ZusammensetzungWirkstoffe
Sotatercept.
Hilfsstoffe
Acidum citricum monohydricum (E330), trinatrii citras dihydricus (E331), polysorbatum 80 (E433), saccharum.
Die rekonstituierte Lösung enthält 0,55 mg Natrium pro ml.
Indikationen/AnwendungsmöglichkeitenWinrevair ist, in Kombination mit einer Standardtherapie für die pulmonale arterielle Hypertonie (PAH), indiziert zur Behandlung von PAH bei erwachsenen Patienten mit WHO-Funktionsklasse (FC) II bis III, zur Verbesserung der körperlichen Leistungsfähigkeit und zur Verzögerung der Krankheitsprogression (siehe «Klinische Wirksamkeit»).
Die Wirksamkeit wurde in einer PAH Population einschliesslich folgender Ätiologien gezeigt: idiopathische und hereditäre PAH, PAH assoziiert mit einer Bindegewebserkrankung, medikamenten- oder toxininduzierte PAH oder PAH in Zusammenhang mit einem angeborenen Herzfehler mit reparierten Shunts (siehe «Klinische Wirksamkeit»).
Dosierung/AnwendungDie Behandlung mit Winrevair sollte nur von einem Arzt mit Erfahrung in der Diagnose und Behandlung der PAH eingeleitet und überwacht werden.
Dosierung
Empfohlene Anfangsdosierung für Erwachsene
Winrevair wird einmal alle 3 Wochen als subkutane (s.c.) Injektion in Abhängigkeit vom Körpergewicht verabreicht. Die Anfangsdosis von Winrevair beträgt 0,3 mg/kg (siehe Tabelle 1).
Vor der ersten Dosis von Winrevair sind der Hämoglobin-(Hb-)Wert und die Thrombozytenzahl zu bestimmen. Bei Patienten mit einer Thrombozytenzahl von < 50'000/mm3 (< 50,0 x 109/l) sollte keine Behandlung mit Winrevair initiiert werden (siehe «Dosierung/Anwendung», «Dosierungsanpassung bei Erwachsenen aufgrund eines Hämoglobin-Anstiegs oder einer Verminderung der Thrombozytenzahl»). Nach Beginn der Behandlung wurden rasche Hb-Anstiege von mehr als 2 g/dl beobachtet.
Tabelle 1: Injektionsvolumen für eine Dosis von 0,3 mg/kg
|
Gewichtsbereich des Patienten (kg)
|
Injektionsvolumen (ml)
|
Set Typ
| |
30,0–40,8
|
0,2
|
45-mg-Set (mit 1 Durchstechflasche zu 45 mg)
| |
40,9–57,4
|
0,3
| |
57,5–74,1
|
0,4
| |
74,2–90,8
|
0,5
| |
90,9–107,4
|
0,6
| |
107,5–124,1
|
0,7
| |
124,2–140,8
|
0,8
| |
140,9–157,4
|
0,9
| |
157,5–174,1
|
1,0
|
60-mg-Set (mit 1 Durchstechflasche zu 60 mg)
| |
174,2–180,0
|
1,1
|
Empfohlene Zieldosierung für Erwachsene
Die Zieldosis von Winrevair beträgt 0,7 mg/kg (siehe Tabelle 2) alle 3 Wochen.
Vor der Erhöhung auf die Zieldosis sind der Hämoglobin-(Hb-)Wert und die Thrombozytenzahl zu bestimmen und zu überprüfen. Die Behandlung wird mit 0,7 mg/kg alle 3 Wochen fortgeführt, sofern keine Dosierungsanpassungen nötig sind (siehe «Dosierung/Anwendung», «Dosierungsanpassung bei Erwachsenen aufgrund eines Hämoglobin-Anstiegs oder einer Verminderung der Thrombozytenzahl»).
Tabelle 2: Injektionsvolumen für eine Dosis von 0,7 mg/kg
|
Gewichtsbereich des Patienten (kg)
|
Injektionsvolumen (ml)
|
Set Typ
| |
30,0–31,7
|
0,4
|
45-mg-Set (mit 1 Durchstechflasche zu 45 mg)
| |
31,8–38,9
|
0,5
| |
39,0–46,0
|
0,6
| |
46,1–53,2
|
0,7
| |
53,3–60,3
|
0,8
| |
60,4–67,4
|
0,9
| |
67,5–74,6
|
1,0
|
60-mg-Set (mit 1 Durchstechflasche zu 60 mg)
| |
74,7–81,7
|
1,1
| |
81,8–88,9
|
1,2
| |
89,0–96,0
|
1,3
|
90-mg-Set (mit 2 Durchstechflaschen zu 45 mg)
| |
96,1–103,2
|
1,4
| |
103,3–110,3
|
1,5
| |
110,4–117,4
|
1,6
| |
117,5–124,6
|
1,7
| |
124,7–131,7
|
1,8
| |
131,8–138,9
|
1,9
|
120-mg-Set (mit 2 Durchstechflaschen zu 60 mg)
| |
139,0–146,0
|
2,0
| |
146,1–153,2
|
2,1
| |
153,3–160,3
|
2,2
| |
160,4–167,4
|
2,3
| |
167,5 und mehr
|
2,4
|
Dosierungsanpassungen bei Erwachsenen aufgrund eines Hämoglobin-Anstiegs oder einer Verminderung der Thrombozytenzahl
Es wurden Hb-Anstiege auf Werte von mehr als 2 g/dl über der oberen Normgrenze (ULN) und Verminderungen der Thrombozytenzahl auf < 50'000/mm3 (< 50,0 x 109/l) beobachtet. Die Hb-Werte und Thrombozytenzahl sind vor jeder Dosis für die ersten 5 Dosen, oder wenn die Werte nicht stabil sind länger, zu kontrollieren. Anschliessend sind die Hb-Werte und die Thrombozytenzahlen regelmässig zu kontrollieren. Bei der Entscheidung, ob eine Dosisanpassung angemessen ist, sollte das Nutzen-Risiko-Verhältnis für den einzelnen Patienten berücksichtigt werden (siehe «Warnhinweise und Vorsichtsmassnahmen», «Erythrozytose», «Schwere Thrombozytopenie»).
Die Behandlung ist um 3 Wochen zu verschieben, wenn eine der folgenden Situationen eintritt:
·Hb-Anstieg > 2,0 g/dl seit der vorherigen Dosis und Hb-Wert über der ULN
·Hb-Anstieg > 4,0 g/dl gegenüber dem Ausgangswert
·Hb-Anstieg > 2,0 g/dl über der ULN
·Verminderung der Thrombozytenzahl auf < 50'000/mm3 (< 50,0 x 109/l)
Im Fall eines Behandlungsunterbruchs von > 9 Wochen ist die Behandlung zunächst mit 0,3 mg/kg wiederaufzunehmen.
