Zusammensetzung
Wirkstoffe
Canagliflozin (als Canagliflozin-Hemihydrat).
Hilfsstoffe
Tablettenkern:
Laktose (39,3 mg pro 100 mg Filmtablette und 117,8 mg pro 300 mg Filmtablette), mikrokristalline Cellulose, Hydroxypropylcellulose, Croscarmellose-Natrium (kann aus gentechnisch veränderten Baumwollsamenkapseln hergestellt sein; 0,84 mg Natrium pro 100 mg Filmtablette und 2,52 mg Natrium pro 300 mg Filmtablette), Magnesiumstearat.
Filmüberzug:
Invokana 100 mg Filmtabletten: Polyvinylalkohol, Titandioxid (E171), Macrogol (3350), Talk, gelbes Eisenoxid (E172).
Invokana 300 mg Filmtabletten: Polyvinylalkohol, Titandioxid (E171), Macrogol (3350), Talk.
Indikationen/Anwendungsmöglichkeiten
Invokana ist in Ergänzung zu Diät und körperlicher Betätigung bei Erwachsenen (ab 18 Jahren) mit unzureichend kontrolliertem Diabetes mellitus Typ 2 indiziert:
·Als Monotherapie
·Als Add-on-Kombinationstherapie mit anderen blutzuckersenkenden Arzneimitteln (siehe «Klinische Wirksamkeit» für Ergebnisse zu den in klinischen Studien untersuchten Kombinationen).
Invokana ist indiziert zur Prävention kardiovaskulärer Ereignisse bei erwachsenen Patienten mit Typ-2 Diabetes mellitus und bereits manifester kardiovaskulärer Erkrankung (siehe «Klinische Wirksamkeit»: Kardiovaskuläre Ergebnisse im CANVAS-Programm).
Invokana ist indiziert zur Senkung des Risikos der Progression einer diabetischen Nierenerkrankung bei erwachsenen Patienten mit Typ-2 Diabetes mellitus und Albuminurie [ACR>300 mg/g] (siehe «Dosierung/Anwendung» und «Klinische Wirksamkeit»).
Dosierung/Anwendung
Die empfohlene Dosis von Invokana beträgt 100 mg einmal täglich. Sollte eine stärkere glykämische Kontrolle erforderlich sein, kann die Dosis auf 300 mg erhöht werden, jedoch nicht bei Patienten mit eingeschränkter Nierenfunktion (eGFR <60 ml/min/1,73 m2) oder einem Risiko für unerwünschte Wirkungen im Zusammenhang mit einem reduzierten intravaskulären Volumen (siehe «Warnhinweise und Vorsichtsmassnahmen»). Bei Patienten mit Anzeichen einer Volumendepletion wird vor Beginn einer Behandlung mit Invokana grundsätzlich eine Korrektur dieses Zustands empfohlen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Tabelle 1: Empfohlene Dosis in Abhängigkeit von der Nierenfunktion
Die blutzuckersenkende Wirkung von Invokana nimmt mit zunehmender Einschränkung der Nierenfunktion ab. Daher sollte rechtzeitig (insbesondere bei Patienten mit einer eGFR <45) die Kombination mit weiteren antihyperglykämischen Wirkstoffen erwogen werden.
|
eGFR (ml/min/1,73 m2) |
Gesamttagesdosis von Canagliflozin |
|
≥60 |
Start der Behandlung mit 100 mg einmal täglich vor der ersten Mahlzeit. Bei unzureichender Senkung der Plasmaglucose und gleichzeitig guter Verträglichkeit, kann die Dosis auf 300 mg erhöht werden. |
|
45 - <60 |
begrenzt auf 100 mg einmal täglich vor der ersten Mahlzeit. |
|
30 - <45 * | |
|
<30 |
Die Behandlung von Patienten mit Albuminurie (ACR >300 mg/g) kann mit 100 mg einmal täglich vor der ersten Mahlzeit fortgeführt werden (es liegen keine Daten zur Initiierung einer Behandlung bei dieser Population vor). |
* Behandlungsstart nur bei Patienten mit Albuminurie (ACR >300 mg/g).
Bei Anwendung von Invokana als Zusatztherapie zu Insulin oder einem Insulin-Sekretagogum (z.B. Sulfonylharnstoff) kann zur Verringerung des Risikos von Hypoglykämien eine Dosisreduktion des Insulins oder des Insulin-Sekretagogums erwogen werden (siehe «Interaktionen» und «Unerwünschte Wirkungen»).
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit leicht oder mässig eingeschränkter Leberfunktion ist keine Dosisanpassung nötig.
Invokana wurde bei Patienten mit schwer eingeschränkter Leberfunktion nicht untersucht. Die Anwendung bei diesen Patienten wird nicht empfohlen (siehe «Pharmakokinetik»).
Ältere Patienten
Die Nierenfunktion und das Risiko für eine Volumendepletion sollten berücksichtigt werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Invokana wurde bei Kindern und Jugendlichen unter 18 Jahren nicht gezeigt. Es sind keine Daten verfügbar.
Verspätete Dosisgabe
Wenn eine Dosis ausgelassen oder vergessen wird, sollte sie am selben Tag eingenommen werden, sobald es dem Patienten einfällt. Es sollte keine doppelte Dosis am selben oder an einem anderen Tag eingenommen werden.
Art der Anwendung
Invokana sollte vor der ersten Mahlzeit des Tages eingenommen werden. Die Filmtabletten müssen als Ganzes geschluckt werden.
Vorübergehende Unterbrechung bei Operationen
Invokana sollte wenn möglich mindestens 3 Tage vor grösseren Operationen oder Eingriffen, die mit längerem Fasten verbunden sind, abgesetzt werden. Die Einnahme von Invokana kann wieder aufgenommen werden, wenn der Patient klinisch stabil ist und orale Nahrung zu sich nimmt (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Kontraindikationen
Überempfindlichkeit gegen Canagliflozin oder einen der Hilfsstoffe.
Warnhinweise und Vorsichtsmassnahmen
Allgemein
Die Sicherheit und Wirksamkeit von Invokana wurde nicht bei Patienten mit Diabetes mellitus Typ 1 untersucht. Bei diesen Patienten sollte Invokana nicht angewendet werden.
Diabetische Ketoazidose
Invokana sollte nicht zur Behandlung einer diabetischen Ketoazidose (DKA) angewendet werden.
In klinischen Studien bei Patienten mit Diabetes mellitus Typ 2 wurden für 0,2% (17/11'078) der Invokana-behandelten Studienteilnehmer Fälle von DKA als Ereignisse mit Hospitalisierung berichtet. Nach Markteinführung wurden schwerwiegende, teils lebensbedrohliche Fälle oder Fälle mit tödlichem Verlauf von DKA bei Patienten mit Diabetes mellitus Typ 2 unter Behandlung mit SGLT2-Inhibitoren, einschliesslich Invokana, berichtet. Auffällig war die Häufung von Fällen, bei denen der Blutzuckerspiegel atypischer Weise nur mässig erhöht war (euglykämische DKA mit Blutzuckerwerten unter 13,9 mmol/l [250 mg/dl]) (siehe «Unerwünschte Wirkungen»).
Deshalb muss bei der Behandlung von Patienten mit Diabetes mellitus Typ 2 mit Invokana eine DKA auch bei Fehlen einer ausgeprägten Hyperglykämie in Betracht gezogen werden, wenn die Patienten Symptome wie Übelkeit, Erbrechen, Appetitlosigkeit, Bauchschmerzen, übermässigen Durst, Atembeschwerden, Verwirrtheit, fruchtig riechender Atem, ungewöhnliche Erschöpfung oder Müdigkeit aufweisen. Bei Invokana-behandelten Patienten mit dieser Symptomatik sollte unabhängig vom Blutzuckerwert umgehend ein Test auf Ketonkörper durchgeführt werden. Die diabetische Ketoazidose kann bei einigen Patienten nach Absetzen von Invokana länger andauern, d.h. sie kann länger andauern als aufgrund der Plasmahalbwertszeit von Canagliflozin zu erwarten ist. Es wurde eine prolongierte Glukosurie zusammen mit einer anhaltenden DKA beobachtet. Die Urin-Glukoseausscheidung hält bis zu 3 Tage nach dem Absetzen von Invokana an; es gibt jedoch Post-Marketing-Berichte über DKA und Glukosurie, die länger als 6 Tage und teilweise bis zu 2 Wochen nach dem Absetzen von SGLT2-Hemmern andauern. Es ist ferner zu beachten, dass zusätzliche Faktoren (z.B. Insulinmangel) unabhängig von Canagliflozin zu einer prolongierten DKA beitragen können.
Bei Vorliegen einer DKA sollte die Behandlung von Patienten mit Diabetes mellitus Typ 2 mit Invokana sofort beendet werden. Bei Patienten mit Diabetes mellitus Typ 2, die wegen eines grösseren chirurgischen Eingriffs oder einer akuten schwerwiegenden Erkrankung stationär behandelt werden, oder für alle Eingriffe, die mit einem längeren Fasten verbunden sind, sollte die Behandlung mit Canagliflozin vorübergehend abgesetzt werden. Es wird empfohlen, diese Patienten im Hinblick auf das Vorliegen einer DKA zu überwachen. Die DKA kann bei einigen Patienten noch längere Zeit fortbestehen. Die Behandlung mit Invokana kann fortgesetzt werden, nachdem sich der Zustand des betreffenden Patienten wieder stabilisiert hat (siehe «Dosierung/Anwendung»).
Ein höheres Risiko für das Auftreten einer DKA unter der Behandlung mit einem SGLT2-Inhibitor (z.B. Invokana) könnte für Patienten bestehen, die sich gleichzeitig sehr kohlenhydratarm ernähren (zusätzliche Begünstigung von Stoffwechselaktivität unter Bildung von Ketonkörpern), für stark dehydrierte Patienten, für Patienten mit einer DKA in der Anamnese und für Patienten mit signifikant verringerter Beta-Zell-Funktion. Invokana sollte bei diesen Patienten mit Vorsicht angewendet werden. Vorsicht ist ebenfalls geboten bei der Reduzierung der Insulindosis von Patienten mit Diabetes mellitus Typ 2, die Invokana zusätzlich zu Insulin erhalten.
Amputation an den unteren Gliedmassen
In klinischen Langzeitstudien mit Invokana bei Patienten mit Diabetes mellitus Typ 2 und bestehender kardiovaskulärer Erkrankung (CVD) oder mindestens zwei Risikofaktoren für CVD war Invokana im Vergleich zum Placebo mit einem erhöhten Risiko für Amputationen der unteren Gliedmassen assoziiert (6,3 gegenüber 3,4 Ereignissen pro 1'000 Patientenjahre), diese Erhöhung betraf in erster Linie die Zehen und den Mittelfussbereich (siehe «Unerwünschte Wirkungen»). In einer klinischen Langzeitstudie mit Patienten mit Diabetes mellitus Typ 2 und chronischer Nierenerkrankung wurde bei Patienten, die mit 100 mg Canagliflozin behandelt wurden, im Hinblick auf das Risiko für Amputationen an den unteren Gliedmassen kein Unterschied zum Placebo beobachtet. Da kein zugrundeliegender Mechanismus identifiziert worden ist, sind Risikofaktoren für eine Amputation, abgesehen von allgemeinen Risikofaktoren, nicht bekannt.
Vor der Einleitung der Behandlung mit Invokana sind Faktoren in der Krankengeschichte des Patienten zu berücksichtigen, die das Amputationsrisiko unter Umständen erhöhen. Vorsichtshalber sollte erwogen werden, Patienten mit erhöhtem Amputationsrisiko aufmerksam zu überwachen und hinsichtlich der Bedeutung einer regelmässigen vorbeugenden Fusspflege und der Aufrechterhaltung einer ausreichenden Flüssigkeitszufuhr zu beraten. Wenn bei Patienten eine Problematik eintritt, die eine Amputation nach sich ziehen könnte, beispielsweise eine Hautulzeration, Infektion, Osteomyelitis oder Gangrän an einer unteren Extremität, sollte auch erwogen werden, die Behandlung mit Invokana zu beenden.
Anwendung bei Patienten mit eingeschränkter Nierenfunktion
Die glukose-senkende Wirksamkeit von Canagliflozin nimmt mit zunehmender Einschränkung der Nierenfunktion ab und ist bei mit milder bis schwerer Einschränkung (eGFR <45 ml/min/1,73 m2) unzureichend. Patienten mit milder bis schwerer Einschränkung der Nierenfunktion können aber von der nephroprotektiven Wirkung von Canagliflozin in einer Dosis 100 mg einmal täglich profitieren (siehe «Dosierung/Anwendung»).
Bei Patienten mit einer eGFR <60 ml/min/1,73 m2 oder einer CrCl <60 ml/min wurde eine höhere Inzidenz von unerwünschten Wirkungen, die mit einem reduzierten intravaskulären Volumen in Zusammenhang stehen (z.B. lageabhängiger Schwindel, orthostatische Hypotonie oder allgemeine Hypotonie) berichtet. Ausserdem wurden bei diesen Patienten häufiger erhöhte Kaliumwerte und stärkere Zunahmen der Kreatininwerte im Serum und der BUN-Werte berichtet (siehe «Unerwünschte Wirkungen»).
Deswegen sollte bei Patienten mit einer eGFR <60 ml/min/1,73 m2 oder einer CrCl <60 ml/min die Canagliflozin-Dosis auf 100 mg pro Tag begrenzt werden.
Diabetische Nierenerkrankung bei Patienten mit Diabetes mellitus Typ 2
Siehe Tabelle 1 für Dosierungsempfehlungen auf der Grundlage der geschätzten glomerulären Filtrationsrate (eGFR).
Unabhängig von der eGFR vor der Behandlung trat bei Patienten, die Canagliflozin erhielten, eine initiale Verringerung der eGFR auf, die sich danach mit der Zeit abschwächte (siehe «Unerwünschte Wirkungen» und «Pharmakokinetische Eigenschaften»).
Bei allen Patienten, die Canagliflozin einnehmen, wird die Überwachung der Nierenfunktion wie folgt empfohlen:
·vor Beginn der Behandlung mit Invokana und danach mindestens jährlich (siehe «Dosierung/Anwendung», «Unerwünschte Wirkungen», «Eigenschaften/Wirkungen» und «Pharmakokinetik»),
·vor Beginn einer Behandlung mit gleichzeitig verabreichten Arzneimitteln, die die Nierenfunktion beeinträchtigen können, und regelmässig danach,
·mindestens 2 Mal bis 4 Mal pro Jahr, wenn die Nierenfunktion sich soweit verringert, dass eine mittelstarke bis starke Beeinträchtigung vorliegt.
Anwendung bei Patienten mit einem Risiko für unerwünschte Ereignisse im Zusammenhang mit einer Volumendepletion
Aufgrund seines Wirkungsmechanismus induziert Invokana über eine Steigerung der Exkretion von Glukose im Urin eine osmotische Diurese, die das intravaskuläre Volumen reduzieren und den Blutdruck senken kann (siehe «Eigenschaften/Wirkungen»). Zu den Patienten, die am anfälligsten für unerwünschte Wirkungen waren, die mit einem reduzierten intravaskulären Volumen in Zusammenhang stehen (z.B. lageabhängiger Schwindel, orthostatische Hypotonie oder allgemeine Hypotonie), zählen Patienten unter Schleifendiuretika, unter ACE-Hemmern oder Angiotensin-Rezeptor-Blockern (ARB), Patienten mit mässig eingeschränkter Nierenfunktion sowie ältere Patienten, z.B. ≥75 Jahre (siehe «Dosierung/Anwendung» und «Unerwünschte Wirkungen»).
In 8 kontrollierten klinischen Studien mit Invokana wurden dosisabhängig (100 mg: 2,3%, 300 mg: 3,4%, Kontrollbehandlungen: 1,5%) unerwünschte Wirkungen im Zusammenhang mit einer Reduktion des intravaskulären Volumens beobachtet, welche sich am häufigsten in den ersten drei Monaten der Behandlung ereigneten (siehe «Unerwünschte Wirkungen»). Aufgrund des reduzierten intravaskulären Volumens wurden innerhalb der ersten sechs Wochen der Behandlung mit Invokana im Allgemeinen geringe mittlere dosisabhängige Anstiege des Serumkreatinins und gleichzeitige Abnahmen der eGFR beobachtet. Bei Patienten, die wie oben beschrieben anfällig für stärkere Reduktionen des intravaskulären Volumens sind, wurden gelegentlich stärkere Reduktionen der eGFR (>30%) beobachtet, die sich im Verlauf meist wieder besserten und nur selten eine Unterbrechung der Behandlung mit Invokana erforderten (siehe «Unerwünschte Wirkungen»).
