ZusammensetzungWirkstoffe
Afliberceptum (durch rekombinante DNA Technologie in chinesischen Hamster Ovar-K1-Zellen (CHO) produziert).
Hilfsstoffe
Polysorbatum 20, Histidinum, Histidini hydrochloridum monohydricum, Trehalosum dihydricum, Acidum hydrochloridum, Natrii hydroxidum, Aqua ad injectabile.
Gesamtmenge an Natrium: 0.03 mg Natrium pro ml, entsprechend 1.73 µg pro Dosis.
Indikationen/AnwendungsmöglichkeitenBehandlung der exsudativen (feuchten) altersbedingten Makuladegeneration (wAMD).
Behandlung des Makulaödems infolge eines retinalen Zentralvenenverschlusses (CRVO).
Behandlung des Makulaödems infolge eines retinalen Venenastverschlusses (BRVO).
Behandlung des diabetischen Makulaödems (DME).
Behandlung von subfovealen und juxtafovealen choroidalen Neovaskularisationen infolge einer pathologischen Myopie (mCNV).
Dosierung/AnwendungDas Präparat darf nur durch einen Ophthalmologen mit Erfahrung in intravitrealen Injektionen angewendet werden. Afqlir wird in den Glaskörper (intravitreal) injiziert.
Exsudative (feuchte) altersbedingte Makuladegeneration (wAMD)
Afqlir 2 mg in 50 µl wird über die ersten drei Monate mit einer 4-wöchentlichen Injektion verabreicht. Basierend auf den Resultaten der Sehschärfeprüfung und den morphologischen Untersuchungsergebnissen kann die Behandlung nach den ersten 3 Monaten mit 8-wöchentlichen Injektionen fortgesetzt oder schrittweise (in Schritten von bis zu 4 Wochen) auf maximal alle 16 Wochen ausgedehnt werden.
Falls sich die Resultate der Sehschärfeprüfung und der morphologischen Untersuchungsergebnisse verschlechtern, soll das Behandlungsintervall entsprechend verkürzt werden.
Im Rahmen jeder Behandlung ist eine Kontrolluntersuchung erforderlich. Zusätzliche Kontrollen werden je nach Zustand des Patienten nach Massgabe des behandelnden Arztes empfohlen.
Behandlungsintervalle von mehr als 4 Monaten (16 Wochen) zwischen den Injektionen wurden nicht untersucht (Siehe «Eigenschaften/Wirkungen»).
Makulaödem infolge eines retinalen Zentralvenenverschlusses (CRVO)
Makulaödem infolge eines retinalen Venenastverschlusses (BRVO)
Nach der Initialinjektion wird Afqlir 2 mg in 50 µl mit 4-wöchentlichen Intervallen intravitreal verabreicht. Drei oder mehr aufeinanderfolgende monatliche (alle 4 Wochen) Injektionen können erforderlich sein, bis der maximale Visus erreicht ist und/oder keine Zeichen von Krankheitsaktivität mehr zu erkennen sind.
Die Behandlung kann fortgesetzt werden und die Intervalle können basierend auf den Resultaten der Sehschärfeprüfung und den morphologischen Untersuchungsergebnissen ausgedehnt werden.
Im Rahmen jeder Behandlung ist eine Kontrolluntersuchung erforderlich. Zusätzliche Kontrollen werden je nach Zustand des Patienten nach Massgabe des behandelnden Arztes empfohlen.
Diabetisches Makulaödem (DME)
Afqlir 2 mg in 50 µl wird über die ersten drei Monate mit 4-wöchentlichen Intervallen intravitreal verabreicht. Basierend auf den Beurteilungsparametern Visus und morphologische Netzhaut-Bildanalyse können die Injektionsintervalle anschliessend bis auf 8 Wochen ausgedehnt werden. Sollte sich bei den weiteren Analysen der individuellen Wirksamkeit wieder eine Verschlechterung der Parameter einstellen, wird das geeignete individuelle Behandlungsintervall empirisch angepasst. Nach dem ersten Behandlungsjahr soll geprüft werden, ob die Injektionsintervalle weiter ausgedehnt werden können.
Im Rahmen jeder Behandlung ist eine Kontrolluntersuchung erforderlich. Zusätzliche Kontrollen werden je nach Zustand des Patienten nach Massgabe des behandelnden Arztes empfohlen.
CNV infolge einer pathologischen Myopie (mCNV)
Afqlir 2 mg in 50 µl wird als einzelne intravitreale Injektion verabreicht.
Zusätzliche Dosen sollten nur verabreicht werden, wenn die Sehschärfeprüfung und morphologische Untersuchungsergebnisse darauf hindeuten, dass die Erkrankung persistiert. Rezidive werden wie eine Neumanifestation der Erkrankung behandelt.
Afqlir darf nicht häufiger als alle vier Wochen angewendet werden.
Patienten mit mCNV wurden in klinischen Studien bis zu einem Jahr untersucht.
Art der Anwendung
Generell müssen eine adäquate Anästhesie und aseptische Bedingungen (geeignete Räumlichkeiten, chirurgische Händedesinfektion, sterile Handschuhe, keimfreie Abdeckung und ein steriles Lidspekulum), inklusive Anwendung eines topischen Breitband-Biozids (z.B. Povidon-Iod), aufgetragen auf die periokuläre Haut, das Augenlid und die Augenoberfläche, sichergestellt werden. Unmittelbar nach der intravitrealen Injektion sollte der Augeninnendruck des Patienten gemessen und die Durchblutung der Sehnervenpapille untersucht werden. Falls nötig sollte eine sterile Parazentese durchgeführt werden können.
Nach der intravitrealen Injektion müssen die Patienten instruiert werden, allfällige Symptome, welche auf eine Endophthalmitis hinweisen (z.B. Augenschmerzen, Augenrötung, Lichtempfindlichkeit verschwommene Sicht), unverzüglich zu melden.
Jede Fertigspritze oder Durchstechflasche darf nur für die Behandlung eines einzigen Auges verwendet werden. Nicht gebrauchte Lösung ist nach der Injektion zu verwerfen.
Die Fertigspritze enthält mehr als die empfohlene Dosis von 2 mg Aflibercept (äquivalent zu 50 µl Injektionslösung). Das entnehmbare Volumen der Spritze darf nicht vollständig genutzt werden. Die überschüssige Menge ist vor der Injektion zu verwerfen.
Eine Injektion des gesamten Volumens der Fertigspritze könnte in einer Überdosierung resultieren. Um alle Bläschen und überschüssiges Arzneimittel auszustossen, die Kolbenstange langsam drücken, um die Grundfläche des kuppelförmigen Kolbens (nicht die Spitze des Kolbens) an der Dosierungslinie der Spritze (entsprechend 50 µl d.h. 2 mg Aflibercept) auszurichten (Siehe Rubriken «Warnhinweise und Vorsichtsmassnahmen», «Überdosierung» und «Hinweise für die Handhabung»).
Spezielle Dosierungsanweisungen
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Afqlir ist in der Pädiatrie nicht untersucht, es besteht keine Indikation.
Ältere Patienten
Die klinischen Studien wurden vornehmlich in älteren Patienten durchgeführt.
Patienten mit Leber- und/oder Nierenfunktionsstörungen
Es wurden keine spezifischen Studien bei Patienten mit Leber- und/oder Nierenfunktionsstörungen durchgeführt. Da die systemische Exposition von Aflibercept nach intravitrealer Applikation sehr gering ist, ist keine Dosisanpassung bei diesen Patienten erforderlich.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Kontraindikationen·Bekannte Überempfindlichkeit gegenüber Aflibercept oder einem der Hilfsstoffe.
·Okuläre oder periokuläre Infektion.
·Aktive oder vermutete intraokuläre Entzündung.
·Schwangerschaft, Stillzeit.
Warnhinweise und VorsichtsmassnahmenIntravitreale Injektionen können eine Endophthalmitis zur Folge haben. Es muss eine aseptische Injektionstechnik angewendet werden. Patienten sollen angewiesen werden, jegliche Symptome, welche auf eine Endophthalmitis hinweisen, unverzüglich zu melden.
Eine Injektion des gesamten Volumens der Fertigspritze könnte in einer Überdosierung resultieren sowie einen Anstieg des Augeninnendrucks verursachen. Für eine korrekte Anwendung der Fertigspritze sind die Rubriken «Dosierung/Anwendung» und «Hinweise zur Vorbereitung und Anwendung der Fertigspritze bzw. der Injektionslösung in der Durchstechflasche» zu beachten.
Erhöhungen des Augeninnendrucks wurden innerhalb von 60 Minuten nach intravitrealen Injektionen beobachtet. Besondere Vorsicht ist bei Patienten mit nicht ausreichend therapiertem Glaukom geboten. Unmittelbar nach der intravitrealen Injektion muss deshalb der Augeninnendruck und die Perfusion der Sehnervenpapille überwacht werden.
