Zusammensetzung
Wirkstoffe
Obeticholsäure
Hilfsstoffe
Mikrokristalline Cellulose (E 460), Natriumstärkeglykolat (Typ A), Magnesiumstearat, Teilhydrolisierter Polyvinylalkohol (E 1203), Macrogol 3350 (E 1521), Talkum (E 553b), Farbstoffe Titandioxid (E 171) und Eisenoxid gelb (E 172).
Indikationen/Anwendungsmöglichkeiten
OCALIVA, ein Farnesoid X-Rezeptor (FXR)-Agonist, ist indiziert für die Behandlung der primären biliären Cholangitis (PBC)
§ohne Zirrhose oder
§mit kompensierter Zirrhose ohne Anzeichen einer portalen Hypertension,
in Verbindung mit Ursodesoxycholsäure (UDCA) bei Erwachsenen, die unzureichend auf UDCA ansprechen oder als Monotherapie bei Erwachsenen, die UDCA nicht tolerieren können.
Diese Indikation wurde aufgrund der Absenkung des Spiegels der alkalischen Phosphatase (ALP) zugelassen.
Eine Verbesserung der Überlebensrate oder krankheitsbedingten Symptomen wurde nicht belegt. Die Aufrechterhaltung der Zulassung für diese Indikation kann vom klinischen Nutzen abhängen, der in Bestätigungsstudien getestet und beschrieben wird.
Dosierung/Anwendung
Anfangsdosis und Dosistitration
Vor dem Beginn der Behandlung mit OCALIVA ist zu bestimmen, ob der Patient eine dekompensierte Leberzirrhose (einschliesslich Child-Pugh-Klassifikation B oder C) hat oder in der Vergangenheit ein Dekompensationsereignis hatte oder eine kompensierte Zirrhose mit Anzeichen einer portalen Hypertension (z. B. Aszites, gastroösophageale Varizen, persistierende Thrombozytopenie <150 x 109/l) aufweist, da OCALIVA bei diesen Patienten kontraindiziert ist (siehe «Kontraindikationen» und «Warnhinweise und Vorsichtsmassnahmen»).
Empfohlenes Dosierungsschema
Nachfolgend finden Sie das empfohlene Dosierungsschema für OCALIVA für PBC-Patienten ohne Zirrhose oder mit kompensierter Zirrhose, die keine Anzeichen einer portalen Hypertension aufweisen, die seit mindestens 1 Jahr kein ausreichendes biochemisches Ansprechen auf eine angemessene Dosierung von UDCA erreicht haben oder eine Unverträglichkeit gegenüber UDCA aufweisen (siehe «Klinische Wirksamkeit»):
§Die empfohlene Anfangsdosis von Obeticholsäure ist einmal täglich 5 mg für die ersten 3 Monate.
§Nach den ersten 3 Monaten kann für Patienten, bei denen keine angemessene Verringerung der alkalischen Phosphatase (ALP) und/oder des Gesamtbilirubins erreicht werden konnte und die Obeticholsäure vertragen, auf eine maximale Dosis von einmal täglich 10 mg erhöht werden.
Monitoring zur Bewertung der Sicherheit, Notwendigkeit des Absetzens von OCALIVA
Überwachen Sie die Patienten während der Behandlung mit OCALIVA routinemässig hinsichtlich des biochemischen Ansprechens, der Verträglichkeit und des Fortschreitens der PBC. Überwachen Sie Patienten mit kompensierter Zirrhose, begleitender Lebererkrankung (z. B. Autoimmunhepatitis, alkoholische Lebererkrankung) und/oder schweren interkurrenten Erkrankungen engmaschig auf neue Anzeichen einer portalen Hypertension (z. B. Aszites, gastroösophageale Varizen, persistierende Thrombozytopenie <150 x 109/l) oder Erhöhungen des Gesamtbilirubins, des direkten Bilirubins oder der Prothrombinzeit über der Obergrenze des Normalbereichs. Bei Patienten, bei denen Labor- oder klinische Nachweise einer hepatischen Dekompensation auftreten, die eine kompensierte Zirrhose aufweisen und Anzeichen einer portalen Hypertension entwickeln, bei denen klinisch signifikante hepatische Nebenwirkungen auftreten oder die einen totalen Gallengangsverschluss entwickeln, muss OCALIVA dauerhaft abgesetzt werden (siehe «Kontraindikationen» und «Warnhinweise und Vorsichtsmassnahmen»).
Management von Patienten mit nicht tolerierbarem Pruritus unter OCALIVA
Bei Patienten mit Unverträglichkeit aufgrund von nicht tolerierbarem Pruritus unter OCALIVA sollten eine oder mehrere der folgenden Massnahmen in Betracht gezogen werden:
§Zusatz von Gallensäure bindenden Harzen oder Antihistaminika.
§Reduktion der Dosis von OCALIVA auf:
o5 mg jeden zweiten Tag, bei Patienten mit Unverträglichkeit von 5 mg einmal täglich.
o5 mg einmal täglich, bei Patienten mit Unverträglichkeit von 10 mg einmal täglich.
§Vorübergehendes Absetzen der OCALIVA-Dosierung für bis zu 2 Wochen und anschliessende Wiederaufnahme mit reduzierter Dosis.
Bei Patienten, deren Behandlung reduziert oder ausgesetzt wird, ist die Dosierung basierend auf dem biochemischen Ansprechen und der Verträglichkeit zu titrieren (siehe «Empfohlenes Dosierungsschema»).
Für Patienten, bei denen weiterhin ein anhaltender, nicht tolerierbarer Pruritus auftritt, ist ein Abbruch der Therapie mit OCALIVA in Betracht zu ziehen.
Gallensäure bindende Harze
Bei Patienten, die Gallensäure bindende Harze einnehmen, sollte Obeticholsäure mindestens 4 bis 6 Stunden vor oder 4 bis 6 Stunden nach der Einnahme eines Gallensäure bindenden Harzes verabreicht werden bzw. in möglichst grossem Abstand dazu (siehe «Interaktionen»).
