ZusammensetzungWirkstoffe
Eplontersen (ein synthetisch hergestelltes chemisch modifiziertes Antisense-Oligonukleotid)
Hilfsstoffe
Natriumdihydrogenphosphat-Dihydrat (corresp. Natrium max 0.1 mg pro Injektion)
Natriummonohydrogenphosphat (wasserfrei) (corresp. Natrium 0.3 mg pro Injektion)
Natriumchlorid (corresp. Natrium 1.9 mg pro Injektion)
Salzsäure (zur pH-Einstellung) (corresp. Natrium max 0.01 mg pro Injektion)
Natriumhydroxid (zur pH-Einstellung)
Wasser für Injektionszwecke ad solutionem pro 0.8 ml.
Indikationen/AnwendungsmöglichkeitenWAINZUA wird angewendet zur Behandlung erwachsener Patienten mit Polyneuropathie im Stadium 1 und 2 im Zusammenhang mit hereditärer Transthyretin-vermittelter Amyloidose (ATTRv).
Dosierung/AnwendungDie Behandlung sollte von einem Arzt mit Erfahrung in der Behandlung von Patienten mit Amyloidose verordnet und beaufsichtigt werden.
Dosierung
Die empfohlene Dosis von WAINZUA beträgt 45 mg und wird als subkutane Injektion verabreicht. Die Dosen sollten einmal monatlich verabreicht werden.
Die Entscheidung, die Behandlung bei den Patienten fortzusetzen, deren Erkrankung zu einer Polyneuropathie des Stadiums 3 fortgeschritten ist, sollte nach Ermessen des Arztes auf der Grundlage der Gesamt-Nutzen-Risiko-Bewertung erfolgen.
Besondere Patientengruppen
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter bis mittelschwerer Nierenfunktionsstörung (geschätzte glomeruläre Filtrationsrate [eGFR] ≥45 bis < 90 ml/min/1,73 m2) ist keine Dosisanpassung erforderlich (siehe «Pharmakokinetik»). WAINZUA wurde bei Patienten mit einer eGFR < 45 ml/min/1,73 m2 oder terminaler Niereninsuffizienz nicht untersucht und sollte bei diesen Patienten nur angewendet werden, wenn der erwartete klinische Nutzen das potenzielle Risiko übersteigt (siehe Rubrik «Pharmakokinetik»).
Patienten mit Leberfunktionsstörungen
Bei Patienten mit leichter Leberfunktionsstörung ist keine Dosisanpassung erforderlich (siehe «Pharmakokinetik»). WAINZUA wurde bei Patienten mit mittelschwerer oder schwerer Leberfunktionsstörung nicht untersucht und sollte bei diesen Patienten nur angewendet werden, wenn der erwartete klinische Nutzen das potenzielle Risiko übersteigt (siehe Rubrik «Pharmakokinetik»).
Ältere Patienten
Bei älteren Patienten (≥65 Jahre) ist keine Dosisanpassung erforderlich (siehe «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von WAINZUA bei Kindern und Jugendlichen unter 18 Jahren ist nicht erwiesen. Es liegen keine Daten vor.
Versäumte Dosis
Wenn eine Dosis Eplontersen versäumt wird, sollte die nächste Dosis so bald wie möglich verabreicht werden. Die Gabe ist in monatlichen Abständen ab dem Datum der letzten Dosis fortzusetzen.
Art der Anwendung
Nur zur subkutanen Anwendung.
Die erste Injektion, die vom Patienten oder einer Betreuungsperson verabreicht wird, sollte unter der Aufsicht einer entsprechend qualifizierten medizinischen Fachperson erfolgen. Patienten und/oder Betreuungspersonen sollten in der subkutanen Verabreichung von WAINZUA geschult werden.
Der Fertigpen sollte mindestens 30 Minuten vor der Anwendung aus der gekühlten Lagerung genommen werden und vor der Injektion Raumtemperatur erreicht haben. Andere Methoden der Erwärmung dürfen nicht verwendet werden.
Die Lösung ist vor Gebrauch per Augenschein zu überprüfen. Die Lösung sollte farblos bis gelb aussehen. Sie darf nicht verwendet werden, wenn eine Trübung, Schwebstoffe oder eine Verfärbung vor der Verabreichung beobachtet wird.
Bei Selbstverabreichung ist WAINZUA am Bauch oder im Bereich der Oberschenkel zu injizieren. Wenn eine Betreuungsperson die Injektion verabreicht, kann auch die Rückseite des Oberarms verwendet werden.
Umfassende Anweisungen zur Verabreichung sind in der «Gebrauchsanweisung» enthalten.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Zusammensetzung.
Warnhinweise und VorsichtsmassnahmenVerringerte Serum-Vitamin-A-Spiegel und empfohlene Supplementation
Aufgrund seines Wirkungsmechanismus ist zu erwarten, dass WAINZUA den Vitamin-A-Spiegel (Retinol) im Serum unter die Normalwerte reduziert (siehe «Eigenschaften/Wirkungen»).
Symptome oder Anzeichen eines Vitamin-A-Mangels sollten vor Beginn der Behandlung mit WAINZUA abgeklärt werden.
Patienten, die WAINZUA erhalten, sollten eine orale Supplementation der empfohlenen täglichen Dosis Vitamin A erhalten, um das potenzielle Risiko für okuläre Symptome infolge von Vitamin-A-Mangel zu reduzieren. Eine Überweisung zur augenärztlichen Untersuchung wird empfohlen, wenn Patienten okuläre Symptome entwickeln, die für einen Vitamin-A-Mangel sprechen, wie eingeschränktes Nachtsehen oder Nachtblindheit sowie anhaltend trockene Augen.
Es ist nicht bekannt, ob eine Vitamin-A-Supplementation in der Schwangerschaft ausreicht, um einem Vitamin-A-Mangel vorzubeugen, wenn die Schwangere weiterhin mit WAINZUA behandelt wird (siehe «Schwangerschaft, Stillzeit»). Eine Steigerung der Vitamin-A-Supplementation auf mehr als die empfohlene tägliche Dosis während der Schwangerschaft korrigiert jedoch die Serum-Retinolspiegel aufgrund des Wirkungsmechanismus von Eplontersen wahrscheinlich nicht und kann für die Mutter und das ungeborene Kind schädlich sein.
Hilfsstoffe von besonderem Interesse
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosiereinheit d.h. es ist nahezu «natriumfrei».
InteraktionenEs wurden keine formalen klinischen Studien zur Erfassung von Arzneimittelwechselwirkungen durchgeführt (siehe «Pharmakokinetik»).
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter / Empfängnisverhütung bei Frauen
WAINZUA verringert die Plasmaspiegel von Vitamin A, das für eine normale fetale Entwicklung von zentraler Bedeutung ist. Es ist nicht bekannt, ob eine Vitamin-A-Supplementation ausreicht, um das Risiko für den Fetus zu verringern. Deshalb sollte vor Beginn der Behandlung mit WAINZUA eine Schwangerschaft ausgeschlossen werden, und Frauen, die schwanger werden können, sollten eine wirksame Schwangerschaftsverhütung praktizieren.
Wenn eine Frau beabsichtigt, schwanger zu werden, sollten WAINZUA und die Vitamin-A-Supplementation abgesetzt werden, und die Serumspiegel von Vitamin A sollten überwacht werden und sich wieder normalisiert haben, bevor eine Empfängnis angestrebt wird. Aufgrund der langen Halbwertszeit von Eplontersen (siehe «Pharmakokinetik») kann sich auch nach Beendigung der Behandlung noch ein Vitamin-A-Defizit entwickeln.
Frauen, die schwanger werden können, sollten eine wirksame Schwangerschaftsverhütung praktizieren.
Schwangerschaft
Bisher liegen keine Daten zur Anwendung von WAINZUA bei Schwangeren vor.
Die Verabreichung von Eplontersen oder eines pharmakologisch aktiven nagerspezifischen Surrogats in bis zu 38-fach höheren Dosen als den empfohlene Dosis beim Menschen in einer kombinierten Studie zur Fertilitäts- und embryofetalen Entwicklungstoxizität an Mäusen ergab keine Wirkungen auf die männliche und weibliche Fertilität oder auf die embryofetale Entwicklung (siehe «Präklinische Daten»).
Aufgrund des potenziellen teratogenen Risikos durch einen unausgeglichenen Vitamin-A-Spiegel darf WAINZUA während der Schwangerschaft nicht angewendet werden. Im Falle einer ungeplanten Schwangerschaft sollten der Fetus und der Vitamin-A-Status engmaschig überwacht werden, insbesondere im ersten Trimenon.
Stillzeit
Humane oder tierexperimentelle Laktationsstudien, um das Vorhandensein von Eplontersen oder dessen Metaboliten in der Muttermilch, die Wirkungen auf den gestillten Säugling oder die Wirkungen auf die Milchbildung bei der Mutter zu untersuchen, wurden nicht durchgeführt. Ein Risiko für den gestillten Säugling kann nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit WAINZUA verzichtet werden soll bzw. die Behandlung abzusetzen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
Es liegen keine Daten zu den Auswirkungen von Eplontersen auf die menschliche Fertilität vor.
Die Verabreichung von Eplontersen oder eines pharmakologisch aktiven nagerspezifischen Surrogats in bis zu 38-fach höheren Dosen als der empfohlenen Exposition beim Menschen ergab bei Mäusen keine Hinweise auf Auswirkung von Eplontersen auf die männliche oder weibliche Fertilität.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenWAINZUA hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die nachfolgend beschriebenen Sicherheitsdaten stammen aus der Exposition von 144 Patienten mit durch ATTRv verursachter Polyneuropathie (ATTRv-PN) gegenüber WAINZUA, die für WAINZUA randomisiert wurden und mindestens eine Dosis WAINZUA erhielten. Von diesen wurden 141 Patienten mindestens 6 Monate lang und 137 Patienten mindestens 12 Monate lang behandelt. Die mittlere Behandlungsdauer betrug 541 Tage (Spannbreite: 57 bis 582 Tage).