Versäumte Dosis, Überdosierung und Unterdosierung
Wenn eine Dosis von Winrevair ausgelassen wurde, sollte die versäumte Dosis so schnell wie möglich verabreicht werden. Sollte die versäumte Dosis von Winrevair nicht innerhalb von 3 Tagen nach dem geplanten Zeitpunkt verabreicht werden, ist das Dosierungsschema so anzupassen, dass das 3-wöchige Dosierungsintervall eingehalten wird. Im Fall einer Über- oder Unterdosierung sollte gegebenenfalls eine erneute Schulung des Patienten bzw. der Betreuungsperson in der korrekten Verabreichung in Erwägung gezogen werden. Im Fall einer Überdosierung ist der Patient auf eine Erythrozytose zu überwachen (siehe «Überdosierung»).
Spezielle Patientengruppen
Patienten mit Leberfunktionsstörungen
Winrevair wurde bei Patienten mit Leberfunktionsstörungen (Child-Pugh-Klasse A bis C) nicht untersucht. Es wird nicht davon ausgegangen, dass eine Leberfunktionsstörung den Metabolismus von Sotatercept beeinflusst, da Sotatercept über den zellulären Katabolismus metabolisiert wird (siehe «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Eine Dosisanpassung von Winrevair aufgrund einer Nierenfunktionsstörung ist nicht erforderlich. Sotatercept wurde bei PAH Patienten mit schwerer Nierenfunktionsstörung (eGFR < 30 ml/min/1,73 m2) nicht untersucht (siehe «Pharmakokinetik»).
Ältere Patienten
Eine altersspezifische Dosisanpassung von Winrevair ist nicht erforderlich (siehe «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Winrevair bei Patienten unter 18 Jahren wurde nicht untersucht.
Art der Anwendung
Winrevair sollte vor der Anwendung rekonstituiert und als subkutane Injektion im Bereich von Bauch (mindestens 5 cm vom Nabel entfernt), Oberarm oder Oberschenkel verabreicht werden.
Das Winrevair-Set ist für die Anwendung unter Anleitung einer medizinischen Fachperson bestimmt. Patienten und Betreuungspersonen können Winrevair verabreichen, wenn sie in der Lage dazu sind und wenn sie von der medizinischen Fachperson in der Rekonstitution, Vorbereitung, Dosierung und Injektion von Winrevair geschult und im weiteren Verlauf unterstützt werden. Ausführliche Hinweise zur sachgerechten Vorbereitung und Verabreichung von Winrevair sind dem Abschnitt «Sonstige Hinweise/Hinweise für die Handhabung» und der dem Set beiliegenden Gebrauchsanweisung zu entnehmen.
Bei Folgeterminen sollte überprüft werden, ob der Patient oder die Betreuungsperson Winrevair korrekt vorbereiten und verabreichen kann:
·wenn sich die Dosis ändert oder der Patient ein anderes Set benötigt
·wenn sich beim Patienten eine Erythrozytose entwickelt (siehe «Warnhinweise und Vorsichtsmassnahmen», «Erythrozytose»)
Auswahl des geeigneten Produkt-Sets
Falls ein Patient aufgrund seines Körpergewichts zwei 45-mg-Durchstechflaschen oder zwei 60-mg-Durchstechflaschen mit Lyophilisat benötigt, ist ein Set mit 2 Durchstechflaschen, statt zwei einzelner Sets mit jeweils 1 Durchstechflasche zu verwenden. Das Set mit 2-Durchstechflaschen enthält Anweisungen für das Kombinieren des Inhalts beider Durchstechflaschen, was die Abmessung der korrekten Dosis erleichtert; zudem entfällt die Mehrfachinjektion (siehe «Sonstige Hinweise/Hinweise für die Handhabung»).
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der unter «Zusammensetzung» aufgelisteten Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenErythrozytose
Bei Patienten, die mit Winrevair behandelt wurden, wurden Anstiege des Hb beobachtet (>2 g/dl bis <4g/dl über ULN in ~15% der Studienteilnehmer). Eine schwere Erythrozytose kann das Risiko thromboembolischer Ereignisse oder eines Hyperviskositätssyndroms erhöhen. Überwachen Sie den Hb-Wert vor jeder Dosis während der ersten 5 Dosen oder länger, wenn die Werte instabil sind, und danach in regelmässigen Abständen, um festzustellen, ob eine Dosisanpassung erforderlich ist (siehe «Dosierung/Anwendung», «Dosierungsanpassungen bei Erwachsenen aufgrund eines Hämoglobin-Anstiegs oder einer Verminderung der Thrombozytenzahl» und «Unerwünschte Wirkungen»).
Schwere Thrombozytopenie
Bei einigen Patienten, die Winrevair anwenden, wurde eine Verminderung der Thrombozytenzahl und eine schwere Thrombozytopenie (Thrombozytenzahl <50'000/mm3 (<50,0 x 109/l)) beobachtet. Thrombozytopenien traten häufiger bei Patienten auf, die auch eine Prostacyclin-Infusion erhielten.
Beginnen Sie die Behandlung nicht, wenn die Thrombozytenzahl <50'000/mm3 (<50 x 109/l) beträgt (siehe «Dosierung/Anwendung»).
Überwachen Sie die Thrombozytenzahl vor jeder Dosis während der ersten 5 Dosen oder länger, wenn die Werte instabil sind, und danach in regelmässigen Abständen, um festzustellen, ob eine Dosisanpassung erforderlich ist (siehe «Dosierung/Anwendung», «Dosierungsanpassungen bei Erwachsenen aufgrund eines Hämoglobin-Anstiegs oder einer Verminderung der Thrombozytenzahl» und «Unerwünschte Wirkungen»).
Schwerwiegende Blutungen
In klinischen Studien wurde bei 4% der Patienten unter Winrevair und bei 1% der Patienten unter Placebo über schwerwiegende Blutungsereignisse (z.B. gastrointestinale, intrakraniale Blutungen) berichtet. Patienten mit schwerwiegenden Blutungsereignissen erhielten häufiger eine Prostacyclin-Backgroundtherapie und/oder Antithrombotika oder hatten eine niedrige Thrombozytenzahl. Informieren Sie die Patienten über Anzeichen und Symptome von Blutverlust. Beurteilen und behandeln Sie Blutungen entsprechend. Verabreichen Sie Winrevair nicht, wenn der Patient ein schwerwiegendes Blutungsereignis erleidet (siehe «Warnhinweise und Vorsichtsmassnahmen/Schwere Thrombozytopenie» und «Unerwünschte Wirkungen»).
Embryofetale Toxizität
Es ist möglich, dass Winrevair angesichts der Ergebnisse aus tierexperimentellen Reproduktionsstudien bei Anwendung in der Schwangerschaft zu einer Schädigung des Fetus führt. Schwangere Frauen sind über das potentielle Risiko für den Fetus aufzuklären. Frauen im gebärfähigen Alter sollten angewiesen werden, während der Behandlung mit Winrevair und für mindestens 4 Monate nach der letzten Dosis eine wirksame Methode der Empfängnisverhütung anzuwenden (siehe «Schwangerschaft, Stillzeit», «Schwangerschaft» und «Präklinische Daten/Reproduktionstoxizität»).
