Zusammensetzung
Wirkstoffe
Tozinameranum (Einzelsträngige Boten-RNA [messenger RNA, mRNA] mit 5'-Cap-Struktur, durch zellfreie In-vitro-Transkription mit entsprechenden DNA-Vorlagen hergestellt und für das Spike [S]-Protein des SARS-CoV-2-Virus [Original] kodierend).
Das Produkt enthält nicht replizierende nukleosidmodifizierte mRNA.
Hilfsstoffe
ALC-0315 (= [(4-Hydroxybutyl)azandiyl]bis(hexan-6,1-diyl)bis(2-hexyldecanoat)), ALC-0159 (=2-[(Polyethylenglycol)-2000]-N,N-ditetradecylacetamid), DSPC (= 1,2-Distearoyl-sn-glycero-3 phosphocholin), cholesterolum, kalii chloridum (entspr. 0.005 mg Kalium pro Dosis), kalii dihydrogenophosphas (entspr. 0.003 mg Kalium pro Dosis), natrii chloridum (entspr. 0.141 mg Natrium pro Dosis), dinatrii phosphas dihydricus (entspr. 0.017 mg Natrium pro Dosis), saccharum, natrii hydroxidum (q.s. ad pH), acidum hydrochloricum (q.s. ad pH), aqua ad iniectabilia.
Natriumgehalt pro Dosis: 0.16 mg.
Kaliumgehalt pro Dosis: 0.01 mg.
Indikationen/Anwendungsmöglichkeiten
Comirnaty 30 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion ist indiziert für die aktive Immunisierung zur Vorbeugung der durch das SARS-CoV-2-Virus hervorgerufenen COVID-19-Erkrankung bei Personen ab 12 Jahren.
Der Comirnaty-Impfstoff sollte gemäss offiziellen Empfehlungen angewendet werden.
Dosierung/Anwendung
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, sollten der Name und die Chargennummer des verabreichten Produkts eindeutig dokumentiert werden.
Personen ab 12 Jahren - Primäre Impfserie
Comirnaty 30 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion für Personen ab 12 Jahren (VIOLETTE Kappe) wird nach Verdünnung in einer Impfserie mit 2 Dosen (je 0.3 ml) intramuskulär verabreicht. Es wird empfohlen, die zweite Dosis 3 Wochen nach der ersten Dosis zu verabreichen (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen» und «Eigenschaften/Wirkungen»).
Stark immungeschwächte Personen ab 12 Jahren
Bei Personen mit stark geschwächtem Immunsystem können im Einklang mit nationalen Empfehlungen zusätzliche Dosen verabreicht werden (siehe Rubrik «Eigenschaften/Wirkungen - Dritte Dosis in Patienten mit geschwächtem Immunsystem»).
Personen ab 12 Jahren - Auffrischimpfung (Boosterdosis)
Bei Personen, die zuvor mit einem COVID-19-Impfstoff geimpft wurden, sollte Comirnaty 30 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion (VIOLETTE Kappe) frühestens 6 Monate nach Abschluss der primären Impfserie oder frühestens 6 Monate nach der letzten COVID-19-Impfstoff-Dosis verabreicht werden.
Austauschbarkeit mit anderen COVID-19 Impfstoffen
Die Austauschbarkeit von Comirnaty mit anderen COVID-19-Impfstoffen ist nicht belegt.
Pädiatrie
Für Kinder <12 Jahren stehen pädiatrische Formulierungen zur Verfügung. Einzelheiten entnehmen Sie bitte den Fachinformationen von Comirnaty 10 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion für Kinder ab 5 bis <12 Jahre (ORANGE Kappe) und Comirnaty 3 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion für Kinder ab 6 Monaten bis <5 Jahre (KASTANIENBRAUNE Kappe).
Die Sicherheit und Wirksamkeit bei Kindern unter 6 Monaten wurde nicht untersucht.
Ältere Personen
Bei älteren Personen ≥65 Jahren ist keine Dosierungsanpassung erforderlich.
Art der Verabreichung
Comirnaty 30 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion für Personen ab 12 Jahren (VIOLETTE Kappe) ist nach dem Verdünnen intramuskulär zu verabreichen (siehe Rubrik «Sonstige Hinweise – Hinweise für die Handhabung»).
Nach dem Verdünnen enthalten die Durchstechflaschen von Comirnaty 30 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion für Personen ab 12 Jahren (VIOLETTE Kappe) sechs Dosen von je 0.3 ml des Impfstoffs. Um 6 Dosen aus einer einzelnen Durchstechflasche zu entnehmen, sollten Spritzen und/oder Nadeln mit geringem Totvolumen verwendet werden. Die Kombination aus Spritze und Nadel mit geringem Totvolumen sollte ein Totvolumen von nicht mehr als 35 Mikrolitern haben. Wenn Standardspritzen und -nadeln verwendet werden, reicht das Volumen möglicherweise nicht aus, um eine sechste Dosis aus einer einzelnen Durchstechflasche zu entnehmen.
