
In den Zulassungsstudien zu Nasenpolypen wurde für den Nachweis der Wirksamkeit das gleiche IgE- und Gewichts-abhängige Dosierungsschema bei Patienten ab 18 Jahren mit einem Körpergewicht von 30 kg bis 150 kg untersucht, wie für allergisches Asthma beschrieben. Das zur Behandlung der chronisch spontanen Urtikaria empfohlene Dosierungsschema wurde für die Behandlung der Polypose nicht untersucht. Bei Patienten mit Nasenpolypen (ab 18 Jahren und Körpergewicht von 30 kg bis 150 kg) wird deshalb das gleiche IgE- und Gewichts-abhängige Dosierungsschema empfohlen, wie bei Patienten mit allergischem Asthma (siehe oben).
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen und Nierenfunktionsstörungen
Es gibt keine Studien zur Auswirkung einer eingeschränkten Leber- oder Nierenfunktion auf die Pharmakokinetik von Xolair. Da die Clearance von Omalizumab bei klinischen Dosen durch das retikuloendotheliale System (RES) bestimmt wird, ist eine Veränderung durch eine Leber- oder Nierenfunktionsstörung unwahrscheinlich. Obgleich keine Dosisanpassung empfohlen wird, sollte Xolair bei diesen Patienten mit Vorsicht angewendet werden (s. «Warnhinweise und Vorsichtsmassnahmen»).
Ältere Patienten
Es liegen nur begrenzte Daten zur Anwendung von Xolair bei 65 jährigen Patienten und älter vor. Es gibt jedoch keine Hinweise dafür, dass bei älteren Patienten eine andere Dosierung als bei jüngeren erwachsenen Patienten erforderlich ist.
Kinder und Jugendliche
Bei allergischem Asthma wurde die Sicherheit und Wirksamkeit bei Patienten unter 6 Jahren nicht untersucht. Daher wird die Anwendung von Xolair bei diesen Patienten nicht empfohlen. Xolair Fertigpen ist nicht für die Anwendung bei Patienten unter 12 Jahren bestimmt (siehe «Übliche Dosierung Erwachsene und Kinder ab 6 Jahren»).
Bei Nasenpolypen wurde die Sicherheit und Wirksamkeit bei Patienten unter 18 Jahren nicht untersucht.
Chronische spontane Urtikaria (CSU)
Die Sicherheit und Wirksamkeit wurde bei Patienten unter 12 Jahren nicht untersucht. Daher wird die Anwendung von Xolair bei diesen Patienten nicht empfohlen.
Art der Anwendung
Nur zur subkutanen Anwendung. Nicht intravenös oder intramuskulär anwenden.
Die Injektionen werden s.c. am Oberarm in der Deltoideus-Region verabreicht. Falls etwas gegen eine Applikation in der Deltoideus-Region spricht, können die Injektionen alternativ dazu in den Oberschenkel verabreicht werden.
Patienten mit keiner bekannten Anaphylaxie in der Vorgeschichte können sich ab der vierten Anwendung Xolair selbst injizieren oder von einer Betreuungsperson injizieren lassen, wenn ein Arzt oder eine Ärztin dies für angemessen hält (siehe «Warnhinweise und Vorsichtsmassnahmen»). Der Patient oder die Betreuungsperson muss in der korrekten Injektionstechnik und dem Erkennen von frühen Anzeichen und Symptomen schwerer allergischer Reaktionen geschult werden.
Patienten oder Betreuungspersonen sollten angewiesen werden, den gesamten Inhalt des Xolair Fertigpens gemäss den Anweisungen in der Packungsbeilage zu injizieren.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Inhaltsstoffe.
Warnhinweise und VorsichtsmassnahmenAllergische Reaktionen, Anaphylaxie
Bei der Anwendung von Omalizumab können lokale oder systemische allergische Reaktionen einschliesslich lebensbedrohlicher Anaphylaxie und anaphylaktischem Schock auftreten. Diese können auch nach einer längeren Behandlungsdauer eintreten. Die meisten dieser Reaktionen traten innerhalb von 2 Stunden nach der ersten und den folgenden Injektionen von Xolair auf, aber manche ereigneten sich nach mehr als 2 Stunden und sogar nach mehr als 24 Stunden nach der Injektion. Die meisten anaphylaktischen Reaktionen traten bei den ersten 3 Anwendungen von Xolair auf. Daher müssen die ersten 3 Anwendungen durch medizinisches Fachpersonal oder unter dessen Aufsicht verabreicht werden. Eine Anaphylaxie in der Vorgeschichte, die nicht im Zusammenhang mit Omalizumab stand, kann einen Risikofaktor für das Auftreten einer Anaphylaxie nach Verabreichung von Xolair darstellen. Aus diesem Grund muss Xolair bei Patienten mit bekannter Anaphylaxie in der Vorgeschichte durch medizinisches Fachpersonal verabreicht werden. Daher sollten immer Arzneimittel für die Behandlung einer anaphylaktischen Reaktion zum sofortigen Einsatz nach der Verabreichung von Xolair vorhanden sein. Die Patienten müssen darüber informiert werden, dass solche Reaktionen möglich sind und sofortige medizinische Behandlung erforderlich ist, wenn allergische Reaktionen auftreten. Bei schweren Reaktionen sollte Xolair sofort abgesetzt werden (s. «Unerwünschte Wirkungen»).
Serumkrankheit und Serumkrankheit-ähnliche Symptome, die verzögerte allergische Typ III Reaktionen sind, wurden selten bei Patienten beobachtet, welche mit humanisierten monoklonalen Antikörpern, einschliesslich Omalizumab, behandelt wurden. Der Beginn war typischerweise 1–5 Tage nach der ersten oder einer späteren Injektion, auch nach einer langen Behandlungsdauer. Symptome, die auf Serumkrankheit hinweisen beinhalten Arthritis/Arthralgie, Rash (Urtikaria oder andere Formen), Fieber und Lymphadenopathie. Antihistamine und Kortikosteroide können zur Vorbeugung oder Behandlung dieser Erkrankung nützlich sein. Patienten sollten darauf hingewiesen werden, verdächtige Symptome zu melden.
Churg-Strauss-Syndrom und hypereosinophiles Syndrom
Patienten mit schwerem allergischem Asthma können selten ein systemisches hypereosinophiles Syndrom oder eine allergische eosinophile granulomatöse Vaskulitis (Churg-Strauss-Syndrom, eosinophilische granulomatöse Polyangiitis) aufweisen, die beide üblicherweise mit systemischen Kortikosteroiden behandelt werden.
Immunogenität
In seltenen Fällen können Patienten gegen Omalizumab Antikörper bilden, wie es bei allen humanisierten Antikörpern aus rekombinierter DNA möglich sein kann.
Tiefe Antikörper-Titer gegen Xolair wurden bei ungefähr 1/1'843 (<0.1%) der mit Xolair behandelten Patienten detektiert. Diese Daten reflektieren die Prozentzahl der Patienten, deren Testresultate mittels eines ELISA-Assays auf Xolair-Antikörper als positiv betrachtet wurden und stark von der Sensitivität und der Spezifität des Assays abhängig sind. Des Weiteren kann die beobachtete Inzidenz der Antikörper-positiven Reaktionen im Assay von verschiedenen Faktoren wie der Handhabung der Proben, Zeitpunkt der Probennahme, gleichzeitige andere Medikation und anderen vorhandenen Krankheiten beeinflusst werden. Daher kann der Vergleich der Inzidenz von Xolair-Antikörpern mit der Inzidenz von Antikörpern gegenüber anderen Produkten irreführen.
Cerebrovaskuläre Erkrankungen
In kontrollierten klinischen Studien mit Erwachsenen und Jugendlichen ab 12 Jahren und älter wurden bei mit Xolair behandelten Patienten cerebrovaskuläre Ereignisse einschliesslich vorübergehendem ischämischen Anfall und ischämischem Schlaganfall häufiger als in der Kontrollgruppe beobachtet (s. «Unerwünschte Wirkungen»).
Parasitäre (Wurm-) Infektionen
IgE kann bei der Immunabwehr gegen manche Wurminfektionen involviert sein. Es wurde eine Studie in Brasilien durchgeführt, bei der Patienten in einem Hochrisikogebiet für Wurminfektionen 1 Jahr mit Omalizumab behandelt wurden. 53% (36/68) der mit Omalizumab behandelten Patienten und 42% (29/69) der mit Placebo behandelten Patienten erlitten eine Wurminfektion, die per Stuhluntersuchung diagnostiziert wurde. Der Unterschied an Wurminfektionen in den beiden Gruppen ist statistisch nicht signifikant. Der Verlauf, der Schweregrad der Erkrankung und das Ansprechen auf die Behandlung der Infektion waren unverändert. Die Rate der Wurmerkrankungen im klinischen Programm, das nicht ausgelegt war, um solche Krankheiten zu erkennen, lag bei weniger als 1 Erkrankung pro 1'000 Patienten. Es ist jedoch Vorsicht geboten bei Patienten mit hohem Risiko für Wurmerkrankungen, speziell bei Reisen in Gebiete mit endemischen Wurminfektionen. Sollten Patienten auf die empfohlene Anti-Wurmbehandlung nicht ansprechen, sollte das Absetzen der Xolair-Therapie in Betracht gezogen werden.
Malignitäten
Während erster klinischer Studien gab es ein numerisches Ungleichgewicht bezüglich Krebs bei der aktiven Behandlungs-Gruppe verglichen zur Kontroll-Gruppe. Die unterschiedlichen Tumortypen, die relativ kurze Behandlungsdauer und die individuellen klinischen Zustände lassen einen kausalen Zusammenhang unwahrscheinlich erscheinen. Die beobachtete Gesamtinzidenz an malignen Erkrankungen im klinischen Entwicklungsprogramm von Xolair war vergleichbar mit derjenigen, die für die Normalbevölkerung berichtet wird. Spätere Studien zeigen, dass das relative Risiko für das Auftreten von malignen Erkrankungen unter Xolair nicht erhöht ist. Die Gesamtdatenlage kann aber weiterhin die Möglichkeit eines leichten Ungleichgewichts nicht komplett ausschliessen (s. «Unerwünschte Wirkungen»).
Allgemein
Xolair ist nicht indiziert zur Behandlung von akuten Asthmaexazerbationen, akuten Bronchospasmen oder Status asthmaticus.
Xolair wurde nicht untersucht bei Patienten mit Hyper-Immunglobulin-E-Syndrom, allergischer bronchopulmonaler Aspergillose oder zur Prävention von allergischen Reaktionen.
Xolair wurde nicht ausreichend untersucht bei atopischer Dermatitis, allergischer Rhinitis oder Lebensmittelallergie.
Die Therapie mit Xolair bei Patienten mit Autoimmunerkrankungen, immunkomplex-vermittelten Zuständen oder mit bereits bestehender Nieren- oder Leberfunktionsstörung wurde nicht untersucht. Wird Xolair bei diesen Patienten angewendet, ist Vorsicht geboten.
Nach Beginn der Therapie mit Xolair wird ein plötzliches Absetzen von systemischen oder inhalativen Kortikosteroiden bei allergischem Asthma oder Nasenpolypen nicht empfohlen. Eine Reduktion der Kortikosteroide sollte unter ärztlicher Aufsicht erfolgen und muss gegebenenfalls stufenweise durchgeführt werden.
InteraktionenCytochrom P450-Enzyme, Austauschpumpen und Protein-bindende Mechanismen sind nicht an der Clearance von Omalizumab beteiligt; daher besteht nur eine geringe Wahrscheinlichkeit für Arzneimittelwechselwirkungen. Es wurden keine speziellen Studien zu Wechselwirkungen mit Arzneimitteln oder Impfstoffen mit Xolair durchgeführt. Aus pharmakologischer Sicht besteht kein Grund, Wechselwirkungen zwischen üblicherweise zur Behandlung von Asthma, Nasenpolypen oder chronischer spontaner Urtikaria (CSU) verschriebenen Arzneimitteln und Omalizumab zu erwarten.