Beeinträchtigte Fertilität
Angesichts der Ergebnisse aus tierexperimentellen Studien ist es möglich, dass Winrevair die weibliche und männliche Fertilität beeinträchtigt. Die Patienten sind über die potentiellen Wirkungen auf die Fertilität aufzuklären (siehe «Schwangerschaft, Stillzeit/Fertilität» und «Präklinische Daten/Reproduktionstoxizität»).
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d.h. es ist nahezu «natriumfrei».
InteraktionenEs wurden keine Interaktionsstudien durchgeführt.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter/Empfängnisverhütung bei Frauen
Bei Frauen im gebärfähigen Alter wird empfohlen, vor Beginn der Behandlung mit Winrevair einen Schwangerschaftstest durchzuführen. Frauen im gebärfähigen Alter sollten während der Behandlung mit Winrevair und für ≥4 Monate nach der letzten Dosis (Ende der Behandlung) eine wirksame Empfängnisverhütung anwenden (siehe «Präklinische Daten»).
Schwangerschaft
Es liegen keine Daten zur Anwendung von Sotatercept bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe «Präklinische Daten»).
Winrevair wird während der Schwangerschaft sowie bei Frauen im gebärfähigen Alter, die nicht verhüten, nicht empfohlen.
Klinische Überlegungen
Bei schwangeren Frauen mit PAH besteht das Risiko von Herzinsuffizienz, Frühgeburt sowie Tod der Mutter oder des Fetus.
Stillzeit
Es ist nicht bekannt, ob Sotatercept oder dessen Metaboliten in die Muttermilch übergehen. Ein Risiko für Neugeborene/Säuglinge kann nicht ausgeschlossen werden.
Das Stillen sollte während der Behandlung mit Winrevair unterbrochen und 4 Monate nach der letzten Behandlungsdosis wieder aufgenommen werden.
Fertilität
Aufgrund der Ergebnisse aus tierexperimentellen Studien ist es möglich, dass Sotatercept die weibliche und männliche Fertilität beeinträchtigt (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenSotatercept hat keinen oder einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit und die Fähigkeit, Maschinen zu bedienen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die Sicherheit von Winrevair wurde in der pivotale Studie STELLAR untersucht, einer placebokontrollierten Langzeitstudie mit 323 PAH Patienten (siehe «Eigenschaften/Wirkungen/Klinische Wirksamkeit»). Nach Abschluss der ersten 24-wöchigen Behandlungsphase setzten die Patienten ihre Behandlung in einer doppelblinden Langzeitstudie unter Beibehaltung ihrer aktuellen Therapie fort, bis alle Patienten die erste Behandlungsphase abgeschlossen hatten. Die mediane Dauer der Behandlung mit Winrevair betrug 252 Tage. Die Gesamtinzidenz von Behandlungsabbrüchen aufgrund von unerwünschten Wirkungen lag bei 4% in der Winrevair-Gruppe bzw. bei 7% in der Placebogruppe. Es gab keine spezifischen, zum Abbruch der Behandlung führenden unerwünschten Wirkungen, die in der Winrevair-Gruppe häufiger und mit einer Häufigkeit von mehr als 1% auftraten.
Unter den beobachteten unerwünschten Wirkungen traten schwerwiegende Ereignisse nur gelegentlich auf (< 1,0%) (siehe Beschreibung ausgewählter unerwünschter Wirkungen). Die am häufigsten gemeldeten unerwünschten Wirkungen waren Kopfschmerzen (24,5%), Epistaxis (22,1%) und Teleangiektasie (16,6%), Diarrhoe (15,3%), Schwindel (14,7%), Ausschlag (12,3%) und Thrombozytopenie (10,4%).
Liste der unerwünschten Wirkungen
Die unter Winrevair in STELLAR berichteten unerwünschten Wirkungen sind in der nachfolgenden Tabelle nach MedDRA Systemorganklasse und Häufigkeit aufgeführt. Die Häufigkeiten sind wie folgt definiert: sehr häufig (≥1/10); häufig (≥1/100, < 1/10); gelegentlich (≥1/1'000, < 1/100); selten (≥1/10'000, < 1/1'000); sehr selten (< 1/10'000).
Tabelle 3: Unerwünschte Wirkungen
|
Systemorganklasse
|
Häufigkeit
|
Unerwünschte Wirkung
| |
Erkrankungen des Blutes und des Lymphsystems
|
Sehr häufig
|
Thrombozytopenie1,2
| |
Häufig
|
Erhöhtes Hämoglobin1,2
| |
Erkrankungen des Nervensystems
|
Sehr häufig
|
Schwindel
Kopfschmerzen
| |
Erkrankungen der Atemwege, des Brustraums und Mediastinums
|
Sehr häufig
|
Epistaxis
| |
Erkrankungen des Gastrointestinaltrakts
|
Sehr häufig
|
Diarrhoe
| |
Erkrankungen der Haut und des Unterhautgewebes
|
Sehr häufig
|
Teleangiektasie2
Ausschlag3
| |
Häufig
|
Erythem3
| |
Untersuchungen
|
Häufig
|
Erhöhter Blutdruck2
|
1 Siehe «Warnhinweise und Vorsichtsmassnahmen»
2 Siehe Beschreibung ausgewählter unerwünschter Wirkungen
3 MedDRA High Level Terms (HLT)
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Die folgenden klinisch relevanten unerwünschten Reaktionen werden an anderer Stelle in der Fachinformation beschrieben:
·Erythrozytose (siehe «Warnhinweise und Vorsichtsmassnahmen/Erythrozytose»)
·Schwere Thrombozytopenie (siehe «Warnhinweise und Vorsichtsmassnahmen/Schwere Thrombozytopenie»)
·Schwerwiegende Blutungen (siehe «Warnhinweise und Vorsichtsmassnahmen/Schwerwiegende Blutungen»)
·Embryofetale Toxizität (siehe «Warnhinweise und Vorsichtsmassnahmen/Embryofetale Toxizität»)
·Beeinträchtigte Fertilität (siehe «Warnhinweise und Vorsichtsmassnahmen/Beeinträchtigte Fertilität»)
Thrombozytopenie
Die meisten Thrombozytopenie-Ereignisse (Thrombozytopenie und Thrombozytenzahl vermindert) waren nicht schwerwiegend, leicht, reversibel und gingen nicht mit einem Behandlungsabbruch einher. Zu einer starken Verminderung der Thrombozytenzahl auf < 50'000/mm3 (< 50,0 x 109/l) kam es bei 1,8% der Patienten unter Winrevair.
