ZusammensetzungWirkstoffe
Inebilizumab.
Inebilizumab ist ein humanisierter monoklonaler Antikörper, der mittels rekombinanter DNA-Technologie in Ovarialzellen des chinesischen Hamsters hergestellt wird.
Hilfsstoffe
Histidin, Histidinhydrochlorid-Monohydrat, Natriumchlorid, Trehalose-Dihydrat, Polysorbat 80 (E433), Wasser für Injektionszwecke.
Dieses Arzneimittel enthält 16,1 mg Natrium pro Durchstechflasche.
Indikationen/AnwendungsmöglichkeitenUplizna ist als Monotherapie zur Behandlung von erwachsenen Patienten mit Neuromyelitis-optica-Spektrum-Erkrankungen (NMOSD) indiziert, die Anti-Aquaporin-4-Immunglobulin-G(AQP4-IgG)-seropositiv sind (siehe Rubrik «Eigenschaften/Wirkungen»).
Dosierung/AnwendungDie Behandlung sollte unter der Aufsicht eines in der Behandlung von NMOSD erfahrenen Arztes eingeleitet werden, der Zugang zu entsprechender medizinischer Versorgung hat, um mögliche schwerwiegende Reaktionen, wie z.B. schwere infusionsbedingte Reaktionen, unter Kontrolle zu bringen.
Der Patient sollte während und für mindestens eine Stunde nach Beendigung der Infusion auf Infusionsreaktionen hin überwacht werden (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Untersuchungen vor der ersten Dosis von Inebilizumab
Vor Beginn der Behandlung sollten folgende Tests durchgeführt werden:
·quantitative Serumimmunglobuline, B-Zellzahl und grosses Blutbild (complete blood count, CBC) einschliesslich Differentialblutbild (siehe Rubriken «Kontraindikationen» und «Warnhinweise und Vorsichtsmassnahmen»)
·Hepatitis-B-Virus (HBV)-Screening (siehe Rubriken «Kontraindikationen» und «Warnhinweise und Vorsichtsmassnahmen»)
·Hepatitis-C-Virus (HCV)-Screening und -Behandlung, die vor Einleitung der Behandlung mit Inebilizumab begonnen wurde (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»)
·Untersuchung auf aktive Tuberkulose und Test auf latente Infektion (siehe Rubriken «Kontraindikationen» und «Warnhinweise und Vorsichtsmassnahmen»)
Sämtliche Impfungen mit Lebendimpfstoffen oder abgeschwächten Lebendimpfstoffen sollten mindestens 4 Wochen vor Beginn der Behandlung mit Inebilizumab gemäss den Impfempfehlungen verabreicht werden (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Wird vermutet, dass ein Verlust der Wirksamkeit auf Immunogenität zurückzuführen ist, sollte der Arzt die B-Zellzahl als direktes Mass für die klinischen Auswirkungen verfolgen (siehe Rubrik «Eigenschaften/Wirkungen»).
Übliche Dosierung
Therapieeinleitung
Die empfohlene Initialdosis ist eine intravenöse Infusion von 300 mg (3 Durchstechflaschen mit je 100 mg), gefolgt von einer zweiten intravenösen Infusion von 300 mg 2 Wochen später.
Erhaltungstherapie
Die empfohlene Erhaltungsdosis beträgt 300 mg als intravenöse Infusion alle 6 Monate. Inebilizumab ist für die langfristige Behandlung bestimmt.
Prämedikation gegen infusionsbedingte Reaktionen
Infektionsbewertung
Vor jeder Infusion von Inebilizumab ist zu prüfen, ob eine klinisch signifikante Infektion vorliegt. Im Falle einer Infektion ist die Infusion von Inebilizumab bis zum Abklingen der Infektion zu verschieben.
Erforderliche Prämedikation
Eine Prämedikation mit einem Kortikosteroid (z.B. Methylprednisolon 80-125 mg intravenös oder gleichwertig) wird etwa 30 Minuten vor jeder Inebilizumab-Infusion verabreicht, ein Antihistaminikum (z.B. Diphenhydramin 25-50 mg oral oder gleichwertig) sowie ein fiebersenkendes Mittel (z.B. Paracetamol 500-650 mg oral oder gleichwertig) werden etwa 30-60 Minuten vor jeder Inebilizumab-Infusion verabreicht (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Spezielle Dosierungsanweisungen
Patienten mit Leber- und Nierenfunktionsstörungen
Inebilizumab wurde nicht an Patienten mit schweren Nieren- oder Leberfunktionsstörungen untersucht. Eine Dosisanpassung auf Grundlage der Nieren- oder Leberfunktion ist jedoch nicht erforderlich, da monoklonale Immunglobulin(Ig)-G-Antikörper nicht primär über die Nieren oder Leber abgebaut werden (siehe Rubrik «Pharmakokinetik»).
Ältere Patienten
Inebilizumab wurde in klinischen Studien 6 älteren Patienten (≥65 Jahre) verabreicht. Auf Basis der begrenzten verfügbaren Daten scheint eine Dosisanpassung bei Patienten über 65 Jahren nicht erforderlich (siehe Rubrik «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Inebilizumab bei Kindern und Jugendlichen im Alter von 0 bis 18 Jahren ist noch nicht erwiesen. Es liegen keine Daten vor.
Verspätete Dosisgabe
Wurde eine Infusion von Inebilizumab verabsäumt, sollte sie so schnell wie möglich nachgeholt und nicht bis zur nächsten geplanten Dosis aufgeschoben werden.
Art der Anwendung
Zur intravenösen Anwendung.
Die Durchstechflaschen nicht schütteln.
Die Durchstechflaschen aufrecht lagern.
Die zubereitete Lösung wird intravenös über eine Infusionspumpe verabreicht, und zwar über eine intravenöse Infusionsleitung mit einem sterilen 0,2- oder 0,22-µm-Inline-Filter mit geringer Proteinbindung gemäss dem Schema in Tabelle 1 mit ansteigender Infusionsrate bis zur vollständigen Gabe (ca. 90 Minuten).
Tabelle 1. Empfohlene Infusionsgeschwindigkeit für die Verabreichung bei Verdünnung in einem 250-ml-Infusionsbeutel
|
Verstrichene Zeit (Minuten)
|
Infusionsrate (ml/Stunde)
| |
0-30
|
42
| |
31-60
|
125
| |
61 – Abschluss der Infusion
|
333
|
Hinweise zur Verdünnung des Arzneimittels vor der Anwendung, siehe Abschnitt «Hinweise für die Handhabung».
Kontraindikationen·Überempfindlichkeit gegen den Wirkstoff oder einen der in Rubrik «Zusammensetzung» genannten Hilfsstoffe
·Schwere aktive Infektion, einschliesslich aktiver chronischer Infektionen wie Hepatitis B
·Aktive oder unbehandelte latente Tuberkulose
·Progressive multifokale Leukoenzephalopathie (PML) in der Anamnese
·Stark immunsupprimierter Zustand
·Aktive Malignome
Warnhinweise und VorsichtsmassnahmenInfusionsbedingte Reaktionen und Überempfindlichkeit
Inebilizumab kann infusionsbedingte Reaktionen und Überempfindlichkeitsreaktionen hervorrufen; diese können Kopfschmerzen, Übelkeit, Somnolenz, Dyspnoe, Fieber, Myalgie, Hautausschlag und andere Symptome umfassen. Infusionsbedingte Reaktionen traten am häufigsten bei der ersten Infusion auf, wurden aber auch bei nachfolgenden Infusionen beobachtet. Obgleich seltend, traten in klinischen Studien mit Inebilizumab schwere Infusionsreaktionen auf (siehe Rubrik «Unerwünschte Wirkungen»).