Unabhängig vom Typ der Spritze und Nadel:
·Jede Dosis MUSS 0.3 ml des Impfstoffes enthalten.
·Wenn die in der Durchstechflasche verbleibende Impfstoffmenge nicht für eine volle Dosis von 0.3 ml ausreicht, entsorgen Sie die Durchstechflasche mit dem überschüssigen Volumen.
·Überschüssiger Impfstoff von mehreren Durchstechflaschen darf nicht zusammengeführt werden.
Die bevorzugte Injektionsstelle ist der Deltamuskel (Musculus deltoideus) am Oberarm.
Der Impfstoff darf nicht intravaskulär, subkutan oder intradermal injiziert werden.
Der Impfstoff darf nicht in derselben Spritze mit anderen Impfstoffen oder Arzneimitteln gemischt werden.
Vorsichtsmassnahmen, die vor der Verabreichung des Impfstoffs zu beachten sind, sind in der Rubrik «Warnhinweise und Vorsichtsmassnahmen» aufgeführt.
Für weitere Hinweise zum Auftauen, zur Handhabung und zur Entsorgung des Impfstoffs siehe Rubrik «Sonstige Hinweise – Hinweise für die Handhabung».
Kontraindikationen
Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Warnhinweise und Vorsichtsmassnahmen
Allgemeine Empfehlungen
Überempfindlichkeit und Anaphylaxie
Es wurden Fälle von Anaphylaxie berichtet. Für den Fall einer anaphylaktischen Reaktion nach der Verabreichung des Impfstoffs sollte immer eine angemessene medizinische Behandlung und Überwachung gewährleistet sein.
Nach der Impfung wird eine engmaschige Beobachtung von mindestens 15 Minuten empfohlen. Es sollte keine weitere Dosis des Impfstoffes an Personen verabreicht werden, bei denen eine Anaphylaxie nach einer vorherigen Dosis von Comirnaty aufgetreten ist.
Myokarditis und Perikarditis
Sehr seltene Fälle von Myokarditis und Perikarditis wurden nach der Impfung mit Comirnaty beobachtet. Diese Fälle traten häufiger bei jüngeren Männern und nach der zweiten Dosis des Impfstoffs auf, in der Regel innerhalb von 14 Tagen nach der Impfung. Die Raten von Myokarditis und Perikarditis bei Auffrischimpfung (Boosterdosis) scheinen nicht höher zu sein als nach der zweiten Dosis in der primären Serie. Die verfügbaren Daten deuten darauf hin, dass sich der Verlauf einer Myokarditis und Perikarditis nach einer Impfung nicht von dem einer Myokarditis oder Perikarditis im Allgemeinen unterscheidet.
Medizinische Fachkräfte sollten in Bezug auf Anzeichen und Symptome von Myokarditis und Perikarditis wachsam sein. Geimpfte Personen sollten angewiesen werden, sofort einen Arzt oder eine Ärztin aufzusuchen, wenn sie nach der Impfung Symptome entwickeln, die auf Myokarditis oder Perikarditis hinweisen, wie (akute und anhaltende) Brustschmerzen, Kurzatmigkeit oder Herzklopfen.
Medizinische Fachkräfte sollten Leitlinien beachten und/oder Spezialisten/Spezialistinnen konsultieren, um diesen Zustand zu diagnostizieren und zu behandeln.
Angstbedingte Reaktionen
Angstbedingte Reaktionen, einschliesslich vasovagaler Reaktionen (Synkope), Hyperventilation oder stressbedingte Reaktionen (z.B. Schwindelgefühl, Palpitationen, Erhöhungen der Herzfrequenz, Blutdruckveränderungen, Parästhesie, Hypoästhesie und Schwitzen) können in Zusammenhang mit dem Impfprozess als solchem auftreten. Stressbedingte Reaktionen sind vorübergehend und klingen von selbst ab. Personen sollten darauf hingewiesen werden, dass sie solche Symptome dem Impfanbieter zur Abklärung mitteilen sollten. Es ist wichtig, Vorsichtsmassnahmen zur Vermeidung von Verletzungen infolge einer Ohnmacht zu treffen.
Gleichzeitige Erkrankung
Bei Personen, die an einer akuten schweren fieberhaften Erkrankung oder einer akuten Infektion leiden, sollte die Impfung verschoben werden.