Die Sicherheit und Wirksamkeit einer gleichzeitigen Behandlung beider Augen mit Afqlir wurden nicht systematisch untersucht.
Patienten mit CNV infolge pathologischer Myopie (mCNV) wurden in klinischen Studien bis zu einem Jahr untersucht.
Systemische Effekte
Systemische Nebenwirkungen inklusive nicht-okularer Hämorrhagien und arterieller thromboembolischer Ereignisse wurden nach intravitrealer Injektion von VEGF-Hemmern berichtet. Es besteht ein theoretisches Risiko, dass diese in Zusammenhang mit der VEGF-Hemmung stehen können. Es gibt begrenzte Daten zur Sicherheit bei der Behandlung von Patienten mit CRVO, BRVO, DME oder mCNV, die innerhalb der letzten 6 Monate einen Schlaganfall oder transitorische ischämische Attacken oder einen Myokardinfarkt in der Vorgeschichte hatten (Siehe «Unerwünschte Wirkungen»). Die Behandlung entsprechender Patienten sollte mit Umsicht erfolgen.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Fertigspritze bzw. pro Durchstechflasche, d.h. es ist nahezu «natriumfrei».
InteraktionenEs wurden keine Interaktionsstudien mit Aflibercept durchgeführt.
Schwangerschaft, StillzeitGebärfähige Frauen sollten während der Behandlung und bis mindestens 3 Monate nach der letzten Behandlung mit Afqlir eine wirksame Methode zur Empfängnisverhütung anwenden.
Schwangerschaft
Es gibt keine Daten zur Anwendung bei Schwangeren.
Tierstudien haben nach systemischer Anwendung eine Reproduktionstoxizität gezeigt («Präklinische Daten»).
Daher sollte Afqlir während der Schwangerschaft nicht angewendet werden.
Stillzeit
Sehr begrenzte Daten beim Menschen weisen darauf hin, dass Aflibercept in geringen Mengen in die Muttermilch übergehen kann. Aflibercept ist ein grosses Proteinmolekül und es ist zu erwarten, dass die Menge an Arzneimittel, die vom Säugling aufgenommen wird, gering ist. Die Auswirkungen von Aflibercept auf gestillte Neugeborene/Kinder sind nicht bekannt.
Als Vorsichtsmassnahme wird das Stillen während der Anwendung von Afqlir nicht empfohlen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenNach der intravitrealen Injektion und den damit verbundenen Augenuntersuchungen kann es zu vorübergehenden Sehstörungen kommen. Patienten mit Sehstörungen dürfen keine Fahrzeuge fahren und keine Maschinen bedienen, bis sich die Sehfunktion erholt hat.
Unerwünschte WirkungenInsgesamt 3102 mit Aflibercept behandelte Patienten bildeten die Sicherheitspopulation in den acht Phase-III-Studien. Von diesen Patienten wurden 2501 mit der empfohlenen Dosis von 2 mg behandelt.
Schwerwiegende unerwünschte Wirkungen im Zusammenhang mit dem Injektionsvorgang traten bei weniger als einer von 2400 intravitrealen Injektionen auf und umfassten Endophthalmitis, Netzhautablösung, traumatische Katarakt, Katarakt, Glaskörperabhebung, und erhöhten Augeninnendruck (siehe Warnhinweise und Vorsichtsmassnahmen).
Die am häufigsten beobachteten Nebenwirkungen (bei mindestens 5% der mit Aflibercept behandelten Patienten) waren Bindehautblutung (25,0 %), Einblutung in die Retina (11,3%), verminderte Sehschärfe (11,1%), Augenschmerzen (10,2%), Katarakt (7,6%), Anstieg des Augeninnendrucks (7,5%), Glaskörperabhebung (7,4%) und Mouches volantes (6,9%).
Integrierte Sicherheitsdaten aus der Patientenpopulation mit feuchter AMD, CRVO, BRVO, DME und mCNV
Die unten aufgeführten Sicherheitsdaten enthalten alle unerwünschten Wirkungen (schwerwiegende und nicht schwerwiegende) aus den acht Phase-III-Studien zur feuchten AMD, CRVO, BRVO, DME und mCNV, oder aus Beobachtungen der Anwendung nach Markteinführung.
Die unerwünschten Wirkungen sind gemäss folgendem Prinzip nach Organklasse und Häufigkeit geordnet: Sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1000, <1/100), selten (≥1/10'000, <1/1000). Innerhalb einer Häufigkeitsklasse sind die unerwünschten Wirkungen ebenfalls nach Häufigkeit geordnet.
Augenerkrankungen
Sehr häufig: Bindehautblutung (25,0%), Einblutung in die Retina (11,3%), verminderte Sehschärfe (11,1%), Augenschmerzen (10,2%).
Häufig: Katarakt, erhöhter Augeninnendruck, Glaskörperabhebung, Mouches Volantes, erhöhter Tränenfluss, okuläre Hyperämie, Fremdkörpergefühl in den Augen, verschwommenes Sehen, Ablösung des retinalen Pigmentepithels, Schmerzen an der Injektionsstelle, Keratitis punctata, Kernkatarakt, subkapsuläre Katarakt, Lidödeme, Hornhautabrasion, kortikale Katarakt, Blutungen an der Injektionsstelle, Hyperämie der Bindehaut, Risse im retinalen Pigmentepithel, Hornhauterosion.
Gelegentlich: Hornhautödem, Linsentrübung, Netzhautablösung, Hornhautepitheldefekt, Vorderkammerreiz, Netzhautriss, Iridocyclitis, Endophthalmitis, Iritis, Uveitis.
Selten: Vitritis, Hypopyon, traumatische Katarakt, Erblindung.
Erkrankungen des Immunsystems
Gelegentlich: Hypersensitivitätsreaktionen (während der post-marketing Phase wurde über Überempfindlichkeit einschliesslich Hautausschlag, Pruritus, Urtikaria und Einzelfälle von schweren anaphylaktischen/ anaphylaktoiden Reaktionen berichtet).
Post-Marketing Beobachtungen
In der Post-Marketing Phase wurde die folgende unerwünschte Wirkung nach intravitrealer Injektion von Aflibercept festgestellt.
Augenerkrankungen: Skleritis (berichtet mit einer Häufigkeit von 0,2 pro 1 Million Injektionen).
Gefässerkrankungen
Arterielle thromboembolische Ereignisse (ATE) sind unerwünschte Wirkungen, die möglicherweise mit der systemischen VEGF-Hemmung in Verbindung stehen. Es besteht ein theoretisches Risiko arterieller thromboembolischer Ereignisse, inklusive Schlaganfall und Myokardinfarkt, nach intravitrealer Anwendung von VEGF-Hemmern.
Eine geringe Inzidenzrate arterieller thromboembolischer Ereignisse wurde in klinischen Studien mit Aflibercept bei Patienten mit wAMD, DME, CRVO, BRVO und mCNV beobachtet. Indikationsübergreifend wurde kein nennenswerter Unterschied zwischen Gruppen, die mit Aflibercept und den jeweiligen Vergleichsgruppen behandelt wurden, beobachtet.
Immunogenität
Antikörper gegen Aflibercept wurden vor der Behandlung bei circa 1-3% der Patienten gemessen. Nach 96 (wAMD), 76 (CRVO), 52 (BRVO), 100 (DME) bzw. 48 (mCNV) Wochen Behandlung, wurden Antikörper gegen Aflibercept bei einem ähnlichen Prozentsatz von Patienten festgestellt. Insgesamt scheint das Risiko für eine klinisch relevante Immunogenität unter Aflibercept sehr niedrig zu sein.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIn klinischen Studien wurden Dosen von bis zu 4 mg in monatlichen Intervallen und einzelne Fälle von Überdosierung mit 8 mg im Allgemeinen gut vertragen.
Die Überdosierung mit einem erhöhten Injektionsvolumen kann den Augeninnendruck erhöhen. Deshalb sollte der Augeninnendruck in Fällen von Überdosierung überwacht werden und es sollte, sofern nötig, eine angemessene Behandlung eingeleitet werden. (Siehe Rubrik «Hinweise für die Handhabung».)
Eigenschaften/WirkungenATC-Code
S01LA05
Afqlir ist ein Biosimilar.
Aflibercept ist ein rekombinantes Fusionsprotein, das aus Teilen der extrazellulären Domänen des humanen VEGF Rezeptors 1 und 2 und dem Fc-Fragment des humanen IgG1 besteht.
Aflibercept wird durch rekombinante DNA Technologie in chinesischen Hamster Ovar-K1-Zellen (CHO) produziert.
Wirkungsmechanismus
Aflibercept bindet alle Isoformen von vaskulärem endothelialem Growth Faktor (VEGF-A und PlGF) mit höherer Affinität als ihre natürlichen Rezeptoren. Es verhindert dadurch die Bindung und Aktivierung der VEGF-Rezeptoren.