Die häufigsten Nebenwirkungen, die während der Behandlung mit WAINZUA bei ≥5 % der Patienten beobachtet wurden, waren Erbrechen und erniedrigtes Vitamin A.
Liste der unerwünschten Wirkungen
Die unerwünschten Wirkungen sind nach Systemorganklasse (SOC) gemäss MedDRA aufgeführt. Innerhalb der einzelnen SOC sind die bevorzugten Bezeichnungen nach abnehmender Häufigkeit und dann nach abnehmendem Schweregrad angeordnet. Die Angaben zur Häufigkeit des Auftretens unerwünschter Wirkungen sind definiert als: sehr häufig (≥1/10); häufig (≥1/100 bis < 1/10), gelegentlich (≥1/1000 bis < 1/100), selten (≥1/10'000 bis < 1/1000), sehr selten (< 1/10'000) und nicht bekannt (auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 1: Zusammenfassung der Nebenwirkungen nach Häufigkeitskategorie
|
Systemorganklasse
|
Nebenwirkung
|
Häufigkeit
| |
Erkrankungen des Gastrointestinaltrakts
|
Erbrechen
|
Häufig
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Erythem an der Injektionsstelle
|
Häufig
| |
Schmerzen an der Injektionsstelle
|
Häufig
| |
Pruritus an der Injektionsstelle
|
Häufig
| |
Untersuchungen
|
Vitamin A erniedrigt
|
Sehr häufig
|
Beschreibung spezifischer unerwünschter Wirkungen
Vitamin A erniedrigt
In der klinischen Studie an Patienten mit ATTRv-PN wurden alle Patienten angewiesen, die empfohlene Tagesdosis Vitamin A einzunehmen. Alle mit WAINZUA behandelten Patienten hatten zu Beginn der Studie normale Vitamin-A-Spiegel, und 96,5 % von ihnen entwickelten im Verlauf der Studie Vitamin-A-Spiegel unter der Untergrenze des Normalbereichs (Lower Limit of Normal, LLN) (siehe «Eigenschaften/Wirkungen»).
Reaktionen an der Injektionsstelle
Erytheme an der Injektionsstelle, Schmerzen an der Injektionsstelle und Pruritus an der Injektionsstelle wurden bei 3,5 %, 3,5 % bzw. 2,1 % der Patienten mit ATTRv-PN unter der Behandlung mit WAINZUA beobachtet.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs gibt keine spezifische Behandlung für eine Überdosierung von Eplontersen. Im Falle einer Überdosierung sollte eine unterstützende medizinische Behandlung durchgeführt und ein Arzt oder eine Ärztin zu Rate gezogen werden.
Eigenschaften/WirkungenATC-Code
N07XX21
Wirkungsmechanismus
Eplontersen ist ein GalNAc-konjugiertes 2′-O-2-Methoxyethyl-(2′-MOE-)modifiziertes chimäres Gapmer-Antisense-Oligonukleotid (ASO) mit einem gemischten Grundgerüst aus Phosphorothioat-(PS-) und Phosphatdiester-(PO-)Internukleotidbindungen. Das GalNAc-Konjugat ermöglicht die zielgerichtete Einschleusung des ASO in Hepatozyten. Die selektive Bindung von Eplontersen an die TTR-Messenger-RNA (mRNA) in den Hepatozyten führt zum Abbau sowohl von mutierter als auch von (normaler) Wildtyp-TTR-mRNA. Dies verhindert die Synthese des TTR-Proteins in der Leber; die Folge ist eine signifikante Reduktion der Mengen von mutiertem und Wildtyp-TTR-Protein, die von der Leber in den Kreislauf sezerniert werden.
TTR ist ein Trägerprotein des retinolbindenden Proteins 4 (RBP4), des Hauptträgers von Vitamin A (Retinol). Deshalb ist zu erwarten, dass eine Reduktion von Plasma-TTR zu einer Senkung der Plasma-Retinolspiegel unter die Untergrenze des Normalbereichs führt.
Pharmakodynamik
In der klinischen Studie an Patienten mit ATTRv-PN, die Eplontersen erhielten, wurde eine Reduktion der TTR-Serumkonzentrationen bei der ersten Untersuchung (Woche 5) beobachtet, und die TTR-Konzentrationen nahmen bis Woche 35 weiter ab. Über die gesamte Behandlungsdauer (85 Wochen) hinweg wurde eine anhaltende Reduktion der TTR-Konzentration beobachtet. Der Mittelwert (SD) der prozentualen Reduktion der TTR-Serumkonzentration gegenüber dem Ausgangswert betrug 82,1 % (11,7) nach 35 Wochen, 83,0 % (10,4) nach 65 Wochen und 81,8 % (13,4) nach 85 Wochen unter Behandlung mit Eplontersen. Unabhängig von Geschlecht, ethnischer Abstammung, Alter, Region, Körpergewicht, Kardiomyopathie-Status, Vorbehandlung, Val30Met-Mutationsstatus, Krankheitsstadium und Diagnose einer familiären Amyloid-Kardiomyopathie (FAC) bei Studienbeginn wurde eine ähnliche Reduktion der TTR-Serumkonzentrationen gegenüber dem Ausgangswert im Vergleich zum Placebo beobachtet (Abbildung 1, a und b).
Abbildung 1: Forest-Plot der LSM-Behandlungsdifferenz für die prozentuale Veränderung vs. Ausgangswert von TTR (g/l) für wichtige Subgruppen (NEURO-TTRansform-Studie) (vollständiges Analysekollektiv)
a) in Woche 35
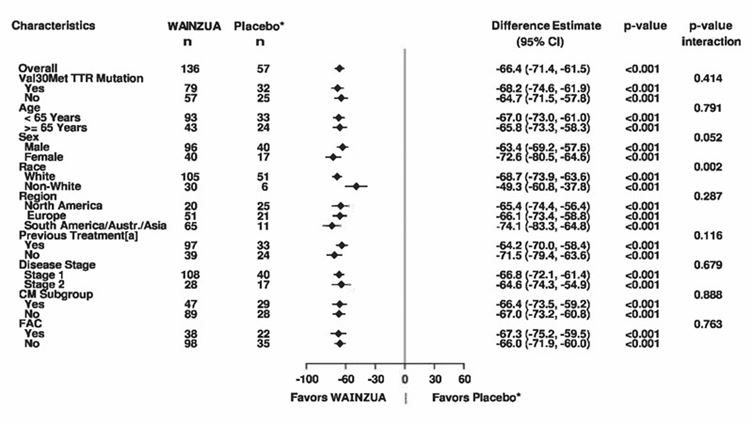
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
[a] Vorbehandelt mit Tafamidis oder Diflunisal.
Basierend auf MMRM adjustiert durch Propensity-Score-Gewichtung mit kategorischen Effekten für Behandlung, Zeit, Behandlung-Zeit-Interaktion und Krankheitsstadium, Val30Met-Mutation, Vorbehandlung sowie festen Kovariablen für die Baseline und die Baseline-Zeit-Interaktion.
Die Subgruppen-Modelle enthielten auch Behandlung-Subgruppe-, Zeit-Subgruppe- und Behandlung-Zeit-Subgruppe-Interaktionen. In die 35-Wochen-Zwischenanalyse wurden nur Daten bis Woche 35 einbezogen.
Die CM-Subgruppe umfasst Patienten mit entweder der Diagnose FAC bei Studieneintritt oder einer interventrikulären (IV) Septumwanddicke ≥13 mm zu Studienbeginn ohne Hypertonie [Anamnese oder Diagnose während der Studie].
Die LSM-Behandlungsdifferenzen nach 35 Wochen (WAINZUA – Placebo) sind mit 95%-KI (unadjustiert) aufgeführt.
KI = Konfidenzintervall, LSM = Least-Squares-Mittelwert, MMRM = Modell mit gemischten Effekten und Messwiederholung, TTR = Transthyretin, CM = Kardiomyopathie, FAC = familiäre Amyloid-Kardiomyopathie.
b) in Woche 65

* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
[a] Vorbehandelt mit Tafamidis oder Diflunisal.
Basierend auf MMRM adjustiert durch Propensity-Score-Gewichtung mit kategorischen Effekten für Behandlung, Zeit, Behandlung-Zeit-Interaktion und Krankheitsstadium, Val30Met-Mutation, Vorbehandlung sowie festen Kovariablen für die Baseline und die Baseline-Zeit-Interaktion.
Die Subgruppen-Modelle enthielten auch Behandlung-Subgruppe-, Zeit-Subgruppe- und Behandlung-Zeit-Subgruppe-Interaktionen.
Die CM-Subgruppe umfasst Patienten mit entweder der Diagnose FAC bei Studieneintritt oder einer interventrikulären Septumwanddicke ≥13 mm zu Studienbeginn ohne Hypertonie [Anamnese oder Diagnose während der Studie].
Die LSM-Behandlungsdifferenzen nach 65 Wochen (WAINZUA – Placebo) sind mit 95%-KI (unadjustiert) aufgeführt.
KI = Konfidenzintervall, LSM = Least-Squares-Mittelwert, MMRM = Modell mit gemischten Effekten und Messwiederholung, TTR = Transthyretin, CM = Kardiomyopathie, FAC = familiäre Amyloid-Kardiomyopathie
Kardiale Elektrophysiologie
Formale QTc-Studien wurden mit WAINZUA nicht durchgeführt. Das Potenzial für eine QTc-Verlängerung durch Eplontersen wurde in einer randomisierten, placebokontrollierten Studie an gesunden Probanden untersucht. Bei einer Dosis in Höhe des 2,7-Fachen der empfohlenen Dosis von 45 mg Eplontersen wurde keine klinisch relevante Wirkung auf das QT-Intervall beobachtet.
