Zusammensetzung
Wirkstoffe
Octocogum alfa (Factor VIII coagulationis humanus (ADNr))
Hilfsstoffe
Mannitolum, Natrii chloridum, Histidinum, Trehalosum, Calcii chloridum, Trometamolum, Polysorbatum 80, Antiox.: Glutathionum 0.4 mg
Lösungsmittel: Aqua ad iniectabilia
Indikationen/Anwendungsmöglichkeiten
Behandlung und Prophylaxe von Blutungen bei Patienten mit Hämophilie A (kongenitaler Faktor VIII-Mangel).
ADVATE enthält keine pharmakologisch wirksamen Mengen des von Willebrand-Faktors und ist daher nicht zur Behandlung des von Willebrand-Jürgens-Syndroms geeignet.
Dosierung/Anwendung
Die Behandlung muss unter Überwachung eines mit der Therapie von Hämophilie A vertrauten Arztes erfolgen.
Dosierung
Dosierung und Dauer der Substitutionstherapie richten sich nach dem Schweregrad des Faktor VIII-Mangels, nach dem Ort und dem Ausmass der Blutung und dem klinischen Zustand des Patienten.
Die verabreichten Faktor VIII-Einheiten werden in Internationalen Einheiten (I.E.) angegeben, entsprechend dem WHO-Standard für Faktor VIII-Produkte. Die Faktor VIII-Aktivität im Plasma wird entweder als Prozentsatz (relativ zum normalen menschlichen Plasma) oder in I.E. (relativ zum Internationalen Standard für Faktor VIII im Plasma) angegeben.
Eine I.E. der Faktor VIII-Aktivität entspricht der Menge an Faktor VIII in einem Milliliter normalem menschlichen Plasma. Die Berechnung der erforderlichen Dosis von Faktor VIII basiert auf dem empirischen Befund, dass 1 I.E. Faktor VIII pro kg Körpergewicht die Faktor VIII-Aktivität im Plasma um 2 I.E./dl erhöht. Die erforderliche Dosis wird mit folgender Formel berechnet:
Erforderliche Einheiten (I.E.) = Körpergewicht (kg) x gewünschter Faktor VIII-Anstieg (%) x 0,5
Bei folgenden hämorrhagischen Ereignissen soll die Faktor VIII-Aktivität im entsprechenden Zeitraum nicht unter die angegebenen Plasmaspiegel (in % der Norm oder in I.E./dl) sinken. Die folgende Tabelle enthält Richtwerte für die Dosierung bei Blutungen und chirurgischen Eingriffen:
Tabelle 1
|
Grad der Blutung/ Art des chirurgischen Eingriffes |
Erforderlicher Faktor VIII Plasmaspiegel (% oder I.E./dl) |
Häufigkeit der Dosierung (Stunden) / Behandlungsdauer (Tage) |
|
Blutung | ||
|
Gelenkblutung im Frühstadium, Muskelblutungen oder Blutungen im Mund |
20 – 40 |
Injektion alle 12 – 24 Stunden für mind. 1 Tag, bis die Blutung – angezeigt durch das Verschwinden der Schmerzen – steht oder Heilung erreicht ist. |
|
Ausgeprägtere Gelenkblutung, Muskelblutung oder Hämatom |
30 – 60 |
Injektion alle 12 – 24 Stunden für 3 – 4 Tage oder länger wiederholen, bis die Schmerzen und die akute Beeinträchtigung beseitigt sind. |
|
Lebensbedrohliche Blutungen |
60 – 100 |
Injektion alle 8 – 24 Stunden wiederholen bis die Gefahr für den Patienten vorüber ist. |
|
Chirurgische Eingriffe | ||
|
Kleinere Eingriffe |
30 – 60 |
Alle 12 Stunden für mind. 1 Tag bis die Wundheilung erreicht ist. |
|
Grössere Eingriffe |
80 – 100 |
Injektion alle 8 – 24 Stunden bis zu angemessener Wundheilung wiederholen, dann Therapie für noch mind. 7 Tage fortsetzen, um eine Faktor VIII-Aktivität von 30 – 60% (I.E./dl) aufrecht zu erhalten. |
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Spezielle Dosierungsanweisungen
Dosis und Häufigkeit der Verabreichung sollen entsprechend der klinischen Wirksamkeit des Produktes im Einzelfall angepasst werden. Unter bestimmten Umständen (Anwesenheit eines «Low-responder»-Inhibitors) können höhere Dosierungen notwendig sein.
Während des Behandlungsverlaufes wird zur Steuerung der zu verabreichenden Dosis und der Häufigkeit der Injektionen eine angemessene Bestimmung der Faktor VIII-Plasmaspiegel empfohlen. Besonders bei grösseren chirurgischen Eingriffen oder bei lebensbedrohlichen Blutungen ist eine genaue Überwachung der Substitutionstherapie durch Bestimmung der Faktor VIII-Aktivität im Plasma unerlässlich. Einzelne Patienten können sich in ihrer Reaktion auf Faktor VIII unterscheiden, verschiedene in vivo Recovery erreichen und unterschiedliche Halbwertszeiten aufweisen.