Allergisches Asthma
In klinischen Studien wurde Xolair im Allgemeinen zusammen mit inhalativen oder oralen Kortikosteroiden, inhalativen kurz- oder langwirksamen Beta-Agonisten, Leukotrien-Rezeptorantagonisten, Theophyllinen und oralen Antihistaminen eingesetzt. Es gab keine Hinweise darauf, dass die Sicherheit von Xolair durch diese anderen im Allgemeinen eingesetzten Asthmamedikamente verändert wurde.
Die Wirksamkeit der Behandlung mit Xolair in Kombination mit spezifischer Immuntherapie wurde nicht nachgewiesen.
Nasenpolypen
In klinischen Studien wurde Xolair gemäss dem Prüfplan in Kombination mit Mometason Nasenpray angewendet. Die anderen häufig verwendeten Begleitmedikamente umfassten andere intranasal angewendete Kortikosteroide, Bronchodilatatoren, Antihistaminika, Leukotrien-Rezeptor-Antagonisten, Adrenergika/Sympathomimetika und intranasal verabreichte Lokalanästhetika. Es gab keine Anzeichen dafür, dass die Sicherheit von Xolair bei gleichzeitiger Anwendung dieser anderen, häufig bei Nasenpolypen angewendeten Arzneimittel beeinträchtigt war.
Chronische spontane Urtikaria (CSU)
In klinischen Studien zu CSU wurde Xolair in Verbindung mit Antihistaminika (anti-H1, anti-H2) und Leukotrien-Rezeptorantagonisten (LTRA) angewendet. In den Phase III-Studien Q4881g und Q4882g erhielten alle Patienten H1-Antihistaminika zusätzlich zu Xolair oder Placebo. In der Phase III-Studie Q4883g erhielten alle Patienten ein oder mehrere H1-Antihistaminika und/oder H2-Antihistaminika und/oder LTRA zusätzlich zu Xolair oder Placebo. Es ergaben sich keine Hinweise darauf, dass durch die gleichzeitige Anwendung mit den genannten Arzneimitteln die Sicherheit von Omalizumab gegenüber dessen bekanntem Sicherheitsprofil bei allergischem Asthma verändert wurde. Zudem zeigte eine populationspharmakokinetische Analyse keinen relevanten Einfluss von H2-Antihistaminika und LTRA auf die Pharmakokinetik von Omalizumab (s. «Eigenschaften/Wirkungen»).
Schwangerschaft, StillzeitSchwangerschaft
Es gibt keine gut kontrollierten klinischen Studien zu Xolair bei schwangeren Frauen. Eine prospektive Schwangerschafts-Registerstudie (EXPECT) an 250 schwangeren Frauen mit Asthma, die mit Xolair behandelt wurden, hat gezeigt, dass die Prävalenz von relevanten kongenitalen Fehlbildungen zwischen Patientinnen, welche mit Xolair behandelt wurden (EXPECT) und Asthmapatientinnen (mittelschweres und schweres Asthma), die kein Xolair erhielten, vergleichbar war (8.1 % vs. 8.9 %). Der Anteil von Kindern mit Geburtsgewicht <2.5 kg war unter Xolair höher (13.7% vs 9.8%), was aber auch durch Unterschiede im Asthmaschweregrad bedingt gewesen sein könnte. Insgesamt kann auch anhand dieser Studie aufgrund von methodischen Einschränkungen einschliesslich eines nicht randomisierten Studiendesigns und möglicher Unterschiede zwischen der Registerpopulation und der Vergleichsgruppe (siehe «Klinische Wirksamkeit») das Risiko kongenitaler Fehlbildungen nicht definitiv beurteilt werden.
Es ist bekannt, dass IgG-Moleküle die Plazentaschranke durchdringen. Tierexperimentelle Studien ergaben keine Hinweise auf eine Reproduktionstoxizität (siehe «präklinische Daten»).
Krankheitsassoziiertes Risiko für die Mutter und/oder den Embryo/Fötus:
Es gibt Evidenz dafür, dass bei Frauen mit schlecht oder mässig kontrolliertem Asthma das Risiko für eine Präeklampsie der Mutter und für eine Frühgeburt, für ein niedriges Geburtsgewicht sowie für eine geringe Körpergrösse des Neugeborenen im Verhältnis zum Gestationsalter erhöht ist. Daher sollte bei schwangeren Frauen die Asthmakontrolle engmaschig überwacht und die Behandlung bei Bedarf angepasst werden, um eine optimale Kontrolle aufrechtzuerhalten.
Stillzeit
Zwar wurde das Vorhandensein von Omalizumab in der Muttermilch nach der Verabreichung von Xolair nicht untersucht, doch gehen die IgG in die Muttermilch über, und es ist daher davon auszugehen, dass Omalizumab in der Muttermilch vorhanden sein wird. Die in der EXPECT-Studie beobachtete Häufigkeit von Infektionen bei Säuglingen wurde als indirektes Mass für die Entwicklung des Immunsystems nach Exposition während der Schwangerschaft oder durch Stillen bewertet. Die Mehrheit der Säuglinge in der primären Analysepopulation (77.5%, 186/240) wurde gestillt.
Schwerwiegende unerwünschte Ereignisse (SUE), die als «Infektionen und parasitäre Erkrankungen» kategorisiert wurden, wurden bei 11.4% (5/44) der nicht gestillten Säuglinge, bei 10,4% (16/154) der Säuglinge, die durch Stillen gegen über Xolair exponiert waren, und bei 12.5% (4/32) der Säuglinge, die ohne Xolair-Exposition gestillt wurden, beobachtet. Die Studie weist methodische Einschränkungen einschliesslich eines nicht randomisierten Studiendesigns auf.
Die Vorteile des Stillens für die Entwicklung und die Gesundheit des Säuglings sollten gemeinsam mit dem klinischen Bedarf der Mutter für Xolair und den möglichen unerwünschten Auswirkungen von Omalizumab oder der zugrunde liegenden Erkrankung der Mutter auf das gestillte Kind abgewogen werden.
Fertilität
Es gibt keine speziellen Empfehlungen für gebärfähige Frauen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine entsprechenden Studien durchgeführt.
Patienten, welche Omalizumab erhalten, müssen darauf aufmerksam gemacht werden, dass falls sie Schwindel, Müdigkeit, Synkope oder Somnolenz erleben, sie nicht am Strassenverkehr teilnehmen oder Maschinen bedienen sollen.
Unerwünschte WirkungenHäufigkeiten
«Sehr häufig» (≥1/10), «häufig» (≥1/100, <1/10), «gelegentlich» (≥1/1'000, <1/100), «selten» (≥1/10'000, <1/1'000), «sehr selten» (<1/10'000).
Allergisches Asthma
In klinischen Versuchen mit Erwachsenen und Jugendlichen ab 12 Jahren und älter waren die am häufigsten beobachteten unerwünschten Wirkungen Kopfschmerzen und Reaktionen an der Injektionsstelle, einschliesslich Schmerzen an der Injektionsstelle, Schwellung, Erythem und Pruritus. In klinischen Versuchen mit Patienten im Alter von 6 Jahren bis weniger als 12 Jahren waren die am häufigsten beobachteten unerwünschten Wirkungen Kopfschmerzen, Pyrexie und Schmerzen im oberen Unterleib. In den meisten Fällen waren diese leicht bis mittelschwer.
Unerwünschte Wirkungen aus den klinischen Studien zu allergischem Asthma
Infektionen und parasitäre Erkrankungen
Gelegentlich: Pharyngitis.
Selten: parasitäre Infektion.
Erkrankungen des Immunsystem
Selten: anaphylaktische und andere allergische Reaktionen wie Serumkrankheit, Grippe-ähnliche Symptome wie Fieber, Gelenkschmerzen, Unwohlsein. Entwicklung von anti-therapeutischen Antikörpern.
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen**.
Gelegentlich: Schwindel, Schläfrigkeit, Parästhesie, Synkope.
Gefässerkrankungen
Gelegentlich: orthostatische Hypotonie, Flush.
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Gelegentlich: Husten, allergische Bronchospasmen.
Selten: Larynxoedem.
Erkrankungen des Gastrointestinaltrakts
Häufig: Schmerzen im oberen Unterleib*.
Gelegentlich: Nausea, Diarrhöe, Dyspepsie.
Erkrankungen der Haut und des Unterhautzellgewebes
Gelegentlich: Urtikaria, Rash, Pruritus, Photosensibilität.
Selten: Angiooedem.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Pyrexie*.
Häufig: Schmerzen, Erythem, Pruritus, Schwellung.
Gelegentlich: Gewichtszunahme, Müdigkeit, Anschwellen der Arme, grippeähnliche Symptome.
In einer offenen Studie an gesunden Erwachsenen, in der der 300 mg/2 ml Fertigpen mit der 300 mg/2 ml Fertigspritze verglichen wurde, wurden Reaktionen an der Injektionsstelle (z.B. Verhärtung, Schmerzen, Erythem, Blutung, Schwellung, Unbehagen, Bluterguss, Hypoästhesie, Ödem, Pruritus) bei 24 % (16/66) der mit dem Fertigpen behandelten Erwachsenen gegenüber 14 % (9/64) der mit der Fertigspritze behandelten Erwachsenen beobachtet.
Die Häufigkeiten der unerwünschten Wirkungen in der Behandlungsgruppe und in der Kontrollgruppe waren vergleichbar.
* in Kindern ab 6 Jahren bis <12 Jahren
** sehr häufig in Kindern ab 6 Jahren bis <12 Jahren
Nasenpolypen
Zusammenfassung des Sicherheitsprofils
Die nachfolgend beschriebenen Daten stammen aus zwei placebokontrollierten Studien an Patienten ab 18 Jahren. In diesen Studien erhielten die Patienten entweder 150 bis 600 mg Xolair alle 2 bzw. 4 Wochen oder Placebo. Alle Patienten erhielten Mometason intranasal als Hintergrundtherapie, wobei das Sicherheitsprofil bei Patienten mit Nasenpolypen mit dem bei allergischem Asthma und CSU übereinstimmte. Die am häufigsten (> 3%) angegebenen Nebenwirkungen, die häufiger auftraten als unter Placebo, sind in Tabelle 4 dargestellt.
In Tabelle 4 sind die unerwünschten Wirkungen, die in klinischen Studien in der mit Xolair behandelten gesamthaft überwachten Population mit Nasenpolypen aufgetreten sind, nach Organklasse und Häufigkeit aufgeführt. Die Häufigkeitskategorien sind wie folgt definiert: sehr häufig (≥1/10), häufig (≥1/100 bis < 1/10), gelegentlich (≥1/1'000 bis < 1/100); selten (≥1/10'000 bis < 1/1'000) und sehr selten (< 1/10'000).
Tabelle 4 Unerwünschte Wirkungen aus den klinischen Studien an Patienten mit Nasenpolypen
|
Unerwünschte Wirkungen
(bezeichnet mit dem bevorzugten Begriff nach MedDRA)
|
Omalizumab Nasenpolypenstudien 1 und 2
gepoolt
|
Häufigkeits-kategorie
| |
Placebo
N=130
|
Omalizumab
N=135
| |
Erkrankungen des Nervensystems
| |
Kopfschmerzen
|
7 (5.4%)
|
11 (8.1%)
|
Häufig
| |
Schwindelgefühl
|
1 (0.8%)
|
4 (3.0%)
|
Häufig
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
| |
Arthralgie
|
2 (1.5%)
|
4 (3.0%)
|
Häufig
| |
Erkrankungen des Gastrointestinaltrakts
| |
Abdominalschmerz
|
1 (0.8%)
|
4 (3.0%)
|
Häufig
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
| |
Reaktionen an der Injektionsstelle (Reaktionen an der Injektionsstelle, injektionsbedingte Reaktion, Schmerzen an der Injektionsstelle)
|
2 (1.5%)
|
7 (5.2%)
|
Häufig
|
Chronische spontane Urtikaria (CSU)
Zusammenfassung des Sicherheitsprofils
Die Sicherheit und Verträglichkeit von Omalizumab in den Dosierungen 75 mg, 150 mg und 300 mg alle 4 Wochen wurden bei 975 Patienten mit CSU untersucht, von denen 242 Placebo erhielten. 733 Patienten wurden bis zu 12 Wochen und 490 Patienten bis zu 24 Wochen lang mit Omalizumab behandelt. 175 bzw. 412 Patienten wurden bis zu 12 Wochen und 87 bzw. 333 Patienten bis zu 24 Wochen lang mit den empfohlenen Dosen von 150 mg bzw. 300 mg behandelt.