Immunogenität
In der klinischen Studie an Patienten mit ATTRv-PN entwickelten nach einer 84wöchigen Behandlungszeit (mediane Behandlungsdauer 561 Tage (80 Wochen), Spannbreite 57 bis 582 Tage) 58 Patienten (40,3 %) behandlungsbedingte Anti-Drug-Antikörper (ADA).
Bei den Patienten, die positiv auf Anti-Eplontersen-Antikörper getestet wurden, zeigte sich keine klinisch bedeutsame Auswirkung auf die Wirksamkeit, Sicherheit, Pharmakokinetik oder Pharmakodynamik von WAINZUA.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von WAINZUA wurden in einer randomisierten, multizentrischen, offenen, extern kontrollierten Studie (NEURO-TTRansform) mit insgesamt 168 Patienten mit ATTRv PN untersucht. Die Patienten wurden im Verhältnis 6:1 für die Behandlung mit einer subkutanen Injektion von 45 mg WAINZUA (N = 144) alle 4 Wochen oder 284 mg Inotersen (N = 24) wöchentlich als Referenzgruppe randomisiert. Von den 144 Patienten, die für Eplontersen randomisiert wurden, durchliefen 140 (97,2 %) die Behandlung bis Woche 35, 135 (93,8 %) bis Woche 65 und 130 (90,3 %) bis Woche 85.
Eine externe Placebokontrolle bestand aus einer Placebokohorte von Patienten aus der zulassungsrelevanten Studie zu Inotersen, einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen klinischen Studie an erwachsenen Patienten mit ATTRv-PN (NEURO-TTR). Diese Kohorte erhielt einmal wöchentlich subkutane Placebo-Injektionen. In beiden Studien galten identische Einschlusskriterien.
Die Merkmale der Eplontersen- und der externen Placebogruppe waren im Allgemeinen vergleichbar, und potenzielle Unausgewogenheiten der wesentlichen Ausgangsmerkmale (Val30Met-Mutationsstatus, Krankheitsstadium und Vorbehandlung) wurden in der vorgegebenen statistischen Analyse berücksichtigt. Die demografischen und krankheitsspezifischen Ausgangsmerkmale sind in Tabelle 2 aufgeführt.
Tabelle 2 Demografische und krankheitsspezifische Ausgangsmerkmale in der Studie NEURO-TTRansform (Sicherheitskollektiv)
|
|
Placebo
*(N = 60)
|
WAINZUA
(N = 144)
| |
Alter, Jahre
|
|
| |
Mittelwert (SD)
|
59,5 (14,1)
|
53,0 (15,0)
| |
Median (min, max)
|
63 (28, 81)
|
51,5 (24, 82)
| |
< 65, n (%)
|
34 (56,7)
|
100 (69,4)
| |
65-74, n (%)
|
17 (28,3)
|
36 (25,0)
| |
≥75, n (%)
|
9 (15,0)
|
8 (5,6)
| |
Männlich, n (%)
|
41 (68,3)
|
100 (69,4)
| |
Ethnie, n (%)
|
|
| |
Asiatisch
|
3 (5,0)
|
22 (15,4)
| |
Schwarz oder Afroamerikanisch
|
1 (1,7)
|
5 (3,5)
| |
Weiss
|
53 (88,3)
|
112 (78,3)
| |
Andere
|
2 (3,3)
|
3 (2,1)
| |
Mehrere
|
1 (1,7)
|
1 (0,7)
| |
Ethnische Zugehörigkeit, n (%)
|
|
| |
m
|
60
|
142
| |
Hispanisch oder Latino
|
7 (11,7)
|
22 (15,5)
| |
Vorbehandlung mit Tafamidis oder Diflunisal, n (%)
|
|
| |
Ja
|
36 (60,0)
|
100 (69,4)
| |
Krankheitsstadium der ATTRv-PN1, n (%)
|
|
| |
Stadium 1
|
42 (70,0)
|
115 (79,9)
| |
Stadium 2
|
18 (30,0)
|
29 (20,1)
| |
Kombinierter mNIS+7-Score, Mittelwert (SD)
|
74,8 (39,0)
|
81,3 (43,4)
| |
Norfolk QoL-DN-Gesamtscore,
|
|
| |
m
|
59
|
137
| |
Mittelwert (SD)
|
48,7 (26,8)
|
44,1 (26,6)
| |
Val30Met-TTR-Mutation, n (%)
|
|
| |
Ja2
|
33 (55,0)
|
85 (59,0)
| |
Nein3
|
27 (45,0)
|
59 (41,0)
| |
Glu89Gln, Glu109Gln
|
0
|
1 (0,7)
| |
Leu58His, Leu78His
|
3 (5,0)
|
4 (2,8)
| |
Phe64Leu, Phe84Leu
|
3 (5,0)
|
5 (3,5)
| |
Ser50Arg, Ser70Arg
|
1 (1,7)
|
2 (1,4)
| |
Ser77Tyr, Ser97Tyr, S97Y
|
5 (8,3)
|
3 (2,1)
| |
Thr49Ala, Thr69Ala
|
0
|
1 (0,7)
| |
Thr60Ala, Thr80Ala
|
8 (13,3)
|
4 (2,8)
| |
Val122Ile, Val142Ile
|
1 (1,7)
|
4 (2,8)
| |
Andere3
|
6 (10,0)
|
35 (24,3)
| |
NYHA-Klasse, n (%)
|
|
| |
I
|
40 (66,7)
|
105 (72,9)
| |
II
|
20 (33,3)
|
39 (27,1)
| |
Krankheitsdauer ab Diagnose der ATTRv-PN (Monate), Mittelwert (SD)
|
39,3 (40,3)
|
46,8 (58,1)
| |
Dauer seit Beginn der ATTRv-PN-Symptome (Monate), Mittelwert (SD)
|
64,0 (52,3)
|
67,7 (50,9)
| |
Diagnose Familiäre Amyloid-Kardiomyopathie (FAC)4, n (%)
|
|
| |
Kriterien zur Dokumentation der klinischen Diagnose einer FAC4, n (%)5
|
22 (36,7)
|
39 (27,1)
| |
Herzbiopsie
|
5 (22,7)
|
1 (2,6)
| |
Ergebnis der Echokardiographie
|
17 (77,3)
|
24 (61,5)
| |
Andere
|
0
|
24 (61,5)
| |
Krankheitsdauer ab der klinischen Diagnose einer FAC4 aus CRF (Monate), Mittelwert (SD)
|
21,0 (22,5)
|
18,5 (21,4)
| |
Dauer seit Beginn der FAC4-Symptome (Monate), Mittelwert (SD)
|
34,1 (29,3)
|
36,3 (63,8)
| |
NT-proBNP (pmol/l), Mittelwert (SD)
|
82,0 (159,2)
|
54,0 (122,6)
| |
Short Form 36 Item Health Survey (SF-36), Gesamtscore der körperlichen Komponente, Mittelwert (SD)
|
37,2 (9,8)
|
39,7 (9,3)
| |
Neuropathy Symptoms and Change (NSC), Gesamtscore, Mittelwert (SD)
|
23,0 (12,6)
|
23,1 (12,4)
| |
Polyneuropathy Disability (PND) Score, n (%)
|
|
| |
I
|
23 (38,3)
|
56 (39,2)
| |
II
|
19 (31,7)
|
61 (42,7)
| |
IIIa
|
15 (25,0)
|
16 (11,2)
| |
IIIb
|
3 (5,0)
|
10 (7,0)
| |
Body-Mass-Index (kg/m2)
|
|
| |
m
|
60
|
138
| |
Mittelwert (SD)
|
24,2 (4,9)
|
24,4 (4,9)
| |
Median (min, max)
|
23,8 (14,5, 39,8)
|
24,1 (15,4, 35,4)
| |
Modifizierter Body-Mass-Index (kg/m2 x g/l),
|
|
| |
m
|
60
|
138
| |
Mittelwert (SD)
|
1049,89 (228,43)
|
1025,78 (235,12)
| |
Median (min, max)
|
1027,55 (668,7, 1710,0)
|
1003,14 (615,7, 1714,0)
| |
Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
1 Das Krankheitsstadium ist definiert als Stadium 1 = benötigt keine Hilfe beim Gehen und Stadium 2 = benötigt Hilfe beim Gehen.
2 Umfasst die Genotypen V30M, V50M, V50M-MUTATION, VAL50MET und P.VAL50MET.
3 Basierend auf klinischer Datenbasis. Die Nicht-Val30Met-Mutationen umfassten: GLU89GLN, LEU58HIS, PHE64LEU, SER50ARG, SER77TYR, THR49ALA, THR60ALA, VAL122LLE und andere (darunter ALA97SER).
4 Familiäre Amyloid-Kardiomyopathie = Hereditäre Transthyretin-vermittelte Amyloidose mit Kardiomyopathie (ATTRv-CM).
5 Der Nenner für die Berechnung des Prozentsatzes ist die Anzahl an Patienten mit der Diagnose FAC.
Vom Datum der Einwilligungserklärung wurden nur Jahr und Monat erhoben, um die Krankheitsdauer ab der Diagnose und ab dem Beginn der Symptome der ATTRv-PN und FAC zu berechnen.
N = Anzahl an Patienten im Sicherheitskollektiv, n = Anzahl an Patienten in einer Subgruppe, m = Anzahl an Patienten mit nicht fehlenden Daten, falls verschieden von N, CRF = Prüfbogen, NT-proBNP = N-terminales natriuretisches Peptid vom proB-Typ, SD = Standardabweichung.
|
Von den 39 Patienten (27,1 %) in der Eplontersen-Gruppe, bei denen bei Studieneintritt eine TTR-Kardiomyopathie diagnostiziert wurde, wurden 41,0 % der Patienten der Klasse I der New York Heart Association (NYHA) und 59,0 % der Patienten der NYHA-Klasse II zugeordnet.