Vor der Infusion
Eine Prämedikation mit einem Kortikosteroid (z.B. Methylprednisolon 80-125 mg intravenös oder gleichwertig), einem Antihistaminikum (z.B. Diphenhydramin 25-50 mg oral oder gleichwertig) und einem fiebersenkenden Mittel (z.B. Paracetamol 500-650 mg oral oder gleichwertig) sollte verabreicht werden (siehe Rubrik «Dosierung/Anwendung»). In der Zulassungsstudie wurde bei Einleitung der Behandlung mit Inebilizumab eine 2-wöchige orale Kortikosteroidtherapie (plus eine 1-wöchige Ausschleichphase) verabreicht (siehe Rubrik «Eigenschaften/Wirkungen»).
Während der Infusion
Der Patient muss hinsichtlich infusionsbedingte Reaktionen überwacht werden. Die Empfehlungen zur Behandlung von Infusionsreaktionen hängen von der Art und dem Schweregrad der Reaktion ab. Bei lebensbedrohlichen Infusionsreaktionen muss die Behandlung unverzüglich und dauerhaft abgebrochen und eine entsprechende zusätzliche Behandlung eingeleitet werden. Bei weniger schweren Infusionsreaktionen kann die Behandlung darin bestehen, die Infusion vorübergehend zu stoppen, die Infusionsrate zu verringern und/oder eine symptomatische Behandlung durchzuführen.
Nach der Infusion
Der Patient muss nach Beendigung der Infusion mindestens eine Stunde lang hinsichtlich Infusionsreaktionen überwacht werden.
Infektionen
Entsprechend dem Wirkmechanismus der B-Zell-Depletion führt Inebilizumab zu einer Verringerung der Lymphozytenzahl und der Ig-Spiegel im peripheren Blut. Auch über eine Verringerung der Neutrophilenzahl wurde berichtet. Daher kann Inebilizumab die Infektionsanfälligkeit erhöhen (siehe Rubrik «Unerwünschte Wirkungen»).
Vor Beginn der Behandlung mit Inebilizumab (d.h. innerhalb von 6 Monaten) müssen ein aktuelles grosses Blutbild (einschliesslich Differentialblutbild) und Immunglobuline bestimmt werden. Es wird empfohlen, das grosse Blutbild (einschliesslich Differentialblutbild) und die Immunglobuline auch während und nach Absetzen der Behandlung bis zur vollständigen Erholung der B-Zellen in regelmässigen Abständen zu bestimmen. Vor jeder Infusion von Inebilizumab ist zu bewerten, ob eine klinisch signifikante Infektion vorliegt. Im Falle einer Infektion muss die Infusion von Inebilizumab so lange verschoben werden, bis die Infektion abgeklungen ist. Die Patienten sind darauf hinzuweisen, dass sie sich bei Symptomen einer Infektion unverzüglich an ihren Arzt wenden. Ein Behandlungsabbruch ist zu erwägen, wenn ein Patient eine schwere opportunistische Infektion oder wiederkehrende Infektionen entwickelt und die Ig-Werte auf eine geschwächte Immunabwehr hinweisen.
Zu den häufigsten Infektionen, die von den mit Inebilizumab behandelten NMOSD-Patienten während der randomisierten kontrollierten Phase (randomised controlled period, RCP) und der Open-Label-Phase (OLP) gemeldet wurden, gehörten Harnwegsinfektionen (26,2 %), Nasopharyngitis (20,9 %), Infektionen der oberen Atemwege (15,6 %), Grippe (8,9 %) und Bronchitis (6,7 %).
Hepatitis B-Virus-Reaktivierung
Das Risiko für eine HBV-Reaktivierung wurde auch bei anderen B-Zell-depletierenden Antikörpern beobachtet. Patienten mit einer chronischen HBV-Erkrankung wurden von klinischen Studien mit Inebilizumab ausgeschlossen. Ein HBV-Screening sollte bei allen Patienten vor Beginn der Behandlung mit Inebilizumab durchgeführt werden. Inebilizumab darf nicht an Patienten verabreicht werden, die an einer durch HBV ausgelösten aktiven Hepatitis leiden und bei denen das Hepatitis-B-Oberflächenantigen (HBsAg) oder die Hepatitis-B-Core-Antikörper (HBcAb) positiv sind. Patienten, die chronische HBV-Träger sind [HBsAg+], sollten vor Beginn und während der Behandlung durch einen Facharzt für Lebererkrankungen betreut werden (siehe Rubrik «Kontraindikationen»).
Hepatitis-C-Virus
HCV-positive Patienten wurden von klinischen Studien mit Inebilizumab ausgeschlossen. Vor Beginn der Inebilizumab-Behandlung ist ein Screening zum Ausgangszeitpunkt auf HCV erforderlich, damit eine Infektion erkannt und ggf. eine Behandlung eingeleitet werden kann.
Tuberkulose
Vor Beginn der Behandlung mit Inebilizumab sind die Patienten auf aktive Tuberkulose zu untersuchen und auf eine latente Infektion zu testen. Bei Patienten mit aktiver Tuberkulose oder positivem Tuberkulose-Screening ohne entsprechende Behandlung in der Anamnese ist ein Facharzt für Infektionskrankheiten zu konsultieren, bevor eine Behandlung mit Inebilizumab begonnen wird.
Progressive multifokale Leukoenzephalopathie (PML)
PML ist eine opportunistische Virusinfektion des Gehirns, die durch das John-Cunningham-Virus (JCV) verursacht wird und typischerweise bei Patienten mit geschwächter Immunabwehr auftritt. Sie kann zum Tod oder zu schwerer Behinderung führen. Eine JCV-Infektion, die zu PML führte, wurde bei Patienten beobachtet, die mit anderen B-Zell-depletierenden Antikörpern behandelt wurden.
In den klinischen Studien mit Inebilizumab verstarb ein Studienteilnehmer an der Entwicklung neuer Hirnläsionen, für die keine definitive Diagnose gestellt werden konnte. Die Differentialdiagnose lautete jedoch auf atypischen NMOSD-Schub, PML oder akute disseminierte Enzephalomyelitis.
Ärzte sollten auf klinische Symptome oder Magnetresonanztomographie (MRT)-Befunde achten, die auf eine PML hindeuten könnten. MRT-Befunde können schon vor dem Auftreten klinischer Anzeichen oder Symptome erkennbar sein. Die typischen Symptome im Zusammenhang mit PML sind vielfältig und können über Tage bis Wochen voranschreiten. Dazu gehören fortschreitende Schwäche auf einer Körperseite oder schwerfälligen Bewegungen der Extremitäten, Sehstörungen sowie Veränderungen des Denkens, des Erinnerungsvermögens und der Orientierung, die zu Verwirrtheit und Persönlichkeitsveränderungen führen.