Teleangiektasie
Teleangiektasie-Ereignisse waren nicht schwerwiegend und nahmen im Lauf der Zeit nicht an Schwere zu. Bei allen Patienten unter Winrevair betrug die mediane Zeit bis zum Auftreten 47,1 Wochen. Die Behandlung wurde aufgrund von Teleangiektasie bei 1% der Patienten in der Winrevair-Gruppe vs. 0% der Patienten in der Placebogruppe abgebrochen. Die Teleangiektasie war nicht mit schwerwiegenden Blutungsepisoden assoziiert.
Erhöhter Blutdruck
Ereignisse mit erhöhtem Blutdruck (Hypertonie, diastolischer Blutdruck erhöht, Blutdruck erhöht) waren nicht schwerwiegend und es wurden keine schweren Ereignisse gemeldet. Bei Patienten, die Winrevair erhielten, war der mittlere systolische Blutdruck nach 24 Wochen um 2,2 mmHg und der diastolische Blutdruck um 4,9 mmHg gegenüber dem Ausgangswert angestiegen. Bei Patienten unter Placebo verringerte sich der mittlere systolische Blutdruck um 1,6 mmHg und der diastolische um 0,6 mmHg gegenüber dem Ausgangswert.
Daten zur langfristigen Sicherheit
Daten zur langfristigen Sicherheit liegen aus einer klinischen Phase-II-Studie vor (PULSAR), die eine 24-wöchige, doppelblinde, placebokontrollierte Behandlungsphase und eine anschliessende 30-monatige, offene Verlängerungsphase umfasste (n = 104). Die meisten dieser Patienten nahmen im Anschluss an einer Langzeit-Folgestudie teil.
In PULSAR und der Langzeit-Folgestudie betrug die mittlere Dauer der Winrevair-Exposition 151 Wochen, mit einer maximalen Exposition von 218 Wochen. Das Sicherheitsprofil war generell mit dem in der pivotale Studie STELLAR beobachteten vergleichbar. Allerdings wurde in der doppelblinden, placebokontrollierten Behandlungsphase von PULSAR keine Teleangiektasie gemeldet. Über Teleangiektasie wurde erstmals in der offenen Verlängerungsphase berichtet, nämlich bei 27% der Patienten bei Studienabschluss, wobei die mediane Zeit bis zum Auftreten 106 Wochen betrug.
In der Studie SOTERIA, einer laufenden, offenen Langzeit-Studie zur Sicherheit und Wirksamkeit von Winrevair, wurde bei 2 Teilnehmern (<0,5%) ein intrapulmonaler Rechts-Links-Shunt berichtet, die trotz verbesserter PAH-Hämodynamik eine Verschlechterung der Hypoxämie entwickelten.
Immunogenität
Die beobachtete Häufigkeit von Anti-Drug-Antikörpern hängt stark von der Sensitivität und Spezifität des Tests ab. Aufgrund von Unterschieden in den Testmethoden ist ein aussagekräftiger Vergleich der Häufigkeit von Anti-Drug-Antikörpern in der unten beschriebenen Studie mit der Häufigkeit von Anti-Drug-Antikörpern in anderen Studien nicht möglich, was auch für Studien mit Winrevair oder anderen Sotatercept Produkten gilt.
Während der 24-wöchigen Behandlungsphase der pivotale Studie (STELLAR) kam es bei 44/163 (27%) der mit Sotatercept behandelten Patienten zur Entwicklung von Antikörpern gegen Sotatercept. Dabei wurden 12 dieser 44 Patienten (27%) positiv auf neutralisierende Antikörper gegen Sotatercept getestet. Die Titer der gegen Sotatercept gerichteten Antikörper waren mit einem medianen Titer von 30 (Bereich < 20 bis 640) generell niedrig.
Während der Behandlungsdauer von 24 Wochen wurden keine klinischen Auswirkungen der gegen Sotatercept gerichteten Antikörper auf die Pharmakokinetik, Pharmakodynamik, Sicherheit oder Wirksamkeit von Sotatercept festgestellt.
Ältere Patienten
Abgesehen von Blutungsereignissen (eine zusammengefasste Gruppe von unerwünschten Ereignissen von klinischem Interesse) gab es zwischen den Untergruppen von Patienten < 65 bzw. ≥65 Jahren keine Unterschiede in der Sicherheit. Blutungsereignisse traten in der älteren Winrevair-Untergruppe häufiger auf; jedoch gab es zwischen den verschiedenen Altersuntergruppen kein nennenswertes Ungleichgewicht in Bezug auf bestimmte Blutungsereignisse.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungBei gesunden Probanden führte die Gabe von Winrevair in einer Dosis von 1 mg/kg zu einem mit Hypertonie assoziierten Hb-Anstieg; beides besserte sich nach Phlebotomie. Im Fall einer Überdosierung sollte der Patient engmaschig auf einen Anstieg des Hämoglobins und des Blutdrucks überwacht und ggf. unterstützende Massnahmen eingeleitet werden. Winrevair ist nicht durch Hämodialyse dialysierbar.
Eigenschaften/WirkungenATC-Code
C02KX06
Wirkungsmechanismus
Sotatercept, ein rekombinantes Fusionsprotein des Activin-Rezeptors Typ IIA-Fc (ActRIIA-Fc), ist ein Activin-Signalinhibitor, der an Activin A und andere Liganden der TGF-β-Superfamilie bindet. Dadurch verbessert Sotatercept das Gleichgewicht zwischen der proproliferativen (ActRIIA/Smad2/3-vermittelten) und der antiproliferativen (BMPRII/Smad1/5/8-vermittelten) Signalübertragung, um die Gefässproliferation zu modulieren.
Pharmakodynamik
In einer klinischen Phase-II-Studie wurde der pulmonale Gefässwiderstand (PVR) bei PAH Patienten nach einer 24-wöchigen Behandlung mit Sotatercept beurteilt. Die Abnahme des PVR gegenüber dem Ausgangswert war in den Gruppen mit 0,7 mg/kg und 0,3 mg/kg Sotatercept signifikant grösser als in der Placebogruppe. Der placebobereinigte mittlere Unterschied (least square, LS) gegenüber dem Ausgangswert betrug -269,4 dyn•s/cm5 (95%-KI: -365,8; -173,0) in der Gruppe mit 0,7 mg/kg Sotatercept und -151,1 dyn•s/cm5 (95%-KI: -249,6; -52,6) in der Gruppe mit 0,3 mg/kg Sotatercept. In STELLAR war die Abnahme des PVR gegenüber dem Ausgangswert in der Gruppe mit 0,7 mg/kg Sotatercept ebenfalls signifikant grösser als in der Placebogruppe (siehe «Klinische Wirksamkeit»).
In PAH Rattenmodellen verringerte ein Sotatercept-Analogon die Expression von proinflammatorischen Markern an der Pulmonalarterienwand, reduzierte die Rekrutierung von Leukozyten, hemmte die Proliferation von Endothel- und glatten Muskelzellen und förderte die Apoptose in den erkrankten Gefässen. Diese zellulären Veränderungen gingen mit dünneren Gefässwänden, einer Umkehrung des arteriellen und rechtsventrikulären Remodellings und einer verbesserten Hämodynamik einher.
