Zusammensetzung
Wirkstoffe
Octocog alfa (Rekombinanter Blutgerinnungsfaktor VIII [rFVIII]), hergestellt aus Baby-Hamster-Nierenzellen, in die das menschliche Faktor-VIII-Gen eingebracht wurde.
Hilfsstoffe
Pulver: Glycinum, Natrii chloridum (enthält 1.9 mg Natrium/Durchstechflasche für 500, 1000 IE / 3.6 mg Natrium/Durchstechflasche für 2000 IE), Calcii chloridum dihydricum, Histidinum, Saccharum, Polysorbatum 80 (aus genetisch verändertem Mais, Zea mays, hergestellt).
Lösungsmittel: 2.5 ml Aqua ad injectabilia (für 500 IE, 1000 IE)/ 5 ml Aqua ad injectabilia (für 2000 IE).
Indikationen/Anwendungsmöglichkeiten
Therapie und Prophylaxe von Blutungen bei Patienten mit Hämophilie A (angeborenem Mangel an Blutgerinnungsfaktor-VIII) für alle Altersstufen.
Kovaltry enthält keinen von Willebrand-Faktor und ist deshalb bei von Willebrand-Jürgens-Syndrom nicht angezeigt.
Dosierung/Anwendung
Die Dosierung und die Dauer der Faktor-VIII-Substitution richten sich nach dem individuellen Bedarf des Patienten (abhängig vom Gewicht des Patienten, dem Grad des Faktor-VIII-Mangels, Ort und Ausmass der Blutung, Hemmkörpertiter und gewünschtem Faktor-VIII-Plasmaspiegel).
Die Berechnung der erforderlichen Faktor-VIII-Dosierung basiert auf dem empirischen Befund, dass die Gabe von 1 IE Faktor-VIII pro kg Körpergewicht die Faktor-VIII-Aktivität im Plasma um 1,5% bis 2,5% – bezogen auf den Normalwert – anhebt.
Die klinische Wirkung von Faktor-VIII ist das wichtigste Element, um die Wirksamkeit der Behandlung zu bewerten.
Unter bestimmten Umständen können höhere Kovaltry-Dosierungen als berechnet notwendig sein, um zufriedenstellende klinische Ergebnisse zu erzielen. Wird der erwartete Faktor-VIII-Spiegel nicht erreicht oder wird die Blutung mit der errechneten Dosis nicht beherrscht, ist an das Vorhandensein eines Faktor-VIII-Hemmkörpers zu denken. Mit geeigneten Labortests sollte das Vorhandensein von Hemmkörpern nachgewiesen und der Hemmkörperspiegel quantifiziert werden. Die notwendige Kovaltry-Dosis ist jedoch bei Vorhandensein von Hemmkörpern höchst variabel und kann nur anhand des klinischen Ansprechens bestimmt werden.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Bedarfsbehandlung
Die erforderliche Dosis wird nach der folgenden Formel berechnet:
Erforderliche Einheiten (IE) = Körpergewicht (kg) × gewünschter Faktor-VIII-Anstieg (% oder IE/dl) × Kehrwert der beobachteten Recovery.
Die übliche Einzeldosis beträgt 10-30 IE/kg Körpergewicht. Bei lebensbedrohlichen oder schweren Blutungen werden höhere Dosierungen empfohlen.
Bei Patienten mit einer Recovery unter 2 [kg/dl] (beispielsweise bei kleinen Kindern) sind unter Umständen höhere Dosen erforderlich.
Die folgende Tabelle (Tabelle 1) enthält Richtwerte für eine Faktor-VIII-Mindestaktivität (in % der Norm) bei Erwachsenen und Kindern, die während des angegebenen Zeitraums nicht unterschritten werden sollten:
|
Schwere der Blutung/Art des chirurgischen Eingriffs |
Benötigter Faktor-VIII-Plasmaspiegel |
Häufigkeit der Dosierung (Stunden)/Behandlungsdauer (Tage) |
|
Blutungen | ||
|
Kleinere Blutungen (Gelenkblutungen im Frühstadium, kleinere Muskelblutungen, Blutungen im Mundbereich) |
20–40 |
Injektion alle 12 bis 24 Stunden; mindestens 1 Tag, bis die (durch Schmerzen erkennbare) Blutung sistiert bzw. Wundheilung erreicht ist. |
|
Mittelschwere bis schwere Blutungen (ausgeprägte Gelenkblutungen, Muskelblutungen oder Hämatome) |
30–60 |
Injektion alle 12 bis 24 Stunden für 3 bis 4 Tage oder länger wiederholen, bis die Schmerzen und Behinderungen beseitigt sind. |
|
Lebensbedrohliche Blutungen |
60–100 |
Injektion alle 8 bis 24 Stunden wiederholen, bis die Gefahr für den Patienten vorüber ist. |
|
Chirurgische Eingriffe | ||
|
Kleinere Eingriffe einschliesslich Zahnextraktionen |
30–60 |
Injektion alle 24 Stunden; mindestens 1 Tag, bis die Wundheilung erreicht ist. |
|
Grössere Eingriffe |
80–100 (prä-und postoperativ) |
wiederholte Gabe alle 8 bis 24 Stunden, bis ausreichende Wundheilung erreicht ist; dann für mindestens weitere 7 Tage einen Faktor-VIII-Spiegel von 30%-60% aufrechterhalten. |
Prophylaxe bei Jugendlichen und Erwachsenen
Zur Langzeitprophylaxe von Blutungen bei Patienten mit schwerer Hämophilie A werden 20-40 IE/kg Körpergewicht zwei- oder dreimal pro Woche empfohlen.