Spezielle Patientengruppen
Ältere Patienten (≥65 Jahre)
Bisher liegen nur begrenzte Daten zu älteren Patienten vor. Bei älteren Patienten ist keine Dosisanpassung erforderlich (siehe «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit Nierenfunktionsstörungen ist keine Dosisanpassung erforderlich (Untersuchung einer Einzeldosis, siehe «Pharmakokinetik»). Die Sicherheit von OCALIVA in der Erhaltungstherapie bei Patienten mit Niereninsuffizienz wurde nicht untersucht.
Patienten mit Leberfunktionsstörungen
Leberdekompensation und Leberversagen, bisweilen mit tödlichem Ausgang oder mit der Folge einer Lebertransplantation, wurden unter der Behandlung mit OCALIVA bei PBC-Patienten mit kompensierter oder dekompensierter Zirrhose gemeldet (siehe «Warnhinweise und Vorsichtsmassnahmen»). OCALIVA ist bei Patienten mit dekompensierter Leberzirrhose (z. B. Child-Pugh-Klassifikation B oder C), mit einem früheren Dekompensationsereignis oder mit kompensierter Zirrhose sowie Anzeichen einer portalen Hypertension (z. B. Aszites, gastroösophageale Varizen, persistierende Thrombozytopenie <150 x 109/l) kontraindiziert (siehe «Kontraindikationen» und «Warnhinweise und Vorsichtsmassnahmen»).
In klinischen Studien zu PBC wurde eine Dosis-Wirkung-Beziehung für das Auftreten hepatischer Nebenwirkungen mit OCALIVA beobachtet (siehe «Warnhinweise und Vorsichtsmassnahmen»). Bei Patienten mit mittelschwerer bis schwerer Leberfunktionsstörung ist die Plasma-Exposition gegenüber Obeticholsäure sowie ihren aktiven Konjugaten signifikant erhöht (siehe «Pharmakokinetik»).
Während der Behandlung mit OCALIVA müssen die Patienten in Bezug auf das Auftreten hepatischer Nebenwirkungen überwacht werden. Bei Patienten, bei denen klinisch signifikante hepatische Nebenwirkungen auftreten, muss OCALIVA dauerhaft abgesetzt werden (siehe oben «Monitoring zur Bewertung der Sicherheit, Notwendigkeit des Absetzens von OCALIVA» sowie «Kontraindikationen», «Warnhinweise und Vorsichtsmassnahmen»).
Kinder und Jugendliche
Es gibt keinen relevanten Nutzen von Obeticholsäure bei Kindern und Jugendlichen zur Behandlung der primären biliären Cholangitis (PBC).
Art der Anwendung
Die Tablette ist oral, mit oder ohne Mahlzeiten, einzunehmen.
Kontraindikationen
§Dekompensierte Leberzirrhose (z. B. Child-Pugh-Klassifikation B oder C) oder ein früheres Dekompensationsereignis (siehe «Warnhinweise und Vorsichtsmassnahmen»).
§Kompensierte Zirrhose sowie Anzeichen einer portalen Hypertension (z. B. Aszites, gastroösophageale Varizen, persistierende Thrombozytopenie <150 x 109/l) (siehe «Warnhinweise und Vorsichtsmassnahmen»).
§Totaler Gallengangsverschluss.
§Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss «Zusammensetzung».
Warnhinweise und Vorsichtsmassnahmen
Leberdekompensation und Leberversagen bei PBC-Patienten mit Zirrhose
Leberdekompensation und Leberversagen, bisweilen mit tödlichem Ausgang oder mit der Folge einer Lebertransplantation, wurden unter der Behandlung mit OCALIVA bei PBC-Patienten mit kompensierter oder dekompensierter Zirrhose gemeldet. Bei den Fällen, die nach der Markteinführung gemeldet wurden, betrug die mediane Zeit bis zur Leberdekompensation (z. B. neu auftretender Aszites) bei Patienten mit kompensierter Zirrhose 4 Monate. Die mediane Zeit bis zu einem neuen Dekompensationsereignis (z. B. hepatische Enzephalopathie) betrug bei Patienten mit dekompensierter Zirrhose 2,5 Monate.
Einige dieser Fälle traten bei Patienten mit dekompensierter Leberzirrhose auf, wenn sie mit einer höheren als der für diese Patientengruppe empfohlenen Dosis behandelt wurden; es wurden jedoch weiterhin Fälle von Leberdekompensation und Leberversagen bei Patienten mit dekompensierter Leberzirrhose berichtet, auch wenn sie die empfohlene Dosis erhielten.
Hepatotoxizität wurde in den klinischen Studien zu OCALIVA beobachtet. Eine Dosis-Wirkung-Beziehung wurde in zwei dreimonatigen, placebokontrollierten klinischen Studien bei Patienten mit vorwiegend frühzeitiger PBC für das Auftreten hepatischer Nebenwirkungen, einschliesslich Ikterus, Verschlechterung des Aszites und Aufflackern der primär biliären Cholangitis, bei OCALIVA-Dosierungen von 10 mg einmal täglich bis 50 mg einmal täglich (bis zum 5-Fachen der höchsten empfohlenen Dosierung) bereits einen Monat nach der Aufnahme der OCALIVA-Behandlung beobachtet (siehe «Überdosierung»). In einer gepoolten Analyse von drei placebokontrollierten klinischen Studien bei Patienten mit PBC im Frühstadium waren die expositionsbereinigten Inzidenzraten für alle schwerwiegenden und anderweitig klinisch signifikanten hepatischen Nebenwirkungen sowie isolierte Erhöhungen der biochemischen Lebertests pro 100 Patientenexpositionsjahre (PEY): 5,2 in der OCALIVA 10 mg-Gruppe (höchste empfohlene Dosierung), 19,8 in der OCALIVA 25 mg-Gruppe (das 2,5-Fache der höchsten empfohlenen Dosierung) und 54,5 in der OCALIVA 50 mg-Gruppe (das 5-Fache der höchsten empfohlenen Dosierung) im Vergleich zu 2,4 in der Placebo-Gruppe.