Patienten sollten angewiesen werden, Symptome eines reduzierten intravaskulären Volumens zu melden. Diese unerwünschten Wirkungen führten in seltenen Fällen zum Abbruch der Therapie mit Invokana und wurden oft unter fortgesetzter Therapie mit Invokana durch eine Anpassung der antihypertensiven Therapie (inklusive Diuretika) behandelt. Bei Patienten mit Volumendepletion wird vor Beginn einer Behandlung mit Invokana eine Korrektur dieses Zustands empfohlen.
Bei Patienten unter Invokana wird im Fall von interkurrenten Erkrankungen, die eine Volumendepletion verursachen können (z.B. gastrointestinale Erkrankungen), eine sorgfältige Überwachung des Volumenstatus einschliesslich Nierenfunktion und der Serumelektrolyte empfohlen. Bei Patienten, die unter der Therapie mit Invokana eine Volumendepletion entwickeln, kann eine zeitweilige Unterbrechung der Behandlung bis zur Korrektur des Zustandes in Erwägung gezogen werden. Wenn die Behandlung unterbrochen wird, sollte ein intensiveres Glukose-Monitoring in Betracht gezogen werden.
Bei Patienten mit bekannter ischämischer Herzkrankheit oder zerebrovaskulärer Erkrankung, für die eine zusätzliche Blutdrucksenkung ein erhöhtes Risiko darstellen kann, sollte vor Beginn der Behandlung mit Invokana eine Anpassung der begleitenden diätetischen Massnahmen (z.B. Salzaufnahme) und/oder der antihypertensiven Behandlung in Betracht gezogen werden.
Ältere Patienten
Ältere Patienten können ein erhöhtes Risiko für eine Volumendepletion aufweisen, werden häufiger mit Diuretika behandelt und weisen oft eine eingeschränkte Nierenfunktion auf. Bei Patienten ≥75 Jahre wurde eine höhere Inzidenz von unerwünschten Wirkungen im Zusammenhang mit einer Reduktion des intravaskulären Volumens (z.B. lageabhängiger Schwindel, orthostatische Hypotonie, allgemeine Hypotonie) berichtet. Ausserdem wurden bei diesen Patienten stärkere Abnahmen der eGFR berichtet (siehe «Dosierung/Anwendung» und «Unerwünschte Wirkungen»).
Hyperkaliämie
Bei Patienten mit mässiger Niereninsuffizienz, die Arzneimittel mit Einfluss auf die Kaliumausscheidung, beispielsweise Kalium-sparende Diuretika, oder Arzneimittel mit Einfluss auf das Renin-Angiotensin-Aldosteronsystem einnehmen, ist die Wahrscheinlichkeit der Entwicklung einer Hyperkaliämie unter einer Behandlung mit Invokana erhöht.
Die Kaliumspiegel sind nach dem Beginn einer Invokana-Therapie bei Patienten mit eingeschränkter Nierenfunktion und bei Patienten, die aufgrund ihrer Medikation oder anderer Erkrankungen für Hyperkaliämie prädisponiert sind, regelmässig zu überprüfen.
Laktoseintoleranz
Die Tabletten enthalten Laktose. Patienten mit hereditärer Galaktose-Unverträglichkeit, Lapp-Laktasemangel oder Glukose-Galaktose-Malabsorptionssyndrom dürfen nicht mit Invokana behandelt werden.
Hypoglykämie bei der Kombinationsbehandlung mit anderen antihyperglykämischen Wirkstoffen
Insulin und Insulin-Sekretagoga (z.B. Sulfonylharnstoffe) können zu Hypoglykämien führen. Wenn Invokana als zusätzliche Therapie zu Insulin oder einem Insulin-Sekretagogum (z.B. einem Sulfonylharnstoff) verwendet wird, liegt eine höhere Inzidenz von Hypoglykämien vor als unter Placebo plus Insulin oder Insulinsekretagogum.
Aus diesem Grund sollte zur Verringerung des Risikos von Hypoglykämien eine Dosisreduktion von Insulin oder des Insulin-Sekretagogums in Erwägung gezogen werden (siehe «Dosierung/Anwendung» und «Unerwünschte Wirkungen»).
Nekrotisierende Fasziitis des Perineums (Fournier-Gangrän)
Aus der Arzneimittelüberwachung nach der Markteinführung wurden bei Patienten mit Diabetes Mellitus, die SGLT2-Inhibitoren, einschliesslich Invokana, erhielten, Fälle von nekrotisierender Fasziitis des Perineums (Fournier-Gangrän) berichtet, eine sehr seltene, aber schwerwiegende und potenziell lebensbedrohliche nekrotisierende Infektion, die ein dringendes chirurgisches Eingreifen erfordert. Es waren sowohl Frauen als auch Männer betroffen. Die schwerwiegenden Ausgänge umfassten Krankenhausaufenthalte, mehrere Operationen und Todesfälle.
Patienten, die mit Invokana behandelt werden und Schmerzen oder Druckempfindlichkeit, Erythem oder Schwellungen im Genital- oder perinealen Bereich sowie Fieber oder Unwohlsein aufweisen, sollten auf nekrotisierende Fasziitis untersucht werden. Liegt ein entsprechender Verdacht vor, soll unverzüglich eine Behandlung mit Breitbandantibiotika und gegebenenfalls mit einem chirurgischen Debridement eingeleitet werden. Invokana sollte abgesetzt und durch eine geeignete Therapiealternative ersetzt werden, dabei sollte der Blutzuckerspiegel engmaschig überwacht werden.
Genitale Pilzinfektionen
Wegen des Wirkmechanismus mit Arzneimittel-bedingter Glykosurie erhöht Invokana das Risiko für genitale mykotische Infektionen. In klinischen Studien wurde bei weiblichen Patienten über vulvovaginale Candidiasis und bei männlichen Patienten über Balanitis oder Balanoposthitis berichtet (siehe «Unerwünschte Wirkungen»). Bei männlichen und weiblichen Patienten mit Genitalmykosen in der Anamnese war die Wahrscheinlichkeit, eine Infektion zu entwickeln, höher. Eine Balanitis oder Balanoposthitis trat vorwiegend bei nicht beschnittenen männlichen Patienten auf. In seltenen Fällen wurde über eine Phimose berichtet und gelegentlich wurden Beschneidungen durchgeführt. Die Mehrzahl der Genitalmykosen wurden mit ärztlich verschriebenen oder selbst gekauften topischen antifungalen Therapien unter Fortsetzung der Therapie mit Invokana behandelt.
Bei Patienten und Patientinnen mit rezidivierenden genitalen Pilzinfektionen muss im Rahmen einer individuellen Nutzen-Risiko-Abwägung erwogen werden, ob eine Fortsetzung der Behandlung mit Invokana angemessen ist.
Überempfindlichkeitsreaktionen
Teilweise als schwerwiegend taxierte Überempfindlichkeitsreaktionen (z.B. generalisierte Urtikaria) wurden unter Invokana beobachtet. Diese Reaktionen traten in der Regel innerhalb von wenigen Stunden oder Tagen nach Beginn der Behandlung auf. Wenn solche Reaktionen auftreten, sollte die Behandlung mit Invokana abgebrochen werden (siehe «Kontraindikationen»).
Natrium
Invokana enthält weniger als 1 mmol (23 mg) Natrium pro Tablette, d.h. sie ist nahezu natriumfrei.
Interaktionen
Pharmakokinetische Interaktionen
In-vitro-Studien
Canagliflozin wird vorrangig über eine durch UDP-Glucuronosyltransferase 1A9 (UGT1A9) und 2B4 (UGT2B4) vermittelte Glucuronid-Konjugation metabolisiert.
In In-vitro-Studien mit humanen Lebermikrosomen wurden die CYP450 Isoenzyme 3A4, 2B6, 2C8 und 2C9 durch Canagliflozin leicht gehemmt. In einer klinischen Studie wurde jedoch keine klinisch relevante Interaktion beobachtet. Andere CYP450-Isoenzyme wurden durch Canagliflozin nicht induziert oder gehemmt. Daher wird nicht erwartet, dass Canagliflozin die metabolische Clearance von gleichzeitig verabreichten Arzneimitteln, die durch diese Enzyme metabolisiert werden, verändert.
Canagliflozin ist ein Substrat des P-Glycoproteins (P-gp) und ein schwacher Inhibitor des P-gp.
Canagliflozin unterliegt einem minimalen oxidativen Metabolismus (siehe «Pharmakokinetik»). Daher sind klinisch relevante Wirkungen anderer Arzneimittel auf die Pharmakokinetik von Canagliflozin mittels des Cytochrom-P450-Systems unwahrscheinlich.
In-vivo-Daten
Wirkung anderer Arzneimittel auf Canagliflozin
In klinischen Studien wurden die Wirkungen anderer Arzneimittel auf Canagliflozin beurteilt. Ciclosporin, Hydrochlorothiazid, orale Kontrazeptiva (Ethinylestradiol und Levonorgestrel), Metformin und Probenecid hatten keine klinisch relevante Auswirkung auf die Pharmakokinetik von Canagliflozin.
Rifampicin: Die gleichzeitige Verabreichung von Rifampicin, einem nichtselektiven Induktor mehrerer UGT-Enzyme und Arzneimitteltransporter inklusive UGT1A9, UGT2B4, P-gp und MRP2, verminderte die Exposition gegenüber Canagliflozin. Diese verminderte Exposition gegenüber Canagliflozin kann die Wirksamkeit reduzieren. Wenn ein Induktor dieser UGT und Arzneimitteltransportsysteme (z.B. Rifampicin, Phenytoin, Barbiturate, Phenobarbital, Ritonavir, Carbamazepin, Johanniskraut (Hypericum perforatum)) gleichzeitig mit Invokana verabreicht werden muss, sollte das HbA1c bei Patienten, die mit Invokana 100 mg einmal täglich behandelt werden, überwacht und eine Erhöhung der Dosis auf 300 mg einmal täglich in Erwägung gezogen werden. Bei Patienten mit einem eGFR-Wert von 45 ml/min/1,73 m2 bis <60 ml/min/1,73 m2 oder einem CrCl-Wert von 45 ml/min bis <60 ml/min die unter einer Behandlung mit einem UGT-Enzyminduktor stehen, sollten andere antihyperglykämische Therapien erwogen werden.
Tabelle 2: Auswirkungen von gleichzeitig verabreichten Arzneimitteln auf die systemische Verfügbarkeit von Canagliflozin
|
Gleichzeitig verabreichtes |
Dosis des gleichzeitig verabreichten Arzneimittels1 |
Dosis von Canagliflozin1 |
Verhältnis der geometrischen Mittelwerte | |
|
AUC2 |
Cmax | |||
|
Eventuell Dosisanpassung notwendig (siehe vorstehende Angaben im Text): | ||||
|
Rifampicin |
600 mg einmal täglich während 8 Tagen |
300 mg |
0,49 |
0,72 |
|
Keine Dosisanpassung von Invokana nötig für: | ||||
|
Ciclosporin |
400 mg |
300 mg einmal täglich während 8 Tagen |
1,23 |
1,01 |
|
Ethinylestradiol und Levonorgestrel |
0,03 mg Ethinylestradiol und 0,15 mg Levonorgestrel |
200 mg einmal täglich während 6 Tagen |
0,91 |
0,92 |
|
Hydrochlorothiazid |
25 mg einmal täglich während 35 Tagen |
300 mg einmal täglich während 7 Tagen |
1,12 |
1,15 |
|
Metformin |
2000 mg |
300 mg einmal täglich während 8 Tagen |
1,10 |
1,05 |
|
Probenecid |
500 mg zweimal täglich während 3 Tagen |
300 mg einmal täglich während 17 Tagen |
1,21 |
1,13 |
1 Einzeldosis, wenn nicht anders angegeben.
2 AUCinf für Arzneimittel, die als Einzeldosis verabreicht werden, und AUC24h für Arzneimittel, die in mehreren Dosen verabreicht werden.
Wirkung von Canagliflozin auf andere Arzneimittel
In Interaktionsstudien mit gesunden Probanden hatte Canagliflozin im Steady-State keine klinisch relevanten Auswirkungen auf die Pharmakokinetik von Metformin, oralen Kontrazeptiva (Ethinylestradiol und Levonorgestrel), Glibenclamid, Simvastatin, Paracetamol, Hydrochlorothiazid.
Digoxin: Die Kombination von Canagliflozin 300 mg einmal täglich während 7 Tagen mit einer Einzeldosis Digoxin 0,5 mg gefolgt von 0,25 mg täglich während 6 Tagen resultierte in einem Anstieg der AUC von Digoxin um 20% und der Cmax von Digoxin um 36%, möglicherweise aufgrund einer Interaktion auf der Ebene von Pgp. Patienten, die Digoxin oder andere Herzglykoside (z.B. Digitoxin) einnehmen, sollten angemessen überwacht werden.
Lithium: Die gleichzeitige Anwendung eines SGLT2-Inhibitors mit Lithium kann die Serum-Lithium-Konzentration verringern. Während der Initiierung der Behandlung mit Canagliflozin und bei Dosierungsänderungen ist die Serum-Lithium-Konzentration häufiger zu überwachen.
Tabelle 3: Auswirkung von Canagliflozin auf die systemische Verfügbarkeit gegenüber gleichzeitig verabreichten Arzneimitteln
|
Gleichzeitig verabreichtes |
Dosis des gleichzeitig verabreichten Arzneimittels1 |
Dosis von Canagliflozin1 |
Verhältnis der geometrischen Mittelwerte | ||
|
|
AUC2 |
Cmax | |||
|
Ad klinische Relevanz siehe Angaben im Text: | |||||
|
Digoxin |
0,5 mg einmal täglich am ersten Tag gefolgt von 0,25 mg einmal täglich während 6 Tagen |
300 mg einmal täglich während 7 Tagen |
Digoxin |
1,20 |
1,36 |
|
Keine Dosisanpassung des gleichzeitig verabreichten Arzneimittels nötig für: | |||||
|
Ethinylestradiol und Levonorgestrel |
0,03 mg Ethinylestradiol und 0,15 mg Levonorgestrel |
200 mg einmal täglich während 6 Tagen |
Ethinylestradiol |
1,07 |
1,22 |
|
Levonorgestrel |
1,06 |
1,22 | |||
|
Glibenclamid |
1,25 mg |
200 mg einmal täglich während 6 Tagen |
Glibenclamid |
1,02 |
0,93 |
|
3-Cis-hydroxy-Glibenclamid |
1,01 |
0,99 | |||
|
4-Trans-hydroxy-Glibenclamid |
1,03 |
0,96 | |||
|
Hydrochlorothiazid |
25 mg einmal täglich während 35 Tagen |
300 mg einmal täglich während 7 Tagen |
Hydrochlorothiazid |
0,99 |
0,94 |
|
Metformin |
2000 mg |
300 mg einmal täglich während 8 Tagen |
Metformin |
1,20 |
1,06 |
|
Paracetamol |
1000 mg |
300 mg zweimal täglich während 25 Tagen |
Paracetamol |
1,063 |
1,00 |
|
Simvastatin |
40 mg |
300 mg einmal täglich während 7 Tagen |
Simvastatin |
1,12 |
1,09 |
|
Simvastatinsäure |
1,18 |
1,26 | |||
|
Warfarin |
30 mg |
300 mg einmal täglich während 12 Tagen |
(R)-Warfarin |
1,01 |
1,03 |
|
(S)-Warfarin |
1,06 |
1,01 | |||
|
INR |
1,00 |
1,05 | |||
1 Einzeldosis, wenn nicht anders beschrieben.
2 AUCinf für Arzneimittel, die als Einzeldosis verabreicht werden, und AUC24h für Arzneimittel, die in mehreren Dosen verabreicht werden.
3 AUC0-12h.
Schwangerschaft, Stillzeit
Schwangerschaft
Unkontrollierter Diabetes während der Schwangerschaft (gestationsbedingt oder vorbestehend) ist mit einem erhöhten Risiko für kongenitale Missbildungen und perinatale Mortalität verbunden.
Es liegen nur begrenzte Erfahrungen mit der Anwendung von Invokana bei Schwangeren vor. Tierexperimentelle Studien mit Canagliflozin allein ergaben Hinweise auf eine Reproduktionstoxizität (siehe «Präklinische Daten»). Invokana sollte während einer Schwangerschaft nicht angewendet werden. Beim Eintritt einer Schwangerschaft sollte Invokana abgesetzt werden.
