ZusammensetzungWirkstoffe
Ruxolitinib (als Ruxolitinibphosphat)
Hilfsstoffe
Butylhydroxytoluol (E321, 1,4 µg/g), Cetylalkohol (30 mg/g), Dimeticon 350, Natriumedetat (E385), Glycerolstearate SE, Macrogol 200, mittelkettige Triglyceride, Methyl-4hydroxybenzoat (E218, 1 mg/g), dünnflüssiges Paraffin, weisses Vaselin (E905), Phenoxyethanol, Polysorbat 20 (E432), Propylenglycol (E1520, 150 mg/g), Propyl-4hydroxybenzoat (0,5 mg/g), gereinigtes Wasser, Stearylalkohol (17,5 mg/g), Xanthangummi (E415)
Indikationen/AnwendungsmöglichkeitenOPZELURA wird angewendet zur Behandlung von nichtsegmentaler Vitiligo mit Beteiligung des Gesichts bei Erwachsenen und Jugendlichen im Alter ab 12 Jahren, wenn nicht-therapeutische Massnahmen nicht ausreichen oder nicht gut vertragen werden. Siehe Rubriken «Warnhinweise und Vorsichtsmassnahmen für die Anwendung» und «Unerwünschte Wirkungen».
Dosierung/AnwendungDie Behandlung mit OPZELURA sollte von Ärzten eingeleitet und überwacht werden, die Erfahrung in der Diagnose und Behandlung von nichtsegmentaler Vitiligo haben.
Übliche Dosierung
Erwachsene
Die empfohlene Dosis ist eine dünne Schicht Creme, die zweimal täglich auf die depigmentierten Hautbereiche bis zu höchstens 10 % der Körperoberfläche aufgetragen wird, wobei zwischen zwei Anwendungen von Ruxolitinib-Creme mindestens 8 Stunden liegen müssen. 10 % der Körperoberfläche entsprechen einer Fläche des 10-Fachen einer Handfläche mit den 5 Fingern. 10 % der Körperoberfläche sollten nicht überschritten werden, da die Sicherheit über 10 % der Körperoberfläche bei Patienten mit Vitiligo nicht nachgewiesen wurde.
Es sollten nicht mehr als zwei 100 g Tuben pro Monat angewendet werden.
Dauer der Behandlung
Für eine zufriedenstellende Repigmentierung kann eine Behandlung über 24 Wochen hinaus erforderlich sein. Wenn in der 52. Woche weniger als 25 % der behandelten Bereiche repigmentiert sind, sollte ein Abbruch der Behandlung in Betracht gezogen werden.
Nach Erreichen einer zufriedenstellenden Repigmentierung kann die Behandlung in diesen Bereichen unterbrochen werden.
Die Behandlung muss nicht ausgeschlichen werden.
Patienten mit Leberfunktionsstörungen
Es wurden keine Studien mit Ruxolitinib-Creme bei Patienten mit eingeschränkter Leberfunktion durchgeführt. Aufgrund der begrenzten systemischen Exposition ist eine Dosisanpassung bei Patienten mit eingeschränkter Leberfunktion jedoch nicht erforderlich.
Patienten mit Nierenfunktionsstörungen
Es wurden keine Studien mit Ruxolitinib-Creme bei Patienten mit eingeschränkter Nierenfunktion durchgeführt. Aufgrund der begrenzten systemischen Exposition ist eine Dosisanpassung bei Patienten mit eingeschränkter Nierenfunktion jedoch nicht erforderlich. Als Vorsichtsmassnahme sollte dieses Arzneimittel nicht von Patienten mit terminaler Niereninsuffizienz angewendet werden, da keine Daten zur Sicherheit vorliegen.
Ältere Patienten
Eine begrenzte Anzahl von Patienten im Alter von 65 Jahren und älter wurde in die klinischen Studien mit OPZELURA bei Vitiligo aufgenommen, um festzustellen, ob sie anders ansprechen als jüngere Patienten (siehe Rubrik «Eigenschaften/Wirkungen»). Bei Patienten ab 65 Jahren ist keine Dosisanpassung erforderlich.
Kinder und Jugendliche
Für Jugendliche (12-17 Jahre) gilt die gleiche Dosierung wie für Erwachsene.
Die Sicherheit und Wirksamkeit von OPZELURA bei Kindern unter 12 Jahren ist nicht erwiesen.
OPZELURA ist für Kinder unter 12 Jahren nicht indiziert.
Art der Anwendung
Die Creme ist nur zur Anwendung auf der Haut bestimmt.
Die behandelten Hautstellen sollten mindestens 2 Stunden nach der Anwendung von OPZELURA nicht gewaschen werden.
Die Creme darf nicht auf die Lippen aufgetragen werden, um ein versehentliches Verschlucken zu vermeiden.
Die Patienten sollten sich nach dem Auftragen der Creme die Hände waschen, es sei denn, die Hände selbst werden behandelt. Wenn eine andere Person dem Patienten die Creme aufträgt, sollte diese sich nach dem Auftragen die Hände waschen.
Die Patienten sollten darauf hingewiesen werden, dass sie im Falle einer vergessenen Dosis die vergessene Dosis auslassen und die nächste Dosis zur gewohnten Zeit anwenden sollten, wenn weniger als 8 Stunden bis zur nächsten geplanten Dosis verbleiben.
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der in der Rubrik «Zusammensetzung» genannten Hilfsstoffe.
Schwangerschaft und Stillzeit (siehe Rubrik «Schwangerschaft, Stillzeit»).