In manchen Fällen, besonders bei jüngeren Patienten, können kürzere Dosierungsabstände oder höhere Einzeldosen erforderlich sein.
Art und Dauer der Anwendung
Kovaltry wird nach dem Auflösen in dem mitgelieferten Lösungsmittel intravenös verabreicht. Die Infusionsgeschwindigkeit sollte sich nach dem Ansprechen jedes einzelnen Patienten richten.
Spezielle Dosierungsanweisungen
Patienten mit Nierenfunktions-/ Leberfunktionsstörung:
Dosisanpassung ist bei Patienten mit Niereninsuffizienz/Leberinsuffizienz in klinischen Studien nicht untersucht worden.
Kinder und Jugendliche:
Kovaltry ist für die Anwendung an Kindern und Jugendlichen geeignet. Sicherheits- und Wirksamkeitsstudien wurden bei Kindern im Alter von 0 bis 12 Jahren durchgeführt. Die empfohlene Dosis zur Prophylaxe beträgt 20-50 IE/kg Körpergewicht 2 Mal wöchentlich, 3 Mal wöchentlich oder alle zwei Tage entsprechend den individuellen Erfordernissen. Die Dosierungsempfehlungen für Patienten über 12 Jahre stimmen mit den Empfehlungen für Erwachsene überein.
Kontraindikationen
Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Bekannte Allergien auf Maus- oder Hamsterproteine.
Warnhinweise und Vorsichtsmassnahmen
Überempfindlichkeit gegen Maus- oder Hamsterproteine
Kovaltry kann Spuren von Mäuse- und Hamstereiweiss enthalten. Diese Proteine können bei manchen Patienten allergische Reaktionen verursachen; allergieartige Überempfindlichkeitsreaktionen sind möglich. Patienten sollten auf mögliche frühe Anzeichen einer Überempfindlichkeitsreaktion oder einer anaphylaktischen Reaktion während der Infusion (Auftreten von Brustbeklemmungen, Schwindel, milder Blutdruckabfall, Übelkeit) hingewiesen werden. Eine symptomatische Behandlung der Überempfindlichkeitsreaktion sollte in angemessener Form eingeleitet werden. Allergische oder anaphylaktische Reaktionen erfordern einen sofortigen Abbruch der Injektion/Infusion. Bei Anaphylaxie sollten die Regeln der aktuellen Standardschocktherapie befolgt werden.
Hemmkörper
Die Bildung von neutralisierenden Antikörpern (Hemmkörpern) gegen Faktor-VIII ist eine bekannte Komplikation bei der Behandlung von Patienten mit Hämophilie A. Diese Hemmkörper sind stets gegen die prokoagulatorische Aktivität von Faktor-VIII gerichtete IgG-Immunglobuline, die in modifizierten Bethesda-Einheiten (B.E.) quantifiziert werden. Das Risiko, Hemmkörper zu entwickeln, ist mit der Exposition gegenüber Blutgerinnungsfaktor-VIII sowie anderen genetischen und umweltbedingten Faktoren korreliert, wobei dieses Risiko innerhalb der ersten 20 Expositionstage am grössten ist. Selten können sich Hemmkörper nach mehr als 100 Expositionstagen entwickeln. Patienten, die mit Faktor-VIII behandelt wurden, sollten sorgfältig klinisch überwacht und mittels geeigneter Labortests auf die Entwicklung von Hemmkörpern untersucht werden.
Katheterbedingte Infektionen
Katheterbedingte Infektionen können auftreten, wenn Kovaltry über einen Zentralkatheter (zentralvenösen Zugang, ZVK) verabreicht wird. Solche Infektionen stehen nicht mit dem Arzneimittel an sich im Zusammenhang.
Kardiovaskuläre Erkrankungen
Hämophilie-Patienten mit kardiovaskulären Risikofaktoren oder kardiovaskulären Erkrankungen haben möglicherweise dasselbe Risiko für ein kardiovaskuläres Ereignis wie Nicht-Hämophilie-Patienten, sobald sich ihre Blutgerinnung mit der Faktor-VIII-Behandlung normalisiert hat. Insofern müssen die kardiovaskulären Risikofaktoren beurteilt werden.