Analysen in Woche 35 (Zwischenanalysen)
Die primären Wirksamkeitsendpunkte waren die Veränderung der Transthyretin-(TTR) Serumkonzentration (siehe Abbildung 2) und des kombinierten modifizierten Neuropathy Impairment Scores + 7 (mNIS+7) bis Woche 35 gegenüber dem Ausgangswert. Der kombinierte mNIS+7-Score ist eine objektive Bewertung der Neuropathie und umfasst die kombinierten NIS- und Modified +7-Scores. In der in der Studie verwendeten Version des kombinierten mNIS+7-Scores misst der NIS objektiv die Defizite der Hirnnervenfunktion, Muskelkraft, Reflexe und Empfindungen, und der Modified +7 bewertet die Herzfrequenzreaktion auf tiefe Atmung, quantitative sensorische Tests (Berührungsdruck und Wärmeschmerz) und die Elektrophysiologie peripherer Nerven.
Die validierte Version des in der Studie verwendeten kombinierten mNIS+7-Scores reichte von -22,3 bis 346,3 Punkten, wobei höhere Scores für einen höheren Schweregrad der Erkrankung stehen.
Der sekundäre Endpunkt war die Veränderung des Gesamtscores des Fragebogens Norfolk Quality of Life – Diabetic Neuropathy (QoL-DN) gegenüber dem Ausgangswert. Die Norfolk QoL-DN-Skala ist eine Patientenselbstbeurteilung der subjektiven Erfahrung von Neuropathie in den folgenden Dimensionen: körperliche Funktionsfähigkeit/Large-Fibre-Neuropathie, Aktivitäten des täglichen Lebens, Symptomatik, Small-Fibre-Neuropathie und autonome Neuropathie. Die Version des in der Studie verwendeten Norfolk QoL-DN-Gesamtscores reichte von -4 bis 136 Punkten, wobei höhere Scores für eine stärkere Beeinträchtigung stehen.
WAINZUA zeigte eine statistisch signifikante Verbesserung im Vergleich zur externen Placebokontrolle nach 35 Wochen im Sinne einer Reduktion der TTR-Serumkonzentration mit einer prozentualen Veränderung von -66,43 % (95%-KI: --71,39 %, -61,47 %; p < 0,0001) (siehe Abbildung 2). WAINZUA zeigte eine statistisch signifikante Verbesserung im Vergleich zur externen Placebokontrolle nach 35 Wochen bezüglich des kombinierten mNIS+7-Scores mit einer LSM-Differenz von -9,0 (95%-KI: --13,5, -4,5; p < 0,0001) (siehe Abbildungen 3, 4a, 7a). WAINZUA zeigte eine statistisch signifikante Verbesserung im Vergleich zur externen Placebokontrolle nach 35 Wochen bezüglich des Norfolk QoL-DN-Gesamtscores mit einer LSM-Differenz von -11,8 (95%-KI: --16,8, -6,8; p < 0,0001) (Tabelle 3 und Abbildungen 5, 6a, 8a).
Analysen in Woche 65/66 (Abschlussanalysen)
Die koprimären Endpunkte für das primäre Studienziel bei der Abschlussanalyse in Woche 66 waren die prozentuale Veränderung der TTR-Serumkonzentration nach 65 Wochen gegenüber dem Ausgangswert, die Veränderung des kombinierten mNIS+7-Scores in Woche 66 gegenüber dem Ausgangswert und die Veränderung des Norfolk QoL-DN-Gesamtscores in Woche 66 gegenüber dem Ausgangswert. Die Reduktion der TTR-Serumkonzentration hielt bis Woche 65 an. Darüber hinaus entsprachen die Ergebnisse in Woche 66 für den kombinierten mNIS+7- und den Norfolk-Gesamtscore den Ergebnissen nach 35 Wochen (siehe Tabelle 3 und Abbildungen 3, 4b, 5, 6b).
Die sekundären Endpunkte waren die Veränderung der Neuropathy Symptoms and Change (NSC) in Woche 66 und Woche 35 gegenüber dem Ausgangswert, die Veränderung des körperlichen Komponenten-Scores (PCS) des Short Form 36 Item Health Survey (Version 2) (SF-36) in Woche 65 gegenüber dem Ausgangswert, die Veränderung des Polyneuropathy Disability (PND) Scores in Woche 65 gegenüber dem Ausgangswert und die Veränderung des modifizierten Body-Mass-Index (mBMI) bis Woche 65 gegenüber dem Ausgangswert.
Der NSC war ein patientenbeantworteter Fragebogen zur Quantifizierung der Art, Verteilung und Schwere der Muskelschwäche, sensorischen Symptome, Schmerzsymptome und autonomen Symptome. Höhere Scores stehen für stärkere Symptome.
Der SF-36 PCS umfasste 4 Skalen zur Bewertung der körperlichen Funktion, Rolleneinschränkungen durch körperliche Probleme, körperlichen Schmerzen und des allgemeinen Gesundheitszustands. Höhere Scores stehen für einen besseren allgemeinen Gesundheitszustand.
Der PND kategorisiert die Behinderung anhand der Mobilität (z.B. Notwendigkeit eines Gehstocks, einer Gehstütze, eines Rollstuhls oder Bettlägerigkeit). Höhere PND-Scores stehen für stärkere Behinderung.
Der modifizierte BMI (BMI × Serumalbumin) ist eine akzeptable Methode zur Bewertung des Ernährungsstatus bei ATTR. Höhere Scores stehen für einen besseren Ernährungsstatus und gelten als Indikator für ein längeres Überleben von Patienten mit ATTRv-PN.
Alle sekundären Endpunkte zeigten eine statistisch signifikante Überlegenheit gegenüber dem externen Placebo (siehe Tabelle 4).
Tabelle 3: Behandlungseffekt in Bezug auf die primären und wichtigsten sekundären Endpunkte (NEURO-TTRansform-Studie) (vollständiges Analysekollektiv)
|
Analyse/Endpunkt
|
Baseline, Mittelwert (SD)
|
LSM-Veränderung/Prozentuale Veränderung vs. Baseline, (SE) [95%-KI]
|
WAINZUA – Externes Placebo* LSM-Differenz
(95%-KI)
|
p-Wert
| |
Externes Placebo*
|
WAINZUA
|
Externes Placebo*
|
WAINZUA
| |
Woche 35
|
N = 59
|
N = 140
|
N = 59
|
N = 140
|
|
| |
Serum-TTR, g/l 1,
|
0,15 (0,04)
|
0,23 (0,08)
|
|
|
|
| |
Prozentuale Veränderung vs. Baseline
|
|
|
-14,8 % (2,0)
[-18,73, -10,80]
|
-81,2 % (1,7)
[-84,55, -77,84]
|
-66,4 %
(-71,39, -61,47)
|
p < 0,0001
| |
Kombinierter mNIS+7-Score 2,3
|
74,1 (39,0)
|
79,6 (42,3)
|
|
|
|
| |
Veränderung vs. Baseline
|
|
|
9,2 (1,9)
[5,54, 12,91]
|
0,2 (1,9)
[-3,46, 3,89]
|
-9,0
(-13,48, -4,54)
|
p < 0,0001
| |
Norfolk QoL-DN-Gesamtscore 2,3
|
48,6
(27,0)
|
43,5
(26,3)
|
|
|
|
| |
Veränderung vs. Baseline
|
|
|
8,7 (2,1)
[4,53, 12,81]
|
-3,1 (2,1)
[-7,19, 0,96]
|
-11,8
(-16,82, -6,76)
|
p < 0,0001
| |
Woche 65/66
|
N = 59
|
N = 141
|
N = 59
|
N = 141
|
|
| |
Serum-TTR, g/l 1
|
0,15 (0,04)
|
0,23 (0,08)
|
|
|
|
| |
Prozentuale Veränderung vs. Baseline
|
|
|
-11,2 % (1,9)
[-15,06, -7,41]
|
-81,7 % (1,6)
[-84,82, -78,48]
|
-70,4 %
(-75,17, -65,66)
|
p < 0,00014
| |
Kombinierter mNIS+7-Score 1
|
74,1 (39,0)
|
79,8 (42,3)
|
|
|
|
| |
Veränderung vs. Baseline
|
|
|
25,1 (2,4)
[20,23, 29,88]
|
0,3 (2,4)
[-4,46, 5,06]
|
-24,8
(-30,96, -18,56)
|
p < 0,00014
| |
Norfolk QoL-DN-Gesamtscore 1
|
48,6 (27,0)
|
43,3 (26,2)
|
|
|
|
| |
Veränderung vs. Baseline
|
|
|
14,2 (2,4)
[9,51, 18,97]
|
-5,5 (2,3)
[-10,03, -0,96]
|
-19,7
(-25,63, -13,84)
|
p < 0,00014
|
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
1 Basierend auf MMRM adjustiert durch Propensity-Score-Gewichtung mit festen kategorischen Effekten für Behandlung, Zeit, Behandlung-Zeit-Interaktion und Krankheitsstadium, Val30M-Mutation, Vorbehandlung sowie festen Kovariablen für den Ausgangswert und die Baseline-Zeit-Interaktion. In die Analyse nach 66 Wochen wurden nur Daten bis Woche 66 einbezogen.
2 Basierend auf einem ANCOVA-Modell adjustiert durch Propensity Score mit den Effekten Behandlung, Krankheitsstadium, Val30M-Mutation, Vorbehandlung und Ausgangswert. In die Zwischenanalyse wurden nur Daten bis Woche 35 einbezogen.