Warnhinweise und VorsichtsmassnahmenUnter der Behandlung mit Opzelura kann die systemische Konzentration von Ruxolitinib auf Werte ansteigen, die zu einer systemischen JAK-Hemmung führen können, sodass das Auftreten von systemischen unerwünschten Wirkungen nicht ausgeschlossen werden kann, einschliesslich der Klasseneffekte der unten beschriebenen oralen JAK-Inhibitoren, die zur Behandlung von chronisch entzündlichen Erkrankungen verschrieben werden. In Einzelfällen wurden nach der Anwendung von Opzelura Ruxolitinib-Konzentrationen beobachtet, die etwa das 10-Fache im Vergleich zur durchschnittlichen Steady-State-Konzentration einer oralen Verabreichung von 2 x 5 mg Ruxolitinib-Tabletten betragen. Die durchschnittliche Bioverfügbarkeit von Opzelura beträgt ca. 12,5 % im Vergleich zur oralen Anwendung. Nutzen und Risiken müssen für jeden Patienten individuell abgewogen werden, bevor eine Behandlung mit Opzelura begonnen oder fortgesetzt wird. Die folgenden unerwünschten Wirkungen wurden bei anderen systemisch verfügbaren JAK-Inhibitoren beobachtet:
Schwere Infektionen
Bei Patienten, denen JAK-Inhibitoren oral verabreicht wurden, wurde über schwerwiegende, manchmal sogar tödliche Infektionen durch Bakterien, Mykobakterien, invasive Pilze, Viren oder andere opportunistische Erreger berichtet.
Die Anwendung von Opzelura sollte bei Patienten mit aktiven schweren Infektionen, einschliesslich lokalisierter Infektionen, vermieden werden.
Der Nutzen und die Risiken der Behandlung müssen vor Beginn der Behandlung mit Opzelura abgewogen werden bei:
§Patienten mit chronischen oder wiederkehrenden Infektionen
§Patienten mit einer Vorgeschichte von schweren oder opportunistischen Infektionen
§Patienten mit erfolgter Tuberkuloseexposition
§Patienten, die sich in Gebieten aufgehalten haben, in denen Tuberkulose oder Pilzerkrankungen endemisch sind, oder die in solche Gebiete gereist sind, oder
§Patienten mit zugrunde liegenden Krankheiten, die sie für eine Infektion prädisponieren könnten.
Überwachen Sie die Patienten während und nach der Behandlung mit Opzelura engmaschig auf Anzeichen oder Symptome einer Infektion.
Brechen Sie die Behandlung mit Opzelura ab, wenn Ihr Patient eine schwerwiegende oder opportunistische Infektion oder eine Sepsis entwickelt.
Beginnen Sie die Behandlung mit Opzelura erst wieder, wenn die Infektion unter Kontrolle ist.
Tuberkulose (TB)
In klinischen Studien mit oralen JAK-Inhibitoren zur Behandlung von chronisch entzündlichen Erkrankungen wurden Fälle von aktiver Tuberkulose berichtet. Sie sollten erwägen, Ihren Patienten vor der Anwendung von Opzelura auf latente oder aktive TB untersuchen zu lassen. Während der Behandlung mit Opzelura müssen die Patienten auf Anzeichen und Symptome von TB überwacht werden.
Virale Reaktivierung
Eine virale Reaktivierung, einschliesslich Fällen von Reaktivierung des Herpesvirus (z. B. Varizella-Zoster-Virus), wurde in klinischen Studien mit JAK-Inhibitoren zur Behandlung von chronisch entzündlichen Erkrankungen berichtet. Sie sollten eine vorübergehende Unterbrechung der Behandlung mit Opzelura bis zum Abklingen der Episode in Betracht ziehen, wenn bei Ihrem Patienten Herpes Zoster auftritt.
Hepatitis B und C
Die Auswirkungen von JAK-Inhibitoren, die zur Behandlung von entzündlichen Erkrankungen eingesetzt werden, auf die Reaktivierung einer chronischen viralen Hepatitis sind nicht bekannt. Patienten mit einer Vorgeschichte von Hepatitis B oder C wurden von den klinischen Studien ausgeschlossen.
Bei Patienten mit chronischer HBV-Infektion, die einen oralen JAK-Inhibitor einnehmen, wurde über Erhöhungen der Hepatitis-B-Viruslast (HBV-DNA-Titer) mit oder ohne damit verbundene Erhöhungen der Alanin-Aminotransferase und der Aspartat-Aminotransferase berichtet.
Die Anwendung von Opzelura bei Patienten mit aktiver Hepatitis B oder C wird nicht empfohlen.
Gesamtmortalität
In einer grossen randomisierten Sicherheitsstudie nach der Markteinführung eines oralen JAK-Inhibitors bei Patienten ab 50 Jahren mit rheumatoider Arthritis (RA) und mindestens einem kardiovaskulären Risikofaktor war die Gesamtmortalitätsrate – einschliesslich plötzlicher kardiovaskulärer Todesfälle – bei Patienten, die mit einem oralen JAK-Inhibitor behandelt wurden, höher als bei Patienten, die mit TNF-Inhibitoren (Tumornekrosefaktor) behandelt wurden.
Maligne Tumore
In einer grossen randomisierten Sicherheitsstudie, die nach der Markteinführung eines oral verabreichten JAK-Inhibitors bei RA-Patienten durchgeführt wurde, wurde bei Patienten, die mit einem oral eingenommenen JAK-Inhibitor im Vergleich zu TNF-Inhibitoren behandelt wurden, eine erhöhte Anzahl von Tumorerkrankungen, insbesondere Lungenkrebs, Lymphome und nichtmelanozytärer Hautkrebs (NMSC) beobachtet. Maligne Tumore, einschliesslich Lymphome, wurden in klinischen Studien mit oralen JAK-Inhibitoren zur Behandlung von entzündlichen Erkrankungen beobachtet. Patienten, die rauchen oder geraucht haben, haben ein erhöhtes zusätzliches Risiko.