Weitere Hinweise
Dieses Arzneimittel enthält weniger als 1mmol Natrium pro Durchstechflasche, d.h. es ist nahezu «natriumfrei».
Interaktionen
Wechselwirkungen von menschlichen Gerinnungsfaktor-VIII-Produkten mit anderen Arzneimitteln wurden nicht berichtet.
Schwangerschaft, Stillzeit
Tierexperimentelle Reproduktionsstudien wurden mit Faktor-VIII nicht durchgeführt.
Aufgrund des seltenen Auftretens von Hämophilie A bei Frauen liegen über die Anwendung von Faktor-VIII während Schwangerschaft und Stillzeit keine Erfahrungen vor. Deshalb sollte Faktor-VIII in Schwangerschaft und Stillzeit nur bei eindeutiger Indikationsstellung angewendet werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Kovaltry hat keinen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte Wirkungen
Die Angaben zum Sicherheitsprofil von Kovaltry basieren auf insgesamt 236 Patienten (193 PTPs, 43 PUPs/MTPs) aus drei Phase-III-Studien. Die mediane Studiendauer der Sicherheitspopulation betrug 558 Tage (Bereich: 14 – 2436 Tage) mit einem Median von 183 Expositionstagen (Bereich: 1 - 1230 Expositionstage). Die Mehrheit der Patienten (n=201 von 236 resp. 85%) akkumulierte ≥100 Expositionstage. Die Gesamtzahl an Expositionstagen für alle Behandlungen (Prophylaxe, Bedarfsbehandlung, perioperatives Management, pharmakokinetische (PK) Studien und Immuntoleranz-Induktionen) betrug 65029 Expositionstage, wovon 59585 Expositionstage auf die Prophylaxe entfallen. Studienteilnehmende, die Kovaltry im Rahmen von perioperativem Management (n=5) mit Behandlungsperioden von 2-3 Wochen, und solche, die Einzeldosen an Kovaltry im Rahmen von PK-Studien erhalten haben (n=6), wurden aus der gepoolten Sicherheitspopulation ausgeschlossen. Die häufigste unerwünschte Wirkung in der gepoolten Population war Fieber (9.3%), Kopfschmerzen (8.5%) und Ausschlag (5.5%). Die häufigste unerwünschte Wirkung bei PUPs/MTPs war FVIII-Inhibition (siehe auch «Beschreibung ausgewählter Nebenwirkungen/Immunogenität»).
Die im Rahmen klinischer Studien aufgetretenen unerwünschten Wirkungen sind nachfolgend nach Organsystem und Häufigkeit zusammengefasst. Es werden folgende Häufigkeitsangaben zugrunde gelegt: «Sehr häufig» (≥1/10), «häufig» (<1/10, ≥1/100), «gelegentlich» (<1/100, ≥1/1000), «selten» (<1/1000, ≥1/10'000), «sehr selten» (<1/10'000).
Erkrankungen des Blutes und des Lymphsystems
Gelegentlich: Lymphadenopathie.
Erkrankungen des Immunsystems
Sehr häufig: Faktor-VIII-Inhibition bei PUPs/MTPs.
Gelegentlich: Überempfindlichkeitsreaktionen, Faktor-VIII-Inhibition bei PTPs.
Psychiatrische Erkrankungen
Häufig: Schlaflosigkeit.
Erkrankungen des Nervensystems
Häufig: Kopfschmerz, Schwindel.
Gelegentlich: Dysgeusie (Störungen des Geschmackssinnes).
Herzerkrankungen
Gelegentlich: Palpitationen (Herzrasen), Sinustachykardie.
Gefässerkrankungen
Gelegentlich: Hitzewallungen.
Erkrankungen des Gastrointestinaltrakts
Häufig: Bauchschmerzen, Abdominalbeschwerden, Dyspepsie.
Erkrankungen der Haut und des Unterhautgewebes
Häufig: Pruritus (Juckreiz), Hautausschlag, Nesselsucht.
Gelegentlich: allergische Dermatitis.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Fieber, lokale Reaktionen an der Injektionsstelle (Paravasation, Bluterguss, Schmerzen an der Injektionsstelle, Juckreiz, Schwellungen).
Gelegentlich: Brustschmerzen, Brustkorbbeschwerden.
Beschreibung ausgewählter Nebenwirkungen
Faktor-VIII-Inhibition bei PTPs (Gepoolte Studienpopulation, n=193).
