ZusammensetzungWirkstoffe
Lutathera: Lutetium (177Lu)-Oxodotreotid 370 MBq/ml Infusionslösung zum Datum und zur Uhrzeit der Kalibrierung. Das 177Lutetium wird aus 176Ytterbium hergestellt und ist non-carrier-added.
Lutathera CA: Lutetium (177Lu)-Oxodotreotid 370 MBq/ml Infusionslösung zum Datum und zur Uhrzeit der Kalibrierung. Das 177Lutetium wird aus 176Lutetium hergestellt und ist carrier-added. Das Präparat enthält die Verunreinigung 177mLutetium.
Hilfsstoffe
Essigsäure, Natriumacetat 0.66 mg/ml, Gentisinsäure, Ascorbinsäure, Pentetinsäure, Natriumchlorid 6.85mg/ml, Natriumhydroxid 0.64 mg/ml, Wasser für Injektionszwecke.
Jeder ml Lösung enthält bis zu 0,14 mmol (3.24 mg) Natrium.
Indikationen/AnwendungsmöglichkeitenLutathera/Lutathera CA ist zur Behandlung von metastatischen oder nicht resezierbaren, progressiven, gut differenzierten (G1 und G2) Somatostatinrezeptor-positiven gastroenteropankreatischen neuroendokrinen Tumoren (GEP-NET) bei Erwachsenen indiziert.
Dosierung/AnwendungDas Arzneimittel ist ausschliesslich zur Verwendung im in Krankenhäusern bestimmt und darf nur von fachärztlichen Personen mit eidgenössischem Weiterbildungstitel in Nuklearmedizin verabreicht werden.
Aufgrund der verabreichten Menge an Radioaktivität muss sich der Patient nach der Behandlung in einem Isolierzimmer aufhalten. Die Vorsichtsmassnahmen zum Strahlenschutz müssen eingehalten werden. Siehe die Rubriken «Warnhinweise und Vorsichtsmassnahmen» sowie «Sonstige Hinweise: Hinweise für die Handhabung/Strahlenschutz».
Bevor eine Behandlung mit Lutathera/Lutathera CA begonnen wird, muss mit Somatostatinrezeptor-Bildgebung (Szintigraphie oder Positronen-Emissions-Tomographie [PET]) die Überexpression dieser Rezeptoren im Tumorgewebe bestätigt werden, wobei die Aufnahme durch Tumorherde mindestens so hoch wie die normale Aufnahme durch die Leber sein muss.
Übliche Dosierung
Erwachsene
In der Regel besteht das empfohlene Behandlungsprotokoll beim Erwachsenen aus 4 Infusionen zu jeweils 7'400 MBq. Zwischen jeder Infusion muss eine Pause von 8 Wochen (± 1 Woche) eingehalten werden (siehe auch «Dosisanpassung»).
Aminosäurenlösung
Zum Schutz der Nierenfunktion muss für 4 Stunden (siehe Tabelle 1 und Tabelle 2) eine Aminosäurenlösung mit L-Lysin und L-Arginin intravenös verabreicht werden. Die Infusion der Aminosäurenlösung sollte 30 Minuten vor Beginn der Verabreichung von Lutathera/Lutathera CA eingeleitet werden.
Die Infusion der Aminosäurenlösung und von Lutathera/Lutathera CA über einen separaten venösen Zugang in je einem Arm des Patienten ist die bevorzugte Methode. Wenn jedoch zwei intravenöse Zugänge aufgrund eines schlechten Venenzugangs oder aufgrund von Präferenzen in der Einrichtung/im Spital nicht möglich sind, kann die Infusion der Aminosäurenlösung und von Lutathera/Lutathera CA über denselben Zugang über einen Dreiwegehahn erfolgen, wobei die Flussrate und die Offenhaltung des Venenzugangs zu überwachen ist. Die Dosis der Aminosäurenlösung sollte nicht verringert werden, auch wenn eine reduzierte Dosis Lutathera/Lutathera CA verabreicht wird.
Angesichts der grossen Menge der Aminosäurelösung und den beträchtlichen Volumina, die handelsübliche Lösungen benötigen, um die oben genannten Spezifikationen zu erfüllen, wird die als Rezepturarzneimittel hergestellte Lösung aufgrund ihres geringeren Infusionsvolumens und der geringeren Osmolalität als Produkt der Wahl angesehen. Die Aminosäurenlösung kann, unter Berücksichtigung der guten Praxis zur Herstellung von sterilen Arzneimitteln des Spitals und gemäss der in Tabelle 1 spezifizierten Zusammensetzung, auf Anfrage hergestellt werden.
Tabelle 1 Zusammensetzung der als Rezepturarzneimittel hergestellten Aminosäurenlösung
|
Zusammensetzung
|
Menge
| |
L-Lysin-HCl
|
25 g (entspricht 20 g Lysin)
| |
L-Arginin-HCl
|
25 g (entspricht 20.7 g Arginin)
| |
Natriumchloridlösung 9 mg/ml (0.9 %) für Injektionszwecke oder Wasser für Injektionszwecke
|
1 l
|
Der pH der als Rezepturarzneimittel hergestellten Aminosäurenlösung gemäss der in Tabelle 1 beschriebenen Zusammensetzung muss mit Natriumhydroxid (NaOH) auf einen Wert von 7.4 ± 0.2 angepasst werden.
Alternativ können einige kommerziell verfügbare Aminosäurenlösungen verwendet werden, wenn diese mit der in Tabelle 2 beschriebenen Spezifikationen übereinstimmen.
Tabelle 2 Spezifikation von kommerziell verfügbaren Aminosäurenlösungen
|
Charakteristika
|
Spezifikation
| |
L-Lysin-HCl
|
Zwischen 18 und 25 g (entspricht 14.4 bis 20 g L-Lysin)
| |
L-Arginin-HCl
|
Zwischen 18 und 25 g (entspricht 14.9 bis 20.7 g L-Arginin)
| |
Volumen
|
1 l bis 2 l
| |
Osmolalität
|
<1'200 mOsmol/kg
|
Überwachung der Behandlung
Vor jeder Anwendung und während der Behandlung müssen Laboruntersuchungen durchgeführt werden, um den Zustand des Patienten neu zu beurteilen und um das Therapieprotokoll, falls nötig, anzupassen (Dosis, Infusionsintervall, Anzahl der Infusionen).
Vor jeder Infusion sind mindestens folgende Laboruntersuchungen erforderlich:
·Hämatologie (Hämoglobin [Hb], weisses Blutbild mit Differenzialblutbild, Thrombozytenzahl)
·Nierenfunktion (Serumkreatinin und Kreatinin-Clearance nach der Cockcroft-Gault-Formel)
·Leberfunktion (Alanin-Aminotransferase [ALT], Aspartat-Aminotransferase [AST], Serumalbumin, internationaler normalisierter Quotient (INR) und Bilirubin).
Diese Untersuchungen müssen mindestens einmal innerhalb von 2 bis 4 Wochen vor der Anwendung sowie unmittelbar vor der Anwendung durchgeführt werden. Es wird auch empfohlen, diese Untersuchungen für mindestens 3 Monate nach der letzten Infusion von Lutathera/Lutathera CA alle 4 Wochen sowie anschliessend alle 6 Monate durchzuführen, um mögliche verzögert auftretende unerwünschte Wirkungen zu erkennen (siehe Rubrik «Unerwünschte Wirkungen»). Die Dosierung muss möglicherweise entsprechend den Laborergebnissen angepasst werden.
Dosisanpassung
Bei schweren oder unerträglichen unerwünschten Wirkungen kann es erforderlich sein, die Dosisgaben vorübergehend auszusetzen (das Dosisintervall von 8 Wochen auf bis zu 16 Wochen zu verlängern), die Dosis zu reduzieren oder die Behandlung mit Lutathera/Lutathera CA ganz abzubrechen (siehe Tabelle 3 und Abbildung 1).
Tabelle 3 Empfohlene Dosisanpassungen bei unerwünschten Wirkungen (UW)
|
UW
|
Schweregrad der UW
|
Dosisanpassung
| |
Thrombozytopenie
|
Erstes Auftreten von:
Grad 2 (Thrombozytenzahl < 75 bis 50 x 109/l)
Grad 3 (Thrombozytenzahl < 50 bis 25 x 109/l)
Grad 4 (Thrombozytenzahl < 25 x 109/l)
|
Nächste Dosis erst nach vollständiger oder teilweiser Besserung (Grad 0 bis 1) verabreichen.
Nach vollständiger oder partieller Besserung Lutathera/Lutathera CA-Gaben mit einer Dosis von 3'700 MBq (100 mCi) wieder aufnehmen. Wenn unter reduzierter Dosis keine Thrombozytopenie Grad 2, 3 oder 4 auftritt, kann Lutathera/Lutathera CA bei der nächsten Gabe mit 7'400 MBq (200 mCi) dosiert werden.
Wenn aufgrund einer Thrombozytopenie Grad ≥2 ein Dosierungsintervall von über 16 Wochen erforderlich wird, ist Lutathera/Lutathera CA dauerhaft abzusetzen.
| |
|
Grad 2, 3 oder 4 mehrfach auftretend
|
Dauerhaftes Absetzen der Lutathera/Lutathera CA-Behandlung.
| |
Anämie und Neutropenie
|
Erstes Auftreten von Anämie:
Grad 3 (Hb < 8.0 g/dl); Transfusion indiziert
Grad 4 (lebensbedrohliche Folgen)
Erstes Auftreten von Neutropenie:
Grad 3 (absolute Neutrophilenzahl (ANC) < 1.0 bis 0.5 x 109/l)
Grad 4 (ANC < 0.5 x 109/l)
|
Nächste Dosis erst nach vollständiger oder teilweiser Besserung (Grad 0, 1 oder 2) verabreichen.
Nach vollständiger oder partieller Besserung Lutathera/Lutathera CA-Gaben mit einer Dosis von 3'700 MBq (100 mCi) wieder aufnehmen. Wenn unter reduzierter Dosis keine Anämie oder Neutropenie Grad 3 oder 4 auftritt, kann Lutathera/Lutathera CA bei der nächsten Gabe mit 7'400 MBq (200 mCi) dosiert werden.
Wenn aufgrund einer Anämie oder Neutropenie Grad ≥3 ein Dosierungsintervall von über 16 Wochen erforderlich wird, ist Lutathera/Lutathera CA dauerhaft abzusetzen.
| |
|
Grad 3 oder 4 mehrfach auftretend
|
Dauerhaftes Absetzen der Lutathera/Lutathera CA-Behandlung.
| |
Nierentoxizität
|
Erstes Auftreten von:
·Kreatinin-Clearance < 40 ml/min, berechnet nach Cockcroft/Gault mit dem aktuellen Körpergewicht, oder
·40 % Anstieg des Serumkreatinin-Ausgangswertes oder
·40 % Abnahme der Kreatinin-Clearance gegenüber dem Ausgangswert, berechnet nach Cockcroft/Gault mit dem aktuellen Körpergewicht.
|
Dosisgaben bis zur
Besserung oder Rückkehr zum Ausgangswert aussetzen.
Nach Besserung oder Rückkehr zum Ausgangswert Lutathera/Lutathera CA-Gaben mit einer Dosis von 3'700 MBq (100 mCi) wieder aufnehmen. Wenn unter reduzierter Dosis keine Nierentoxizität auftritt, kann Lutathera/Lutathera CA bei der nächsten Gabe mit 7'400 MBq (200 mCi) dosiert werden.
Wenn aufgrund von Nierentoxizität ein Dosierungsintervall von über 16 Wochen erforderlich wird, ist Lutathera/Lutathera CA dauerhaft abzusetzen.
| |
|
Wiederholte Nierentoxizität
|
Dauerhaftes Absetzen von Lutathera/Lutathera CA.
| |
Hepatotoxizität
|
Definiert als:
·Bilirubinämie > dem dreifachen oberen Normgrenzwert (Grad 3 oder 4) oder
·Albuminämie < 30 g/l mit INR > 1.5
|
Dosisgaben bis zur Besserung oder Rückkehr zum Ausgangswert aussetzen.
Nach vollständiger Besserung oder Rückkehr zum Ausgangswert Lutathera/Lutathera CA-Gaben mit einer Dosis von 3'700 MBq (100 mCi) wieder aufnehmen. Wenn unter reduzierter Dosis keine Hepatotoxizität auftritt, kann Lutathera/Lutathera CA bei der nächsten Gabe mit 7'400 MBq (200 mCi) dosiert werden.
Wenn aufgrund von Hepatotoxizität ein Dosierungsintervall von über 16 Wochen erforderlich wird, ist Lutathera/Lutathera CA dauerhaft abzusetzen.
| |
|
Wiederholte Hepatotoxizität
|
Dauerhaftes Absetzen von Lutathera/Lutathera CA
| |
Jede andere UAW vom Grad 3 oder Grad 4 gemäss CTCAE*
|
Erstes Auftreten von Grad 3 oder 4
|
Dosisgaben bis zur vollständigen oder partiellen Rückbildung (auf Grad 0 bis 2) aussetzen.