Die im Rahmen der klinischen Studien bei erwachsenen und jugendlichen Patienten (12 Jahre und älter) am häufigsten berichteten unerwünschten Wirkungen waren Kopfschmerzen und Nasopharyngitis.
|
Tabelle 5: Tabellarische Zusammenfassung der unerwünschten Wirkungen von den gepoolten Phase-III-Studien mit der empfohlenen Dosis von 150 mg und 300 mg (Tag 1 bis Woche 12)
| |
Unterwünschte Wirkungen
(MedDRA)
|
Omalizumab Studien Q4881g, Q4882g und Q4883g gepoolt
|
Häufigkeiten
| |
Placebo
N=242
|
150 mg
N=175
|
300 mg
N=412
|
| |
Infektionen und parasitäre Erkrankungen
| |
Nasopharyngitis
|
17 (7.0%)
|
16 (9.1%)
|
27 (6.6%)
|
Häufig
| |
Sinusitis
|
5 (2.1%)
|
2 (1.1%)
|
20 (4.9%)
|
Häufig
| |
Virale Entzündung des oberen Atmungstraktes
|
0
|
4 (2.3%)
|
2 (0.5%)
|
Häufig
| |
Erkrankungen des Nervensystems
| |
Kopfschmerzen
|
7 (2.9%)
|
21 (12.0%)
|
25 (6.1%)
|
Sehr häufig
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
| |
Gelenkschmerzen
|
1 (0.4%)
|
5 (2.9%)
|
12 (2.9%)
|
Häufig
|
Weitere Ereignisse, die zu irgendeinem Zeitpunkt während der Behandlungsperiode von Tag 1 bis Woche 24 (Studien Q4881g und Q4883g) berichtet wurden und die Kriterien für unerwünschte Wirkungen erfüllten:
Infektionen und parasitäre Erkrankungen: Infektionen der oberen Atemwege (Placebo 3.1%, 150 mg 3.4%, 300 mg 5.7%), Harnwegsinfektion (Placebo 1.8%, 150 mg 4.6%, 300 mg 2.4%)
Erkrankungen des Nervensystems: Sinus-Kopfschmerzen (Placebo 0%, 150 mg 2.3%, 300 mg 0.3%)
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen: Myalgie (Placebo 0%, 150 mg 2.3%, 300 mg 0.9%), Gliederschmerzen (Placebo 0%, 150 mg 3.4%, 300 mg 0.9%), muskuloskelettale Schmerzen (Placebo 0%, 150 mg 2.3%, 300 mg 0.9%)
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort: Pyrexie (Placebo 1.2%, 150 mg 3.4%, 300 mg 0.9%),Reaktionen an der Injektionsstelle traten in den Studien bei den mit Omalizumab behandelten Patienten häufiger als bei den Patienten unter Placebo auf (2.7% unter 300 mg, 0.6% unter 150 mg, 0.8% unter Placebo). Diese Reaktionen umfassten: Schwellung, Erythem, Schmerzen, Bluterguss, Juckreiz, Blutung und Urtikaria.
In einer 48-wöchigen Studie erhielten 81 Patienten mit CSU alle 4 Wochen Omalizumab 300 mg (siehe Klinische Wirksamkeit – CSU). Das Sicherheitsprofil bei Langzeitanwendung war ähnlich dem Sicherheitsprofil, das in CSU-Studien über bis zu 24 Wochen beobachtet wurde.
Unerwünschte Wirkungen nach Markteinführung
Die folgenden Reaktionen wurden primär durch spontane Berichte identifiziert:
Immunsystem: Anaphylaxie und anaphylaktoide Reaktionen wurden sowohl nach der ersten wie auch nach späteren Verabreichungen berichtet, (s. «Warnhinweise und Vorsichtsmassnahmen»). Serumkrankheit (s. «Warnhinweise und Vorsichtsmassnahmen»).
Haut: Alopezie.
Blut und Lymphsystem: idiopathische schwere Thrombozytopenie.
Atmungsorgane: Churg Strauss Syndrom (d.h. eosinophilische granulomatöse Polyangiitis).
Muskelskelettsystem: Arthralgie, Myalgie, Gelenkschwellung.
Beschreibung ausgewählter Nebenwirkungen
Thrombozytopenie
In klinischen Versuchen traten bei wenigen Patienten Thrombozytenzahlen unterhalb des unteren Normwertes auf. Keine dieser Veränderungen wurden mit Blutungen oder einer Abnahme von Hämoglobin assoziiert. Es gibt keine Anzeichen einer anhaltenden Abnahme der Thrombozytenzahl beim Menschen (Patienten älter als 6 Jahre), wie dies bei Primatenaffen beobachtet wurde (s. «Präklinische Daten»). Über Thrombozytopenie wurde nach Markteinführung berichtet.
Parasitäre Infektionen
Bei allergischen Patienten mit einer chronisch erhöhten Neigung zu Wurminfektionen zeigte eine Placebo-kontrollierte Studie einen leichten Anstieg der Infektionsrate unter Omalizumab. Der Verlauf und der Schweregrad der Infektion sowie das Ansprechen auf die Behandlung waren nicht beeinflusst (s. «Warnhinweise und Vorsichtsmassnahmen»).
Malignitäten
Während erster klinischer Studien bei Erwachsenen und Jugendlichen ab 12 Jahren und älter gab es ein numerisches Ungleichgewicht bezüglich Krebs bei der aktiven Behandlungs-Gruppe verglichen zur Kontroll-Gruppe. Sowohl in der aktiven als auch in der Kontrollgruppe trat Krebs gelegentlich (<1/100) auf. In einer späteren Beobachtungsstudie über 5 Jahre zum Vergleich von 5007 mit Xolair behandelten und 2829 nicht mit Xolair behandelten Patienten ist das relative Risiko für das Auftreten von malignen Erkrankungen unter Xolair nicht erhöht. Die Inzidenzrate primärer Malignitäten pro 1000 Patientenjahre betrug 16.01 (295/18426 Patientenjahre) bzw. 19.07 (190/9963 Patientenjahre), was einem Verhältnis der Häufigkeiten von 0.84 entspricht (95% Konfidenzintervall, 0.62-1.13). In einer prospektiven Analyse randomisierter, doppelblinder, placebokontrollierter klinischer Studien bei 4254 Patienten unter Behandlung mit Xolair und 3178 Patienten mit Placebogabe war die Behandlung mit Xolair, ausgehend von den Inzidenzraten pro 1000 Patientenjahren von 4.14 (14/3382 Patientenjahre) bei mit Xolair behandelten Patienten und 4.45 (11/2474 Patientenjahre) bei den Patienten in der Placebogruppe, nicht mit einem erhöhten Malignitätsrisiko verbunden (Verhältnis der Häufigkeiten 0.93, 95% Konfidenzintervall 0.39-2.27). Die Gesamtdatenlage kann aber weiterhin die Möglichkeit eines leichten Ungleichgewichts nicht komplett ausschliessen.
Die gesamthaft beobachtete Inzidenzrate an Malignitäten während der Xolair- Studie mit Erwachsenen und Jugendlichen ab 12 Jahren und älter war vergleichbar mit derjenigen in der breiten Bevölkerung.
In Folgestudien war das relative Risiko für Malignitäten in der mit Xolair behandelten Gruppe nicht erhöht (s. «Warnhinweise und Vorsichtsmassnahmen»).
Arterielle thromboembolische Ereignisse (ATE)
In kontrollierten klinischen Studien und bei Zwischenanalysen einer Beobachtungsstudie wurde ein numerisches Ungleichgewicht für ATEs beobachtet. ATE umfasste Schlaganfall, vorübergehende ischämische Attacken, Myokardinfarkt, instabile Angina und kardiovaskulärer Tod (einschliesslich Tod mit unbekannter Ursache). In der Abschlussanalyse der Beobachtungsstudie betrug die ATE-Rate pro 1000 Patientenjahren bei mit Xolair behandelten Patienten 7.52 (115/15286 Patientenjahre) und bei den Patienten in der Kontrollgruppe 5.12 (51/9963 Patientenjahre). In einer multivariaten Analyse mit Berichtigung nach vorliegenden kardiovaskulären Risikofaktoren zum Baseline-Zeitpunkt betrug das Hazard-Ratio 1.32 (95% Konfidenzintervall 0.91-1.91).
In einer prospektiven Analyse gepoolter klinischer Studien, einschliesslich aller randomisierter, doppelblinder, placebokontrollierter klinischer Studien mit einer Dauer von mindestens 8 Wochen, betrug die ATE-Rate pro 1000 Patientenjahren bei mit Xolair behandelten Patienten 2.69 (5/1856 Patientenjahre) und bei den Patienten in der Placebogruppe 2.38 (4/1680 Patientenjahre) (Verhältnis der Häufigkeiten 1.13, 95% Konfidenzintervall 0.24-5.71).
Labordaten
Nach Applikation von Xolair stiegen die Gesamt-IgE-Werte im Serum aufgrund der Bildung von Xolair-IgE-Komplexen an (s. «Pharmakokinetik», «Dosierung/Anwendung»).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungDie maximal verträgliche Dosis von Xolair wurde nicht ermittelt. Intravenöse Einzeldosen bis zu 4'000 mg wurden Patienten ohne Hinweise auf dosislimitierende Toxizität verabreicht. Die höchste kumulierte Dosis, die einem Patienten verabreicht wurde betrug 44'000 mg (über einen Zeitraum von 20 Wochen), ohne dass durch diese Dosis unerwünschte akute Effekte auftraten.
Eigenschaften/WirkungenATC-Code
R03DX05
Wirkungsmechanismus
Omalizumab ist ein rekombinanter, humanisierter monoklonaler Antikörper, der selektiv an das menschliche Immunglobulin E (IgE) bindet. Es handelt sich um einen IgG1-kappa-Antikörper mit einem humanen Grundgerüst, dessen komplementaritäts-bestimmende Region muriner Herkunft ist und an humanes IgE bindet.
Pharmakodynamik
Patienten mit allergischem Asthma
Durch die Bindung an freies IgE verhindert Omalizumab somit die Bindung von IgE an den hochaffinen FcεRI- Rezeptor (hochaffiner IgE-Rezeptor). Die Menge an freiem IgE, das zum Auslösen der allergischen Kaskade verfügbar ist, wird reduziert. Die Behandlung atopischer Patienten mit Omalizumab reduzierte die Anzahl an FcεRI-Rezeptoren auf den Basophilen. Ausserdem wurde die in vitro Histaminausschüttung aus Basophilen, welche von mit Xolair behandelten Patienten gewonnen wurden, nach Allergenstimulation im Vergleich zu den Werten vor der Behandlung um ca. 90% gesenkt.
Durch die Behandlung mit Xolair kommt es zu einer verminderten Eosinophilenzahl im Blut und Gewebe sowie zu einem Rückgang der Entzündungsmediatoren. Zu diesen zählen auch Interleukine (IL-4, IL-5 und IL-13). In klinischen Studien wurden die freien IgE-Werte im Serum innerhalb 1 h nach der ersten Dosis dosisabhängig reduziert und zwischen den einzelnen Dosen behalten.