Zur Dauersubstitution von Blutungen bei Patienten mit schwerer Hämophilie A sollen Dosen zwischen 20 – 40 I.E. von Faktor VIII pro kg Körpergewicht im Abstand von 2 – 3 Tagen gegeben werden. In manchen Fällen, besonders bei jüngeren Patienten, können kürzere Dosierungsabstände oder höhere Dosen erforderlich sein.
Patienten mit Inhibitoren
Patienten sollten regelmässig auf die Bildung von Inhibitoren gegen Faktor VIII überwacht werden. Falls die erwarteten Faktor VIII-Plasmaaktivitäten nicht erreicht werden oder die Blutung mit einer angemessenen Dosis nicht beherrscht wird, muss ein Inhibitortest durchgeführt werden. Bei Patienten mit hohen Inhibitorwerten, wenn die Faktor VIII-Therapie nicht effektiv ist, müssen andere therapeutische Massnahmen erwogen werden. Diese Therapien sollten nur von Ärzten durchgeführt werden, die über Erfahrung in der Behandlung von Patienten mit Hämophilie verfügen. Siehe auch «Warnhinweise und Vorsichtsmassnahmen».
Art der Anwendung
ADVATE wird intravenös verabreicht. Im Allgemeinen erfolgt die intravenöse Injektion als Bolus über einen Zeitraum von ≤5 Minuten. Die Verabreichungsgeschwindigkeit richtet sich nach dem Befinden des Patienten, wobei eine maximale Injektionsrate von 10 ml/min nicht überschritten werden sollte.
Anweisungen zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt «Hinweise für die Handhabung».
Kontraindikationen
Überempfindlichkeit gegenüber dem arzneilich wirksamen Bestandteil, einem der Hilfsstoffe sowie Maus- oder Hamsterproteinen.
Warnhinweise und Vorsichtsmassnahmen
Überempfindlichkeit
Wie bei allen intravenösen Substanzen können allergische Überempfindlichkeitsreaktionen auftreten. Das Produkt enthält Spuren von Maus- und Hamsterproteinen. Es sind Fälle von allergischen Überempfindlichkeitsreaktionen, auch Anaphylaxie, nach Anwendung von ADVATE bekannt, welche sich durch Schwindel, Parästhesien, Hautausschlag, Hitzegefühl, Schwellungen des Gesichts, Urtikaria und Juckreiz bemerkbar machen. Die Patienten sollen über die Anzeichen einer Überempfindlichkeitsreaktion vom Soforttyp, wie Ausschlag, Juckreiz, generalisierte Urtikaria, Angioödem, Hypotonie (z.B. Schwindel oder Synkope), Schock und akute respiratorische Beschwerden (z.B. Engegefühl in der Brust, Keuchatmung) aufgeklärt sein. Wenn diese Symptome auftreten, sollen die Patienten die Behandlung sofort abbrechen und ihren Arzt kontaktieren. Im Fall eines anaphylaktischen Schocks soll eine Schocktherapie nach aktuellem medizinischem Standard durchgeführt werden.
Inhibitoren
Die Bildung von neutralisierenden Antikörpern (Inhibitoren) gegen Faktor VIII ist eine bekannte Komplikation bei der Behandlung von Patienten mit Hämophilie A. Diese Inhibitoren sind stets gegen die prokoagulatorische Aktivität von Faktor VIII gerichtete IgG Immunglobuline, die in Bethesda Einheiten (B.E.) pro ml Plasma mittels modifiziertem Bethesda Assay quantifiziert werden. Bei Patienten, die Inhibitoren gegen Faktor VIII bilden, manifestiert sich dieser Zustand als eine unzureichende klinische Antwort. In diesen Fällen wird empfohlen, ein Hämophilie-Zentrum zu besuchen. Das Risiko, Inhibitoren zu entwickeln, korreliert mit dem Ausmass der Exposition gegenüber dem Faktor VIII, wobei das Risiko innerhalb der ersten 20 Expositionstage am grössten ist und von genetischen und sonstigen Faktoren abhängt. In seltenen Fällen können sich Inhibitoren nach den ersten 100 Expositionstagen bilden.
Bei zuvor unbehandelten pädiatrischen Patienten (PUPs) die mit Faktor VIII Produkten behandelt wurden, liegt die Gesamtinzidenz von Inhibitorenbildung bei 3% bis 13% bei leichter bis mittelschwerer Hämophilie und etwa bei 30% in Patienten mit schwerer Hämophilie.
Bei vorbehandelten Patienten (PTPs) mit mehr als 100 Expositionstagen und anamnestisch bekannter Inhibitorenwirkung wurde, nach Umstellung von einem rekombinanten Faktor VIII-Produkt auf ein anderes, das Wiederauftreten von (niedrigtitrigen) Inhibitoren beobachtet. Es wird deshalb empfohlen, Patienten, die mit ADVATE behandelt wurden, sollten sorgfältig klinisch und mit geeigneten Labortests hinsichtlich der Entwicklung von Inhibitoren zu überwachen.