Nichtmelanozytärer Hautkrebs (NMSC)
Bei Patienten, die mit Opzelura behandelt wurden, wurde das Auftreten von nichtmelanozytärem Hautkrebs (NMSC), einschliesslich Basalzell- und Plattenepithelkarzinomen, berichtet. Sie sollten während und gegebenenfalls nach der Behandlung mit Opzelura regelmässig Hautuntersuchungen durchführen. Die Exposition gegenüber der Sonne und UV-Strahlen sollte durch das Tragen von entsprechender Kleidung mit UV-Schutz und die Verwendung eines Breitband-Sonnenschutzmittels begrenzt werden.
Schwere unerwünschte kardiovaskuläre Ereignisse (MACE)
In einer grossen randomisierten Sicherheitsstudie nach der Markteinführung eines oralen JAK-Hemmers bei RA-Patienten im Alter von 50 Jahren und älter mit mindestens einem kardiovaskulären Risikofaktor wurde eine höhere Rate an MACEs (kardiovaskulärer Tod, nicht tödlicher Myokardinfarkt und nicht tödlicher Schlaganfall) beobachtet als bei einer Behandlung mit TNF-Inhibitoren. Patienten, die rauchen oder geraucht haben, hatten ein erhöhtes zusätzliches Risiko.
Die Patienten müssen über die Symptome von schweren kardiovaskulären Ereignissen und die Massnahmen, die im Falle eines solchen Ereignisses zu ergreifen sind, informiert werden. Beenden Sie die Behandlung mit Opzelura bei Patienten, die einen Myokardinfarkt oder Schlaganfall in der Vorgeschichte hatten.
Thromboembolische Komplikationen
Thrombosen, einschliesslich tiefer Venenthrombosen (TVT), Lungenembolien (PE) und arterieller Thrombosen, wurden bei Patienten berichtet, die orale JAK-Inhibitoren zur Behandlung von entzündlichen Erkrankungen erhalten haben. Viele dieser unerwünschten Ereignisse waren schwerwiegend und einige führten zum Tod der Patienten.
In einer grossen randomisierten Sicherheitsstudie nach Markteinführung eines oralen JAK-Inhibitors bei RA-Patienten ab 50 Jahren mit mindestens einem kardiovaskulären Risikofaktor wurden im Vergleich zu Patienten, die mit TNF-Inhibitoren behandelt wurden, höhere Raten von Thrombosen aller Art, TVT und PE beobachtet.
Sie sollten Opzelura nicht an Patienten mit einem erhöhten Thromboserisiko verschreiben. Wenn Symptome einer Thrombose auftreten, muss die Behandlung mit Opzelura abgebrochen werden und der Patient muss untersucht und entsprechend behandelt werden.
Thrombozytopenie, Anämie und Neutropenie
In den klinischen Studien mit anderen JAK-Inhibitoren wurden Thrombozytopenie, Anämie und Neutropenie berichtet. Wenn es klinisch angezeigt ist, muss das Blutbild überwacht werden. Wenn Anzeichen und/oder Symptome einer klinisch relevanten Thrombozytopenie, Anämie oder Neutropenie auftreten, muss die Behandlung mit Opzelura abgebrochen werden.
Die Creme ist nicht für die ophthalmische, orale oder intravaginale Verabreichung bestimmt (siehe Rubrik «Art der Anwendung»). Wenn die Creme versehentlich in die Augen oder auf Schleimhäute gelangt, muss sie sorgfältig abgewischt und/oder mit Wasser abgespült werden.
Das Sicherheitsprofil der Langzeitanwendung von OPZELURA bei Vitiligo ist nicht bekannt. OPZELURA sollte auf einer möglichst kleinen Hautfläche angewendet werden und die Dosierungsempfehlungen (Rubrik «Übliche Dosierung») dürfen nicht überschritten werden.
Bei Patienten, die mit Ruxolitinib topisch behandelt wurden, wurden Fälle von nichtmelanozytärem Hautkrebs (NMSC) berichtet, und zwar hauptsächlich Fälle von Basalzellkarzinomen. Die meisten dieser Patienten wiesen Risikofaktoren auf, wie z. B. eine Vorgeschichte mit Phototherapie oder NMSC. Ein Kausalzusammenhang mit Ruxolitinib wurde nicht festgestellt. Eine regelmässige Untersuchung der Haut wird bei allen Patienten empfohlen, insbesondere bei Patienten mit Risikofaktoren für Hautkrebs.
Propylenglykol
Dieses Arzneimittel enthält 150 mg Propylenglykol (E1520) pro Gramm Creme.
Cetylalkohol und Stearylalkohol
Dieses Arzneimittel enthält Cetylalkohol und Stearylalkohol, die örtlich begrenzt Hautreizungen (z. B. Kontaktdermatitis) hervorrufen können.
4-hydroxybenzoate
Dieses Arzneimittel enthält Methyl-4hydroxybenzoat (E218) und Propyl-4hydroxybenzoat, die allergische Reaktionen (möglicherweise auch Spätreaktionen) hervorrufen können.
Butylhydroxytoluol
Dieses Arzneimittel enthält Spuren von Butylhydroxytoluol (E321), das örtlich begrenzt Hautreizungen (z. B. Kontaktdermatitis), Reizungen der Augen und der Schleimhäute hervorrufen kann.
InteraktionenEs wurden keine Studien zur Erfassung von Interaktionen mit topisch angewendetem OPZELURA durchgeführt.
Vorsicht ist geboten bei der gleichzeitigen Anwendung von OPZELURA mit starken CYP3A4-Inhibitoren, da dies das Risiko von unerwünschten Wirkungen von OPZELURA erhöhen könnte.
Basierend auf In-vitro-Daten wird Ruxolitinib vorwiegend durch Cytochrom P450 3A4 (CYP3A4) verstoffwechselt. Das Potenzial für Interaktionen mit oral angewendetem Ruxolitinib wurde in speziellen klinischen Pharmakologiestudien untersucht, in denen starke oder mässige CYP3A4-Inhibitoren oder ein starker CYP3A4-Induktor gegeben wurden. Die Fläche unter der Kurve (AUC) im Plasma wird bei gleichzeitiger Gabe eines starken CYP3A4-Inhibitors fast verdoppelt, während bei gleichzeitiger Gabe eines mässigen CYP3A4-Inhibitors nur ein leichter Anstieg zu beobachten war.