Bei Patienten, die Obeticholsäure einnehmen, wurden erhöhte Konzentrationen an Alaninaminotransferase (ALT) und Aspartataminotransferase (AST) festgestellt. Ausserdem wurden klinische Anzeichen und Symptome einer Leberdekompensation festgestellt. Diese Ereignisse traten teilweise bereits im ersten Behandlungsmonat auf.
Nach Beginn der Behandlung müssen alle Patienten routinemässig durch Labortests und klinische Bewertungen hinsichtlich eines Fortschreitens der PBC, einschliesslich hepatischer unerwünschter Ereignisse, überwacht werden, um zu bestimmen, ob ein Abbruch der Behandlung mit Obeticholsäure notwendig ist (siehe «Dosierung/Anwendung»).
Überwachen Sie Patienten mit kompensierter Zirrhose, begleitender Lebererkrankung (z. B. Autoimmunhepatitis, alkoholische Lebererkrankung) und/oder schweren interkurrenten Erkrankungen engmaschig auf neue Anzeichen der portalen Hypertension (z. B. Aszites, gastroösophageale Varizen, persistierende Thrombozytopenie <150 x 109/l) oder Erhöhungen des Gesamtbilirubins, des direkten Bilirubins oder der Prothrombinzeit über Obergrenze des Normalbereichs, um festzustellen, ob ein Absetzen des Arzneimittels erforderlich ist (siehe «Dosierung/Anwendung»).
Setzen Sie OCALIVA dauerhaft ab bei Patienten, die:
§Labor- oder klinische Anzeichen einer hepatischen Dekompensation entwickeln (z. B. Aszites, Ikterus, Varizenblutung, hepatische Enzephalopathie, Child-Pugh-Klassifikation B oder C) (siehe «Kontraindikationen»),
§kompensierte Zirrhose haben und Anzeichen einer portalen Hypertension entwickeln (z. B. Aszites, gastroösophageale Varizen, persistierende Thrombozytopenie <150 x 109/l) (siehe «Kontraindikationen»),
§klinisch signifikante hepatische Nebenwirkungen erfahren,
§einen totalen Gallengangsverschluss entwickeln (siehe «Kontraindikationen»).
Die Behandlung mit Obeticholsäure ist bei schweren interkurrenten Erkrankungen zu unterbrechen und die Leberfunktion des Patienten muss überwacht werden. Nach Abklingen der interkurrenten Erkrankung müssen die potenziellen Risiken und der potenzielle Nutzen einer Wiederaufnahme der Behandlung mit Obeticholsäure abgewogen werden.
Starker Pruritus
In der klinischen Studie der Phase III für OCALIVA (POISE) wurde starker Pruritus bei 23% der Patienten im mit OCALIVA 10 mg behandelten Arm gemeldet, bei 19% der Patienten im OCALIVA-Titrationsarm und bei 7% der Patienten im Placebo-Arm. Die mediane Zeitdauer bis zum Einsetzen von starkem Pruritus betrug 11, 158 bzw. 75 Tage bei Patienten der Arme OCALIVA 10 mg, OCALIVA-Titrierung und Placebo. Die Managementstrategien umfassen den Zusatz von Gallensäure bindenden Harzen oder Antihistaminika, Dosisreduzierung, reduzierte Dosierungshäufigkeit und/oder vorübergehendes Aussetzen der Dosis (siehe «Dosierung/Anwendung» sowie «Unerwünschte Wirkungen»).
Interaktionen
Wirkung anderer Arzneimittel auf Obeticholsäure
Gallensäure bindende Harze
Gallensäure bindende Harze, wie Cholestyramin, Colestipol oder Colesevelam adsorbieren und reduzieren die Gallensäureabsorption und können die Wirksamkeit von Obeticholsäure reduzieren. Bei gleichzeitiger Verabreichung von Gallensäure bindenden Harzen hat die Einnahme der Obeticholsäure mindestens 4 bis 6 Stunden vor oder 4 bis 6 Stunden nach der Einnahme eines Gallensäure bindenden Harzes zu erfolgen bzw. in möglichst grossem Abstand dazu.
Wirkung von Obeticholsäure auf andere Arzneimittel
CYP1A2-Substrate mit geringer therapeutischer Breite
Obeticholsäure kann die Exposition gegenüber Begleitmedikamenten erhöhen, bei denen es sich um CYP1A2-Substrate handelt. Bei CYP1A2-Substraten mit geringer therapeutischer Breite (z.B. Theophyllin und Tizanidin) wird eine Therapieüberwachung empfohlen.
Schwangerschaft, Stillzeit
Schwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Obeticholsäure bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte gesundheitsschädigende Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe «Präklinische Daten»). Als Vorsichtsmassnahme darf Obeticholsäure während der Schwangerschaft nicht angewendet werden, es sei denn, der klinische Zustand der werdenden Mutter erfordert eine Behandlung.
Stillzeit
Es ist nicht bekannt, ob Obeticholsäure in die Muttermilch übergeht. Ausgehend von tierexperimentellen Studien und der intendierten Pharmakologie wird nicht erwartet, dass Obeticholsäure das Stillen oder das Wachstum oder die Entwicklung eines gestillten Kindes beeinträchtigt. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit Obeticholsäure verzichtet werden soll / die Behandlung mit Obeticholsäure zu unterbrechen ist. Dabei soll sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau berücksichtigt werden (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Obeticholsäure hat keinen oder einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte Wirkungen
Insgesamt sind 432 Patienten mit PBC in drei doppelblinden, placebokontrollierten Studien beurteilt worden. Von diesen Patienten wurden 290 mindestens 6 Monate lang mit OCALIVA behandelt, 232 wurden mindestens 12 Monate lang und 70 mindestens 2 Jahre lang behandelt. Es gab 131 Patienten, die OCALIVA 10 mg einmal täglich, und 70 Patienten, die OCALIVA 5 mg einmal täglich erhielten.