Nach vollständiger oder partieller Besserung Lutathera/Lutathera CA-Gaben mit einer Dosis von 3'700 MBq (100 mCi) wieder aufnehmen. Wenn unter reduzierter Dosis keine Toxizität Grad 3 oder 4 auftritt, kann Lutathera/Lutathera CA bei der nächsten Gabe mit 7'400 MBq (200 mCi) dosiert werden.
Wenn aufgrund einer UAW vom Grad ≥3 ein Dosierungsintervall von über 16 Wochen erforderlich wird, ist Lutathera/Lutathera CA dauerhaft abzusetzen.
| |
|
Wiederkehrende Toxizität Grad 3 oder 4
|
Dauerhaftes Absetzen von Lutathera/Lutathera CA.
| |
Keine Dosisanpassung erforderlich bei hämatologischen Toxizitäten des Grades 3 oder 4, die ausschliesslich auf eine Lymphopenie zurückzuführen sind
*CTCAE: Common Terminology Criteria for Adverse Events (Gemeinsame Terminologiekriterien für unerwünschte Ereignisse), National Cancer Institute
|
Abbildung 1 Anweisungsschemata für Dosisanpassungen
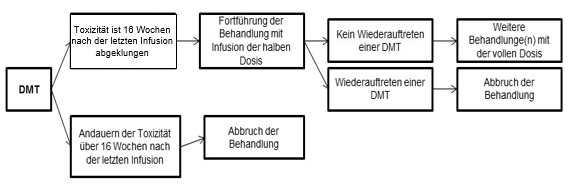
DMT: Dosis-modifizierende Toxizität
Weitere Gründe, eine vorübergehende Unterbrechung der Lutathera/Lutathera CA-Behandlung zu erwägen, sind interkurrente Erkrankungen (z.B. eine Harnwegsinfektion), die nach Einschätzung des Arztes die mit der Verabreichung von Lutathera/Lutathera CA verbundenen Risiken verstärken könnten und die daher vor einer Wiederaufnahme der Behandlung abgeklungen oder stabil sein müssen, sowie grössere Operationen, in diesem Fall sollte die Behandlung mit Lutathera/Lutathera CA für 12 Wochen nach dem Eingriff ausgesetzt werden.
Spezielle Populationen
Ältere Patienten
In den Ergebnissen klinischer Studien konnten keine Unterschiede bezüglich des Ansprechens zwischen älteren und jüngeren Patienten beobachtet werden. Weil aber bei älteren Patienten (≥70 Jahre alt) ein erhöhtes Risiko für das Auftreten einer Hämatotoxizität beschrieben wurde, ist in dieser Population eine engmaschige Beobachtung mit der Möglichkeit einer raschen Dosisanpassung empfohlen.
Patienten mit Nierenfunktionsstörungen
Die zu verabreichende Aktivität muss bei Patienten mit Nierenfunktionsstörung sorgfältig erwogen werden, weil eine erhöhte Strahlenbelastung auftreten kann. Das pharmakokinetische Profil und die Sicherheit von Lutetium (177Lu)-Oxodotreotid bei Patienten mit einer vorbestehenden schweren oder terminalen Niereninsuffizienz wurden nicht untersucht. Die Behandlung mit Lutathera/Lutathera CA ist bei Patienten mit schwerer Niereninsuffizienz und einer Kreatinin-Clearance unter 30 ml/min kontraindiziert (siehe Rubrik «Kontraindikationen»). Wenn der Ausgangswert der Kreatinin-Clearance (nach Cockcroft/Gault) unter 40 ml/min liegt, wird die Behandlung mit Lutathera/Lutathera CA nicht empfohlen. Für Patienten mit eingeschränkter Nierenfunktion und einer vorbestehenden Kreatinin-Clearance ≥40 ml/min wird keine Dosisanpassung empfohlen. Weil dieses Arzneimittel hauptsächlich über die Nieren ausgeschieden wird, sollte die Nierenfunktion jedoch während der Behandlung häufiger überwacht werden, denn es besteht bei diesen Patienten ein erhöhtes Toxizitätsrisiko.
Für weitere Informationen zur Behandlung von Patienten mit Nierentoxizität, siehe die Rubriken «Dosierung/Anwendung» (Tabelle 3) und «Warnhinweise und Vorsichtsmassnahmen».
Patienten mit Leberfunktionsstörungen
Die zu verabreichende Aktivität muss bei Patienten mit einer Leberinsuffizienz sorgfältig erwogen werden, weil eine erhöhte Strahlenbelastung auftreten kann. Das pharmakokinetische Profil und die Sicherheit von Lutetium (177Lu)-Oxodotreotid bei Patienten mit vorbestehender schwerer Leberinsuffizienz (Gesamtbilirubin oberhalb des dreifachen oberen Normgrenzwerts, unabhängig vom AST-Wert) wurden nicht untersucht. Infolgedessen wird die Behandlung dieser Patienten mit Lutathera/Lutathera CA nicht empfohlen. Patienten mit einer vorbestehenden Leberfunktionsstörung, bei denen entweder das Gesamtbilirubin mehr als das 3-Fache der oberen Grenze des Normalwerts oder die Albuminämie < 30 g/l und die INR > 1,5 beträgt, sollten nur nach einer sorgfältigen Nutzen-Risiko-Abwägung mit Lutathera/Lutathera CA behandelt werden.
Für das bei Patienten mit Hepatotoxizität anzuwendende Verfahren, siehe Tabelle 3 in der Rubrik «Dosierung/Anwendung» und der Rubrik «Warnhinweise und Vorsichtsmassnahmen».
Pädiatrische Population
Es gibt im Anwendungsgebiet der GEP-NET (mit Ausnahme von Neuroblastom, Neuroganglioblastom und Phäochromozytom) keinen relevanten Nutzen von Lutathera/Lutathera CA bei Kindern und Jugendlichen. Lutathera/Lutathera CA ist für die Anwendung in der pädiatrischen Population nicht zugelassen.
Prämedikation
Antiemetika
Eine Prämedikation mit Antiemetika muss mit ausreichender Vorlaufzeit vor Beginn der Infusion der Aminosäurenlösung verabreicht werden. Hinweise zur Verabreichung entnehmen Sie bitte der ausführlichen Fachinformation der Antiemetika.
Tritt während der Infusion der Aminosäurenlösung trotz der vorherigen Verabreichung eines Antiemetikums starke Übelkeit oder Erbrechen auf, kann ein Antiemetikum einer anderen pharmakologischen Klasse verabreicht werden.
Gleichzeitige Anwendung von Somatostatinanaloga
Vor Beginn der Behandlung mit Lutathera/Lutathera CA: Die Verabreichung von Somatostatinanaloga mit verzögerter Freisetzung (wie z.B. Octreotid mit verlängerter Wirkstoffabgabe [LAR]) ist mindestens 4 bis 6 Wochen vor Beginn der Behandlung mit Lutathera/Lutathera CA zu unterbrechen. Gegebenenfalls kann Octreotid mit kurzer Wirkungsdauer bis 24 Stunden vor der Anwendung von Lutathera/Lutathera CA verabreicht werden (siehe Rubrik «Interaktionen»).
Während der Behandlung mit Lutathera/Lutathera CA: In den 4 bis 6 Wochen vor jeder Infusion von Lutathera/Lutathera CA darf Octreotid LAR nicht verabreicht werden. Zum Management der Krankheitssymptome während der Behandlung mit Lutathera/Lutathera CA kann dem Patienten Octreotid mit kurzer Wirkungsdauer verabreicht werden; diese Verabreichung muss spätestens 24 Stunden vor jeder Infusion von Lutathera/Lutathera CA unterbrochen werden.
Nach der Behandlung mit Lutathera/Lutathera CA: Fortführen der Verabreichung von 30 mg Octreotid LAR intramuskulär alle 4 Wochen nach Ende der Behandlung mit Lutathera/Lutathera CA, falls klinisch angezeigt.
Art der Anwendung
Lutathera/Lutathera CA ist zur intravenösen Anwendung vorgesehen. Es handelt sich um ein gebrauchsfertiges Radiopharmazeutikum zum Einmalgebrauch.
Anweisungen für die Verabreichung
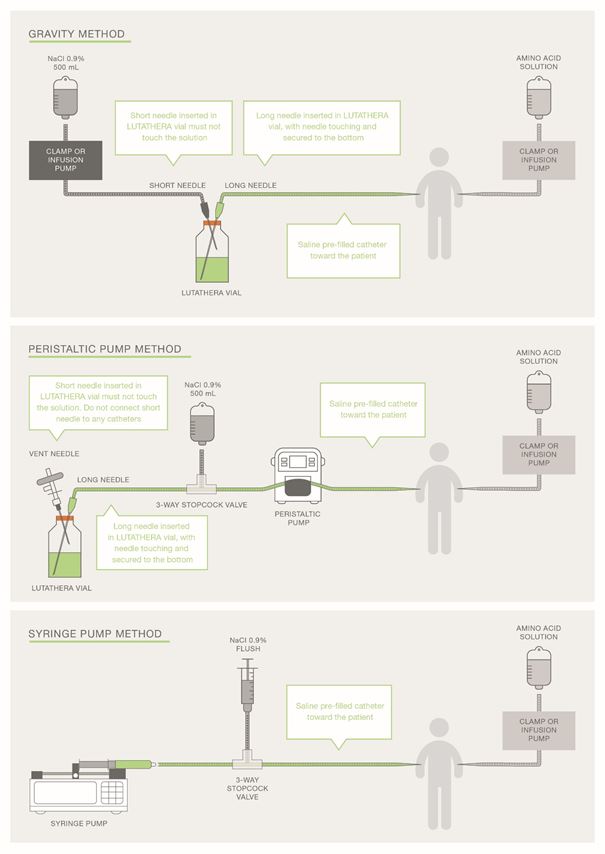
Für die Verabreichung der empfohlenen Dosis kann die Infusionsmethode mittels Schwerkraft, die Schlauchpumpenmethode (Peristaltikpumpe) oder die Spritzenpumpenmethode verwendet werden. Das behandelnde medizinische Fachpersonal kann andere als geeignet und sicher geltende Methoden anwenden, besonders dann, wenn eine Dosisreduktion erforderlich ist.
Bei Anwendung der Schwerkraftmethode oder der Schlauchpumpenmethode sollte Lutathera/Lutathera CA direkt aus dem Originalbehältnis infundiert werden. Die Schlauchpumpenmethode oder die Spritzenpumpenmethode sollte verwendet werden, wenn eine reduzierte Dosis von Lutathera/Lutathera CA nach einer Dosisänderung aufgrund einer unerwünschten Reaktion verabreicht wird (siehe Tabelle 3). Die Verwendung der Schwerkraftmethode zur Verabreichung einer reduzierten Dosis von Lutathera/Lutathera CA kann zur Abgabe einer falschen Menge Lutathera/Lutathera CA führen, wenn die Dosis vor der Verabreichung nicht angepasst wird. Während der Verabreichung sollten die üblichen Vorsichtsmassnahmen zum Strahlenschutz unabhängig von der Infusionsmethode getroffen werden (s. Rubrik «Hinweise für die Handhabung/den Strahlenschutz»).
Lutathera/Lutathera CA darf nicht als Bolus infundiert werden.
Kurz nach Beginn der Infusion sollte die vom Patienten abgegebene Radioaktivität mit einem kalibrierten System zur Messung der Radioaktivität überwacht werden, um sicherzustellen, dass die Dosis abgegeben wird. Während der Infusion sollte die vom Patienten abgegebene Radioaktivität stetig ansteigen, während die von der Lutathera/Lutathera CA-Durchstechflasche abgegebene Radioaktivität abnehmen sollte.
Es wird empfohlen, die Vitalzeichen des Patienten während der Infusion sorgfältig zu überwachen.
Tabelle 4 fasst die im Verlauf einer Behandlung mit Lutathera/Lutathera CA notwendigen Verfahren zusammen.
Tabelle 4 Verfahren zur Verabreichung der Antiemetika, der Aminosäurenlösung und von Lutathera/Lutathera CA
|
Verabreichte Mittel
|
Startzeit
(min)
|
Infusionsrate
(ml/h)
|
Dauer
| |
Antiemetika
|
mit ausreichender Vorlaufzeit vor der Aminosäurenlösung
|
nach Angaben in der Fachinformation
|
nach Angaben in der Fachinformation
| |
Aminosäurenlösung: Auf Anfrage zubereitete Lösung (1 l) oder kommerzielle Lösung (1 bis 2 l).
|
0
|
250–500
je nach Volumen
|
4 Stunden
| |
Lutathera/Lutathera CA mit Natriumchloridlösung 9 mg/ml (0.9 %) für Injektionszwecke
|
30
|
bis 400
|
30 ± 10 Minuten
|
Hinweise zur Handhabung des Arzneimittels vor der Anwendung, siehe Rubrik «Sonstige Hinweise».