Thrombozytopenie und Gerinnungsstörungen
Wie bei anderen intramuskulären Injektionen sollte der Impfstoff bei Personen, die eine Therapie mit Antikoagulantien erhalten, oder bei Personen mit Thrombozytopenie oder einer Gerinnungsstörung (z.B. Hämophilie) mit Vorsicht verabreicht werden, da bei diesem Personenkreis nach einer intramuskulären Verabreichung Blutungen oder Blutergüsse auftreten können.
Immungeschwächte Personen
Die Wirksamkeit, Sicherheit und Immunogenität des Impfstoffs wurden bei immungeschwächten Personen, einschliesslich Personen unter immunsuppressiver Behandlung, nicht untersucht. Die Wirksamkeit von Comirnaty kann bei immunsupprimierten Personen verringert sein.
Dauer des Schutzes
Die Dauer des durch den Impfstoff induzierten Schutzes ist nicht bekannt, da sie noch in laufenden klinischen Studien ermittelt wird.
Einschränkungen der Effektivität des Impfstoffs
Wie bei jedem Impfstoff schützt die Impfung mit Comirnaty möglicherweise nicht jeden Geimpften. Personen sind möglicherweise erst 7 Tage nach ihrer zweiten Impfdosis vollständig geschützt.
Hilfsstoffe von besonderem Interesse
Dieses Arzneimittel enthält Kalium, jedoch weniger als 1 mmol (39 mg) Kalium pro Dosis, d.h., es ist nahezu «kaliumfrei».
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d.h., es ist nahezu «natriumfrei».
Interaktionen
Es wurden keine Interaktionsstudien durchgeführt.
Die gleichzeitige Verabreichung von Comirnaty mit anderen Impfstoffen wurde nicht untersucht.
Schwangerschaft, Stillzeit
Schwangerschaft
Es liegen nur begrenzte Erfahrungen zur Anwendung von Comirnaty bei Schwangeren vor. Tierexperimentelle Studien weisen nicht auf direkte oder indirekte schädliche Wirkungen in Bezug auf Schwangerschaft, embryonale/fötale Entwicklung, Geburt oder postnatale Entwicklung hin (siehe Rubrik «Präklinische Daten»). Die Verabreichung von Comirnaty in der Schwangerschaft sollte nur in Betracht gezogen werden, wenn der potenzielle Nutzen die möglichen Risiken für Mutter und Fötus überwiegt.
Stillzeit
Es ist nicht bekannt, ob Comirnaty in die Muttermilch übergeht.
Fertilität
Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe Rubrik «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Comirnaty hat keinen oder einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Jedoch können einige der in der Rubrik «Unerwünschte Wirkungen» aufgeführten Wirkungen die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen, vorübergehend beeinträchtigen.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
Comirnaty (Original, 30 Mikrogramm/Dosis)
Die Sicherheit von Comirnaty (Original, 30 Mikrogramm/Dosis) wurde bei Teilnehmenden ab einem Alter von 12 Jahren in 2 klinischen Studien beurteilt, in die 23'157 Teilnehmende (bestehend aus 22'026 Teilnehmenden ab 16 Jahren und 1'131 Jugendlichen im Alter von 12 bis 15 Jahren) eingeschlossen waren, die mindestens 1 Dosis Comirnaty (Original, 30 Mikrogramm/Dosis) erhielten.
Das allgemeine Sicherheitsprofil von Comirnaty (Original, 30 Mikrogramm/Dosis) bei Teilnehmenden im Alter von 5 bis 15 Jahren war ähnlich wie bei Teilnehmenden ab 16 Jahren.
Des Weiteren erhielten 306 bisherige Teilnehmende der Phase 3 im Alter von 18 bis 55 Jahren etwa 6 Monate nach der zweiten Dosis Comirnaty (Original, 30 Mikrogramm/Dosis) eine Auffrischimpfung (Boosterdosis) Comirnaty im nicht placebokontrollierten Teil zur Boosterdosis in Studie 2. Das allgemeine Sicherheitsprofil für die Auffrischimpfung (Boosterdosis) war ähnlich wie das nach 2 Dosen beobachtete.
In Studie 4, einer placebokontrollierten Studie zur Auffrischimpfung, wurden 5'081 Teilnehmende ab 16 Jahren aus Studie 2 rekrutiert, um mindestens 6 Monate nach der zweiten Dosis eine Auffrischimpfung (Boosterdosis) Comirnaty (Original, 30 Mikrogramm/Dosis) zu erhalten. Das allgemeine Sicherheitsprofil für die Auffrischimpfung (Boosterdosis) war ähnlich wie das nach 2 Dosen beobachtete.