3 Bei Teilnehmenden mit fehlendem mNIS+7 oder Norfolk QoL-DN in Woche 35 wurde der Wert unter Verwendung eines Imputationsmodells nach dem Verfahren der der multiplen Imputation erzeugt. Jeder von 500 imputierten Datensätzen wurde mittels einfachem ANCOVA-Modell analysiert, und die 500 Ergebnisse des ANCOVA-Modells wurden unter Verwendung der Rubin-Regeln kombiniert.
4 Nicht formal getestet aufgrund von statistisch signifikanten Ergebnissen in Woche 35.
Die Analyse basierte auf Daten, die bis zu 52 Tage nach der letzten Dosis des Studienpräparates erfasst wurden. Daten aus der Zwischenanalyse in Woche 35 und Daten aus Woche 65/66 aus der Analyse in Woche 66. Im vollständigen Analysekollektiv umfasste die Eplontersen-Gruppe 140 Teilnehmende nach 35 Wochen und 141 Teilnehmende nach 66 Wochen. Ein Teilnehmer hatte keine mNIS+7- oder Norfolk QoL-DN-Bewertung in Woche 35, aber eine Bewertung für mindestens einen dieser Parameter in Woche 66.
ANCOVA = Kovarianzanalyse, KI = Konfidenzintervall, LSM = Least-Squares-Mittelwert, MMRM = Modell mit gemischten Effekten und Messwiederholung, mNIS+7 = modifizierter Neuropathy Impairment Score +7, N = Anzahl an Teilnehmenden in der Gruppe, Norfolk QoL-DN = Norfolk Quality of Life – Diabetic Neuropathy Questionnaire, SD = Standardabweichung, SE = Standardfehler, TTR = Transthyretin.
Tabelle 4: Hierarchische Prüfung der sekundären Endpunkte (NEURO-TTRansform-Studie)
|
|
|
|
Vergleich WAINZUA vs. externes Placebo*
| |
Sekundärer Endpunkt/
Behandlungsgruppe (N)
|
n
|
Veränderung vs. Baseline
LSM (95%-KI)
|
Schätzung
|
95%-KI
|
p-Wert
| |
LSM-Veränderung des NSC vs. Baseline in Woche 66
| |
WAINZUA (N = 141)
|
132
|
0,0 (-1,92, 1,86)
|
-8,2
|
-10,65, -5,76
|
< 0,0001
| |
Externes Placebo* (N = 59)
|
52
|
8,2 (6,24, 10,12)
| |
LSM-Veränderung des NSC vs. Baseline in Woche 35
| |
WAINZUA (N = 141)
|
141
|
0,8 (-0,92, 2,50)
|
-3,9
|
-6,08, -1,80
|
0,0005
| |
Externes Placebo* (N = 59)
|
56
|
4,7 (2,98, 6,48)
| |
LSM-Veränderung des SF-36 PCS vs. Baseline in Woche 65
| |
WAINZUA (N = 141)
|
136
|
0,85 (-0,711, 2,412)
|
5,31
|
3,195, 7,416
|
< 0,0001
| |
Externes Placebo* (N = 59)
|
50
|
-4,46 (-6,139, -2,770)
| |
LSM-Veränderung des PND-Scores vs. Baseline in Woche 65
| |
WAINZUA (N = 141)
|
134
|
0,1 (0,0, 0,2)
|
-0,2
|
-0,4, 0,0
|
0,0241
| |
Externes Placebo* (N = 59)
|
51
|
0,3 (0,2, 0,4)
| |
LSM-Veränderung des mBMI vs. Baseline in Woche 65
| |
WAINZUA (N = 141)
|
130
|
-8,1 (-28,55, 12,42)
|
82,7
|
54,64, 110,76
|
< 0,0001
| |
Externes Placebo* (N = 59)
|
49
|
-90,8 (-112,84, -68,69)
|
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
N = Anzahl an Patienten im vollständigen Analysekollektiv in Woche 66.
n = Anzahl an Patienten mit nicht fehlenden Daten für Baseline-Kovariablen und Veränderung vs. Baseline zum betreffenden Zeitpunkt.
Die Analyse basierte auf Daten, die bis zu 28 Tage nach der letzten Dosis des Studienpräparates erfasst wurden. Das Analyse-Besuchszeitfenster von Woche 65 erstreckt sich von Tag 419 bis Tag 479.
Basierend auf einem Modell mit gemischten Effekten und Messwiederholung (MMRM) adjustiert durch Propensity-Score-Gewichtung mit festen kategorischen Effekten für Behandlung, Zeit, Behandlung-Zeit-Interaktion und Krankheitsstadium, Val30M-Mutation, Vorbehandlung sowie festen Kovariablen für den Ausgangswert und die Baseline-Zeit-Interaktion. In die Abschlussanalyse nach 66 Wochen wurden nur Daten bis Woche 65 einbezogen.
KI = Konfidenzintervall, LSM = Least-Squares-Mittelwert, mBMI = modifizierter Body-Mass-Index, NSC = Neuropathy Symptoms and Change, PND = Polyneuropathy Disability, PCS = körperlicher Komponenten-Score, SF-36 PCS = Short Form-36 Health Survey Questionnaire Physical Component Score.
Abbildung 2: Prozentuale LSM-Veränderung der TTR-Serumkonzentration von Baseline bis Woche 65, WAINZUA vs. externes Placebo* bis Woche 65 (NEURO-TTRansform-Studie) (vollständiges Analysekollektiv)
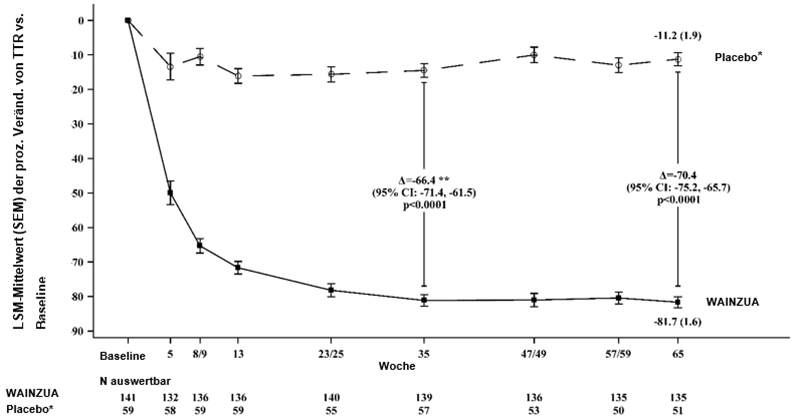
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
** Behandlungsdifferenz entspricht den Ergebnissen der formalen Zwischenanalyse in Woche 35. In die Zwischenanalyse in Woche 35 wurden nur Daten bis Woche 35 einbezogen.
Basierend auf MMRM adjustiert durch Propensity-Score-Gewichtung mit festen kategorischen Effekten für Behandlung, Zeit, Behandlung-Zeit-Interaktion und Krankheitsstadium, Val30Met-Mutation, Vorbehandlung sowie festen Kovariablen für den Ausgangswert und die Baseline-Zeit-Interaktion.
Die Analyse basierte auf Daten, die bis zu 28 Tage nach der letzten Dosis des Studienpräparates erfasst wurden. Daten bis Woche 65 sind einbezogen. Placebo wurde zur Baseline sowie in den Wochen 5, 8, 13 23, 35, 47, 59 und 65 bewertet; WAINZUA wurde zur Baseline sowie in den Wochen 5, 9, 13, 25, 35, 49, 57 und 65 bewertet.
Die LS-Mittelwert-Behandlungsdifferenzen nach 35 Wochen und 65 Wochen (WAINZUA – Placebo) sind mit 95%-KI (unadjustiert) aufgeführt.
KI = Konfidenzintervall, LSM = Least-Squares-Mittelwert, SEM = Standardfehler des Mittelwertes, MMRM = Modell mit gemischten Effekten und Messwiederholung, TTR = Transthyretin.
Abbildung 3: LSM-Veränderung des kombinierten mNIS+7-Scores vs. Baseline (NEURO-TTRansform-Studie) (vollständiges Analysekollektiv)
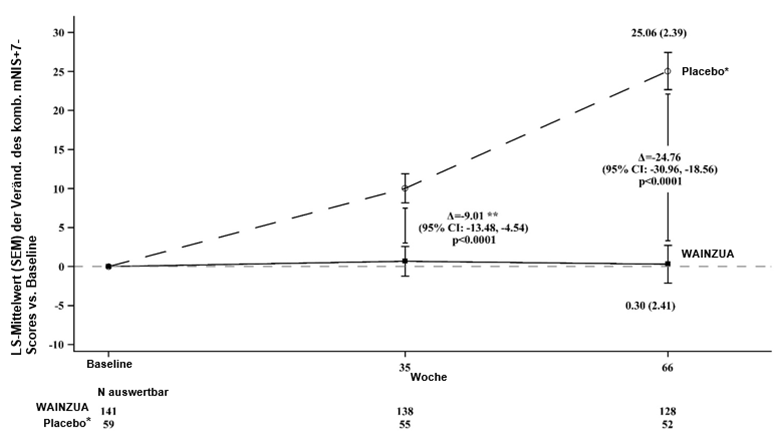
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
** Behandlungsdifferenz entspricht den Ergebnissen der formalen Zwischenanalyse in Woche 35. Basierend auf MI ANCOVA adjustiert durch Propensity-Score-Gewichtung mit festen kategorischen Effekten für Behandlung, Krankheitsstadium, Val30Met-Mutation, Vorbehandlung sowie festen Kovariablen für den Ausgangswert. In die Zwischenanalyse in Woche 35 wurden nur Daten bis Woche 35 einbezogen.
Die Analyse in Woche 66 basierte auf MMRM adjustiert durch Propensity Score-Gewichtung mit kategorischen Effekten für Behandlung, Zeit, Behandlung-Zeit-Interaktion und Krankheitsstadium, Val30Met-Mutation, Vorbehandlung sowie festen Kovariablen für die Baseline und die Baseline-Zeit-Interaktion.