Immunogenität
Die Immunogenität von Kovaltry wurde an vorbehandelten Patienten (193 PTPs) und 37 PUPs/6 MTPs untersucht. In klinischen Studien mit Kovaltry an etwa 200 pädiatrischen und erwachsenen Patienten mit schwerer Hämophilie A (Faktor-VIII <1%), die an ≥50 Expositionstagen mit Faktor-VIII-Konzentraten vorbehandelt waren, ist ein Fall von transient niedrigem Hemmkörpertiter aufgetreten. Bei einem 13-jährigen PTP wurde nach 549 Expositionstagen ein Peak Titer von 1.0 B.E./ml gemessen, zeitgleich mit einer akuten Infektion und positiven IgG-Anticardiolipin-Antikörpern. Die Faktor-VIII-Recovery war normal (2.7 IE/dl und IE/kg).
In der klinischen Studie mit nicht vorbehandelten und minimal vorbehandelten Patienten (PUPs/MTPs ) (definiert als ≤ 3 Expositionstage Behandlung mit einem Faktor-VIII Präparat zum Zeitpunkt des Studieneinschlusses) wurden bei 23 von 42 Patienten Faktor-VIII-Inhibitoren festgestellt mit einem Median (Bereich) von 9 (4-42) Expositionstagen zum Zeitpunkt des ersten positiven Inhibitorbefunds. 6 von diesen 23 Patienten hatten einen tiefen Inhibitortiter (≤ 5.0 B.E.) während 17 Patienten einen hohen (≥5.0 B.E.) aufwiesen. In den Patienten, die hohe Inhibitortiter entwickelten, konnten mittels verfügbaren FVIII-Mutationsdaten bei 12 von 14 Patienten Hochrisiko-Mutationen für das Auftreten von Inhibitoren festgestellt werden.
In klinischen Studien mit 51 pädiatrischen vorbehandelten Patienten (PTPs) wurden hinsichtlich der Häufigkeit, Art und Schwere der unerwünschten Wirkungen keine Unterschiede zwischen Kindern und Erwachsenen festgestellt.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Symptome durch Überdosierung von Kovaltry sind bisher nicht bekannt.
Eigenschaften/Wirkungen
ATC-Code
B02BD02
Wirkungsmechanismus
Kovaltry ist ein steriles, stabiles, aufgereinigtes, pyrogenfreies, gefriergetrocknetes Konzentrat, hergestellt aus Baby-Hamster-Nierenzellen, in die das menschliche Blutgerinnungsfaktor-VIII-Gen eingebracht wurde.
Die Aktivität (IE) wird unter Verwendung eines chromogenen Tests bestimmt; die spezifische Aktivität von Kovaltry beträgt 4000 IE/mg Protein. Durch die Gabe von Kovaltry werden die Faktor-VIII-Plasmaspiegel erhöht, wodurch eine vorübergehende Korrektur des Faktor-VIII-Mangels und der Blutungsneigung erreicht wird.
Die aktivierte partielle Thromboplastinzeit (aPTT) ist bei Hämophilie verlängert. Die konventionelle Bestimmung der biologischen Aktivität von Faktor-VIII im Patientenplasma (one stage clotting assay) basiert auf der aPTT. Die Behandlung mit rekombinantem Faktor-VIII bewirkt eine Normalisierung der aPTT, die vergleichbar ist mit der aPTT, die mit aus Plasma gewonnenen Faktor-VIII erzielt wird.
Pharmakodynamik
Siehe Rubrik «Wirkungsmechanismus».
Klinische Wirksamkeit
Therapie und Prophylaxe von Blutungen bei Patienten mit Hämophilie A (alle Altersstufen)
Es wurden drei internationale, multizentrische Phase III Studien mit pädiatrischen, jugendlichen und erwachsenen vorbehandelten Patienten mit schwerer Hämophilie A (Faktor-VIII <1%) durchgeführt.
Insgesamt wurden 247 Patienten (204 PTPs / 37 PUPs/6 MTPs) im Rahmen des Studienprogramms behandelt: 153 Patienten ≥12 Jahre und 94 Patienten <12 Jahre. 208 Patienten (174 PTPs, 31 PUPs/3MTPs) wurden mindestens 12 Monate lang behandelt und 98 dieser Patienten (78 PTPs, 19 PUPs/1MTP) 24 Monate lang.
Studie 1:
In dieser multizentrischen, offenen, unkontrollierten, randomisierten Studie wurden Pharmakokinetik, Sicherheit und Wirksamkeit von Prophylaxebehandlung und Hämostase während chirurgischen Eingriffen an 73 Patienten im Alter von 12-61 Jahren untersucht. Die Studiendauer betrug ein Jahr mit einer optionalen Verlängerung um ein weiteres Jahr. Die Prophylaxe-Patienten (n=62) erhielten eine mediane Dosis von 35 IE/kg (24-43 IE/kg) (2× wöchentlich) bzw. 31 IE/kg (21-42 IE/kg) (3× wöchentlich). Die mediane jährliche Blutungsrate bei Patienten bei prophylaktischer Behandlung war 1.0 (2× wöchentlich) bzw. 2.0 (3× wöchentlich). 86,3% der Blutungen konnten mit ≤2 Infusionen behandelt werden. In der Patientengruppe mit chirurgischen Interventionen wurde die hämostatische Wirksamkeit bei allen Eingriffen (12 grössere, 26 kleinere) mit ausgezeichnet oder gut bewertet.