Stillzeit
Es ist nicht bekannt, ob Canagliflozin und/oder seine Metaboliten in die Muttermilch ausgeschieden werden. Tierexperimentelle Studien haben gezeigt, dass Canagliflozin/Metaboliten in die Muttermilch übergehen (siehe «Präklinische Daten»). Die zur Verfügung stehenden limitierten Daten schliessen das theoretische Risiko von Hypoglykämien beim gestillten Kind nicht aus. Die Entscheidung, Invokana abzusetzen oder abzustillen, sollte unter Berücksichtigung des Nutzens des Medikaments für die Mutter und des potentiellen Risikos für das Kind getroffen werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Es wurden keine entsprechenden Studien durchgeführt.
Patienten sollten jedoch auf das Risiko einer Hypoglykämie hingewiesen werden, wenn Invokana als zusätzliche Therapie zu Insulin oder einem Insulin-Sekretagogum verabreicht wird. Zudem sollten sie auf das erhöhte Risiko unerwünschter Wirkungen in Zusammenhang mit reduziertem intravaskulärem Volumen, wie z.B. lageabhängiger Schwindel (siehe «Dosierung/Anwendung», «Warnhinweise und Sicherheitsmassnahmen» und «Unerwünschte Wirkungen») aufmerksam gemacht werden.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
Die Sicherheit von Canagliflozin wurde an 22'645 Patienten mit Diabetes mellitus Typ 2 in 15 doppelblinden, kontrollierten klinischen Phase-3 und Phase-4 Studien untersucht. Diese beinhalteten 13'278 Patienten, die mit Canagliflozin, und 9'367 Patienten, die mit einem Vergleichspräparat behandelt wurden. In zwei gezielten kardiovaskulären Studien wurden insgesamt 10'134 Patienten über eine mittlere Behandlungsdauer von 149 Wochen behandelt (in CANVAS 223 Wochen und in CANVAS-R 94 Wochen), und in 12 doppelblinden, kontrollierten, klinischen Studien der Phasen 3 und 4 wurden 8'114 Patienten über eine mittlere Behandlungsdauer von 49 Wochen behandelt. In einer Studie, in der speziell die nierenbezogenen Therapieergebnisse untersucht wurden, betrug die mittlere Dauer der Exposition von insgesamt 4'397 Patienten mit Diabetes mellitus Typ 2 und diabetischer Nierenerkrankung 115 Wochen.
Die primäre Beurteilung der Sicherheit und Verträglichkeit wurde mittels einer gepoolten Analyse (n=2'313) der gepoolten Daten von vier 26-wöchigen, Placebo-kontrollierten, klinischen Studien durchgeführt. Die am häufigsten berichteten unerwünschten Wirkungen, die während der Behandlung auftraten, waren Hypoglykämie in Kombination mit Insulin oder Sulfonylharnstoffen, vulvovaginale Candidiasis, Harnwegsinfektionen und Polyurie oder Pollakisurie. Unerwünschte Wirkungen, die zu einem Behandlungsabbruch bei ≥0,5% aller in diesen Studien mit Invokana behandelten Patienten führten, waren vulvovaginale Candidiasis (0,7% aller Frauen) und Balanitis oder Balanoposthitis (0,5% aller Männer).
Auflistung der unerwünschten Wirkungen
Nachfolgend sind die unerwünschten Wirkungen aufgeführt, die in den oben beschriebenen gepoolten, Placebo- und aktiv-kontrollierten klinischen Studien auftraten. Die unten aufgeführten unerwünschten Wirkungen sind nach der Häufigkeit und nach Systemorganklassen (SOC) aufgelistet. Die Häufigkeitskategorien wurden in Anlehnung an folgende Konvention definiert: sehr häufig (≥1/10), häufig (≥1/100 bis <1/10), gelegentlich (≥1/1000 bis <1/100), selten (≥1/10'000 bis <1/1'000), sehr selten (<1/10'000), unbekannt (kann anhand der zur Verfügung stehenden Daten nicht abgeschätzt werden).
Häufigkeit unerwünschter Wirkungen (MedDRA) in Placebo- und aktiv-kontrollierten Studiena und aus Erfahrung nach Markteinführung
Infektionen und parasitäre Erkrankungen
Sehr häufig: Vulvovaginale Candidiasis**,f.
Häufig: Balanitis oder Balanoposthitis**,g, Harnwegsinfektione (Pyelonephritis und Urosepsis wurden nach Markteinführung berichtet).
Selten: Fournier-Gangrän (nekrotisierende Fasziitis des Perineums)i.
Erkrankungen des Blutes und des Lymphsystems
Häufig: Erhöhte Hämatokritwerte.
Erkrankungen des Immunsystems
Selten: Anaphylaktische Reaktioni.
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: Hypoglykämie in Kombination mit Insulin oder Sulfonylharnstoffen.
Häufig: Dyslipidämie.
Gelegentlich: Dehydrierung*, erhöhte Kaliumwerte im Blut, erhöhte Phosphatwerte im Blut.
Selten: Diabetische Ketoazidoseh.
Erkrankungen des Nervensystems
Gelegentlich: Lageabhängiger Schwindel*, Synkope*.
Gefässerkrankungen
Gelegentlich: Arterielle Hypotonie*, Orthostatische Hypotonie*.
Erkrankungen des Gastrointestinaltrakts
Häufig: Obstipation, Durstb, Nausea.
Erkrankungen der Haut und des Unterhautzellgewebes
Gelegentlich: Ausschlagc, Photosensibilität, Urtikaria.
Selten: Angioödemi.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Gelegentlich: Knochenfrakturen.
Erkrankungen der Nieren und Harnwege
Häufig: Polyurie oder Pollakisuried.
Gelegentlich: Erhöhte Kreatininwerte im Blut, erhöhte Harnstoffwerte im Blut, Nierenversagen (vorwiegend im Zusammenhang mit einem Volumenverlust).
Chirurgische und medizinische Eingriffe
Gelegentlich: Amputationen der unteren Gliedmassen (hauptsächlich der Zehen und des Mittelfusses), insbesondere bei Patienten mit hohem kardiovaskulärem Risiko.
* In Zusammenhang mit einem reduzierten intravasalen Volumen; siehe «Warnhinweise und Vorsichtsmassnahmen».
** Siehe «Warnhinweise und Vorsichtsmassnahmen».
a Die Sicherheitsdatenprofile aus den individuellen pivotalen Studien (einschliesslich Studien an Patienten mit mässig eingeschränkter Nierenfunktion, älteren Patienten und Patienten mit erhöhtem kardiovaskulärem und renalem Risiko) stimmten im Allgemeinen mit den oben aufgelisteten unerwünschten Wirkungen überein.
b «Durst» beinhaltet die Termini Durst, trockener Mund und Polydipsie.
c «Ausschlag» beinhaltet die Termini erythematöser Ausschlag, generalisierter Ausschlag, makulärer Ausschlag, makulopapulöser Ausschlag, papulöser Ausschlag, pruritischer Ausschlag, pustulöser Ausschlag und vesikulöser Ausschlag.
d «Polyurie oder Pollakisurie» beinhaltet die Termini Polyurie, Pollakisurie, Harndrang, Nykturie und erhöhte Harnmenge.
e «Harnwegsinfektion» beinhaltet die Termini Harnwegsinfektion, Zystitis, Niereninfektion und Urosepsis. Es bestand kein Ungleichgewicht zwischen Invokana 100 mg, Invokana 300 mg und Placebo in Bezug auf Niereninfektionen oder Urosepsis.
f «Vulvovaginale Candidiasis» beinhaltet die Termini vulvovaginale Candidiasis, vulvovaginale Pilzinfektion, Vulvovaginitis, vaginale Infektion, Vulvitis und genitale Pilzinfektion.
g «Balanitis oder Balanoposthitis» beinhaltet die Termini Balanitis, Balanoposthitis, Candida-Balanitis und genitale Pilzinfektion.
h Phase 3 und Phase 4 klinische Studien, inklusive Nicht-CANVAS/Nicht-CREDENCE Studien und CANVAS Programm und Spontanmeldungen
i Klinische Studien der Phasen 3 und 4, einschliesslich CANVAS- und CREDENCE-Programm
Beschreibung ausgewählter Nebenwirkungen
Diabetische Ketoazidose
In einer Langzeitstudie, in der speziell die nierenbezogenen Therapieergebnisse bei Patienten mit Diabetes mellitus Typ 2 und diabetischer Nierenerkrankung untersucht wurden, betrug die Rate der Inzidenz adjudizierter Fälle diabetischer Ketoazidose (DKA) 2,1 (0,5%, 12/2'200) pro 1'000 Patientennachbeobachtungsjahre bei Patienten, die mit Canagliflozin 100 mg behandelt wurden, und 0,3 (0,1%, 2/2'197) pro 1'000 Patientennachbeobachtungsjahre bei Patienten, die ein Placebo erhielten; von den 14 Patienten mit DKA wiesen 8 (7 in der Canagliflozin 100 mg und 1 in der Placebogruppe) vor der Behandlung eine eGFR von 30 bis <45 ml/min/1,73 m2 auf (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Amputation an den unteren Gliedmassen
Wie im integrierten CANVAS-Programm festgestellt wurde, das aus den beiden randomisierten placebokontrollierten Langzeitstudien CANVAS und CANVAS-R mit insgesamt 10'134 Patienten bestand, war die Anwendung von Invokana bei Patienten mit Diabetes mellitus Typ 2 und bestehender kardiovaskulärer Erkrankung oder mindestens zwei Risikofaktoren für eine kardiovaskuläre Erkrankung mit einem ungefähr um das 2-Fache erhöhten Risiko einer Amputation an den unteren Gliedmassen verbunden. Unter Invokana waren 2,4% der Patienten betroffen vs. 1,1% der Patienten unter Placebo; pro 100 Patientenjahre betrugen die entsprechenden Inzidenzraten 0,63 und 0,34 (Hazard Ratio HR=1,97 (95%-KI 1,41–2,75). Das Ungleichgewicht begann sich bereits in den ersten 26 Therapiewochen zu zeigen. Die Patienten in der CANVAS- und in der CANVAS-R-Studie wurden durchschnittlich 5,7 bzw. 2,1 Jahre nachbeobachtet. Unabhängig von der Behandlung mit Invokana oder dem Placebo war das Amputationsrisiko bei Patienten mit vorgängiger Amputation, peripherer Gefässkrankheit und Neuropathie in der Krankengeschichte zum Studienbeginn am höchsten. Das Risiko einer Amputation an den unteren Gliedmassen war nicht dosisabhängig. In der CREDENCE-Studie, einer Langzeitstudie, in der speziell die nierenbezogenen Therapieergebnisse bei 4'397 Patienten mit Diabetes mellitus Typ 2 und diabetischer Nierenerkrankung untersucht wurden, wurde kein unterschiedliches Risiko für Amputationen an den unteren Gliedmassen im Zusammenhang mit der Anwendung von Canagliflozin 100 mg im Vergleich zum Placebo festgestellt (12 gegenüber 11 Ereignissen pro 1'000 Patientenjahre [HR: 1,11; 95%-KI 0,79, 1,56]) (siehe «Warnhinweise und Vorsichtsmassnahmen»).
In anderen Studien mit Invokana bei Diabetes mellitus Typ 2, in die eine allgemeine Population von insgesamt 8'114 diabetischen Patienten aufgenommen worden war, wurde kein Unterschied bezüglich des Risikos einer Amputation an den unteren Gliedmassen im Vergleich zu den Kontrollgruppen festgestellt (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Unerwünschte Wirkungen in Zusammenhang mit einem reduzierten intravasalen Volumen
In der Analyse der gepoolten Daten der vier 26-wöchigen, Placebo-kontrollierten Studien betrug die Inzidenz aller unerwünschten Wirkungen in Zusammenhang mit reduziertem intravasalem Volumen (z.B. lageabhängiger Schwindel, orthostatische Hypotonie, Dehydrierung und Synkope) 1,2% für Invokana 100 mg, 1,3% für Invokana 300 mg und 1,1% für Placebo. Die Inzidenzen in den zwei aktiv kontrollierten Studien unter Invokana ähnelten denjenigen der Vergleichssubstanzen.
In einer der gezielten Herz-Kreislauf-Langzeitstudien (CANVAS), in welcher die Patienten in der Regel älter waren mit einer höheren Prävalenz von Komorbiditäten, betrug die Inzidenzrate unerwünschter Wirkungen in Zusammenhang mit reduziertem intravasalem Volumen 23,4 unter Invokana 100 mg, 28,7 unter Invokana 300 mg und 18,5 unter Placebo, jeweils Ereignisse pro 1000 Patientenexpositionsjahre.
Zur Beurteilung der Risikofaktoren für diese unerwünschten Wirkungen wurde eine grössere Analyse (n=12'441) von gepoolten Daten von Patienten aus 13 kontrollierten Phase-3 und Phase-4 Studien unter Einbezug beider Dosen von Invokana durchgeführt. In dieser gepoolten Analyse wiesen Patienten unter Schleifendiuretika, Patienten mit einer Ausgangs-eGFR 30 bis <60 ml/min/1,73 m2 sowie Patienten ≥75 Jahre grundsätzlich eine höhere Inzidenz für diese unerwünschten Wirkungen auf. Bei Patienten unter Schleifendiuretika betrugen die Inzidenzraten 49,8 unter Invokana 100 mg und 56,7 unter Invokana 300 mg, verglichen mit 41,5 Ereignissen pro 1000 Patientenexpositionsjahre in der Kontrollgruppe. Für Patienten mit einer Ausgangs-eGFR von 30 bis <60 ml/min/1,73 m2 betrugen die Inzidenzraten 52,4 unter Invokana 100 mg und 53,5 unter Invokana 300 mg, verglichen mit 31,1 Ereignissen pro 1000 Patientenexpositionsjahre in der Kontrollgruppe. Für Patienten ≥75 Jahre betrugen die Inzidenzraten 52,7 unter Invokana 100 mg und 60,8 unter Invokana 300 mg, verglichen mit 24,1 Ereignissen pro 1000 Patientenexpositionsjahre in der Kontrollgruppe (siehe «Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»).
In der zugehörigen kardiovaskulären Studie und der grösseren gepoolten Analyse, wie auch in einer Studie, in der speziell die nierenbezogenen Therapieergebnisse untersucht wurden, waren Abbrüche aufgrund von Nebenwirkungen und schwerwiegenden, mit einem Volumenmangel assoziierten Nebenwirkungen unter Canagliflozin nicht erhöht.
Hypoglykämie
Bei der Anwendung von Invokana als Monotherapie oder als zusätzliche Therapie zu Metformin war die Häufigkeit von Hypoglykämien in den Behandlungsgruppen mit ungefähr 4% eher niedrig versus ca. 2% unter Placebo. Bei zusätzlicher Verabreichung von Invokana mit Insulin wurden Hypoglykämien bei 49,3%, 48,2% bzw. 36,8% der Patienten unter Invokana 100 mg, unter Invokana 300 mg bzw. Placebo berichtet. Zu schweren Hypoglykämien kam es bei 1,8%, 2,7% bzw. 2,5% der Patienten unter Invokana 100 mg, Invokana 300 mg bzw. Placebo. Bei zusätzlicher Verabreichung von Invokana mit Sulfonylharnstoffen wurden hypoglykämische Episoden bei 4,1%, 12,5% bzw. 5,8% der Patienten unter Invokana 100 mg, Invokana 300 mg bzw. Placebo beobachtet (siehe «Dosierung/Anwendung» und «Interaktionen»).
Nekrotisierende Fasziitis des Perineums (Fournier-Gangrän)
Auf der Grundlage spontan gemeldeter unerwünschter Ereignisse wurde Fournier-Gangrän als klassenspezifische unerwünschte Wirkung von SGLT2-Inhibitoren identifiziert. Im Rahmen des klinischen Canagliflozin-Entwicklungsprogramms der Phasen 3 und 4 (einschliesslich des CANVAS- und des CREDENCE-Programms) wurde diese unerwünschte Wirkung nur bei vier Patienten beobachtet (zwei im Canagliflozin Arm und zwei im Vergleichsarm). Alle diese vier Fälle von Fournier-Gangrän wurden als schwerwiegende unerwünschte Ereignisse bewertet.