Ganz allgemein sollten alle Patienten, die mit Blutgerinnungsfaktor VIII behandelt wurden, sorgfältig mittels klinischer Befunde und mit geeigneten Labortests hinsichtlich der Entwicklung von Inhibitoren überwacht werden.
Katheterbedingte Komplikationen bei der Behandlung
Falls ein zentralvenöser Zugang erforderlich sein sollte, ist auf Komplikationen, z.B. lokale Infektionen, Bakteriämie und Katheterthrombose zu achten.
Hinweis zu den sonstigen Bestandteilen
Dieses Arzneimittel enthält nach der Rekonstitution 0,45 mmol Natrium (10 mg) pro Durchstechflasche. Dies muss bei Patienten, die einer natriumkontrollierten Diät unterliegen, berücksichtigt werden.
Interaktionen
Wechselwirkungen von ADVATE mit anderen Arzneimitteln sind nicht bekannt.
Schwangerschaft, Stillzeit
Reproduktionsstudien an Tieren wurden mit ADVATE nicht durchgeführt. Aufgrund des seltenen Auftretens der Hämophilie A bei Frauen liegen über die Anwendung von ADVATE während der Schwangerschaft und Stillzeit keine Erfahrungen vor. Deshalb muss in der Schwangerschaft und Stillzeit der Nutzen der ADVATE-Behandlung gegen das mögliche Risiko für Mutter und Kind sorgfältig abgewogen werden und ADVATE darf nur bei eindeutiger Indikationsstellung angewendet werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
ADVATE hat einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit Maschinen zu bedienen.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
Während klinischer Studien mit ADVATE wurden insgesamt 93 Nebenwirkungen (ADRs) bei 45 von 418 behandelten Patienten angezeigt. Die bei der grössten Zahl Patienten auftretenden Nebenwirkungen waren Inhibitoren gegen Faktor VIII, Kopfschmerzen und Fieber. Von den insgesamt 93 kausal mit ADVATE zusammenhängenden Nebenwirkungen wurden keine bei Neugeborenen beobachtet, 30 wurden bei 20/60 Kleinkindern, 7 bei 3/68 Kindern, 10 bei 5/38 Jugendlichen und 46 bei 17/147 Erwachsenen gemeldet.
Häufigkeit der Nebenwirkungen in klinischen Studien sowie aus spontanen Meldungen
Die Häufigkeit wurde ermittelt unter Verwendung der folgenden Kriterien: sehr häufig (≥10%), häufig (≥1% bis <10%), gelegentlich (≥0.1% bis<1%), selten (≥0.01% bis <0.1%), sehr selten (<0.01%), Einzelfälle (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Infektionen und parasitäre Erkrankungen
Gelegentlich: Influenza, Laryngitis
Erkrankungen des Blutes und des Lymphsystems
Sehr häufig: Faktor VIII Hemmung 29% (in PUPs)
Gelegentlich: Lymphangitis, Faktor VIII Hemmung (in PTPs)
Herzerkrankungen
Gelegentlich: Palpitationen
Erkrankungen des Immunsystems
Einzelfälle: anaphylaktische Reaktion, Überempfindlichkeit, Schwellung des Gesichts
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen
Gelegentlich: Schwindelgefühl, Geschmacksstörung, Migräne, Erinnerungsvermögen eingeschränkt, Tremor, Synkope
Augenerkrankungen
Gelegentlich: Augenentzündung
Gefässerkrankungen
Gelegentlich: Hitzewallung, Hämatom, Blässe
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Gelegentlich: Dyspnoe
Erkrankungen des Gastrointestinaltrakts
Gelegentlich: Diarrhoea, Übelkeit, Oberbauchschmerzen, Erbrechen
Erkrankungen der Haut und des Unterhautgewebes
Gelegentlich: Hyperhidrosis, Pruritus, Ausschlag, Urtikaria
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Fieber
Gelegentlich: Brustkorbschmerz, Brustkorbbeschwerden, Periphere Ödeme, Schüttelfrost, Anomales Gefühl, Hämatom an der Gefässpunkturstelle
Einzelfälle: Reaktionen an der Injektionsstelle, Ermüdung, Unwohlsein
Untersuchungen
Gelegentlich: Haematokritabfall, auffällige Laborwerte, Blutgerinnungsfaktor VIII Abfall, Monozytenzahl erhöht (Der unerwartete Abfall des Blutgerinnungsfaktor VIII-Spiegels trat postoperativ (10. – 14. postoperativer Tag) bei einem Patienten unter kontinuierlicher ADVATE Infusion auf. Die Blutgerinnung wurde während der ganzen Zeit aufrechterhalten und sowohl die Faktor VIII-Spiegel im Plasma als auch die Clearance Rate zeigten am 15. postoperativen Tag wieder ausreichende Werte. Nach Beendigung der kontinuierlichen Infusion wurden Tests auf Faktor VIII-Inhibitoren durchgeführt und waren am Ende der Studie negativ.)