Die durchschnittliche Reduktion des freien IgE war bei Anwendung der empfohlenen Dosen grösser als 96%. Die Gesamt-IgE-Werte im Serum (d.h. gebunden und ungebunden) stiegen nach der ersten Dosis aufgrund der Bildung von Omalizumab-IgE-Komplexen an. Die Omalizumab-IgE-Komplexe haben im Vergleich zum freien IgE eine langsamere Eliminationsrate. 16 Wochen nach der ersten Dosis waren die Gesamt-IgE-Werte 5 mal höher als die Werte bei der Vorbehandlung, wobei Standard-Assays zur Bestimmung verwendet wurden. Nach Abbruch der Xolair-Therapie waren der Anstieg des Gesamt-IgE und die Reduktion des freien IgE reversibel, ohne dass ein Rebound-Effekt der IgE-Werte nach Auswaschen von Omalizumab beobachtet wurde. Gesamt-IgE-Werte wie sie vor der Therapie waren, wurden innerhalb eines Jahres nach Abbruch der Therapie mit Xolair nicht erreicht.
Die Auswirkungen von Omalizumab auf IgE-tragende B-Zellen und auf die langfristige Regulation der allergenspezifischen IgE-Synthese sind unklar.
Patienten mit Nasenpolypen
In klinischen Studien an Patienten mit Nasenpolypen führte die Behandlung mit Xolair zu einer Reduktion der Werte für das freie IgE im Serum und einer Zunahme der Werte für das Gesamt-IgE im Serum, ähnlich den Beobachtungen an Patienten mit allergischem Asthma.
Patienten mit chronischer spontaner Urtikaria (CSU)
Es bestehen mehrere Theorien für die Ätiologie der CSU, darunter eine Theorie, die einen autoimmunen Ursprung annimmt. Bei einigen Patienten mit CSU wurden Autoimmunantikörper gegen IgE und dessen Rezeptor, FcεRI, aus dem Serum isoliert. Diese Autoantikörper können basophile Granulozyten oder Mastzellen aktivieren und dadurch die Freisetzung von Histamin auslösen.
Eine Hypothese über den Wirkungsmechanismus von Omalizumab bei CSU besagt, dass Omalizumab die Spiegel an freiem IgE im Blut und dadurch auch in der Haut erniedrigt. Dies führt zu einer Herunterregulation von IgE-Oberflächenrezeptoren, wodurch die Downstream-Signalübertragung über den FcεRI-Signalweg vermindert wird, was eine Hemmung der Zellaktivierung und der Entzündungsreaktion bewirkt. In der Folge werden die Häufigkeit und der Schweregrad der Symptome der CSU vermindert. Eine andere Hypothese besagt, dass die Verminderung der Spiegel an freiem IgE eine rasche und unspezifische Desensibilisierung kutaner Mastzellen zur Folge hat. Die Herunterregulation von FcεRI könnte die Aufrechterhaltung dieser Reaktion unterstützen.
In klinischen Studien bei Patienten mit CSU bewirkte die Behandlung mit Omalizumab eine dosisabhängige Reduktion der Spiegel an freiem IgE und eine Zunahme der Gesamt-IgE-Werte im Serum, ähnlich den Beobachtungen bei Patienten mit allergischem Asthma. Die maximale Suppression des freien IgE wurde 3 Tage nach der ersten subkutanen Dosis beobachtet. Nach Mehrfachdosierung einmal alle 4 Wochen blieben die Spiegel an freiem IgE im Serum vor Dosisgabe zwischen den Wochen 12 und 24 der Behandlung stabil. Der Gesamt-IgE-Wert im Serum stieg nach der ersten Dosis infolge einer Bildung von Omalizumab-IgE-Komplexen, welche einer langsameren Eliminationsrate als freies IgE unterliegen. Nach Mehrfachdosierung von 75 mg bis 300 mg einmal alle 4 Wochen betrugen die mittleren Gesamt-IgE-Werte im Serum vor Dosisgabe in Woche 12 das Zwei- bis Dreifache der Werte vor Beginn der Behandlung und blieben zwischen Woche 12 und 24 der Behandlung stabil. Nach Beendigung der Behandlung mit Xolair, während einer 16-wöchigen behandlungsfreien Nachbeobachtungsphase, stiegen die Spiegel an freiem IgE während Gesamt-IgE-Werte abnahmen, beide in Richtung der Werte vor der Behandlung.
Klinische Wirksamkeit
Allergisches Asthma
Erwachsene und Jugendliche (≥12 Jahre)
Wirksamkeit und Sicherheit von Xolair wurden in einer 28-wöchigen Placebo-kontrollierten pivotalen Studie (Studie 5) bei 419 Patienten mit schwerem allergischem Asthma im Alter zwischen 12 und 79 Jahren nachgewiesen, die eine eingeschränkte Lungenfunktion (angenommener Forced Expiratory Volume/1 second: FEV1 40-80%) aufwiesen und deren Symptomatik auf Behandlung mit >1'000 µg Beclomethason-Dipropionat (oder gleichwertig) und langwirkenden Beta2-Agonisten schlecht ansprach. Die zur Studie zugelassenen Patienten hatten im Verlauf des letzten Jahres mehrere Asthma-Exazerbationen erlitten, deren Behandlung eine systemische Gabe von Kortikosteroiden erforderte, wurden stationär im Spital behandelt oder hatten wegen schwerer Asthma-Exazerbation trotz kontinuierlicher Behandlung mit hochdosierten inhalierten Kortikosteroiden und langwirkenden Beta2-Agonisten eine Notfallbehandlung beansprucht. Xolair oder Placebo wurden subkutan als Zusatztherapie zu >1'000 µg Beclomethason-Dipropionat (oder gleichwertig) und langwirkenden Beta2-Agonisten verabreicht. Weiterhin erhielten die Patienten eine Dauertherapie von oralen Kortikosteroiden (22%), Theophyllin (27%) und Leukotrienantagonisten (35%). Während der Behandlungsphase wurde die begleitende Asthmatherapie nicht geändert.
Den primären Endpunkt stellt die Rate der Asthma-Exazerbationen dar, bei denen eine Akut-Behandlung mit systemischen Kortikosteroiden nötig war. Omalizumab reduzierte die Rate der Asthma-Exazerbationen um 19% (p=0.153). Weitere Auswertungen, die statistische Signifikanz (p=0.05) zu Gunsten von Xolair zeigten, beinhalten die Reduzierung von schweren Exazerbationen (bei denen die Lungenfunktion des Patienten auf weniger als 60% des persönlichen Bestwertes reduziert war und systemische Kortikosteroide benötigt wurden) und Asthma-bedingtes Aufsuchen einer Notfallambulanz (einschliesslich Hospitalisierung, Notfallambulanz und nicht geplante Arztbesuche) sowie Verbesserung der ärztlichen Gesamtbewertung der Wirksamkeit der Behandlung, der Lebensqualität bezüglich Asthma (AQL), der Asthmasymptome und der Lungenfunktion.
In einer Subgruppen-Analyse bei Patienten mit einem IgE-Wert von ≥76 I.E./ml vor der Behandlung war ein klinisch bedeutsamer Nutzen von Xolair wahrscheinlicher. Bei diesen Patienten der Studie 1 reduzierte Xolair die Anzahl der Asthma-Exazerbationen um 40% (p=0.002). Zusätzlich zeigten im Studienprogramm zu Xolair bei schwerem Asthma in der Population mit einem IgE-Gesamtwert ≥76 I.E./ml mehr Patienten ein klinisch relevantes Ansprechen (s. Tabelle 6).
In vier weiteren grossen Placebo-kontrollierten unterstützenden Studien mit einer Dauer von 28 bis 52 Wochen mit 1'722 Erwachsenen und Jugendlichen (Studien 3, 4, 5, 6) wurden die Wirksamkeit und Verträglichkeit von Xolair bei Patienten mit schwerem persistierenden Asthma untersucht. Die meisten Patienten waren ungenügend kontrolliert, erhielten jedoch weniger Begleitmedikation für Asthma als Patienten in Studie 1 oder 2. Die Studien 3–5 hatten Exazerbationen als primären Endpunkt, wogegen Studie 6 primär das Einsparen von inhalativen Kortikosteroiden ermittelte.
In der Studie 2 wurden Sicherheit und Wirksamkeit von Omalizumab an 312 Patienten mit schwerem allergischem Asthma, welche der Population der Studie 1 entsprachen, nachgewiesen. Behandlung mit Xolair in dieser open-label-Studie führte zu 61% Reduktion der klinisch signifikanten Asthma-Exazerbations-Rate verglichen mit der aktuellen Asthma-Therapie alleine.
In den Studien 3, 4 und 5 hatten die mit Xolair behandelten Patienten jeweils Reduktionen der Asthma-Exazerbations-Rate um 37.5% (p=0.027), 40.3% (p<0.001) und 57.6% (p<0.001) im Vergleich zu Placebo.
In Studie 6 waren signifikant mehr Patienten mit schwerem allergischem Asthma in der Lage ohne Verschlechterung der Asthma-Kontrolle mit Xolair ihre Fluticason-Dosis auf ≤500 µg/Tag zu reduzieren (60.3%), im Vergleich zur Placebo-Gruppe (45.8%, p<0.05).
|
Tabelle 6: Resultate der Studie
| |
|
Gesamtpopulation der Studie
| |
|
Xolair
N=209
|
Placebo
N=210
| |
Asthma-Exazerbationen
|
|
| |
Häufigkeit pro 28 Wochen
|
0.74
|
0.92
| |
% Reduktion, p-Wert für Verhältnis der Häufigkeiten
|
19.4%, p = 0.153
| |
Schwere Asthma-Exazerbationen
|
|
| |
Häufigkeit pro 28 Wochen
|
0.24
|
0.48
| |
% Reduktion, p-Wert für Verhältnis der Häufigkeiten
|
50.1%, p = 0.002
| |
Notfallambulanzbesuche
|
|
| |
Häufigkeit pro 28 Wochen
|
0.24
|
0.43
| |
% Reduktion, p-Wert für Verhältnis der Häufigkeiten
|
43.9%, p = 0.038
| |
Ärztliche Gesamtbewertung
|
|
| |
% Responder*
|
60.5%
|
42.8%
| |
p-Wert**
|
<0.001
| |
AQL***-Verbesserungen
|
|
| |
% Patienten mit einer Verbesserung ≥0.5
|
60.8%
|
47.8%
| |
p-Wert
|
0.008
| |
* merkliche Verbesserung oder vollständige Kontrolle
** p-Wert für die allgemeine Verteilung der Bewertung
*** Asthma Quality of Life
|
Kinder im Alter von 6 bis <12 Jahren
Die grundlegenden Daten für die Sicherheit und Wirksamkeit von Xolair in der Altersgruppe von 6 bis <12 Jahren stammen aus einer randomisierten, doppelblinden, Placebo-kontrollierten multizentrischen Studie (Studie 7).
Studie 7 war eine Placebo-kontrollierte Studie mit einer spezifischen Subgruppe (N=235) von Patienten nach derzeitiger Indikation, die mit hoch dosierten inhalativen Kortikosteroiden (≥500 µg Fluticason-Äquivalent/Tag) und langwirksamen Beta-Agonisten behandelt wurden.
Eine klinisch signifikante Exazerbation wurde definiert als eine vom Prüfarzt klinisch beurteilte Verschlechterung der Asthmasymptome, die eine Verdopplung der Ausgangsdosis des inhalativen Kortikosteroids für mindestens 3 Tage und/oder einer Notfallbehandlung mit systemischem (oral oder intravenös) Kortikosteroid für mindestens 3 Tage erforderte.
Bei der spezifischen Subgruppe von Patienten, die hoch dosierte inhalative Kortikosteroide erhielten, zeigte die Omalizumab-Gruppe eine statistisch signifikant niedrigere Rate an Asthma-Exazerbationen als die Placebo-Gruppe. Nach 24 Wochen wurde bei der Betrachtung der Differenzen der Raten für die Omalizumab-Patienten eine um 34% (Verhältnis der Raten 0.662, p=0.047) geringere Rate im Verhältnis zu Placebo erzielt. Im zweiten doppelblinden, 28-wöchigen Behandlungszeitraum wurde bei der Betrachtung der Differenzen der Raten für die Omalizumab-Patienten eine um 63% (Verhältnis der Raten 0.37, p<0.001) geringere Rate im Verhältnis zu Placebo erzielt.