Die Anwendung von Ruxolitinib-Creme in Kombination mit anderen topischen Arzneimitteln zur Behandlung von Vitiligo wurde nicht untersucht und die gleichzeitige Anwendung auf denselben Hautbereichen wird nicht empfohlen.
Andere topische Arzneimittel, die zur Behandlung anderer Erkrankungen auf denselben Hautbereichen angewendet werden, sollten mindestens 2 Stunden nach der Anwendung von OPZELURA aufgetragen werden. Dies gilt auch für die Verwendung von Sonnenschutzmitteln oder Hautpflegemitteln (Emollienzien).
Schwangerschaft, StillzeitEmpfängnisverhütung bei Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter müssen während der gesamten Behandlungsdauer und bis zu 4 Wochen nach Beendigung der Behandlung eine zuverlässige Empfängnisverhütung anwenden.
Schwangerschaft
Bisher liegen keine oder nur sehr begrenzte Erfahrungen mit der Anwendung von Ruxolitinib bei Schwangeren vor. Es liegen keine ausreichenden Daten über die systemische Absorption von topischem Ruxolitinib während der Schwangerschaft vor. Bestimmte individuelle Faktoren (z. B. beschädigte Hautbarriere, übermässiger Gebrauch) können zu einer erhöhten systemischen Exposition beitragen.
Tierexperimentelle Studien haben gezeigt, dass Ruxolitinib nach oraler Verabreichung embryotoxisch und fötotoxisch ist. Bei Ratten oder Kaninchen wurde keine Teratogenität beobachtet (siehe Rubrik «Präklinische Daten»). OPZELURA ist während der Schwangerschaft kontraindiziert (siehe Rubrik «Kontraindikationen»).
Stillzeit
Es liegen keine Daten über das Vorhandensein von Ruxolitinib in der Muttermilch, über die Auswirkungen auf das gestillte Kind oder die Auswirkungen auf die Milchproduktion nach einer topischen Anwendung von OPZELURA vor. Nach der oralen Verabreichung von Ruxolitinib an säugende Ratten lag die Konzentration von Ruxolitinib und/oder seinen Metaboliten in der Milch um das 13-Fache höher als die maternale Plasmakonzentration. In Studien mit juvenilen Ratten führte die orale Verabreichung von Ruxolitinib zu Auswirkungen auf das Wachstum und die Knochenmasse (siehe Rubrik «Präklinische Daten»). OPZELURA ist während der Stillzeit kontraindiziert (siehe Rubrik «Kontraindikationen») und die Behandlung muss etwa 4 Wochen vor Beginn der Stillzeit abgebrochen werden.
Fertilität
Es gibt keine Daten über die Wirkung von Ruxolinitib auf die menschliche Fertilität. In Tierstudien wurden keine Auswirkungen auf die Fertilität bei der oralen Verabreichung von Ruxolinitib beobachtet.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenOPZELURA hat keinen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die Sicherheit wurde hauptsächlich in den Zulassungsstudien für den Zeitraum von bis zu einem Jahr bewertet. Die häufigste unerwünschte Wirkung ist Akne an der Applikationsstelle (5,8 %).
Auflistung der unerwünschten Wirkungen
Die unerwünschten Wirkungen (jede Kausalität) sind nachstehend nach Organsystemklassen und Häufigkeit aufgeführt. Die Häufigkeitskategorien sind folgendermassen definiert: sehr häufig (≥1/10), häufig (≥1/100 bis < 1/10), gelegentlich (≥1/1000 bis < 1/100), selten (≥1/10‘000 bis < 1/1000), sehr selten (< 1/10‘000) oder Häufigkeit nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Tabelle 1: Unerwünschte Wirkungen, die während der Behandlung bei Studienteilnehmern auftraten, die in den Studien TRuE-V1 (INCB 18424-306) und TRuE-V2 (INCB 18424-307) bis Woche 24 mit OPZELURA gegen Vitiligo behandelt wurden.
|
Organsystemklasse Häufigkeitskategorie
Unerwünschtes Ereignis
|
Häufigkeit
OPZELURA
N=449
|
Häufigkeit
Vehikel
N=224
| |
Infektionen und Infektionen
| |
Häufig (≥1/100 bis < 1/10)
| |
Rhinopharyngitis
|
4,2 %
|
2,2 %
| |
Infektion der oberen Atemwege
|
2,9 %
|
2,2 %
| |
Grippe
|
1,3 %
|
0,4 %
| |
Harnwegsinfektion
|
1,3 %
|
0,4 %
| |
Gelegentlich (≥1/1000 bis < 1/100)
| |
Follikulitis an der Applikationsstelle
|
0,7 %
|
NR
| |
Erkrankungen des Nervensystems
| |
Häufig (≥ 1/100 bis < 1/10)
| |
Kopfschmerzen
|
3,8 %
|
2,7 %
| |
Gefässerkrankungen
| |
Gelegentlich (≥1/1000 bis < 1/100)
| |
Hypertonie
|
0,9 %
|
NR
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
| |
Häufig (≥ 1/100 bis < 1/10)
| |
Akne an der Applikationsstelle
|
5,8 %
|
1,3 %
| |
Pruritus an der Applikationsstelle
|
5,1 %
|
2,7 %
| |
Erythem an der Applikationsstelle
|
1,6 %
|
0,4 %
| |
Ausschlag an der Applikationsstelle
|
1,6 %
|
0,9 %
| |
Fieber
|
1,3 %
|
NR
| |
Exfoliation an der Applikationsstelle
|
1,1 %
|
0,4 %
| |
Gelegentlich (≥ 1/1000 bis < 1/100)
| |
Dermatitis an der Applikationsstelle
|
0,9 %
|
NR
| |
Verfärbung an der Applikationsstelle
|
0,7 %
|
NR
| |
Trockenheit an der Applikationsstelle
|
0,7 %
|
0,4 %
| |
Schmerzen an der Applikationsstelle
|
0,4 %
|
NR
| |
Blauer Fleck an der Applikationsstelle
|
0,2 %
|
NR
| |
Ekzem an der Applikationsstelle
|
0,2 %
|
NR
| |
Urtikaria an der Applikationsstelle
|
0,2 %
|
NR
| |
Untersuchungen
| |
Häufig (≥ 1/100 bis < 1/10)
| |
Alaninaminotransferase erhöht
|
1,1 %
|
0,4 %
|
N: Anzahl der Studienteilnehmer
NR: Nicht berichtet
Einzelne Fälle von spezifischen schwerwiegenden unerwünschten Wirkungen, die bei einzelnen Studienteilnehmern (0,2 %) auftraten, die während der Doppelblindphase bis Woche 24 in den Studien TRuE-V1 und TRuE-V2 mit OPZELURA behandelt wurden: Analfistel, Appendizitis, Stenose der Koronararterie, Epstein-Barr-Virus-Hepatitis (Hepatitis durch infektiöse Mononukleose), Myokarditis.