Teilnehmende ab einem Alter von 16 Jahren
In Studie 2 erhielten insgesamt 22'026 Teilnehmende ab 16 Jahren mindestens 1 Dosis Comirnaty (Original, 30 Mikrogramm/Dosis) und insgesamt 22'021 Teilnehmende ab 16 Jahren Placebo (darunter 138 Jugendliche im Alter von 16 und 17 Jahren in der Impfstoffgruppe und 145 Jugendliche im Alter von 16 und 17 Jahren in der Placebo-Gruppe). Insgesamt 20'519 Teilnehmende ab 16 Jahren erhielten 2 Dosen Comirnaty (Original, 30 Mikrogramm/Dosis).
Die Teilnehmenden in Studie 2 wurden bis zur Aufhebung ihrer Verblindung nachbeobachtet (placebokontrollierte, verblindete Nachbeobachtungsphase). Bis zum Stichtag 13. März 2021 wurden dabei insgesamt 25'651 (58.2%) Teilnehmende (13'031 in der Comirnaty [Original, 30 Mikrogramm/Dosis]- und 12'620 in der Placebo-Gruppe) ab einem Alter von 16 Jahren über ≥4 Monate nach der zweiten Dosis nachbeobachtet. Darunter waren insgesamt 15'111 (7'704 Comirnaty [Original, 30 Mikrogramm/Dosis] und 7'407 Placebo) Teilnehmende im Alter von 16 bis 55 Jahren und insgesamt 10'540 (5'327 Comirnaty [Original, 30 Mikrogramm/Dosis] und 5'213 Placebo) Teilnehmende ab 56 Jahren.
Die häufigsten unerwünschten Wirkungen bei Teilnehmenden ab 16 Jahren waren Schmerzen an der Injektionsstelle (>80%), Ermüdung (>60%), Kopfschmerzen (>50%), Myalgie (>40%), Schüttelfrost (>30%), Arthralgie (>20%), Fieber und Schwellung an der Injektionsstelle (>10%), die in der Regel von leichter bis mittelstarker Intensität waren und sich innerhalb weniger Tage nach der Impfung zurückbildeten. Eine etwas geringere Häufigkeit von Reaktogenitätsereignissen war mit höherem Alter assoziiert.
In Studie 2 waren auch 200 Teilnehmende mit bekannter stabiler Infektion mit Humanem Immundefizienz-Virus (HIV) eingeschlossen. Das Sicherheitsprofil bei den Teilnehmenden mit stabiler HIV-Infektion, die Comirnaty (Original, 30 Mikrogramm/Dosis) erhielten (n=100), ähnelte dem der Allgemeinpopulation.
Das Sicherheitsprofil bei 545 Teilnehmenden ab 16 Jahren, die Comirnaty (Original, 30 Mikrogramm/Dosis) erhielten und zu Studienbeginn seropositiv für SARS-CoV-2 waren, war ähnlich wie in der Allgemeinbevölkerung.
Jugendliche zwischen 12 bis 15 Jahren
In einer Analyse der Langzeitnachbeobachtung der Sicherheit in Studie 2, basierend auf Daten bis zum Stichtag 13. März 2021, waren 2'260 Jugendliche (1'131 Comirnaty [Original, 30 Mikrogramm/Dosis] und 1'129 Placebo) 12 bis 15 Jahre alt. Davon wurden 1'559 Jugendliche (786 Comirnaty [Original, 30 Mikrogramm/Dosis] und 773 Placebo) nach der zweiten Comirnaty-Dosis >4 Monate lang beobachtet. Die Sicherheitsbewertung in Studie 2 wird fortgeführt.
Die häufigsten unerwünschten Wirkungen bei Jugendlichen im Alter von 12 bis 15 Jahren waren Schmerzen an der Injektionsstelle (>90%), Ermüdung und Kopfschmerzen (>70%), Myalgie und Schüttelfrost (>40%), Arthralgie und Fieber (>20%).
Teilnehmende ab einem Alter von 16 Jahren – nach Auffrischimpfung (Boosterdosis)
Eine Untergruppe von 306 erwachsenen Teilnehmenden der Phase 2/3 der Studie 2 im Alter von 18 bis 55 Jahren, welche die ursprüngliche Impfserie mit 2 Dosen Comirnaty (Original, 30 Mikrogramm/Dosis) abgeschlossen hatten, erhielt etwa 6 Monate (Spanne von 4.8 bis 8.0 Monaten) nach Erhalt der 2. Dosis eine Auffrischimpfung (Boosterdosis) Comirnaty (Original, 30 Mikrogramm/Dosis). Davon wurden 301 Teilnehmende für ≥4 Monate nach der Auffrischimpfung (Boosterdosis) Comirnaty (Original, 30 Mikrogramm/Dosis) nachbeobachtet. Das allgemeine Sicherheitsprofil für die Auffrischimpfung (Boosterdosis) war ähnlich wie das nach 2 Dosen beobachtete. Bei Teilnehmenden, welche eine Auffrischimpfung (dritte Dosis) mit Comirnaty (Original, 30 Mikrogramm/Dosis) erhielten, wurde eine höhere Häufigkeit von Lymphadenopathie beobachtet als bei Teilnehmenden, die 2 Dosen erhielten (2.8% gegenüber 0.4%).