Hinweise zur Vorbereitung des Patienten, siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen».
Hinweise zu Empfehlungen im Falle einer Paravasation, siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen».
Intravenöse Verabreichungsverfahren
Anweisungen für die Schwerkraftinfusion (mit einer Schlauchklemme oder einer Infusionspumpe)
1.Eine 2,5 cm lange 20-Gauge-Nadel (Kurznadel) in die Lutathera/Lutathera CA-Durchstechflasche einführen und über einen Katheter mit 500 ml steriler 0,9%iger Natriumchloridlösung verbinden (die zum Transport der Lutathera/Lutathera CA-Lösung während der Infusion verwendet wird). Darauf achten, dass die kurze Nadel die Lutathera/Lutathera CA-Lösung in der Durchstechflasche nicht berührt. Die Kurznadel darf nicht direkt an den Patienten angeschlossen werden. Die Natriumchloridlösung darf nicht in die Lutathera/Lutathera CA-Durchstechflasche fliessen, bevor die Lutathera/Lutathera CA-Infusion eingeleitet wird. Die Lutathera/Lutathera CA-Lösung darf nicht direkt in die Natriumchloridlösung injiziert werden.
2.Eine 9 cm lange 18-Gauge-Nadel (Langnadel) in die Lutathera/Lutathera CA-Durchstechflasche einführen und darauf achten, dass diese lange Nadel während der gesamten Infusion den Boden der Lutathera/Lutathera CA-Durchstechflasche berührt und dort fixiert ist. Die Langnadel über einen intravenösen Katheter, der mit 0,9%iger steriler Natriumchloridlösung verbunden ist und für die Lutathera/Lutathera CA-Infusion in den Patienten verwendet wird, mit dem Patienten verbinden.
3.Eine Rollenklemme oder eine Infusionspumpe verwenden, um den Fluss der Natriumchloridlösung über die Kurznadel in die Lutathera/Lutathera CA-Durchstechflasche zu regulieren. Die Natriumchloridlösung, die über die Kurznadel in die Durchstechflasche gelangt, transportiert die Lutathera/Lutathera CA-Lösung aus der Durchstechflasche über den an die Langnadel angeschlossenen intravenösen Katheter über eine Gesamtdauer von 30±10 Minuten mit einer Infusionsgeschwindigkeit von bis zu 400 ml/h zum Patienten. Die Infusion sollte in den ersten 5 bis 10 Minuten mit einer niedrigeren Infusionsgeschwindigkeit von <100 ml/h beginnen und dann in Abhängigkeit vom Venenstatus des Patienten erhöht werden. Der Druck in der Durchstechflasche sollte während der gesamten Infusion konstant gehalten werden.
4.Während der Infusion sicherstellen, dass das Niveau der Lösung in der Lutathera/Lutathera CA-Flasche konstant bleibt. Dafür wiederholt eine direkte Sichtkontrolle durchführen, wenn ein transparenter, abgeschirmter Behälter verwendet wird, oder die Durchstechflache mit einer Zange handhaben, wenn ein Bleiversandbehälter verwendet wird.
5.Der Fluss von Lutathera/Lutathera CA von der Durchstechflasche zum Patienten muss während der gesamten Infusion überwacht werden.
6.Die Infusion muss abgebrochen werden (die Durchstechflasche von der Schlauchleitung mit der Langnadel und die Kochsalzlösungsleitung abklemmen), sobald der Radioaktivitätswert während mindestens fünf Minuten stabil bleibt.
7.Im Anschluss an die Infusion werden dem Patienten 25 ml sterile 0,9%ige Natriumchloridlösung über den Venenkatheter intravenös zugeführt.
Anleitung für die apparategestützte Infusion mit Peristaltikpumpe (Schlauchpumpe)
1.Eine gefilterte 2,5 cm lange 20-Gauge-Nadel (kurze Entlüftungsnadel) in die Lutathera/Lutathera CA-Durchstechflasche einführen. Darauf achten, dass die Kurznadel die Lutathera/Lutathera CA-Lösung in der Durchstechflasche nicht berührt. Die Kurznadel darf nicht direkt an den Patienten oder an die Peristaltikpumpe angeschlossen werden.
2.Eine 9 cm lange 18-Gauge-Nadel (Langnadel) in die Lutathera/Lutathera CA-Durchstechflasche einführen und darauf achten, dass diese Langnadel während der gesamten Infusion den Boden der Lutathera/Lutathera CA-Durchstechflasche berührt und dort fixiert ist. Die Langnadel über einen geeigneten Schlauch an einen Dreiwegehahn und eine 0,9%ige sterile Natriumchloridlösung anschliessen.
3.Den Ausgang des Dreiwegehahns mit den Schläuchen verbinden, die an der Eingangsseite der Peristaltikpumpe angebracht sind. Dabei die Anweisungen des Pumpenherstellers beachten.
4.Den Schlauch vorbefüllen, indem der Dreiwegehahn geöffnet und die Lutathera/Lutathera CA-Lösung durch den Schlauch gepumpt wird, bis sie den Ausgang des Ventils erreicht.
5.Den intravenösen Katheter vorbefüllen, der an den Patienten angeschlossen wird, indem der Dreiwegehahn geöffnet wird, damit die 0,9%ige sterile Natriumchloridlösung fliessen kann. Die 0,9%ige sterile Natriumchloridlösung wird gepumpt, bis sie am Ende des Katheterschlauchs austritt.
6.Den vorbefüllten intravenösen Katheter an den Patienten anschliessen und den Dreiwegehahn so einstellen, dass die Lutathera/Lutathera CA-Lösung in einer Linie mit der Peristaltikpumpe steht.
7.Ein angemessenes Volumen der Lutathera/Lutathera CA-Lösung über einen Zeitraum von 30±10 Minuten als Infusion verabreichen, um die gewünschte Radioaktivität zu liefern.
8.Sobald die gewünschte Lutathera/Lutathera CA-Radioaktivität abgegeben wurde, die Peristaltikpumpe stoppen und dann die Position des Dreiwegehahns ändern, sodass die Peristaltikpumpe mit der 0,9%igen sterilen Natriumchloridlösung in Verbindung steht. Die Peristaltikpumpe wieder starten und dem Patienten eine intravenöse Spülung mit 25 ml 0,9%iger steriler Natriumchloridlösung über den Venenkatheter zuführen.
Anleitung für die apparategestützte Infusion mit Spritzenpumpe
1.Ein angemessenes Volumen der Lutathera/Lutathera CA-Lösung aufziehen, um die gewünschte Radioaktivität zu verabreichen, dafür eine Einwegspritze mit einem Spritzenschutz und einer sterilen 9 cm langen 18-Gauge-Einwegnadel (Langnadel) verwenden. Zur Erleichterung der Entnahme der Lösung kann eine gefilterte 2,5 cm lange 20-Gauge-Nadel (kurze Entlüftungsnadel) verwendet werden, um den Widerstand der unter Druck stehenden Durchstechflasche zu verringern. Darauf achten, dass die kurze Nadel die Lutathera/Lutathera CA-Lösung in der Durchstechflasche nicht berührt.
2.Die Spritze in die abgeschirmte Pumpe einsetzen und einen Dreiwegehahn zwischen der Spritze und einem intravenösen Katheter einsetzen, der mit 0,9%iger steriler Natriumchloridlösung gefüllt ist und für die Verabreichung von Lutathera/Lutathera CA an den Patienten verwendet wird.
3.Ein angemessenes Volumen der Lutathera/Lutathera CA-Lösung über einen Zeitraum von 30±10 Minuten als Infusion verabreichen, um die gewünschte Radioaktivität zu liefern.
4.Sobald die gewünschte Lutathera/Lutathera CA-Radioaktivität abgegeben wurde, die Spritzenpumpe stoppen und dann die Position des Dreiwegehahns ändern, um die Spritze mit 25 ml steriler 0,9%iger Natriumchloridlösung zu spülen. Die Spritzenpumpe erneut starten.
5.Nachdem die Spülung der Spritze abgeschlossen ist, eine intravenöse Spülung mit 25 ml steriler 0,9%iger Natriumchloridlösung durch den intravenösen Katheter zum Patienten durchführen.
Abbildung 2 Überblick über die Verabreichungsmethoden
STRAHLENEXPOSITION
Die während den klinischen Studien zu Lutathera/Lutathera CA durchgeführten dosimetrischen Analysen haben zu folgenden Schlussfolgerungen geführt:
·Das kritische Organ ist das Knochenmark. Allerdings wurden mit der empfohlenen kumulativen Dosis von 29'600 MBq (4 Gaben von je 7'400 MBq) weder in der Erasmus-Studie der Phase I/II noch in der NETTER-1-Studie der Phase III eine Korrelation zwischen der Hämatotoxizität und der insgesamt verabreichten Radioaktivität oder vom Knochenmark absorbierten Dosis beobachtet.
·Die Niere ist kein kritisches Organ, wenn eine begleitende Infusion einer adäquaten Aminosäurenlösung durchgeführt wird.
Insgesamt stimmen die Ergebnisse der in der NETTER-1- Studie der Phase III und der Erasmus-Studie der Phase I/II durchgeführten dosimetrischen Analysen überein und weisen darauf hin, dass das Dosisregime von Lutathera/Lutathera CA (4 Gaben von 7'400 MBq) sicher ist.
Tabelle 5 Absorbierte Dosisschätzungen für Lutetium (177Lu)-Oxodotreotid aus der NETTER-1- Studie der Phase III (Olinda-Output)
|
Organ
|
Vom Organ absorbierte Dosis (mGy/MBq)
(n = 20)
| |
|
Mittelwert
|
SD
| |
Nebennieren
|
0.037
|
0.016
| |
Gehirn
|
0.027
|
0.016
| |
Brust**
|
0.027
|
0.015
| |
Gallenblasenwand
|
0.042
|
0.019
| |
Wand des unteren Dickdarms
|
0.029
|
0.016
| |
Dünndarm
|
0.031
|
0.015
| |
Magenwand
|
0.031
|
0.015
| |
Wand des oberen Dickdarms
|
0.032
|
0.015
| |
Herzwand
|
0.032
|
0.015
| |
Nieren
|
0.654
|
0.295
| |
Leber*
|
0.199
|
0.226
| |
Lunge
|
0.031
|
0.015
| |
Muskel
|
0.029
|
0.015
| |
Ovarien***
|
0.031
|
0.013
| |
Pankreas
|
0.038
|
0.016
| |
Rotes Knochenmark
|
0.035
|
0.029
| |
Osteogene Zellen
|
0.151
|
0.268
| |
Haut
|
0.027
|
0.015
| |
Milz
|
0.846
|
0.804
| |
Testikel**
|
0.026
|
0.018
| |
Thymus
|
0.028
|
0.015
| |
Schilddrüse
|
0.027
|
0.016
| |
Blasenwand
|
0.437
|
0.176
| |
Gebärmutter***
|
0.032
|
0.013
| |
Gesamter Organismus
|
0.052
|
0.027
|
* n=18 (zwei Patienten wurden ausgeschlossen, weil die von der Leber absorbierte Dosis durch die Aufnahme durch Lebermetastasen verzerrt war)
**n=11 (nur männliche Patienten)
***n=9 (nur weibliche Patienten)
Die Strahlendosis für bestimmte Organe, die nicht notwendigerweise Zielorgane der Therapie sind, kann durch pathophysiologische Veränderungen infolge des Krankheitsprozesses stark beeinflusst werden. Dies muss bei Verwendung dieser Informationen berücksichtigt werden.
Kontraindikationen·Hinweis auf Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Rubrik «Zusammensetzung».
·Festgestellte oder vermutete Schwangerschaft oder wenn eine Schwangerschaft nicht ausgeschlossen werden kann (siehe Rubrik «Schwangerschaft, Stillzeit»).
·Schwere Niereninsuffizienz mit einer Kreatinin-Clearance <30 ml/min.
Warnhinweise und VorsichtsmassnahmenRisiken im Zusammenhang mit der Strahlenexposition
Lutathera/Lutathera CA trägt langfristig zur kumulierten Gesamtstrahlenexposition des Patienten mit ionisierenden Strahlen bei. Die langfristige kumulierte Strahlenexposition kann mit einem erhöhten Krebsrisiko in Zusammenhang gebracht werden.