Fälle von Neoplasmen, einschliesslich Brustkrebs, Eierstockkrebs (Ovarialkrebs), papilläres Schilddrüsenkarzinom, Prostatakrebs und nicht-melanozytärer Hautkrebs wurden während der Behandlung mit OPZELURA beobachtet oder symptomatisch.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEine Überdosierung nach Anwendung auf der Haut ist unwahrscheinlich. Wenn zu viel Creme aufgetragen wurde, kann die überschüssige Menge abgewischt werden.
Bei versehentlichem Kontakt mit den Augen, der Mundschleimhaut oder der Vagina sollte die Creme gründlich abgewischt und/oder mit Wasser abgespült werden (siehe Rubriken «Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»).
Eigenschaften/WirkungenATC-Code
D11AH09
Wirkungsmechanismus / Pharmakodynamik
Ruxolitinib ist ein Inhibitor der Januskinasen (JAK) mit Selektivität für die Isoformen JAK1 und JAK2. Die intrazelluläre JAK-Signalgebung umfasst die Rekrutierung von STAT-Proteinen (Signaltransducer und Transkriptionsaktivatoren) an Zytokinrezeptoren und die anschliessende Modulation der Genexpression. Es wird angenommen, dass autoimmune, IFNγ-produzierende zytotoxische T-Lymphozyten direkt für die Zerstörung von Melanozyten bei Vitiligo beim Menschen verantwortlich sind. Die Rekrutierung zytotoxischer Lymphozyten in die Hautläsionen wird durch IFNγ-abhängige Chemokine wie CXCL10 vermittelt. Die nachgeschaltete Signalgebung von IFNγ ist JAK1/2-abhängig und die Behandlung mit Ruxolitinib senkt die CXCL10-Spiegel bei Vitiligo-Patienten.
Klinische Wirksamkeit
Es wurden insgesamt 674 Patienten mit Vitiligo mit Beteiligung des Gesichts und einer Gesamtkörper-Vitiligofläche (Gesicht und ausserhalb des Gesichts) von nicht mehr als 10 % der Körperoberfläche, mit einem Krankheitsausmass zu Beginn von 3,2% bis 10,1% der Körperoberfläche, in zwei identisch angelegte doppelblinde, randomisierte, vehikelkontrollierte Studien (TRuE-V1, INCB 18424-306 und TRuE-V2, INCB 18424-307) aufgenommen. Die Patienten waren 12 Jahre und älter (10,7 % der Patienten waren 12 bis 17 Jahre alt und 6,7 % waren 65 Jahre oder älter). Der Frauenanteil betrug 53,1 %, 81,9 % der Patienten waren weisser Hautfarbe, 4,7 % waren schwarzer Hautfarbe und 4,2 % waren asiatischer Herkunft. Die Mehrheit der Patienten hatte die Fitzpatrick-Hauttypen III, IV, V oder VI (67,5 %).
In beiden Studien wurden Patienten mit einer betroffenen Körperoberfläche von nicht mehr als 10 % im Verhältnis 2:1 randomisiert und erhielten 24 Wochen lang zweimal täglich entweder Ruxolitinib-Creme oder Vehikel, wenn die betroffene Körperoberfläche nicht mehr als 10 % betrug. Auf diese erste Phase folgt eine Behandlungsphase von weiteren 28 Wochen, während der alle Patienten zweimal täglich mit Ruxolitinib-Creme behandelt werden. Der primäre Endpunkt war der Anteil der Patienten, die in Woche 24 eine Repigmentierung von 75 % erreichten, bewertet mit dem Vitiligo Area Scoring Index (F-VASI75) für das Gesicht. Zu den wichtigsten sekundären Endpunkten gehörten der Anteil der Patienten, die eine 90 %ige Repigmentierung F-VASI (F-VASI90), eine 50 %ige Verbesserung des Gesamtkörper-Vitiligo-Area-Scoring-Index (T-VASI50) und einen Wert von 4 oder 5 auf der Vitiligo Noticeability Scale (VNS) (Vitiligo «deutlich unauffälliger» oder «nicht mehr wahrnehmbar») erreichten.
In beiden Studien wurden eine Repigmentierung der behandelten Vitiligo-Läsionen und eine Überlegenheit der Ruxolitinib-Creme gegenüber der Vehikel-Creme beobachtet, was durch statistisch signifikante Unterschiede in den Ansprechraten für F-VASI75/90, T-VASI50 und den VNS-Score von 4 oder 5 in Woche 24 belegt wurde (Tabelle 2).