Während der 52-wöchigen, doppelblinden Behandlung (bestehend aus der 24-wöchigen Phase mit konstanter Steroid-Dosis und der 28-wöchigen Phase mit angepasster Steroid-Dosis) zeigten die Differenzen der Raten zwischen den Behandlungsgruppen eine 50%ige (Verhältnis der Raten 0.504, p<0.001) Abnahme der Exazerbationen für Omalizumab-Patienten.
Die Omalizumab-Gruppe zeigte am Ende der 52-wöchigen Behandlung eine grössere Abnahme des Gebrauchs von Beta-Agonisten als Notfallmedikation als die Placebo-Gruppe, auch wenn der Unterschied zwischen den Behandlungsgruppen nicht statistisch signifikant war. In der Gesamtauswertung der Wirksamkeit nach 52-wöchiger, doppelblinder Behandlung war in der Untergruppe der schwer erkrankten Patienten mit hoch dosierten inhalativen Kortikosteroiden und gleichzeitigen langwirksamen Beta-Agonisten der Anteil der Patienten mit «exzellentem» Behandlungserfolg bei der Omalizumab-Gruppe höher als bei der Placebo-Gruppe. Die Anteile der Patienten mit «moderatem» oder «schlechtem» Behandlungserfolg waren in der Omalizumab-Gruppe geringer als bei der Placebo-Gruppe. Die Unterschiede zwischen den Gruppen waren statistisch signifikant (p <0.001). Bei den subjektiven Patientenbewertungen ihrer Lebensqualität gab es keine Unterschiede zwischen der Omalizumab-Gruppe und der Placebo-Gruppe.
Nasenpolypen
Die Sicherheit und die Wirksamkeit von Xolair wurden in zwei randomisierten, multizentrischen, doppelblinden, placebokontrollierten klinischen Studien (Studie 1, N=138; Studie 2, N=127) an Patienten mit chronischer Rhinosinusitis und Nasenpolypen bewertet. Die Patienten erhielten Xolair oder Placebo subkutan alle 2 oder 4 Wochen, wobei die Dosierung und die Anwendungshäufigkeit den Angaben in den Tabellen 7 und 8 entsprachen (siehe «Dosierung/Anwendung»). Ausserdem erhielten alle Patienten während der gesamten Studie eine Hintergrundtherapie mit Mometason intranasal. Eine vorangegangene sinonasale Operation bzw. eine vorherige systemische Behandlung mit Kortikosteroiden waren für die Aufnahme in die Studien nicht erforderlich. Die Studienteilnehmer erhielten 24 Wochen lang Xolair oder Placebo und darauf folgte eine 4-wöchige behandlungsfreie Nachbeobachtungsphase. Die demografischen Angaben und die Baseline-Charakteristika einschliesslich der allergischen Komorbiditäten sind in Tabelle 7 dargestellt.
Tabelle 7 Demografische Angaben und Baseline-Charakteristika in den Nasenpolypen-Studien
|
Parameter
|
Nasenpolypenstudie 1
N=138
|
Nasenpolypenstudie 2
N=127
| |
Mittleres Alter in Jahren (SD)
|
51.0 (13.2)
|
50.1 (11.9)
| |
% männlich
|
63.8
|
65.4
| |
Patienten mit Anwendung systemischer Kortikosteroide im Vorjahr (%)
|
18.8
|
26.0
| |
Mittlerer bilateraler endoskopischer NPS-Score* (SD), Spanne: 0-8
|
6.2 (1.0)
|
6.3 (0.9)
| |
Mittlerer Score für die Nasenkongestion (NC)* (SD), Spanne: 0-3
|
2.4 (0.6)
|
2.3 (0.7)
| |
Mittlerer Score für den Geruchssinn* (SD) Spanne: 0-3
|
2.7 (0.7)
|
2.7 (0.7)
| |
Mittlerer SNOT-22-Gesamtscore* (SD) Spanne: 0-110
|
60.1 (17.7)
|
59.5 (19.3)
| |
Mittlere Eosinophilenzahl im Blut (Zellen/µl) (SD)
|
346.1 (284.1)
|
334.6 (187.6)
| |
Mittleres Gesamt-IgE in IU/ml (SD)
|
160.9 (139.6)
|
190.2 (200.5)
| |
Asthma (%)
|
53.6
|
60.6
| |
Leicht (%)
|
37.8
|
32.5
| |
Mittelschwer (%)
|
58.1
|
58.4
| |
Schwer (%)
|
4.1
|
9.1
| |
Aspirin-exazerbierte Atemwegserkrankung (%)
|
19.6
|
35.4
| |
Allergische Rhinitis
|
43.5
|
42.5
|
SD= Standardabweichung; NPS= Nasenpolypenscore; SNOT-22 = Fragebogen zum Sino-Nasal Outcome Test mit 22 Fragen; IgE = Immunoglobulin E; IU= internationale Einheiten.
Beim NPS, beim NCS und bei den Scores für den Geruchssinn, das Postnasal-Drip-Syndrom und das Naselaufen sowie beim SNOT-22-Score weisen höhere Punktwerte auf eine stärkere Ausprägung der Erkrankung hin.
Die ko-primären Endpunkte waren der bilaterale Nasenpolypenscore (NPS) und der gemittelte tägliche Score für die Nasenkongestion (NCS), jeweils bestimmt in Woche 24. Der NPS wurde mittels Endoskopie zur Baseline und zu vorab festgelegten Zeitpunkten bestimmt (Spanne: 0-4 pro Nasenloch), und aus diesen Werten wurde der Gesamt-NPS berechnet (Spanne: 0 = bester Wert bis 8 = schlechtester Wert). Die Nasenkongestion wurde täglich anhand der NCS-Skala bewertet (Spanne: 0 = bester Wert bis 3 = schlechtester Wert). Die Patienten mussten vor der Randomisierung trotz der Verwendung von Mometason intranasal einen NPS ≥5 und einen Wochenmittelwert für den NCS > 1 aufweisen. Der mittlere NPS zur Baseline war in beiden Studien zwischen den beiden Behandlungsgruppen ausgeglichen.
Sowohl in Studie 1 als auch in Studie 2 der Nasenpolypenstudien zeigten die Patienten, die Xolair erhielten, in Woche 24 sowohl beim NPS als auch beim über die Woche gemittelten NCS eine statistisch signifikant stärkere Verbesserung gegenüber der Baseline als die Patienten, die Placebo erhielten (siehe Tabelle 8).
Die stärkeren Verbesserungen beim NPS und beim NCS in der Xolair-Gruppe im Vergleich zur Placebo-Gruppe wurden in beiden Studien bereits bei der ersten Beurteilung in Woche 4 beobachtet, wie aus Abbildung 1 ersichtlich ist. In Woche 4 betrug die Differenz der Kleinste-Quadrate- (LS-) Mittelwerte für die Veränderung gegenüber der Baseline beim NPS in der Xolair-Gruppe im Vergleich zur Placebo-Gruppe -0.92 (95% KI: -1.37, -0.48) in Studie 1 und -0.52 (95% KI: -0.94, -0.11) in Studie 2. Beim NCS betrug die Differenz der LS-Mittelwerte in der Xolair-Gruppe für die Veränderung gegenüber der Baseline in Woche 4 im Vergleich mit der Placebo-Gruppe -0.25 (95% KI: -0.46, -0.04) in Studie 1 und -0.26 (95% KI: -0.45, -0.07) in Studie 2. Die statistischen Tests waren zu diesem Zeitpunkt jedoch nicht präspezifiziert.
Tabelle 8 Veränderung gegenüber der Baseline in Woche 24 beim Nasenpolypenscore und beim über 7 Tage gemittelten Score für die Nasenkongestion in den Nasenpolypenstudien 1 und 2
|
|
Nasenpolypenstudie 1
|
Nasenpolypenstudie 2
| |
|
Placebo
|
Xolair
|
Placebo
|
Xolair
| |
N
|
66
|
72
|
65
|
62
| |
Nasenpolypenscore
|
|
|
|
| |
Baseline Mittelwert
|
6.32
|
6.19
|
6.09
|
6.44
| |
LS-Mittelwert für die Veränderung gegenüber der Baseline bis Woche 24
|
0.06
|
-1.08
|
-0.31
|
-0.90
| |
Differenz der LS-Mittelwerte
vs. Placebo
|
-1.14
|
-0.59
| |
95% KI der Differenz
|
-1.59, -0.69
|
-1.05, -0.12
| |
p-Wert
|
<0.0001
|
0.0140
| |
Über 7 Tage gemittelter Wert des täglichen Scores für die Nasenkongestion
|
|
|
|
| |
Baseline Mittelwert
|
2.46
|
2.40
|
2.29
|
2.26
| |
LS-Mittelwert für die Veränderung gegenüber der Baseline bis Woche 24
|
-0.35
|
-0.89
|
-0.20
|
-0.70
| |
Differenz der LS-Mittelwerte
vs. Placebo
|
-0.55
|
-0.50
| |
95% KI für die Differenz
|
-0.84, -0.25
|
-0.80, -0.19
| |
p-Wert
|
0.0004
|
0.0017
|
LS = kleinste Quadrate (Bestimmung des Mittelwerts nach der Methode der Least Squares = kleinste Quadrate)
Abbildung 1 Mittlere Veränderung gegenüber der Baseline beim Score für die nasale Kongestion und dem Nasenpolypenscore nach Behandlungsgruppe in den Nasenpolypenstudien 1 und 2
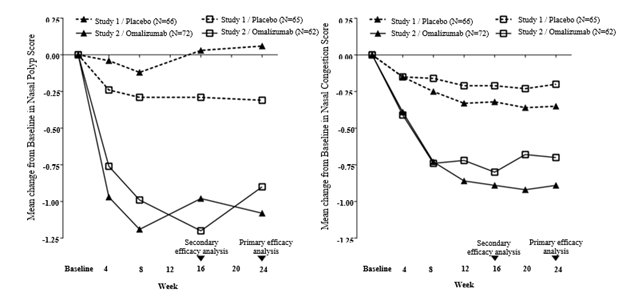
Ein sekundärer Hauptendpunkt war die Bewertung der Veränderung des Gesamtscores der nasalen Symptome (Total nasal symptom score, TNSS) in Woche 24 gegenüber der Baseline. Bei dem patientenberichteten TNSS handelte es sich um einen Score, der der Summe von 4 gleich gewichteten täglichen Symptom-Scores entsprach. Dies waren: der NCS, der Geruchssinn-Score, der posteriore Rhinorrhö-Score und der anteriore Rhinorrhö-Score. Die Spanne beim TNSS reichte von 0 = bester Wert bis 12 = schlechtester Wert. Unter Xolair kam es zu einer signifikanten Verbesserung des mittleren täglichen TNSS im Vergleich mit Placebo. Die Differenz der LS-Mittelwerte für die Veränderung gegenüber Baseline bis Woche 24 betrug -1.91 Punkte (95% KI: -2.85, -0.96; p = 0.0001) in Studie 1 und -2.09 Punkte (95% KI: -3.00, -1.18; p < 0.0001) in Studie 2.
Unter Xolair kam es zu einer signifikanten Verbesserung beim SNOT-22 (Sino-Nasal OutcomeTest), bei dem Fragen aus den Bereichen sinonasale Symptome, Psychologie und Schlafqualität kombiniert sind. Der SNOT-22 lag innerhalb einer Spanne von 0 bis 110 (0 = bester Wert, 110 = schlechtester Wert). Die Differenz der LS-Mittelwerte für die Veränderung gegenüber der Baseline bis Woche 24 im SNOT-22 unter Xolair im Vergleich mit Placebo lag bei -16.12 (95% KI: -21.86, -10.38; p < 0.0001) in Studie 1 und -15.04 (95% KI: -21.26, -8.82; p < 0.0001) in Studie 2.