Studie 2:
In einer weiteren multizentrischen, offenen, unkontrollierten, randomisierten Studie wurde die Prophylaxebehandlung mit der Bedarfsbehandlung während eines einjährigen Behandlungszeitraums bei 80 Patienten im Alter von 14-59 Jahren verglichen, die zuvor nach Bedarf behandelt worden waren. Die Patienten wurden in Gruppen mit Bedarfsbehandlung (n=21; Dosierung nach Einschätzung des Prüfarztes) bzw. Prophylaxe (wöchentlich 2× 20-30 IE/kg, n= 28 oder 3× 30-40 IE/kg, n= 31) randomisiert.
Zur Prophylaxe erhielten die Patienten eine mediane Dosis von 30 IE/kg (21-34 IE/kg, low dose, 2× wöchentlich) oder 37 IE/kg (30-42 IE/kg, high dose, 3× wöchentlich) und zur Bedarfsbehandlung 28 IE/kg (11-49 IE/kg).
Die mediane jährliche Blutungsrate bei Patienten mit prophylaktischer Anwendung war 4.0 (2× wöchentlich) bzw. 2.0 (3× wöchentlich) verglichen mit 60.0 bei Patienten mit Bedarfsbehandlung. 95,5% der Blutungen konnten mit ≤2 Infusionen behandelt werden. Bei allen chirurgischen Interventionen (1 grössere, 20 kleinere) wurde die hämostatische Wirksamkeit mit ausgezeichnet oder gut bewertet.
Studie 3
In dieser multizentrischen, offenen, unkontrollierten Studie wurde die Sicherheit, Wirksamkeit, Pharmakokinetik sowie das perioperative Management der Hämostase während chirurgischen Eingriffen bei 51 vorbehandelten Kindern (≥50 Expositionstage) unter 12 Jahren (n=25 im Alter von 1-5 [mittleres Alter 3,8]; n= 26 im Alter von 6-11 [mittleres Alter 8,8]) untersucht (Studie 3, Teil A). Die mediane Dosierung betrug 36 IE/kg (Alter <6) und 32 IE/kg (Alter ≥6). In der Altersgruppe <6 und ≥6 Jahre wurde bei 97,8% bzw. 81,0% der behandelten Blutungen die Wirksamkeit von Kovaltry als ausgezeichnet oder gut bewertet, bei 0% bzw. 18,9% als mässig und bei 2,3 bzw. 0% als schlecht.
In der klinischen Studie mit PUPs und MTPs unter 6 Jahren (Studie 3, Teil B, wurden insgesamt 43 Patienten (37 PUPs/6 MTPs) untersucht. Die mediane Dosierung betrug 29IE/kg. Die Wirksamkeit bei der Behandlung von Blutungen wurde von 79.0% als ausgezeichnet oder gut bewertet, bei 15.2% als mässig bzw, bei 5.7% als schlecht.
Verlängerungs-Studie zur Studie 3
Von den 94 Patienten der Studie 3 haben 82 (46 aus Teil A, 36 aus Teil B) die Behandlung in der Verlängerungsstudie fortgesetzt über einen medianen Zeitraum von 3.1 Jahren (0.3 - 6.4). Insgesamt (Studie 3 plus Verlängerung) betrug die mediane Studiendauer 3.8 Jahre (0.8 - 6.7). Die Mehrheit der Teilnehmenden (76 von 82, 92.7%) akkumulierten mindestens 100 Expositionstage. In der Verlängerungsstudie erhielten 67 Patienten eine Prophylaxe-Behandlung mit einer medianen Dosierung von 34 IE/kg (16 – 57 IE/kg). Die Häufigkeit der Behandlung erfolgte mehrheitlich 2 Mal pro Woche bei einer Mehrheit der Patienten (61 von 67, 91%). Die mediane jährliche Blutungsrate beobachtet innert 48 Stunden bei Patienten mit prophylaktischer Anwendung war 0.7 (0.0 – 1.9), die von der Injektionszeit unabhängige mediane jährliche Blutungsrate betrug 1.9 (0.3 - 3.9).
Die Mehrheit der insgesamt 472 Blutungen (394 von 472, 83.5%) wurden mit 1-2 Kovaltry-Infusionen behandelt. Die Wirksamkeit wurde in den meisten Fällen (415 von 472, 87.9%) als ausgezeichnet oder gut bewertet.