Die Analyse basierte auf Daten, die bis zu 52 Tage nach der letzten Dosis des Studienpräparates erfasst wurden. Daten bis Woche 66 sind einbezogen.
Die LS-Mittelwert-Behandlungsdifferenzen nach 35 Wochen und 66 Wochen (WAINZUA – Placebo) sind mit 95%-KI (unadjustiert) aufgeführt.
KI = Konfidenzintervall, LS-Mittelwert = Least-Squares-Mittelwert, SEM = Standardfehler des Mittelwertes, MI ANCOVA = Kovarianzanalyse mit multipler Imputation, MMRM = Modell mit gemischten Effekten und Messwiederholung.
Abbildung 4: Histogramm der Veränderung des kombinierten mNIS+7-Scores vs. Baseline (NEURO-TTRansform-Studie) (Sicherheitsanalysekollektiv)
a) in Woche 35
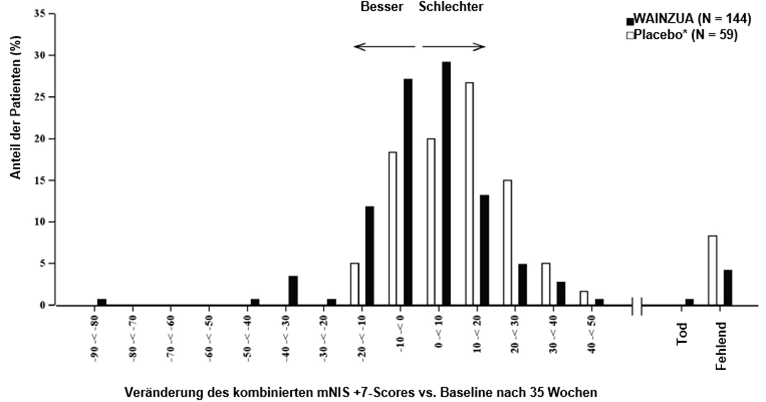
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
b) in Woche 66
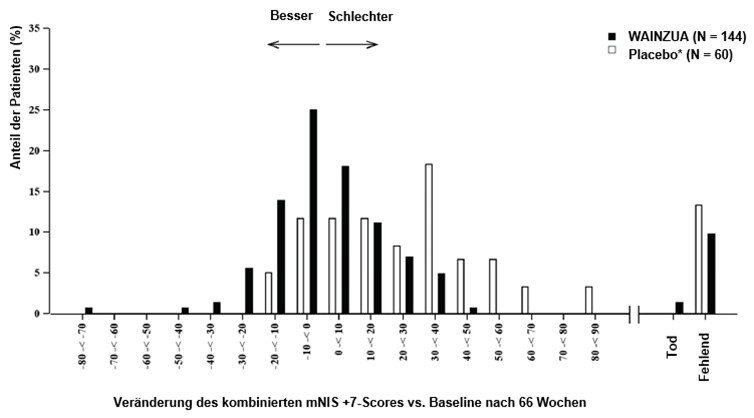
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
Abbildung 5: LSM-Veränderung des Norfolk QoL-DN-Gesamtscores vs. Baseline (NEURO-TTRansform-Studie)
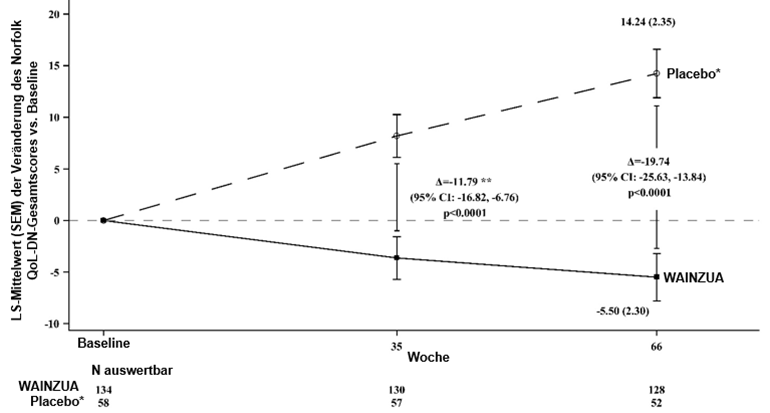
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
** Behandlungsdifferenz entspricht den Ergebnissen der formalen Zwischenanalyse in Woche 35. Basierend auf MI ANCOVA adjustiert durch Propensity-Score-Gewichtung mit festen kategorischen Effekten für Behandlung, Krankheitsstadium, Val30Met-Mutation, Vorbehandlung sowie festen Kovariablen für den Ausgangswert. In die Zwischenanalyse in Woche 35 wurden nur Daten bis Woche 35 einbezogen.
Die Analyse in Woche 66 basierte auf MMRM adjustiert durch Propensity-Score-Gewichtung mit kategorischen Effekten für Behandlung, Zeit, Behandlung-Zeit-Interaktion und Krankheitsstadium, Val30Met-Mutation, Vorbehandlung sowie festen Kovariablen für die Baseline und die Baseline-Zeit-Interaktion.
Die Analyse basierte auf Daten, die bis zu 52 Tage nach der letzten Dosis des Studienpräparates erfasst wurden. Daten bis Woche 66 sind einbezogen.
Die LS-Mittelwert-Behandlungsdifferenzen nach 35 Wochen und 66 Wochen (WAINZUA – Placebo) sind mit 95%-KI (unadjustiert) aufgeführt.
KI = Konfidenzintervall, LS-Mittelwert = Least-Squares-Mittelwert, SEM = Standardfehler des Mittelwertes, MI ANCOVA = Kovarianzanalyse mit multipler Imputation, MMRM = Modell mit gemischten Effekten und Messwiederholung.
Abbildung 6: Histogramm der Veränderung des Norfolk QoL-DN-Gesamtscores vs. Baseline (NEURO-TTRansform-Studie) (Sicherheitsanalysekollektiv)
a) in Woche 35
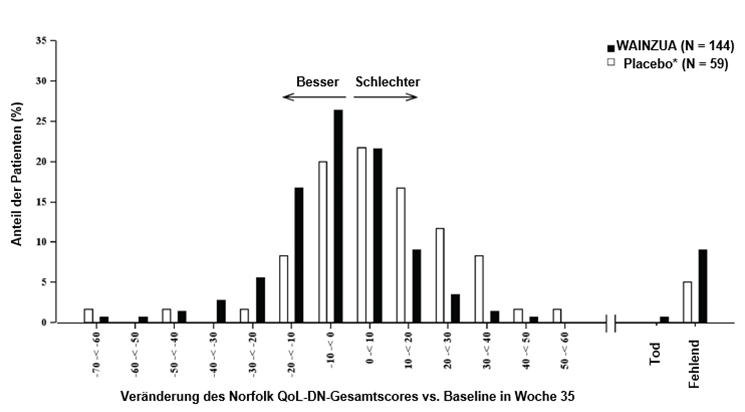
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
b) in Woche 66
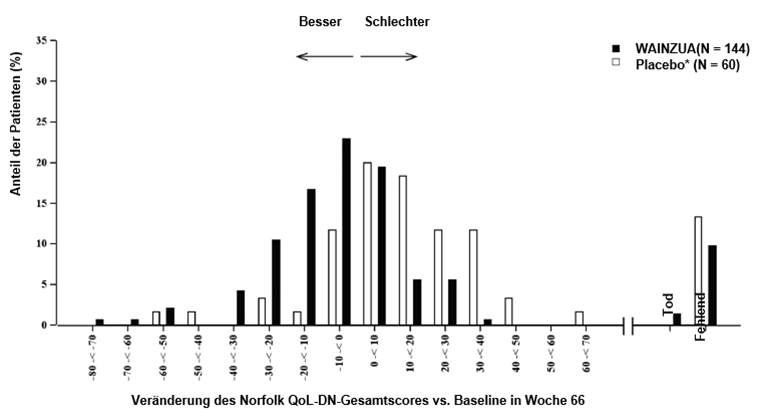
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
Sowohl in Woche 35 als auch in Woche 65/66 zeigten Patienten unter WAINZUA ähnliche Verbesserungen gegenüber Placebo in Bezug auf die Reduktion der TTR-Serumkonzentration, den kombinierten mNIS+7- und den Norfolk QoL-DN-Gesamtscore in allen Subgruppen einschliesslich Alter, Geschlecht, Ethnie, Region, Val30Met-Mutationsstatus, Kardiomyopathie-Status, Baseline-FAC-Diagnose und Krankheitsstadium (Abbildungen 1a und b, 7a und b sowie 8a und b).
Abbildung 7: Forest-Plot der LSM-Behandlungsdifferenz bezüglich der Veränderung des kombinierten mNIS+7-Scores vs. Baseline für wichtige Subgruppen (NEURO-TTRansform-Studie) (vollständiges Analysekollektiv)
a) in Woche 35
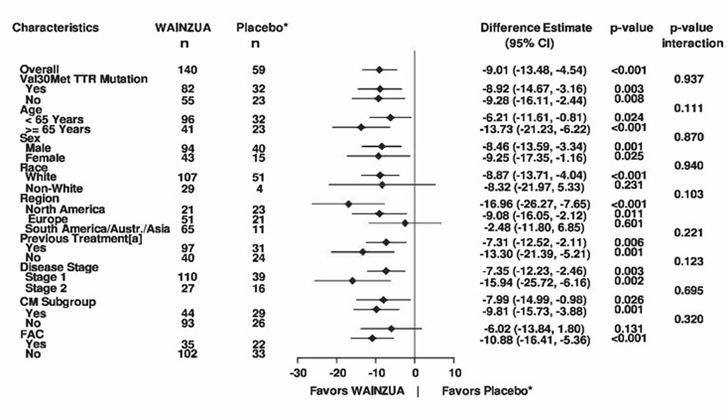
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
[a] Vorbehandelt mit Tafamidis oder Diflunisal.