Aufgrund der in klinischen Studien beobachteten Häufigkeit wird diese unerwünschte Wirkung als «selten» eingestuft (≥1/10'000 bis < 1/1'000) [≥0,01% und <0,1%] (siehe Auflistung der unerwünschten Wirkungen weiter oben).
Genitale Pilzinfektionen
Eine vulvovaginale Pilzinfektion (z.B. Vulvovaginitis, vulvovaginale Candidiasis) wurde bei 10,4% bzw. 11,4% der mit Invokana 100 mg bzw. Invokana 300 mg behandelten Patientinnen berichtet, verglichen mit 3,2% unter der Gabe von Placebo. Die meisten Berichte über vulvovaginale Candidiasis stammten aus den ersten vier Monaten der Behandlung mit Canagliflozin. Von den mit Invokana behandelten Patientinnen berichteten 2,3% über mehr als eine Infektion. Insgesamt brachen 0,7% aller Patientinnen die Behandlung mit Invokana aufgrund einer vulvovaginalen Candidiasis ab (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Eine Balanitis oder Balanoposthitis mit Candida wurde von 4,2% bzw. 3,7% der mit Invokana 100 mg bzw. Invokana 300 mg behandelten männlichen Patienten berichtet, verglichen mit 0,6% unter der Gabe von Placebo. Von den mit Invokana behandelten männlichen Patienten berichteten 0,9% über mehr als eine Infektion. Insgesamt brachen 0,5% aller männlichen Patienten die Behandlung mit Invokana aufgrund einer Balanitis oder Balanoposthitis mit Candida ab. Die Phimose-Inzidenzrate betrug in einer gepoolten Analyse von 10 kontrollierten Studien 5,6 Ereignisse pro 1000 Patientenexpositionsjahre bei nicht beschnittenen Männern. In dieser gepoolten Analyse betrug die Beschneidungsinzidenzrate 3,8 Ereignisse pro 1000 Patientenexpositionsjahre bei männlichen Patienten unter der Behandlung mit Invokana (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Harnwegsinfektionen
Unter der Gabe von Invokana 100 mg bzw. 300 mg traten Harnwegsinfektionen häufiger auf als unter Placebo (5,9% bzw. 4,3%, verglichen mit 4,0%). Die meisten Infektionen waren leicht bis mittelschwer; es kam zu keinem vermehrten Auftreten schwerer unerwünschter Wirkungen. Die Patienten sprachen auf die übliche Behandlung an, während sie die Therapie mit Canagliflozin fortsetzten. Die Therapie mit Canagliflozin wurde nur selten abgesetzt. Die Inzidenz wiederholter Infektionen war unter Canagliflozin nicht erhöht.
Knochenfrakturen
In einer kardiovaskulären Studie (CANVAS) mit 4327 behandelten Patienten mit manifestierter kardiovaskulärer Erkrankung oder mindestens zwei Risikofaktoren für eine kardiovaskuläre Erkrankung betrugen die Inzidenzraten aller adjudizierten Knochenfrakturen 15,9 pro 1000 Patientennachbeobachtungsjahre bei Invokana 100 mg, bei Invokana 300 mg 17,9 pro 1000 Patientennachbeobachtungsjahre sowie 10,9 pro 1000 Patientennachbeobachtungsjahre bei Placebogabe. Die entsprechende Hazard RatioCanagliflozin 100 mg/Placebo betrug 1,45 [1,10; 1,92] 95%CI resp. HRCanagliflozin 300 mg/Placebo 1,64 [1,25; 2,15] 95%CI. Das Ungleichgewicht der Frakturen trat bereits innerhalb der ersten 26 Wochen der Behandlung auf. Ein Zusammenhang mit vermehrten Stürzen konnte nicht sicher bestätigt werden. In zwei weiteren Langzeitstudien und in Studien mit einer allgemeinen Diabetespopulation wurde kein Unterschied zwischen Canagliflozin und der Kontrollsubstanz im Hinblick auf das Frakturrisiko beobachtet.
In einer zweiten kardiovaskulären Studie (CANVAS-R) mit 5'807 behandelten Patienten mit manifestierter kardiovaskulärer Erkrankung oder mindestens zwei Risikofaktoren für eine kardiovaskuläre Erkrankung betrugen die Inzidenzraten aller adjudizierten Knochenfrakturen bei Invokana-Gabe 11,4 Ereignisse pro 1000 Patientennachbeobachtungsjahre und unter Placebogabe 13,2 Ereignisse pro 1000 Patientennachbeobachtungsjahre.
In einer Langzeitstudie, in der speziell die nierenbezogenen Therapieergebnisse bei 4'397 Patienten mit Diabetes mellitus Typ 2 und diabetischer Nierenerkrankung untersucht wurden, betrugen die Inzidenzraten aller adjudizierten Knochenfrakturen sowohl bei Gabe von Canagliflozin 100 mg als auch in der Placebogruppe 12,1 Ereignisse pro 1'000 Patientennachbeobachtungsjahre.
In anderen Studien welche eine allgemeine Diabetespopulation von 7'729 Patienten umfassten und in denen Knochenfrakturen adjudiziert waren, wurde kein Unterschied des Frakturrisikos unter Invokana gegenüber der Kontrollgruppe beobachtet. Die Inzidenzraten aller adjudizierten Knochenfrakturen betrugen bei Invokana-Gabe 11,8 Ereignisse pro 1000 Patientennachbeobachtungsjahre und in der Kontrollgruppe 10,8 Ereignisse pro 1000 Patientennachbeobachtungsjahre. Nach 104-wöchiger Behandlung zeigte Canagliflozin keine unerwünschte Wirkung auf die Knochenmineraldichte.
Photosensibilität
Unerwünschte Reaktionen in Zusammenhang mit Photosensibilität (einschliesslich Photosensibilitätsreaktionen, polymorpher Lichtdermatose und Sonnenbrand) traten bei Anwendung der Vergleichspräparate und bei Anwendung von Invokana 100 mg bzw. Invokana 300 mg bei 0,1%, 0,2% bzw. 0,2% der Patienten auf.
Beeinträchtigung der Nierenfunktion
Aufgrund unzureichend eingestelltem Diabetes mellitus Typ 2 behandelte Patienten
Invokana ist mit einem dosisabhängigen Anstieg des Serumkreatinins und einem begleitenden Abfall der geschätzten GFR verbunden (Tabelle 4). Bei Patienten mit mässiger Beeinträchtigung der Nierenfunktion zum Studienbeginn (Baseline) waren die mittleren Veränderungen ausgeprägter.
Tabelle 4: Veränderungen des Serum-Kreatinins und des eGFR-Werts in Zusammenhang mit Invokana in insgesamt vier placebokontrollierten Studien und einer Studie mit Patienten mit mässiger Beeinträchtigung der Nierenfunktion
|
|
Placebo |
Invokana |
Invokana | ||
|
Pool aus vier placebokontrollierten |
Baseline |
Kreatinin (µmol/l) |
74,5 |
72,9 |
72,6 |
|
eGFR (ml/min/1,73 m2) |
87,0 |
88,3 |
88,8 | ||
|
Veränderung in Woche 6 |
Kreatinin (µmol/l) |
1,05 |
2,93 |
4,08 | |
|
eGFR (ml/min/1,73 m2) |
-1,6 |
-3,8 |
-5,0 | ||
|
Veränderung zu Behandlungsende* |
Kreatinin (µmol/l) |
0,91 |
1,96 |
2,99 | |
|
eGFR (ml/min/1,73 m2) |
-1,6 |
-2,3 |
-3,4 | ||
|
|
|
Placebo |
Invokana |
Invokana | |
|
Studie bei mässiger Beeinträchtigung |
Baseline |
Kreatinin (µmol/l) |
142,0 |
143,5 |
144,2 |
|
eGFR (ml/min/1,73 m2) |
40,1 |
39,7 |
38,5 | ||
|
Veränderung in Woche 3 |
Kreatinin (µmol/l) |
2,91 |
16,21 |
25,10 | |
|
eGFR (ml/min/1,73 m2) |
-0,7 |
-4,6 |
-6,2 | ||
|
Veränderung zu Behandlungsende* |
Kreatinin (µmol/l) |
6,04 |
14,04 |
16,22 | |
|
eGFR (ml/min/1,73 m2) |
-1,5 |
-3,6 |
-4,0 | ||
* Woche 26 beim mITT LOCF-Kollektiv.
In dem Pool der vier placebokontrollierten Studien bei Patienten mit normaler oder leicht eingeschränkter Nierenfunktion zum Studienbeginn (Baseline) betrug der Anteil der Patienten mit mindestens einem Ereignis eines signifikanten Abfalls der Nierenfunktion (definiert als eGFR unter 80 ml/min/1,73 m2 und 30% niedriger als der Baseline-Wert) in der Placebogruppe 2,1%, unter Invokana 100 mg 2,0% und unter Invokana 300 mg 4,1%. Zu Behandlungsende hatten in der Placebogruppe 0,5%, unter Invokana 100 mg 0,7% und unter Invokana 300 mg 1,4% eine signifikant reduzierte Nierenfunktion.
In einer Studie mit Patienten mit mässiger Niereninsuffizienz und einer Baseline-eGFR von 30 bis <50 ml/min/1,73 m2 (mittlere Baseline-eGFR 39 ml/min/1,73 m2) (siehe «Eigenschaften/Wirkungen, Spezifische Populationen») betrug der Anteil der Patienten mit mindestens einem Ereignis eines signifikanten Abfalls der Nierenfunktion (definiert als um 30% niedrigere eGFR verglichen mit dem Baseline-Wert) in der Placebogruppe 6,9%, unter Invokana 100 mg 18% und unter Invokana 300 mg 22,5%. Zu Behandlungsende hatten in der Placebogruppe 4,6% und unter Invokana 100 mg oder 300 mg jeweils 3,4% eine signifikant reduzierte Nierenfunktion.
In einem gepoolten Kollektiv von Patienten mit mässiger Niereninsuffizienz (N=1'087) mit einer Baseline eGFR von 30 bis <60 ml/min/1,73 m2 (mittlere Baseline-eGFR 48 ml/min/1,73 m2), war die Gesamthäufigkeit dieser Ereignisse niedriger als in der dedizierten Studie. Dennoch wurde ein dosisabhängiger Anstieg der aufgetretenen Episoden mit signifikantem Abfall der Nierenfunktion im Vergleich zu Placebo festgestellt.
Die Anwendung von Invokana war mit einer erhöhten Häufigkeit renal bedingter unerwünschter Ereignisse (z.B. erhöhter Serum-Kreatininwert, verringerte glomeruläre Filtrationsrate, Niereninsuffizienz und akutes Nierenversagen) verbunden, insbesondere bei Patienten mit mässiger Beeinträchtigung der Nierenfunktion.
In der Analyse der gepoolten Daten von Patienten mit mässiger Einschränkung der Nierenfunktion betrug die Häufigkeit renal bedingter unerwünschter Ereignisse in der Placebogruppe 3,7%, unter Invokana 100 mg 8,9% und unter Invokana 300 mg 9,3%. Ein Behandlungsabbruch aufgrund renal bedingter unerwünschter Ereignisse erfolgte bei 1,0% der Patienten in der Placebogruppe, bei 1,2% der mit Invokana 100 mg und bei 1,6% der mit Invokana 300 mg behandelten Patienten (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Aufgrund einer diabetischen Nierenerkrankung mit Albuminurie (ACR>300 mg/g) behandelte Patienten mit Diabetes mellitus Typ 2
In einer Langzeitstudie, in der speziell die nierenbezogenen Therapieergebnisse bei Patienten mit Diabetes mellitus Typ 2 und chronischer Nierenerkrankung untersucht wurden, waren die Inzidenzraten unerwünschter Reaktionen im Zusammenhang mit Volumendepletion in der Subgruppe der Patienten mit einer eGFR vor der Behandlung von 45 bis <60 ml/min/1,73 m2 [CrCl 45 bis <60 ml/min] ähnlich: 23 Ereignisse pro 1'000 Patientenjahre bei Canagliflozin 100 mg Gabe und 26 Ereignisse pro 1'000 Patientenjahre bei Placebo-Gabe. In derselben Studie war die Inzidenzrate bei Patienten mit einer eGFR vor der Behandlung von 30 bis <45 ml/min/1,73 m2 [CrCl 30 bis <45 ml/min] bei Gabe von Canagliflozin 100 mg (49 Ereignisse pro 1'000 Patientenjahre) höher als bei Placebo-Gabe (26 Ereignisse pro 1'000 Patientenjahre).
In der Langzeitstudie, in der speziell die nierenbezogenen Therapieergebnisse untersucht wurden, wurde bei Gabe von Canagliflozin 100 mg im Vergleich zum Placebo kein Unterschied im Hinblick auf die Kaliumwerte im Serum, keine Erhöhung der unerwünschten Hyperkaliämie-Ereignisse und keine absoluten (>6,5 mEq/l) oder relativen (> obere Grenze des Normbereichs und >15% Erhöhung gegenüber dem Ausgangswert) Erhöhungen des Serumkaliums beobachtet.
Im Allgemeinen wurde kein Ungleichgewicht zwischen den Behandlungsgruppen im Hinblick auf abnorme Phosphat-Werte beobachtet, und zwar insgesamt und in den einzelnen eGFR-Kategorien (45 bis <60 oder 30 bis <45 ml/min/1,73 m2 [CrCl 45 bis <60 oder 30 bis <45 ml/min]).
Bei Patienten, die das Placebo erhielten, trat eine progressive Abnahme der eGFR auf, während bei Patienten, die mit Canagliflozin 100 mg behandelt wurden, zunächst eine Verringerung der mittleren eGFR auftrat, die sich danach allmählich abschwächte (bis zu 3,5 Jahre). Am Ende der Behandlung war die mittlere eGFR in der Placebogruppe um 1,6 ml/min/1,73 m2 geringer als in der Canagliflozin-100 mg-Gruppe.
Laboruntersuchungen
Die unten beschriebenen Laborergebnisse stammen aus der Analyse der gepoolten Daten der 26-wöchigen, Placebo-kontrollierten klinischen Studien (sofern nicht anders angegeben).
Erhöhte Kaliumwerte im Serum
Die mittleren prozentualen Veränderungen gegenüber den Ausgangswerten für Serumkalium betrugen 0,5% unter Invokana 100 mg bzw. 1,0% unter Invokana 300 mg, verglichen mit 0,6% unter Placebo. Episoden mit erhöhten Kaliumwerten im Serum (>5,4 mmol/l und 15% oberhalb des Ausgangswertes) wurden bei 4,4% der Patienten unter Invokana 100 mg, bei 7,0% der Patienten unter Invokana 300 mg und bei 4,8% der Patienten unter Placebo beobachtet. Im Allgemeinen waren die Erhöhungen leicht ausgeprägt (<6,5 mmol/l), vorübergehend und bedurften keiner spezifischen Therapie.
In einer Studie mit Patienten mit mässiger Einschränkung der Nierenfunktion (eGFR 30 bis <50 ml/min/1,73 m2) (siehe «Eigenschaften/Wirkungen, Spezifische Populationen») wurden bereits früh nach Behandlungsbeginn (d.h. innerhalb von 3 Wochen) dosisabhängige, vorübergehende Erhöhungen des Serumkaliums festgestellt. In dieser Studie traten bei 16,1% der Patienten in der Placebogruppe, bei 12,4% der mit Invokana 100 mg und bei 27,0% der mit Invokana 300 mg behandelten Patienten Erhöhungen des Serumkaliums über 5,4 mEq/L und 15% über dem Baseline-Wert auf. Bei 1,1% der Patienten in der Placebogruppe und bei jeweils 2,2% der mit Invokana 100 mg bzw. 300 mg behandelten Patienten waren die Erhöhungen ausgeprägter (d.h. mindestens 6,5 mEq/L). Bei Patienten mit mässiger Beeinträchtigung der Nierenfunktion waren die Erhöhungen des Serumkaliums häufiger, wenn bereits der Baseline-Kaliumwert erhöht war sowie bei Anwendung von Arzneimitteln, welche die Kaliumausscheidung reduzieren, z.B. Kalium-sparende Diuretika, ACE-Hemmer und Angiotensinrezeptorblocker (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Auswirkungen auf die Lipide
Die mittleren Veränderungen (Veränderungen in Prozent) gegenüber den Ausgangswerten für LDL-C betrugen im Vergleich zu Placebo +0,11 mmol/l (4,5%) unter Invokana 100 mg bzw. +0,21 mmol/l (8,0%) unter Invokana 300 mg. In Bezug auf das Gesamtcholesterin wurden relativ zu Placebo geringere Zunahmen von 2,5% bzw. 4,3% unter Invokana 100 mg bzw. 300 mg beobachtet. Die Zunahmen des High-Density-Lipoprotein-Cholesterins (HDL-C) betrugen im Vergleich zu Placebo 5,4% unter Invokana 100 mg bzw. 6,3% unter Invokana 300 mg. Die Zunahmen des Non-HDL-C betrugen im Vergleich zu Placebo 0,05 mmol/l (1,5%) unter Invokana 100 mg bzw. 0,13 mmol/l (3,6%) unter Invokana 300 mg. Das LDL-C/HDL-C-Verhältnis veränderte sich unter keiner der Invokana-Dosen im Vergleich zu Placebo. Die Konzentrationen der ApoB- und LDL-C-Partikelzahlen (in zwei Studien gemessen) sowie der Non-HDL-C-Partikelzahlen stiegen im Vergleich zu den LDL-C-Veränderungen in einem geringeren Ausmass an.