Pharmakokinetik
Absorption
Durch die i.v. Applikation ist das Präparat sofort und vollständig bioverfügbar.
Distribution
Siehe «Absorption».
Metabolismus
Siehe «Elimination».
Elimination
Nach Gabe von Kovaltry verläuft die Elimination von Faktor-VIII aus dem Plasma biphasisch mit einer klinisch relevanten Halbwertzeit, die mit der mittleren terminalen Halbwertzeit von Plasma-Faktor-VIII (ca. 13 Stunden) vergleichbar ist.
Kinetik spezieller Patientengruppen
Die pharmakokinetischen (PK) Eigenschaften von Kovaltry wurden bei vorbehandelten Erwachsenen, Jugendlichen und Kindern untersucht (siehe Tabelle 2). Wiederholte PK-Messungen nach 6 bis 12-monatiger Prophylaxe mit Kovaltry zeigten keine relevanten Veränderungen der pharmakokinetischen Parameter nach Langzeit-Behandlung.
Tabelle 2: Pharmakokinetische Parameter (Geometrisches Mittel [%CV]) für Kovaltry basierend auf den Resultaten mit dem Chromogenic Assay
|
Parameter |
0 bis <6 Jahre |
6 bis <12 Jahre |
12 bis <18 Jahre |
≥18 Jahre |
|
AUC [IE*h/dl] a |
1499.3 (27.6) |
1010.5 (59.3) |
1519.5 (30.1) |
1989.8 (35.9) |
|
Cmax [IE/dl] |
85.3 (36.6) |
82.2 (24.5) |
124.0 (46.4) |
131.6 (15.8) |
|
t½ [h] a |
11.8 (20.6) |
11.0 (31.2) |
13.7 (35.9) |
13.8 (27.0) |
|
CL [dl/h/kg] a |
0.032 (28.6) |
0.049 (59.5) |
0.033 (30.1) |
0.025 (35.9) |
|
MRTIV [h] a |
17.4 (19.3) |
15.9 (37.6) |
19.2 (28.4) |
19.3 (27.2) |
|
Vss [dl/kg] a |
0.56 (23.3) |
0.79 (29.6) |
0.63 (57.6) |
0.49 (21.1) |
a n=7 für PTPs 0 bis <6 Jahre
Anhand der PK- und Recovery-Daten von 183 Patienten aus 3 klinischen Studien wurde eine Populationsanalyse für Kovaltry durchgeführt. Die Analyse aller erfassten in-vivo-Recoverys (IVR) an erwachsenen/jugendlichen vorbehandelten Patienten zeigte im Median einen Anstieg von Faktor-VIII: C von >2 IE/dl pro IE/kg Körpergewicht (siehe Tabelle 3). Dieser Wert ist ähnlich den berichteten Werten für aus menschlichem Plasma gewonnenem Faktor-VIII. Für Kinder und Jugendliche betrugen die medianen Werte der in-vivo-Recovery 1,6 kg/dl (Alter 0 bis <6 Jahre) bzw. 1,8 kg/dl (Alter 6 bis 12 Jahre). Im Lauf der 6- bis 12-monatigen Behandlungsphase waren keine relevanten Veränderungen zu verzeichnen.
Tabelle 3: In-vivo-Recovery (kg/dl)
|
|
Leopold Kids Teil A |
Leopold I + II | |
|
Altersgruppe |
0-<6 Jahre |
6-12 Jahre |
≥12 Jahre |
|
Anzahl Patienten |
N=24 |
N=25 |
N=115 |
|
Chromogenic assay Werte |
Starta 1.6 (1.3; 1.9) |
Starta 1.7 (1.4; 2.0) |
2.3 (1.8; 2.6) |
|
One-stage assay Werte |
-- |
-- |
2.2 (1.8; 2.4) |
a Start und Ende der 6- Monats Studien Periode
Präklinische Daten
Mit nicht-klinischen Studien zur Beurteilung der Wirksamkeit von Kovaltry an Hämophilie-A-Mausmodellen wurde die Wiederherstellung der Hämostase nachgewiesen. Aus den präklinischen Untersuchungen zur Sicherheitspharmakologie, akuten Toxizität, Toxizität nach wiederholter Gabe und Genotoxizität ergaben sich keine Sicherheitsbedenken für den Menschen.
Die embryonale/fötale Entwicklung wurde nicht tierexperimentell bewertet, da es sich bei Faktor-VIII um ein endogenes Ersatzprotein handelt und zudem die Patientenpopulation vorwiegend männlich ist.
Bei Studien zur Toxizität bei wiederholter Verabreichung wurde keine Auswirkung auf die männlichen Fortpflanzungsorgane beobachtet. Beim Menschen wurden keine Auswirkungen dieses endogenen Proteins auf die Fertilität festgestellt.