Einzelfälle: Erhöhte Anzahl eosinophiler Granulozyten
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Gelegentlich: Komplikationen nach der Behandlung, Blutung nach der Behandlung, Reaktion am Verabreichungsort
Beschreibung einzelner Nebenwirkungen
Inhibitorenentwicklung
In einer abgeschlossenen unkontrollierten Studie bildeten 16 von 45 (35.6%) nicht vorbehandelte Patienten mit schwerer Hämophilie A (FVIII < 1 %) nach mindestens 25 Tagen Exposition gegenüber FVIII FVIII-Hemmkörper: 7 Probanden (15,6 %) entwickelten hochtitrige, 9 (20 %) niedrigtitrige Inhibitoren, von denen einer als transienter Inhibitor eingestuft wurde.
Riskofaktoren für die Bildung von Inhibitoren waren in dieser Studie z.B. nicht-kaukasische Abstammung, häufiges Auftreten von Inhibitoren in der Familie und intensive Behandlung mit hohen Dosen an den ersten 20 Expositionstagen. Bei den 20 Probanden, die keines dieser erhöhten Risiken hatten, traten keine Inhibitoren auf.
Die Neoantigenität von ADVATE wurde an zuvor behandelten Patienten beurteilt. Bei klinischen Studien mit ADVATE an 276 Patienten, davon Kinder (Alter: ≤2 Jahre bis < 12 Jahre), Jugendliche (≤12 Jahre bis < 16 Jahre) und Erwachsene (Alter: ≥16 Jahre) mit diagnostizierter schwerer bis mittelschwerer Hämophilie A (FVIII ≤2 %) und vorheriger Exposition gegenüber Faktor-VIII-Konzentraten (≥150 Tage bei Erwachsenen und ältere Kinder und > 50 Tage bei Kinder mit einem Alter < 6 Jahre) zeigte nur ein Proband nach 26-tägiger ADVATE-Exposition einen niedrigen Inhibitortiter (2,4 B.E. im modifizierten Bethesda-Assay). Nachfolgende Inhibitortests an diesem Probanden nach Ausschluss aus der Studie waren negativ.
Reaktionen auf herstellungsbedingte Rückstände
Die Immunantwort der Probanden auf Spuren von kontaminierenden Proteinen wurde durch die Untersuchung der Antikörpertiter gegen diese Proteine, Laborparameter und gemeldete Nebenwirkungen analysiert. Von den 182 behandelten, auf Antikörper gegen CHO-Zellprotein getesteten Probanden zeigten 3 in der linearen Regressionsanalyse einen statistisch signifikanten Aufwärtstrend der Titer. 4 dieser Patienten wiesen anhaltende Peaks oder vorübergehende Spitzen auf. 1 Proband zeigte sowohl einen statistisch signifikanten Aufwärtstrend als auch einen anhaltenden Peak des Antikörperspiegels gegen CHO-Zellprotein, ansonsten traten jedoch keinerlei Anzeichen oder Symptome auf, die auf eine allergische Reaktion oder eine Überempfindlichkeit hinwiesen. Von den 182 behandelten, auf Antikörper gegen murines IgG getesteten Probanden zeigten 10 in der linearen Regressionsanalyse einen statistisch signifikanten Aufwärtstrend der Titer. 2 der Patienten wiesen einen anhaltenden Peak oder eine vorübergehende Spitze auf. 1 Proband zeigte sowohl einen statistisch signifikanten Aufwärtstrend als auch einen anhaltenden Peak der Antikörperspiegel gegen murines IgG. Bei vier der Probanden wurde vereinzelt über das Auftreten von Urtikaria, Pruritus, Hautausschlag und erhöhte Anzahl eosinophiler Granulozyten bei mehreren wiederholten Produktexpositionen im Rahmen der Studie berichtet.
Überempfindlichkeitsreaktionen
Überempfindlichkeitsreaktionen vom allergischen Typ einschliesslich Anaphylaxie äussern sich in Schwindel, Parästhesien, Hautausschlag, Hitzegefühl, Schwellungen des Gesichts, Urtikaria und Juckreiz.
Kinder und Jugendliche
Ausser bei der Entwicklung von Inhibitoren bei zuvor unbehandelten pädiatrischen Patienten (PUPs) und katheterbedingten Komplikationen wurden in den klinischen Studien keine altersspezifischen Unterschiede bei den ADRs beobachtet.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Es wurden keine Fälle von Überdosierung berichtet.
Eigenschaften/Wirkungen
ATC-Code
B02BD02 (Gerinnungsfaktor VIII)
Der Faktor VIII / von-Willebrand-Faktor-Komplex besteht aus zwei Molekülen (Faktor VIII und von Willebrand Faktor) mit unterschiedlichen physiologischen Funktionen.