Bei den ersten Anzeichen oder Symptomen, die auf eine PML hindeuten, ist die Behandlung mit Inebilizumab so lange auszusetzen, bis eine PML ausgeschlossen wurde. Weitere Untersuchungen sollten in Erwägung gezogen werden, einschliesslich einer neurologischen Konsultation, einer MRT-Untersuchung, möglichst mit Kontrastmittel, einer Liquoruntersuchung auf JC-Virus-DNA sowie wiederholten neurologischen Tests. Bei Bestätigung ist die Behandlung mit Inebilizumab abzubrechen.
Späte Neutropenie
Es wurde über Fälle von spät einsetzender Neutropenie berichtet (siehe Rubrik «Unerwünschte Wirkungen»). Obwohl einige Fälle mit Grad 3 eingestuft wurden, war die Mehrzahl der Fälle vom Grad 1 oder 2. Es wurde über Fälle von spät einsetzender Neutropenie berichtet, die mindestens 4 Wochen nach der letzten Infusion von Inebilizumab auftraten. Bei Patienten, die Anzeichen und Symptome einer Infektion aufweisen, wird eine Messung der neutrophilen Granulozyten im Blut empfohlen.
Behandlung von schwer immungeschwächten Patienten
Patienten mit einer stark eingeschränkten Immunabwehr dürfen so lange nicht behandelt werden, bis der Zustand abgeklungen ist (siehe Rubrik «Kontraindikationen»).
Inebilizumab wurde nicht zusammen mit anderen Immunsuppressiva untersucht. Bei Kombination mit einer anderen immunsuppressiven Therapie ist das Risiko einer verstärkten immunsuppressiven Wirkung zu beachten.
Patienten mit einer bekannten angeborenen oder erworbenen Immunschwäche, einschliesslich HIV-Infektion oder Splenektomie, wurden nicht untersucht.
Impfungen
Sämtliche Impfungen sollten gemäss den Impfempfehlungen mindestens 4 Wochen vor Beginn der Behandlung mit Inebilizumab verabreicht werden. Die Wirksamkeit und Sicherheit einer Immunisierung mit Lebendimpfstoffen oder abgeschwächten Lebendimpfstoffen im Anschluss an eine Inebilizumab-Therapie wurde nicht untersucht. Eine Impfung mit abgeschwächten Lebendimpfstoffen oder Lebendimpfstoffen wird während der Behandlung und bis zur vollständigen Erholung der B-Zellen nicht empfohlen.
Säuglingen von Müttern, die während der Schwangerschaft Inebilizumab erhalten haben, dürfen keine Lebendimpfstoffe oder abgeschwächte Lebendimpfstoffe verabreicht werden, bevor nicht die Erholung der B-Zellzahlen des Säuglings bestätigt wurde. Die B-Zell-Depletion bei diesen exponierten Säuglingen kann die Risiken von Lebendimpfstoffen oder abgeschwächten Lebendimpfstoffen erhöhen. Nicht-Lebendimpfstoffe können je nach Indikation auch vor der Erholung von der B-Zell- und Ig-Depletion verabreicht werden. Es sollte jedoch ein qualifizierter Facharzt zu Rate gezogen werden, um zu beurteilen, ob eine schützende Immunantwort aufgebaut wurde.
B-Zell-Repletionszeit
Die Zeit bis zur vollständigen Erholung der B-Zellen nach der Verabreichung von Inebilizumab ist nicht bekannt. Eine B-Zell-Depletion unterhalb der unteren Normgrenze blieb bei 94 % der Patienten für mindestens 6 Monate nach der Behandlung bestehen.
Schwangerschaft
Aus Vorsichtsgründen soll eine Anwendung von Inebilizumab während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, vermieden werden (siehe Rubrik «Schwangerschaft, Stillzeit»). Patientinnen sind darauf hinzuweisen, dass sie ihren Arzt informieren, falls sie schwanger sind oder planen, während der Anwendung von Inebilizumab schwanger zu werden. Frauen im gebärfähigen Alter sollten während der Behandlung mit Uplizna und bis 6 Monate nach der letzten Verabreichung von Uplizna eine wirksame Methode (d.h. eine Methode mit einer Schwangerschaftsrate unter 1 %) zur Empfängnisverhütung anwenden.
Malignome
Immunmodulatorische Arzneimittel können das Risiko einer malignen Erkrankung erhöhen. Auf der Grundlage der begrenzten Erfahrungen mit Inebilizumab bei NMOSD (siehe Rubrik «Unerwünschte Wirkungen») scheinen die derzeitigen Daten nicht auf ein erhöhtes Risiko für Malignome hinzudeuten. Ein mögliches Risiko für die Entwicklung solider Tumoren kann jedoch derzeit nicht ausgeschlossen werden.
Natriumgehalt
Dieses Arzneimittel enthält 48,3 mg Natrium pro Dosis, entsprechend 2 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
InteraktionenEs wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Der primäre Ausscheidungsweg für therapeutische Antikörper ist die Elimination durch das retikuloendotheliale System. Cytochrom-P450-Enzyme, Efflux-Pumpen und Proteinbindungsmechanismen sind an der Ausscheidung therapeutischer Antikörper nicht beteiligt. Aus diesem Grund ist das potenzielle Risiko für pharmakokinetische Wechselwirkungen zwischen Inebilizumab und anderen Arzneimitteln gering.
Impfungen
Die Wirksamkeit und Sicherheit einer Immunisierung mit Lebendimpfstoffen oder abgeschwächten Lebendimpfstoffen nach einer Inebilizumab-Therapie wurde nicht untersucht. Die Reaktion auf eine Impfung könnte beeinträchtigt sein, wenn die B-Zellen erschöpft sind. Es wird empfohlen, dass die Patienten erforderliche Impfungen vor Beginn der Inebilizumab-Therapie abschliessen (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Immunsuppressiva
Inebilizumab wurde als Monotherapie für diese Indikation getestet und soll auch als solche eingesetzt werden. Es liegen keine Daten über die Sicherheit oder Wirksamkeit von Inebilizumab in Kombination mit anderen Immunsuppressiva vor. In der Zulassungsstudie erhielten alle Teilnehmer nach der ersten Verabreichung von Inebilizumab eine zweiwöchige Behandlung mit oralen Kortikosteroiden (plus einer einwöchigen Ausschleichphase).
Die gleichzeitige Anwendung von Inebilizumab mit Immunsuppressiva, einschliesslich systemischer Kortikosteroide, kann das Infektionsrisiko erhöhen. Die Auswirkungen von Inebilizumab auf B-Zellen und Immunglobuline können noch 6 Monate oder länger nach der Verabreichung anhalten.
Bei der Einleitung von Inebilizumab im Anschluss an andere immunsuppressive Behandlungen mit verlängerten immunologischen Wirkungen oder bei der Einleitung anderer immunsuppressiver Therapien mit verlängerten immunologischen Wirkungen im Anschluss an Inebilizumab müssen die Wirkungsdauer und der Wirkmechanismus dieser Arzneimittel wegen der potenziellen zusätzlichen immunsuppressiven Wirkung berücksichtigt werden (siehe Rubrik «Eigenschaften/Wirkungen»).