Zusammenfassung des Sicherheitsprofils
Die am häufigsten gemeldeten unerwünschten Wirkungen waren Pruritus (63%) und Müdigkeit (22%). Gesamthaft betrug die Therapieabbruchrate 12% im OCALIVA 10 mg-Arm, 10% im OCALIVA-Titrationsarm und 4% im Placeboarm in der OCALIVA-Phase III Studie POISE. Die häufigste zu einem Abbruch führende unerwünschte Wirkung war Pruritus. Die meisten Fälle von Pruritus traten im ersten Behandlungsmonat auf und zeigten bei Fortsetzung der Dosierung eine Tendenz zum Abklingen im Verlauf der Zeit.
Liste der unerwünschten Wirkungen
Die in der klinischen Studie der Phase III für OCALIVA gemeldeten unerwünschten Wirkungen sind in der folgenden Tabelle nach MedDRA-Systemorganklasse und Häufigkeit aufgeführt. Die Häufigkeiten sind definiert als: sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1‘000, <1/100), selten (≥1/10‘000, <1/1‘000), sehr selten (<1/10‘000) und nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Innerhalb jeder Häufigkeitskategorie sind die unerwünschten Reaktionen absteigend nach ihrem Schweregrad aufgeführt.
Tabelle 1: Häufigkeit von unerwünschten Wirkungen bei PBC-Patienten
|
Systemorganklasse |
Sehr häufig |
Häufig |
Nicht bekannt |
|
Endokrine Erkrankungen |
|
Schilddrüsenfunktionsstörung* |
|
|
Erkrankungen des Nervensystems |
|
Schwindel* |
|
|
Herzerkrankungen |
|
Herzklopfen* |
|
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
|
Schmerzen im Mund- und Rachenraum* |
|
|
Erkrankungen des Gastrointestinaltrakts |
Schmerzen und Beschwerden im Abdomen* (14%) |
Verstopfung* |
|
|
Leber- und Gallenerkrankungen |
|
|
Leberversagen#, Bilirubin-Erhöhung#, Ikterus#, Zirrhose# |
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Pruritus* (63%) |
Ekzem*, Hautausschlag* |
|
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
|
Gelenkschmerz* |
|
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Müdigkeit* (22%) |
Peripheres Ödem*, Fieber* |
|
* Unerwünschte Wirkungen sind definiert als Ereignisse, die bei Patienten des Obeticholsäure-Behandlungsarms mit einer Häufigkeit von ≥5% auftreten und deren Inzidenz ≥1% höher ist als im Placebo-Behandlungsarm.
# In der Postmarketingphase identifiziert, siehe auch nachfolgend unter «Unerwünschte Wirkungen aus der Postmarketingphase».
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Pruritus
Bei der Aufnahme in die Phase-III-Studie wiesen circa 60% der Patienten in der Vorgeschichte Pruritus auf. Behandlungsbezogener Pruritus begann in der Regel innerhalb des erstens Monats nach Behandlungsbeginn.
Im Vergleich zu Patienten, deren Anfangsdosis im OCALIVA-10mg-Arm einmal täglich 10 mg betrug, zeigten Patienten im OCALIVA-Titrationsarm eine niedrigere Pruritus-Inzidenz (70% bzw. 56%) sowie eine niedrigere Häufigkeit von Abbrüchen infolge von Pruritus (10% bzw. 1%).
Die Anzahl der Patienten mit einem Pruritus, der eine Intervention erforderlich machte (z. B. Dosisanpassung, Therapieunterbruch oder zusätzliche Gabe von Gallensäurebindern oder Antihistaminika) lag bei 30 von 51 Patienten (59%) im Arm mit Ocaliva 10 mg, bei 24 von 39 Patienten (62%) im Ocaliva -Titrationsarm und bei 14 von 28 Patienten (50%) im Placebo-Arm.
Unerwünschte Wirkungen aus der Postmarketingphase
Leber- und Gallenerkrankungen: Leberversagen teilweise mit tödlichem Ausgang, Bilirubin-Erhöhung, Ikterus, Zirrhose (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
In klinischen Studien, kam es bei PBC-Patienten, die Ocaliva 25 mg einmal täglich (2,5-faches der empfohlenen Höchstdosis) oder 50 mg einmal täglich (5-faches der empfohlenen Höchstdosis) erhalten hatten, zu einer dosisabhängigen Zunahme der Häufigkeit von hepatischen Nebenwirkungen, wie erhöhte Werte in den biochemischen Lebertests, Aszites, Ikterus, portale Hypertonie und Aufflackern der primär biliären Cholangitis (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Im Falle einer Überdosierung sind die Patienten sorgsam zu überwachen und unterstützend zu versorgen.
Eigenschaften/Wirkungen
ATC-Code
A05AA04
Pharmakotherapeutische Gruppe: Gallensäuren und Derivate
Wirkungsmechanismus
Obeticholsäure ist ein selektiver und potenter Agonist für den Farnesoid X-Rezeptor (FXR), ein Kernrezeptor, der in hohen Konzentrationen in Leber und Darm exprimiert wird. Man nimmt an, dass FXR ein wichtiger Regulator für Gallensäure-, Entzündungs-, Fibrose- und Stoffwechsel-Wege ist. Die FXR-Aktivierung senkt die intrazelluläre Gallensäurekonzentration in den Hepatozyten durch Unterdrückung der de-novo-Synthese aus Cholesterin sowie durch Erhöhen des Transports von Gallensäuren aus den Hepatozyten. Diese Mechanismen begrenzen die Gesamtgrösse des zirkulierenden Gallensäure-Pools und fördern gleichzeitig die Cholerese, wodurch die Leberexposition gegenüber Gallensäuren reduziert wird.
Pharmakodynamik
Dosistitration
In der randomisierten, doppelblinden, placebokontrollierten, 12-monatigen Parallelgruppen-Studie der Phase III (POISE) pendelte sich bei den meisten mit OCALIVA 5 mg einmal täglich behandelten Patienten die ALP-Reduktion nach ungefähr 3 Monaten ein. Eine abhängig von der Verträglichkeit und dem Ansprechen vorgenommene Erhöhung der OCALIVA-Dosis auf 10 mg einmal täglich führte bei der Mehrzahl der Patienten zu einer weiteren Reduktion von ALP (siehe «Dosierung/Anwendung»).