Vaskulärer endothelialer Wachstumsfaktor A (VEGF-A) und plazentaler Wachstumsfaktor (PlGF) sind Mitglieder der VEGF Familie. VEGF wirkt über zwei Rezeptor-Tyrosinkinasen VEGFR-1 und VEGFR-2, welche auf der Oberfläche von Endothelzellen vorkommen. PlGF bindet nur an VEGFR-1, der auch auf der Oberfläche von Leukozyten vorkommt. Die übermässige Aktivierung dieser Rezeptoren durch VEGF-A kann in einer pathologischen Neovaskularisation und einer erhöhten Durchlässigkeit der Gefässe resultieren. PlGF kann in diesen Prozessen mit VEGF-A synergistisch wirken und ist verantwortlich für die Förderung der Leukozyteninfiltration und Gefässentzündung. Verschiedene Augenerkrankungen, inklusive die feuchte AMD, gehen mit einer pathologischen Neovaskularisation und einem Flüssigkeitsaustritt aus den Gefässen einher, wodurch eine Verdickung und Ödeme der Retina resultieren können. Dies trägt vermutlich zum Verlust der Sehfähigkeit bei.
In Tierstudien konnte Aflibercept pathologische Neovaskularisationen und damit verbundene Gefässdurchlässigkeit in verschiedenen Modellen von Augenerkrankungen verhindern. Zum Beispiel verhinderte die intravitreale Anwendung von Aflibercept bei Affen die Entwicklung einer signifikanten choroidalen Neovaskularisation (CNV) nach Laserverletzung und machte Gefässdurchlässigkeiten von bestehenden CNV Läsionen rückgängig.
Exsudative (feuchte) altersbedingte Makuladegeneration (wAMD)
Die feuchte AMD wird durch eine pathologische choroidale Neovaskularisation (CNV) charakterisiert. Der Austritt von Blut und Flüssigkeit durch CNV kann retinale Ödeme und/oder sub-/intra-retinale Blutungen bewirken, was den Verlust der Sehschärfe zur Folge hat.
Bei Patienten, welche mit Aflibercept behandelt wurden, nahmen die zentrale Netzhautdicke und die mittlere Grösse der CNV-Läsionen bald nach Behandlungsbeginn ab. Diese Abnahme war bis zu einer Behandlungsdauer von 2 Jahren weitgehend stabil.
Die maximale Plasmakonzentration von freiem Aflibercept liegt ungefähr 50- bis 500-mal niedriger als die Aflibercept Konzentration, die nötig ist, um die biologische Aktivität von systemischem VEGF in Tiermodellen um 50% zu hemmen. In diesen Tiermodellen wurden Änderungen des Blutdrucks bei einem Plasmaspiegel von 10 µg/ml zirkulierendem freien Aflibercept beobachtet. Nachdem die Plasmaspiegel wieder unter 1 µg/ml abfielen, gingen die Blutdruckwerte wieder auf den Ausgangswert zurück. Es wird geschätzt, dass bei Patienten nach intravitrealer Anwendung von 2 mg die mittlere maximale Plasmakonzentration von freiem Aflibercept mehr als 100-fach tiefer ist als die Konzentration von Aflibercept, die nötig ist, um systemischen VEGF halb-maximal zu binden. Daher sind systemische pharmakodynamische Wirkungen, wie Veränderungen des Blutdrucks, unwahrscheinlich.
Makulaödem infolge eines retinalen Zentralvenenverschlusses (CRVO)
Bei einem CRVO tritt eine retinale Ischämie ein, welche die Freisetzung von VEGF signalisiert, welches wiederum die Tight Junctions (Zona occludens) destabilisiert und die Proliferation von Endothelzellen fördert. Die Hochregulierung des VEGF geht mit dem Abbau der Blut-Retina-Schranke einher. Diese erhöhte Durchlässigkeit der Gefässe wiederum führt zu Ödemen der Retina, Stimulierung des endothelialen Zellwachstums und einer Neovaskularisation.
Bei Patienten, welche mit Aflibercept behandelt wurden (eine Injektion monatlich während sechs Monaten), kam es zu einer schnellen Verbesserung der zentralen retinalen Schichtdicke (CRT, bewertet mit OCT). Die Verbesserungen der mittleren CRT wurden bis Woche 24 beibehalten.
Die retinale Schichtdicke mit OCT in Woche 24 im Vergleich zum Ausgangswert war sowohl in der COPERNICUS als auch der GALILEO Studie ein sekundärer Endpunkt für die Wirksamkeit. In beiden Studien war die mittlere Veränderung der retinalen Schichtdicke zwischen dem Ausgangswert und Woche 24 statistisch signifikant zu Gunsten von Aflibercept, nämlich -145 µm in der Kontrollgruppe und -457 µm in der mit Aflibercept 2Q4 behandelten Gruppe (COPERNICUS) und -169 µm in der Kontrollgruppe und -449 µm in der mit Aflibercept 2Q4 behandelten Gruppe (GALILEO).
Makulaödem infolge eines retinalen Venenastverschlusses (BRVO)
Patienten, welche mit Aflibercept behandelt wurden (eine Injektion monatlich während sechs Monaten), zeigten ein übereinstimmendes, rasch einsetzendes und stabiles morphologisches Ansprechen (zentrale retinale Schichtdicke, CRT, bewertet mit OCT).
Die mittels OCT gemessene Retinadicke in Woche 24 im Vergleich zum Ausgangswert war ein sekundärer Endpunkt für die Wirksamkeit in der VIBRANT-Studie.
In der mit Aflibercept 2Q4 behandelten Gruppe kam es in Woche 24 zu einer statistisch signifikanten Verbesserung im Vergleich zur Kontrollgruppe (-280 µm gegenüber -128 µm). In Woche 24 wurde das Dosierungsintervall auf alle zwei Monate erweitert. Die Abnahme der CRT im Vergleich zum Ausgangswert blieb bis Woche 52 aufrechterhalten (nämlich -284 µm gegenüber -249 µm, zugunsten von Aflibercept).
Diabetisches Makulaödem (DME)
Das diabetische Makulaödem zeichnet sich durch eine erhöhte Gefässpermeabilität und Schädigung der Netzhautkapillaren aus, die zu einem Verlust der Sehschärfe führen können.
Bei Patienten, die mit Aflibercept behandelt wurden, zeigte sich kurz nach Therapieeinleitung morphologisch ein rasches und robustes Ansprechen (zentrale Retinadicke [CRT]), gemessen mittels OCT (optische Kohärenztomographie).
Die retinale Schichtdicke mit OCT in Woche 52 war in beiden Phase III Studien ein sekundärer Endpunkt für die Wirksamkeit. In beiden Studien war die mittlere Veränderung der retinalen Schichtdicke zwischen dem Ausgangswert und Woche 52 statistisch signifikant zu Gunsten von Aflibercept, nämlich -66,2 µm in der Laserkontrollgruppe und -195,0 µm bzw. -192,4 µm in der mit Eylea 2Q4 bzw. 2Q8 behandelten Gruppe (VIVIDDME) und -73,3 µm in der Laserkontrollgruppe und -185,9 µm bzw. -183,1 µm in der mit Aflibercept 2Q4 bzw. 2Q8 behandelten Gruppe (VISTADME). Diese Ergebnisse konnten bis Woche 100 beibehalten werden, nämlich -85,7 µm in der Laserkontrollgruppe und -211,8 µm bzw. -195.8 µm in der mit Aflibercept 2Q4 bzw. 2Q8 behandelten Gruppe (VIVIDDME) und -83,9 µm in der Laserkontrollgruppe und -191,4 µm bzw. -191,1 µm in der mit Aflibercept 2Q4 bzw. 2Q8 behandelten Gruppe (VISTADME).
CNV infolge einer pathologischen Myopie (mCNV)
Die CNV infolge einer pathologischen Myopie (mCNV) stellt eine häufige Ursache für den Verlust des Sehvermögens bei Erwachsenen mit pathologischer Myopie dar. Bei pathologischer Myopie sind die Augen – oftmals übermässig stark – elongiert und weisen pathologische Gewebeveränderungen auf, wie Verdünnung und Defekte des retinalen Pigmentepithels, «Lacksprünge» (Lacquer Cracks) und Risse in der Bruch'schen Membran, choroidale Neovaskularisation, subretinale Blutung und choroidale Atrophie. Infolge von Rissen in der Bruch'schen Membran entwickelt sich als Wundheilungsmechanismus eine CNV, was zugleich das bedrohlichste Ereignis für das Sehvermögen bei pathologischer Myopie darstellt.