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Frauen im gebärfähigen Alter sollten während der Behandlung mit Uplizna und bis 6 Monate nach der letzten Verabreichung von Uplizna eine wirksame Methode zur Empfängnisverhütung anwenden (d.h. eine Methode mit einer Schwangerschaftsrate von weniger als 1 %).
Schwangerschaft
Es liegen nur begrenzte Daten über die Anwendung von Inebilizumab bei Schwangeren vor. Inebilizumab ist ein humanisierter monoklonaler IgG1-Antikörper und es ist bekannt, dass Immunglobuline die Plazentaschranke passieren können. Bei Säuglingen von Müttern, die während der Schwangerschaft mit anderen B-Zell-depletierenden Antikörpern behandelt wurden, wurde eine vorübergehende periphere B-Zell-Depletion und Lymphozytopenie berichtet.
In Bezug auf eine Reproduktionstoxizität deuten tierexperimentelle Untersuchungen nicht auf eine direkte oder indirekte gesundheitsschädliche Wirkung hin. Sie haben jedoch eine Verminderung der B-Zellen in der fetalen Leber der Nachkommen gezeigt (siehe Rubrik «Präklinische Daten»).
Eine Behandlung mit Inebilizumab während der Schwangerschaft ist zu vermeiden, es sei denn, der voraussichtliche Nutzen für die Mutter überwiegt das potenzielle Risiko für den Fötus.
Im Falle einer Exposition während der Schwangerschaft ist aufgrund der pharmakologischen Eigenschaften des Präparats und der Ergebnisse aus tierexperimentellen Untersuchungen mit einer B-Zell-Depletion beim Neugeborenen zu rechnen (siehe Rubrik «Präklinische Daten»). Die mögliche Dauer der B-Zell-Depletion bei Säuglingen, die Inebilizumab in utero ausgesetzt waren, ist nicht bekannt. Auch die Auswirkungen der B-Zell-Depletion auf die Sicherheit und Wirksamkeit von Impfstoffen sind nicht bekannt (siehe Rubriken «Warnhinweise und Vorsichtsmassnahmen» und «Eigenschaften/Wirkungen»). Folglich sollten Neugeborene auf eine B-Zell-Depletion hin überwacht werden, und Impfungen mit Lebendvirus-Impfstoffen, wie z.B. Bacillus-Calmette-Guérin (BCG)-Impfstoff, sollten so lange verschoben werden, bis sich die B-Zellzahl des Säuglings erholt hat (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Stillzeit
Die Anwendung von Inebilizumab bei stillenden Frauen wurde nicht untersucht. Es ist nicht bekannt, ob Inebilizumab in die Muttermilch übertritt. Beim Menschen kommt es in den ersten Tagen nach der Geburt zur Ausscheidung von IgG-Antikörpern in die Muttermilch, die bald darauf auf geringe Konzentrationen zurückgeht.
Daher kann ein Risiko für das gestillte Kind während dieser kurzen Zeit nicht ausgeschlossen werden. Danach kann Uplizna auch während der Stillzeit angewendet werden, wenn dies aus klinischer Sicht notwendig ist. Wenn die Patientin jedoch bis in die letzten Monate der Schwangerschaft mit Uplizna behandelt wurde, kann unmittelbar nach der Geburt mit dem Stillen begonnen werden.
Fertilität
Es liegen nur begrenzte Daten über die Auswirkungen von Inebilizumab auf die Fortpflanzungsfähigkeit beim Menschen vor. Untersuchungen an Tieren haben jedoch eine verminderte Fruchtbarkeit gezeigt. Die klinische Bedeutung dieser präklinischen Befunde ist nicht bekannt (siehe Rubrik «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDie pharmakologische Wirkung und die bisher gemeldeten Nebenwirkungen deuten darauf hin, dass Inebilizumab keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen hat.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Zu den häufigsten unerwünschten Wirkungen, die von den mit Inebilizumab behandelten Patienten während der randomisierten klinischen Phase (RCP) und der Open-Label-Phase (OLP) gemeldet wurden, gehörten Harnwegsinfektionen (26,2 %), Nasopharyngitis (20,9 %), Infektionen der oberen Atemwege (15,6 %), Arthralgie (17,3 %) und Rückenschmerzen (13,8 %).
Die am häufigsten gemeldeten schwerwiegenden unerwünschten Wirkungen bei den mit Inebilizumab behandelten Patienten in der RCP und OLP waren Infektionen (11,1 %) (darunter Harnwegsinfektionen (4,0 %), Pneumonie (1,8 %) und NMOSD (1,8 %)).
Liste der unerwünschten Wirkungen
Die in den klinischen Studien von Inebilizumab bei NMOSD gemeldeten unerwünschten Wirkungen sind in Tabelle 2 nach den folgenden Häufigkeitskategorien aufgeführt: sehr häufig (≥1/10), häufig (≥1/100 bis < 1/10), gelegentlich (≥1/1'000 bis < 1/100), selten (≥1/10'000 bis < 1/1'000), sehr selten (< 1/10'000), nicht bekannt (auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 2. Unerwünschte Wirkungen
|
MedDRA-Systemorganklasse
|
Unerwünschte Wirkung
|
Häufigkeit
| |
Infektionen und parasitäre Erkrankungen
|
Harnwegsinfektion (26,2 %),
Atemwegsinfektion (15,6 %),
Nasopharyngitis (20,9 %),
|
Sehr häufig
| |
|
Pneumonie, Grippe, Zellulitis, Herpes zoster, Sinusitis
|
Häufig
| |
|
Sepsis, subkutaner Abszess, Bronchiolitis
|
Gelegentlich
| |
Erkrankungen des Blutes und des Lymphsystems
|
Lymphopenie, Neutropenie, Neutropenie mit später Manifestation
|
Häufig
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
|
Arthralgie (17,3 %), Rückenschmerzen (13,8 %)
|
Sehr häufig
| |
Untersuchungen
|
Immunglobuline erniedrigt (3,8% - 29,3%)
|
Sehr häufig
| |
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
|
Infusionsbedingte Reaktion (12,9 %)
|
Sehr häufig
|
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Infusionsbedingte Reaktionen
Inebilizumab kann infusionsbedingte Reaktionen hervorrufen, darunter Kopfschmerzen, Übelkeit, Somnolenz, Dyspnoe, Fieber, Myalgie, Hautausschlag und andere Symptome. Alle Patienten erhielten eine Prämedikation. Infusionsreaktionen wurden bei 9,2 % der NMOSD-Patienten während des ersten Behandlungszyklus mit Inebilizumab beobachtet, gegenüber 10,7 % der mit Placebo behandelten Patienten. Infusionsbedingte Reaktionen traten am häufigsten bei der ersten Infusion auf, wurden aber auch bei nachfolgenden Infusionen beobachtet. Die Mehrzahl der infusionsbedingten Reaktionen, die bei mit Inebilizumab behandelten Patienten gemeldet wurden, war leicht oder mittelschwer.