Klinische Wirksamkeit
Die Wirksamkeit von Winrevair wurde in der Studie STELLAR bei erwachsenen Patienten mit PAH beurteilt. STELLAR war eine globale, doppelblinde, placebokontrollierte, multizentrische klinische Parallelgruppenstudie, in der 323 Patienten mit PAH (WHO-Gruppe 1, Funktionsklasse II oder III) randomisiert im Verhältnis 1:1 einer Behandlung mit Winrevair (Zieldosis 0,7 mg/kg) (n = 163) oder Placebo (n = 160) subkutan einmal alle 3 Wochen erhielten.
Die demographischen und klinischen Baseline-Charakteristika waren in der Winrevair- und der Placebogruppe generell vergleichbar. Studienteilnehmende waren Erwachsene mit einem medianen Alter von 48,0 Jahren (Bereich: 18 bis 82 Jahre); das mediane Gewicht betrug 68 kg (Bereich: 38,0 bis 141,3 kg); 89,2% waren weiss und 79,3% waren nicht hispanisch oder lateinamerikanisch; 79,3% waren weiblich. Die häufigsten PAH Ätiologien waren: idiopathische PAH (58,5%), hereditäre PAH (18,3%) und PAH assoziiert mit Bindegewebserkrankungen (14,9 %). Die mediane Zeit zwischen PAH Diagnose und Screening betrug 8,76 Jahre. Die meisten Teilnehmenden erhielten entweder eine dreifache (61,3%) oder eine zweifache (34,7%) PAH Backgroundtherapie, und mehr als ein Drittel (39,9%) erhielten Prostacyclin-Infusionen. Der Anteil von Teilnehmenden mit PAH der WHO-Funktionsklasse II (48,6%) bzw. WHO-Funktionsklasse III (51,4%) war in beiden Gruppen ähnlich. Von der Teilnahme an der STELLAR-Studie ausgeschlossen waren Patienten mit diagnostizierter PAH in Zusammenhang mit einer Infektion mit dem Humanen Immundefizienz-Virus (HIV), PAH in Zusammenhang mit portaler Hypertonie, Schistosomiasis-assoziierter PAH und pulmonaler venookklusiver Erkrankung.
Primärer Wirksamkeitsendpunkt war die Veränderung der 6-Minuten-Gehstrecke (6MWD) in Woche 24 im Vergleich zum Ausgangswert. In der Winrevair-Behandlungsgruppe betrug die placebobereinigte Veränderung der 6MWD in Woche 24 gegenüber dem Ausgangswert im Median 40,8 Meter (95%-KI: 27,5; 54,1; p < 0,001). Der Median der placebobereinigten Veränderungen der 6MWD in Woche 24 wurde auch in Subgruppen beurteilt (siehe Abbildung 1).
Abbildung 1: Veränderung der 6-Minuten-Gehstrecke (Meter) in Woche 24 gegenüber dem Ausgangswert in Subgruppen
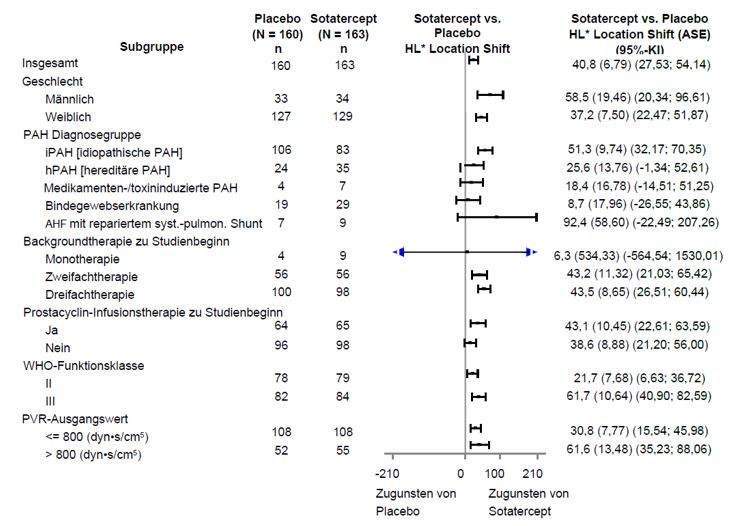
AHF = angeborener Herzfehler
* Hodges-Lehmann-Schätzer des Location Shift gegenüber Placebo (Median aller gepaarten Unterschiede). ASE = asymptotischer Standardfehler. Bei Teilnehmenden, die verstarben, wurde der Veränderung der 6MWD in Woche 24 gegenüber dem Ausgangswert ein Wert von bis zu -2000 Metern zugeordnet, um den schlechtesten Rang zu erhalten. Bei Teilnehmenden mit fehlenden Daten aufgrund einer nicht tödlichen klinischen Verschlechterung wurde für die Veränderung der 6MWD in Woche 24 gegenüber dem Ausgangswert ein Wert von bis zu -1000 Metern imputiert, um den zweitschlechtesten Rang zu erhalten.
Die klinische Verbesserung war ein vordefinierter Endpunkt, der anhand des Anteils der Patienten gemessen wurde, die in Woche 24 im Vergleich zum Ausgangszeitpunkt alle drei der folgenden Kriterien erfüllten: Verbesserung der 6MWD (Zunahme um ≥30 m), Verbesserung des N-terminalen pro-B-Typ-natriuretischen Peptids (NT-proBNP) (Abnahme des NT-proBNP um ≥30% oder Aufrechterhaltung/Erreichen eines NT-proBNP-Spiegels < 300 ng/l) und Verbesserung der WHO-Funktionsklasse oder Erhaltung der WHO-Funktionsklasse II. Das Fortschreiten der Erkrankung wurde gemessen als Zeit bis zum Tod oder bis zum ersten Auftreten eines Ereignisses einer klinischen Verschlechterung. Zu den Ereignissen einer klinischen Verschlechterung zählten eine Listung zur Lungen- und/oder Herztransplantation infolge einer Verschlechterung, die Notwendigkeit der Einleitung einer Rescue-Therapie mit einer zugelassenen PAH Backgroundtherapie oder die Notwendigkeit einer Dosiserhöhung der Prostacyclin-Infusionen um ≥10%, die Notwendigkeit einer Vorhofseptostomie, eine Hospitalisierung wegen einer sich verschlechternden PAH (≥24 Stunden) oder eine Verschlechterung der PAH (Verschlechterung der WHO-Funktionsklasse und Abnahme der 6MWD um ≥15%, wobei beide Ereignisse gleichzeitig oder zu unterschiedlichen Zeitpunkten eintreten können). Ereignisse einer klinischen Verschlechterung sowie Todesfälle wurden erfasst, bis der letzte Patient die Visite in Woche 24 absolviert hatte (Daten bis zum data cutoff; mediane Dauer der Exposition 33,6 Wochen).