Myelosuppression
In der NETTER-1-Studie wurden Fälle von Myelosuppression bei Patienten, die Lutathera/Lutathera CA zusammen mit Octreotid LAR erhielten, häufiger beobachtet als bei Patienten, die Octreotid LAR in hoher Dosierung erhielten (alle Grade/Grad 3 oder 4): Anämie (81 %/0) versus (54 %/1 %); Thrombozytopenie (53 %/1 %) versus (17 %/0); und Neutropenie (26 %/3 %) versus (11 %/0). Die mediane Zeit bis zum Erreichen der niedrigsten Thrombozytenkonzentration (Thrombozyten-Nadir) betrug in der NETTER-1- Studie 5.1 Monate nach der ersten Dosis. Unter den 59 Patienten, die eine Thrombozytopenie entwickelten, erholte sich die Thrombozytenzahl bei 68 % der Betroffenen auf den Ausgangswert oder den Normalwert. Die mediane Zeit bis zur Erholung der Thrombozytenzahl betrug 2 Monate. Fünfzehn der neunzehn Patienten, für die keine Erholung der Thrombozytenzahl dokumentiert wurde, wiesen einen Thrombozytenwert nach dem Thrombozyten-Nadir auf. Von diesen 15 Patienten erreichten 5 eine Erholung auf Grad 1, 9 eine solche auf Grad 2 und 1 auf Grad 3.
Für Patienten mit beeinträchtigter Knochenmarksfunktion sowie Patienten, die zuvor eine Chemotherapie oder eine perkutane Strahlentherapie erhalten haben, besteht möglicherweise ein erhöhtes Risiko für hämatotoxische Wirkungen einer Lutathera/Lutathera CA-Behandlung. Bei Patienten, die bereits vor und während der Anwendung von Lutathera/Lutathera CA eine schwere Beeinträchtigung der hämatologischen Funktionen aufweisen, wird die Einleitung der Behandlung nicht empfohlen (z.B. bei Hb < 4.9 mmol/l bzw. 8 g/dl, Thrombozytenzahl < 75 G/l, oder Leukozyten < 2 G/l) es sei denn, sie sind ausschliesslich auf eine Lymphopenie zurückzuführen.
Eine Überwachung des Blutbildes zu Beginn der Behandlung sowie vor jeder Dosis Lutathera/Lutathera CA ist erforderlich. Je nach Schwere der unerwünschten Wirkungen ist die Behandlung vorübergehend zu unterbrechen, die Dosierung anzupassen oder die Behandlung endgültig abzubrechen (siehe Rubrik: «Dosierung/Anwendung: Anpassung der Behandlung»).
Sekundäres myelodysplastisches Syndrom und akute Leukämie
Nach der Behandlung mit Lutathera/Lutathera CA wurden verzögert auftretende myelodysplastische Syndrome (MDS) und akute Leukämien (AL) beobachtet (siehe Rubrik «Unerwünschte Wirkungen»). In der NETTER-1- Studie wurden nach einer medianen Nachbeobachtungszeit von 76 Monaten in der Hauptstudie bei 3 Patienten (2 Patienten aus der Hauptstudie und 1 Patient aus der Dosimetrie-Teilstudie; 2.3 %), die Lutathera/Lutathera CA und Octreotid LAR erhalten hatten, Fälle eines myelodysplastischen Syndroms berichtet, nicht aber bei den Patienten, die Octreotid LAR in hohen Dosen erhalten hatten. In der Erasmus-Studie entwickelten 16 Patienten (2.0 %) ein MDS und 4 Patienten (0.5 %) eine akute Leukämie. Die mediane Zeit bis zum Auftreten betrug 29 Monate (9 bis 45 Monate) für das MDS, und 55 Monate (32 bis 125 Monate) für die AL. Die Ätiologie dieser therapiebedingten sekundären myeloiden Neoplasien ist nicht geklärt. Faktoren wie Alter >70 Jahre, Niereninsuffizienz, vorbestehende Zytopenien, die Anzahl der vorangehenden Behandlungen, eine frühere Exposition gegenüber Chemotherapeutika (insbesondere Alkylanzien) und eine frühere Strahlentherapie werden als mögliche Risiken und/oder prädiktive Faktoren für MDS/AL angesehen.
Nierentoxizität
In der Erasmus-Studie entwickelten 8 Patienten (< 1 %) 3 bis 36 Monate nach der Verabreichung von Lutathera/Lutathera CA eine Niereninsuffizienz. Zwei dieser Patienten hatten eine zugrundeliegende Niereninsuffizienz oder Risikofaktoren für eine Niereninsuffizienz (insbesondere Diabetes oder Hypertonie) und waren dialysepflichtig.
Die Aminosäurenlösung muss vor, während und nach der Anwendung von Lutathera/Lutathera CA (siehe Rubrik «Dosierung/Anwendung: Aminosäurenlösung») verabreicht werden, um die Rückresorption von Lutetium (177Lu)-Oxodotreotid durch die proximalen Tubuli und damit die Strahlenbelastung für die Nieren zu verringern. Der Patient sollte aufgefordert werden, hydriert zu bleiben und seine Blase vor, am Tag der Verabreichung von Lutathera/Lutathera CA und am Tag danach so oft wie möglich zu entleeren. Serumkreatinin und die berechnete Kreatinin-Clearance sind zu überwachen.
Je nach Schweregrad der unerwünschten Wirkungen ist die Behandlung vorrübergehend zu unterbrechen, die Dosierung anzupassen oder endgültig abzubrechen (siehe Rubrik: «Dosierung/Anwendung: Anpassung der Behandlung»).
Bei Patienten mit vorbestehender Einschränkung der Nierenfunktion oder Anomalien der Nieren oder Harnwege ist das Risiko toxischer Wirkungen möglicherweise erhöht.
Bei Patienten mit einer Kreatinin-Clearance unter 50 ml/min ist zudem das erhöhte Risiko für passagere Hyperkaliämien zu berücksichtigen (siehe Warnhinweise und Vorsichtsmassnahmen zur gleichzeitig verabreichten nephroprotektiven Aminosäurenlösung).
Hepatotoxizität
In der Erasmus-Studie wurden bei 2 Patienten (0.25 %) tumorale Leberblutungen, ein Ödem oder eine Nekrose beobachtet, und einer von ihnen (0.12 %) entwickelte eine Stauung und intrahepatische Cholestase. Da Lutathera/Lutathera CA bei vielen Patienten mit Lebermetastasen indiziert ist, weisen viele dieser Patienten eine veränderte Grundfunktion der Leber auf. Ein erhöhtes Risiko für Hepatotoxizität kann bei Exposition gegenüber ionisierenden Strahlen bei diesen Patienten beobachtet werden.
Die Konzentration der Transaminasen, von Bilirubin und Serumalbumin muss während der Behandlung überwacht werden (siehe Rubrik «Dosierung/Anwendung»).
Je nach Schweregrad der unerwünschten Wirkungen ist die Behandlung vorrübergehend zu unterbrechen, die Dosierung anzupassen oder endgültig abzubrechen, (siehe Rubrik: «Dosierung/Anwendung: Anpassung der Behandlung»).
Überempfindlichkeit
Fälle von Überempfindlichkeitsreaktionen (einschliesslich isolierter Angioödeme) wurden in der Zeit nach der Markteinführung bei Patienten berichtet, die mit Lutathera/Lutathera CA behandelt wurden (s. Rubrik «Unerwünschte Wirkungen»). Beim Auftreten schwerwiegender Überempfindlichkeitsreaktionen muss die Behandlung mit Lutathera/Lutathera CA sofort abgebrochen werden. Geeignete Arzneimittel und Ausrüstung zur Behandlung solcher Reaktionen sollten zur sofortigen Anwendung zur Verfügung stehen.
Neuroendokrine hormonelle Krisen
Neuroendokrine hormonelle Krisen aufgrund der übermässigen Sekretion von Hormonen oder von bioaktiven Substanzen, die sich durch Hitzewallungen, Durchfall, Bronchospasmus und Hypotonie äussern, traten bei 2 Patienten (0.25 %) in der Erasmus-Studie typischerweise während der Behandlung mit Lutathera/Lutathera CA oder in den 24 Stunden nach dessen Verabreichung auf. Bei 2 Patienten (0.25 %) wurden ausserdem Fälle einer Hyperkalzämie berichtet. Deshalb muss in gewissen Fällen (z.B. Patienten mit einer schwachen pharmakologischen Kontrolle der Symptome) eine Hospitalisierung über Nacht zur Beobachtung der Patienten in Betracht gezogen werden.
Falls Hitzewallungen, Durchfall, Hypotonie, Bronchokonstriktion oder anderen Zeichen oder Symptome im Zusammenhang mit einer neuroendokrinen Tumorerkrankung auftreten sind die Patienten zu überwachen. Je nach Bedarf sind hohe Dosen von Somatostatinanaloga, Kortikoiden und intravenöser Rehydratationslösung zu verabreichen.
Übelkeit und Erbrechen
Zur Prävention von Übelkeit und Erbrechen im Zusammenhang mit der Behandlung müssen mit ausreichender Vorlaufzeit vor Beginn der Infusion der Aminosäurenlösung Antiemetika intravenös verabreicht werden (siehe Rubrik «Dosierung/Anwendung»).
Gleichzeitige Anwendung von Somatostatinanaloga
Eine gleichzeitige Anwendung von kalten Somatostatinanaloga kann zur Symptomkontrolle der Erkrankung erforderlich sein (siehe Rubrik «Dosierung/Anwendung»).
Tumor-Lyse-Syndrom
Nach der Behandlung mit Lutetium (177Lu) enthaltenden Arzneimitteln wurde über ein Tumorlyse-Syndrom berichtet. Patienten mit einer Niereninsuffizienz in der Vorgeschichte und einer grossen Tumormasse können ein erhöhtes Risiko aufweisen und müssen mit besonderer Vorsicht behandelt werden. Die Nierenfunktion und das Elektrolytgleichgewicht sollten vor und während der Behandlung überprüft werden.
Strahlenschutzrichtlinien
Die Therapie muss in einer Einrichtung erfolgen, die für die therapeutische Verwendung von offenen Strahlenquellen eine Bewilligung des BAG hat.
In Übereinstimmung mit der guten Praxis des Strahlenschutzes der Einrichtung und mit den Verfahren für das Patientenmanagement sind die Strahlenexposition von Patienten und Gesundheitspersonal während und nach der Behandlung mit Lutathera/Lutathera CA zu minimieren und die Kontakte mit anderen Personen zu begrenzen.
Während der Verabreichung von Lutathera/Lutathera CA muss die Unterbringung der behandelten Person in einem separaten, speziell ausgerüsteten Zimmer erfolgen. Die Hospitalisierung und die Entlassung nach der Therapie mit radioaktiven Stoffen ist in Übereinstimmung mit der Strahlenschutzverordnung, der Verordnung des EDI über den Umgang mit radioaktivem Material sowie den Wegleitungen des Bundesamts für Gesundheit durchzuführen.
Patienten müssen instruiert werden, am Tag vor der Infusion, am Tag der Infusion und am Folgetag beträchtliche Mengen Wasser zu trinken (z.B. mindestens 1 Glas Wasser pro Stunde), um die Ausscheidung über den Urin zu fördern. Der Patient muss ebenfalls angehalten werden, täglich Stuhlgang zu haben und gegebenenfalls ein Abführmittel zu verwenden. Urin und Fäzes müssen gemäss den nationalen Vorgaben beseitigt werden (Verordnungen und Wegleitungen).
Sofern die Haut des Patienten nicht durch ein Leck der Infusion oder durch Harninkontinenz kontaminiert wurde, ist auf der Haut oder im Erbrochenen keine radioaktive Kontaminierung zu erwarten. Bei der Standardversorgung oder bei Behandlungen mit Medizinprodukten oder anderen Geräten, die in Kontakt mit der Haut gelangen (z.B. Elektrokardiogramm [EKG]), sind trotzdem grundlegende Schutzvorkehrungen zu treffen, wie das Tragen von Handschuhen, das Anlegen von Geräten und Elektroden vor Beginn der radioaktiven Infusion, das Wechseln der Geräte und der Elektroden nach der Messung und eventuelles Messen der Radioaktivität der Geräte nach der Anwendung.
Der Facharzt für Nuklearmedizin ist in Übereinstimmung mit den Anforderungen der Strahlenschutzverordnung verpflichtet, den Patienten vor Entlassung im Einzelgespräch die Verhaltensregeln im Umgang mit Angehörigen sowie Dritten bezüglich des Strahlenschutzes sowie die allgemeinen Vorsichtsmassnahmen zu erläutern, die bei den täglichen Aktivitäten nach der Behandlung zu beachten sind, um die Strahlenbelastung der Personen in seiner Umgebung so gering wie möglich zu halten.
Nach jeder Verabreichung sollen die folgenden allgemeinen Empfehlungen sowie die nationalen, lokalen und institutionellen Verfahren und Vorschriften berücksichtigt werden:
·Der nähere Kontakt (näher als 1 Meter) mit anderen Personen ist während 7 Tagen einzuschränken.