Infektionen
Eine Infektion wurde von 74,7 % der mit Inebilizumab behandelten NMOSD-Patienten in der RCP und OLP gemeldet. Zu den häufigsten Infektionen gehörten Harnwegsinfektionen (26,2 %), Nasopharyngitis (20,9 %), Infektionen der oberen Atemwege (15,6 %), Grippe (8,9 %) und Bronchitis (6,7 %). Schwerwiegende Infektionen, die bei mehr als einem mit Inebilizumab behandelten Patienten auftraten, waren Harnwegsinfektionen (4,0 %) und Pneumonie (1,8 %). Siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen» bezüglich des Vorgehens im Falle einer Infektion.
Opportunistische und schwerwiegende Infektionen
Während der RCP traten in keiner der beiden Behandlungsgruppen opportunistische Infektionen auf, und bei einem mit Inebilizumab behandelten Patienten trat eine einzige infektiöse unerwünschte Wirkung von Grad 4 (atypische Pneumonie) auf. Während der OLP traten bei 2 mit Inebilizumab behandelten Patienten (0,9 %) opportunistische Infektionen auf (von denen eine nicht bestätigt wurde) und bei 3 mit Inebilizumab behandelten Patienten (1,4 %) wurde eine infektiöse unerwünschte Wirkung von Grad 4 festgestellt. Siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen» bezüglich des Vorgehens im Falle einer Infektion.
Laboranomalien
Erniedrigte Immunglobulinspiegel
In Einklang mit dem Wirkmechanismus des Arzneimittels verringerten sich die durchschnittlichen Immunglobulinspiegel bei der Anwendung von Inebilizumab. Am Ende der 6,5-monatigen RCP war der Anteil der Patienten mit Werten unterhalb des unteren Normwerts wie folgt: IgA 9,8 % Inebilizumab und 3,1 % Placebo, IgE 10,6 % Inebilizumab und 12,5 % Placebo, IgG 3,8 % Inebilizumab und 9,4 % Placebo und IgM 29,3 % Inebilizumab und 15,6 % Placebo. Es wurde eine einzige unerwünschte Wirkung einer IgG-Verringerung gemeldet (Grad 2, während der OLP). Der Anteil der mit Inebilizumab behandelten Patienten mit IgG-Spiegeln unterhalb des unteren Normwerts lag im ersten Jahr bei 7,4 % und im zweiten Jahr bei 9,9 %. Bei einer medianen Exposition von 3,2 Jahren betrug die Häufigkeit einer moderaten IgG-Senkung (300 bis < 500 mg/dl) bei 14,2 % und die Häufigkeit einer gravierenden IgG-Senkung (< 300 mg/dl) bei 3,6 %.
Erniedrigte Neutrophilenzahl
Nach einer 6,5-monatigen Behandlung wurden Neutrophilenzahlen von 1,0-1,5 x 109/l (Grad 2) bei 7,5 % der mit Inebilizumab behandelten Patienten beobachtet, gegenüber 1,8 % bei den mit Placebo behandelten Patienten. Neutrophilenzahlen von 0,5-1,0 x 109/l (Grad 3) wurden bei 1,7 % der mit Inebilizumab behandelten Patienten beobachtet, gegenüber 0 % bei jenen mit Placebo. Die Neutropenie war im Allgemeinen vorübergehend und nicht mit schweren Infektionen assoziiert.
Erniedrigte Lymphozytenzahl
Nach einer 6,5-monatigen Behandlung wurde bei Patienten, die mit Inebilizumab behandelt wurden, häufiger eine Abnahme der Lymphozytenzahl beobachtet als bei Patienten, die Placebo erhielten: Lymphozytenzahlen zwischen 500 und < 800/mm3 (Grad 2) wurden bei 21,4 % der mit Inebilizumab behandelten Patienten beobachtet, gegenüber 12,5 % bei jenen mit Placebo. Lymphozytenzahlen zwischen 200 und < 500/mm3 (Grad 3) wurden bei 2,9 % der mit Inebilizumab behandelten Patienten beobachtet, gegenüber 1,8 % der mit Placebo behandelten Patienten. Dieses Ergebnis passt zum Wirkmechanismus der B-Zell-Depletion, da B-Zellen eine Untergruppe der Lymphozytenpopulation sind.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungDie höchste Dosis von Inebilizumab, die bei Autoimmunpatienten untersucht wurde, betrug 1200 mg, verabreicht als zwei intravenöse Infusionen mit 600 mg im Abstand von zwei Wochen. Die Nebenwirkungen waren vergleichbar mit jenen, die in der klinischen Zulassungsstudie mit Inebilizumab beobachtet wurden.
Es gibt kein spezifisches Gegenmittel im Falle einer Überdosierung. Die Infusion muss sofort unterbrochen und der Patient auf infusionsbedingte Reaktionen überwacht werden (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»). Der Patient ist engmaschig auf Anzeichen oder Symptome von Nebenwirkungen zu überwachen, und bei Bedarf muss eine entsprechende unterstützende Behandlung eingeleitet werden.
Eigenschaften/WirkungenATC-Code
L04AA47
Wirkungsmechanismus
Inebilizumab ist ein monoklonaler Antikörper, der spezifisch an CD19, ein Zelloberflächenantigen auf Prä-B- und reifen B-Zell-Lymphozyten (einschliesslich Plasmablasten und einigen Plasmazellen) bindet. Nach der Bindung an die Zelloberfläche von B-Lymphozyten unterstützt Inebilizumab die Antikörper-abhängige zelluläre Zytolyse (antibody-dependent cellular cytolysis, ADCC) und die Antikörper-abhängige zelluläre Phagozytose (antibody-dependent cellular phagocytosis, ADCP). Man geht davon aus, dass B-Zellen eine zentrale Rolle bei der Pathogenese von NMOSD spielen. Der genaue Mechanismus, durch den Inebilizumab seine therapeutische Wirkung bei NMOSD entfaltet, ist nicht bekannt, aber es wird vermutet, dass er mit einer B-Zell-Depletion einhergeht und möglicherweise die Unterdrückung der Antikörpersekretion, Antigenpräsentation, B-Zell-T-Zell-Interaktion sowie der Produktion von Entzündungsmediatoren umfasst.
Pharmakodynamik
Die Pharmakodynamik von Inebilizumab wurde mit einem Assay für CD20+-B-Zellen untersucht, da Inebilizumab mit dem Assay für CD19+-B-Zellen interferieren kann. Die Behandlung mit Inebilizumab verringert bis 8 Tage nach der Infusion die Anzahl der CD20+-B-Zellen im Blut. In einer klinischen Studie an 174 Patienten wurde die Anzahl der CD20+-B-Zellen bei 100 % der mit Inebilizumab behandelten Patienten innerhalb von 4 Wochen auf Werte unterhalb des unteren Normwerts gesenkt und blieb bei 94 % der Patienten 28 Wochen nach Behandlungsbeginn unterhalb des unteren Normwerts. Die Zeit bis zur vollständigen Erholung der B-Zellen nach der Verabreichung von Inebilizumab ist nicht bekannt.