Die CM-Subgruppe umfasst Patienten mit der Diagnose einer FAC bei Studieneintritt oder einer IV-Septumwanddicke ≥13 mm zu Studienbeginn ohne Hypertonie [Anamnese oder Diagnose während der Studie].
Die LS-Mittelwert-Differenz, Konfidenzintervalle und p-Werte basieren auf einem ANCOVA-Modell adjustiert durch Propensity Score mit den Effekten Behandlung, Subgruppenfaktoren, Krankheitsstadium, Val30Met-Mutation, Vorbehandlung, Behandlung-Subgruppe-Interaktion und Ausgangswert. In die 35-Wochen-Analyse wurden Daten bis Woche 35 einbezogen.
b) in Woche 66
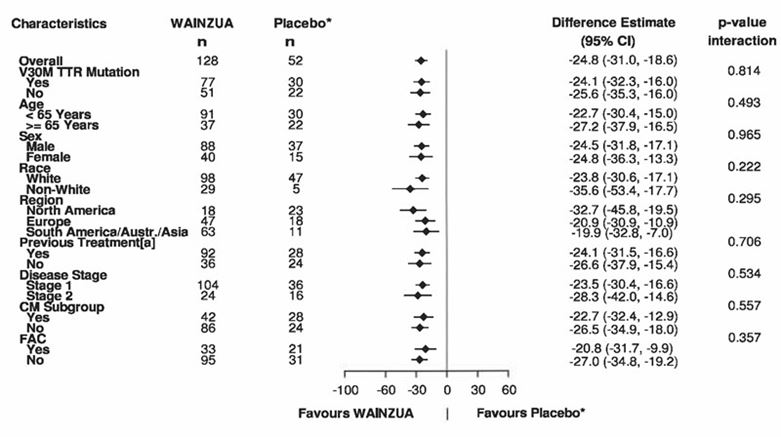
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
[a] Vorbehandelt mit Tafamidis oder Diflunisal.
Basierend auf MMRM adjustiert durch Propensity-Score-Gewichtung mit kategorischen Effekten für Behandlung, Zeit, Behandlung-Zeit-Interaktion und Krankheitsstadium, Val30Met-Mutation, Vorbehandlung sowie festen Kovariablen für die Baseline und die Baseline-Zeit-Interaktion.
Die Subgruppen-Modelle enthielten auch Behandlung-Subgruppe-, Zeit-Subgruppe- und Behandlung-Zeit-Subgruppe-Interaktionen. Daten bis Woche 66 sind einbezogen.
Die CM-Subgruppe umfasst Patienten mit der Diagnose einer FAC bei Studieneintritt oder einer IV-Septumwanddicke ≥13 mm zu Studienbeginn ohne Hypertonie [Anamnese oder Diagnose während der Studie].
Die LSM-Behandlungsdifferenzen nach 66 Wochen (WAINZUA – Placebo) sind mit 95%-KI (unadjustiert) aufgeführt.
KI = Konfidenzintervall, LSM = Least-Squares-Mittelwert, MMRM = Modell mit gemischten Effekten und Messwiederholung, CM = Kardiomyopathie, FAC = familiäre Amyloid-Kardiomyopathie.
Abbildung 8: Forest-Plot der LSM-Behandlungsdifferenz bezüglich der Veränderung des Norfolk QoL-DN-Gesamtscores vs. Baseline für wichtige Subgruppen (NEURO-TTRansform-Studie) (vollständiges Analysekollektiv)
a) in Woche 35
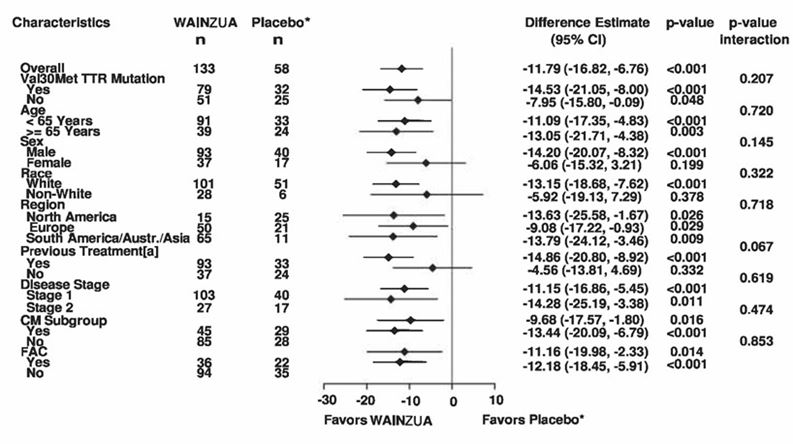
* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
[a] Vorbehandelt mit Tafamidis oder Diflunisal.
Die CM-Subgruppe umfasst Patienten mit der Diagnose einer FAC bei Studieneintritt oder einer IV-Septumwanddicke ≥13 mm zu Studienbeginn ohne Hypertonie [Anamnese oder Diagnose während der Studie].
Die LS-Mittelwert-Differenz, Konfidenzintervalle und p-Werte basieren auf einem ANCOVA-Modell adjustiert durch Propensity Score mit den Effekten Behandlung, Subgruppenfaktoren, Krankheitsstadium, Val30Met-Mutation, Vorbehandlung, Behandlung-Subgruppe-Interaktion und Ausgangswert. In die 35-Wochen-Zwischenanalyse wurden nur Daten bis Woche 35 einbezogen.
b) in Woche 66

* Externe Placebogruppe aus einer anderen randomisierten, kontrollierten Studie (NEURO-TTR).
[a] Vorbehandelt mit Tafamidis oder Diflunisal.
Basierend auf MMRM adjustiert durch Propensity-Score-Gewichtung mit kategorischen Effekten für Behandlung, Zeit, Behandlung-Zeit-Interaktion und Krankheitsstadium, Val30Met-Mutation, Vorbehandlung sowie festen Kovariablen für die Baseline und die Baseline-Zeit-Interaktion.
Die Subgruppen-Modelle enthielten auch Behandlung-Subgruppe-, Zeit-Subgruppe- und Behandlung-Zeit-Subgruppe-Interaktionen. Daten bis Woche 66 sind einbezogen.
Die CM-Subgruppe umfasst Patienten mit der Diagnose einer FAC bei Studieneintritt oder einer IV-Septumwanddicke ≥13 mm zu Studienbeginn ohne Hypertonie [Anamnese oder Diagnose während der Studie].
Die LSM-Behandlungsdifferenzen nach 66 Wochen (WAINZUA – Placebo) sind mit 95%-KI (unadjustiert) aufgeführt.
KI = Konfidenzintervall, LSM = Least-Squares-Mittelwert, MMRM = Modell mit gemischten Effekten und Messwiederholung, CM = Kardiomyopathie, FAC = familiäre Amyloid-Kardiomyopathie.
In einer explorativen Analyse der kardialen Untersuchungen mittels serieller Echokardiogramme zeigte WAINZUA eine Verbesserung im E/e'-Verhältnis (ein Mass für die linksventrikuläre diastolische Funktion) nach 65-wöchiger Behandlung in der Kardiomyopathie-Subgruppe (adjustierte placebokontrollierte LS-Mittelwert-Differenz: 3,94 [95%-KI 6,46, 1,42]). Veränderungen in Richtung eines Nutzens von WAINZUA gegenüber Placebo in Woche 66 wurden auch beobachtet bezüglich der vorgegebenen explorativen kardialen Endpunkten mittlere LV-linksventrikuläre (LV) Wanddicke (LSM-Differenz -0,04 cm [95%-KI -0,12, 0,04]), Wanddicke des Ventrikelseptums (LSM-Differenz -0,05 cm [95%-KI -0,16, 0,06]) und NT proBNP, ein prognostischer Biomarker der kardialen Dysfunktion (geometrischer LSM 0,88, [95%-KI 0,68, 1,14]) beobachtet. Trotz dieser beobachteten Werte ist ein klinischer Nutzen bei Kardiomyopathie noch zu bestätigen.
Analyse in Woche 85 (Analyse bei Behandlungsende)
Daten aus Woche 85 liegen für die externe Placebogruppe nicht vor, da die Behandlungsdauer in der Studie NEURO-TTR nur 66 Wochen betrug.
Der beobachtete Effekt in der mit WAINZUA behandelten Gruppe bezüglich des kombinierten mNIS+7-Scores war gleichbleibend und hielt bis zum Behandlungsende in Woche 85 an. Die mittlere (SD) Veränderung des kombinierten mNIS+7-Scores vs. Baseline betrug -0,04 % (16,2) in Woche 35, -0,21 % (17,6) in Woche 66 und -2,9 % (20,5) in Woche 85. Der mittlere Norfolk QoL-DN-Gesamtscore blieb bis Woche 85 stabil. In der Eplontersen-Gruppe betrug die mittlere (SD) Veränderung des Norfolk QoL-DN-Gesamtscores vs. Baseline -4,8 (16,5) in Woche 35, -7,2 (18,5) in Woche 66 und 6,2 (18,0) in Woche 85.
Die Parameter NSC, PND und mBMI blieben bis Woche 85 stabil, während der SF-36 weiterhin eine Tendenz zur Verbesserung zeigte.
PharmakokinetikDie pharmakokinetischen (PK) Eigenschaften von WAINZUA wurden nach subkutaner Verabreichung von Einzel- und Mehrfachdosen (einmal alle 4 Wochen) an gesunde Probanden und Mehrfachdosen (einmal alle 4 Wochen) an Patienten mit ATTRv-PN untersucht.
Absorption
Nach subkutaner Verabreichung wird Eplontersen rasch in den systemischen Kreislauf resorbiert, wobei die Zeit bis zum Erreichen der maximalen Plasmakonzentration basierend auf Populationsschätzungen ca. 2 Stunden beträgt.