Mit dem Mauslymphomtest wurde keine Genotoxizität von Kovaltry nachgewiesen.
Karzinogenizitätsstudien wurden nicht durchgeführt, da Faktor-VIII ein endogenes Ersatzprotein ist und rekombinanter Faktor-VIII keinerlei genotoxisches oder karzinogenes Potenzial zeigt.
In Einzel- und Mehrfachdosisstudien an Ratten, Kaninchen und Hunden wurde bei Dosen, die um ein Mehrfaches höher als die empfohlene klinische Dosis (bezogen auf das Körpergewicht) waren, keine Toxizität aufgezeigt.
Sonstige Hinweise
Inkompatibilitäten
Kovaltry darf nicht mit anderen Arzneimitteln und Infusionslösungen gemischt werden.
Beeinflussung diagnostischer Methoden
Siehe Rubrik «Wirkungsmechanismus».
Haltbarkeit
Das auf der Packung unter «EXP» angegebene Datum darf auf keinen Fall überschritten werden. Die gebrauchsfertige Lösung muss innerhalb von 3 Stunden verwendet werden.
Besondere Lagerungshinweise
In der Originalpackung im Kühlschrank (2-8 °C) aufbewahren. Vor Licht schützen. Nicht einfrieren.
Das lyophilisierte Pulver kann für eine begrenzte Periode von 12 Monaten bei Raumtemperatur (15–25 °C) aufbewahrt werden. Wenn Kovaltry ausserhalb des Kühlschranks gelagert wird, sollte das Datum der Herausnahme notiert und Packung und Durchstechflasche mit dem neuen Verfalldatum versehen werden. In diesem Fall verfällt das Produkt am Ende der 12-Monats Periode. Dieses neue Verfalldatum darf jedoch auf keinen Fall länger sein als das auf der Packung unter «EXP» angegebene Datum. Wenn Kovaltry bei Raumtemperatur gelagert wurde, darf es nicht erneut im Kühlschrank gelagert werden.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Sollte eine der Komponenten der Packung geöffnet oder beschädigt sein, darf diese Komponente nicht verwendet werden.
Bei der Herstellung und anschliessenden intravenösen Gabe der Lösung ist darauf zu achten, dass aseptische Bedingungen eingehalten werden.
Parenteral anzuwendende Arzneimittel sollten vor Verwendung auf Partikel und Verfärbungen geprüft werden. Kovaltry nicht verwenden, wenn nach der Zubereitung der Lösung Partikel oder eine Trübung bemerkt werden.
Kovaltry sollte mit den Komponenten, die mitgeliefert werden, aufbereitet und angewendet werden, da als Folge einer Adsorption von humanem Gerinnungsfaktor-VIII an inneren Oberflächen mancher Infusionssets die injizierte Faktor-VIII-Dosis reduziert sein kann.
Vor der Anwendung muss die gebrauchsfertige Lösung durch Verwendung des mitgelieferten Adapters mit integriertem Filter filtriert werden, um allfällige Partikel zurückzuhalten.
Das mitgelieferte Injektionsbesteck darf nicht verwendet werden, um Blutentnahmen zu machen, da es einen in-line-Filter enthält. Falls eine Blutentnahme vorgängig zu einer Kovaltry-Infusion nötig ist, muss ein Injektionsbesteck ohne Filter verwendet werden.
Handhabung des Adapters und des Injektionsbesteck (Adapter)
Nicht alle Schritte wurden bebildert. Bitte Text unbedingt beachten.
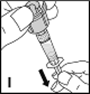

1.Waschen Sie Ihre Hände gründlich mit Wasser und Seife. Arbeiten Sie auf einem sauberen Untergrund.
2.Erwärmen Sie die ungeöffnete Produktflasche und die mit Lösungsmittel gefüllte Spritze in Ihren Händen annähernd auf Körpertemperatur (nicht mehr als 37 °C). Wischen Sie sichtbare Feuchtigkeit von der Produktflasche ab.
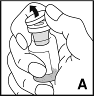
3.Entfernen Sie die Schutzkappe von der Produktflasche (A) und reinigen Sie den Gummistopfen mit einem Alkoholtupfer (oder verwenden Sie einen Desinfektionsspray).
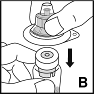
4.Stellen Sie die Flasche auf eine feste, rutschsichere Unterlage. Ziehen Sie die Papierfolie von der Verpackung des Adapters, aber nehmen Sie den Adapter nicht aus der Verpackung. Halten Sie die Verpackung des Adapters fest, platzieren Sie diese über die Durchstechflasche mit Pulver und drücken sie den Adapter fest auf die Flasche (B). Der Flaschenadapter rastet über der Flaschenkappe ein. Entfernen Sie zu diesem Zeitpunkt noch nicht die Verpackung des Adapters.