Pharmakodynamische Marker
In der POISE-Studie war von Therapiebeginn bis Monat 12 die Anwendung von OCALIVA 10 mg einmal täglich mit einer 173%igen Erhöhung der Konzentrationen von FGF-19 verbunden, einem durch FXR induzierten Enterokin, das an der Homöostase der Gallensäure beteiligt ist. Von Therapiebeginn bis Monat 12 waren die Konzentrationen der Cholsäure und der Chenodesoxycholsäure reduziert (1,4 μM bzw. 2,7 μM). Die klinische Bedeutung dieser Befunde ist unklar.
Kardiale Elektrophysiologie
Bei einer Dosis, die das 10-fache der empfohlenen Höchstdosis betrug, verlängerte OCALIVA das QT-Intervall in keinem klinisch relevanten Ausmass.
Klinische Wirksamkeit
Eine randomisierte, doppelblinde, placebokontrollierte, 12-monatige Parallelgruppen-Studie der Phase III (POISE) beurteilte die Sicherheit und Wirksamkeit von OCALIVA bei 216 Patienten mit PBC, die mindestens 12 Monate lang UDCA einnahmen (stabile Dosis über ≥3 Monate vor Studieneinschluss) bzw. die UDCA nicht tolerieren konnten und UDCA in ≥3 Monaten vor Studieneinschluss nicht erhielten. Die Patienten wurden in die Studie aufgenommen, wenn die Konzentration an alkalischer Phosphatase (ALP) ≥1,67× Obergrenze des Normalbereichs (ULN) betrug und/oder wenn der Gesamtbilirubin-Wert >1× ULN, jedoch <2× ULN betrug.
Die Patienten erhielten nach Randomisierung (1:1:1) einmal täglich ein Placebo, 10 mg OCALIVA oder eine OCALIVA-Titrierung (5 mg, titriert auf 10 mg nach 6 Monaten, je nach dem Ansprechen/der Verträglichkeit der Behandlung). Die Mehrheit (93%) der Patienten erhielt eine mit UDCA kombinierte Behandlung, und eine kleine Anzahl von Patienten (7%), die UDCA nicht tolerieren konnten, erhielt ein Placebo, OCALIVA (10 mg) oder eine OCALIVA-Titrierung (5 mg auf 10 mg) als Monotherapie.
Die ALP- und Gesamtbilirubin-Werte wurden als kategorische Variablen zum primären kombinierten Endpunkt sowie als fortlaufende Variablen im Verlauf der Zeit beurteilt.
Die Studienpopulation war überwiegend weiblich (91%) und weiss (94%). Das mittlere Alter betrug 56 Jahre, und die Mehrheit der Patienten war weniger als 65 Jahre alt. Die mittleren Baseline-ALP-Werte reichten von 316 U/l bis 327 U/l. Die mittleren Baseline-Gesamtbilirubin-Werte reichten über die Behandlungsarme hinweg von 10 μmol/l bis 12 μmol/l, wobei 92% der Patienten innerhalb des Normalbereichs lagen.
Die Behandlung mit 10 mg OCALIVA oder einer OCALIVA-Titrierung (5 mg auf 10 mg) resultierte in klinisch und statistisch signifikanten Zunahmen (p <0,0001) im Vergleich zum Placebo bei der Anzahl der Patienten, die den primären kombinierten Endpunkt zu allen Studienzeitpunkten erreichten (siehe Tabelle 2). Das Ansprechen erfolgte teilweise bereits nach 2 Wochen und war dosisabhängig (5 mg OCALIVA im Vergleich zu 10 mg nach 6 Monaten, p = 0,0358).
Tabelle 2. Prozentsatz der PBC-Patienten, die den primären kombinierten Endpunkta zum 6-Monate- und zum 12-Monate-Zeitpunkt erreichten (mit oder ohne UDCA)b
|
|
OCALIVA |
OCALIVA |
Placebo |
|
Monat 6 |
|
|
|
|
Responder, n (%) |
37 (51) |
24 (34) |
5 (7) |
|
p-Wertd |
<0,0001 |
<0,0001 |
NA |
|
Monat 12 |
|
|
|
|
Responder, n (%) |
35 (48) |
32 (46) |
7 (10) |
|
p-Wert d |
<0,0001 |
<0,0001 |
NA |
|
Komponenten des primären Endpunktse | |||
|
ALP-Wert von weniger als dem 1,67-fachen des ULN, n (%) |
40 (55) |
33 (47) |
12 (16) |
|
Reduzierung des ALP-Werts um mindestens 15%, n (%) |
57 (78) |
54 (77) |
21 (29) |
|
Gesamtbilirubin von ≤1× ULNf, n (%) |
60 (82) |
62 (89) |
57 (78) |
a Prozentsatz der Patienten, bei denen ein Ansprechen erfolgte, definiert als ALP-Wert von weniger als dem 1,67-fachen des ULN, Gesamtbilirubin-Wert im Normalbereich und Reduzierung des ALP-Werts um mindestens 15%. Fehlende Werte galten als Nichtansprechen. Die Berechnung der 95%-Konfidenzintervalle (KI) erfolgte anhand des exakten Tests nach Fisher.
b Bei dieser Studie hatten 16 Patienten (7%) eine Unverträglichkeit und erhielten kein UDCA als Begleitmedikament: 6 Patienten (8%) im OCALIVA 10 mg-Arm, 5 Patienten (7%) im OCALIVA-Titrationsarm und 5 Patienten (7%) im Placebo-Arm.
c Die Patienten erhielten nach Randomisierung (1:1:1) einmal täglich 10 mg OCALIVA über die gesamten 12 Monate der Studie hinweg oder eine OCALIVA-Titrierung (einmal täglich 5 mg während den ersten 6 Monaten, mit der Möglichkeit einer Steigerung auf einmal täglich 10 mg während den letzten 6 Monaten, wenn der Patient OCALIVA tolerierte, aber ALP-Werte von mindestens dem 1,67-fachen der ULN und/oder Gesamtbilirubin-Werte über der ULN oder eine ALP-Reduzierung von weniger als 15% hatte) oder Placebo.