In der Zulassungsstudie mit NMOSD-Patienten wurden bei 14,7 % der Patienten am Ende der OLP Anti-Arzneimittel-Antikörper (anti-drug antibodies, ADA) nachgewiesen; die Gesamthäufigkeit der unter der Behandlung auftretenden ADA lag bei 7,1 % (16 von 225) und das Auftreten und der Titer zu ADA-positiven Messzeitpunkten nahm im Laufe der Inebilizumab-Behandlung ab. Der positive ADA-Status hatte offenbar keine klinisch relevanten Auswirkungen auf die PK- und PD-(B-Zell)-Parameter und keinen Einfluss auf das langfristige Sicherheitsprofil. Es gab keine erkennbaren Auswirkungen des ADA-Status auf das Wirksamkeitsergebnis. Angesichts der geringen Inzidenz von ADA im Zusammenhang mit der Inebilizumab-Behandlung kann die Auswirkung jedoch nicht vollständig bewertet werden.
Klinische Wirksamkeit
Die Wirksamkeit von Inebilizumab zur Behandlung von NMOSD wurde in einer randomisierten (3:1), doppelblinden, placebokontrollierten klinischen Studie an Erwachsenen mit AQP4-IgG-seropositiver oder -seronegativer NMOSD untersucht. An der Studie nahmen Patienten teil, die im Jahr zuvor mindestens einen akuten NMOSD-Schub oder in den letzten zwei Jahren mindestens zwei Schübe erlitten hatten, der/die eine Notfallbehandlung (z.B. Steroide, Plasmaaustausch, intravenöses Immunglobulin) erforderte(n), und die einen EDSS (Expanded Disability Severity Scale)-Score von ≤7,5 aufwiesen (Patienten mit einem Score von 8,0 waren teilnahmeberechtigt, sofern die Patienten in angemessener Weise zur Teilnahme in der Lage waren). Patienten waren von der Studienteilnahme ausgeschlossen, wenn sie zuvor innerhalb eines für jede der entsprechenden Therapien festgelegten Intervalls mit Immunsuppressiva behandelt worden waren. Immunsuppressive Hintergrundtherapien zur Vorbeugung von NMOSD-Schüben waren nicht zulässig. In der Zulassungsstudie wurde bei Einleitung der Behandlung mit Inebilizumab eine 2-wöchige orale Kortikosteroidtherapie (plus eine 1-wöchige Ausschleichphase) verabreicht.
Die Patienten erhielten Inebilizumab 300 mg oder das entsprechende Placebo als intravenöse Infusion an Tag 1 und Tag 15 und wurden anschliessend über einen Zeitraum von bis zu 197 Tagen oder bis zu einem bestätigten Schub beobachtet; dies wurde als randomisiert-kontrollierte Phase (RCP) bezeichnet. Alle potenziellen Schübe wurden von einem verblindeten, unabhängigen Beurteilungsausschuss (Adjudication Committee, AC) bewertet, der feststellte, ob der Schub die im Prüfplan definierten Kriterien erfüllte. Die Schubkriterien berücksichtigten Schübe in allen von der NMOSD betroffenen Bereichen (Optikusneuritis, Myelitis, Gehirn und Hirnstamm). Sie enthielten Kriterien, die sich ausschliesslich auf wesentliche klinische Manifestationen stützen, sowie Kriterien, bei denen leichtere klinische Befunde durch den Einsatz von MRT ergänzt wurden (siehe Tabelle 3).
Tabelle 3. Überblick über die im Prüfplan definierten Kriterien für einen NMOSD-Schub
|
Bereich
|
Repräsentative Symptome
|
Rein klinische Befunde
|
Klinische PLUS radiologische Befunde
| |
Sehnerv
|
Verschwommenes Sehen
Sehverlust
Augenschmerzen
|
8 Kriterien auf Grundlage von Veränderungen der Sehschärfe oder des relativen afferenten Pupillendefekts (RAPD)
|
3 Kriterien auf Grundlage von Veränderungen der Sehschärfe oder des RAPD plus Vorliegen entsprechender MRT-Befunde des Sehnervs
| |
Rückenmark
|
Tiefer oder Wurzelschmerz
Parästhesien der Extremitäten
Schwäche
Funktionsstörung des Schliessmuskels
Lhermitte-Zeichen (nicht isoliert)
|
2 Kriterien auf Grundlage von Veränderungen in den Funktionswerten des pyramidalen Systems, von Blase/Darm oder Sensorik
|
2 Kriterien auf Grundlage von Veränderungen in den Funktionswerten des pyramidalen Systems, von Blase/Darm oder Sensorik PLUS entsprechender MRT-Befunde
| |
Hirnstamm
|
Übelkeit
Unbehandelbares Erbrechen
Hartnäckiger Schluckauf
Sonstige neurologische Anzeichen (z.B. Doppeltsehen, Dysarthrie, Dysphagie, Vertigo, okulomotorische Lähmung, Schwäche, Nystagmus, andere Hirnnervenanomalien)
|
Keine
|
2 Kriterien auf Grundlage von Symptomen oder Veränderungen der Funktionswerte von Hirnstamm/ Kleinhirn PLUS entsprechender MRT-Befunde des Hirnstamms
| |
Gehirn
|
Enzephalopathie
Hypothalamus-Funktionsstörung
|
Keine
|
1 Kriterium auf Grundlage von Veränderungen der zerebralen/sensorischen/ pyramidalen Funktionswerte PLUS entsprechender MRT-Befunde des Gehirns
|
Patienten, die während der RCP einen durch den AC bestätigten Schub erlitten oder die den Besuch an Tag 197 ohne Schub abschlossen, verliessen die RCP und hatten die Möglichkeit, in eine Open-Label-Phase (OLP) aufgenommen zu werden und die Behandlung mit Inebilizumab zu beginnen bzw. fortzusetzen.
Insgesamt wurden 230 Patienten in die Studie aufgenommen: 213 Patienten waren AQP4-IgG-seropositiv und 17 waren -seronegativ; 174 Patienten wurden in der RCP der Studie mit Inebilizumab behandelt und 56 Patienten erhielten Placebo. Von den 213 AQP4-IgG-seropositiven Patienten wurden 161 mit Inebilizumab behandelt und 52 erhielten Placebo in der RCP behandelt. Die Daten zum Ausgangszeitpunkt und Wirksamkeitsergebnisse werden für die AQP4-IgG-seropositiven Patienten dargestellt.
Die demografischen Daten zum Ausgangszeitpunkt und die Krankheitsmerkmale waren in beiden Behandlungsgruppen ausgeglichen (siehe Tabelle 4).