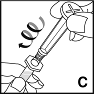
5.Entnehmen Sie die Spritze mit der Verdünnungslösung und halten Sie die wassergefüllte Spritze aufrecht. Nehmen Sie den Kolbenstempel wie in der Abbildung gezeigt und schrauben Sie den Stempel durch Drehen im Uhrzeigersinn in den Gewindestopfen (C).
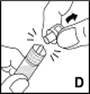
6.Halten Sie den Spritzenzylinder fest und entfernen Sie die Spritzenkappe von der Spitze (D). Berühren Sie die Spitze der Spritze nicht mit Ihrer Hand oder irgendeiner Oberfläche. Legen Sie die Spritze zur späteren Benutzung zur Seite.
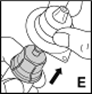
7.Entfernen Sie jetzt die Verpackung des Adapters und entsorgen Sie diese (E).
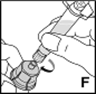
8.Verbinden Sie nun durch eine Drehung im Uhrzeigersinn die Spritze mit dem Gewinde des Adapters (F).
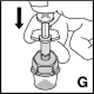
9.Drücken Sie das Lösungsmittel langsam in die Produktflasche (G).
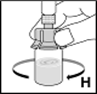
10.Lösen Sie das Pulver mit kreisenden Bewegungen (H). Nicht schütteln! Stellen Sie sicher, dass das Pulver vollständig gelöst ist. Verwenden Sie keine Lösungen mit sichtbaren Partikeln oder Trübungen.
11.Halten Sie die Flasche mit Adapter und Spritze nach oben (I) und füllen Sie die Spritze durch langsamen, gleichmässigen Zug des Kolbens. Stellen Sie sicher, dass der gesamte Inhalt der Flasche in die Spritze überführt wurde. Entlüften Sie die Spritze.
12.Falls Sie mehr als eine Dosis benötigen, bereiten Sie die gewünschte Produktmenge vor. Wiederholen Sie dazu die Schritte 2.–11.
13.Legen Sie einen Stauschlauch an. Lokalisieren Sie die Injektionsstelle, reinigen Sie diese mit einem Alkoholtupfer und desinfizieren Sie die Injektionsstelle. Punktieren Sie die Vene und fixieren Sie das Injektionsbesteck mit einem Pflaster.
14.Schrauben Sie die Spritze vom Flaschenadapter, ohne den Kolben zu bewegen (der Flaschenadapter soll auf der Durchstechflasche verbleiben).
15.Verbinden Sie das Injektionsbesteck mit einer Rechtsdrehung mit der Spritze und vermeiden Sie Bluteintritt in die Spritze (J).
16.Lösen Sie den Stauschlauch!
17.Lösung über einen Zeitraum von mehreren Minuten intravenös injizieren, dabei Nadelposition kontrollieren! Die Injektionsgeschwindigkeit richtet sich nach persönlichem Befinden des Patienten (maximale Injektionsgeschwindigkeit: 2 ml/Minute).
18.Wenn eine weitere Dosis erforderlich ist, entfernen Sie mit einer Linksdrehung die leere Spritze und verbinden Sie eine neue, vorbereitete Spritze mit dem Injektionsbesteck.
19.Wenn keine weitere Dosis erforderlich ist, entfernen Sie das Injektionsbesteck zusammen mit der Spritze. Drücken Sie einen Tupfer ca. 2 Minuten fest auf die Injektionsstelle und halten Sie dabei den Arm gestreckt. Schliesslich versorgen Sie die Injektionsstelle mit einem kleinen Druckverband und allenfalls mit einem Pflaster falls nötig.
Zulassungsnummer
65773 (Swissmedic).
Packungen
Kovaltry 500 IE (B)
1 Durchstechflasche mit Vial-Adapter und Pulver mit 500 IE Octocog alfa.
1 Fertigspritze mit Lösungsmittel (2,5 ml Wasser für Injektionszwecke) und separat beigefügtem Spritzenstempel.
1 Injektionsbesteck
Kovaltry 1000 IE (B)
1 Durchstechflasche mit Vial-Adapter und Pulver mit 1000 IE Octocog alfa.
1 Fertigspritze mit Lösungsmittel (2,5 ml Wasser für Injektionszwecke) und separat beigefügtem Spritzenstempel.
1 Injektionsbesteck
Kovaltry 2000 IE (B)
1 Durchstechflasche mit Vial-Adapter und Pulver mit 2000 IE Octocog alfa.
1 Fertigspritze mit Lösungsmittel (5 ml Wasser für Injektionszwecke) und separat beigefügtem Spritzenstempel.
1 Injektionsbesteck
Zulassungsinhaberin
Bayer (Schweiz) AG, 8045 Zürich.
Stand der Information
Januar 2022.