In Studie 4, einer placebokontrollierten Studie zur Auffrischimpfung, erhielten aus Studie 2 rekrutierte Teilnehmende ab 16 Jahren mindestens 6 Monate nach der zweiten Dosis mit Comirnaty (Original, 30 Mikrogramm/Dosis) eine Auffrischimpfung (Boosterdosis) Comirnaty (Original, 30 Mikrogramm/Dosis) (5'081 Teilnehmende), oder Placebo (5'044 Teilnehmende). Insgesamt hatten Teilnehmende, die eine Auffrischimpfung mit Comirnaty (Original, 30 Mikrogramm/Dosis) erhielten, bis zum Stichtag (8. Februar 2022) eine mediane Nachbeobachtungszeit von 2.8 Monaten (Spanne von 0.3 bis 7.5 Monaten) nach der Auffrischimpfung (Boosterdosis) in der verblindeten placebokontrollierten Nachbeobachtungszeit. Davon wurden 1'281 Teilnehmende (895 Comirnaty und 386 Placebo) für ≥4 Monate nach der Auffrischimpfung (Boosterdosis) Comirnaty nachbeobachtet.
Liste der unerwünschten Wirkungen aus klinischen Studien und aus der Postmarketingphase bei Personen ab 12 Jahren
Die unerwünschten Wirkungen sind nach MedDRA-Systemorganklassen und Häufigkeit gemäss folgender Konvention geordnet: «sehr häufig» (≥1/10), «häufig» (≥1/100 bis <1/10), «gelegentlich» (≥1/1'000 bis <1/100), «selten» (≥1/10'000 bis <1/1'000), «sehr selten» (<1/10'000), «nicht bekannt» (kann auf Grundlage der verfügbaren Daten nicht geschätzt werden).
Erkrankungen des Blutes und des Lymphsystems
Gelegentlich: Lymphadenopathiea.
a In Studie 4 wurde bei Teilnehmenden, welche eine Auffrischimpfung (Boosterdosis) erhielten, eine höhere Häufigkeit von Lymphadenopathie beobachtet als bei Teilnehmenden, die 2 Dosen erhielten (2.8% gegenüber 0.4%).
Erkrankungen des Immunsystems
Gelegentlich: Überempfindlichkeitsreaktionen (z.B. Ausschlag, Pruritus, Urtikaria, Angioödemb).
Nicht bekannt: Anaphylaxie.
b Angioödem wurden mit der Häufigkeit «selten» gemeldet.
Stoffwechsel- und Ernährungsstörungen
Gelegentlich: Appetit vermindert.
Psychiatrische Erkrankungen
Gelegentlich: Schlaflosigkeit.
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (Altersgruppe ≥16 Jahre 57.1% / Altersgruppe 12-15 Jahre 75.5%).
Gelegentlich: Lethargie, Schwindelgefühld.
Selten: akute periphere Fazialisparesec.
Nicht bekannt: Parästhesied, Hypoästhesied.
c Während des Sicherheitsnachbeobachtungszeitraums bis 14. November 2020 wurde von vier Teilnehmenden in der Comirnaty-Gruppe eine akute periphere Fazialisparese (oder Gesichtslähmung) berichtet. Der Beginn war bei einem Teilnehmenden am Tag 37 nach Dosis 1 (der Teilnehmende erhielt keine zweite Dosis) und bei den anderen Teilnehmenden an den Tagen 3, 9 und 48 nach Dosis 2. In der Placebo-Gruppe wurden keine Fälle von akuter peripherer Fazialisparese (oder Gesichtslähmung) berichtet.
d In der Postmarketingphase von Comirnaty gemeldet.
Herzerkrankungen
Sehr selten: Myokarditisd, Perikarditisd.
d In der Postmarketingphase von Comirnaty gemeldet.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Diarrhoed (Altersgruppe ≥16 Jahre 15.4% / Altersgruppe 12-15 Jahre 12.5%).
Häufig: Übelkeit, Erbrechend.
d In der Postmarketingphase von Comirnaty gemeldet.
Erkrankungen der Haut und des Unterhautgewebes
Gelegentlich: Hyperhidrosis, nächtliche Schweissausbrüche.
Nicht bekannt: Erythema multiformed.
d In der Postmarketingphase von Comirnaty gemeldet.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Arthralgie (Altersgruppe ≥16 Jahre 25.0% / Altersgruppe 12-15 Jahre 20.2%), Myalgie (Altersgruppe ≥16 Jahre 40.2% / Altersgruppe 12-15 Jahre 42.2%).