Bei Patienten unter Behandlung mit Aflibercept (eine Injektion zu Therapiebeginn und eine weitere Injektion bei persistierender oder rezidivierender Erkrankung) verminderte sich die Retinadicke, gemessen mit OCT und die mittlere CNV-Läsionsgrösse ging zurück. Die mittlere Veränderung der CRT vom Ausgangswert bis Woche 24 war statistisch signifikant zu Gunsten von Aflibercept.
Pharmakodynamik
Siehe auch unter «Wirkungsmechanismus».
Klinische Wirksamkeit
Exsudative (feuchte) altersbedingte Makuladegeneration
Die Sicherheit und Wirksamkeit von Aflibercept wurden in zwei randomisierten, multizentrischen, doppelt verblindeten, aktiv kontrollierten Studien bei Patienten mit feuchter AMD untersucht. 2412 Patienten wurden in den beiden Studien (VIEW1 und VIEW2) behandelt und bezüglich der Wirksamkeit ausgewertet (1817 mit Aflibercept). In beiden Studien wurden die Patienten randomisiert und in einem Verhältnis von 1:1:1:1 einem der 4 Dosierungsschemen zugewiesen.
1.Initial während 3 Monaten Aflibercept 2 mg alle 4 Wochen, danach Aflibercept 2 mg alle 8 Wochen (Aflibercept 2Q8),
2.Aflibercept 2 mg alle 4 Wochen (Aflibercept 2Q4),
3.Aflibercept 0,5 mg alle 4 Wochen (Aflibercept 0,5Q4) und
4.Ranibizumab 0,5 mg alle 4 Wochen (Ranibizumab 0,5Q4).
Das Alter der Patienten lag zwischen 49 und 99 Jahren mit einem durchschnittlichen Alter von 76 Jahren.
Im zweiten Studienjahr erhielten die Patienten weiterhin die Dosierungsstärke für die sie ursprünglich randomisiert waren, jedoch mit einem modifizierten Dosierungsschema, welches an die visuellen und anatomischen Resultate angepasst war, mit einem per Protokoll definierten maximalen Dosierungsintervall von 12 Wochen. Während des zweiten Studienjahres erhielten 90% der Patienten, die ursprünglich mit Aflibercept 2Q8 behandelt wurden und das zweite Studienjahr beendeten, 6 Dosen oder weniger und 72% erhielten 4 Dosen oder weniger.
In beiden Studien war der primäre Endpunkt für die Wirksamkeit der Anteil an Patienten im Per Protocol Set, welcher keinen Sehverlust erlitt, definiert als ein Verlust der Sehschärfe um weniger als 15 Buchstaben in Woche 52 im Vergleich zum Ausgangswert.
In der VIEW1 Studie behielten in Woche 52 95,1% der Patienten in der Aflibercept 2Q8 Behandlungsgruppe und 95,1% der Patienten in der Aflibercept 2Q4 Behandlungsgruppe ihre Sehschärfe im Vergleich zu 94,4% der Patienten in der Ranibizumab 0,5Q4 Gruppe bei. Alle Aflibercept Behandlungsgruppen erwiesen sich als nicht unterlegen und klinisch äquivalent zur Ranibizumab 0,5Q4 Gruppe.
In der VIEW2 Studie behielten in Woche 52 95,6% der Patienten in der Aflibercept 2Q8 Behandlungsgruppe und 95,6% der Patienten in der Aflibercept 2Q4 Behandlungsgruppe ihre Sehschärfe im Vergleich zu 94,4% der Patienten in der Ranibizumab 0,5Q4 Gruppe bei. Alle Aflibercept Behandlungsgruppen erwiesen sich als nicht unterlegen und klinisch äquivalent zur Ranibizumab 0,5Q4 Gruppe.
Die Auswertung der sekundären Endpunkte der kombinierten Analysen von beiden Studien zeigten folgende Resultate:
Die durchschnittlichen Veränderungen der BCVA gegenüber dem Ausgangswert, gemessen mittels ETDRS*-Letter Score (* Early Treatment Diabetic Retinopathy Study) brachten die folgenden Ergebnisse: 8,40 in der Aflibercept 2Q8 Gruppe (n= 607), 9,26 in der Aflibercept 2Q4 Gruppe (n= 613 und 8,74 in der Ranibizumab-Gruppe (n= 595).
Der Anteil der Patienten, die gegenüber dem Ausgangswert mindestens 15 Buchstaben an Sehschärfe gewannen, lag in der Aflibercept 2Q8 Behandlungsgruppe bei 30,97% und in der Aflibercept 2Q4 Behandlungsgruppe bei 33,44%, im Vergleich zu 32,44% in der Ranibizumab 0,5Q4 Gruppe.
In beiden Studien zeigte sich in allen Dosierungsgruppen eine Abnahme der mittleren CNV-Fläche.
Bei der kombinierten Datenanalyse der VIEW1 und VIEW2 Studien zeigte Aflibercept beim vorab definierten sekundären Wirksamkeitsendpunkt (National Eye Institute Visual Function Questionnaire, NEI VFQ-25) klinisch bedeutsame Veränderungen in Bezug auf den Ausgangswert. Das Ausmass dieser Änderungen war mit denjenigen aus veröffentlichten Studien vergleichbar und entsprach einem Gewinn von 15 Buchstaben bei der bestmöglich korrigierten Sehschärfe (BCVA).
In Woche 52 wurden keine klinisch bedeutsamen Unterschiede zwischen Aflibercept und der Referenzsubstanz Ranibizumab betreffend Veränderungen der NEI VFQ-25 Gesamtzahl und Teilskalen (nahe Aktivitäten, entfernte Aktivitäten und sichtspezifische Abhängigkeit) gegenüber dem Ausgangswert gefunden.
In beiden Studien sowie in der kombinierten Analyse stimmten die Resultate zur Wirksamkeit in allen auswertbaren Untergruppen (z.B. Alter, Geschlecht, ethnische Zugehörigkeit, Ausgangswert der Sehschärfe, Läsionstyp, Läsionsgrösse) mit den Resultaten der gesamten Patientenpopulation überein.
Im zweiten Studienjahr blieb die Wirksamkeit bis zur letzten Auswertung in Woche 96 erhalten. Über den Zeitraum von 2 Jahren, erhielten die Patienten in der Aflibercept 2Q8 Gruppe im Durchschnitt 11,2 Dosen und die Patienten in der Ranibizumab Gruppe erhielten im Durchschnitt 16,5 Dosen.
Die ALTAIR-Studie war eine 2-armige multizentrische, randomisierte, unverblindete Phase-4-Studie zur Wirksamkeit und Sicherheit von Aflibercept an japanischen Patienten mit behandlungsnaiver feuchter AMD unter Verwendung von Treat-and-Extend Behandlungsschemata mit zwei unterschiedlichen Anpassungsintervallen (2 Wochen [2W] und 4 Wochen [4W]). Die in die ALTAIR Studie eingeschlossenen 247 Patienten erhielten initial 3 Injektionen Aflibercept 2 mg alle 4 Wochen, gefolgt von einer weiteren Injektion im Abstand von 8 Wochen. Ab Woche 16 wurden die Patienten randomisiert (1:1) gemäss einem der beiden Schemata, [2W] bzw. [4W], weiterbehandelt. Dabei wurden die Behandlungsintervalle im jeweiligen Arm basierend auf im Prüfplan definierten visuellen und anatomischen Kriterien um 2 bzw. 4 Wochen ausgedehnt oder verkürzt, mit einem maximalen Behandlungsintervall von 16 Wochen. Bis Woche 52 ergaben sich daraus 6 bis 8 Injektionen.
Primärer Wirksamkeitsendpunkt war die mittlere Änderung der BCVA in Woche 52 gegenüber dem Ausgangswert. Nach 52 Wochen hatten die Patienten des [2W]-Behandlungsarms im Mittel 9,0 Buchstaben und die Patienten des [4W]-Behandlungsarms im Mittel 8,4 Buchstaben gegenüber dem Ausgangswert gewonnen [Differenz der KQ-Mittelwerte für Buchstaben (95%-KI): -0,4 (-3,8; 3,0), Kovarianzanalyse]. Die mittleren Gewinne bei der BCVA waren bei Patienten, die 6, 7 oder 8 Injektionen erhielten, vergleichbar. Der Anteil der Patienten, bei denen das Behandlungsintervall auf 12 Wochen oder mehr ausgedehnt wurde, betrug 42,3% im [2W]-Arm und 49,6% im [4W]-Arm. Die initial erreichten Verbesserungen der BCVA waren in beiden Behandlungsarmen über den Beobachtungszeitraum von 2 Jahren weitgehend stabil. In Woche 96 betrug der mittlere Gewinn im Vergleich zum Wert bei Studienbeginn 7,6 Buchstaben [2W] bzw. 6,1 Buchstaben [4W]. Der Anteil der Patienten, bei denen das Behandlungsintervall bis Woche 96 auf 12 Wochen oder mehr ausgedehnt wurde, betrug 56,9% ([2W]-Arm) und 60,2% ([4W]-Arm). Im zweiten Behandlungsjahr erhielten Patienten in der Gruppe mit Anpassung um jeweils 2 Wochen durchschnittlich 3,6 Injektionen und in der Gruppe mit Anpassung um jeweils 4 Wochen durchschnittlich 3,7 Injektionen. Über den 2jährigen Behandlungszeitraum erhielten die Patienten durchschnittlich 10,4 Injektionen.