Unter Xolair kam es ausserdem zu einer signifikanten Verbesserung des mittleren täglichen UPSIT- (University of Pennsylvania Smell Identification Test-)Scores im Vergleich mit Placebo. Der UPSIT-Score lag innerhalb einer Spanne von 0 bis 40 (0 = schlechtester Wert, 40 = bester Wert). Die Differenz der LS-Mittelwerte für die Veränderung gegenüber der Baseline bis Woche 24 betrug unter Xolair im Vergleich mit Placebo 3.81 Punkte (95% KI: 1.38, 6.24; p = 0.0024) in Studie 1 und 3.86 Punkte (95% KI: 1.57, 6.15; p= 0.0011) in Studie 2.
Die Auswirkung auf den TNSS und den SNOT-22 wurde in beiden Studien bereits bei der ersten Beurteilung in Woche 4 beobachtet. Darüber hinaus wurde in beiden Studien die Auswirkung auf den UPSIT-Score bei der ersten Beurteilung in Woche 8 beobachtet.
Zusätzliche sekundäre Endpunktanalysen umfassten Beurteilungen des NPS und des NCS in Woche 16. Unter Xolair kam es zu einer signifikanten Verbesserung des NPS in Woche 16 (0 = schlechtester Wert, 8= bester Wert) im Vergleich mit Placebo. Die Differenz der LS-Mittelwerte für die Veränderung gegenüber der Baseline bis Woche 16 betrug unter Xolair im Vergleich mit Placebo -1.01 (95% KI: -1.43, -0.60; p < 0.0001) in Studie 1 und -0.91 (95% KI: -1.39, -0.44; p= 0.0002) in Studie 2. Unter Xolair kam es zu einer signifikanten Verbesserung des NCS in Woche 16 (0 = bester Wert, 3= schlechtester Wert) im Vergleich mit Placebo. Die Differenz der LS-Mittelwerte für die Veränderung gegenüber Baseline bis Woche 16 beim mittleren täglichen NCS betrug unter Xolair im Vergleich mit Placebo -0.57 (95% KI: -0.83, -0.31; p < 0.0001) in Studie 1 und -0.59 (95% KI: -0.87, -0.30; p < 0.0001) in Studie 2.
Chronische spontane Urtikaria (CSU)
Das klinische Phase-III-Entwicklungsprogramm bei CSU umfasste drei randomisierte, doppelblinde, Placebo-kontrollierte, multizentrische Parallelgruppenstudien: Q4881g, Q4882g und Q4883g.
Untersucht wurden Erwachsene und Jugendliche (ab 12 Jahren) mit CSU während ≥6 Monaten (6 Monate bis 66 Jahre, durchschnittliche 6 Jahre) mit ständigen Schüben trotz Antihistaminika in zugelassenen Maximaldosen (UAS 7≥16/42 während ≥8 nacheinander folgenden Tagen).
Studien Q4881g und Q4882g dienten zur Beurteilung der Wirksamkeit und Sicherheit der Verabreichung von 75 mg, 150 mg oder 300 mg Xolair alle 4 Wochen während 24 bzw. 12 Wochen mit einer 16-wöchigen behandlungsfreien Nachbeobachtungsphase bei Patienten (12-75 Jahre) mit refraktärer CSU trotz Behandlung mit H1-Antihistaminen.
Studie Q4883g diente zur Beurteilung der Sicherheit und Wirksamkeit von 300 mg Xolair, verabreicht alle 4 Wochen, während 24 Wochen mit einer 16-wöchigen behandlungsfreien Nachbeobachtungsphase bei Patienten (12-75 Jahre) mit refraktärer CSU trotz Behandlung mit H1- und/oder H2-Antihistaminen und/oder Leukotrien-Rezeptorantagonisten (LTRA).
|
Tabelle 9
|
Endpunkte betreffend der Wirksamkeit
| |
Veränderung des wöchentlich erhobenen Itch Severity Scores (ISS, Bereich 0-21) in Woche 12 gegenüber Baseline
|
Primärer Endpunkt in Studien Q4881g und Q4882g
Sekundärer Endpunkt in der Sicherheitsstudie Q4883g
| |
Zeit bis zu einem MID a-Ansprechen (Verringerung um ≥5 Punkte gegenüber Baseline) des wöchentlich erhobenen ISS bis Woche 12
|
Sekundäre Endpunkte in allen drei Studien Q4881g, Q4882g und Q4883g
| |
Veränderung des über einen Zeitraum von 7 Tagen gemessenen Urtikaria-Aktivitäts-Scores (UAS7 b, Bereich 0-42) in Woche 12 gegenüber Baseline
| |
Anteil der Patienten mit einem über einen Zeitraum von 7 Tagen gemessenen Urtikaria-Aktivitäts-Score ≤6 (UAS7 b ≤6) in Woche 12
| |
Anteil der Patienten mit einem über einen Zeitraum von 7 Tagen gemessenen Urtikaria-Aktivitäts-Score = 0 (UAS7 b = 0) in Woche 12 c
| |
Veränderung des wöchentlich erhobenen Scores für die Anzahl der Quaddeln in Woche 12 gegenüber Baseline
| |
Veränderung des Gesamtscores des Dermatologischen Lebensqualitäts-Index (DLQI) in Woche 12 gegenüber Baseline
| |
Anteil der Patienten mit Angioödem-freien Tagen zwischen Woche 4 und Woche 12 d
| |
a
MID: Geringster relevanter Unterschied (Minimally Important Difference)
b UAS7: Zusammengesetzt aus dem Schweregrad des Juckreizes und der Anzahl der Quaddeln; Summe der an 7 aufeinanderfolgenden Tagen gemessenen Scores
c Post-hoc-Analyse für Studie Q4882g
d Der mittlere Anteil der Angioödem-freien Tage zwischen Woche 4 und Woche 12 wurde für die gesamte Studienpopulation einschliesslich der Patienten ohne Symptome eines Angioödems berechnet.
|
Die 75 mg-Dosis erreichte in den Studien Q4881g und Q4882g weder konsistent den primären Wirksamkeitsendpunkt (Veränderung des wöchentlich erhobenen Itch Severity Scores (ISS) in Woche 12 gegenüber Baseline) noch mehrere sekundäre Endpunkte. Daher wurde diese Dosis als nicht wirksam erachtet und wird daher nicht weiter dargestellt.
Der primäre Wirksamkeitsendpunkt, die Veränderung des wöchentlich erhobenen Itch Severity Scores in Woche 12 gegenüber Baseline, wurde in den Studien Q4881g und Q4882g sowohl mit der 150 mg-Dosis als auch mit der 300 mg-Dosis und in Studie Q4883g mit der 300 mg-Dosis erreicht (sekundärer Endpunkt; siehe Tabelle 10).
|
Tabelle 10: Veränderung des wöchentlich erhobenen Itch Severity Scores in Woche 12 gegenüber Baseline, Studien Q4881g, Q4882g und Q4883g (mITT-Population*)
| |
|
Placebo
|
Omalizumab
150 mg
|
Omalizumab
300 mg
| |
Studie Q4881g
|
|
|
| |
N
|
80
|
80
|
81
| |
Mittelwert (SD)
|
-3.63 (5.22)
|
-6.66 (6.28)
|
-9.40 (5.73)
| |
Difference in LS means vs. placebo1
|
-
|
-2.95
|
-5.80
| |
95% CI for difference
|
-
|
−4.72,−1.18
|
−7.49,−4.10
| |
P-value vs. placebo2
|
-
|
0.0012
|
<0.0001
| |
Studie Q4882g
|
|
|
| |
N
|
79
|
82
|
79
| |
Mittelwert (SD)
|
-5.14 (5.58)
|
-8.14 (6.44)
|
-9.77 (5.95)
| |
Difference in LS means vs. placebo1
|
-
|
-3.04
|
-4.81
| |
95% CI for difference
|
-
|
−4.85,−1.24
|
−6.49,−3.13
| |
P-value vs. placebo2
|
-
|
0.0011
|
<0.0001
| |
Studie Q4883g
|
|
|
| |
N
|
83
|
-
|
252
| |
Mittelwert (SD)
|
-4.01 (5.87)
|
-
|
-8.55 (6.01)
| |
Difference in LS means vs. placebo1
|
-
|
-
|
-4.52
| |
95% CI for difference
|
-
|
-
|
−5.97, −3.08
| |
P-value vs. placebo2
|
-
|
-
|
<0.0001
| |
* Modifizierte Intent-to-Treat (mITT)-Population: umfasst alle randomisierten Patienten, die mindestens eine Dosis des Prüfmedikaments erhalten hatten
Zur Kalkulation fehlender Daten wurde das BOCF (Baseline Observation Carried Forward)-Verfahren angewendet.
1 Der LS-Mittelwert wurde unter Anwendung eines ANCOVA-Modells berechnet. Stratifizierungsfaktoren waren der Ausgangswert des wöchentlich erhobenen Itch Severity Scores (< 13 vs. ≥13) und das Ausgangsgewicht (< 80 kg vs. ≥80 kg).
2 p-Werte entstammen dem ANCOVA t-Test.
|
In Abbildung 2 ist der mittlere wöchentlich erhobene ISS im Zeitverlauf in Studie Q4881g dargestellt. Der mittlere wöchentlich erhobene Itch Severity Score nahm in beiden Behandlungsgruppen signifikant ab. Die maximale Wirkung wurde ungefähr in Woche 12 erreicht und blieb über die 24-wöchige Behandlungsphase bestehen. In den Studien Q4883g (300 mg über eine 24-wöchige Behandlungsphase) und Q4882g (150 mg oder 300 mg über eine 12-wöchige Behandlungsphase) waren die Resultate ähnlich wie in Studie Q4881g.
In allen drei Studien (siehe Abbildung 2 für Studie Q4881g) stieg der mittlere wöchentlich erhobene Itch Severity Score für beide Dosierungen während der 16-wöchigen behandlungsfreien Phase allmählich an, parallel zum Wiederauftreten der Symptome. Am Ende der Nachbeobachtungsphase waren die Mittelwerte mit denen der Placebo-Gruppe vergleichbar, jedoch niedriger als die entsprechenden mittleren Ausgangswerte.
Abbildung 2: Mittlerer wöchentlich erhobener Itch Severity Score im Zeitverlauf, Studie Q4881g (BOCF, mITT-Population)
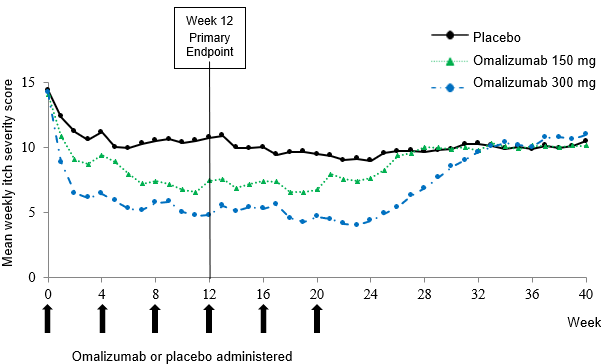
BOCF = Baseline Observation Carried Forward; mITT = modifizierte Intent-to-Treat-Population
Zeit bis zu einem MID-Ansprechen des wöchentlich erhobenen ISS bis Woche 12
In den Studien Q4881g und Q4882g betrug die mediane Zeit bis zum Erreichen eines MID des wöchentlich erhobenen Itch Severity Scores von 5 Punkten bei Patienten in der Behandlungsgruppe mit 150 mg 2 Wochen (p=0.0301 in Studie Q4881g; p=0.0101 in Studie Q4882g) und bei Patienten in der Behandlungsgruppe mit 300 mg 1 Woche (p<0.0001), verglichen mit 4 Wochen bei Patienten in den Placebo-Gruppen. Vergleichbare Resultate wurden in Studie Q4883g mit einer medianen Zeit bis zum Erreichen eines MID von 2 Wochen in der Behandlungsgruppe mit 300 mg (p<0.0001) vs. 5 Wochen in der Placebo-Gruppe beobachtet.