Gelegentlich: Schmerzen in den Extremitätene.
e Bezieht sich auf den geimpften Arm. Bei Teilnehmenden, die in Studie 4 eine Auffrischimpfung (Boosterdosis) erhielten, wurde eine höhere Häufigkeit von Schmerzen in den Extremitäten (1.1% vs. 0.8%) beobachtet, verglichen mit Teilnehmenden, die 2 Dosen erhielten.
Erkrankungen der Geschlechtsorgane und der Brustdrüse
Nicht bekannt: Menstruationsstörungenf.
f In der Postmarketingphase von Comirnaty gemeldet. Die meisten Fälle von starken Menstruationsblutungen wurden als nicht schwerwiegend und von vorübergehender Natur berichtet.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Schmerzen an der Injektionsstelle (Altersgruppe ≥16 Jahre 84.3% / Altersgruppe 12-15 Jahre 90.5%), Ermüdung (Altersgruppe ≥16 Jahre 64.7% / Altersgruppe 12-15 Jahre 77.5%), Schüttelfrost (Altersgruppe ≥16 Jahre 34.7% / Altersgruppe 12-15 Jahre 49.2%), Fieberg (Altersgruppe ≥16 Jahre 15.2% / Altersgruppe 12-15 Jahre 24.3%), Rötung an der Injektionsstelle (Altersgruppe ≥16 Jahre 9.9% / Altersgruppe 12-15 Jahre 8.5%), Schwellung an der Injektionsstelle (Altersgruppe ≥16 Jahre 11.1% / Altersgruppe 12-15 Jahre «häufig»).
Gelegentlich: Asthenie, Unwohlsein, Juckreiz an der Injektionsstelle.
Nicht bekannt: ausgedehnte Schwellung der geimpften Gliedmassed, Anschwellen des Gesichtsd.
d In der Postmarketingphase von Comirnaty gemeldet.
g Im Vergleich zur 1. Dosis wurde nach der 2. Dosis eine höhere Häufigkeit von Fieber beobachtet.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Daten zur Überdosierung liegen von 52 Studienteilnehmenden vor, die aufgrund eines Verdünnungsfehlers in der klinischen Studie 58 µg Comirnaty erhalten haben. Die Geimpften berichteten weder über eine Zunahme der Reaktogenität noch über unerwünschte Reaktionen.
Im Falle einer Überdosierung wird eine Überwachung der Vitalfunktionen und gegebenenfalls eine symptomatische Behandlung empfohlen.
Eigenschaften/Wirkungen
ATC-Code
J07BN01
Wirkungsmechanismus
Die nukleosidmodifizierte messenger RNA in diesem Impfstoff ist in Lipid-Nanopartikel verpackt, welche die Aufnahme der nicht replizierenden RNA in Wirtszellen gestatten, und regelt so die transiente Expression des SARS-CoV-2-S-Antigens. Die mRNA kodiert für membranverankertes Spike-(S)-Antigen in voller Länge mit zwei Punktmutationen innerhalb der zentralen Helix. Die Mutation dieser beiden Aminosäuren zu Prolinen fixiert das S-Antigen in einer antigenisch bevorzugten Prä-Fusions-Konformation. Der Impfstoff löst sowohl die Produktion neutralisierender Antikörper als auch eine zelluläre Immunantwort gegen das S-Antigen aus und könnte auf diese Weise zu einem Schutz vor COVID-19 beitragen.
Pharmakodynamik
Keine weiteren Angaben.
Klinische Wirksamkeit
Wirksamkeit
Comirnaty (Original, 30 Mikrogramm/Dosis)
Bei Studie 2 handelt es sich um eine multizentrische, multinationale, randomisierte, placebokontrollierte, beobachterverblindete Phase-I/II/III-Studie zur Dosisfindung, zur Auswahl des Impfstoffkandidaten und zur Untersuchung der Wirksamkeit bei Teilnehmenden ab einem Alter von 12 Jahren. Die Randomisierung erfolgte stratifiziert nach Alter: 12 bis 15 Jahre, 16 bis 55 Jahre bzw. 56 Jahre und älter. Mindestens 40% der Teilnehmenden befanden sich im Stratum ≥56 Jahre. Aus der Studie ausgeschlossen waren immungeschwächte Personen und Personen mit früherer klinischer oder mikrobiologischer COVID-19-Diagnose. Personen mit vorbestehender stabiler Erkrankung, definiert als Erkrankung, für die in den 6 Wochen vor der Aufnahme in die Studie keine erhebliche Therapieumstellung oder Hospitalisierung aufgrund einer Krankheitsverschlechterung erforderlich war, wurden ebenso eingeschlossen wie Personen mit bekannter stabiler Infektion mit HIV, Hepatitis-C-Virus (HCV) oder Hepatitis-B-Virus (HBV).