Die okulären und systemischen Sicherheitsprofile waren mit denjenigen in den pivotalen Studien VIEW1 und VIEW2 beobachteten vergleichbar.
Sicherheit und Wirksamkeit bei älteren Patienten
In den pivotalen wAMD Studien waren ca. 89% (1616/1817) der für die Behandlung mit Aflibercept randomisierten Patienten 65 Jahre alt oder älter und ca. 63% (1139/1817) waren 75 Jahre alt oder älter.
Makulaödem infolge eines retinalen Zentralvenenverschlusses (CRVO)
Die Sicherheit und Wirksamkeit von Aflibercept wurden in zwei randomisierten, multizentrischen, doppelt verblindeten, mit Scheininjektionen kontrollierten Studien bei Patienten mit Makulaödem infolge eines CRVO beurteilt. Insgesamt 358 Patienten wurden in den beiden Studien COPERNICUS und GALILEO behandelt und bezüglich der Wirksamkeit ausgewertet (217 mit Aflibercept). In beiden Studien wurden die Patienten randomisiert und in einem Verhältnis von 3:2 folgenden Behandlungen zugewiesen: entweder 2 mg Aflibercept alle 4 Wochen (2Q4) oder Scheininjektionen alle 4 Wochen in insgesamt 6 Injektionen (Kontrollgruppe).
Nach 6 monatlichen Injektionen erhielten die Patienten nur dann eine Behandlung, wenn sie die vorspezifizierten Kriterien für eine erneute Behandlung erfüllten, mit Ausnahme der Patienten in der Kontrollgruppe in der GALILEO Studie, die auch weiterhin eine Scheininjektion erhielten (Kontrolle an Kontrolle).
Das Alter der Patienten lag zwischen 22 und 89 Jahren mit einem durchschnittlichen Alter von 64 Jahren.
In beiden Studien war der primäre Endpunkt für die Wirksamkeit der Anteil an Patienten, die mindestens 15 Buchstaben in der bestmöglich korrigierten Sehschärfe (BCVA) in Woche 24 im Vergleich zum Ausgangswert gewannen.
In der COPERNICUS-Studie betrug der Anteil Patienten mit einem Gewinn von mindestens 15 Buchstaben in der bestkorrigierten Sehschärfe gegenüber dem Ausgangswert nach 24 Wochen 12% in der Kontrollgruppe und 56% in der mit Aflibercept 2Q4 behandelten Gruppe. Diese Werte unterschieden sich signifikant voneinander. Die mittlere Veränderung der bestkorrigierten Sehschärfe anhand des ETDRS-Letter Scores betrug -4,0 in der Kontrollgruppe und 17,3 in der mit Aflibercept 2Q4 behandelten Gruppe. Nach der sich anschliessenden, bis Woche 52 dauernden Phase, während der beide Gruppen Aflibercept 2 mg nach Bedarf erhielten, betrug der Anteil der Patienten mit einem Gewinn von mindestens 15 Buchstaben 55% in der von Beginn an mit Aflibercept behandelten Gruppe, im Gegensatz zu 30% in der ursprünglich mit Scheininjektionen behandelten Gruppe. Die mittlere Veränderung der bestkorrigierten Sehschärfe anhand des ETDRS-Letter Scores betrug nach 52 Wochen 16,2 in der zuvor mit Aflibercept 2Q4 behandelten Gruppe und unterschied sich damit von der ursprünglich mit Scheininjektionen behandelten Gruppe, mit einem ETDRS-Letter Score von 3,8.
In der GALILEO-Studie betrug der Anteil Patienten mit einem Gewinn von mindestens 15 Buchstaben in der bestkorrigierten Sehschärfe gegenüber dem Ausgangswert nach 24 Wochen 22% in der Kontrollgruppe und 60% in der mit Aflibercept 2Q4 behandelten Gruppe. Die mittlere Veränderung der bestkorrigierten Sehschärfe anhand des ETDRS-Letter Scores betrug 3,3 in der Kontrollgruppe und 18,0 in der mit Aflibercept 2Q4 behandelten Gruppe. Nach der sich anschliessenden, bis Woche 52 dauernden Phase, während derer die Patienten nach Bedarf behandelt wurden, betrug der Anteil Patienten mit einem Gewinn von mindestens 15 Buchstaben in der bestkorrigierten Sehschärfe gegenüber dem Ausgangswert 60% in der mit Aflibercept 2 mg behandelten Gruppe, im Gegensatz zu 32% in der mit Scheininjektionen behandelten Gruppe. Die mittlere Veränderung der bestkorrigierten Sehschärfe anhand des ETDRS-Letter Scores betrug nach 52 Wochen 16,9 in der mit Aflibercept 2Q4 behandelten Gruppe und unterschied sich damit von der Kontrollgruppe, mit einem ETDRS-Letter Score von 3,8.
Sicherheit und Wirksamkeit bei älteren Patienten
In den CRVO-Studien waren ungefähr 52% (112/217) der für die Behandlung mit Aflibercept randomisierten Patienten 65 Jahre alt oder älter und ungefähr 18% (38/217) waren 75 Jahre alt oder älter.
Makulaödem infolge eines retinalen Venenastverschlusses (BRVO)
Die Sicherheit und Wirksamkeit von Aflibercept wurde in einer randomisierten, multizentrischen, doppelt verblindeten, mit Scheininjektionen kontrollierten Studie bei Patienten mit Makulaödem infolge BRVO einschliesslich retinalen Hemizentralvenenverschlusses beurteilt. Insgesamt 181 Patienten wurden in der VIBRANT-Studie behandelt und waren hinsichtlich der Wirksamkeit auswertbar (91 mit Aflibercept). In der Studie wurden die Patienten im Verhältnis 1:1 entweder einer Behandlung mit 2 mg Aflibercept alle 4 Wochen (2Q4) bei insgesamt 6 Injektionen oder einer Behandlung mittels Laserphotokoagulation bei Studienbeginn (Laser-Kontrollgruppe) randomisiert zugewiesen. Ab Woche 12 konnten Patienten der Laserkontrollgruppe bei Bedarf zusätzliche Laserphotokoagulationen (sogenannte «Bedarfs-Laser-Behandlung») erhalten, falls mindestens ein vorabdefiniertes Kriterium für die Bedarfs-Laser-Behandlung erfüllt war. Das kürzeste Intervall zwischen den Behandlungen mit Laserphotokoagulation war 12 Wochen. Ab Woche 24 erhielten die Patienten der Aflibercept Gruppe 2 mg alle 8 Wochen bis Woche 48 und es war möglich, Patienten der Laserkontrollgruppe eine Bedarfsbehandlung mit 2 mg Aflibercept zukommen zu lassen, sofern mindestens ein vorabdefiniertes Kriterium für die Bedarfs-Behandlung erfüllt war. Dabei wurde 2 mg Aflibercept alle 4 Wochen (2Q4) für 3 Behandlungsintervalle, gefolgt von intravitrealen Injektionen alle 8 Wochen, verabreicht.
Das Alter der Patienten lag zwischen 42 und 94 Jahren mit einem Mittel von 65 Jahren.
Primärer Wirksamkeitsendpunkt der VIBRANT-Studie war der Anteil der Patienten, die in Woche 24 eine Verbesserung der bestkorrigierten Sehschärfe (BCVA) um mindestens 15 Buchstaben im Vergleich zum Ausgangswert erreichten hatten.
Der Anteil Patienten mit einem Gewinn von mindestens 15 Buchstaben in der bestkorrigierten Sehschärfe gegenüber dem Ausgangswert betrug nach 24 bzw. 52 Wochen 26,7% bzw. 41,1% in der Kontrollgruppe und 52,7% bzw. 57,1% in der mit Aflibercept 2Q4 behandelten Gruppe. Diese Werte unterschieden sich signifikant voneinander.
Die Änderung der Sehschärfe in Woche 24 im Vergleich zum Ausgangswert war eine sekundäre Wirksamkeitsvariable in der VIBRANT Studie.
Die mittlere Veränderung der BCVA anhand des ETDRS-Letter Scores betrug nach 24 bzw. 52 Wochen 6,9 bzw. 12,2 in der Kontrollgruppe und 17,0 bzw. 17,1 in der Aflibercept 2 mg Gruppe. Differenz nach 24 Wochen: 10,5 [7,1; 14,0]95%CI; Differenz nach 52 Wochen: 5,2 [1,7; 8,7]95%CI.