Bei den mit Winrevair behandelten Patienten kam es im Vergleich zu den Patienten der Placebogruppe zu einer statistisch signifikanten klinischen Verbesserung, einer Verbesserung der WHO-Funktionsklasse und zu einem verzögerten Fortschreiten der Erkrankung, einschliesslich eines geringeren Risikos von Tod und Hospitalisierung (siehe Tabelle 4 und Abbildung 2).
In Woche 24 zeigten 38,9% der Patienten unter Sotatercept eine Verbesserung der MCI gegenüber 10,1% in der Placebogruppe (p<0,001). Der mediane Behandlungsunterschied im PVR zwischen der Sotatercept- und der Placebogruppe betrug -234,6 dyn*s/cm5 (95% KI: -288,4, -180,8; p<0,001). Der mediane Behandlungsunterschied im NT-proBNP zwischen der Sotatercept- und der Placebogruppe betrug -441,6 pg/ml (95% KI: -573,54, -309,61; p<0,001). Eine Verbesserung der Funktionsklasse gegenüber dem Ausgangswert trat bei 29% der Patienten in der Sotatercept-Gruppe gegenüber 13,8% in der Placebogruppe auf (p<0,001).
Tabelle 4: Tod oder Ereignisse einer klinischen Verschlechterung
|
|
Placebo
(N = 160)
|
Winrevair
(N = 163)
| |
Gesamtanzahl der Patienten, die verstarben oder mindestens ein Ereignis einer klinischen Verschlechterung erlitten, n (%)
|
42 (26,3)
|
9 (5,5)
| |
Bewertung von Tod oder erstem Auftreten von Ereignissen einer klinischen Verschlechterung*, n (%)
|
|
| |
Tod
|
6 (3,8)
|
2 (1,2)
| |
Verschlechterungsbedingte Listung zur Lungen- und/oder Herztransplantation
|
1 (0,6)
|
1 (0,6)
| |
Notwendigkeit der Einleitung einer Rescue-Therapie mit einer zugelassenen PAH
Therapie oder Notwendigkeit einer Dosiserhöhung der Prostacyclin-Infusionen um
≥10 %
|
17 (10,6)
|
2 (1,2)
| |
Notwendigkeit einer Vorhofseptostomie
|
0 (0,0)
|
0 (0,0)
| |
PAH spezifische Hospitalisierung (≥24 Stunden)
|
7 (4,4)
|
0 (0,0)
| |
Verschlechterung der PAH†
|
15 (9,4)
|
4 (2,5)
| |
* Bei einem Patienten können für das erste Ereignis einer klinischen Verschlechterung mehr als eine Bewertung dokumentiert sein. Bei 3 Patienten unter Placebo und 0 Patienten unter Sotatercept wurden für ihr erstes Ereignis einer klinischen Verschlechterung mehr als eine Bewertung dokumentiert.
† Verschlechterung der PAH ist definiert als Auftreten beider folgender Ereignisse zu einem beliebigen Zeitpunkt, auch wenn diese zu verschiedenen Zeiten auftraten, verglichen mit den Ausgangswerten: (a) Verschlechterung der WHO-Funktionsklasse (II zu III, III zu IV, II zu IV usw.); und (b) Abnahme der 6MWD um ≥15% (bestätigt durch zwei 6-Minuten-Gehtests im Abstand von mindestens 4 Stunden, aber höchstens einer Woche).
N = Anzahl Patienten in der FAS-Population; n = Anzahl Patienten in der Kategorie. Prozentanteile berechnet als (n/N)•100.
|
Abbildung 2: Kaplan-Meier-Kurve der Zeit bis zum Tod oder bis zum ersten Auftreten von Ereignissen einer klinischen Verschlechterung
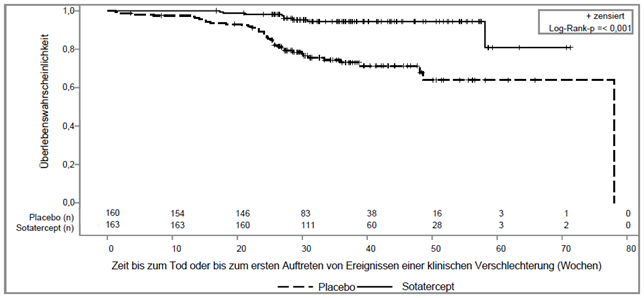
n = Anzahl subjects at risk
PharmakokinetikBei PAH Patienten betrugen die geometrischen Mittelwerte (%CV) der AUC im Steady State und der maximalen Konzentration (Cmax) im Steady State bei einer Dosis von 0,7 mg/kg alle 3 Wochen (Q3W) 171,3 μg•d/ml (34,2 %) bzw. 9,7 μg/ml (30 %CV). AUC und Cmax von Sotatercept steigen nach Gabe von subkutanen Einzeldosen zwischen 0.1 mg/kg und 1.0 mg/kg dosisproportional an. Der Steady State wird bei mehrfacher Q3W-Gabe nach ungefähr 15 Wochen erreicht. Das Akkumulationsverhältnis für die AUC von Sotatercept beträgt ungefähr 2,2.
Absorption
Bei der subkutanen Formulierung beläuft sich die absolute Bioverfügbarkeit auf etwa 66 %. Die maximale Sotatercept Konzentration wurde nach mehrfachen subkutanen Dosen (0,1 mg/kg alle 4 Wochen) bei postmenopausalen Frauen nach einer medianen Zeit (Tmax) von ungefähr 7 Tagen erreicht (Bereich 2 bis 8 Tage).
Distribution
Das zentrale Distributionsvolumen (%CV) von Sotatercept liegt bei ungefähr 3,6 l (24,7 %). Das periphere Distributionsvolumen (%CV) beläuft sich auf ungefähr 1,7 l (73,3 %).
Metabolismus
Sotatercept wird über allgemeine Protein-Abbauprozesse katabolisiert.
Elimination
Die Clearance von Sotatercept beträgt ungefähr 0,18 l/Tag. Der geometrische Mittelwert der terminalen Halbwertszeit (%CV) liegt bei ungefähr 21 Tagen (33,8 %).
Kinetik spezieller Patientengruppen
Es wurden keine klinisch signifikanten Unterschiede in der Pharmakokinetik (PK) von Sotatercept auf Grundlage von Alter (18 bis 81 Jahre), Geschlecht oder ethnischer Zugehörigkeit festgestellt.
Die Clearance (CL) und das zentrale Distributionsvolumen (Vc) von Sotatercept stiegen mit zunehmendem Körpergewicht an. Das empfohlene gewichtsabhängige Dosierungsschema führt zu einer konsistenten Sotatercept-Exposition, unabhängig vom Körpergewicht.
Leberfunktionsstörungen
Es wird nicht davon ausgegangen, dass eine Leberfunktionsstörung (ermittelt anhand der Child-Pugh-Klassifikation) den Metabolismus von Sotatercept beeinflusst, da Sotatercept über den zellulären Katabolismus metabolisiert wird. Sotatercept wurde bei PAH Patienten mit Leberfunktionsstörungen (Child-Pugh-Klasse A bis C) nicht untersucht.