Der Unterschied der therapeutischen Wirkung im Vergleich zum Vehikel zeigt sich numerisch ab Woche 12. Eine anhaltende Repigmentierung, die anhand der VASI- und VNS-Scores bewertet wurde, wurde bis Woche 52 bei den Patienten beobachtet, die Ruxolitinib-Creme seit Beginn der Studie zweimal täglich angewendet hatten. Der Anteil der Patienten, die während des 52-wöchigen Behandlungszeitraums in den konsolidierten Daten der Studien TRuE-V1 und TRuE-V2 den F-VASI75-Score erreichten, ist in Abbildung 1 dargestellt.
Ein ähnliches Ansprechen auf die Behandlung wurde in Woche 52 bei den Patienten beobachtet, die von der Behandlung mit dem Vehikel auf Ruxolitinib umgestellt wurden (Abbildung 1).
Tabelle 2: Prozentualer Anteil der Patienten mit Vitiligo, die die primären und wichtigsten sekundären Endpunkte in Woche 24 erreichten (intent-to-treat)a
|
|
TRuE-V1 (INCB 18424-306)
|
TRuE-V2 (INCB 18424-307)
| |
OPZELURA
|
Vehikel
|
OPZELURA
|
Vehikel
| |
(N = 221)
|
(N = 109)
|
(N = 222)
|
(N = 109)
| |
F-VASI75 (%)
|
29,8
|
7,4
|
30,9
|
11,4
| |
Unterschied in der Antwortrate (95 %-KI)
|
22,3b
(14.214, 30.471)
|
-
|
19,5c
(10.537, 28.420)
|
-
| |
F-VASI90 (%)
|
15,3
|
2,2
|
16,3
|
1,3
| |
Unterschied in der Antwortrate (95 %-KI)
|
13,2d
(7.497, 18.839)
|
-
|
15,0e
(9.250, 20.702)
|
-
| |
T-VASI50 (%)
|
20,6
|
5,1
|
23,9
|
6,8
| |
Unterschied in der Antwortrate (95 %-KI)
|
15,5d
(8.339, 22.592)
|
-
|
17,1c
(9.538, 24.721)
|
-
| |
VNS 4 oder 5 (%)
|
24,5
|
3,3
|
20,5
|
4,9
| |
Unterschied in der Antwortrate (95 %-KI)
|
21,2c
(14.271, 28.143)
|
-
|
15,5d
(8.515, 22.561)
|
-
|
a Die primären und wichtigsten sekundären Endpunkte wurden nach der Methode der multiplen Imputation korrigiert.
b p-Wert < 0,0001
c p-Wert < 0,001
d p-Wert < 0,005
e p-Wert < 0,01
Abbildung 1: Anteil der Patienten, die während des 52-wöchigen Behandlungszeitraums einen F-VASI75-Score erreichen (Intent-to-treat) – gepoolte Daten der StudienTRuE-V1 (INCB 18424-306) und TRuE-V2 (INCB 18424-307)
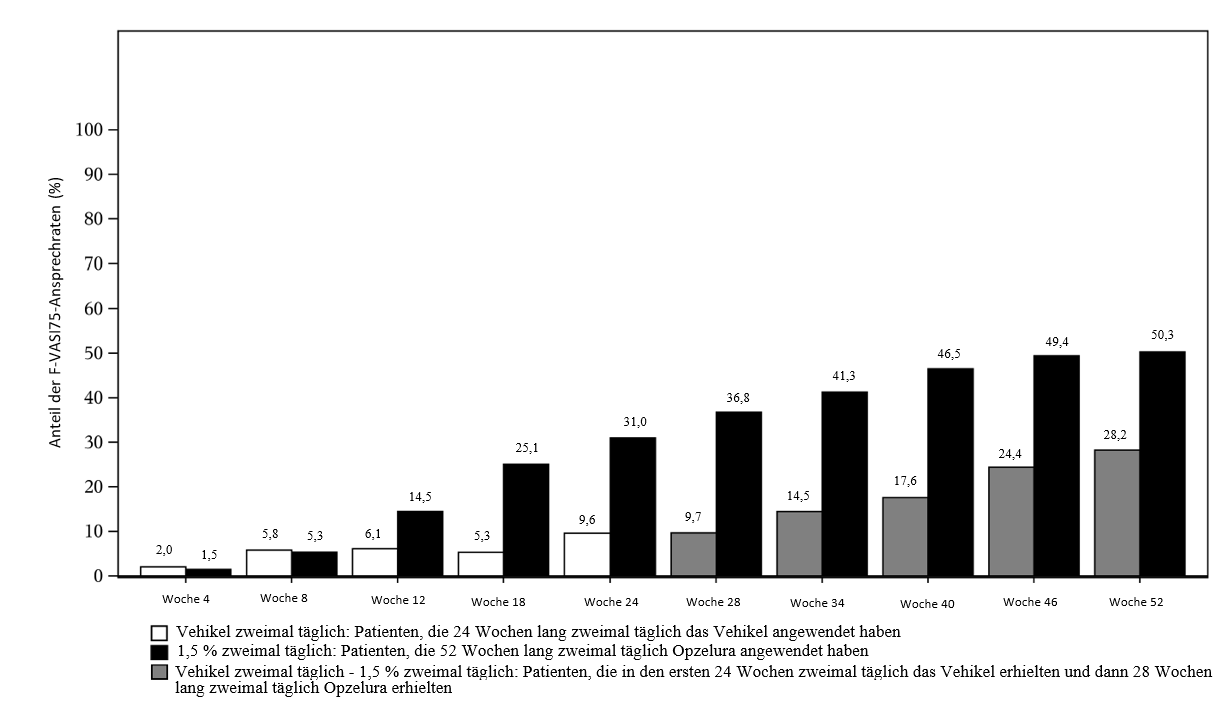
In Woche 52 betrug die beobachtete Ansprechrate für F-VASI90, T-VASI50 und VNS 30,3 %, 51,1 % bzw. 36,3 % für die gepoolte ITT-Population.