Erhöhte Hämoglobinwerte
Unter Invokana 100 mg und 300 mg wurden im Vergleich zu den Ausgangswerten geringfügige Erhöhungen der Hämoglobinkonzentrationen beobachtet (+3,5% oder +4,7 g/l bzw. +3,8% oder 5,1 g/l), verglichen mit einer leichten Abnahme unter Placebo (-1,1% oder -0,18 g/l). Entsprechend wurden bei der Anzahl der Erythrozyten und dem Hämatokrit minimale Erhöhungen der mittleren prozentualen Veränderungen gegenüber den Ausgangswerten beobachtet.
Am Ende der Behandlung wiesen 4,0% der Patienten unter Invokana 100 mg, 2,7% der Patienten unter Invokana 300 mg bzw. 0,8% der Patienten unter Placebo Hämoglobinwerte oberhalb des Normbereichs auf.
Erhöhungen des Serummagnesiums
Kurz (innerhalb von 6 Wochen) nach Einleitung einer Behandlung mit Invokana wurden dosisabhängige Erhöhungen des Serummagnesiums festgestellt, die während der Behandlung fortbestanden. In dem Pool der vier placebokontrollierten Studien betrug die mittlere Veränderung des Serummagnesiumspiegels unter Invokana 100 mg +8,1% und unter Invokana 300 mg +9,3%, verglichen mit -0,6% in der Placebogruppe. In einer Studie bei Patienten mit mässiger Einschränkung der Nierenfunktion (siehe «Eigenschaften/Wirkungen, Spezifische Populationen») erhöhten sich die Magnesium-Serumkonzentrationen in der Placebogruppe um 0,2%, unter Invokana 100 mg um 9,2% und unter Invokana 300 mg um 14,8%.
Erhöhungen des Serumphosphats
Unter Invokana wurden dosisabhängige Erhöhungen des Serumphosphatspiegels festgestellt. In dem Pool der vier placebokontrollierten Studien betrug die mittlere prozentuale Erhöhung des Serumphosphatspiegels unter Invokana 100 mg 3,6% und unter Invokana 300 mg 5,1%, verglichen mit 1,5% in der Placebogruppe. In einer Studie mit Patienten mit mässiger Beeinträchtigung der Nierenfunktion (siehe «Eigenschaften/Wirkungen, Spezifische Populationen») erhöhten sich die Phosphat-Serumkonzentrationen in der Placebogruppe um 1,2%, unter Invokana 100 mg um 5,0% und unter Invokana 300 mg um 9,3%.
Reduzierte glomeruläre Filtrationsrate
Zu Beginn der Behandlung mit Canagliflozin in Langzeitstudien zum kardiovaskulären Ergebnis kam es zu einem Abfall der eGFR. Dieser war nach Absetzen der Canagliflozin Behandlung reversibel. Bei fortgesetzter Canagliflozin-Behandlung stieg die eGFR im weiteren Studienverlauf (bis zu 6,5 Jahre) allmählich wieder an und erreichte einen mittleren eGFR-Wert von >70 ml/min/1,73 m2 am Ende der Studie. Demgegenüber wurde bei Patienten der Placebo-Kontrolle eine progrediente Abnahme der eGFR beobachtet.
Unerwünschte Wirkungen bei spezifischen Populationen
Ältere Patienten
Das Sicherheitsprofil bei älteren Patienten entsprach grundsätzlich demjenigen jüngerer Patienten. Patienten im Alter von ≥75 Jahre wiesen jedoch eine höhere Inzidenz von unerwünschten Wirkungen auf, die mit einem reduzierten intravasalen Volumen in Zusammenhang standen (wie z.B. lageabhängiger Schwindel, arterielle Hypotonie, orthostatische Hypotonie). Die Inzidenzraten für diese Ereignisse betrugen 52,7 bzw. 60,8 bzw. 24,1 Ereignisse pro 1000 Patientenexpositionsjahre unter Invokana 100 mg bzw. Invokana 300 mg bzw. Placebo. Eine Abnahme der eGFR wurde unter Invokana 100 mg um -3,41 ml/min/1,73 m2, unter Invokana 300 mg um -4,67 ml/min/1,73 m2 und unter Placebo um -4,15 ml/min/1,73 m2 festgestellt (siehe «Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»).
Patienten mit verminderter Nierenfunktion
Patienten mit einer Ausgangs-eGFR <60 ml/min/1,73 m2 oder CrCl <60 ml/min hatten eine höhere Inzidenz von unerwünschten Wirkungen, die mit einem reduzierten intravasalen Volumen in Zusammenhang standen (z.B. lageabhängiger Schwindel, orthostatische Hypotonie, Hypotonie). Die Inzidenzraten für diese Ereignisse betrugen 53 bzw. 51 bzw. 31 Ereignisse pro 1000 Patientenexpositionsjahre unter Invokana 100 mg bzw. Invokana 300 mg bzw. Placebo (siehe «Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»).
Die Gesamtinzidenzraten von erhöhten Kaliumwerten im Serum war bei Patienten mit mässig verminderter Nierenfunktion höher. Die Inzidenzraten für diese Ereignisse betrugen 49 bzw. 61 bzw. 54 Ereignisse pro 1000 Patientenexpositionsjahre unter Invokana 100 mg bzw. Invokana 300 mg bzw. Placebo. Die Erhöhungen waren zumeist vorübergehend und bedurften keiner spezifischen Behandlung.
Bei Patienten mit mittelschwerer Nierenfunktionsstörung wurden unter beiden Dosen von Invokana Erhöhungen der Kreatininwerte im Serum um 9,2 µmol/l und von Blut-Harnstoff-Stickstoff um ungefähr 1,0 mmol/l beobachtet. Die Inzidenzraten mit einer stärker ausgeprägten Abnahme der eGFR (>30%) zu einem beliebigen Zeitpunkt der Behandlung betrug 73 unter Invokana 100 mg und 81 unter Invokana 300 mg, verglichen mit 65 Ereignissen pro 1000 Patientenexpositionsjahre unter Placebo. Bei der letzten Messung nach Beginn der Studie betrugen die Inzidenzraten solcher Abnahmen 33 Ereignisse unter Invokana 100 mg, 27 Ereignisse unter Invokana 300 mg und 37 Ereignisse pro 1000 Patientenexpositionsjahre unter Placebo (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Anzeichen und Symptome
Einzeldosen von bis zu 1600 mg Invokana bei gesunden Probanden und Invokana 300 mg zweimal täglich während 12 Wochen bei Patienten mit Diabetes mellitus Typ 2 waren im Allgemeinen gut verträglich.
Behandlung
Im Fall einer Überdosierung ist es angemessen, die üblichen unterstützenden Massnahmen zu ergreifen, z.B. die Entfernung nicht absorbierter Substanzen aus dem Gastrointestinaltrakt, klinische Überwachung und unterstützende Therapie in Abhängigkeit vom klinischen Zustand des Patienten. Canagliflozin wurde während einer vierstündigen Hämodialyse in vernachlässigbarem Ausmass aus dem Körper entfernt. Es ist nicht zu erwarten, dass Canagliflozin mittels Peritonealdialyse dialysierbar ist.
Eigenschaften/Wirkungen
ATC-Code
A10BK02
Wirkungsmechanismus
Der Natrium-Glukose-Co-Transporter 2 (SGLT2), in den proximalen renalen Tubuli exprimiert, ist für einen Grossteil der Resorption der filtrierten Glukose aus dem Tubuluslumen verantwortlich. Canagliflozin ist ein oral aktiver Inhibitor von SGLT2. Durch die Inhibition von SGLT2 reduziert Canagliflozin die Reabsorption der filtrierten Glukose und senkt die renale Schwelle für Glukose (RTG), was zu einer erhöhten Glukoseausscheidung im Urin (UGE: urinary glucose excretion) führt. Hierdurch wird bei Patienten mit Diabetes mellitus Typ 2 der Glukosespiegel im Plasma gesenkt. Dieser Mechanismus ist nicht abhängig von Insulin. Die erhöhte Glukoseausscheidung im Urin (UGE) durch die Inhibition von SGLT2 führt darüber hinaus zu einer osmotischen Diurese, und über einen diuretischen Effekt zu einer Senkung des systolischen Blutdrucks. Die vermehrte Glukoseausscheidung im Urin (UGE) verursacht einen Kalorienverlust. Die UGE erhöht das Risiko für Harnwegsinfektionen und für genitale mykotische Infektionen.
In den Studien konnte unter Canagliflozin keine Glukose-Malabsorption beobachtet werden.
Canagliflozin erhöht die Zufuhr von Natrium in den distalen Tubulus durch Blockade der SGLT2-abhängigen Glukose- und Natrium-Reabsorption, wodurch das tubuloglomeruläre Feedback steigt; dies ist in präklinischen Diabetes-Modellen und klinischen Studien mit einer Verringerung des intraglomerulären Drucks sowie der Hyperfiltration und potenziell mit einer nierenprotektiven Wirkung assoziiert.
Pharmakodynamik
Nach ein- und mehrfachen oralen Dosen von Canagliflozin bei Patienten mit Diabetes mellitus Typ 2 wurden dosisabhängige Senkungen der RTG und Steigerungen der Glukoseausscheidung im Urin beobachtet. Bei einem Ausgangswert der RTG von ungefähr 13 mmol/l wurde unter der Dosis von 300 mg täglich bei Patienten mit Diabetes mellitus Typ 2 in Phase-1-Studien eine maximale Suppression der mittleren RTG über 24 Stunden auf ungefähr 4 bis 5 mmol/l beobachtet, was auf ein geringes Risiko für eine behandlungsinduzierte Hypoglykämie hindeutet. Die Reduktion der RTG führte bei Patienten mit Diabetes mellitus Typ 2, die entweder mit Canagliflozin 100 mg oder 300 mg behandelt wurden, zu einer erhöhten Glukoseausscheidung im Urin im Bereich zwischen 77 und 119 g/Tag über alle Phase-1-Studien hinweg. Die beobachtete Glukoseausscheidung im Urin entspricht einem Verlust zwischen 308 und 476 kcal/Tag. Die Senkungen der RTG und die Steigerungen der Glukoseausscheidung im Urin blieben bei Patienten mit Diabetes mellitus Typ 2 über eine 26-wöchige Behandlungsperiode hinweg erhalten. Nach einer leichten Zunahme (meist <400-500 ml) der täglichen Urinmenge nahm diese im Verlauf der ersten Tage nach Behandlungsbeginn wieder ab. Durch Canagliflozin erhöhte sich vorübergehend die Ausscheidung von Harnsäure im Urin (im Vergleich zum Ausgangswert am ersten Tag um 19% gesteigert, Abschwächung auf 6% an Tag 2 und 1% an Tag 13). In diesem Rahmen kam es zu einer nachhaltigen Senkung der Harnsäurespiegel im Serum von ungefähr 20%.
In einer Einzeldosisstudie an Patienten mit Diabetes mellitus Typ 2 führte die Verabreichung von 300 mg vor einer gemischten Mahlzeit zu einer Verzögerung der intestinalen Glukoseabsorption und zu einer Reduktion der postprandialen Blutzuckerwerte.
Kardiale Elektrophysiologie
In einer randomisierten, doppelblinden, Placebo-kontrollierten Studie mit positiver Kontrolle (Moxifloxacin) erhielten 60 gesunde Probanden eine orale Einzeldosis von Canagliflozin 300 mg, Canagliflozin 1'200 mg (das Vierfache der empfohlenen Maximaldosis), Moxifloxacin und Placebo. Es wurden weder unter der empfohlenen Dosis von 300 mg noch unter der Dosis von 1'200 mg bedeutsame Veränderungen des QTc-Intervalls beobachtet. Unter der Dosis von 1'200 mg betrugen die Spitzenkonzentrationen von Canagliflozin im Plasma ungefähr das 1,4-fache der Steady-State-Spitzenkonzentrationen unter einer einmal täglichen Dosis von 300 mg.
Klinische Wirksamkeit
Glykämische Wirksamkeit und Sicherheit
Insgesamt nahmen 10'285 Patienten mit Diabetes mellitus Typ 2 an neun doppelblinden, kontrollierten klinischen Wirksamkeits- und Sicherheitsstudien teil, in denen die Wirkung von Invokana auf die glykämische Kontrolle beurteilt wurde. Die Verteilung der ethnischen Zugehörigkeit entsprach 72% Weissen, 16% Asiaten, 4% Schwarzen und 8% Angehörigen anderer Gruppen. 16% der Patienten waren hispanischer Herkunft. Ungefähr 58% der Patienten waren männlich. Das Durchschnittsalter der Patienten betrug 59,6 Jahre (Bandbreite 21 bis 96 Jahre). 3'082 Patienten waren 65 Jahre und älter und 510 Patienten waren ≥75 Jahre. 58% der Patienten wiesen einen Body Mass Index (BMI) von 30 kg/m2 oder höher auf. Für die Untergruppe der Patienten mit mässiger Niereninsuffizienz mit einer Anfangs-eGFR von 30 bis <60 ml/min/1,73 m2 wurden Daten von 1'087 Patienten aus dem klinischen Entwicklungsprogramm gepoolt und analysiert.
Placebo-kontrollierte Studien
Invokana wurde als Monotherapie, als duale Therapie mit Metformin, als duale Therapie mit Sulfonylharnstoff, als dreifache Therapie mit Metformin und Sulfonylharnstoff, sowie als zusätzliche Therapie zu Insulin untersucht (Tabelle 5). Zusammenfassend führte die Behandlung mit Invokana im Vergleich zu Placebo zu klinisch und statistisch signifikanten (p<0,001) Ergebnissen bei der glykämischen Kontrolle, einschliesslich HbA1c, Anteil der Patienten mit einem HbA1c <7%, Veränderungen der Nüchternglukose (FPG) gegenüber dem Ausgangswert sowie postprandiale Glukosewerte (PPG) (nach 2 Stunden). Zusätzlich wurden relativ zu Placebo auch Abnahmen des Körpergewichts und des systolischen Blutdrucks beobachtet.