Nierenfunktionsstörungen
Die PK von Sotatercept war bei PAH Patienten mit gering- bis mittelgradiger Nierenfunktionsstörung (eGFR zwischen 30 und 89 ml/min/1,73 m2) und bei Patienten mit normaler Nierenfunktion (eGFR ≥90 ml/min/1,73 m2) vergleichbar. Die PK von Sotatercept war auch bei Patienten ohne PAH mit terminaler Niereninsuffizienz (ESKD) und bei Patienten mit normaler Nierenfunktion ähnlich.
Winrevair ist nicht durch Hämodialyse dialysierbar. Für Patienten mit Nierenfunktionsstörungen wird keine Dosisanpassung empfohlen. Sotatercept wurde bei PAH Patienten mit hochgradiger Nierenfunktionsstörung (eGFR < 30 ml/min/1,73 m2) nicht untersucht.
Präklinische DatenToxizität bei wiederholter Gabe
Bei Ratten und Affen dauerten die längsten subkutanen Toxizitätsstudien 3 Monate bzw. 9 Monate. Bei Ratten, denen über einen Zeitraum von 3 Monaten einmal wöchentlich Dosen von 0,3; 3 und 30 mg/kg verabreicht wurden, traten unerwünschte Ereignisse wie Degeneration der Ductuli efferentes/Hoden, Kongestion/Nekrose der Nebennieren sowie membranoproliferative Glomerulonephritis und tubulointerstitielle Nephritis in den Nieren auf. Die Veränderungen in Nebennieren und Nieren waren nach einer 1-monatigen Erholungsphase teilweise reversibel. Bei Affen, denen eine Dosis von 1; 2,6 und 10 mg/kg einmal alle 4 Wochen und von 10 mg/kg einmal alle 2 Wochen verabreicht wurde, beinhalteten die unerwünschten Veränderungen Glomerulonephritis und tubulointerstitielle Nephritis in den Nieren. Die Veränderungen in den Nieren bei Affen waren nach einer 3-monatigen Erholungsphase teilweise reversibel. Bei Affen waren bei der klinischen Exposition entzündliche Infiltrate im Plexus choroideus vorhanden. Beim No Observed Adverse Effect Level (NOAEL) bei Ratten und Affen betrug die Sotatercept-Exposition das ≤2- fache der klinischen Exposition bei der maximal empfohlenen Humandosis (MRHD).
Genotoxizität und Kanzerogenität
Mit Sotatercept wurden keine Studien zur Kanzerogenität oder Mutagenität durchgeführt.
Reproduktionstoxizität
In einer Studie zur Fertilität und frühen embryonalen Entwicklung bei weiblichen Ratten wurde Sotatercept einmal wöchentlich in Dosen von 5, 15 und 50 mg/kg subkutan verabreicht, beginnend 2 Wochen vor der Paarung und bis zum Tag 7 der Trächtigkeit. Bei Dosen von ≥15 mg/kg (Expositionen ab dem 9-Fachen der MRHD, basierend auf der geschätzten AUC) sanken die Trächtigkeitsraten, und es kam zu einem Anstieg der Prä- und Postimplantationsverluste sowie zu einer Grössenabnahme der Lebendwürfe. Eine verlängerte Zyklusdauer trat nur bei 50 mg/kg auf (entspricht dem 21-Fachen der MRHD, basierend auf der geschätzten AUC).
In einer Studie zur Fertilität bei männlichen Ratten wurde Sotatercept 13 Wochen lang einmal wöchentlich in Dosen von 0,3; 3 und 30 mg/kg subkutan verabreicht (beginnend 10 Wochen vor der Paarung). Eine Teilgruppe der Tiere wurde nach einer 13-wöchigen Erholungsphase untersucht. Bei einer Dosis von ≥0,3 mg/kg (entspricht dem 0,5-Fachen der MRHD, basierend auf der geschätzten AUC) traten irreversible histologische Veränderungen in den Ductuli efferentes, Hoden und Nebenhoden auf. Eine reversible Verminderung der Fertilität wurde bei einer Dosis von 30 mg/kg beobachtet (entspricht dem 20-Fachen der MRHD, basierend auf der geschätzten AUC).
In Studien zur embryofetalen Entwicklungstoxizität wurden trächtige Tiere während der Organogenese subkutan mit Sotatercept behandelt. Ratten wurden an den Trächtigkeitstagen 6 und 13 mit einer Dosis von 5, 15 oder 50 mg/kg und Kaninchen an den Trächtigkeitstagen 7 und 14 mit einer Dosis von 0,5; 1,5 oder 5 mg/kg Sotatercept behandelt. Bei beiden Spezies waren eine verringerte Anzahl lebender Feten und ein verringertes Fetalgewicht, eine verzögerte Ossifikation sowie eine Zunahme von Resorptionen und Postimplantationsverlusten zu verzeichnen. Bei Ratten und Kaninchen wurden diese Effekte bei Expositionen (basierend auf der Fläche unter der Kurve [AUC]) beobachtet, die etwa dem 4- bzw. 0,6-Fachen der MRHD entsprachen. Ausschliesslich bei Ratten traten bei einer Exposition in Höhe des 15-Fachen der Humanexposition der MRHD-Skelettveränderungen auf (erhöhte Anzahl überzähliger Rippen und Veränderung der Anzahl von Brust- oder Lendenwirbeln).
In einer Studie mit Ratten zur prä- und postnatalen Entwicklung wurde Sotatercept subkutan in Dosen von 1,5 und 5 mg/kg an den Trächtigkeitstagen 6 und 13 oder in Dosen von 1,5; 5 oder 10 mg/kg während der Laktation an den Tagen 1, 8 und 15 verabreicht. Bei Jungtieren der ersten Filialgeneration (F1) von Muttertieren, die während der Trächtigkeit behandelt wurden (geschätzte Exposition bis zum 2-Fachen der MRHD), traten keine unerwünschten Wirkungen auf. Bei F1-Jungtieren von Muttertieren, die während der Laktation behandelt wurden, korrelierte die Abnahme des Gewichts der Jungtiere bei einer geschätzten Exposition (basierend auf der AUC) ab dem 2-Fachen der MRHD mit einer Verzögerung der Geschlechtsreife.
Sonstige HinweiseInkompatibilitäten
Das Arzneimittel darf nur mit den unter «Hinweise für die Handhabung» aufgeführten Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Aus mikrobiologischer Sicht sollte das Arzneimittel unmittelbar bzw. nicht später als 4 Stunden nach Rekonstitution verwendet werden.
Bei nicht unmittelbarer Verwendung liegen Aufbrauchfristen und Lagerungsbedingungen vor Verwendung in der Verantwortung des Anwenders.
Besondere Lagerungshinweise
Im Kühlschrank (2–8 °C) lagern.
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe «Haltbarkeit».