Pädiatrie
Insgesamt wurden 72 Jugendliche (12 bis < 18 Jahre; n = 55 Ruxolitinib-Creme, n = 17 Vehikel) in die Zulassungsstudien aufgenommen. Jugendliche zeigten bei der Behandlung mit Ruxolitinib-Creme nach 24 Wochen die gleichen Ansprechraten bei primären und wichtigen sekundären Endpunkten wie Erwachsene im Alter von 18 bis 65 Jahren.
PharmakokinetikAbsorption
Die Pharmakokinetik von Ruxolitinib-Creme wurde bei 429 Studienteilnehmern mit Vitiligo im Alter von 12 Jahren und älter (12,6 % waren 12-17 Jahre alt) mit einer mittleren betroffenen Körperoberfläche ± SD von 7,31 ± 2,02 % (Bereich von 3,2 % bis 10,0 %) untersucht. Die Patienten trugen 24 Wochen lang zweimal täglich etwa 1,58 mg/cm2 Ruxolitinib-Creme auf dieselben Hautstellen auf (der Dosisbereich lag zwischen ca. 0,18 g und 8,4 g Ruxolitinib-Creme pro Anwendung).
Die mittleren minimalen Plasmakonzentrationen ± SD im Steady State betrugen 56,9 ± 62,6 nM mit einer projizierten AUC0-12h von 683 ± 751 h*nM, was etwa 25 % der beobachteten mittleren AUC0-12h im Steady-State (2716 h*nM) nach zweimal täglicher oraler Gabe von 15 mg bei gesunden Teilnehmern entspricht. Die topische Bioverfügbarkeit von Ruxolitinib-Creme im Vergleich zur mittleren oralen Gabe von Ruxolitinib bei Teilnehmern mit Vitiligo in den gepoolten Daten der beiden Phase-3-Studien betrug 12,5 %, wobei jedoch die Variabilität der geschätzten Bioverfügbarkeit hoch war.
Distribution
In einer In-vitro-Studie wurde festgestellt, dass Ruxolitinib zu 97 % an menschliche Plasmaproteine, hauptsächlich an Albumin, gebunden wird.
Metabolismus
Ruxolitinib wird durch CYP3A4 und in geringerem Masse durch CYP2C9 verstoffwechselt.
Elimination
Die mittlere Eliminationshalbwertszeit von oral verabreichtem Ruxolitinib beträgt etwa 3 Stunden. Die mittlere scheinbare terminale Halbwertszeit von Ruxolitinib nach topischer Anwendung von OPZELURA wurde bei 9 erwachsenen und jugendlichen Patienten mit einer Beteiligung von ≥ 25 % der Körperoberfläche mit atopischer Dermatitis geschätzt und beträgt etwa 116 Stunden, was eher die langsame Absorption des Arzneimittels als seine Ausscheidungsgeschwindigkeit widerspiegelt.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Obwohl die AUC nach oraler Verabreichung von Ruxolitinib bei Patienten mit Leberfunktionsstörung erhöht war, gab es keinen klaren kausalen Zusammenhang zwischen dem Schweregrad der Leberinsuffizienz und dem Anstieg der AUC. Eine Dosierungsempfehlung für Patienten mit Leberinsuffizienz ist nicht erforderlich.
Nierenfunktionsstörungen
Die geschätzte AUC, die um die pharmakologische Aktivität von Ruxolitinib und den Metaboliten bereinigt ist, steigt im Falle einer terminalen Niereninsuffizienz um etwa das Zweifache. Als Vorsichtsmassnahme und aufgrund der unzureichenden Sicherheitsdaten in diesem Bereich sollte OPZELURA nicht bei Patienten mit terminaler Niereninsuffizienz angewendet werden.
Präklinische DatenRuxolitinib wurde in Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität, Reproduktionstoxizität und Karzinogenität nach oraler Verabreichung untersucht. Weitere Studien wurden nach dermaler Verabreichung an Minischweinen und Mäusen durchgeführt. Zu den Zielorganen, die mit der pharmakologischen Wirkung von Ruxolitinib in Studien zur Toxizität mit wiederholter oraler Gabe in Verbindung gebracht werden, gehören Knochenmark, peripheres Blut und lymphatisches Gewebe. Bei Hunden wurden Infektionen beobachtet, die im Allgemeinen mit einer Immunsuppression in Verbindung gebracht werden. Die Sicherheitsspannen (basierend auf der ungebundenen AUC) bei nicht schädlichen Konzentrationen in Studien zur chronischen Toxizität lagen bei männlichen und weiblichen Ratten bei etwa dem 6- bis 200-Fachen und bei Hunden etwa beim 10-Fachen, bezogen auf die systemische Exposition, die bei Vitiligo-Patienten beobachtet wurde, die zweimal täglich die 1,5 %ige Ruxolitinib-Creme aufgetragen hatten. In einer Telemetriestudie an Hunden wurde eine unerwünschte Senkung des Blutdrucks zusammen mit einem Anstieg der Herzfrequenz beobachtet, und in einer Studie zur Funktion der Atemwege bei Ratten wurde eine unerwünschte Senkung des Atemminutenvolumens festgestellt. Die Sicherheitsspannen (basierend auf dem ungebundenen Cmax des Arzneimittels) bei nicht schädlichen Konzentrationen lagen in den Studien mit Hunden und Ratten bei etwa dem 300-Fachen bzw. dem 100-Fachen bezogen auf die systemische Exposition, die bei Vitiligo-Patienten beobachtet wurde, die zweimal täglich die 1,5 %ige Ruxolitinib-Creme angewendet hatten. Bei der Bewertung der neuropharmakologischen Wirkungen von Ruxolitinib bei Ratten wurden keine unerwünschten Wirkungen beobachtet.