d OCALIVA-Titrierung und OCALIVA 10 mg im Vergleich zu Placebo. P-Werte werden anhand des Cochran-Mantel-Haenszel General Association-Tests mit Stratifizierung nach UDCA-Intoleranz und ALP-Werten vor der Behandlung von mehr als dem 3-fachen der ULN und/oder AST-Werten von mehr als dem 2-fachen der ULN und/oder Gesamtbilirubin von mehr als der ULN erhalten.
e Die Berechnung der Ansprechraten erfolgte auf der Grundlage der Fallbeobachtungsanalyse (d. h., [n = beobachteter Responder]/[N = Intention to Treat [ITT]-Kollektiv]); Prozentsätze der Patienten mit Monat-12-Werten betragen 86%, 91% bzw. 96% für die Arme OCALIVA 10 mg, OCALIVA-Titrierung bzw. Placebo.
f Die mittleren Baseline-Gesamtbilirubin-Werte reichten über die Behandlungsarme hinweg von 10 μmol/l bis 12 μmol/l, wobei 92% der eingeschlossenen Patienten innerhalb des Normalbereichs lagen (d. h. ≤ ULN).
Mittlere ALP-Reduktion
Abbildung 1 zeigt die durchschnittliche Reduktion der ALP bei den mit OCALIVA behandelten Patienten im Vergleich zu Placebo. Eine Reduktion wurde bereits in Woche 2 beobachtet. Bis zum Monat 3 pendelte sie sich ein und blieb bei den Patienten, die über die gesamten 12 Monate die gleiche Dosierung erhalten hatten, bis zum Monat 12 erhalten. Obwohl in der POISE-Studie eine Titration nach 6 Monaten beurteilt worden ist, unterstützt die Datenlage eine Titration von OCALIVA bereits nach 3 Monaten. Bei den Patienten des OCALIVA -Titrationsarms, deren OCALIVA -Dosis von 5 mg einmal täglich auf 10 mg einmal täglich erhöht worden war, wurde im Monat 12 mehrheitlich eine weitere Reduktion der ALP beobachtet (siehe «Pharmakokinetik»).
Abbildung 1: Mittlerer ALP-Wert über 12 Monate in der POISE-Studie, nach Therapiearm, mit oder ohne UDCAa
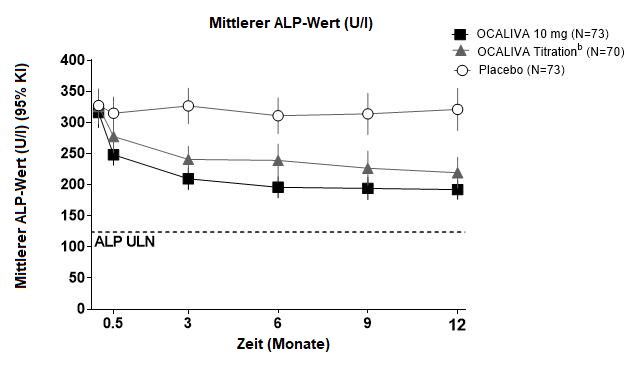
a In der Studie gab es 16 Patienten (7%), die wegen Unverträglichkeit keine begleitende UDCA-Gabe erhielten: 6 Patienten (8%) im OCALIVA 10 mg-Arm, 5 Patienten (7%) im OCALIVA-Titrationsarm und 5 Patienten (7%) im Placebo-Arm.
b Die dem OCALIVA-Titrationsarm randomisiert zugewiesenen Patienten erhielten während den ersten 6 Monaten OCALIVA 5 mg einmal täglich. Ab Monat 6 kamen diejenigen Patienten, die OCALIVA vertrugen, deren ALP jedoch beim 1,67-fachen der Normobergrenze (ULN) oder höher lag und/oder deren Gesamtbilirubin über ULN lag, oder die eine ALP-Reduktion von weniger als 15% aufwiesen, für eine Titration von 5 mg einmal täglich auf 10 mg einmal täglich in den letzten 6 Monaten der Studie in Frage.
Mittlere GGT-Reduktion
Die mittlere (95% KI) Reduktion der Gamma-Glutamyltransferase (GGT) betrug 178 (137, 219) U/l im Arm mit OCALIVA 10 mg, 138 (102, 174) U/l im OCALIVA-Titrationsarm und 8 (-32, 48) U/l im Placebo-Arm.
OCALIVA als Monotherapie
Einundfünfzig PBC-Patienten mit Baseline-ALP-Werten von ≥1,67× ULN und/oder Gesamtbilirubin-Werten über der ULN wurden im Hinblick auf biochemisches Ansprechen auf die OCALIVA-Monotherapie beurteilt (24 Patienten erhielten einmal täglich 10 mg OCALIVA und 27 Patienten erhielten Placebo); dies erfolgte im Rahmen einer gepoolten Analyse der Daten der randomisierten, doppelblinden, placebokontrollierten 12-monatigen Phase-III-Studie (POISE) und einer randomisierten, doppelblinden, placebokontrollierten 3monatigen klinischen Studie. Zum Monat 3-Zeitpunkt war bei 9 (38%) der mit OCALIVA behandelten Patienten ein Ansprechen auf den kombinierten Endpunkt erreicht, im Vergleich zu 1 (4%) mit Placebo behandelten Patienten. Die mittlere (95%-KI) Reduzierung des ALP-Werts bei den mit OCALIVA behandelten Patienten betrug 246 (165, 327) U/l im Vergleich zu einem Anstieg von 17 (-7, 42) U/l bei den mit Placebo behandelten Patienten.
Pharmakokinetik
Absorption
Obeticholsäure wird mit Plasma-Spitzenkonzentrationen (Cmax) nach einer medianen Zeitdauer (tmax) von circa 1,5 Stunden resorbiert. Die gemeinsame Verabreichung mit Mahlzeiten ändert das Ausmass der Resorption von Obeticholsäure nicht.