·Für Kinder und/oder schwangere Frauen istenger Kontakt (näher als 1 Meter) 7 Tage lang auf unter 15 Minuten täglich zu beschränken.
·Patienten müssen während 7 Tagen getrennt von anderen Personen in einem separaten Schlafzimmer schlafen; sie sollten während 15 Tagen getrennt von Kindern und/oder schwangeren Frauen in einem separaten Schlafzimmer schlafen.
Die Radioaktivität kann im Urin bis zu 30 Tage nach der Verabreichung von Lutathera/Lutathera CA nachgewiesen werden.
Empfohlene Massnahmen im Falle einer Paravasation
Tragen von wasserdichten Wegwerfhandschuhen. Die Infusion des Arzneimittels muss unverzüglich gestoppt und das zur Anwendung verwendete Gerät (Katheter, usw.) entfernt werden. Der Facharzt für Nuklearmedizin und die Strahlenschutz verantwortliche Person müssen informiert werden.
Das Verabreichungsmaterial muss aufbewahrt werden, damit die verbleibende Radioaktivität gemessen und die effektiv verabreichte Aktivität und eventuell die absorbierte Dosis bestimmt werden kann. Der Bereich der Paravasation muss mit einem dokumentenechten Stift markiert werden; nach Möglichkeit sollte ein Foto gemacht werden. Es wird auch empfohlen, den Zeitpunkt der Paravasation und das geschätzte Paravasationsvolumen zu dokumentieren.
Zur Weiterführung der Infusion von Lutathera/Lutathera CA muss in jedem Fall ein neuer Katheter in einer gegenüberliegenden Vene eingelegt werden, und auf derselben Seite, an der die Paravasation aufgetreten ist, darf kein weiteres Arzneimittel verabreicht werden.
Zur Beschleunigung der Dispersion des Arzneimittels und zur Verhinderung der Stockung im Gewebe sollte der Blutfluss durch Hochlagern des betroffenen Armes erhöht werden. Je nach Fall sollte die Aspiration des Paravasats, eine Spülinjektion mit Natriumchloridlösung 9 mg/ml (0.9 %) oder die Anwendung warmer Kompressen oder eines Heizkissens am Ort der Infusion in Betracht gezogen werden, um die Vasodilatation zu beschleunigen.
Symptome, insbesondere Entzündung und/oder Schmerzen müssen behandelt werden. Je nach Situation muss der Facharzt für Nuklearmedizin den Patienten über die Risiken im Zusammenhang mit einer Paravasation aufklären und ihn zu möglichen Behandlungen sowie den weiteren zu befolgenden Schritten beraten. Der Bereich der Paravasation muss überwacht werden, bis der Patient aus dem Krankenhaus entlassen wird. Je nach Schweregrad muss dieses Ereignis als unerwünschte Wirkung gemeldet werden.
Patienten mit Harninkontinenz
Es wird empfohlen, Patienten mit Harninkontinenz während der Behandlung häufiger zu überwachen. Bei Patienten mit Harninkontinenz empfiehlt es sich, nach der Verabreichung von Lutathera/Lutathera CA spezielle Vorkehrungen zu treffen, um eine radioaktive Kontaminierung zu vermeiden. Dazu gehört das Hantieren mit allem möglicherweise durch Urin kontaminierten Material.
Patienten mit Hirnmetastasen
Zur Wirksamkeit bei Patienten mit Hirnmetastasen liegen keine Daten vor. Das Nutzen-Risiko-Verhältnis muss bei diesen Patienten infolgedessen individuell beurteilt werden.
Sekundäre maligne Neoplasmen
Die Exposition gegenüber ionisierenden Strahlen kann die Entwicklung von Krebs und Erbleiden begünstigen. Die Strahlenbelastung durch eine therapeutische Exposition kann zu einer erhöhten Inzidenz von Krebs und von Genmutationen führen. Auf jeden Fall muss sichergestellt werden, dass die Risiken im Zusammenhang mit der Strahlenbelastung niedriger als die Risiken der Erkrankung selbst sind.
Für zu treffende Vorsichtsmassnahmen im Zusammenhang mit Umweltrisiken, siehe Rubrik «Sonstige Hinweise».
Sonstige Patienten mit Risikofaktoren
Bei Patienten mit einer der folgenden Erkrankungen besteht ein erhöhtes Risiko für das Auftreten von unerwünschten Wirkungen. Diese Patienten sollten während der Behandlung deshalb häufiger überwacht werden. Für den Fall von dosisabhängiger Toxizität, siehe Tabelle 3.
·Knochenmetastasen;
·Vorausgegangene onkologische radiometabolische Behandlungen mit 131I markierten Radiopharmaka oder andere Therapien mit nicht abgeschirmten radioaktiven Quellen;
·Vorgeschichte anderer bösartiger Tumore, es sei denn, der Patient befindet sich seit mindestens 5 Jahren in Remission.
Warnhinweise und Vorsichtsmassnahmen zur nephroprotektiven Aminosäurenlösung
Hyperkaliämie im Zusammenhang mit der Aminosäurenlösung
Bei Patienten, die Arginin und Lysin erhalten, kann der Kaliumspiegel vorübergehend ansteigen, kehrt jedoch in der Regel innerhalb von 24 Stunden ab dem Beginn der Infusion mit der Aminosäurenlösung wieder in den Normalbereich zurück. Bei Patienten mit verminderter Kreatinin-Clearance kann ein erhöhtes Risiko für eine vorübergehende Hyperkaliämie bestehen (s. Rubrik «Nierentoxizität»).
Vor jeder Verabreichung von Aminosäurenlösung muss der Serumkaliumspiegel des Patienten bestimmt werden. Im Falle einer Hyperkaliämie sind die Begleitmedikation und die Vorgeschichte des Patienten bezüglich Hyperkaliämien zu überprüfen. Eine bestehende Hyperkaliämie muss vor Beginn der Infusion ausgeglichen werden.
Bei einer klinisch relevanten vorbestehenden Hyperkaliämie muss durch eine zweite Kontrolle vor der Infusion der Aminosäurenlösung bestätigt werden, dass der Kaliumhaushalt erfolgreich ausgeglichen wurde. Der Patient ist engmaschig auf Zeichen und Symptome einer Hyperkaliämie – z.B. Dyspnoe, Schwäche, Taubheitsgefühl, Schmerzen im Thorax und kardiale Manifestationen (Erregungsleitungsstörungen und Arrhythmien) – zu überwachen. Vor der Entlassung des Patienten sollte ein Elektrokardiogramm (EKG) durchgeführt werden.
Unabhängig vom Ausgangswert des Serumkaliumspiegels sind während der Infusion die Vitalzeichen zu überwachen. Die Patienten sollten angehalten werden, am Tag vor der Infusion, am Infusionstag selbst und am Folgetag viel zu trinken (d.h. mindestens 1 Glas Wasser stündlich), um ausreichend hydriert zu sein und die Ausscheidung von überschüssigem Kalium aus dem Serum zu unterstützen.
Falls sich während der Infusion der Aminosäurenlösung Symptome einer Hyperkaliämie entwickeln, müssen geeignete Gegenmassnahmen getroffen werden. Bei einer schweren, symptomatischen Hyperkaliämie ist der Abbruch der Aminosäureninfusion zu erwägen, wobei eine Nutzen-Risiko-Abwägung zwischen der Nephroprotektion und der akuten Hyperkaliämie zu treffen ist.
Herzinsuffizienz
Da das Risiko klinischer Komplikationen durch eine Volumenüberlastung besteht, ist bei der Anwendung von Arginin und Lysin bei Patienten mit schwerer Herzinsuffizienz (Klasse III oder IV nach der Klassifikation der New York Heart Association [NYHA]) besondere Vorsicht erforderlich. Patienten mit schwerer Herzinsuffizienz (NYHA-Klasse III oder IV) dürfen nur nach sorgfältiger Nutzen-Risiko-Abwägung unter Berücksichtigung von Volumen und Osmolalität der Aminosäurenlösung behandelt werden.
Metabolische Azidose
Bei Verabreichung komplexer Aminosäurenlösungen im Rahmen einer total parenteralen Ernährung wurde eine metabolische Azidose beobachtet. Verschiebungen des Säure-Base-Gleichgewichtes verändern das Gleichgewicht zwischen extra- und intrazellulärem Kalium und eine entstehende Azidose kann mit einem raschen Anstieg des Plasmakaliumspiegels einhergehen.
Natriumgehalt
Dieses Arzneimittel enthält bis zu 3,5 mmol (81,1 mg) Natrium pro Dosis, entsprechend 4 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
InteraktionenSomatostatin und seine Analoga binden kompetitiv an Somatostatinrezeptoren und können die Wirksamkeit von Lutathera/Lutathera CA beeinträchtigen. Die Behandlung mit lang wirksamen Somatostatinanaloga muss infolgedessen 4 bis 6 Wochen vor der Behandlung mit Lutathera/Lutathera CA unterbrochen werden. Nötigenfalls können Patienten bis zu 24 Stunden vor der Infusion von Lutathera/Lutathera CA mit kurz wirkenden Somatostatinanaloga behandelt werden.
Es gibt Hinweise darauf, dass Glukokortikoide eine Herabregulierung der Somatostatin-Rezeptoren vom Subtyp 2 (SSTR2) bewirken können. Daher sollte aus Vorsichtsgründen während der Behandlung mit Lutathera/Lutathera CA auf die wiederholte Verabreichung hoher Dosen von Glukokortikoiden verzichtet werden. Bei Patienten, die über längere Zeit mit Glukokortikoiden behandelt wurden, sollte sorgfältig geprüft werden, ob eine ausreichende Expression von Somatostatinrezeptoren vorliegt. Es ist nicht bekannt, ob die intermittierende Anwendung von Glukokortikoiden zur Vorbeugung von Übelkeit und Erbrechen während der Verabreichung von Lutathera/Lutathera CA zu einer SSTR2-Herabregulierung führen könnte. Deshalb sind als Vorsichtsmassnahme Glukokortikosteroide als präventive antiemetische Behandlung zu vermeiden. Sollte sich die zur Vorbeugung von Übelkeit und Erbrechen vor der Infusion der Aminosäurelösung verabreichte Behandlung als unzureichend erweisen, kann eine einzelne Glukokortikoid-Dosis gegeben werden, sofern dies nicht vor oder innerhalb einer Stunde ab Ende der Lutathera/Lutathera CA-Infusion geschieht.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Vor der Verwendung von Lutathera/Lutathera CA muss eine Schwangerschaft mit einem adäquaten/validierten Test ausgeschlossen werden.
Schwangerschaftsverhütung bei Männern und Frauen
Lutathera/Lutathera CA kann den Fötus schädigen, wenn es bei einer schwangeren Frau angewendet wird. Während der Behandlung mit Lutathera/Lutathera CA und für mindestens 7 Monate ab Ende der Behandlung müssen geeignete Massnahmen getroffen werden, um eine Schwangerschaft bei Patientinnen zu verhindern, Während der Behandlung mit Lutathera/Lutathera CA und für mindestens die folgenden 4 Monate nach Beendigung der Behandlung müssen geeignete Massnahmen getroffen werden, um eine Schwangerschaft bei Partnerinnen männlicher Patienten zu vermeiden.
Schwangerschaft
Es wurden keine Tierversuche zur Bestimmung der Wirkungen von Lutetium (177Lu)-Oxodotreotid auf die Reproduktion bei Frauen und die embryofetale Entwicklung durchgeführt.
Nuklearmedizinische Untersuchungen bei Schwangeren beinhalten auch eine Bestrahlung des Fötus. Die Verwendung von Lutathera/Lutathera CA während einer bestätigten oder vermuteten Schwangerschaft oder wenn eine solche nicht ausgeschlossen wurde, ist aufgrund des Risikos im Zusammenhang mit ionisierender Strahlung kontraindiziert (siehe Rubrik «Kontraindikationen»). Schwangere Frauen sollten auf das Risiko für den Fötus hingewiesen werden.
Stillzeit
Es ist nicht erwiesen, ob Lutetium (177Lu)-Oxodotreotid in die Muttermilch übergeht.
Ein Risiko für das gestillte Kind im Zusammenhang mit ionisierender Strahlung kann nicht ausgeschlossen werden. Das Stillen sollte während der Behandlung mit diesem Arzneimittel vermieden werden. Wenn eine Behandlung von Lutathera/Lutathera CA während der Stillzeit notwendig ist, muss das Kind abgestillt und das Stillen unterbrochen werden.