Eine 3-monatige Studie mit wiederholter Verabreichung über die Haut ergab eine verringerte Lymphozytenzahl bei Mäusen. Die Sicherheitsspannen (basierend auf der ungebundenen AUC) bei nicht schädlichen Werten lagen bei männlichen Mäusen bei etwa dem 10-Fachen und bei weiblichen Mäusen bei etwa dem 24-Fachen, bezogen auf die systemische Exposition, die bei Vitiligo-Patienten beobachtet wurde, die die 1,5 %ige Ruxolitinib-Creme zweimal täglich angewendet hatten. In einer 9-monatigen Studie zur dermalen Toxizität wurde bei Minischweinen ebenfalls eine nicht schädliche Verringerung der peripheren Lymphozytenzahl beobachtet. Die Sicherheitsspannen (basierend auf der ungebundenen AUC) lagen bei nicht schädlichen Konzentrationen bei Minischweinen bei etwa dem 3-Fachen, bezogen auf die systemische Exposition, die bei Vitiligo-Patienten beobachtet wurde, die zweimal täglich die 1,5 %ige Ruxolitinib-Creme angewendet hatten. Diese Auswirkung wurde in einer 3-monatigen Studie zur dermalen Toxizität bei Minischweinen nicht beobachtet. Bei Göttinger Minischweinen wurden nach topischer Verabreichung der 1,5%igen Ruxolitinib-Creme zweimal täglich über einen Zeitraum von bis zu 9 Monaten keine Anzeichen systemischer Toxizität beobachtet.
In Studien zur embryofötalen Entwicklung führte die orale Verabreichung von Ruxolitinib an Ratten und Kaninchen während der Trächtigkeit zu einem verringerten Gewicht des Fötus und einem erhöhten Postimplantationsverlust bei Dosen, die mit einer maternalen Toxizität in Verbindung gebracht werden. Bei Ratten und Kaninchen gab es keine Hinweise auf eine teratogene Wirkung. Die Sicherheitsspannen (basierend auf der ungebundenen AUC) lagen bei nicht schädlichen Konzentrationen für die Entwicklungstoxizität bei Ratten bei etwa dem 25-Fachen, bezogen auf die systemische Exposition, die bei Vitiligo-Patienten beobachtet wurde, die zweimal täglich die 1,5 %ige Ruxolitinib-Creme angewendet hatten. Es wurden keine Auswirkungen von oral verabreichtem Ruxolitinib auf die Fertilität von männlichen oder weiblichen Ratten festgestellt. In einer Studie zur prä- und postnatalen Entwicklung wurde eine leichte Verlängerung der Trächtigkeitsdauer, eine geringere Anzahl der Einnistungsstellen und eine geringere Anzahl geborener Jungtiere beobachtet. Bei den Rattenjungen wurden ein geringeres mittleres Ausgangskörpergewicht und eine kurzzeitig geringere mittlere Körpergewichtszunahme beobachtet. Bei laktierenden Ratten wurden Ruxolitinib und/oder seine Metaboliten mit einer Konzentration in die Milch ausgeschieden, die um das 13-Fache höher war als die mütterliche Plasmakonzentration. Ruxolitinib war weder mutagen noch klastogen. Ruxolinitib zeigte nach topischer Verabreichung bei Mäusen und nach oraler Verabreichung bei Sprague-Dawley-Ratten und Tg.rasH2-transgenen Mäusen kein karzinogenes Potential.
Toxizitätstests bei juvenilen Tieren
In Studien an juvenilen Ratten hatte die orale Verabreichung von Ruxolitinib Auswirkungen auf das Wachstum und die Knochenmasse. Ein vermindertes Knochenwachstum wurde bei Dosen ≥ 5 mg/kg/Tag beobachtet, wenn die Behandlung am 7.Tag nach der Geburt begann (vergleichbar mit einem menschlichen Neugeborenen) und bei Dosen ≥ 15 mg/kg/Tag, wenn die Behandlung am 14. oder 21. Tag nach der Geburt begann (vergleichbar mit dem menschlichen Kleinkind im Alter von 1 bis 3 Jahren). Knochenbrüche und ein frühzeitiges Absterben der Ratten wurde bei Dosen ≥ 30 mg/kg/Tag beobachtet, wenn die Behandlung am 7. Tag nach der Geburt eingeleitet wurde. Auf der Grundlage der ungebundenen AUC betrug die NOAEL-Exposition (Dosis ohne beobachtete Nebenwirkungen) bei juvenilen Ratten, die ab dem 7.Tag nach der Geburt behandelt wurden, etwa das 20-Fache der Exposition von erwachsenen Vitiligo-Patienten, während ein vermindertes Knochenwachstum und Knochenbrüche bei Expositionen auftraten, die dem 22- bis 150-Fachen der Exposition von erwachsenen Vitiligo-Patienten entsprachen. Die Auswirkungen waren im Allgemeinen stärker bei männlichen Tieren und traten auf, wenn die Verabreichung früher nach der Geburt begann. Abgesehen von der Knochenentwicklung waren die Auswirkungen von Ruxolitinib auf juvenile Ratten ähnlich wie bei adulten Ratten. Juvenile Ratten sind empfindlicher gegenüber der Toxizität von Ruxolinitib als adulte Ratten.
Sonstige HinweiseInkompatibilitäten
Nicht zutreffend.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Nach Anbruch innerhalb von 6 Monaten zu verwenden.
Besondere Lagerungshinweise
Nicht über 30°C lagern.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer69443 (Swissmedic)
PackungenOpzelura ist in zwei Arten von Tuben erhältlich:
·Aluminiumtube mit 100 g Creme (B).
·Laminierte Tube mit 100 g Creme (B).
Der Inhalt ist für beide Tubenvarianten identisch.
ZulassungsinhaberinIncyte Biosciences International Sàrl, 1110 Morges
Stand der InformationJuni 2024
|