ADVATE enthält rekombinanten Blutgerinnungsfaktor VIII, ein Glycoprotein, mit einer dem menschlichen Faktor VIII vergleichbaren Aminosäuresequenz und mit posttranslationellen Modifikationen, die mit denen der aus Plasma hergestellten Produkte vergleichbar sind.
Der rekombinante Blutgerinnungsfaktor VIII wird aus genetisch veränderten Ovarialzellen des chinesischen Hamsters (CHO), die das menschliche Blutgerinnungsfaktor VIII-Gen enthalten, hergestellt. ADVATE enthält Spuren von murinem IgG, CHO-Zellprotein und rekombinantem von Willebrand-Faktor (siehe «Kontraindikationen»).
Die Aktivität (I.E.) wird unter Verwendung eines chromogenen Tests gegen einen hausinternen Standard, der auf den 8. WHO-Standard referenziert, ermittelt. Die spezifische Aktivität beträgt etwa 4520 – 11300 I.E./mg Protein.
ADVATE ist ein steriles, pyrogenfreies und lyophilisiertes Präparat, das keine Konservierungsstoffe, tierische oder humane Zusatzstoffe enthält.
ADVATE ist ein Glycoprotein, das aus 2332 Aminosäuren mit einem Molekulargewicht von ca. 280 kD besteht. Wird einem Hämophilie A Patienten Faktor VIII injiziert, so bindet sich dieser im Blutkreislauf an den von Willebrand-Faktor. Der aktivierte Faktor VIII wirkt als Cofaktor für den aktivierten Faktor IX und beschleunigt die Bildung von aktiviertem Faktor X aus Faktor X. Der aktivierte Faktor X wandelt Prothrombin in Thrombin um. Dieses setzt dann Fibrin aus Fibrinogen frei, und die Gerinnselbildung kann erfolgen.
Hämophilie A ist eine geschlechtsgebundene erbliche Störung der Blutgerinnung aufgrund erniedrigter Faktor VIII Spiegel. Dies führt, entweder spontan oder in Folge unfallbedingter oder chirurgischer Traumata zu starken Blutungen in Gelenken, Muskeln oder inneren Organen. Durch die Substitutionstherapie werden die Faktor VIII-Plasmaspiegel erhöht, wodurch eine vorübergehende Korrektur des Faktor VIII-Mangels und der Blutungsneigung erfolgt.
Hämostatische Wirksamkeit
In der Schlüsselstudie der Phase II/III wurden insgesamt 107 mit FVIII vorbehandelte Patienten (mindestens 150 Expositionstage mit verschiedenen aus Plasma gewonnenen und rekombinanten FVIII-Präparaten) mit schwerer oder mittelschwerer Hämophilie A im Alter von mehr als 10 Jahren eingeschlossen und prophylaktisch behandelt. Unter einer Standardprophylaxe (25-40 IU/kg, 3-4x/Woche) zeigte sich eine jährliche Blutungsrate von 4.1 (spontan) respektive 4.7 (traumabedingt). Bei Therapiecompliance betrug die jährliche Spontanblutungsrate 3.3 (vs. 5.2), die traumabedingte Blutungsrate 3.4 (vs. 10). Insgesamt wurden 510 Blutungsepisoden mit ADVATE behandelt. Die hämostatische Wirksamkeit wurde bei 86% (439/510) als ausgezeichnet oder gut bewertet. Insgesamt 93% (473) der Blutungsepisoden wurden mit 1-2 Injektionen behandelt. Bei einer mittleren Expositionsdauer von 117 Tagen wurde ein transienter Low-Titer-Inhibitor (nach 26 Expositionstagen) festgestellt.
In der Phase II/III Fortsetzungsstudie mit 82 Studienteilnehmern, die die Schlüsselstudie abgeschlossen hatten, traten bei 70 von 81 Patienten 837 Blutungsepisoden auf. Die hämostatische Wirksamkeit von ADVATE wurde bei 673 Blutungen (80.4%) mit ausgezeichnet oder gut bewertet. 23 Blutungsepisoden waren nicht auswertbar (keine Angabe bzw. keine Behandlung notwendig). Bei 737 Blutungsepisoden (88%) genügten 1-2 Injektionen. Unter der Standardprophylaxe (n = 54, mindestens 1 Infusion) zeigte sich eine jährliche Blutungsrate von 1.74 (spontan) respektive 3 (traumabedingt); unter modifizierter Prophylaxe (n = 53, mind. 1 Infusion) betrug die jährliche Blutungsrate 1.45 (spontan) respektive 2 (traumabedingt). Die jährliche Blutungsrate bei «on-demand» Regime (n = 9) betrug 18.47. Wie in der Schlüsselstudie war die Blutungsrate bei Compliance niedriger als bei Noncompliance. Bei einer mittleren Expositionsdauer von 246 Tagen wurden keine Inhibitoren festgestellt.