Distribution
Basierend auf tierexperimentellen Studien (Maus, Ratte, Affe) wird Eplontersen nach subkutaner Gabe hauptsächlich in die Leber und die Nierenrinde verteilt. Eplontersen wird hochgradig an humane Plasmaproteine gebunden (> 98 %). Die Populationsschätzungen für das apparente zentrale Verteilungsvolumen betragen 12,9 l, und das apparente periphere Verteilungsvolumen beträgt 11,100 l.
Metabolismus
Eplontersen wird in der Leber durch Endo- und Exonukleasen zu kurzen Oligonukleotidfragmenten unterschiedlicher Grösse verstoffwechselt. Es wurden keine zirkulierenden Hauptmetaboliten bei Menschen festgestellt. Oligonukleotid-Therapeutika wie Eplontersen werden typischerweise nicht von CYP-Enzymen metabolisiert.
Elimination
Eplontersen wird hauptsächlich durch Verstoffwechselung und anschliessende renale Ausscheidung der kurzen Oligonukleotid-Metaboliten eliminiert. Die mittlere Fraktion von im Urin ausgeschiedenem unverändertem ASO betrug weniger als 1 % der verabreichten Dosis innerhalb von 24 Stunden. Die terminale Eliminationshalbwertszeit beträgt ungefähr 3 Wochen auf Basis von Populationsschätzungen.
Linearität/Nichtlinearität
Die Cmax und AUC von Eplontersen zeigten einen etwas stärkeren als dosisproportionalen Anstieg nach subkutanen Einzeldosen im Bereich von 45 bis 120 mg (d.h. das 1- bis 2,7-Fache der empfohlenen Dosis) bei gesunden Probanden.
Populationsschätzungen der Maximalkonzentrationen (Cmax), Talkonzentrationen (Ctrough) und der Fläche unter der Kurve (AUCτ) im Steady State betrugen 0,218 μg/ml, 0,000200 μg/ml bzw. 1,95 μg h/ml nach Gabe von 45 mg alle 4 Wochen bei Patienten mit ATTRv-PN. Nach wiederholter Gabe (einmal alle 4 Wochen) wurde keine Kumulation von Eplontersen (Cmax und AUC) im Plasma beobachtet. Eine Kumulation wurde im Ctrough beobachtet, und der Steady State wird nach ungefähr 17 Wochen erreicht.
Kinetik spezieller Patientengruppen
Basierend auf der populationspharmakokinetischen und -pharmakodynamischen Analyse haben Körpergewicht, Geschlecht, Ethnie und Val30Met-Mutationsstatus keine klinisch bedeutsame Auswirkung auf die Exposition gegenüber Eplontersen oder die Serum-TTR-Reduktion im Steady State. Die endgültige Beurteilung war in einigen Fällen nur eingeschränkt möglich, weil die Kovariablen durch die insgesamt niedrigen Zahlen begrenzt waren.
Ältere Patienten
Es wurden insgesamt keine Unterschiede der Pharmakokinetik zwischen jüngeren und älteren Patienten (≥65 Jahre) beobachtet.
Nierenfunktionsstörungen
Es wurden keine formalen klinischen Studien durchgeführt, um den Einfluss einer eingeschränkten Nierenfunktion auf die PK von Eplontersen zu untersuchen. Eine populationspharmakokinetische und -pharmakodynamische Analyse zeigte keine klinisch bedeutsamen Unterschiede der Pharmakokinetik oder Pharmakodynamik von Eplontersen bei leichter und mittelschwerer Nierenfunktionsstörung (eGFR ≥45 bis < 90 ml/min). Eplontersen wurde bei Patienten mit schwerer Nierenfunktionsstörung oder Patienten mit terminaler Niereninsuffizienz nicht untersucht.
Leberfunktionsstörungen
Es wurden keine formalen klinischen Studien durchgeführt, um den Einfluss einer eingeschränkten Leberfunktion auf Eplontersen zu untersuchen. Eine populationspharmakokinetische und -pharmakodynamische Analyse zeigte keine klinisch bedeutsamen Unterschiede der Pharmakokinetik oder Pharmakodynamik von Eplontersen bei leichter Leberfunktionsstörung (Gesamtbilirubin ≤1 x ULN und AST > 1 x ULN oder Gesamtbilirubin > 1,0 bis 1,5 x ULN und jeglicher AST-Wert). Eplontersen wurde bei Patienten mit mittelschwerer oder schwerer Leberfunktionsstörung und bei Patienten mit früherer Lebertransplantation nicht untersucht.
Arzneimittelwechselwirkungen
Es wurden keine formalen klinischen Studien zur Erfassung von Arzneimittelwechselwirkungen durchgeführt. In-vitro-Studien zeigen, dass Eplontersen kein Substrat oder Inhibitor von Transportern ist, nicht mit stark an Plasmaproteine gebundenen Arzneimitteln interagiert und kein Inhibitor oder Induktor von CYP-Enzymen ist. Oligonukleotid-Therapeutika wie Eplontersen sind typischerweise keine Substrate von CYP-Enzymen. Daher ist nicht zu erwarten, dass Eplontersen Arzneimittelwechselwirkungen verursacht oder durch Arzneimittelwirkungen, die durch Wirkstofftransporter, die Plasmaproteinbindung oder CYP-Enzyme vermittelt werden, beeinflusst wird.
Präklinische DatenNichtklinische Toxizität/Toxizität bei wiederholter Gabe
Die wiederholte Verabreichung von Eplontersen oder nagerspezifischem Surrogat führte zur Reduktion der hepatischen TTR-mRNA-Konzentrationen (Reduktionen von bis zu ~ 62 % und 82 % bei Affen bzw. Mäusen) mit nachfolgenden Abnahmen der TTR-Plasmaproteinspiegel (Abnahme von bis zu 70 % bei Affen). Es ergaben sich keine toxikologisch relevanten Befunde im Zusammenhang mit dieser pharmakologischen Hemmung der TTR-Expression.
Die meisten Befunde, die nach wiederholter Gabe über bis zu 6 Monate bei Mäusen und 9 Monate bei Affen beobachtet wurden, betrafen die Aufnahme und Kumulation von Eplontersen und wurden nicht als schädlich betrachtet. Durch die Aufnahme von Eplontersen bedingte mikroskopische Befunde wurden in verschiedenen Zelltypen mehrerer Organe bei allen getesteten Tierarten beobachtet, unter anderem in Monozyten/Makrophagen, proximalen Tubulusepithelien der Nieren, Kupffer-Zellen der Leber und Histiozyteninfiltraten in Lymphknoten und Injektionsstellen.
Stark erniedrigte Thrombozytenzahlen in Verbindung mit Spontanblutungen wurden in einer Studie zur subchronischen Toxizität an einem Affen bei der höchsten getesteten Dosis (24 mg/kg/Woche) beobachtet. Bei Affen, die eine mittlere Dosis von 6 mg/kg/Woche erhielten, was dem 73 Fachen der humanen AUC bei der empfohlenen therapeutischen Dosis von Eplontersen entspricht, wurden keine derartigen Befunde beobachtet.
Mutagenität und Karzinogenität
Eplontersen zeigte in vitro und in vivo kein genotoxisches Potenzial und war bei transgenen ras.H2-Mäusen nicht karzinogen.
In In-vitro-Tests (bakterielle Mutagenität, Chromosomenaberration in der Lunge des chinesischen Hamster) und In-vivo-Tests (Mikronukleus-Assay im Knochenmark der Maus) war Eplontersen negativ für Genotoxizität.
In einer subkutanen Karzinogenitätsstudie an transgenen ras.H2-Mäusen wurde Eplontersen über 26 Wochen in Dosen von 250, 500 und 1500 mg/kg/Monat verabreicht. Nach der 26-wöchigen Behandlung der Mäuse ergaben sich keine Hinweise auf eine Karzinogenität von Eplontersen.
Reproduktionstoxizität
Embryofetale Toxizität/Entwicklungstoxizität/Fertilität
Eplontersen hatte keine Wirkungen auf die Fertilität oder die embryofetale Entwicklung bei Mäusen im bis zu 38-Fachen der empfohlenen monatlichen Dosis von 45 mg beim Menschen. Eplontersen ist bei Mäusen nicht pharmakologisch aktiv. Mit einem mausspezifischen Analogon von Eplontersen, das mit einer Hemmung der TTR-mRNA-Expression um > 90 % bei Mäusen assoziiert war, wurde jedoch ebenfalls keine Wirkung auf die Fertilität oder die embryofetale Entwicklung festgestellt.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern.
WAINZUA kann in der Originalverpackung ungekühlt für bis zu 6 Wochen bei maximal 30 °C aufbewahrt werden. Falls es nicht innerhalb von 6 Wochen aufgebraucht wird, sollte es verworfen werden.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Nicht einfrieren. Keiner Hitze aussetzen.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
WAINZUA sollte vor der Anwendung per Augenschein inspiziert werden. Die Lösung muss klar und farblos bis gelb sein. WAINZUA darf nicht angewendet werden, wenn die Lösung Trübungen aufweist, sichtbare Schwebstoffe enthält oder verfärbt ist.
Der vorgefüllte Fertigpen zum Einmalgebrauch sollte in einem durchstichsicheren Abwurfbehälter entsorgt werden.
Weitere Informationen und Hinweise zur Zubereitung und Verabreichung von WAINZUA mithilfe der des Fertigpens finden sich in der Packungsbeilage und in der «Gebrauchsanweisung».
Zulassungsnummer69332 (Swissmedic)
PackungenWAINZUA Injektionslösung: Packung mit 1 Fertigpen zum Einmalgebrauch [B]
ZulassungsinhaberinAstraZeneca AG, 6340 Baar
Stand der InformationMai 2025
|