Ab Woche 24 erhielten 67 Patienten der Lasergruppe die Bedarfsbehandlung mit Aflibercept (aktive Kontrolle/Aflibercept 2 mg Gruppe). In dieser Behandlungsgruppe verbesserte sich die Sehschärfe um ca. 5 Buchstaben von Woche 24 zu 52.
Diabetisches Makulaödem (DME)
Die Sicherheit und Wirksamkeit von Aflibercept wurden in zwei randomisierten, multizentrischen, doppelt maskierten, aktiv kontrollierten Studien an Patienten mit DME beurteilt. Insgesamt 862 randomisierte und behandelte Patienten wurden in die Wirksamkeitsanalyse eingeschlossen; 576 wurden in den beiden Studien (VIVIDDME und VISTADME) mit Aflibercept behandelt. In jeder Studie wurden die Patienten per Randomisierung im Verhältnis 1:1:1 einem von drei Dosierungsschemata zugeteilt:
1.Aflibercept 2 mg alle acht Wochen im Anschluss an fünf initiale monatliche Injektionen (Aflibercept 2Q8);
2.Aflibercept 2 mg alle vier Wochen (Aflibercept 2Q4); und
3.Laserphotokoagulation der Makula (aktive Kontrolle).
Ab Woche 24 konnten Patienten, die entsprechend einem vordefinierten Grenzwert für den Verlust des Sehvermögens geeignet waren, eine zusätzliche Behandlung erhalten: Patienten in den Aflibercept-Gruppen konnten sich einer Laserbehandlung unterziehen, und Patienten in der Lasergruppe konnten mit Aflibercept behandelt werden.
Das Alter der Patienten lag zwischen 23 und 87 Jahren, mit einem durchschnittlichen Alter von 63 Jahren.
In beiden Studien war der primäre Endpunkt für die Wirksamkeit die mittlere Veränderung der bestmöglich korrigierten Sehschärfe (BCVA) in Woche 52 gegenüber Studienbeginn, gemessen anhand des ETDRS-Letter-Scores. Sowohl in der Aflibercept-2Q8- als auch in der Aflibercept-2Q4-Gruppe hat sich eine gegenüber der Laserkontrollgruppe statistisch überlegene Wirksamkeit gezeigt.
In der VIVIDDME Studie zeigten in Woche 52 (bzw. 100) die Patienten in der 2Q8 Behandlungsgruppe einen mittleren Buchstabengewinn von 10,7 (bzw. 9,4), in der 2Q4 Behandlungsgruppe einen mittleren Buchstabengewinn von 10,5 (bzw. 11,4), im Vergleich zur Laserkontrollgruppe mit einem mittleren Buchstabengewinn von 1,2 (bzw. 0,7).
In der VISTADME Studie zeigten in Woche 52 (bzw. 100) die Patienten in der 2Q8 Behandlungsgruppe einen mittleren Buchstabengewinn von 10.7 (bzw. 11,1), in der 2Q4 Behandlungsgruppe einen mittleren Buchstabengewinn von 12,5 (bzw. 11,5), im Vergleich zur Laserkontrollgruppe mit einem mittleren Buchstabengewinn von 0,2 (bzw. 0,9).
Zusammenfassend zeigen die 2-Jahres-Daten, dass die klinische Wirksamkeit über den gesamten Studienzeitraum erhalten bleibt.
Alle Aflibercept Behandlungsgruppen erwiesen sich bezüglich des primären Endpunkts für Wirksamkeit als gegenüber der Laserkontrollgruppe statistisch signifikant überlegen.
Die Auswertung der sekundären Endpunkte zeigte folgende Resultate:
In der VIVIDDME Studie lag der Anteil der Patienten, die gegenüber dem Ausgangswert mindestens 15 Buchstaben an Sehschärfe gewannen, in den 2Q8 und 2Q4 Behandlungsgruppen in Woche 52 (bzw. 100) bei 33,3% (bzw. 31,1%) und 32,4% (bzw. 38,2%), im Vergleich zu 9,1% (bzw. 12,1%) in der Laserkontrollgruppe.
In der VISTADME Studie lag der Anteil der Patienten, die gegenüber dem Ausgangswert mindestens 15 Buchstaben an Sehschärfe gewannen, in den 2Q8 und 2Q4 Behandlungsgruppen in Woche 52 (bzw. 100) bei 31,1% (bzw. 33,1%) und 41,6% (bzw. 38,3%), im Vergleich zu 7,8% (bzw. 13,0%) in der Laserkontrollgruppe.
In der VIVIDDME Studie war in Woche 52 (bzw. in Woche 100) bei 33,3% (bzw. 29,3%) der 2Q4 Patienten und bei 27,7% (bzw. 32,6%) der 2Q8 Patienten und bei 7,5% (bzw. 8,2%) der Laserkontrollpatienten eine Besserung der Schwere der diabetischen Retinopathie nachweisbar, gemessen als Verbesserung um ≥2 Stufen auf der DRSS-Skala (Diabetic Retinopathy Severity Scale; DRSS).
In der Studie VISTADME Studie waren es in Woche 52 (bzw. in Woche 100) 33,8% (bzw. 37,0%) der 2Q4-Patienten, 29,1% (bzw. 37,1%) der 2Q8-Patienten und 14,3% (bzw. 15,6%) der Laserkontrollpatienten.
In der VIVIDDME- bzw. der VISTADME-Studie hatten 36 (8,9%) bzw. 197 (42,9%) Patienten eine vorherige Anti-VEGF-Therapie erhalten, wobei die Auswaschphase mindestens drei Monate betrug. Die Behandlungseffekte in der Subgruppe von Patienten, die vor der Studienteilnahme bereits mit einem VEGF-Inhibitor behandelt wurden, waren mit den Behandlungseffekten bei Patienten ohne vorherige VEGF-Inhibitor-Therapie vergleichbar.
Patienten mit bilateraler Erkrankung konnten eine Anti-VEGF-Behandlung im kontralateralen Auge erhalten. In der VISTADME-Studie erhielten 217 (70,7%) Aflibercept-Patienten bilaterale Aflibercept-Injektionen bis Woche 100, in der VIVIDDME-Studie dagegen wurden 97 (35,8%) Aflibercept-Patienten am kontralateralen Auge mit einer anderen Anti-VEGF-Therapie behandelt.
Die Behandlungseffekte in den auswertbaren Subgruppen (z.B. Alter, Geschlecht, ethnische Abstammung, Ausgangs-HbA1c, Sehschärfe zu Studienbeginn, vorherige Anti-VEGF-Therapie) der einzelnen Studien sowie in der kombinierten Analyse standen generell mit den Ergebnissen der Gesamtpopulationen in Einklang.
CNV infolge einer pathologischen Myopie (mCNV)
Die Sicherheit und die Wirksamkeit von Aflibercept wurden in einer randomisierten, multizentrischen, doppelt maskierten, mit Scheininjektionen kontrollierten Studie an Patienten mit CNV infolge einer pathologischen Myopie (mCNV) beurteilt (MYRROR-Studie). Insgesamt 121 Patienten wurden behandelt und im Hinblick auf die Wirksamkeit ausgewertet (90 mit Aflibercept). Die Patienten stammten alle aus asiatischen Ländern, 76% waren Frauen, das Alter betrug im Mittel 58 Jahre (in einem Bereich von 27 bis 83 Jahren). Die aktive CNV lag subfoveal oder juxtafoveal und umfasste im Durchschnitt 0,3894 (SD 0.4666) Papillenflächen. Die zentrale Retinadicke betrug im Mittel 350,9 (SD 95,2) μm, der Visus (BCVA) 56,5 (SD 9,5) Buchstaben. Bei 80% der Patienten wurde die Diagnose mCNV vor <2 Monaten gestellt und das Studienauge hatte noch keine Vorbehandlung mit VEGF-Rezeptorblockern, PDT, Laser, Steroiden oder Chirurgie. Die Patienten wurden per Randomisierung im Verhältnis 3:1 entweder einer Behandlung mit 2 mg Aflibercept (einmalige Injektion zu Studienbeginn und gegebenenfalls zusätzliche Injektionen bei persistierender oder wieder auftretender Erkrankung) zugewiesen, oder der Kontrollgruppe, die Scheininjektionen erhielt. Bis zum Beurteilungszeitpunkt für den primären Endpunkt in Woche 24 konnten insgesamt sechs Injektionen mit einem minimalen Intervall von 4 Wochen angewendet werden.
Nach den ersten sechs Monaten konnten Patienten, die ursprünglich den Scheininjektionen zugeteilt wurden, eine erste Dosis Aflibercept in Woche 24 erhalten. Danach konnten diese Patienten der ursprünglichen Scheininjektionsgruppe sowie Patienten, die ursprünglich per Randomisierung der aktiven Behandlung zugeteilt wurden, weiterhin zusätzliche Injektionen bei persistierender oder wieder auftretender Erkrankung erhalten.