In einer Studie mit 53 vorbehandelten Kindern (mindestens 50 Expositionstage mit verschiedenen aus Plasma gewonnenen und rekombinanten FVIII-Präparaten) unter 6 Jahren (24 davon < 3 J.) wurden 430 Blutungsepisoden in 47 Kindern erfasst. 57 (13.3%) dieser Episoden bedurften keiner Infusion; bei 345 der behandelten Blutungen (93.8%) wurde die Wirksamkeit von ADVATE als ausgezeichnet oder gut bewertet, bei 18 (4.9%) als mässig und bei 5 (1.4%) Episoden liegen keine Angaben vor.
Unter einer Standard-Prophylaxe (n=21; 25-50 IU/kg, 3-4x/Wo) respektive einer modifizierten Prophylaxe (n=37) zeigte sich eine jährliche Blutungsrate um 4 (Median) im Vergleich zu einem Regime «on demand» (n=5) mit um 24 Blutungen (Median). Bei 89% der 368 behandelten Blutungsepisoden genügten 1 oder 2 Injektionen (Dauer ≤5 min) zur Erreichung der Hämostase. Auch bei 7 meist kleineren operativen Eingriffen bei 7 Patienten war die Wirksamkeit intraoperativ und postoperativ zufriedenstellend. Bei einer mittleren Expositionsdauer von 156 Tagen wurde bei keinem dieser 53 behandelten Kindern ein Inhibitor festgestellt.
Pharmakokinetik
Alle Pharmakokinetikstudien mit ADVATE wurden an Patienten mit schwerer oder mittelschwerer Hämophilie A (Faktor-VIII-Aktivität ≤2%) durchgeführt. Die pharmakokinetischen Parameter stammen von insgesamt 260 Patienten und sind in untenstehender Tabelle aufgelistet.
|
Zusammenfassung der pharmakokinetischen Parameter von Advate pro Altersgruppe | ||||
|
PK-Parameter (Mittel ± Standardabweichung (SD) |
Kleinkinder (1 Monat bis < 2 Jahre) |
Kinder (2 bis < 12 Jahre) |
Jugendliche (12 bis < 16 Jahre) |
Erwachsene (16 Jahre und älter) |
|
Total AUC (I.E. * h/dl) |
1240 ± 330 |
1263 ± 471 |
1300 ± 469 |
1555 ± 508 |
|
Korrigierte Inkremental-Recovery bei Cmax (I.E./dl/I.E./kg)* |
2.1 ± 0.5 |
1.9 ± 0.5 |
2.1± 0.5 |
2.2 ± 0.6 |
|
Halbwertszeit (h) |
8.7 ± 1.4 |
10.2 ± 2.7 |
12.0 ± 2.9 |
13.0 ± 4.0 |
|
Cmax (I.E./dl) |
104 ± 27 |
97.2 ± 27.1 |
103 ± 25 |
112.4 ± 30.3 |
|
MRT (h) |
10.4 ± 2.5 |
12.9 ± 3.7 |
14.9 ± 4.6 |
16.4 ± 5.8 |
|
Vss (dl/kg) |
0.4 ± 0.1 |
0.6 ± 0.2 |
0.6 ± 0.1 |
0.6 ± 0.2 |
|
Clearance (ml/kg*h) |
4.3 ± 1.0 |
4.5 ± 1.5 |
4.2 ± 1.2 |
3.6 ± 1.2 |
*Berechnet als (Cmax – baseline Faktor VIII) geteilt durch die Dosis in IU/kg, bei der Cmax die maximale post-Faktor VIII-Infusion Messung ist
Kinetik spezieller Patientengruppen
Kinder und Jugendliche
Es wurden keine Unterschiede der pharmakokinetischen Parameter zwischen den Altersgruppen bei Erwachsenen (16 Jahre und älter verglichen mit 18 Jahre und älteren) beobachtet. Zwischen den Kindern (2 bis <12 Jahre), hatten ältere Kinder (5 bis < 12 Jahre ) höhere Werte als jüngere Kinder (2 bis < 5 Jahre) bei den pharmakokinetischen Parametern total AUC, inkremental Recovery bei Cmax, t½, Cmax und mittlere Residenzzeit. Der pharmakokinetische Parameter Vss war für beide Kinder-Subgruppen vergleichbar, und CI war tiefer bei älteren Kindern (5 bis < 12 Jahre) als bei jüngeren Kindern (2 bis < 5 Jahre).
Die korrigierte Recovery sowie die Halbwertszeit waren um ca. 20% niedriger als bei Erwachsenen.
Derzeit gibt es keine Daten zur Pharmakokinetik von ADVATE bei zuvor unbehandelten Patienten.
Präklinische Daten
Toxikologische Untersuchungen an Ratten und Kaninchen haben bei Dosen, die die maximale humantherapeutische deutlich überschritten, keine behandlungsabhängigen Effekte gezeigt. Nach peri- oder intravenöser Verabreichung von ADVATE an Kaninchen zeigten sich keine lokalen Reaktionen.