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Aufmachung als Set
Ausführliche Hinweise für die sachgerechte Vorbereitung und Verabreichung von Winrevair entnehmen Sie bitte der Gebrauchsanweisung.
Hinweise für die Rekonstitution
·Nehmen Sie das Injektionsset aus dem Kühlschrank und warten Sie 15 Minuten, damit die vorgefüllte(n) Spritze(n) und das Arzneimittel vor der Vorbereitung Raumtemperatur annehmen können.
·Prüfen Sie die Durchstechflasche, um sicherzustellen, dass das Produkt nicht abgelaufen ist. Das Pulver sollte weiss bis gebrochen weiss sein und kann wie ein ganzer oder ein zerbrochener Kuchen aussehen.
·Nehmen Sie die Verschlusskappe der Winrevair-Durchstechflasche mit dem lyophilisierten Pulver ab und reinigen Sie den Gummistopfen mit einem Alkoholtupfer.
·Setzen Sie den Durchstechflaschen Adapter auf die Durchstechflasche.
·Überprüfen Sie visuell die vorgefüllte Spritze auf Beschädigungen oder undichte Stellen und das darin befindliche sterile Wasser, um sicherzustellen, dass keine sichtbaren Partikel vorhanden sind.
·Brechen Sie den Verschluss der vorgefüllten Spritze ab und verbinden Sie die Spritze mit dem Durchstechflaschen Adapter.
·Injizieren Sie das sterile Wasser vollständig aus der befestigten Spritze in die Durchstechflasche mit dem lyophilisierten Pulver. Dadurch ergibt sich eine endgültige Konzentration von 50 mg/ml. Die Durchstechflaschen enthalten ein überschüssiges Volumen an Sotatercept, damit nach Rekonstitution mit 1 ml bzw. 1,3 ml die nominal entnehmbare Menge 45 mg/0,9 ml bzw. 60 mg/1,2 ml beträgt.
·Die Durchstechflasche vorsichtig schwenken, um das Arzneimittelpulver zu rekonstituieren. NICHT schütteln oder heftig bewegen.
·Lassen Sie die Durchstechflasche bis zu 3 Minuten stehen, damit die Bläschen verschwinden.
·Überprüfen Sie die rekonstituierte Lösung visuell. Wenn Winrevair richtig gemischt wurde, sollte es klar bis opaleszierend und farblos bis leicht bräunlich-gelb sein und weder Klumpen noch Pulver enthalten.
·Drehen Sie die Spritze vom Durchstechflaschen Adapter ab und werfen Sie die leere Spritze in einen durchstichsicheren Abfallbehälter.
·Wurde ein Set mit 2 Durchstechflaschen verordnet, wiederholen Sie die Schritte in diesem Abschnitt mit der zweiten Durchstechflasche.
·Verwenden Sie die rekonstituierte Lösung so schnell wie möglich, spätestens aber innerhalb von 4 Stunden nach der Rekonstitution.
Vorbereitung der Spritze
·Wischen Sie den Durchstechflaschen Adapter mit einem Alkoholtupfer ab.
·Nehmen Sie die Dosierspritze aus der Verpackung und befestigen Sie die Spritze am Durchstechflaschen Adapter.
·Drehen Sie die Spritze und die Durchstechflasche auf den Kopf und entnehmen Sie das entsprechende Injektionsvolumen, basierend auf dem Gewicht des Patienten.
·Wenn aufgrund der Dosismenge zwei Durchstechflaschen verwendet werden müssen, entnehmen Sie den gesamten Inhalt der ersten Durchstechflasche und überführen Sie diesen langsam und vollständig in die zweite Durchstechflasche.
·Drehen Sie die Spritze und die Durchstechflasche auf den Kopf und entnehmen Sie die erforderliche Menge des Arzneimittels.
·Falls erforderlich, drücken Sie den Kolben hinein, um überschüssiges Arzneimittel oder Luft aus der Spritze zu entfernen.
·Trennen Sie die Spritze von der Durchstechflasche und setzen Sie die Nadel auf.
Hinweise für die Verabreichung
Winrevair ist zur subkutanen Injektion bestimmt.
·Wählen Sie die Injektionsstelle im Bereich von Bauch (mindestens 5 cm vom Nabel entfernt), Oberschenkel oder Oberarm und reinigen Sie sie mit einem Alkoholtupfer. Wählen Sie für jede Injektion einen neuen Bereich, der nicht vernarbt oder gereizt ist und keinen Bluterguss aufweist.
·Erfolgt die Verabreichung durch den Patienten oder die Betreuungsperson, soll nur der Bauch oder Oberschenkel als Injektionsstelle gewählt werden (siehe Gebrauchsanweisung).
·Nehmen Sie die subkutane Injektion vor.
·Entsorgen Sie die leere Spritze in einem durchstichsicheren Abfallbehälter. Die Spritze nicht wiederverwenden.
Zulassungsnummer69787 (Swissmedic)
PackungenDurchstechflasche aus Typ-I-Glas, verschlossen mit einem Stopfen aus Brombutyl-Kautschuk und einer Aluminiumversiegelung mit einer limettengrünen Flip-off-Kappe aus Polypropylen sowie vorgefüllter Spritze (Spritzenkörper aus Typ-I-Glas, verschlossen mit einem Stopfen aus Brombutyl-Kautschuk) mit 1 ml Wasser für Injektionszwecke (WFI) für Sets mit 45 mg/Durchstechflasche bzw. mit 1,3 ml WFI für Sets mit 60 mg/Durchstechflasche.
Winrevair 45 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
·Packung mit 1 Durchstechflasche mit Pulver, 1 vorgefüllten Spritze mit Lösungsmittel, 1 Dosierspritze mit 0,1-ml-Graduierung, 1 Durchstechflaschen Adapter (13 mm), 1 Injektionsnadel und 4 Alkoholtupfern. [B]
·Packung mit 2 Durchstechflaschen mit Pulver, 2 vorgefüllten Spritzen mit Lösungsmittel, 1 Dosierspritze mit 0,1-ml-Graduierung, 2 Durchstechflaschen Adaptern (13 mm), 1 Injektionsnadel und 8 Alkoholtupfern. [B]
Winrevair 60 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
·Packung mit 1 Durchstechflasche mit Pulver, 1 vorgefüllten Spritze mit Lösungsmittel, 1 Dosierspritze mit 0,1-ml-Graduierung, 1 Durchstechflaschen Adapter (13 mm), 1 Injektionsnadel und 4 Alkoholtupfern. [B]
·Packung mit 2 Durchstechflaschen mit Pulver, 2 vorgefüllten Spritzen mit Lösungsmittel, 1 Dosierspritze mit 0,1-ml-Graduierung, 2 Durchstechflaschen Adaptern (13 mm), 1 Injektionsnadel und 8 Alkoholtupfern. [B]
ZulassungsinhaberinMSD MERCK SHARP & DOHME AG, Luzern
Stand der InformationOktober 2025
MK7962-kit-S-CCDS-I-042025a/RCN000028056-CH
|