Tabelle 5: Wirksamkeitsresultate aus Placebo-kontrollierten klinischen Studiena
|
Monotherapie (26 Wochen) | |||
|
|
Invokana |
Placebo | |
|
100 mg |
300 mg | ||
|
HbA1c (%) | |||
|
Ausgangswert (Mittelwert) |
8,06 |
8,01 |
7,97 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-0,77 |
-1,03 |
0,14 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-0,91b |
-1,16b |
N/Ac |
|
Patienten (%), die ein HbA1c <7% erreichten |
44,5b |
62,4b |
20,6 |
|
Nüchtern-Plasmaglukose (mmol/l) | |||
|
Ausgangswert (Mittelwert) |
9,57 |
9,57 |
9,20 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-1,51 |
-1,94 |
0,46 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-1,97b |
-2,41b |
N/Ac |
|
2-Stunden postprandiale Glukose (mmol/l) | |||
|
Ausgangswert (Mittelwert) |
13,87 |
14,10 |
12,74 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-2,38 |
-3,27 |
0,29 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-2,67b |
-3,55b |
N/Ac |
|
Körpergewicht | |||
|
Ausgangswert (Mittelwert) in kg |
85,9 |
86,9 |
87,5 |
|
% Abweichung vom Ausgangswert (LS-Mittelwert) |
-2,8 |
-3,9 |
-0,6 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-2,2b |
-3,3b |
N/Ac |
|
Duale Therapie mit Metformin (26 Wochen) | |||
|
|
Invokana + Metformin |
Placebo + | |
|
100 mg |
300 mg | ||
|
HbA1c (%) | |||
|
Ausgangswert (Mittelwert) |
7,94 |
7,95 |
7,96 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-0,79 |
-0,94 |
-0,17 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-0,62b |
-0,77b |
N/Ac |
|
Patienten (%), die ein HbA1c <7% erreichten |
45,5 |
57,8 |
29,8 |
|
Nüchtern-Plasmaglukose (mmol/l) | |||
|
Ausgangswert (Mittelwert) |
9,36 |
9,59 |
9,12 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-1,52 |
-2,10 |
0,14 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-1,65b |
-2,23b |
N/Ac |
|
2-Stunden postprandiale Glukose (mmol/l) | |||
|
Ausgangswert (Mittelwert) |
14,30 |
14,54 |
13,81 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-2,66 |
-3,17 |
-0,55 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-2,12b |
-2,62b |
N/Ac |
|
Körpergewicht | |||
|
Ausgangswert (Mittelwert) in kg |
88,7 |
85,4 |
86,7 |
|
% Abweichung vom Ausgangswert (LS-Mittelwert) |
-3,7 |
-4,2 |
-1,2 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-2,5b |
-2,9b |
N/Ac |
|
Dreifache Therapie mit Metformin und Sulfonylharnstoff (26 Wochen) | |||
|
|
Invokana + Metformin und |
Placebo + Metformin | |
|
100 mg |
300 mg | ||
|
HbA1c (%) | |||
|
Ausgangswert (Mittelwert) |
8,13 |
8,13 |
8,12 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-0,85 |
-1,06 |
-0,13 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-0,71b |
-0,92b |
N/Ac |
|
Patienten (%), die ein HbA1c <7% erreichten |
43,2b |
56,6b |
18,0 |
|
Nüchtern-Plasmaglukose (mmol/l) | |||
|
Ausgangswert (Mittelwert) |
9,60 |
9,34 |
9,42 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-1,01 |
-1,69 |
0,23 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-1,24b |
-1,92b |
N/Ac |
|
Körpergewicht | |||
|
Ausgangswert (Mittelwert) in kg |
93,5 |
93,5 |
90,8 |
|
% Abweichung vom Ausgangswert (LS-Mittelwert) |
-2,1 |
-2,6 |
-0,7 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-1,4b |
-2,0b |
N/Ac |
|
Zusatztherapie zu Insulind (18 Wochen) | |||
|
|
Invokana + Insulin |
Placebo + Insulin | |
|
100 mg |
300 mg | ||
|
HbA1c (%) | |||
|
Ausgangswert (Mittelwert) |
8,33 |
8,27 |
8,20 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-0,63 |
-0,72 |
0,01 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-0,65b |
-0,73b |
N/Ac |
|
Patienten (%), die ein HbA1c <7% erreichten |
19,8 |
24,7 |
7,7 |
|
Nüchtern-Plasmaglukose (mmol/l) | |||
|
Ausgangswert (Mittelwert) |
9,43 |
9,33 |
9,38 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-1,03 |
-1,39 |
0,22 |
|
Unterschied zu Placebo (LS-Mittelwert) (95% KI) |
-1,25b |
-1,61b |
N/Ac |
|
Körpergewicht | |||
|
Ausgangswert (Mittelwert) in kg |
96,9 |
96,7 |
97,7 |
|
% Abweichung vom Ausgangswert (LS-Mittelwert) |
-1,8 |
-2,3 |
0,1 |
|
Unterschied zu Placebo (LS-Mittelwert) (97.5% KI) |
-1,9b |
-2,4b |
N/Ac |
a Intenttotreat-Population unter Anwendung der letzten Beobachtung in der Studie vor Einsatz einer glykämischen Rettungstherapie.
b p<0.001 im Vergleich zu Placebo.
c Nicht anwendbar.
d Invokana als Zusatztherapie zu Insulin (mit oder ohne andere antihyperglykämische Arzneimittel).
Zusatztherapie: Canagliflozin in Kombination mit Metformin und einem Dipeptidylpeptidase-4-Inhibitor
Canagliflozin wurde als Add-on bei Patienten, die unter vorheriger Behandlung mit Metformin und Sitagliptin keine adäquate glykämische Kontrolle erzielten, untersucht und dabei gemäss Titrationsplan (Anfangsdosis 100 mg; Erhöhung auf 300 mg nach 6 Wochen bei Patienten mit unzureichender glykämischer Kontrolle, angemessener eGFR und guter Verträglichkeit für 100 mg Canagliflozin) dosiert.
Bei diesen Patienten verbesserte sich die glykämische Kontrolle (siehe Tabelle 6) nach Gabe von Canagliflozin im Vergleich zu Placebo. Zusätzlich reduzierte Canagliflozin gegenüber Placebo das Körpergewicht und den systolischen Blutdruck.
Tabelle 6: Ergebnisse aus der 26-wöchigen placebokontrollierten klinischen Studie mit Canagliflozin in Kombination mit Metformin und Sitagliptin*
|
Wirksamkeitsparameter |
Placebo + |
Canagliflozin + |
|
HbA1c (%) | ||
|
Ausgangswert (Mittelwert) |
8,38 |
8,53 |
|
Abweichung vom Ausgangswert (angepasster Mittelwert) |
-0,01 |
-0,91 |
|
Unterschied zu Placebo (angepasster Mittelwert) (95% KI)† |
|
-0,89‡ |
|
Patienten (%), die ein HbA1c <7% erreichten |
12 |
32§ |
|
Nüchtern-Plasmaglukose (mg/dl) | ||
|
Ausgangswert (Mittelwert) |
180 |
186 |
|
Abweichung vom Ausgangswert (angepasster Mittelwert) |
-3 |
-30 |
|
Unterschied zu Placebo (angepasster Mittelwert) (95% KI)† |
|
-27‡ |
|
Körpergewicht | ||
|
Ausgangswert (Mittelwert) in kg |
89,9 |
93,8 |
|
% Abweichung vom Ausgangswert (angepasster Mittelwert) |
-1,6 |
-3,4 |
|
Unterschied zu Placebo (angepasster Mittelwert) (95% KI)† |
|
-1,8‡ |
* Intent-to-treat-Population
† Der angepasste Mittelwert und das KI wurden mit einem Mischmodell für Messwiederholungen berechnet
‡ p<0,001
§ p<0,01
Aktiv-kontrollierte Studien
Invokana wurde als duale Therapie (zusammen mit Metformin) mit Glimepirid sowie als dreifache Therapie (zusammen mit Metformin und Sulfonylharnstoff) mit Sitagliptin verglichen (Tabelle 7).
Tabelle 7: Wirksamkeitsresultate aus den aktiv-kontrollierten Studiena
|
Im Vergleich zu Glimepirid als duale Therapie zusammen mit Metformin (52 Wochen) | |||
|
|
Invokana + Metformin |
Glimepirid (titriert) | |
|
100 mg |
300 mg | ||
|
HbA1c (%) | |||
|
Ausgangswert (Mittelwert) |
7,78 |
7,79 |
7,83 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-0,82 |
-0,93 |
-0,81 |
|
Unterschied zu Glimepirid (LS-Mittelwert) (95% KI) |
-0,01b |
-0,12b |
N/Ac |
|
Patienten (%), die ein HbA1c <7% erreichten |
53,6 |
60,1 |
55,8 |
|
Nüchtern-Plasmaglukose (mmol/l) | |||
|
Ausgangswert (Mittelwert) |
9,18 |
9,09 |
9,20 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-1,35 |
-1,52 |
-1,02 |
|
Unterschied zu Glimepirid (LS-Mittelwert) (95% KI) |
-0,33 |
-0,51 |
N/Ac |
|
Körpergewicht | |||
|
Ausgangswert (Mittelwert) in kg |
86,8 |
86,6 |
86,6 |
|
% Abweichung vom Ausgangswert (LS-Mittelwert) |
-4,2 |
-4,7 |
1,0 |
|
Unterschied zu Glimepirid (LS-Mittelwert) (95% KI) |
-5,2d |
-5,7d |
N/Ac |
|
Im Vergleich zu Sitagliptin als Dreifachtherapie zusammen mit Metformin und Sulfonylharnstoff (52 Wochen) | ||
|
|
Invokana 300 mg + |
Sitagliptin 100 mg + |
|
HbA1c (%) | ||
|
Ausgangswert (Mittelwert) |
8,12 |
8,13 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-1,03 |
-0,66 |
|
Unterschied zu Sitagliptin (LS-Mittelwert) (95% KI) |
-0,37e |
N/Ac |
|
Patienten (%), die ein HbA1c <7% erreichten |
47,6 |
35,3 |
|
Nüchtern-Plasmaglukose (mmol/l) | ||
|
Ausgangswert (Mittelwert) |
9,42 |
9,09 |
|
Abweichung vom Ausgangswert (LS-Mittelwert) |
-1,66 |
-0,32 |
|
Unterschied zu Sitagliptin (LS-Mittelwert) (95% KI) |
-1,34 |
N/Ac |
|
Körpergewicht | ||
|
Ausgangswert (Mittelwert) in kg |
87,6 |
89,6 |
|
% Abweichung vom Ausgangswert (LS-Mittelwert) |
-2,5 |
0,3 |
|
Unterschied zu Sitagliptin (LS-Mittelwert) (95% KI) |
-2,8d |
N/Ac |
a Intenttotreat-Population LOCF in der Studie vor Einsatz einer glykämischen Rettungstherapie.
b Invokana + Metformin wird gegenüber Glimepirid+Metformin als nicht unterlegen angesehen, da die obere Grenze des Konfidenzintervalls kleiner ist als die vordefinierte Nicht-Unterlegenheitsgrenze von <0,3%.
c Nicht anwendbar.
d p<0.01.
e Invokana+Metformin+Sulfonylharnstoff wird gegenüber Sitagliptin+Metformin+Sulfonylharnstoff als nicht unterlegen angesehen, da die obere Grenze des Konfidenzintervalls kleiner ist als die vordefinierte Nicht-Unterlegenheitsgrenze von <0,3%.
Canagliflozin in Kombination mit Metformin als Initialtherapie
Die Kombination von Canagliflozin und Metformin wurde bei nicht vorbehandelten Patienten mit Diabetes mellitus Typ 2 untersucht, welche mit Ernährungsmassnahmen und Bewegung allein keine adäquate glykämische Kontrolle erzielen konnten. Die initiale Kombinationstherapie (Canagliflozin 100 mg bzw. Canagliflozin 300 mg mit Metformin XR) war der jeweiligen Monotherapien mit Canagliflozin (100 mg oder 300 mg) und der Metformin-Monotherapie hinsichtlich der Verbesserung des HbA1c überlegen (siehe Tabelle 8).
Tabelle 8: Ergebnisse aus der 26-wöchigen aktiv-kontrollierten klinischen Studie mit Canagliflozin in Kombination mit Metformin als Initialtherapie*
|
Wirksamkeitsparameter |
Metformin XR |
Canagliflozin 100 mg |
Canagliflozin 300 mg |
Canagliflozin 100 mg + |
Canagliflozin 300 mg + |
|
HbA1c (%) | |||||
|
Ausgangswert (Mittelwert) |
8,81 |
8,78 |
8,77 |
8,83 |
8,90 |
|
Abweichung vom Ausgangswert (angepasster Mittelwert) |
-1,30 |
-1,37 |
-1,42 |
-1,77 |
-1,78 |
|
Unterschied zu 100 mg Canagliflozin (angepasster Mittelwert) (95% KI) † |
|
|
|
-0,40‡ |
|
|
Unterschied zu 300 mg Canagliflozin (angepasster Mittelwert) (95% KI) † |
|
|
|
|
-0,36‡ |
|
Unterschied zu Metformin XR (angepasster Mittelwert) (95% KI) † |
|
-0,06‡ |
-0,11‡ |
-0,46‡ |
-0,48‡ |
|
Patienten (%), die ein HbA1c <7% erreichten |
43 |
39 |
43 |
50§§ |
57§§ |
|
Körpergewicht | |||||
|
Ausgangswert (Mittelwert) in kg |
92,1 |
90,3 |
93,0 |
88,3 |
91,5 |
|
% Abweichung vom Ausgangswert (angepasster Mittelwert) |
-2,1 |
-3,0 |
-3,9 |
-3,5 |
-4,2 |
|
Unterschied zu Metformin XR (angepasster Mittelwert) (95% KI)† |
|
-0,9§§ |
-1,8§ |
-1,4‡ |
-2,1‡ |
* Intent-to-treat-Population
† Least squares mean (LS-Mittelwert), angepasst bzgl. Covariaten einschliesslich Ausgangswert und Stratifizierungsfaktor
‡ Angepasst p=0,001
§ Angepasst p<0,01
§§ Angepasst p<0,05
Spezifische Populationen
In drei Studien an spezifischen Populationen (ältere Patienten, Patienten mit eGFR von 30 bis <50 ml/min/1,73 m2 und Patienten mit kardiovaskulärer Erkrankung oder einem erhöhten Risiko für eine solche) wurde Invokana der aktuellen Diabetesbehandlung der Patienten (Diät, Monotherapie, Kombinationstherapie) hinzugefügt.
Ältere Patienten
An einer doppelblinden, Placebo-kontrollierten Studie über 26 Wochen nahmen insgesamt 714 Patienten im Alter von ≥55 bis ≤80 Jahren (davon 227 Patienten zwischen 65 und <75 Jahren und 46 Patienten zwischen 75 und <85 Jahren) mit ungenügender glykämischer Kontrolle unter der aktuellen Diabetes-Behandlung (entweder Diät und körperliche Betätigung alleine oder in Kombination mit oralen oder parenteralen antihyperglykämischen Wirkstoffen) teil. Unter Invokana 100 mg bzw. 300 mg wurden jeweils im Vergleich zu Placebo statistisch signifikante (p<0,001) Veränderungen des HbA1c gegenüber dem Ausgangswert von -0,57% und -0,70% beobachtet. Darüber hinaus kam es zu einer statistisch signifikanten Senkung der FPG und ein höherer Prozentsatz der Patienten erreichte ein HbA1c von <7,0%, verglichen mit Placebo (siehe «Dosierung/Anwendung» und «Unerwünschte Wirkungen»).
Patienten mit einer eGFR von 45 bis <60 ml/min/1,73 m2
In einer gepoolten Analyse der Daten von Patienten (n=722) mit einer Ausgangs-eGFR von 45 ml/min/1,73 m2 bis <60 ml/min/1,73 m2 bewirkte Canagliflozin verglichen mit dem Placebo eine Reduzierung des HbA1c um -0,47% unter Canagliflozin 100 mg. Patienten mit einer Ausgangs-eGFR von 45 ml/min/1,73 m2 bis <60 ml/min/1,73 m2 zeigten unter Canagliflozin 100 mg eine mittlere Abnahme des Körpergewichts um -1,8% verglichen mit Placebo.
Eine Mehrheit der Patienten mit einer Ausgangs-eGFR von 45 bis <60 ml/min/1,73 m2 wurde mit Insulin und/oder Sulfonylharnstoffen therapiert (85% [614/722]). In dieser Analyse von gepoolten Daten hatten 27,3% der Patienten unter Placebo ≥1 Hypoglykämie, versus 39,8% unter 100 mg Invokana und 41,8% unter Invokana 300 mg. Diese erhöhte Inzidenz von Hypoglykämien ist erklärbar durch die gleichzeitige Behandlung mit Insulin und/oder Sulfonylharnstoffen (siehe «Unerwünschte Wirkungen»).
Blutdruck
In den Placebo-kontrollierten Studien wurden relativ zu Placebo folgende mittlere Senkungen des systolischen Blutdrucks beobachtet: unter Invokana 100 mg -3,9 mmHg, unter Invokana 300 mg -5,3 mmHg und unter Placebo -0,1 mmHg. In der gleichen Population war die Wirkung auf den diastolischen Blutdruck geringer ausgeprägt, mit mittleren Veränderungen von -2,1 mmHg unter Invokana 100 mg, -2,5 mmHg unter Invokana 300 mg und -0,3 mmHg unter Placebo. Es kam zu keinen merklichen Veränderungen der Herzfrequenz.