Veränderung des UAS7 in Woche 12 gegenüber Baseline
In den Phase-III-Studien wiesen die Behandlungsgruppen mit 150 mg und 300 mg Omalizumab im Vergleich zu Placebo einen statistisch signifikanten Unterschied der mittleren Veränderung des UAS7 in Woche 12 gegenüber Baseline auf (Abbildung 3 für Studie Q4881g). Statistische Signifikanz (p<0.0001) wurde in allen drei Studien in der Behandlungsgruppe mit 300 mg sowie in den Studien Q4881g (p=0.0008) und Q4882g (p=0.0001) in der Behandlungsgruppe mit 150 mg erreicht.
In Abbildung 3 ist der mittlere UAS7 im Zeitverlauf in Studie Q4881g dargestellt, der in beiden Behandlungsgruppen eine signifikante Abnahme gegenüber Baseline bei einer maximalen Wirkung um Woche 12 herum aufwies. Die Stärke dieser Wirkung blieb über die 24-wöchige Behandlungsphase bestehen. In den Studien Q4882g (150 mg und 300 mg während einer 12-wöchigen Behandlungsphase) und Q4883g (300 mg während einer 24-wöchigen Behandlungsphase) waren die Resultate mit denen von Studie Q4881g vergleichbar.
In allen drei Studien (siehe Abbildung 3 für Studie Q4881g) stieg der UAS7 in beiden Omalizumab-Behandlungsgruppen während der 16-wöchigen behandlungsfreien Nachbeobachtungsphase allmählich an, parallel zum Wiederauftreten der Symptome. Am Ende der Nachbeobachtungsphase waren die Mittelwerte mit denen der Placebo-Gruppe vergleichbar, jedoch niedriger als die entsprechenden Ausgangswerte.
Abbildung 3: Mittlerer UAS7 im Zeitverlauf, Studie Q4881g (BOCF, mITT-Population)
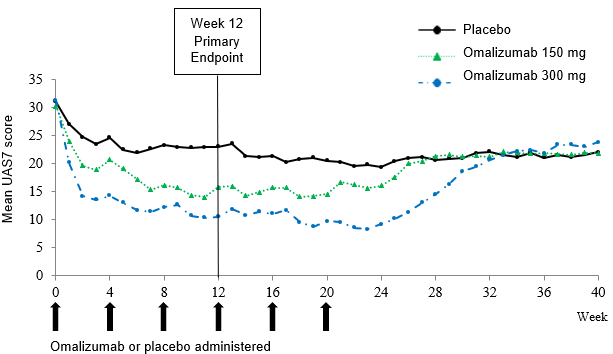
BOCF = Baseline Observation Carried Forward; mITT = modifizierte Intent-to-Treat-Population; UAS7 = Über einen Zeitraum von 7 Tagen gemessener Urtikaria-Aktivitäts-Score
Anteil der Patienten mit einem UAS7 ≤6 in Woche 12
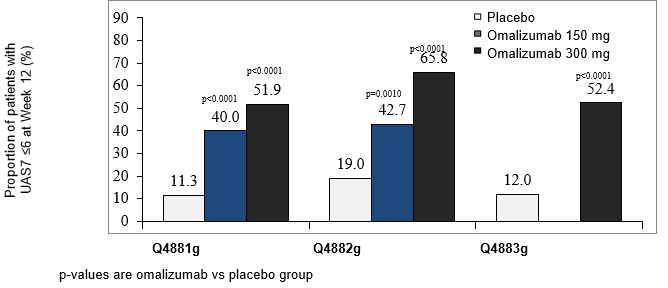
Der Anteil der Patienten mit einem UAS7 ≤6 in Woche 12 ist in Abbildung 4 dargestellt. Die Ansprechraten waren alle statistisch signifikant und lagen zwischen 52% und 66% (300 mg-Dosis; p<0.0001) bzw. zwischen 40% und 43% (150 mg-Dosis; p<0.001) im Vergleich zu 11-19% in der Placebo-Gruppe.
Abbildung 4: Anteil der Patienten mit einem UAS7 ≤6 in Woche 12, Studien Q4881g, Q4882g und Q4883g
Anteil der Patienten mit einem UAS7 = 0 in Woche 12
Der Anteil der Patienten mit einem vollständigen Ansprechen gemäss einem UAS7 = 0 in Woche 12 betrug 34-44% (300 mg-Dosis, statistisch signifikant, alle p<0.0001) bzw. 15-22% (150 mg-Dosis) im Vergleich zu 5-9% in der Placebo-Gruppe (Abbildung 5).
Abbildung 5: Anteil der Patienten mit einem UAS7 = 0 in Woche 12, Studien Q4881g, Q4882g und Q4883g

Prospektive Analyse in den Studien Q4881g und Q4883g und als Post-hoc-Analyse in Studie Q4882g
Veränderung des wöchentlich erhobenen Scores für die Anzahl der Quaddeln in Woche 12 gegenüber Baseline
Die mittlere Veränderung des wöchentlich erhobenen Scores für die Anzahl der Quaddeln in Woche 12 gegenüber Baseline war in allen drei Phase-III-Studien in der Behandlungsgruppe mit 300 mg statistisch signifikant (p<0.001) und zeigte eine Verminderung der Anzahl der Quaddeln im Vergleich zu Placebo (-11.35 in Q4881g, -11.97 in Q4882g und -10.46 in Q4883g versus -4.37, -5.22 bzw. -4.49 in den entsprechenden Placebo-Gruppen). In der Behandlungsgruppe mit 150 mg betrug die mittlere Veränderung -7.78 (p=0.0017) in Q4881g und -9.75 (p<0.0001) in Q4882g.
Anteil der Angioödem-freien Tage zwischen Woche 4 und 12
In allen drei Phase-III-Studien erreichten die Behandlungsgruppen mit 300 mg durchgängig den höchsten mittleren Anteil der Angioödem-freien Tage zwischen Woche 4 und 12 (91-96%). Die Zunahme des Anteils der Angioödem-freien Tage war im Vergleich zu Placebo statistisch signifikant (p<0.001) (Abb. 6). In der Behandlungsgruppe mit 150 mg betrug der mittlere Anteil der Angioödem-freien Tage über den gleichen Zeitraum 89.6% in Studie Q4881g und 91.6% in Studie Q4882g. Die entsprechenden Placebo-Werte der betreffenden Studien betrugen 88.2% bzw. 89.2%. In beiden Studien erreichten die Unterschiede zwischen der 150 mg-Dosis und Placebo keine statistische Signifikanz.
Abbildung 6: Anteil der Angioödem-freien Tage zwischen Woche 4 und 12, Studien Q4881g, Q4882g und Q4883g
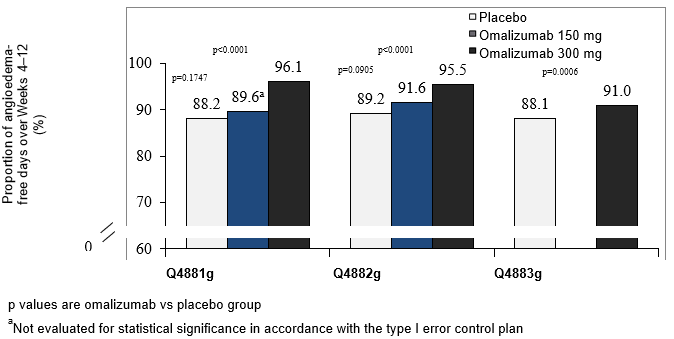
Der mittlere Anteil der Angioödem-freien Tage zwischen Woche 4 und Woche 12 wurde für die gesamte Studienpopulation einschliesslich der Patienten ohne Symptome eines Angioödems berechnet.
Veränderung des Gesamtscores des Dermatologischen Lebensqualitäts-Index (DLQI) in Woche 12 gegenüber Baseline
In allen drei Phase-III-Studien war die mittlere Veränderung des DLQI-Gesamtscores in Woche 12 gegenüber Baseline in der Behandlungsgruppe mit 300 mg statistisch signifikant (p<0.001) grösser als unter Placebo. Die Behandlungsgruppe mit 150 mg Omalizumab wies in Studie Q4882g einen statistisch signifikanten (p=0.022) Unterschied gegenüber Placebo auf (Abbildung 7).
Abbildung 7: Veränderung des Gesamtscores des Dermatologischen Lebensqualitäts-Index in Woche 12 gegenüber Baseline, Studien Q4881g, Q4882g und Q4883g
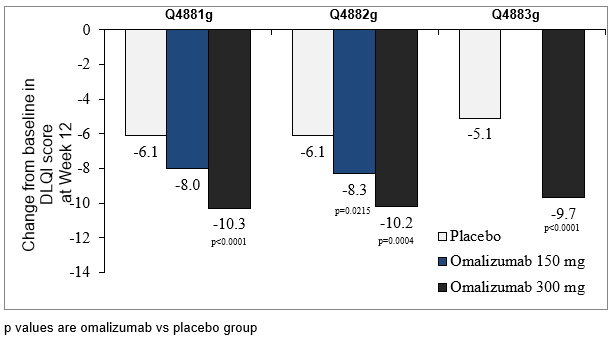
DLQI=Dermatology Life Quality Index
Wirksamkeit nach 24-wöchiger Behandlung
In Tabelle 11 sind die Resultate nach 24-wöchiger Behandlung dargestellt. Die Grössenordnungen des Ansprechens sind ähnlich denen, die nach 12 Wochen beobachtet wurden.
|
Tabelle 11: Wirksamkeitsergebnisse nach 24-wöchiger Behandlung, Studien Q4881g und Q4883g (mITT-Population*)
| |
Studienparameter
|
Woche
|
Placebo
|
Omalizumab
150 mg
|
Omalizumab
300 mg
| |
Änderung gegenüber Baseline des wöchentlichen Itch Severity Scores (BOCF), Mittelwert
| |
Studie Q4881g
|
Woche 24
|
−5.41
|
−6.47
|
−9.84**
| |
Studie Q4883g
|
Woche 24
|
−4.03
|
NA
|
−8.60**
| |
Änderung gegenüber Baseline des UAS7 (BOCF), Mittelwert
| |
Studie Q4881g
|
Woche 24
|
−11.73
|
−14.21
|
−22.11**
| |
Studie Q4883g
|
Woche 24
|
−8.85
|
NA
|
−19.15**
| |
Anteil der Patienten mit UAS7 ≤6, % Patienten
| |
Studie Q4881g
|
Woche 24
|
25.0
|
36.3
|
61.7**
| |
Studie Q4883g
|
Woche 24
|
16.9
|
NA
|
55.6**
| |
Anteil der Patienten mit UAS7 = 0, % Patienten
| |
Studie Q4881g
|
Woche 24
|
12.5
|
20.0
|
48.1**
| |
Studie Q4883g
|
Woche 24
|
3.6
|
NA
|
42.5**
| |
* Modifizierte Intent-to-Treat (mITT)-Population: umfasst alle randomisierten Patienten, die mindestens eine Dosis des Prüfmedikaments erhalten hatten.
** p-Wert ≤0.0001 im jeweiligen statistischen Test zwischen der Behandlung und Placebo
NA: Nicht zutreffend (Not Applicable).
BOCF: Baseline Observation Carried Forward.
|
Wirksamkeit nach 48 Behandlungswochen
In einer 48-wöchigen Studie nahmen 206 Patienten mit CSU, welche mit einer H1 Antihistaminika-Therapie nicht kontrolliert werden konnte, im Alter zwischen 12 und 75 Jahren an einer 24-wöchigen offenen Behandlungsphase mit Omalizumab 300 mg alle 4 Wochen als Zusatztherapie teil. Patienten, die auf die Behandlung in dieser offenen Phase ansprachen, erhielten anschliessend verblindet nach dem Zufallsprinzip Omalizumab 300 mg (81 Patienten) oder Placebo (53 Patienten) alle 4 Wochen für weitere 24 Wochen.