Distribution
Die Human-Plasmaproteinbindung von Obeticholsäure und deren Konjugaten beträgt mehr als 99%. Das Verteilungsvolumen von Obeticholsäure beträgt 618 l. Das Verteilungsvolumen von Glyko- und Tauro-Obeticholsäure wurde noch nicht ermittelt.
Metabolismus
Obeticholsäure wird in der Leber mit Glycin oder Taurin konjugiert und in die Galle ausgeschieden. Diese Glycin- und Taurin-Konjugate von Obeticholsäure werden im Dünndarm resorbiert, was eine enterohepatische Rezirkulation zur Folge hat. Die Konjugate können im Ileum und im Kolon von Darmbakterien dekonjugiert werden, was zur Umwandlung in Obeticholsäure führt, die erneut resorbiert oder im Stuhl, dem Hauptausscheidungsweg, ausgeschieden werden kann.
Nach täglicher Verabreichung von Obeticholsäure kam es zu einer Anhäufung der Glycin- und Taurin-Konjugate der Obeticholsäure, die in vitro ähnliche pharmakologische Aktivität zeigen, wie die Muttersubstanz. Die Metabolit-Muttersubstanz-Verhältnisse der Glycin- und Taurin-Konjugate von Obeticholsäure betrugen nach täglicher Verabreichung 13,8 bzw. 12,3. Es wird ein weiterer, dritter, Obeticholsäure-Metabolit, 3-Glukuronid, gebildet, dessen pharmakologische Aktivität jedoch als minimal angesehen wird.
Elimination
Nach Verabreichung von radioaktiv markierter Obeticholsäure werden mehr als 87% im Stuhl ausgeschieden. Die Harnausscheidung beträgt weniger als 3%.
Dosis/Zeit-Proportionalität
Nach Verabreichung mehrerer Dosen von einmal täglich 5, 10 oder 25 mg über 14 Tage hinweg, erhöhte sich die systemische Obeticholsäure-Exposition proportional zur Dosis. Glyko- und Tauro-Obeticholsäuresowie die Gesamt-Obeticholsäure-Exposition nahmen überproportional mit der Dosis zu. Die an Tag 14 erreichte systemische Steady-State-Exposition (AUC0-24h) der Gesamt-Obeticholsäure betrug das 4,2-, 6,6- bzw. 7,8-Fache der an Tag 1 erreichten systemischen Exposition (AUC0-24h) nach einmal täglicher Gabe von 5, 10 bzw. 25 mg.
Kinetik spezieller Patientengruppen
Körpergewicht
Basierend auf einer Analyse der Populationspharmakokinetik war das Körpergewicht ein signifikanter Prädiktor für die Pharmakokinetik von Obeticholsäure, wobei eine geringere Obeticholsäure-Exposition im Plasma bei höherem Körpergewicht erwartet wurde. Der Effekt des Körpergewichts hat voraussichtlich keinen bedeutenden Einfluss auf die Wirksamkeit.
Ältere Patienten
Es liegen nur begrenzte pharmakokinetische Daten zu älteren Patienten (≥65 Jahre) vor. Die Analyse der Populationspharmakokinetik anhand der Daten von bis zu 65 Jahre alten Patienten wies darauf hin, dass das Alter die Obeticholsäure-Clearance aus dem Kreislauf voraussichtlich nicht signifikant beeinflusst.
Kinder und Jugendliche
Bei Patienten unter 18 Jahren wurden keine pharmakokinetischen Studien mit Obeticholsäure durchgeführt.
Geschlecht
Die Analyse der Populationspharmakokinetik zeigte, dass das Geschlecht die Obeticholsäure-Pharmakokinetik nicht beeinflusst.
Rasse
Die Analyse der Populationspharmakokinetik zeigte, dass kein Einfluss der Rasse auf die Obeticholsäure-Pharmakokinetik zu erwarten ist.
Nierenfunktionsstörungen
In einer speziellen pharmakokinetischen Einzeldosis-Studie mit 25 mg Obeticholsäure war die Plasmaexposition gegenüber Obeticholsäure und dessen Konjugaten bei Probanden mit leichter (Modification of Diet in Renal Disease [MDRD] eGFR ≥60 und <90 ml/min/1,73 m2), mittelschwerer (MDRD eGFR ≥30 und <60 ml/min/1,73 m2) und schwerer (MDRD eGFR ≥15 und <30 ml/min/1,73 m2) Nierenfunktionsstörung im Vergleich zu Probanden mit normaler Nierenfunktion um etwa das 1,4bis 1,6-Fache erhöht. Die Sicherheit von Ocaliva in der Erhaltungstherapie bei Patienten mit Niereninsuffizienz wurde nicht untersucht.
Leberfunktionsstörungen
Obeticholsäure wird in der Leber und im Darm metabolisiert. Die systemische Exposition von Obeticholsäure, deren aktiven Konjugaten und endogenen Gallensäuren ist bei Patienten mit mittelschweren und schweren Leberfunktionsstörungen im Vergleich zu gesunden Kontrollpersonen erhöht. Die Anwendung von Obeticholsäure ist bei Patienten mit dekompensierter Leberzirrhose (z. B. Child-Pugh-Klassifikation B oder C), mit einem früheren Dekompensationsereignis oder einer kompensierten Zirrhose mit Anzeichen einer portalen Hypertension (z. B. Aszites, gastroösophageale Varizen, persistierende Thrombozytopenie <150 x 109/l) kontraindiziert (siehe «Kontraindikationen» und «Warnhinweise und Vorsichtsmassnahmen»).
Die Auswirkungen einer leichten Leberfunktionsstörung (Child-Pugh-Klassifikation A) auf die Pharmakokinetik von Obeticholsäure waren vernachlässigbar.
Bei Patienten mit leichten, mittelschweren und schweren Leberfunktionsstörungen (Child-Pugh-Klassifikation A, B bzw. C) nahmen der mittlere AUC-Wert (Fläche unter der Kurve) für Gesamt-Obeticholsäure, die Summe von Obeticholsäure und deren beiden aktiven Konjugaten nach Verabreichung einer Einzeldosis von 10 mg Obeticholsäure im Vergleich zu Patienten mit normaler Leberfunktion um das 1,13-fache, 4-fache bzw. 17-fache zu.