Kardiovaskuläre Ergebnisse im CANVAS-Programm
Die Auswirkung von Canagliflozin auf das kardiovaskuläre Risiko bei Erwachsenen mit Typ-2-Diabetes mellitus und manifestierter kardiovaskulärer (KV) Erkrankung oder mit KV-Risiko (zwei oder mehr KV-Risikofaktoren) wurde im CANVAS-Programm (Studie CANVAS und Studie CANVAS-R) untersucht. Dabei handelte es sich um multizentrische, multinationale, randomisierte, doppelblinde Studien mit Parallelgruppen und ähnlichen Ein- und Ausschlusskriterien und Patientenkollektiven. In den Studien wurde das Risiko des Auftretens eines gravierenden unerwünschten kardiovaskulären Ereignisses (MACE; definiert als Tod infolge eines kardiovaskulären Ereignisses, nicht-tödlicher Myokardinfarkt oder nicht-tödlicher Schlaganfall) zwischen Canagliflozin und einem Placebo vor dem Hintergrund einer Standardbehandlung gegen Diabetes und atherosklerotische kardiovaskuläre Erkrankung verglichen.
In der CANVAS-Studie wurden die Patienten im Zufallsverfahren 1:1:1 randomisiert und der Behandlung mit Canagliflozin 100 mg, Canagliflozin 300 mg oder einem passenden Placebo zugeteilt. In der Studie CANVAS-R wurden die Patienten im Zufallsverfahren 1:1 randomisiert und der Behandlung mit Canagliflozin 100 mg oder einem passenden Placebo zugeteilt. Nach Ermessen des Prüfarztes war nach Woche 13 je nach Verträglichkeit und glykämischem Bedarf eine Dosiserhöhung auf 300 mg zulässig. Zur Sicherstellung, dass die Teilnehmer dem Versorgungsstandard entsprechend eine Behandlung gegen Diabetes und Atherosklerose erhielten, war eine Anpassung der entsprechenden Begleittherapien nach Ermessen des Prüfarztes zulässig.
Es wurden insgesamt 10'134 Patienten (4'327 in der CANVAS-Studie und 5'807 in der Studie CANVAS-R; insgesamt wurden 4'344 im Zufallsverfahren dem Placebo zugewiesen und 5'790 der Behandlung mit Canagliflozin) über 149 Wochen behandelt (Mittelwert; in der CANVAS-Studie betrug die Behandlungsdauer 223 Wochen und in der CANVAS-R 94 Wochen). Bei 99,6% der Teilnehmer beider Studien wurde der Vitalstatus erfasst. Ungefähr 78% der Studienteilnehmer waren Kaukasier, 13% waren Asiaten und 3% waren Schwarze. Das mittlere Alter betrug 63 Jahre und ungefähr 64% waren männlich.
Alle Patienten in der Studie hatten zum Screening-Zeitpunkt einen unzureichend kontrollierten Typ-2-Diabetes mellitus (HbA1c ≥7,0% bis ≤10,5%). Der mittlere HbA1c zum Studienbeginn betrug 8,2% und die mittlere Diabetesdauer betrug 13,5 Jahre. Bei ungefähr 31%, 21% und 18% lag eine Vorgeschichte mit Neuropathie, Retinopathie bzw. Nephropathie vor. Die Nierenfunktion zum Studienbeginn war bei 80% der Patienten normal oder leicht beeinträchtigt und bei 20% der Patienten moderat beeinträchtigt (mittlere eGFR: 77 ml/min/1,73 m2). Zum Studienbeginn standen die Patienten unter Behandlung mit mindestens einer Antidiabetesmedikation, einschliesslich Metformin (77%), Insulin (50%) und Sulfonylharnstoff (43%).
Sechsundsechzig Prozent der Teilnehmer hatten eine manifestierte kardiovaskuläre Erkrankung in der Vorgeschichte, davon hatten 56% eine Koronarerkrankung, 19% eine zerebrovaskuläre Erkrankung und 21% eine periphere Gefässerkrankung; 14% hatten eine Herzinsuffizienz in der Vorgeschichte. Zum Studienbeginn betrug der mittlere systolische Blutdruck 137 mmHg und der mittlere diastolische Blutdruck 78 mmHg, der mittlere LDL-Wert betrug 89 mg/dl, der mittlere HDL-Wert 46 mg/dl und die mittlere Urin-Albumin-Kreatinin-Ratio (UACR) 115 mg/g. Ungefähr 80% der Patienten standen zum Studienbeginn unter Behandlung mit Renin-Angiotensin-System-Hemmern, 54% unter Behandlung mit Betablockern, 13% unter Behandlung mit Schleifendiuretika, 36% unter Behandlung mit anderen Diuretika, 75% unter Behandlung mit Statinen und 74% unter Behandlung mit Thrombozytenaggregationshemmern (überwiegend Acetylsalicylsäure).
Der primäre Endpunkt im CANVAS-Programm war die Dauer des Zeitraums bis zum ersten Auftreten eines MACE. Die MACE-HR bei Patienten unter Behandlung mit Canagliflozin im Vergleich zu denen in der Placebogruppe und das dazugehörige 95%-KI wurden mit einem stetigen Cox-Regressionsmodell mit Stratifizierung nach Studie und manifestierter kardiovaskulärer Erkrankung geschätzt.
Canagliflozin bewirkte eine signifikante Reduktion des Risikos des Erstauftretens des primären kombinierten Endpunktes MACE (HR: 0,86; 95%-KI 0,75, 0,97), zu welcher jede einzelne MACE-Komponente beitrug. Die Ergebnisse für die beiden empfohlenen Canagliflozin-Dosierungen (100 mg und 300 mg) waren konsistent mit dem Ergebnis für die gepoolten Daten. Die Wirksamkeit von Canagliflozin in Bezug auf MACE unterschied sich abhängig davon, ob Patienten zu Studienbeginn bereits eine manifeste kardiovaskuläre Erkrankung (HR: 0,82; 95%-KI 0,72, 0,95) oder nur kardiovaskuläre Risikofaktoren (HR: 0,98; 95%-KI 0,74, 1,30) aufwiesen.
Es gab 2'011 Patienten mit einer eGFR von 30 bis <60 ml/min/1,73 m2. Die MACE-Ergebnisse in dieser Teilgruppe korrelierten mit den Gesamtbefunden.
Tabelle 9: Effekt der Behandlung für den primären kombinierten Endpunkt und seinen Komponenten
|
|
Placebo |
Canagliflozin |
Hazard Ratio |
|
Kombination aus Tod infolge eines kardiovaskulären Ereignisses, nicht-tödlicher Myokardinfarkt, oder nicht-tödlicher Schlaganfall (Dauer des Zeitraums bis zum ersten Auftreten; Intent-to-Treat-Analysegruppe)1 |
31,48 |
26,93 |
0,86 (0,75-0,97) |
|
Tod infolge eines kardiovaskulären Ereignisses |
12,84 |
11,60 |
0,87 (0,72-1,06) |
|
nicht-tödlicher Myokardinfarkt |
11,61 |
9,74 |
0,85 (0,69-1,05) |
|
nicht-tödlicher Schlaganfall |
8,39 |
7,12 |
0,90 (0,71-1,15) |
1 P Wert für die Überlegenheit (zweiseitig) = 0,0158
Ergebnisse nach Subgruppen Vorliegen oder Abwesenheit einer manifestierten kardiovaskulären Erkrankung:
Die primäre Wirksamkeitsanalyse basierte auf der Gesamtpopulation. Eine vordefinierte Subgruppenanalyse für Patienten mit bzw. ohne manifeste kardiovaskuläre Vorerkrankung erbrachte zwar keinen eindeutigen Hinweis auf eine Heterogenität der Wirksamkeit in diesen beiden Gruppen (Interaktion p-Wert = 0,1803); allerdings war nur bei Patienten mit manifester Vorerkrankung eine klinisch relevante Reduktion des kardiovaskulären Risikos (HR: 0,82; 95% KI 0,72, 0,95) zu beobachten, nicht bei Patienten ohne manifeste Vorerkrankung (HR: 0,98; 95% KI: 0,74, 1,30).
Ausgehend von dem Kaplan-Meier-Plot des ersten Auftretens eines MACE war eine Reduzierung des MACE in der Canagliflozin-Gruppe bereits in Woche 26 festzustellen und blieb für den Rest der Studie erhalten.
Gesamtmortalität:
In der kombinierten Canagliflozin-Gruppe betrug die HR für die Gesamtmortalität gegenüber Placebo 0,87 (0,74; 1,01).
Stationär behandelte Herzinsuffizienz
Canagliflozin reduzierte im Vergleich mit Placebo das Risiko für eine Herzinsuffizienz, die einen Krankenhausaufenthalt erforderte (HR: 0,67; 95% KI [0,52; 0,87]).
Renale Endpunkte im CANVAS Programm
Im CANVAS-Programm lag die HR für die Zeit bis zur ersten bestätigten Nephropathie (Verdoppelung des Serum-Kreatinins, Notwendigkeit einer Nierenersatztherapie, Nierentod) bei 0,53 (95% CI: 0,33; 0,84) für Canagliflozin (1,5 Ereignisse pro 1000 Patientenjahre) im Vergleich zu Placebo (2,8 Ereignisse pro 1000 Patientenjahre). Zusätzlich reduzierte Canagliflozin das Fortschreiten der Albuminurie bei Patienten mit einer Normo- oder Mikroalbuminurie vor Behandlungsbeginn um 25,8% gegenüber 29,2% unter Placebo (HR: 0,73; 95% KI: 0,67, 0,79). Obwohl die Behandlung mit ACEi und/oder ARB im CANVAS-Programm kein Einschlusskriterium darstellte, nahmen 80% der Teilnehmer zum Zeitpunkt der Randomisierung einen ACEi und/oder ARB ein.
Nierenbezogene Therapieergebnisse in der CREDENCE-Studie
Die Wirkung von Canagliflozin 100 mg auf die renalen Ereignisse bei Erwachsenen mit Diabetes mellitus Typ 2 und diabetischer Nierenerkrankung (DKD) mit einer geschätzten glomerulären Filtrationsrate (eGFR) von 30 bis <90 ml/min/1,73 m2 oder einer CrCl von 30 bis <90 ml/min und Albuminurie (>300 bis 5'000 mg/g Kreatinin) wurde in der Studie «Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation Trial» (CREDENCE) bewertet. Dabei handelte es sich um eine multizentrische, internationale, randomisierte (1:1), doppelblinde, ereignisgesteuerte, placebokontrollierte Parallelgruppenstudie. Insgesamt 4'401 Probanden wurden randomisiert, um den ITT (Intent-To-Treat) Analysedatensatz zu bilden, welcher für die wichtigsten Wirksamkeitsanalysen verwendet wurde, während 4'397 behandelte Probanden für die Bewertung der Sicherheit zur Verfügung standen. In der CREDENCE-Studie wurde das Risiko des Auftretens einer DKD (definiert als zusammengesetzter Endpunkt aus terminaler Niereninsuffizienz, Verdopplung des Serumkreatinins und Tod durch renale oder kardiovaskuläre Ursachen) bei Gabe von Canagliflozin 100 mg bzw. eines Placebos vor dem Hintergrund einer Standardbehandlung gegen DKD (einschliesslich Angiotensinkonversionsenzym-Hemmer [ACE-Hemmer] und Angiotensin-Rezeptorblocker [ARB]) untersucht und verglichen.
In der CREDENCE-Studie wurden die Probanden entweder mit Canagliflozin 100 mg oder Placebo, stratifiziert nach der Screening-eGFR (30 bis <45, 45 bis <60, 60 bis <90 ml/min/1,73 m2) oder der CrCl (30 bis <45, 45<60, 60<90 ml/min) bis zum Beginn einer Dialyse oder bis zur Nierentransplantation behandelt.
Insgesamt wurden 4'397 Probanden behandelt, die mittlere Expositionsdauer betrug 115 Wochen. Der Vitalstatus wurde während der Studie von 99,9% der Probanden erhoben. Das Durchschnittsalter betrug 63 Jahre, 66% waren Männer.
Der mittlere HbA1c-Ausgangswert betrug 8,3%, der mediane Ausgangswert des Albumin-Kreatinin-Quotienten im Urin lag bei 927 mg/g. Die zu Studienbeginn am häufigsten verwendeten antihyperglykämischen Wirkstoffe waren Insulin (65,5%), Biguanid-Derivate (57,8%) und Sulfonylharnstoffe (28,8%). Beinahe alle Probanden (99,9%) erhielten zum Zeitpunkt der Randomisierung ACE-Hemmer oder ARB. Rund 92% der Probanden erhielten zu Studienbeginn eine kardiovaskuläre Therapie (abgesehen von ACE-Hemmern und ARB), etwa 60% wurden mit einem antithrombotischen Wirkstoff (einschliesslich Acetylsalicylsäure) und 69% mit Statinen behandelt.
Zu Studienbeginn betrug die mittlere eGFR 56,2 ml/min/1,73 m2 (rund 60% der Studienpopulation hatten eine eGFR von <60 ml/min/1,73 m2 ). Die Probanden litten durchschnittlich seit rund 16 Jahren an Diabetes. Der Anteil der Probanden mit einer kardiovaskulären Erkrankung in der Vergangenheit betrug 50,4%; bei 14,8% war in der Vergangenheit Herzinsuffizienz aufgetreten. Zusätzlich zur diabetischen Nierenerkrankung wiesen etwa 64% der Population mindestens eine weitere mikrovaskuläre Komplikation auf.
HRs und zugehörige 95%-KI für den primären und die sekundären Endpunkte wurden mittels eines stratifizierten Proportional-Hazards-Regressionsmodell nach Cox geschätzt (mit der Therapie als der erklärenden Variable und stratifiziert nach der Screening-eGFR [30 bis <45, 45 bis <60, 60 bis <90 ml/min/1,73 m2] oder CrCl [30 bis <45, 45 bis <60, 60 bis <90 ml/min].
Der zusammengesetzte primäre Endpunkt der CREDENCE-Studie bestand aus dem Zeitraum bis zum/zur a) ersten Auftreten einer terminalen Niereninsuffizienz (ESKD; definiert durch einen Abfall der eGFR unter 15 ml/min/1,73 m2 bzw. der CrCl unter 15 ml/min) oder Beginn einer chronischen Dialyse oder einer Nierentransplantation, b) Verdopplung des Serumkreatinins, c) Tod durch renale Ursachen, oder d) Tod durch kardiovaskuläre Ursachen.
Canagliflozin 100 mg verringerte das Risiko des ersten Auftretens des zusammengesetzten primären Endpunkts (ESKD, Verdopplung des Serumkreatinins und Tod durch renale oder kardiovaskuläre Ursachen) signifikant [p <0,0001; HR: 0,70; 95%-KI: 0,59, 0,82] (siehe Abbildung 1). Der Behandlungseffekt war in allen Subgruppen vergleichbar, darunter in den drei aufgrund der eGFR definierten Subgruppen und bei Probanden mit einer kardiovaskulären Erkrankung in der Anamnese oder ohne diese.
Basierend auf dem Kaplan-Meier-Plot der Zeit bis zum ersten Auftreten des primären zusammengesetzten Endpunkts war die Wirkung auf die Behandlung von 100 mg Canagliflozin ab Woche 52 erkennbar und blieb bis zum Ende der Studie erhalten.
Die Effekte von Canagliflozin 100 mg auf sekundäre Endpunkte sind in Abbildung 1 zusammengefasst.
Abbildung 1: Behandlungseffekt im Hinblick auf die zusammengesetzten primären und sekundären Endpunkte und ihre Bestandteile
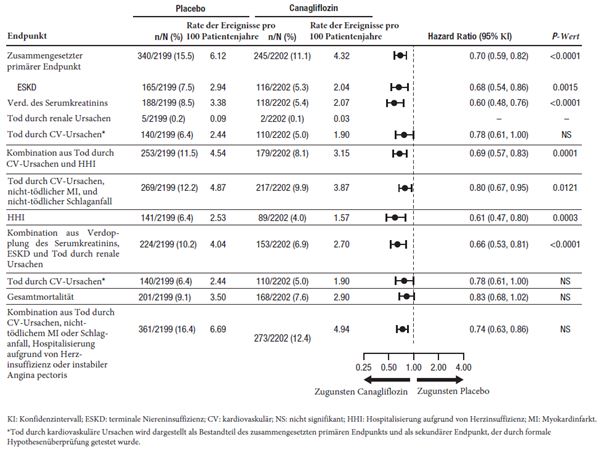
Wie in Abbildung 2 dargestellt, sank die eGFR bei placebobehandelten Patienten allmählich auf progressive und lineare Weise; in der Canagliflozin-Gruppe dagegen war eine abrupte Verringerung in Woche 3 zu beobachten, die sich danach mit der Zeit abschwächte; die nach der Methode der kleinsten Quadrate errechnete mittlere Verringerung der eGFR war nach Woche 52 in der Canagliflozin-Gruppe geringer als in der Placebogruppe, der Behandlungseffekt blieb bis zum Behandlungsende erhalten.