Da die ausgedehnte klinische Erfahrung mit rekombinantem Faktor VIII keinen Hinweis auf ein tumorigenes oder mutagenes Potential erkennen liess, wurden Langzeituntersuchungen zur Bestimmung des kanzerogenen Potentials nicht durchgeführt.
Sonstige Hinweise
Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf ADVATE nicht mit anderen Arzneimitteln oder Lösungsmitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden (siehe «Lagerungshinweise»).
Haltbarkeit nach Anbruch
Nach Auflösen bei Raumtemperatur (15 – 25 °C) lagern und innerhalb 3 Stunden verabreichen.
Besondere Lagerungshinweise
Im Kühlschrank (2 – 8 °C) lagern. Nicht einfrieren.
ADVATE mit BAXJECT II Gerät: In der Originalverpackung vor Licht geschützt aufbewahren.
ADVATE mit BAXJECT III System: Den verschlossenen Blister in der Originalverpackung vor Licht geschützt aufbewahren.
Innerhalb des aufgedruckten Verfallsdatums darf das Produkt für den ambulanten Einsatz einmalig aus dem Kühlschrank entnommen werden und dann maximal 6 Monate vor Gebrauch unterhalb 25 °C gelagert werden. In diesem Fall sollte das Datum der Entnahme aus dem Kühlschrank auf der Originalpackung vermerkt werden. Nach Ablauf der 6-Monatsfrist darf ADVATE nicht erneut im Kühlschrank gelagert werden, sondern ist zu vernichten.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Das Präparat wird nach dem Auflösen intravenös verabreicht. Restmengen sachgemäss entsorgen.
Die rekonstituierte Lösung ist vor der Verabreichung immer auf Schwebeteilchen oder Verfärbung zu überprüfen. Nur klare, farblose Lösungen verwenden.
ADVATE wird entweder mit einem BAXJECT II Gerät zur Rekonstitution oder mit einem gebrauchsfertigem BAXJECT III System in einem verschlossenen Blister (das Lyophilisat und das Wasser für Injektionszwecke sind vormontiert mit einem System zur Rekonstitution) geliefert.
Rekonstitution mit dem BAXJECT II Gerät
Zur Rekonstitution nur das beigepackte sterilisierte Wasser für Injektionszwecke und das beigepackte Gerät zur Rekonstitution verwenden.
Das BAXJECT II Gerät nicht verwenden, wenn seine sterile Barriere oder seine Verpackung beschädigt ist.
Auflösen: Auf aseptische Arbeitsweise achten.
1.ADVATE (Lyophilisat) und das Lösungsmittel (Aqua ad iniectabilia) auf 15 – 25 °C bringen.
2.Hände sorgfältig mit Seife und warmen Wasser waschen.
3.Schutzkappen von den Fläschchen mit Lyophilisat und Aqua ad iniectabilia entfernen.
4.Gummistopfen mit Alkoholtupfern reinigen. Die Durchstechflaschen auf eine ebene Oberfläche stellen.
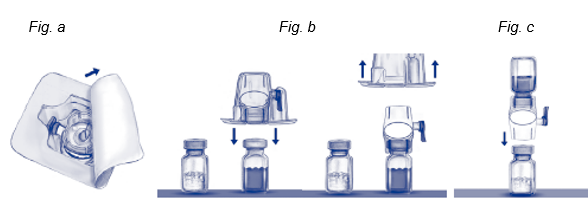
5.Die Verpackung des BAXJECTs II öffnen, indem die Schutzfolie abgezogen wird, ohne dabei den Packungsinhalt zu berühren (Fig. a).
6.Das Gerät zur Rekonstitition nicht aus der Verpackung nehmen. Die Öffnung nach unten drehen und den durchsichtigen Plastikdorn durch den Gummistopfen des Lösungsmittelfläschchens drücken. Nun die Verpackung vom BAXJECT II abnehmen (Fig. b). Die blaue Schutzkappe nicht vom BAXJECT II entfernen.
7.Das System, bestehend aus dem BAXJECT II und dem Lösungsmittelfläschchen, nun wenden, so dass sich das Lösungsmittelfläschchen oben befindet.
8.Den weissen Dorn des BAXJECTs II durch den Gummistopfen des ADVATE-Fläschchens drücken. Durch das Vakuum wird das Lösungsmittel in das ADVATE-Fläschchen gezogen (Fig. c).
9.Vorsichtig schwenken bis ADVATE vollständig gelöst ist, da sonst wirksame Substanz durch den Filter im BAXJECT II zurückgehalten wird. Das Präparat löst sich schnell auf (normalerweise in weniger als 1 Minute). Nach der Rekonstitition sollte die Lösung klar, farblos und frei von fremden Partikeln sein.
Rekonstitution mit dem BAXJECT III System
Nicht verwenden, falls die Schutzfolie des Blisters nicht vollständig versiegelt ist.