Wirksamkeit bei Teilnehmenden ab 16 Jahren
Für den Phase-II/III-Studienabschnitt der Studie 2, basierend auf den bis zum 14. November 2020 gesammelten Daten, wurden etwa 44'000 Teilnehmende zu gleichen Teilen randomisiert und sollten 2 Dosen Comirnaty (Original, 30 Mikrogramm/Dosis) oder Placebo erhalten. Die Wirksamkeitsanalysen umfassten Teilnehmende, die ihre zweite Impfung innerhalb von 19 bis 42 Tagen nach ihrer ersten Impfung erhielten. Die Mehrheit (93.1%) der Geimpften erhielt die zweite Dosis 19 bis 23 Tage nach Dosis 1. Eine Nachbeobachtung der Teilnehmenden über einen Zeitraum von bis zu 24 Monaten nach Dosis 2 ist geplant, um die Sicherheit und Wirksamkeit des Impfstoffs gegen COVID-19 zu beurteilen. In der klinischen Studie mussten die Teilnehmenden, um entweder Placebo oder Comirnaty (Original, 30 Mikrogramm/Dosis) zu erhalten, ein Mindestintervall von 14 Tagen vor und nach der Verabreichung eines Influenza-Impfstoffs einhalten. In der klinischen Studie war es Teilnehmern innerhalb 60 Tagen vor Anmeldung und bis zum Abschluss der Studie nicht gestattet Blut-/Plasmaprodukte oder Immunglobuline zu erhalten.
Die Studienpopulation für die Auswertung des primären Wirksamkeitsendpunkts für Comirnaty (Original, 30 Mikrogramm/Dosis) umfasste 36'621 Teilnehmende ab einem Alter von 12 Jahren (18'242 in der Comirnaty [Original, 30 Mikrogramm/Dosis]- und 18'379 in der Placebo-Gruppe), bei denen bis 7 Tage nach Erhalt der zweiten Dosis keine vorbestehende Infektion mit SARS-CoV-2 nachgewiesen wurde. Ausserdem waren 134 Teilnehmende im Alter von 16 bis 17 Jahren (66 in der Comirnaty [Original, 30 Mikrogramm/Dosis]- und 68 in der Placebo-Gruppe) und 1'616 Teilnehmende 75 Jahre und älter (804 in der Comirnaty [Original, 30 Mikrogramm/Dosis]- und 812 in der Placebo-Gruppe).
Tabelle 1: Demografische Daten (Population für den primären Wirksamkeitsendpunkt)a
|
|
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
|
Geschlecht | ||
|
Männlich |
9'318 (51.1) |
9'225 (50.2) |
|
Weiblich |
8'924 (48.9) |
9'154 (49.8) |
|
Alter (Jahre) | ||
|
Mittelwert (SD) |
50.6 (15.70) |
50.4 (15.81) |
|
Median |
52.0 |
52.0 |
|
Min.; Max. |
(12; 89) |
(12; 91) |
|
Altersgruppe | ||
|
≥12 bis 15 Jahre |
46 (0.3) |
42 (0.2) |
|
≥16 bis 17 Jahre |
66 (0.4) |
68 (0.4) |
|
≥16 bis 64 Jahre |
14'216 (77.9) |
14'299 (77.8) |
|
≥65 bis 74 Jahre |
3'176 (17.4) |
3'226 (17.6) |
|
≥75 Jahre |
804 (4.4) |
812 (4.4) |
|
75 bis 85 Jahre |
799 (4.4) |
807 (4.4) |
|
>85 Jahre |
5 (0.0) |
5 (0.0) |
|
Hautfarbe | ||
|
Weiss |
15'110 (82.8) |
15'301 (83.3) |
|
Schwarz oder Afro-Amerikanisch |
1'617 (8.9) |
1'617 (8.8) |
|
Indigene Amerikaner oder Alaskaner |
118 (0.6) |
106 (0.6) |
|
Asiatisch |
815 (4.5) |
810 (4.4) |
|
Indigene Hawaiianer oder andere pazifische Inselbewohner |
48 (0.3) |
29 (0.2) |
|
Sonstigeb |
534 (2.9) |
516 (2.8) |
|
Ethnizität | ||
|
Hispanisch oder Latino |
4'886 (26.8) |
4'857 (26.4) |
|
Nicht hispanisch oder Latino |
13'253 (72.7) |
13'412 (73.0) |
|
Keine Angabe |
103 (0.6) |
110 (0.6) |
|
Komorbiditätenc | ||
|
Ja |
8'432 (46.2) |
8'450 (46.0) |
|
Nein |
9'810 (53.8) |
9'929 (54.0) |
|
a.