Abbildung 2: Nach der Methode der kleinsten Quadrate berechnete mittlere Veränderung des eGFR-Ausgangswerts im zeitlichen Verlauf (Analyseset der weiterhin behandelten Patienten)
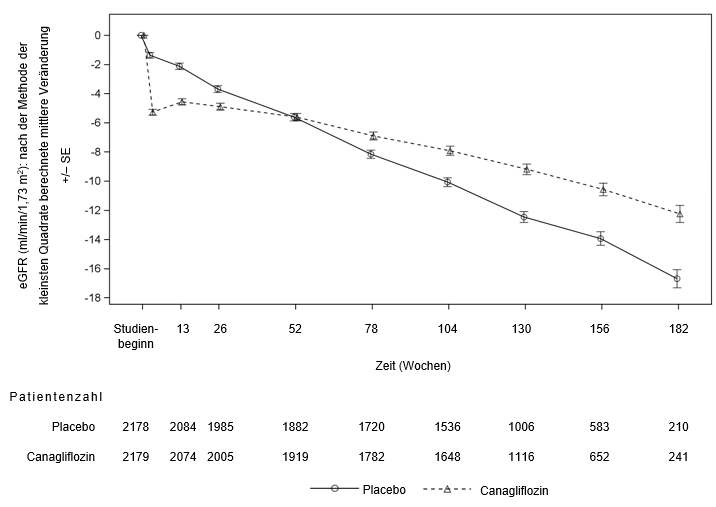
In der CREDENCE-Studie war die Inzidenzrate nierenbezogener unerwünschter Ereignisse in der Canagliflozin-100-mg-Gruppe geringer als in der Placebogruppe (57 pro 1'000 Patientenjahre in der Canagliflozin- bzw. 79 pro 1'000 Patientenjahre in der Placebogruppe).
Pharmakokinetik
Die Pharmakokinetik von Canagliflozin ist bei gesunden Probanden und Patienten mit Diabetes mellitus Typ 2 im Wesentlichen vergleichbar.
Absorption
Die mittlere absolute orale Bioverfügbarkeit von Canagliflozin beträgt ungefähr 65%. Die gleichzeitige Verabreichung von Canagliflozin mit einer fettreichen Mahlzeit hatte keine Auswirkung auf die Pharmakokinetik von Canagliflozin; daher kann Invokana mit oder ohne Mahlzeit eingenommen werden. Aufgrund des Potentials für eine Reduktion postprandialer Glukoseexkursionen durch die verzögerte intestinale Glukoseabsorption wird jedoch empfohlen, Invokana vorzugsweise vor der ersten Mahlzeit des Tages einzunehmen (siehe «Dosierung/Anwendung»).
Nach Verabreichung einer oralen Einmaldosis von 100 mg bzw. 300 mg an gesunde Probanden wurde Canagliflozin rasch absorbiert, wobei Spitzenkonzentrationen im Plasma (mediane Tmax) 1 bis 2 Stunden nach der Einnahme gemessen wurden. Die mittlere AUC betrug 6,8 µg.h/ml bzw. 22,9 µg.h/ml und die maximale Konzentration (Cmax) 1,1 µg.h/ml bzw. 2,8 µg.h/ml nach Dosen von 100 mg bzw. 300 mg. Die Cmax im Plasma und die AUC von Canagliflozin stiegen zwischen 50 mg und 300 mg dosisproportional. Der Steady State wurde unter einmal täglicher Dosierung mit Canagliflozin 100 mg und 300 mg nach 4 bis 5 Tagen erreicht. Canagliflozin zeigt keine zeitabhängige Pharmakokinetik und akkumulierte im Plasma bis zu 36% nach multiplen Dosen von 100 mg und 300 mg.
Distribution
Das mittlere Verteilungsvolumen von Canagliflozin im Steady State nach einer einzelnen intravenösen Infusion bei gesunden Probanden betrug 83,5 L, was auf eine extensive Gewebeverteilung hinweist. Canagliflozin wird extensiv an Plasmaproteine gebunden (99%), hauptsächlich an Albumin. Die Proteinbindung ist unabhängig von den Plasmakonzentrationen von Canagliflozin. Die Plasmaproteinbindung ist bei Patienten mit eingeschränkter Nieren- oder Leberfunktion nicht relevant verändert. Der enterohepatische Kreislauf von Canagliflozin war schwach ausgeprägt.
Metabolismus
Die O-Glukuronidierung ist der wichtigste metabolische Pfad für die Elimination von Canagliflozin, welches hauptsächlich von UGT1A9 und UGT2B4 zu zwei inaktiven O-Glukuronid-Metaboliten glukuronidiert wird. Bei Trägern des UGT1A9*3-Allels bzw. des UGT2B4*2-Allels wurden Anstiege der AUC von Canagliflozin (um 26% bzw. 18%) beobachtet. Diese Steigerungen der Exposition gegenüber Canagliflozin werden nicht als klinisch relevant angesehen. Der CYP3A4-vermittelte (oxidative) Metabolismus von Canagliflozin ist beim Menschen minimal (ungefähr 7%).
Elimination
Nach Gabe einer oralen Einzeldosis von [14C] Canagliflozin an gesunde Probanden wurden insgesamt ungefähr 61% der radioaktiven Dosis in den Faezes ausgeschieden, 41,5%, 7,0% bzw. 3,2% als Canagliflozin, ein hydroxylierter Metabolit bzw. ein O-Glukuronid-Metabolit. Ungefähr 33% der verabreichten radioaktiven Dosis wurde im Urin ausgeschieden, hauptsächlich als O-Glukuronid-Metaboliten (30,5%). Weniger als 1% der Dosis wurde als unverändertes Canagliflozin im Urin ausgeschieden. Die renale Clearance für die Dosen zu 100 mg und 300 mg reichte von 1,30 bis 1,55 ml/min.
Canagliflozin ist ein Arzneimittel mit niedriger Clearance, d.h. mit einer mittleren systemischen Clearance von ungefähr 192 ml/min nach intravenöser Verabreichung an gesunde Probanden.
Die apparente terminale Halbwertszeit (t½) betrug für die Dosis von 100 mg 10,6 Stunden und für die Dosis von 300 mg 13,1 Stunden.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Nach Gabe einer Einzeldosis von 300 mg Canagliflozin betrug das Verhältnis der geometrischen Mittelwerte der Cmax und der AUC∞ von Canagliflozin bei Patienten mit leicht eingeschränkter Leberfunktion (Child-Pugh-Klasse A) im Vergleich zu den Werten bei Probanden mit normaler Leberfunktion 107% bzw. 110% und bei Patienten mit mässig eingeschränkter Leberfunktion (Child-Pugh-Klasse B) 96% bzw. 111%.
Diese Unterschiede werden als klinisch nicht bedeutsam beurteilt. Bei Patienten mit leicht oder mässig eingeschränkter Leberfunktion ist keine Dosisanpassung nötig. Es liegen keine klinischen Erfahrungen bezüglich der Anwendung bei Patienten mit Child-Pugh Klasse C (schwer eingeschränkte Leberfunktion) vor, und aus diesem Grund wird Invokana für diese Patientengruppe nicht empfohlen.
Nierenfunktionsstörungen
Die Pharmakokinetik von Canagliflozin 200 mg wurde in einer offenen Einzeldosisstudie an Probanden mit unterschiedlich schwer eingeschränkter Nierenfunktion [klassifiziert nach der Modification-of-Diet-in-Renal-Disease (MDRD)-eGFR-Formel] im Vergleich zu gesunden Probanden untersucht. An der Studie nahmen 3 Probanden mit normaler Nierenfunktion (eGFR ≥90 ml/min/1,73 m2), 10 Probanden mit leicht eingeschränkter Nierenfunktion (eGFR 60 bis <90 ml/min/1,73 m2), 9 Probanden mit mässig eingeschränkter Nierenfunktion (eGFR 30 bis <60 ml/min/1,73 m2) und 10 Probanden mit schwer eingeschränkter Nierenfunktion (eGFR 15 bis <30 ml/min/1,73 m2) sowie 8 Probanden mit einer terminalen Niereninsuffizienz (ESRD) unter Hämodialyse teil.
Die Cmax von Canagliflozin war bei Probanden mit leicht eingeschränkter Nierenfunktion um 13%, bei Probanden mit mässig eingeschränkter Nierenfunktion um 29% und bei Probanden mit schwer eingeschränkter Nierenfunktion um 29% erhöht, jedoch nicht bei Probanden unter Hämodialyse. Im Vergleich zu gesunden Probanden war die AUC von Canagliflozin im Plasma bei Probanden mit leicht, mässig bzw. schwer eingeschränkter Nierenfunktion um 17%, 63% und 50% erhöht, jedoch bei Probanden mit ESRD und gesunden Probanden ähnlich (siehe «Dosierung/Anwendung», «Warnhinweise und Vorsichtsmassnahmen» sowie «Unerwünschte Wirkungen»).
Canagliflozin wurde durch Hämodialyse in vernachlässigbarem Ausmass entfernt.
Ältere Patienten
Das Alter hatte, entsprechend einer populationsbasierten pharmakokinetischen Analyse, keine klinisch bedeutsame Auswirkung auf die Pharmakokinetik von Canagliflozin (siehe «Dosierung/Anwendung», «Warnhinweise und Vorsichtsmassnahmen» sowie «Unerwünschte Wirkungen»).
Kinder und Jugendliche
Eine Phase-1-Studie untersuchte die Pharmakokinetik und Pharmakodynamik von Canagliflozin bei Kindern und Jugendlichen im Alter von ≥10 bis <18 Jahren mit Typ-2-Diabetes-mellitus. Das beobachtete pharmakokinetische und pharmakodynamische Ansprechen stimmte mit dem Erwachsener überein.
Andere Populationen
Es ist keine Dosisanpassung basierend auf Geschlecht, Ethnizität oder Body Mass Index (BMI) nötig. Diese Charakteristika hatten, entsprechend einer populationsbasierten pharmakokinetischen Analyse, keinen klinisch bedeutsamen Einfluss auf die Pharmakokinetik von Canagliflozin.
Präklinische Daten
Präklinische Daten ergaben keine besondere Gefahr für den Menschen, basierend auf konventionellen Studien zur Sicherheitspharmakologie, Toxizität von wiederholten Dosen, Genotoxizität und Reproduktionstoxizität. In Studien zur Entwicklungstoxizität konnten minimale Störungen in der Entwicklung der Nieren und der Reifung der Föten (Knochenbildung) nicht ausgeschlossen werden. Eine Studie bei juvenilen Ratten, an welche Canagliflozin von Tag 21 postnatal bis Tag 90 verabreicht wurde, ergab keine erhöhte Empfindlichkeit im Vergleich zu den Wirkungen, die bei adulten Ratten beobachtet wurden.
Karzinogenität
Canagliflozin führte in einer Zweijahresstudie unter Dosen von 10, 30 und 100 mg/kg zu keiner Zunahme der Inzidenz von Tumoren bei männlichen und weiblichen CDI-Mäusen. Die höchste Dosis von 100 mg/kg entsprach dem bis zu 14-Fachen der Exposition (gemäss AUC) unter der klinischen Dosis von 300 mg. Unter Canagliflozin kam es bei allen getesteten Dosen von 10, 30 und 100 mg/kg zu einer erhöhten Inzidenz von testikulären Leydigzelltumoren bei männlichen Sprague-Dawley-Ratten; die niedrigste Dosis von 10 mg/kg entsprach ungefähr dem 1,5-Fachen der Exposition (gemäss AUC) unter der klinischen Dosis von 300 mg. Die höheren Dosen von Canagliflozin (100 mg/kg) führten bei männlichen und weiblichen Sprague-Dawley-Ratten zu einer Zunahme der Inzidenz von Phäochromozytomen und renalen tubulären Tumoren; auf der Basis der Exposition gemäss AUC entspricht der No-Observable-Effect-Level (NOEL) von 30 mg/kg/Tag für Phäochromozytome und renale tubuläre Tumoren etwa dem 4,5-Fachen der Exposition unter der klinischen Tagesdosis von 300 mg. Basierend auf präklinischen und klinischen mechanistischen Studien sind die Leydigzell-Tumoren, die renalen tubulären Tumoren und Phäochromozytome spezifisch für Ratten. Die Ursache für Canagliflozin-induzierte renale tubuläre Tumoren und Phäochromozytome bei Ratten scheint in einer Kohlehydrat-Malabsorption als Folge der inhibitorischen Wirkung von Canagliflozin auf intestinales SGLT1 im Darm der Ratte zu liegen; mechanistische klinische Studien haben beim Menschen unter Dosen von Canagliflozin von bis zum Doppelten der maximalen empfohlenen klinischen Dosis keine Kohlehydrat-Malabsorption nachgewiesen. Die Leydigzell-Tumoren sind mit einer Zunahme des luteinisierenden Hormons (LH) verbunden, einem bekannten Entstehungsmechanismus von Leydigzell-Tumoren bei Ratten. In einer 12-wöchigen klinischen Studie kam es zu keiner Erhöhung des basalen LH bei mit Canagliflozin behandelten Männern.
Mutagenität
Canagliflozin war im Ames-Assay mit oder ohne metabolische Aktivierung nicht mutagen. Canagliflozin war im in vitro Maus-Lymphom-Assay mutagen, jedoch nur nach metabolischer Aktivierung. Canagliflozin war in einem oralen in vivo Mikronukleus-Assay sowie einem oralen in vivo Comet-Assay bei Ratten weder mutagen noch klastogen.
Reproduktionstoxizität
In einer prä- und postnatalen Entwicklungsstudie führte Canagliflozin, welches von Tag 6 der Gestation bis Tag 20 der Laktation an weibliche Ratten verabreicht wurde, in maternal toxischen Dosen >30 mg/kg/Tag (entsprechend dem ≥5,9-Fachen der menschlichen Exposition gegenüber Canagliflozin unter der maximalen empfohlenen Humandosis [MRHD]) zu vermindertem Körpergewicht bei männlichen und weiblichen Nachkommen. Die maternale Toxizität war auf verminderte Körpergewichtszunahme beschränkt.
In Studien mit Ratten hatte Canagliflozin bis zu den höchsten Dosen von 100 mg/kg [dem bis zu 19-Fachen der Exposition (gemäss AUC) unter der klinischen Dosis von 300 mg] keine nachteilige Wirkung auf die frühe embryonale Entwicklung, das Paarungsverhalten und die Fertilität.
Die vorliegenden pharmakodynamischen/toxikologischen Daten aus tierexperimentellen Studien zeigten eine Exkretion von Canagliflozin/Metaboliten in die Muttermilch und pharmakologisch vermittelte Wirkungen in gestillten Jungtieren und juvenilen Ratten, denen Canagliflozin verabreicht wurde.
Sonstige Hinweise
Beeinflussung diagnostischer Methoden
Interferenzen des Arzneimittels mit Laboruntersuchungen
1,5-AG-Assay
Die Steigerung der Glukoseausscheidung im Urin durch Canagliflozin kann die renale 1,5-Anhydroglucitol (1,5-AG) Rückresorption vermindern und zu einer Unzuverlässigkeit der 1,5-AG-Messungen bei der Beurteilung der glykämischen Kontrolle führen. Daher sollten 1,5-AG-Assays bei Patienten unter Canagliflozin nicht zur Beurteilung der glykämischen Kontrolle verwendet werden. Es kann ratsam sein, sich für genauere Informationen an den Hersteller des 1,5-AG-Assays zu wenden.
Harnglukosetest
Aufgrund des Wirkungsmechanismus fällt bei Patienten, die Invokana einnehmen, der Harnglukosetest positiv aus.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Bei 15-30°C lagern.
In der Originalverpackung aufbewahren.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer
62956 (Swissmedic).
Packungen
Invokana Filmtabletten zu 100 mg: 30, 100 Filmtabletten (B)
Invokana Filmtabletten zu 300 mg: 30, 100 Filmtabletten (B)
Invokana ist in perforierten PVC/Aluminium Einzeldosisblistern mit 10 Tabletten in jedem Streifen erhältlich.
Zulassungsinhaberin
Janssen-Cilag AG, Zug, ZG.
Stand der Information
Mai 2024