Der primäre Wirksamkeitsendpunkt bestand in der Veränderung der Sehschärfe in Woche 24 gegenüber Studienbeginn.
Der konfirmatorische sekundäre Wirksamkeitsendpunkt war der Anteil von Patienten, die in Woche 24 in der BCVA mindestens 15 Buchstaben gegenüber Studienbeginn hinzugewonnen hatten.
Die mittlere Veränderung der bestkorrigierten Sehschärfe (BCVA) anhand des ETDRS-Letter Scores betrug in Woche 24 -2,0 in der Kontrollgruppe und +12,1 in der mit Aflibercept behandelten Gruppe. Dieser Unterschied war statistisch signifikant zugunsten von Aflibercept. Der Anteil Patienten mit einem Gewinn von mindestens 15 Buchstaben in der BCVA gegenüber dem Ausgangswert betrug nach 24 Wochen 9,7% in der Kontrollgruppe und 38,9% in der mit Aflibercept behandelten Gruppe. Diese Werte unterschieden sich signifikant voneinander.
Die mittlere Veränderung der BCVA anhand des ETDRS-Letter Scores betrug nach 48 Wochen 3,9 in der Kontrollgruppe und 13,5 in der mit Aflibercept behandelten Gruppe (Analyse aller Patienten mit LOCF). 29,0% der Patienten der Kontrollgruppe und 50,0% der Patienten unter Aflibercept hatten einen Gewinn von mindestens 15 Buchstaben in der BCVA.
Die Behandlungseffekte in allen auswertbaren Subgruppen standen allgemein mit den Ergebnissen der Gesamtpopulationen in Einklang.
Sicherheit und Wirksamkeit bei älteren Patienten
In der mCNV-Studie waren ungefähr 36% (33/91) der Patienten, die für eine Behandlung mit Aflibercept randomisiert wurden, 65 Jahre alt oder älter, rund 10% (9/91) waren 75 Jahre alt oder älter.
PharmakokinetikAbsorption
Aflibercept wird nach intravitrealer Anwendung langsam vom Auge in den systemischen Kreislauf absorbiert und wird im systemischen Kreislauf vorwiegend als inaktiver, stabiler Komplex mit VEGF vorgefunden.
Distribution
In einer pharmakokinetischen Teilstudie waren die maximalen Plasmaspiegel von freiem Aflibercept innerhalb von 1 bis 3 Tagen nach einer intravitrealen Injektion von 2 mg tief und lagen bei ungefähr 0,02 µg/ml (Bereich 0 bis 0,054). Zwei Wochen nach der Verabreichung war bei fast allen Patienten kein freies Aflibercept mehr nachweisbar.
Metabolismus
Nicht zutreffend.
Elimination
Freies Aflibercept bindet VEGF und bildet einen stabilen, inerten Komplex. Wie bei anderen grossen Proteinen wird angenommen, dass sowohl freies wie gebundenes Aflibercept durch proteolytischen Abbau eliminiert wird.
Kinetik spezieller Patientengruppen
Nierenfunktionsstörungen
Mit Aflibercept wurden keine speziellen Studien bei Patienten mit eingeschränkter Nierenfunktion durchgeführt.
Die pharmakokinetische Untersuchung von Patienten der klinischen Studien, in welcher 40% eine eingeschränkte Nierenfunktion aufwiesen (24% mild, 15% mässig und 1% schwerwiegend), zeigte keine Unterschiede in Bezug auf die Plasmakonzentrationen des Wirkstoffs nach intravitrealer Anwendung in 4- oder 8-wöchentlichen Abständen.
Leberfunktionsstörungen
Mit Aflibercept wurden keine speziellen Studien bei Patienten mit eingeschränkter Leberfunktion durchgeführt.
Präklinische DatenIn nicht-klinischen Studien mit wiederholter Dosierung wurde eine toxische Wirkung nur bei einer systemischen Exposition beobachtet, die deutlich über der maximalen Exposition bei Menschen nach intravitrealer Verabreichung der empfohlenen Dosierung liegt. Dies weist auf eine geringe Relevanz für die klinische Anwendung hin.
Bei adulten Affen, die intravitreal mit Aflibercept behandelt wurden, wurden Erosionen und Ulzerationen des respiratorischen Epithels der Nasenmuscheln beobachtet bei einer systemischen Exposition von ungefähr 200- bzw. 700-mal höher (basierend auf Cmax und AUC), als die Exposition bei erwachsenen Patienten nach einer intravitrealen Dosis von 2 mg.
Die systemische Exposition beim No Observed Adverse Effect Level (NOAEL) in Affen, war (basierend auf Cmax und AUC), 42-bzw. 56-mal höher als die Exposition bei erwachsenen Patienten.
Es wurden keine Studien bezüglich mutagenem oder karzinogenem Potential von Aflibercept durchgeführt.
Systemisch verabreichtes Aflibercept bewirkte glomeruläre Veränderungen, schädigte die Nebennieren und die Gonaden. Aflibercept zeigte embryo-fetale Toxizität (Missbildungen und Aborte) in einer Entwicklungsstudie mit intravenöser (3 bis 60 mg/kg) sowie subkutaner (0,1 bis 1 mg/kg) Applikation bei trächtigen Kaninchen. Das mütterliche NOAEL lag bei einer Dosis von 3 mg/kg bzw. 1 mg/kg. Ein NOAEL für die embryo-fetale Entwicklung wurde nicht identifiziert. Bei der Dosis von 0,1 mg/kg war die systemische Exposition bei den Muttertieren ungefähr 17- bzw. 10-mal höher (basierend auf Cmax und kumulativer AUC) als die Exposition bei erwachsenen Patienten nach einer intravitrealen Dosis von 2 mg.
Die Wirkung auf die männliche und weibliche Fertilität wurde als Teil einer 6-monatigen Studie bei Affen mit intravenöser Anwendung von Aflibercept mit Dosen von 3 bis 30 mg/kg untersucht. Ausbleibende oder unregelmässige Menstruationen einhergehend mit verändertem Spiegel der weiblichen Reproduktionshormone sowie Änderungen in der Morphologie und Beweglichkeit der Spermien wurden bei allen Dosierungen beobachtet. Basierend auf Cmax und AUC waren die systemischen Expositionen bei der intravenösen Dosis von 3 mg/kg ungefähr 4900- bzw. 1500-mal höher als die Exposition, die bei Menschen nach einer intravitrealen Dosis von 2 mg beobachtet wurde. Alle Veränderungen waren reversibel.
Sonstige HinweiseInkompatibilitäten
Dieses Arzneimittel darf nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden. Vor Gebrauch kann die ungeöffnete Durchstechflasche oder die ungeöffnete Blisterpackung für bis zu 14 Tage unter 30°C gelagert werden.
Haltbarkeit nach Anbruch
Nach dem Öffnen der Durchstechflasche oder der Blisterpackung muss unter aseptischen Bedingungen weitergearbeitet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern. In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen. Ausser Reichweite von Kindern lagern. Nicht einfrieren.
Hinweise für die Handhabung
Die Fertigspritze enthält mehr als die empfohlene Dosis von 2 mg Aflibercept in 50 µl. Siehe «Anwendung der Fertigspritze». Das entnehmbare Volumen der Spritze darf nicht vollständig genutzt werden.
Eine Injektion des gesamten Volumens der Fertigspritze könnte in einer Überdosierung resultieren sowie einen Anstieg des Augeninnendrucks verursachen (Siehe Rubrik «Überdosierung»).
Die Fertigspritze und die Durchstechflasche sind nur für den einmaligen Gebrauch an einem Auge bestimmt. Die Entnahme mehrerer Dosen aus einer Fertigspritze oder einer Durchstechflasche kann das Risiko einer Kontamination und nachfolgender Entzündung erhöhen.
Sterile Injektionslösung: klar, farblos bis leicht bräunlich-gelbe Lösung. Vor der Anwendung muss die Injektionslösung visuell geprüft werden. Die Durchstechflasche oder Fertigspritze darf nicht verwendet werden, wenn Partikel, Trübungen oder Verfärbungen sichtbar sind.
Nicht verwenden, wenn ein Teil der Fertigspritze beschädigt oder lose ist, oder wenn die Spritzenkappe sich vom Luer-Lock abgelöst hat.
Für die Entnahme des Produktes aus der Durchstechflasche ist eine Nadel mit integriertem Filter zu verwenden.
Für die intravitreale Injektion sollte eine 0,3× 13 mm (30G x ½ Zoll) Injektionsnadel verwendet werden.
Betreffend Vorbereitung der Fertigspritzen bzw. der Injektionslösung in der Durchstechflasche sind die Hinweise am Ende der Fachinformation zu berücksichtigen.