1.Der verschlossene Blister (enthält Fläschchen mit Lyophilisat und Aqua ad iniectabilia vormontiert mit einem System zur Rekonstitution) auf 15 – 25 °C bringen.
2.Hände sorgfältig mit Seife und warmen Wasser waschen.
3.Die ADVATE Blisterpackung öffnen, indem die Schutzfolie abgezogen wird. Das BAXJECT III System aus dem Blister entnehmen.
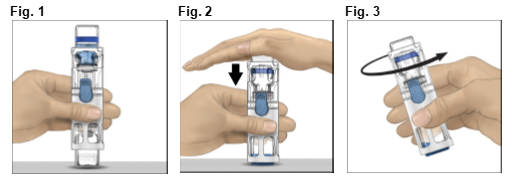
4.Das ADVATE auf eine flache Oberfläche stellen, so dass sich das Lösungsmittelfläschchen oben befindet (Fig. 1). Das Lösungsmittelfläschchen hat einen blauen Streifen. Die blaue Schutzkappe nicht entfernen, bis dies in einem späteren Schritt angewiesen wird.
5.Während Sie mit einer Hand das ADVATE im BAXJECT III System halten, drücken Sie mit der anderen Hand fest auf das Lösungsmittelfläschchen, bis das System vollständig zusammengedrückt ist und das Lösungsmittel in das ADVATE Fläschchen hinunterfliesst (Fig. 2). Kippen Sie das System nicht um, bis der Transfer abgeschlossen ist.
6.Überprüfen Sie, ob der Transfer des Lösungsmittels abgeschlossen ist. Vorsichtig schwenken bis ADVATE vollständig gelöst ist, da sonst wirksame Substanz durch den Filter zurückgehalten wird. Das Präparat löst sich schnell auf (normalerweise in weniger als 1 Minute). Nach der Rekonstitition sollte die Lösung klar, farblos und frei von fremden Partikeln sein.
Verabreichung: Auf aseptische Arbeitsweise achten.
1.Die blaue Schutzkappe des BAXJECTs II/BAXJECTs III entfernen. KEINE LUFT IN DIE SPRITZE AUFZIEHEN: Die Spritze an den BAXJECT II/BAXJECT III anschliessen.
2.Das System umdrehen (mit dem ADVATE-Fläschchen nach oben). Die ADVATE-Lösung durch Zurückziehen des Kolbens in die Spritze aufziehen.
3.Die Spritze entfernen.
4.Das Perfusionsbesteck an die Spritze anschliessen und das Präparat intravenös injizieren. Es kann mit einer Geschwindigkeit von bis zu 10 ml/min verabreicht werden. Der Puls des Patienten sollte vor und während der Verabreichung von ADVATE gemessen werden. Eine deutliche Erhöhung der Pulsfrequenz kann durch Verlangsamen oder zeitweiliges Unterbrechen der Injektion meist sofort wieder gesenkt werden (siehe «Warnhinweise und Vorsichtsmassnahmen» und «Unerwünschte Wirkungen»).
Zulassungsnummer
56352 (Swissmedic)
Packungen
Packungen mit BAXJECT II Gerät
Jede Packung ADVATE enthält: 1 Durchstechflasche Lyophilisat, 1 Durchstechflasche mit Wasser für Injektionszwecke als Lösungsmittel, 1 BAXJECT II zur Auflösung, 1 Mini – Infusionsset, 1 sterile 10 ml Einmalspritze zur Verabreichung, 2 Alkoholtupfer und 2 Pflaster
Packungen mit BAXJECT III System
Jede Packung ADVATE enthält: ein gebrauchsfertiges BAXJECT III System in einem verschlossenen Blister (das Lyophilisat und das Wasser für Injektionszwecke sind vormontiert mit dem System zur Rekonstitution), 1 Mini – Infusionsset, 1 sterile 10 ml Einmalspritze zur Verabreichung, 2 Alkoholtupfer und 2 Pflaster
ADVATE 250 [B] Lyophilisat zur Injektion mit 250 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 2 ml Wasser für Injektionszwecke.
ADVATE 250 [B] Lyophilisat zur Injektion mit 250 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
ADVATE 500 [B] Lyophilisat zur Injektion mit 500 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 2 ml Wasser für Injektionszwecke.
ADVATE 500 [B] Lyophilisat zur Injektion mit 500 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
ADVATE 1000 [B] Lyophilisat zur Injektion mit 1000 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 2 ml Wasser für Injektionszwecke.
ADVATE 1000 [B] Lyophilisat zur Injektion mit 1000 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
ADVATE 1500 [B] Lyophilisat zur Injektion mit 1500 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 2 ml Wasser für Injektionszwecke.
ADVATE 1500 [B] Lyophilisat zur Injektion mit 1500 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
ADVATE 2000 [B] Lyophilisat zur Injektion mit 2000 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
ADVATE 3000 [B] Lyophilisat zur Injektion mit 3000 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
Zulassungsinhaberin
Takeda Pharma AG, 8152 Opfikon
Stand der Information
März 2024