Von den Patienten, die insgesamt 48 Wochen lang weiter mit Omalizumab behandelt wurden, zeigten 21 % eine klinische Verschlechterung (UAS7-Wert von 12 oder mehr für mindestens 2 aufeinanderfolgende Wochen nach der Randomisierung zwischen Woche 24 und 48), während dies bei 60,4 % der mit Placebo behandelten Patienten in Woche 48 der Fall war (Der Unterschied lag bei -39,4 %, p < 0,0001, 95%-KI: -54,5 %; -22,5 %).
Die prospektive Schwangerschaftsregisterstudie (EXPECT)
Eine von 2006 bis 2018 in den USA durchgeführte prospektive Schwangerschaftsregisterstudie (EXPECT) umfasste 250 schwangere Frauen mit Asthma, die mit Xolair behandelt wurden. 246 der Frauen waren im ersten Schwangerschaftstrimester mit Xolair behandelt worden, und 78.4% (196/250) der Frauen waren in jedem der 3 Schwangerschaftstrimester mindestens einmal mit Xolair behandelt worden, wobei die gesamte Expositionsdauer im Median 8,7 Monate betrug. Die EXPECT-Ergebnisse bei den relevanten Mutter- und Säuglingsuntergruppen wurden mit den altersangepassten Häufigkeiten in einer krankheitsangepassten externen Kohorte von 1'153 schwangeren Frauen mit Asthma (ohne Behandlung mit Xolair) verglichen, die aus Gesundheitsdatenbanken von Einwohnern der kanadischen Provinz Quebec ermittelt und als Quebec External Comparator Cohort (QECC) bezeichnet wurde.
Bei den Säuglingen aus EXPECT, die mit Säuglingen aus der QECC (n=223) verglichen wurden, lag die Prävalenz von relevanten kongenitalen Fehlbildungen bei 8.1% und war damit vergleichbar mit der bei den Säuglingen aus der QECC (8.9%). Bei den Schwangerschaften aus EXPECT, die zum Vergleich mit der QECC (n=230) herangezogen wurden, führten 99.1% zu Lebendgeburten; ähnlich den 99.3% aus den QECC Schwangerschaften.
In einer Substudie von EXPECT wurden die Spiegel der Blutplättchen bei 51 Säuglingen untersucht, die von Frauen, mit Exposition gegenüber Xolair, geboren wurden; diese Spiegel lagen alle im Normbereich.
PharmakokinetikAbsorption
Nach subkutaner Injektion beträgt die mittlere absolute Bioverfügbarkeit von Omalizumab 62%. Nach einmaliger subkutaner Verabreichung bei erwachsenen und jugendlichen Asthmapatienten wurde Omalizumab langsam absorbiert. Die maximale Serumkonzentration wurde nach durchschnittlich 7-8 Tagen erreicht. Bei Dosierungen über 0.5 mg/kg verläuft die Pharmakokinetik von Omalizumab linear.
Nach Mehrfachdosierung von Omalizumab war die AUC (im steady state zwischen Tag 0 und Tag 14) bis zu 6-fach höher als nach der ersten Verabreichung.
Distribution
In vitro bildet Omalizumab mit IgE Komplexe mit begrenztem Molekulargewicht. Ausfallende Komplexe oder Komplexe mit einem Molekulargewicht von mehr als 1'000'000 Da konnten weder in vitro noch in vivo beobachtet werden. Das apparente Verteilungsvolumen bei Patienten nach subkutaner Verabreichung betrug 78±32 ml/kg.
Metabolismus
Es liegen für die Anwendung keine relevanten Daten vor.
Elimination
Die Clearance von Omalizumab schliesst sowohl IgG-Clearanceprozesse als auch Clearance durch spezifische Bindung und Komplexbildung mit dem Zielliganden, IgE, ein. Die Ausscheidung von IgG über die Leber beinhaltet den Abbau im retikuloendothelialen System (RES) und in Endothelialzellen. Intaktes IgG wird auch über die Galle ausgeschieden. Bei Asthmapatienten liegt die durchschnittliche Halbwertszeit für die Elimination von Omalizumab aus dem Serum bei 26 Tagen, wobei die durchschnittliche apparente Clearance 2.4±1.1 ml/kg/Tag beträgt.
Eine Verdoppelung des Körpergewichtes führte zu einer ungefähren Verdoppelung der apparenten Clearance.
Patienten mit Nasenpolypen
Die populationsbezogenen pharmakokinetischen Analysen zu Omalizumab deuteten darauf hin, dass die Pharmakokinetik von Omalizumab bei Nasenpolypen mit der bei Asthma übereinstimmt. Es wurden grafische Kovariatenanalysen durchgeführt, um die Auswirkungen von demografischen Merkmalen und sonstigen Faktoren auf die Omalizumab-Exposition und das klinische Ansprechen zu bewerten. Diese Analysen zeigen, dass keine Dosisanpassungen auf der Grundlage von Alter (18 bis 75 Jahre) oder Geschlecht erforderlich sind. Die vorliegenden Daten zu Rasse und ethnischer Zugehörigkeit sind für Patienten mit Nasenpolypen zu begrenzt, um Aussagen über eine eventuell erforderliche Dosisanpassung treffen zu können.
Linearität/Nicht Linearität
Bei Dosierungen über 0.5 mg/kg verläuft die Pharmakokinetik von Omalizumab linear.
Kinetik spezieller Patientengruppen
Analysen der Pharmakokinetik in Abhängigkeit von Alter, Geschlecht, ethnischer Zugehörigkeit oder Body Mass Index ergaben, dass bei diesen Patientengruppen keine Dosisanpassung (6-76 Jahre) vorgenommen werden muss.
Bei Patienten mit eingeschränkter Nieren- oder Leberfunktion liegen keine pharmakokinetischen oder pharmakodynamischen Daten vor (s. «Warnhinweise und Vorsichtsmassnahmen»).
Alter, ethnische Zugehörigkeit, Geschlecht, Körpergewicht, Body Mass Index, IgE-Basiswert, anti-FcεRI-Autoantikörper, Begleitmedikationen
Die Auswirkungen demographischer Kovariaten und anderer Faktoren auf die Exposition gegenüber Omalizumab wurden mittels populationspharmakokinetischer Verfahren beurteilt. Zusätzlich wurden die Auswirkungen der Kovariaten durch die Analyse des Verhältnisses zwischen den Omalizumab-Konzentrationen und dem klinischen Ansprechen beurteilt. Diese Analysen lassen darauf schliessen, dass bei Patienten mit CSU keine Dosisanpassungen in Abhängigkeit von Alter (12 bis 75 Jahre), ethnischer Zugehörigkeit, Geschlecht, Körpergewicht, Body Mass Index, IgE-Basiswert, anti-FcεRI-Autoantikörper oder gleichzeitiger Anwendung von H2-Antihistaminen oder Leukotrien-Rezeptorantagonisten (LTRA) erforderlich sind.
Präklinische DatenDie Verabreichung von Xolair bei adulten und juvenilen Cynomolgusaffen gab keinerlei Anzeichen einer systemischen anaphylaktoiden Reaktion infolge von Mastozyten-Degranulation.
In allen Studien an Affen wurden zirkulierende Omalizumab-IgE-Antikörperkomplexe gefunden, es gab jedoch in keinem Organ (auch nicht in der Niere) Anzeichen für eine immunkomplex-vermittelte Reaktion nach Gabe von Omalizumab. Omalizumab-IgE-Komplexe können weder Komplement binden noch komplement-abhängige Zytotoxizität vermitteln.
Bei einigen Affen wurden nach subkutaner oder intravenöser Verabreichung Antikörper gegen Omalizumab gefunden. Nach Verabreichung eines heterologen Proteins konnte ein solcher Befund erwartet werden. Einige Tiere konnten wegen hoher Omalizumab-Serumkonzentrationen, hohen IgE-Werten oder beidem nicht ausgewertet werden. Die Tiere wiesen jedoch über die ganze Behandlungsdauer der Studien hohe Omalizumab-Serumkonzentrationen auf, und es traten keine offensichtlichen Toxizitäten infolge der Anti-Omalizumab-Antikörper auf.
Gewebsdistributionsstudien an Cynomolgusaffen zeigten keine spezifische Aufnahme von 125I-Omalizumab durch irgendein Organ oder Gewebe.
In Studien an Mäusen und Affen wurden die Omalizumab-IgE-Komplexe durch Interaktion mit den Fcγ-Rezeptoren im RES eliminiert mit Abbauraten, die im Allgemeinen schneller als die IgG-Clearance waren.
Langzeittoxizität (bzw. Toxizität bei wiederholter Verabreichung)
Langzeitverabreichung von Omalizumab bei Dosierungen von bis zu 250 mg/kg (maximal zulässige klinische Dosis in Asthma oder CSU gemäss den Empfehlungen 15 mg/kg) wurde von Primatenaffen (sowohl adulte als auch juvenile Tiere) gut vertragen. Eine Ausnahme bildete eine dosisabhängige Abnahme der Blutplättchenzahl bei mehreren Primatenaffen mit Serumkonzentrationen, die im Allgemeinen über den maximalen Expositionswerten beim Menschen in den pivotalen klinischen Studien lagen. Juvenile Affen waren für Blutplättcheneffekte empfindlicher als adulte Affen. Zusätzlich wurden an den Einstichstellen bei Cynomolgusaffen akute Blutungen und Entzündungen beobachtet; diese Erscheinungen sind vereinbar mit einer lokalisierten Immunantwort auf wiederholte subkutane Verabreichung eines heterologen Proteins.
Karzinogenität
Es wurden keine eigentlichen Studien zur Karzinogenität von Omalizumab durchgeführt.
Reproduktionstoxizität
Es wurden Reproduktionsstudien mit Omalizumab bei Cynomolgusaffen durchgeführt. Subkutane Dosen von Omalizumab bis zu 75 mg/kg pro Woche (das ungefähr 17-fache Expositionsverhältnis gegenüber der MRHD über einen Zeitraum von 4 Wochen) während der Organogenese verabreicht, zeigten keine Toxizität bei den Muttertieren, Embryotoxizität oder Teratogenität. Ausserdem wurden bei einer Verabreichung während der Gestation, Geburt oder des Stillens keine nachteiligen Effekte auf das foetale oder neonatale Wachstum beobachtet.
Die Exkretion von Omalizumab in die Milch wurde bei weiblichen Cynomolgusaffen untersucht, welche subkutane Dosen von 75 mg/kg/Woche verabreicht erhielten. Die Serum Omalizumabspiegel beim Neugeborenen nach in utero-Exposition und 28-tägigem Stillen lagen zwischen 11% und 94% des Serumspiegels der Mutter. Omalizumabspiegel in der Milch betrugen 0.15% der mütterlichen Serumkonzentration.
Fertilität
Es gibt keine Daten für Omalizumab zur Beeinflussung der Fertilität beim Menschen. Bei spezifisch angelegten nicht-klinischen Studien zur Fertilität, einschliesslich Paarungsuntersuchungen, wurde nach wiederholter Gabe von Omalizumab bei Dosen von bis zu 75 mg/kg keine Störung der männlichen oder weiblichen Fertilität festgestellt.
Sonstige HinweiseInkompatibilitäten
Xolair darf nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern.
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht (und/oder Feuchtigkeit) zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Der Fertigpen darf nicht länger als total 48 Stunden bei Raumtemperatur (25°C) aufbewahrt werden.
Anweisungen zur Verwendung und Handhabung
Jede Xolair Packung enthält einen Fertigpen.
Eine detaillierte Gebrauchsanweisung ist in der Patienteninformation vorhanden.
Zulassungsnummer69272 (Swissmedic)
Packungen1 Injektionslösung in Fertigpen zu 75 mg Omalizumab zu 0.5 ml [B]
1 Injektionslösung in Fertigpen zu 150 mg Omalizumab zu 1.0 ml [B]
1 Injektionslösung in Fertigpen zu 300 mg Omalizumab zu 2.0 ml [B]
ZulassungsinhaberinNovartis Pharma Schweiz AG, Risch; Domizil: 6343 Rotkreuz
Stand der InformationMärz 2025
|