Filternadel (steril, im Lieferumfang der Durchstechflasche enthalten):
BD stumpfe sterile 5 Mikrometer-Filter (Füll)-Nadel, nicht zur Injektion ins Auge.
Die BD stumpfe Filter (Füll)-Nadel nicht autoklavieren.
Die Filternadel ist nicht pyrogen. Bei Beschädigung von Einzelverpackungen nicht verwenden.
Verwendete BD stumpfe Filter (Füll)-Nadel in einem geprüften Sicherheitsbehälter entsorgen.
Vorsicht: Erneute Verwendung der Filternadel kann zu Infektionen oder anderen Erkrankungen/Verletzungen führen.
Zulassungsnummer70126, 70127 (Swissmedic)
PackungenPackung mit 1 Fertigspritze (B)
Packung mit 1 Durchstechflasche und einer Filternadel (B)
ZulassungsinhaberinSandoz Pharmaceuticals AG, Risch; Domizil: Rotkreuz
Stand der InformationMai 2025.
Hinweise zur Vorbereitung und Anwendung der Fertigspritzen bzw. der Injektionslösung in der Durchstechflasche
Anwendung der Fertigspritze:
Vorbereitung und Verabreichung
Die Fertigspritze enthält mehr als die empfohlene Dosis von 2 mg Aflibercept (entsprechend 0,05 ml). Die überschüssige Menge muss vor der Anwendung verworfen werden.
Jede Fertigspritze sollte nur zur Behandlung eines einzigen Auges verwendet werden. Die sterile Blisterpackung der Fertigspritze nicht ausserhalb des sauberen Behandlungsraums öffnen.
Für die intravitreale Injektion sollte eine 0,3× 13 mm (30G x ½ Zoll) Injektionsnadel verwendet werden.
Unter sterilen Bedingungen sind folgende Schritte zu befolgen
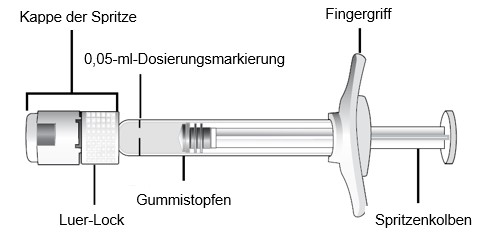
Beschreibung der Fertigspritze
|
1.
|
Erst vor der Anwendung von Afqlir den Umkarton öffnen und die sterile Bilsterpackung entnehmen. Die sterile Blisterpackung vorsichtig öffnen, so dass der Inhalt weiterhin steril bleibt. Die Spritze bis zum weiteren Gebrauch in der sterilen Ablage liegen lassen.
| |
2.
|
Unter sterilen Bedingungen die Spritze aus der sterilen Blisterpackung nehmen.
| |
3.
|
Um die Kappe von der Spritze zu entfernen, mit einer Hand die Spritze halten, während Daumen und Zeigefinger der anderen Hand die Kappe der Spritze festhalten. Die Kappe der Spritze muss abgeknickt werden (nicht abschrauben oder abbrechen).
Hinweis: Um die Sterilität des Arzneimittels nicht zu gefährden, darf der Spritzenkolben nicht herausgezogen werden.
|
|

| |
4.
|
Unter sterilen Bedingungen eine 0,3× 13 mm (30G x ½ Zoll) Injektionsnadel fest auf die Spitze des Luer-Lock-Adapters aufschrauben.
|
|

| |
5.
|
Die Spritze mit der Nadel nach oben halten und auf Bläschen hin prüfen.
Wenn Bläschen zu sehen sind, leicht mit dem Finger gegen die Spritze klopfen, bis die Bläschen nach oben steigen.
Die Nadelkappe durch gerades Abziehen vorsichtig entfernen.
|
|

| |
6.
|
Um alle Bläschen und überschüssiges Arzneimittel zu entfernen, den Spritzenkolben langsam soweit eindrücken, bis die Grundfläche des kuppelförmigen Kolbens auf derselben Höhe ist wie die Dosierungslinie der Spritze (entsprechend 50 Mikroliter).
Hinweis: Injektion sofort nach dem Vorbereiten der Spritze.
|
|

| |
7.
|
Langsam injizieren, bis der Gummistopfen das Ende der Spritze erreicht, um das Volumen von 0,05 ml zu verabreichen. Um sicherzustellen, dass die gesamte Dosis verabreicht wurde, überprüfen, ob der Gummistopfen das Ende des Spritzenkörpers erreicht hat.
| |
8.
|
Fertigspritze nur für einmaligen Gebrauch.
Die Entnahme von mehr als einer Dosis aus der Fertigspritze kann das Risiko einer Kontamination und nachfolgender Infektion erhöhen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
|
Anwendung der Durchstechflasche:
Vorbereitung und Verabreichung
Die Durchstechflasche enthält mehr als die empfohlene Dosis von 2 mg Aflibercept (entsprechend 0,05 ml). Die überschüssige Menge muss vor der Anwendung verworfen werden.
Jede Durchstechflasche sollte nur zur Behandlung eines einzigen Auges verwendet werden.
Für die Vorbereitung und intravitreale Injektion werden die folgenden Gebrauchsartikel für den einmaligen Gebrauch benötigt:
·Eine stumpfe 5 µm-Filternadel (18G x 1½ Zoll, steril, im Lieferumfang der Durchstechflasche enthalten)
·Eine 1-ml-Spritze mit einer 0,05-ml Dosierungsmarkierung, steril
·Eine 0,3× 13 mm (30G x ½ Zoll) Injektionsnadel, steril (nicht im Lieferumfang enthalten)
Unter aseptischen Bedingungen sind folgende Schritte zu befolgen.
|
1.
|
Die Kunststoffkappe von der Durchstechflasche entfernen.
|
|

| |
2.
|
Den oberen Bereich der Durchstechflasche mit einem alkoholgetränkten Tupfer reinigen.
|
|

| |
3.
|
Die 18 G x 1½ Zoll-, 5 µm Filternadel an einer mit einem Luer-Lock-Adapter ausgestatteten sterilen Spritze befestigen.
|
|

| |
4.
|
Die Filternadel durch die Mitte des Durchstechflaschen-Stopfens stechen, bis die Nadel vollständig in die Durchstechflasche eingeführt ist und die Spitze den Boden oder die Unterkante der Durchstechflasche berührt.
| |
5.
|
Unter aseptischen Bedingungen den gesamten Inhalt der Afqlir-Durchstechflasche in die Spritze aufnehmen, indem die Durchstechflasche aufrecht in einer leicht geneigten Position gehalten wird, um das vollständige Entleeren zu erleichtern. Um das Aufziehen von Luft zu verhindern, sollte darauf geachtet werden, dass die abgeschrägte Kante der Filternadel in die Lösung eintaucht. Um dies auch während der Entnahme zu gewährleisten, ist die Durchstechflasche schräg zu halten.
|
|

| |
6.
|
Bitte beachten, dass der Spritzenkolben beim Entleeren der Durchstechflasche ausreichend zurückgezogen wird, damit auch die Filternadel vollständig entleert wird.
| |
7.
|
Die Filternadel von der Spritze entfernen und diese vorschriftsmässig entsorgen.
Hinweis: Die Filternadel darf nicht für die intravitreale Injektion verwendet werden.
| |
8.
|
Die 0,3× 13 mm (30G x ½ Zoll) Injektionsnadel an der Spritze befestigen, indem Sie die Injektionsnadel fest auf die Luer-Lock-Spitze der Spritze schrauben.
Die Nadelkappe durch gerades Abziehen vorsichtig entfernen.
|
|

| |
9.
|
Die Spritze mit der Nadel nach oben halten und auf Bläschen hin prüfen. Wenn Bläschen zu sehen sind, leicht mit dem Finger gegen die Spritze klopfen, bis die Bläschen nach oben steigen.
|
|

| |
10.
|
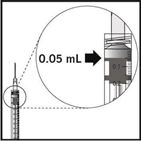
Um alle Bläschen und überschüssiges Arzneimittel zu entfernen, den Spritzenkolben langsam so weit eindrücken, bis die Kolbenspitze auf derselben Höhe ist wie die 0,05 ml-Linie der Spritze.
Hinweis: Injektion sofort nach dem Vorbereiten der Spritze.
|
|
| |
11.
|
Langsam injizieren, bis der Gummistopfen das Ende der Spritze erreicht, um das Volumen von 0,05 ml zu verabreichen. Um sicherzustellen, dass die gesamte Dosis verabreicht wurde, überprüfen, ob der Gummistopfen das Ende des Spritzenkörpers erreicht hat.
| |
12.
|
Durchstechflasche nur für einmaligen Gebrauch. Die Entnahme von mehr als einer Dosis aus der Durchstechflasche kann das Risiko einer Kontamination und nachfolgender Infektion erhöhen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
|
|