Tabelle 4. Demografische Daten und Ausgangsmerkmale der AQP4-IgG-seropositiven NMOSD-Patienten
|
Merkmal
|
Placebo
N = 52
|
Inebilizumab
N = 161
|
Insgesamt
N = 213
| |
Alter (Jahre): Mittelwert (Standardabweichung [standard deviation, SD])
|
42,4 (14,3)
|
43,2 (11,6)
|
43,0 (12,3)
| |
Alter ≥65 Jahre, n (%)
|
4 (7,7)
|
6 (3,7)
|
10 (4,7)
| |
Geschlecht: männlich, n (%)
|
3 (5,8)
|
10 (6,2)
|
13 (6,1)
| |
Geschlecht: weiblich, n (%)
|
49 (94,2)
|
151 (93,8)
|
200 (93,9)
| |
Erweiterte Skala zur Einstufung einer Behinderung (EDSS): Mittelwert (SD)
|
4,35 (1,63)
|
3,81 (1,77)
|
3,94 (1,75)
| |
Dauer der Erkrankung (in Jahren): Mittelwert (SD)
|
2,92 (3,54)
|
2,49 (3,39)
|
2,59 (3,42)
| |
Anzahl der vorherigen Rückfälle: ≥2, n (%)
|
39 (75,0)
|
137 (85,1)
|
176 (82,6)
| |
Annualisierte Rückfallrate: Mittelwert (SD)
|
1,456 (1,360)
|
1,682 (1,490)
|
1,627 (1,459)
|
Bei auftretenden NMOSD-Schüben wurde je nach Bedarf eine Notfalltherapie eingeleitet. Alle Patienten erhielten vor der Verabreichung des Prüfpräparats eine Prämedikation, um das Risiko infusionsbedingter Reaktionen zu verringern.
Der primäre Wirksamkeitsendpunkt war die Zeit (in Tagen) von Tag 1 bis zum Auftreten eines durch den AC bestätigten NMOSD-Schubs an oder vor Tag 197. Weitere wesentliche sekundäre Endpunkte waren die Verschlechterung des EDSS beim letzten Besuch während der RCP gegenüber dem Ausgangszeitpunkt, die Veränderung des binokularen Sehschärfe-Scores im Niedrigkontrastbereich gegenüber dem Ausgangszeitpunkt (gemessen mithilfe des kontrastarmem Landholtrings) beim letzten Besuch während der RCP, die kumulative Gesamtzahl aktiver MRT-Läsionen (neue Gadolinium-anreichernde oder neue/sich vergrössernde T2-Läsionen) während der RCP sowie die Anzahl der NMOSD-bedingten stationären Krankenhausaufenthalte. Eine Verschlechterung des EDSS-Scores lag vor, wenn eines der folgenden Kriterien erfüllt war: (1) Verschlechterung des EDSS-Scores um 2 oder mehr Punkte bei einem Ausgangswert von 0; (2) Verschlechterung des EDSS-Scores um 1 oder mehr Punkte bei Patienten mit einem Ausgangswert von 1 bis 5; (3) Verschlechterung des EDSS-Scores um 0,5 Punkte oder mehr bei Patienten mit einem Ausgangswert von 5,5 oder höher. Obwohl während der OLP keine Vergleichsgruppe zur Verfügung stand, wurde die annualisierte Schubrate sowohl für die randomisierte als auch für die unverblindete Behandlung ermittelt.
Die Ergebnisse der AQP4-IgG-seropositiven Patienten sind in Tabelle 5 und Abbildung 1 dargestellt. In dieser Studie reduzierte die Behandlung mit Inebilizumab statistisch signifikant das Risiko eines durch den AC bestätigten NMOSD-Schubs gegenüber Placebo (Hazard Ratio: 0,227, p < 0,0001; Verringerung des Risikos eines durch den AC bestätigten NMOSD-Schubs um 77,3 %) bei AQP4-IgG-seropositiven Patienten. Bei AQP4-IgG-seronegativen Patienten wurde kein Behandlungsnutzen festgestellt.
In der Inebilizumab-Gruppe war die Verschlechterung des EDDS signifikant geringer als in der Placebo-Gruppe (14,9 % gegenüber 34,6 % der Teilnehmer). Es gab keine Unterschiede in der binokularen Sehschärfe bei geringem Kontrast zwischen den Studienarmen. Die mittlere Gesamtanzahl der aktiven MRT-Läsionen (1,7 gegenüber 2,3) und die mittlere Gesamtanzahl der NMOSD-bedingten Krankenhausaufenthalte (1,0 gegenüber 1,4) waren in der Inebilizumab-Gruppe geringer.
Tabelle 5. Wirksamkeitsergebnisse der Zulassungsstudie bei NMOSD mit seropositivem AQP4-IgG
|
|
Behandlungsgruppe
| |
|
Placebo
N = 52
|
Inebilizumab
N = 161
| |
Zeit bis zu einem vom Beurteilungsausschuss (AC) festgestellten Schub (primärer Wirksamkeitsendpunkt)
| |
Anzahl (%) der Patienten mit Schub
|
22 (42,3 %)
|
18 (11,2 %)
| |
Hazard Ratio (95 %-KI)a
|
0,227 (0,1214; 0,4232)
| |
p-Werta
|
< 0,0001
|
a Cox-Regressionsmethode, mit Placebo als Referenzgruppe.
Abbildung 1. Kaplan-Meier-Diagramm der Zeit bis zum ersten durch den AC bestätigten NMOSD-Schub während der RCP bei AQP4-IgG-seropositiven Patienten
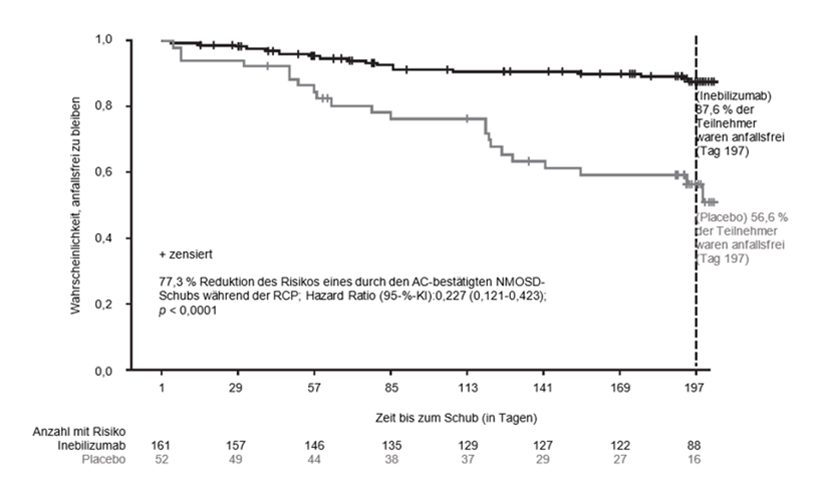
AC: Beurteilungsausschuss; AQP4-IgG: Anti-Aquaporin-4-Immunglobulin G; KI: Konfidenzintervall; NMOSD: Neuromyelitis-optica-Spektrum-Erkrankungen; RCP: Randomisiert-kontrollierte Phase.
Die annualisierte, durch den Beurteilungsausschuss (AC) bestätigte NMOSD-Schubrate wurde im Rahmen der RCP und OLP als sekundärer Endpunkt analysiert. Bei den AQP4-IgG-seropositiven Patienten, die mit Inebilizumab behandelt wurden, lag das Ergebnis bei 0,09.
PharmakokinetikAbsorption
Inebilizumab wird als intravenöse Infusion verabreicht.
Distribution
Auf der Grundlage einer populationspharmakokinetischen Analyse wurde das typische zentrale und periphere Verteilungsvolumen von Inebilizumab auf 2,95 l bzw. 2,57 l geschätzt.