Fertilität
Es wurden keine Tierstudien zur Bestimmung der Wirkungen von Lutetium (177Lu)-Oxodotreotid auf die Fertilität bei beiden Geschlechtern durchgeführt. Die ionisierende Strahlung von Lutetium (177Lu)-Oxodotreotid kann toxische Wirkungen auf die weiblichen und männlichen Gonaden haben und zu einer vorübergehenden oder endgültigen Infertilität führen. Wenn der Patient bzw. die Patientin nach der Behandlung Kinder haben möchte, ist eine genetische Beratung angezeigt. Als Option kann den Patienten vor der Behandlung die Kryokonservierung von Spermien oder Eizellen vorgeschlagen werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenLutathera/Lutathera CA hat keinen Einfluss oder einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen. Trotzdem müssen vor dem Lenken von Fahrzeugen oder der Bedienung von Maschinen der allgemeine Gesundheitszustand des Patienten und mögliche unerwünschte Wirkungen im Zusammenhang mit der Behandlung berücksichtigt werden.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Das Gesamt-Sicherheitsprofil von Lutathera/Lutathera CA basiert auf gepoolten Daten von Patienten aus klinischen Studien (NETTER-1-Studie der Phase III und niederländischen Patienten aus der Erasmus-Studie der Phase I/II) und aus dem Härtefallprogramm («Compassionate Use Program»).
Die häufigsten unerwünschten Wirkungen bei Patienten, denen Lutathera/Lutathera CA verabreicht wurde, waren Übelkeit und Erbrechen; dies trat bei Beginn der Infusion bei 58.9 % bzw. bei 45.5 % der Patienten auf. Die Bestimmung der Kausalität von Übelkeit und Erbrechen wird durch die Übelkeit auslösenden Wirkungen der zeitgleichen Infusion von Aminosäurelösungen zum Schutz der Nieren erschwert.
Aufgrund der Knochenmarkstoxizität von Lutathera/Lutathera CA hingen die am häufigsten erwarteten unerwünschten Wirkungen mit der hämatologischen Toxizität zusammen: Thrombozytopenie (25 %), Lymphopenie (22.3 %), Anämie (13.4 %) und Panzytopenie (10.2 %).
Müdigkeit (27.7 %) und Appetitverlust (13.4 %) wurden als weitere sehr häufig auftretende unerwünschte Wirkungen berichtet.
Zum Zeitpunkt der abschliessenden Analyse der NETTER-1-Studie, nach einer medianen Nachbeobachtungszeit von 76 Monaten in jedem Studienarm, blieb das Sicherheitsprofil konsistent mit dem zuvor berichteten.
Zusammenfassende Tabelle der unerwünschten Wirkungen
Die unerwünschten Wirkungen werden in Tabelle 6 nach Häufigkeit und der Klassifikation der Organsysteme (MedDRA) aufgeführt. Die Häufigkeiten wurden gemäss der folgenden Konvention eingeteilt: sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1'000, <1/100), selten (≥1/10'000, <1/1'000), sehr selten (<1/10'000) und unbestimmte Häufigkeit (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Tabelle 6 Aus klinischen Studien und Nachbeobachtung nach Markteinführung berichtete Häufigkeit von unerwünschten Wirkungen
|
Systemorganklassen gemäss MedDRA
|
Sehr häufig
|
Häufig
|
Gelegentlich
|
Häufigkeit nicht bekannt
| |
Infektionen und parasitäre Erkrankungen
|
|
|
Konjunktivitis
Infektion der Atemwege
Zystitis
Pneumonie
Herpes zoster
Ophthalmischer Herpes zoster
Grippe
Staphylokokken-Infektionen
Streptokokken-Bakteriämien
|
| |
Gutartige, bösartige und nicht spezifizierte Neubildungen (einschl. Zysten und Polypen)
|
|
Refraktäre Zytopenie mit multilinearer Dysplasie (Myelodysplastisches Syndrom)
|
Akute myeloide Leukämie
Akute Leukämie
Chronisch myelomonozytäre Leukämie
|
| |
Erkrankungen des Blutes und des Lymphsystems
|
Thrombozytopenie (25 %)2
Lymphopenie (22.3 %)3
Anämie (13.4 %)4
Panzytopenie (10.2 %)
|
Leukopenie5
Neutropenie6
|
Refraktäre Zytopenie mit multilinearer Dysplasie
Nephrogene Anämie
Knochenmarkinsuffizienz
Thrombozytopenische Purpura
|
| |
Erkrankungen des Immunsystems
|
|
|
Hypersensitivität
|
Angioödem12
| |
Endokrine Erkrankungen
|
|
Sekundäre Hypothyreose
|
Hypothyreose
Diabetes mellitus
Karzinoide Krise
Hyperparathyreose
|
| |
Stoffwechsel- und Ernährungsstörungen
|
Verminderter Appetit (13,4 %)
|
Hyperglykämie
Dehydrierung
Hypomagnesiämie
Hyponatriämie
|
Hypoglykämie
Hypernatriämie
Hypophosphatämie
Tumorlyse-Syndrom
Hyperkalzämie
Hypokalzämie
Hypoalbuminämie
Metabolische Azidose
|
| |
Psychiatrische Erkrankungen
|
|
Schlafstörungen
|
Angst
Halluzinationen
Desorientiertheit
|
| |
Erkrankungen des Nervensystems
|
|
Schwindel
Dysgeusie
Kopfschmerzen10
Lethargie
Synkope
|
Ameisenlaufen
Hepatische Enzephalopathie
Parästhesie
Parosmie
Somnolenz
Rückenmarkskompression
|
| |
Augenerkrankungen
|
|
|
Augenerkrankungen
|
| |
Erkrankungen des Ohrs und des Labyrinths
|
|
|
Vertigo
|
| |
Herzerkrankungen
|
|
QT-Verlängerung im Elektrokardiogramm
|
Vorhofflimmern
Palpitationen
Myokardinfarkt
Angina pectoris
Kardiogener Schock
|
| |
Gefässerkrankungen
|
|
Hypertonie7
Erröten
Hitzewallungen
Hypotonie
|
Vasodilatation
Peripheres Kältegefühl
Blässe
Orthostatische Hypotonie
Phlebitis
|
| |
Erkrankungen der Atemwege, des Brustraums und Mediastinums
|
|
Dyspnoe
|
Schmerzen im Oropharynx
Pleuraerguss
Vermehrtes Sputum
Druckgefühl
|
| |
Erkrankungen des Gastrointestinaltrakts
|
Übelkeit (58.9 %)
Erbrechen (45.5 %)
|
Aufgetriebener Bauch
Diarrhö
Abdominale Schmerzen
Obstipation
Oberbauchschmerzen
Dyspepsie
Gastritis
|
Mundtrockenheit
Flatulenz
Aszites
Gastrointestinale Schmerzen
Stomatitis
Hämatochezie
Darmbeschwerden
Intestinale Obstruktion
Colitis
Akute Pankreatitis
Rektale Blutung
Meläna
Unterbauchschmerzen
Hämatemesis
Hämorrhagischer Aszites
Ileus
|
| |
Leber- und Gallenerkrankungen
|
|
Hyperbilirubinämie9
|
Verminderte Pankreasenzyme
Leberzellschädigung Cholestase
Leberstauung
Leberversagen
|
| |
Erkrankungen der Haut und des Unterhautgewebes
|
|
Alopezie
|
Hautausschlag
Trockene Haut
Schwellung im Gesicht
Hyperhidrose
Generalisierter Pruritus
|
| |
Skelettmuskulatur, Bindegewebs- und Knochenerkrankungen
|
|
Schmerzen des Muskel- und Skelettsystems8
Muskelspasmen
|
|
| |
Erkrankungen der Nieren und Harnwege
|
|
Akute Nierenschädigung
Hämaturie
Nierenversagen
Proteinurie
|
Leukozyturie
Harninkontinenz
Verminderte glomeruläre Filtrationsrate
Erkrankung der Niere
Akutes prärenale
Insuffizienz
Nierenschädigung
|
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Müdigkeit (27.7 %)1
|
Reaktion an der Injektionsstelle11
Peripheres Ödem
Schmerzen an der Verabreichungsstelle
Schüttelfrost
Grippeähnliche Erkrankung
|
Raumforderung an der Injektionsstelle
Unwohlsein im Brustkorb
Brustschmerzen
Fieber
Unwohlsein
Schmerzen
Tod
Gefühlsstörung
|
| |
Untersuchungen
|
|
Erhöhung des Kreatinins im Blut [Erkrankungen der Nieren und der Harnwege]
Erhöhung der GGT* [Leber- und Gallenerkrankungen]
Erhöhung der ALT** [Leber- und Gallenerkrankungen]
Erhöhung der AST*** [Leber- und Gallenerkrankungen]
Erhöhung der ALP**** [Leber- und Gallenerkrankungen]
|
Erniedrigtes Kalium im Serum [Erkrankungen der Nieren und der Harnwege]
Erhöhung des Harnstoffs im Blut [Erkrankungen der Nieren und Harnwege]
Erhöhung des glykosylierten Hämoglobins [Stoffwechsel- und Ernährungsstörungen]
Erniedrigter Hämatokrit [Erkrankungen des Blutes und des Lymphsystems]
Proteinurie [Erkrankungen der Nieren und Harnwege]
Gewichtsverlust [Allgemeine Erkrankungen und Beschwerden am Verabreichungsort]
Konzentrationserhöhung der Kreatinin-Phosphokinase im Serum [Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen]
Konzentrationserhöhung der Lactat-Dehydrogenase im Serum [Muskuloskelettale und systemische Erkrankungen]
Erhöhung der Blutkatecholamine [Hormonelle Erkrankungen]
Erhöhung des C-reaktiven Proteins [Infektionen und parasitäre Erkrankungen]
|
| |
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
|
|
|
Fraktur des Schlüsselbeins
|
| |
Chirurgische und medizinische Eingriffe
|
|
Transfusion
|
Drainage der Bauchhöhle
Dialyse
Einführung einer Magensonde
Stentanlage
Abszessdrainage
Knochenmarksgewinnung
Polypektomie
|
| |
Soziale Umstände
|
|
|
Körperliche Behinderung
|
|
1 Inklusive Asthenie und Erschöpfung
2 Inklusive Thrombozytopenie und verminderte Thrombozytenzahl
3 Inklusive Lymphopenie und verminderte Lymphozytenzahl
4 Inklusive Anämie und vermindertes Hämoglobin
5 Inklusive Leukopenie und Verminderung der Zahl weisser Blutkörperchen
6 Inklusive Neutropenie und verminderte Neutrophilenzahl
7 Inklusive Hypertonie und hypertensive Krise
8 Inklusive Arthralgie, Schmerzen in den Extremitäten, Rückenschmerzen, Knochenschmerzen, Flankenschmerzen, muskuloskelettale Brustschmerzen und Nackenschmerzen
9 Inklusive erhöhtes Blutbilirubin und Hyperbilirubinämie
10 Inklusive Kopfschmerzen und Migräne
11 Inklusive Reaktion an der Injektionsstelle, Überempfindlichkeit an der Injektionsstelle, Verhärtung an der Injektionsstelle, Schwellung an der Injektionsstelle
12 Meldung aus Postmarketing-Phase
* Erhöhung der Gamma-Glutamyltransferase
**Alanin-Amino-Transferase
***Aspartat-Amino-Transferase
****Alkalin-Phosphatase
Unerwünschte Wirkungen aus Spontanmeldungen (Häufigkeit nicht bekannt)
Die folgenden unerwünschten Wirkungen wurden aus Erfahrungen mit Lutathera/Lutathera CA nach der Markteinführung durch spontane Fallberichte abgeleitet (siehe Tabelle 6). Da diese Reaktionen freiwillig aus einer Population von ungewisser Grösse gemeldet werden, ist es nicht möglich, ihre Häufigkeit zuverlässig abzuschätzen, weshalb dieses als nicht bekannt eingestuft wird. Unerwünschte Wirkungen werden in MedDRA nach Systemorganklassen aufgeführt. Innerhalb jeder Systemorganklasse werden unerwünschte Wirkungen in der Reihenfolge ihres abnehmenden Schweregrades aufgeführt.
Erkrankungen des Immunsystems
Angioödem.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurden keine Fälle einer Überdosierung berichtet.
Im Falle einer Überdosierung ist eine Erhöhung der Häufigkeit von unerwünschten Wirkungen mit Bezug zur Radiotoxizität zu erwarten.
Im Falle der Verabreichung einer Überdosis von Lutathera/Lutathera CA sollte die vom Patienten aufgenommene Strahlendosis durch Erhöhung der Ausscheidung des Radionuklids aus dem Körper durch forcierte Diurese und häufige Blasenentleerung sowie durch erhöhte Flüssigkeitszufuhr während der ersten 48 Stunden nach der Infusion so weit wie möglich reduziert werden. Die verabreichte effektive Dosis ist abzuschätzen.