Präklinische Daten
Die orale Verabreichung von Obeticholsäure über dem NOAEL (No Observed Adverse Effect Level) an Mäusen, Ratten und Hunden in pivotalen Toxizitätsstudien mit wiederholter Dosierung hatte primär Auswirkungen auf das hepatobiliäre System. Dazu zählten erhöhtes Lebergewicht, Veränderungen der Serumwerte (ALT, AST, LDH, ALP, GGT und/oder Bilirubin) sowie makroskopische/mikroskopische Veränderungen. Alle Veränderungen waren nach Abbruch der Dosierung reversibel und zeigen Konsistenz und eine Vorhersagekraft im Hinblick auf die dosislimitierende Toxizität beim Menschen.
Das karzinogene Potenzial von Obeticholsäure wurde in Karzinogenitätsstudien von bis zu 2 Jahren Dauer in Mäusen und Ratten untersucht. In Mäusen ergaben Dosierungen bis zu 25 mg/kg/Tag Obeticholsäure keine Arzneimittel-abhängigen neoplastischen Befunde. Diese Dosierungen erzielten in Mäusen systemische Expositionen, die etwa das 12-fache der höchsten empfohlenen Dosis von 10 mg beim Menschen betragen. Obeticholsäure wurde Ratten in den Dosen von 2, 7 und 20 mg/kg/Tag verabreicht. Bei 20 mg/kg/Tag (etwa das 12-fache der menschlichen Exposition bei der MRHD) verursachte Obeticholsäure eine Zunahme der Inzidenz von gutartigen granulären Zelltumoren in den Eierstöcken und gutartigen granulären Zelltumoren im Gebärmutterhals und der Vagina bei weiblichen Ratten. Es gab keine arzneimittelabhängigen neoplastischen Befunde bei männlichen Ratten.
Obeticholsäure war im Ames-Test, in einem menschlichen peripheren Blut-Lymphozyten-Chromosomen-Aberrationstest und einem Maus-Mikronukleustest nicht genotoxisch. Auch das Glycin-Konjugat von Obeticholsäure war im Ames-Test und im menschlichen peripheren Blut-Lymphozyten-Chromosomen-Aberrationstest nicht genotoxisch. Im Ames-Test war das Taurin-Konjugat von Obeticholsäure nicht genotoxisch und war in Gegenwart einer metabolischen Aktivierung in einem menschlichen peripheren Blut-Lymphozyten-Chromosomen-Aberrationstest negativ; die Ergebnisse des Chromosomen-Aberrationstests waren in Abwesenheit der metabolischen Aktivierung nicht schlüssig.
Obeticholsäure wurde männlichen Ratten in oralen Dosen von 5, 25 und 50 mg/kg/Tag für 28 Tage vor der Paarung und während der Paarungsperiode verabreicht, sowie an weibliche Ratten 14 Tagen vor der Paarung, bei der Paarung und bis zu Tag 7 der Schwangerschaft. Unter keiner Dosis (die 50 mg/kg/Tag Dosis beträgt etwa das 13-fache der menschlichen Exposition bei der MRHD) wurde die männliche oder weibliche Fruchtbarkeit oder frühe embryonale Entwicklung verändert.
In einer Studie zur embryofetalen Entwicklung an Ratten wurde Obeticholsäure während der Organogenese oral in Dosen von 5, 25 und 75 mg/kg/Tag verabreicht. Bei 25 mg/kg/Tag (einer Dosis, die einer systemischen Exposition des ca. 13-fachen der menschlichen Exposition bei der MRHD von 10 mg entsprach) gab es keine maternale Toxizität oder Entwicklungstoxizität. Bei 75 mg/kg/Tag (das ca. 40-fache der menschlichen Exposition bei der MRHD) waren die fetalen Körpergewichte verringert und es wurde eine höhere Zahl an frühen oder späten Resorptionen und nicht lebensfähigen Feten beobachtet. Bei Muttertieren wurden bei 75 mg/kg/Tag Mortalität, Verlust von Feten, geringeres Körpergewicht und verminderte Nahrungsaufnahme sowie eine verminderte Zunahme des Körpergewichts beobachtet. Demnach kann es sich bei der bei dieser Dosis beobachteten Entwicklungstoxizität um eine Sekundärerscheinung der maternalen Toxizität handeln. Bei Kaninchen wurde Obeticholsäure während der Organogenese oral in Dosen von 3, 9 und 20 mg/kg/Tag verabreicht. Wurde Obeticholsäure in Dosen von ≤20 mg/kg/Tag verabreicht (das ca. 6-fache der menschlichen Exposition bei der MRHD), war es nicht teratogen und es gab keine Hinweise auf eine Schädigung der Feten.
In einer Studie der prä- und postnatalen Entwicklung an Ratten wurde Obeticholsäure in Dosen von 5, 25 und 40 mg/kg/Tag von der Organogenese bis zur Laktation verabreicht. Keine der Dosen hatte Auswirkungen auf Trächtigkeit, Geburt oder postnatale Entwicklung (die Dosis von 40 mg/kg/Tag entspricht etwa dem 21-fachen der menschlichen Exposition bei MRHD). In der Studie wurde das Tauro-Konjugat von Obeticholsäure bei Jungtieren gefunden, die von mit Obeticholsäure-behandelten Muttertieren gesäugt wurden.
Sonstige Hinweise
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren. In der Originalverpackung bei Raumtemperatur (15-25°C) lagern.
Zulassungsnummer
66530 (Swissmedic)
Packungen
OCALIVA 5 mg: 30 Filmtabletten in HDPE-Flasche mit kindersicherem Verschluss (B)
OCALIVA 10 mg: 30 Filmtabletten in HDPE-Flasche mit kindersicherem Verschluss (B)
Zulassungsinhaberin
Advanz Pharma Specialty Medicine Switzerland GmbH, Zürich
Stand der Information
März 2022