Metabolismus
Inebilizumab ist ein humanisierter monoklonaler IgG1-Antikörper, der durch proteolytische Enzyme abgebaut wird, die im gesamten Körper verteilt sind.
Elimination
Bei erwachsenen Patienten mit NMOSD betrug die terminale Eliminationshalbwertszeit etwa 18 Tage. Aus der populationspharmakokinetischen Analyse ergab sich eine geschätzte systemische Clearance von Inebilizumab über den primären Eliminationsweg von 0,19 l/Tag. Bei niedrigen pharmakokinetischen Expositionswerten unterlag Inebilizumab wahrscheinlich der (CD19)-rezeptorvermittelten Clearance, die mit der Zeit abnahm, vermutlich aufgrund der Depletion von B-Zellen durch die Behandlung mit Inebilizumab.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Es wurden keine formellen klinischen Studien durchgeführt, um die Auswirkungen einer Leberfunktionsstörung auf Inebilizumab zu untersuchen. In klinischen Studien wurden keine Patienten mit schweren Leberfunktionsstörungen mit Inebilizumab behandelt. Monoklonale IgG-Antikörper werden nicht primär über die Leber abgebaut. Es ist daher nicht zu erwarten, dass Veränderungen der Leberfunktion die Clearance von Inebilizumab beeinflussen. Eine populationspharmakokinetische Analyse ergab, dass die Biomarker für die Leberfunktion (Aspartat-Aminotransferase (AST), (ALP) und Bilirubin) zum Ausgangszeitpunkt keinen klinisch relevanten Einfluss auf die Clearance von Inebilizumab hatten.
Nierenfunktionsstörungen
Es wurden keine formellen klinischen Studien durchgeführt, um die Auswirkungen einer Nierenfunktionsstörung auf Inebilizumab zu untersuchen. Aufgrund des hohen Molekulargewichts und der hydrodynamischen Grösse eines monoklonalen IgG-Antikörpers ist nicht zu erwarten, dass Inebilizumab durch den Glomerulus gefiltert wird. Aus der populationspharmakokinetischen Analyse ging hervor, dass die Inebilizumab-Clearance von Patienten mit unterschiedlich stark ausgeprägten Nierenfunktionsstörungen mit der von Patienten vergleichbar war, die eine normale geschätzte glomeruläre Filtrationsrate hatten.
Ältere Patienten
Eine populationspharmakokinetische Analyse ergab, dass das Alter keinen Einfluss auf die Clearance von Inebilizumab hat.
Kinder und Jugendliche
Inebilizumab wurde bei Kindern und Jugendlichen nicht untersucht.
Geschlecht, ethnische Abstammung
Eine populationspharmakokinetische Analyse ergab, dass das Geschlecht und die ethnische Abstammung keinen signifikanten Einfluss auf die Clearance von Inebilizumab haben.
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität und zum kanzerogenen Potential lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Inebilizumab wurde in einer kombinierten Fertilitäts- und embryofetalen Entwicklungsstudie an weiblichen und männlichen huCD19Tg-Mäusen in intravenösen Dosen von 3 mg/kg und 30 mg/kg untersucht. Es wurden keine Auswirkungen auf die embryonale und fetale Entwicklung festgestellt, jedoch gab es eine behandlungsbedingte Verringerung des Fruchtbarkeitsindexes bei beiden getesteten Dosen. Die Bedeutung dieser Ergebnisse für den Menschen ist unbekannt. Darüber hinaus kam es bei Mäuseföten, die von mit Inebilizumab behandelten Muttertieren geboren wurden, im Vergleich zu den Nachkommen von Kontrolltieren zu einem Rückgang der B-Zell-Populationen am Ort der B-Zell-Entwicklung, was darauf hindeutet, dass Inebilizumab die Plazenta passiert und die B-Zellen depletiert.
In der kombinierten Fertilitäts- und embryofetalen Entwicklungsstudie wurden nur wenige toxikokinetische Proben entnommen. Auf der Grundlage der maximalen Konzentration der ersten Dosis (Cmax) lagen die Expositionsmultiplikatoren von 3 mg/kg und 30 mg/kg bei weiblichen huCD19-Tg-Mäusen bei der klinischen therapeutischen Dosis von 300 mg beim 0,4-Fachen bzw. 4-Fachen.
In einer prä- und postnatalen Entwicklungsstudie an transgenen Mäusen führte die Verabreichung von Inebilizumab an die Muttertiere vom 6. Tag der Trächtigkeit bis zum 20. Tag der Laktation bei den Nachkommen am 50. postnatalen Tag zu einer Depletion der B-Zellpopulation. Die B-Zell-Populationen der Nachkommen erholten sich bis zum 357. postnatalen Tag. Die Immunantwort auf das Neoantigen war bei den Nachkommen der mit Inebilizumab behandelten Tiere im Vergleich zu jenen der Kontrolltiere vermindert, was auf eine Beeinträchtigung der normalen B-Zell-Funktion hindeutet.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Verdünnung
Die zubereitete Infusionslösung sollte sofort verabreicht werden. Wird sie nicht sofort verabreicht, kann sie vor Beginn der Infusion bis zu 24 Stunden im Kühlschrank bei 2 °C bis 8 °C oder 4 Stunden bei Raumtemperatur gelagert werden.
Besondere Lagerungshinweise
Im Kühlschrank lagern (2-8°C).
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Verdünnung des Arzneimittels, siehe Abschnitt «Haltbarkeit nach Verdünnung».
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Herstellung der Infusionslösung
Vor Beginn der intravenösen Infusion sollte die zubereitete Infusionslösung eine Raumtemperatur zwischen 20 °C und 25 °C haben.
Das Konzentrat sollte per Sichtprobe auf Schwebeteilchen und Verfärbungen untersucht werden. Die Durchstechflasche ist zu verwerfen, wenn die Lösung trüb oder verfärbt ist oder wenn sie sichtbare Fremdpartikel enthält.
·Die Durchstechflasche nicht schütteln.
·Die Durchstechflasche aufrecht lagern.
·Einen intravenösen Infusionsbeutel mit 250 ml 0,9%iger Natriumchlorid-Infusionslösung (9 mg/ml) bereithalten. Keine anderen Lösungen zur Verdünnung von Inebilizumab verwenden, da deren Einsatz nicht untersucht wurde.
·Entnehmen Sie aus jeder der 3 im Karton enthaltenen Durchstechflaschen 10 ml Uplizna und füllen Sie insgesamt 30 ml in den 250-ml-Infusionsbeutel. Die verdünnte Lösung durch vorsichtiges Drehen mischen. Die Lösung nicht schütteln.
Beseitigung
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Zulassungsnummer69322 (Swissmedic)
Packungen10 ml Konzentrat in einer Durchstechflasche aus Glas (Typ 1) mit einem Elastomerstopfen und einer nebelgrauen Flip-off-Aluminiumdichtung.
Packungsgrösse mit 3 Durchstechflaschen. (A)
ZulassungsinhaberinAmgen Switzerland AG, Risch; Domizil: 6343 Rotkreuz
Stand der InformationOktober 2023
|