Es ist zu empfehlen, die folgenden Laboruntersuchungen während der folgenden 10 Wochen wöchentlich durchzuführen:
·Hämatologische Überwachung: weisses Blutbild, Plättchen und Hämoglobin;
·Überwachung der Blutchemie: Serumkreatinin und Glykämie.
Eigenschaften/WirkungenATC-Code
V10XX04
Physikalische Eigenschaften
Lutathera/Lutathera CA mit Lutetium (177Lu) zerfällt mit einer Halbwertszeit von 6.647 Tagen zu stabilem Hafnium (177Hf) und emittiert hauptsächlich β—Strahlung —mit einer maximalen Energie von 0.498 MeV. Die durchschnittliche β-Energie beträgt 0.13 MeV. Es wird auch Photonen-Strahlung (γ) von 0.113 MeV (6.2 %) und 0.208 MeV (11 %) emittiert.
Lutathera CA wird unter Verwendung von 176Lu hergestellt, das eine kleine Menge des metastabilen Lutetium-Kernisomers (177mLu) enthält. Das Isomer 177mLu hat eine Halbwertszeit von 160.44 Tagen. Das Isomer 177mLu zerfällt teilweise (22.8 %) durch isomeren Übergang unter Emission von Gammastrahlung und Konversionselektronen zum Grundzustand von Lu-177, und teilweise (77.2 %) durch Abgabe von Betastrahlung (40.8 keV) zu metastabilem Hafnium-177 (177mHf), das durch mehrfache Gammaemission und Konversionselektronen sofort zu stabilem 177Hf zerfällt. Bezüglich allen aus der Verwendung von Lutetium (177Lu)-Oxodotreotid entstehenden radioaktiven Abfällen muss für eine sachgerechte Entsorgung das Vorhandensein und die Menge dieses speziellen Isomers beachtet werden.
Wirkungsmechanismus
Lutetium (177Lu)-Oxodotreotid hat eine hohe Affinität zu Somatostatin-Subtyp-2-Rezeptoren (sst2). Es bindet spezifisch an maligne Zellen, die sst2-Rezeptoren überexprimieren.
Lutetium-177 ist ein β-minus emittierendes Radionuklid mit einem maximalen Penetrationsbereich im Gewebe von etwa 2.2 mm (mittlerer Penetrationsbereich von 0.67 mm), das den Tod der anvisierten Tumorzellen herbeiführt bei gleichzeitig begrenzter Wirkung auf benachbarte normale Zellen.
Pharmakodynamik
Bei der verwendeten Konzentration (insgesamt ca. 10 µg/ml, sowohl für die freie als auch die radiomarkierte Form), übt das Peptid Oxodotreotid keine klinisch relevante pharmakodynamische Wirkung aus.
Klinische Wirksamkeit
NETTER-1-Studie
Die NETTER-1-Studie der Phase III war eine randomisierte, multizentrische, offene aktiv-kontrollierte Vergleichsstudie, in der die Behandlung mit Lutathera CA (4 Dosen mit jeweils 7'400 MBq, eine Dosis alle 8 Wochen [±1 Woche]) bei gleichzeitiger Verabreichung einer Aminosäurenlösung und bestmöglicher unterstützender Behandlung (Octreotid mit verlängerter Wirkstoffabgabe [LAR] 30 mg nach jeder Lutathera CA-Dosis und alle 4 Wochen nach Abschluss der Behandlung mit Lutathera CA zur Symptomkontrolle, ersetzt durch Octreotid mit verkürzter Wirkstoffabgabe in den 4 bis 6 Wochen vor der Verabreichung von Lutathera CA) mit hochdosiertem Octreotid mit LAR (60 mg alle 4 Wochen) bei Patienten mit inoperablen, progressiven, Somatostatinrezeptor-positiven karzinoiden Tumoren des Mitteldarms verglichen wurde. Der primäre Endpunkt der Studie war das progressionsfreie Überleben (PFS), beurteilt anhand der RECIST-Kriterien (RECIST V.1.1) und basierend auf einer verblindeten unabhängigen radiologischen Beurteilung. Sekundäre Wirksamkeitsendpunkte schlossen die objektive Ansprechrate (ORR), das Gesamtüberleben (OS), die Zeit bis zur Tumorprogression (TTP), die Sicherheit und Verträglichkeit des Arzneimittels sowie die gesundheitsbezogene Lebensqualität (HRQoL) mit ein.
Zum Zeitpunkt der Primäranalyse waren 229 Patienten randomisiert und erhielten entweder Lutathera CA (n = 116) oder eine hohe Dosis von 60 mg Octreotid LAR (n = 113). Die Randomisierung erfolgte stratifiziert nach dem Score der Octreoscan®-Szintigraphie (Grad 2, 3 und 4) und der längsten Dauer der vom Patienten zuletzt vor der Randomisierung erhaltenen konstanten Octreotid-Dosis (d.h. ≤6 oder > 6 Monate). Demografische Kriterien sowie Patienten- und Krankheitscharakteristika waren zwischen den beiden Studienarmen ausgeglichen, mit einem medianen Alter von 64 Jahren und einem Anteil von 82.1 % Patienten weisser Hautfarbe in der allgemeinen Population.
Die Ergebnisse der endgültigen Per-Protokoll-Analyse (Stichtag 24. Juli 2015) sind in Tabelle 7 dargestellt.
Tabelle 7 Beobachtetes PFS in der Phase-III-Studie NETTER 1 bei Patienten mit progressiven karzinoiden Mitteldarmtumoren – (volles Analyseset [FAS], n = 229)
|
|
Behandlung
| |
|
Lutathera CA und Octreotid LAR
|
Hochdosiertes Octreotid LAR
| |
N
|
116
|
113
| |
Patienten mit Ereignissen
|
21
|
70
| |
Ausgeschlossene Patienten
|
95
|
43
| |
Median Monate (95 %-CI)
|
Nicht erreicht
|
8.5 (5.8; 9.1)
| |
p-Wert des Log-Rank-Tests
|
< 0.0001
| |
Hazard ratio (95 %-CI)
|
0.177 (0.108; 0.289)
|
N: Anzahl Patienten, CI: Konfidenzintervall LAR = verlängerte Wirkstoffabgabe.
Die Kaplan-Meier-Grafik des PFS für das volle Analyseset (FAS) ist in Abbildung 3 dargestellt.
Abbildung 3 Kaplan-Meier-Kurven des PFS für Patienten mit progressiven karzinoiden Mitteldarmtumoren – (NETTER-1- Studie der Phase III; FAS, n = 229)

Die OS Ergebnisse der Zwischenanalyse (Stichtag 24. Juli 2015) und der endgültigen Analyse (Stichtag 18. Januar 2021) sind in Tabelle 8 dargestellt.
Zum Zeitpunkt der endgültigen OS-Analyse, die 5 Jahre nach der Randomisierung des letzten Patienten stattfand (N=231, Stichtag 24. Juli 2015) betrug die mediane Nachbeobachtungszeit in jedem Studienarm 76 Monate. Die endgültigen OS-Ergebnisse erreichten keine statistische Signifikanz.
Im hochdosierten Octreotid-LAR-Arm erhielten 22.8 % der Patienten innerhalb von 24 Monaten nach der Randomisierung eine anschliessende Radioliganden-Therapie (einschliesslich Lutetium (177Lu)-Oxodotreotid) und 36 % der Patienten bis zum endgültigen OS-Stichtag, was zusammen mit anderen Faktoren das OS in dieser Untergruppe von Patienten beeinflusst haben könnte.
Tabelle 8 Ergebnisse der NETTER-1-Phase-III-Studie zum Gesamtüberleben von Patienten mit progressiven karzinoiden Mitteldarmtumoren (FAS)
|
|
LUTATHERA CA und Octreotid LAR
|
Hochdosiertes Octreotid LAR
| |
Zwischenanalyse des Gesamtüberlebens (24. Juli 2015) - N=229*
| |
Todesfälle (%)
|
17 (14.7 %)
|
31 (27.4 %)
| |
Median in Monaten (95 % CI)
|
NR (NE, NE)
|
27.4 (20.1, NE)
| |
Hazard Ratioa,b (99,9915 % CI)
|
0.46 (0.14, 1.51)
| |
Endgültige Analyse des Gesamtüberlebens (18. Januar 2021) - N=231**
| |
Todesfälle (%)
|
73 (62.4 %)
|
69 (60.5 %)
| |
Median in Monaten (95 % CI)
|
48.0 (37.4, 55.2)
|
36.3 (25.9, 51.7)
| |
Hazard Ratioa,b,c (95 % CI)
|
0.84 (0.60, 1.17)
| |
Endgültige Analyse des Gesamtüberlebens nach eingeschränkter mittlerer Überlebenszeit (RMST = Restricted Mean Survival Time) nach 60 Monaten (18. Januar 2021) – N=231**)
| |
Todesfälle (%)
|
65 (55.6)
|
63 (55.3)
| |
RMST (95% CI)
|
41.2 (37.6, 44.9)
|
36.1 (31.9, 40.4)
| |
Differenz (95% CI)
|
5.1 (-0.5, 10.7)
| |
a: Hazard Ratio basierend auf einem nicht-stratifizierten Cox-Modell
b: Nach den vorgegebenen Signifikanzkriterien statistisch nicht signifikant
c: HR auf der Grundlage nichtproportionaler Hazards
*: Die Analyse wurde bei 116 Patienten in der Lutathera CA-Gruppe und 113 Patienten in der Gruppe mit hochdosiertem Octreotid LAR durchgeführt (N=229).
**: Die Analyse wurde bei 117 Patienten in der Lutathera CA-Gruppe und 114 Patienten in der Gruppe mit hochdosiertem Octreotid LAR durchgeführt (N=231).
NR=Nicht erreicht
NE=Nicht schätzbar
|
Das OS-Kaplan-Meier-Diagramm für den vollständigen Analysesatz (FAS) zum Stichtag 18. Januar 2021 ist in Abbildung 4 dargestellt.
Abbildung 4 OS-Kaplan-Meier-Kurven für Patienten mit progressiven karzinoiden Mitteldarmtumoren - Stichtag 18. Januar 2021 (NETTER-1-Studie der Phase III; FAS, N=231)
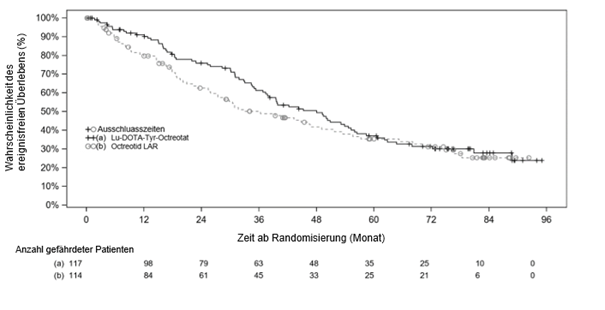
Bei Vorliegen nichtproportionaler Hazards wurde zum Zeitpunkt der endgültigen OS-Analyse eine zusätzliche Sensitivitätsanalyse (Restricted Mean Survival Time) durchgeführt, um den Behandlungseffekt weiter abzuschätzen (siehe Tabelle 8).
Die gesundheitsbezogene Lebensqualität (Health Related Quality of Life, HRQoL) wurde unter Verwendung des Fragebogens zur Lebensqualität der Europäische Organisation zur Erforschung und Behandlung von Krebs (EORTC QLQ-C30) (allgemeines Instrument) und seinem Modul für neuroendokrine Tumoren (EORTC QLQ-GI.NET-21) beurteilt.
Die Ergebnisse weisen für Patienten im Lutathera CA-Behandlungsarm im Vergleich zum Behandlungsarm mit hochdosiertem Octreotid LAR auf eine Verbesserung der allgemeinen gesundheitsbezogenen Lebensqualität bis Woche 84 hin.
Somatostatinrezeptor-exprimierende gastroenteropankreatische neuroendokrine Tumoren (GEP-NET)
Die Wirksamkeit von Lutathera CA bei Patienten mit Somatostatinrezeptor-exprimierenden gastroenteropankreatischen (GEP) und bronchialen neuroendokrinen Tumoren (NETs), wurde in der klinischen, monozentrischen, nicht vergleichenden, offenen Erasmus-Studie untersucht. Das Behandlungsprotokoll für Lutathera CA bestand hier aus vier intravenösen Verabreichungen von jeweils 7'400 MBq zusammen mit einer Aminosäurenlösung bestand. Lutathera CA wurde zunächst im Rahmen eines in einem einzigen Zentrum in den Niederlanden durchgeführten Härtefallprogramms («Compassionate Use Program») gemäss einem allgemeinen Peptidrezeptor-Radionuklidtherapie-Protokoll verabreicht. Acht Jahre nach dem Beginn dieses Programms wurde dann ein konkret auf Lutathera CA bezogenes Protokoll erstellt, das eine retrospektive Datenerfassung ermöglichte, auch wenn die Gesamtzahl der Patienten und die geprüfte Hypothese nicht genau beschrieben sind. Insgesamt wurden 360 Patienten langfristig nachbeobachtet, die zu Beginn der Behandlung einen gastroenteropankreatischen und bronchialen neuroendokrinen Tumor aufwiesen (ein Tumor des Mitteldarms 183, Pankreas 133, Bronchien 19, Hinterdarm 13, Vorderdarm ohne Bronchien und Pankreas 12). Das mittlere Alter betrug 60 Jahre, 51 % waren Männer, 99.4 % wiesen beim Octreoscan einen Anreicherungsscore von ≥2 (5.6 % (2)/62.8 % (3)/31.1 % (4)) auf, 71.4 % hatten einen Karnofsky-Index ≥90 und 52 % eine Begleitmedikation mit Somatostatin-Analoga. Die durch den Prüfarzt bestimmte objektive Ansprechrate als Hauptkriterium der Wirksamkeit betrug 45 % (95 % CI: 40, 50). Der Median der Ansprechdauer lag bei 16.3 Monaten (95 % CI: 12.2, 17.8). Die höchste objektive Ansprechrate wurde bei Patienten mit neuroendokrinen Pankreastumoren festgestellt (61 %, 95 % CI: 52, 69), die geringste Ansprechrate bei neuroendokrinen Tumoren des Mitteldarms (33 %, 95 % CI: 27, 41).
PharmakokinetikAbsorption
Das Arzneimittel wird intravenös verabreicht und ist sofort und vollständig bioverfügbar.
Distribution
Eine mit menschlichem Plasma zur Bestimmung des Ausmasses der Plasmaproteinbindung der nicht-radioaktiven Verbindung (Lutetium (175Lu)-Oxodotreotid) durchgeführte Analyse zeigte, dass ungefähr 50 % der Verbindung an Plasmaproteine gebunden wird.
Es konnte keine Transchelatierung von Lutetium aus Lutetium (175Lu)-Oxodotreotid auf Serumproteine beobachtet werden.
Aufnahme in die Organe
Innerhalb von 4 Stunden nach Verabreichung zeigt das Verteilungsmuster von Lutetium (177Lu)-Oxodotreotid eine rasche Aufnahme in Nieren, Tumorläsionen, Leber und Milz sowie bei einigen Patienten in die Hypophyse und die Schilddrüse. Die gleichzeitige Verabreichung einer Aminosäurenlösung verringert die Aufnahme in die Niere und begünstigt damit die Eliminierung des radioaktiven Produkts (siehe «Warnhinweise und Vorsichtsmassnahmen»). Biodistributionsstudien zeigen, dass Lutetium (177Lu)-Oxodotreotid rasch aus der Blutzirkulation eliminiert wird.
Metabolismus
Biotransformation
In der Analyse von Urinproben von 20 Patienten aus der Substudie zu Dosimetrie, Pharmakokinetik und EKG der NETTER-1-Studie der Phase III wurde nachgewiesen, dass Lutetium (177Lu)-Oxodotreotid schwach metabolisiert und im Wesentlichen in unveränderter Form über die Niere ausgeschieden wird.
An bis zu 48 Stunden nach Infusion gewonnenen Urinproben durchgeführte HPLC-Analysen zeigten unverändertes Lutetium (177Lu)-Oxodotreotid von nahezu 100 % in den meisten analysierten Proben (mit einem Tiefstwert grösser als 92 %), was darauf hinweist, dass das Produkt grösstenteils in unveränderter Form im Urin ausgeschieden wird.
Diese Ergebnisse bestätigen die in der vorgängigen Erasmus-Studie der Phase I/II gewonnenen Erkenntnisse, in der die HPLC-Analyse von 1 Stunde nach Verabreichung von 1.85 MBq Lutetium (177Lu)-Oxodotreotid von einem Patienten entnommenen Urinproben nachwies, dass der Hauptanteil (91 %) unverändert ausgeschieden wurde.
Diese Befunde werden durch in vitro-Metabolismusdaten in menschlichen Hepatozyten unterstützt, in denen kein metabolischer Abbau von Lutetium (175Lu)-Oxodotreotid beobachtet wurde.
Elimination
Basierend auf den während der Erasmus-Studie der Phase I/II und der NETTER-1-Studie der Phase III erhobenen Daten wird Lutetium (177Lu)-Oxodotreotid primär durch die Niere ausgeschieden: In den ersten 24 Stunden nach der Verabreichung wird ungefähr 60 %, in den ersten 48 Stunden ungefähr 65 % des Arzneimittels über den Urin eliminiert.
In vitro-Bewertung des Interaktionspotenzials
Das Fehlen einer Inhibition oder einer signifikanten Induktion humaner CYP450-Enzyme, die Absenz einer spezifischen Interaktion mit P-Glycoprotein (Efflux-Transporter) sowie mit OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3 und BCRP-Transportern in präklinischen Studien legen nahe, dass das Risiko von anderen Arzneimittel-Interaktionen mit Lutathera/Lutathera CA wenig wahrscheinlich ist.
Präklinische DatenToxikologische Studien an Ratten haben gezeigt, dass eine einzelne intravenöse Injektion von bis zu 4'550 MBq/kg gut toleriert wurde; es gab keine Todesfälle. Bei Prüfung der nicht-radioaktiven Verbindung (nicht-radioaktives Lutetium (175Lu)-Oxodotreotid) als einzelne intravenöse Injektion an Ratten und Hunden bei Dosierungen von bis zu 20'000 µg/kg (Ratten) und 3'200 µg/kg (Hunde) zeigte sich, dass die nicht-radioaktive Verbindung (nicht-radioaktives Lutetium (175Lu) Oxodotreotid) bei beiden Spezies gut verträglich war und es keine Todesfälle gab. Bei der wiederholten Verabreichung von vier Dosen der nicht-radioaktiven Verbindung alle zwei Wochen (1'250 µg/kg bei der Ratte und 80 µg/kg beim Hund) wurde keinerlei Toxizität beobachtet. Dieses Arzneimittel ist nicht zur regelmässigen oder fortgesetzten Verabreichung gedacht.
Es wurden keine Mutagenitätsstudien und Langzeitstudien zur Kanzerogenität durchgeführt.
Die präklinischen Daten der nicht-radioaktiven Verbindung (nicht-radioaktives Lutetium (175Lu)-Oxodotreotid) lassen, basierend auf konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe und Genotoxizität, keine besonderen Gefahren für den Menschen erkennen.
Sonstige HinweiseInkompatibilitäten
Das Arzneimittel darf nur mit den unter Rubrik «Dosierung/Anwendung» aufgeführten Arzneimitteln gemischt werden.
Haltbarkeit
Dieses Arzneimittel darf ab dem Zeitpunkt der Kalibrierung höchstens während 72 Stunden gelagert werden.
Besondere Lagerungshinweise
Nicht über 25 °C lagern. Nicht einfrieren.
In der Originalverpackung aufbewahren, um vor ionisierender Strahlung zu schützen (Bleiabschirmung).
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Die Lagerung von Radiopharmaka muss in Übereinstimmung mit den nationalen Vorschriften über radioaktive Produkte erfolgen.
Hinweise für die Handhabung/Strahlenschutz
Die Anwendung von radioaktiven Stoffen am Menschen wird durch die Strahlenschutzverordnung geregelt. Für die Verwendung radioaktiver Stoffe ist eine Umgangsbewilligung des Bundesamtes für Gesundheit erforderlich.
Lutathera/Lutathera CA darf nur in zugelassenen Einrichtungen von autorisierten Personen in Empfang genommen, verwendet und verabreicht werden. Die Entgegennahme, Lagerung, Anwendung, der Transport und die Entsorgung von Lutathera/Lutathera CA unterliegen den strahlenschutzrechtlichen Bestimmungen und/oder entsprechenden Genehmigungen der zuständigen Aufsichtsbehörden.
Lutathera/Lutathera CA muss mit geeigneten Sicherheitsmassnahmen zur Minimierung der Strahlenexposition angewandt werden Es sind geeignete aseptische Vorsichtsmassnahmen zu treffen.
Die Lösung muss vor der Verwendung visuell auf Partikel, Verfärbungen und Verunreinigungen überprüft werden. Es dürfen nur klare und partikelfreie Lösungen verwendet werden. Das Vial sollte verworfen werden, wenn Partikel/Verfärbungen/Verunreinigungen vorhanden sind. Die Sichtprüfung der Lösung muss zum Schutz vor Strahlung hinter einer Abschirmung durchgeführt werden.
Die Verpackung muss auf Beschädigungen untersucht werden, und das Vorhandensein einer radioaktiven Kontamination sollte mit einem geeigneten Aktivitätsmessgerät festgestellt werden. Wenn die Unversehrtheit des Fläschchens oder des Bleibehälters beeinträchtigt ist, darf das Produkt nicht verwendet werden. Lutathera/Lutathera CA darf nicht direkt in eine andere intravenöse Lösung injiziert werden.
Der Gehalt an Radioaktivität in der Durchstechflasche muss vor und nach jeder Infusion mit einem kalibrierten Aktivitätsmessgerät gemessen werden, um sicherzustellen, dass die Menge an zu verabreichender Radioaktivität der geplanten Menge zum Zeitpunkt der Infusion entspricht.
Die Anwendung ist so durchzuführen, dass das Risiko einer Kontaminierung des Arzneimittels und einer Strahlenexposition des Anwenders auf ein Minimum beschränkt wird. Eine ausreichende Abschirmung ist zwingend erforderlich und es müssen undurchlässige Handschuhe getragen werden, wenn dieses Arzneimittel gehandhabt wird.
Die Anwendung von Radiopharmaka stellt aufgrund der vom Patienten ausgehenden Strahlung oder durch Kontaminierung durch Körperflüssigkeiten, wie Urin, Stuhl, Erbrochenem usw. einen Risikofaktor für andere Personen dar. Daher sind entsprechende Vorsichtsmassnahmen zu treffen.
Das medizinische Personal ist angehalten, die Dauer des engen Kontakts mit Patienten, die Lutathera/Lutathera CA erhalten haben, zu begrenzen. Die Verwendung von Überwachungsbildschirmen zur Patientenbeobachtung wird empfohlen. Aufgrund der langen Halbwertszeit von 177Lu muss insbesondere eine interne Kontaminierung vermieden werden. Zur Vermeidung von direktem Kontakt mit dem radioaktiven Arzneimittel (Durchstechflasche, Spritze) müssen qualitativ hochwertige (Latex/Nitril) Schutzhandschuhe getragen werden. Zur Minimierung der Strahlenexposition müssen immer die Prinzipien Zeit, Distanz und Abschirmung (indem die Handhabung der Durchstechflasche so gering wie möglich gehalten und die vom Hersteller bereitgestellten Geräte verwendet werden) befolgt werden.
Die Verabreichung kann zu einer bedeutenden Strahlenexposition für die Umgebung führen.
Dies kann für die engen Familienmitglieder der behandelten Personen oder für die Öffentlichkeit bedeutsam sein. Die Strahlenschutzanforderungen müssen dementsprechend zwingend befolgt werden (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»). Bezüglich der von Patienten eliminierten Aktivität müssen in Übereinstimmung mit nationalen Vorschriften geeignete Vorsichtsmassnahmen getroffen werden, um jegliche Kontaminierung zu verhindern.
Entsorgung der Abfälle
Ungebrauchte radioaktive Produkte oder Abfälle dürfen nur nach den geltenden schweizerischen Strahlenschutzvorschriften entsorgt werden.
Lutathera
Lutetium-177 für Lutathera wird unter Verwendung des stabilen Isotops Ytterbium-176 («non-carrier added») hergestellt.
Lutathera CA
Lutetium-177 für Lutathera CA wird unter Verwendung des stabilen Isotops Lutetium-176 («carrier added») hergestellt und erfordert aufgrund des Vorhandenseins von metastabilem Lutetium-177 (177mLu) besondere Aufmerksamkeit bei der Abfallentsorgung.
Zulassungsnummer66580, 69776 (Swissmedic)
PackungenKlare, farblose Durchstechflasche aus Glas Typ I, die mit einem Bromobutyl-Gummipropfen verschlossen und mit einer Aluminiumkapsel versiegelt ist.
Jede Durchstechflasche enthält ein variierendes Volumen von 20.5 bis 25.0 ml Lösung, entsprechend einer Aktivität von 7'400 MBq am Tag und zum Zeitpunkt der Infusion.
Die Durchstechflasche ist zur Abschirmung von einem Bleibehältnis umschlossen.
Abgabekategorie A.
ZulassungsinhaberinNovartis Pharma Schweiz AG. Risch; Domizil: 6343 Rotkreuz
Stand der InformationJuni 2024
|