Alle geeigneten randomisierten Teilnehmenden, die alle Impfungen wie randomisiert innerhalb des vordefinierten Zeitfensters erhalten, nach Ermessen des Klinikers ansonsten keine wichtigen Protokollabweichungen aufweisen und bei denen bis Ablauf von 7 Tagen nach Dosis 2 kein Nachweis einer SARS-CoV-2-Infektion vorliegt. | ||
Zum Zeitpunkt der primären Wirksamkeitsanalyse der primären Immunisierung mit Comirnaty (Original, 30 Mikrogramm/Dosis) wurden die Teilnehmenden für insgesamt 2'214 Personenjahre in der Comirnaty-Gruppe (Original, 30 Mikrogramm/Dosis) und für insgesamt 2'222 Personenjahre in der Placebo-Gruppe auf symptomatisches COVID-19 untersucht.
Es wurden keine bedeutsamen klinischen Unterschiede in Bezug auf die Gesamtwirksamkeit der primären Immunisierung mit Comirnaty (Original, 30 Mikrogramm/Dosis) bei Teilnehmenden mit Risiko eines schweren Verlaufs von COVID-19 festgestellt, einschliesslich Teilnehmenden mit mindestens 1 Begleiterkrankung, die das Risiko für einen schweren Verlauf von COVID-19 erhöht (z.B. Asthma, BMI ≥30 kg/m2, chronische Lungenerkrankung, Diabetes mellitus, Hypertonie).
Die Informationen zur Wirksamkeit des Impfstoffs sind in Tabelle 2 aufgeführt.
Tabelle 2: Wirksamkeit von Comirnaty (Original, 30 Mikrogramm/Dosis) – Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2, nach Altersuntergruppen – Teilnehmende ohne Nachweis einer Infektion vor Ablauf von 7 Tagen nach Dosis 2 – auswertbare Wirksamkeitspopulation (7 Tage)
|
Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2 bei Teilnehmenden ohne Nachweis einer früheren SARS-CoV-2-Infektion* | |||
|
Untergruppe |
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
Wirksamkeit des Impfstoffs |
|
Alle Teilnehmenden |
8 |
162 |
95.0 (90.0; 97.9) |
|
16 bis 64 Jahre |
7 |
143 |
95.1 (89.6; 98.1) |
|
65 Jahre und älter |
1 |
19 |
94.7 (66.7; 99.9) |
|
65 bis 74 Jahre |
1 |
14 |
92.9 (53.1; 99.8) |
|
75 Jahre und älter |
0 |
5 |
100.0 (-13.1, 100.0) |
|
Hinweis: Bestätigte Fälle wurden bestimmt durch Reverse-Transkriptase-Polymerase-Kettenreaktion (RT-PCR) und mindestens 1 auf COVID-19 hindeutendes Symptom (*Falldefinition: [mindestens 1 Symptom der Folgenden:] Fieber, neu aufgetretener oder verstärkter Husten, neu aufgetretene oder verstärkte Atemnot, Schüttelfrost, neu aufgetretene oder verstärkte Muskelschmerzen, neu aufgetretener Verlust des Geschmacks- oder Geruchssinns, Halsschmerzen, Durchfall oder Erbrechen.) | |||
Die Wirksamkeit der primären Immunisierung mit Comirnaty (Original, 30 Mikrogramm/Dosis) in der Verhinderung des ersten Auftretens von COVID-19 ab 7 Tagen nach der zweiten Dosis im Vergleich zu Placebo betrug 94.6% (95%-Konfidenzintervall von 89.6 % bis 97.6 %) bei Teilnehmenden ab 16 Jahren mit oder ohne Nachweis einer früheren Infektion mit SARS-CoV-2.
Darüber hinaus zeigten Untergruppenanalysen des primären Wirksamkeitsendpunkts ähnliche Wirksamkeitspunktschätzungen für alle Geschlechter, Hautfarben und ethnischen Gruppen sowie für Teilnehmende mit medizinischen Komorbiditäten, die mit einem hohen Risiko für einen schweren COVID-19-Verlauf assoziiert sind.
Aktualisierte Wirksamkeitsanalysen der primären Immunisierung mit Comirnaty (Original, 30 Mikrogramm/Dosis) wurden mit zusätzlichen bestätigten COVID-19-Fällen durchgeführt, die während der verblindeten, placebokontrollierten Nachbeobachtung bis zum 13. März 2021 auftraten. Dies entsprach einer Nachbeobachtungszeit von bis zu 6 Monaten nach Dosis 2 für Teilnehmende in der Wirksamkeitspopulation.
Die aktualisierten Informationen zur Wirksamkeit des Impfstoffs sind in Tabelle 3 aufgeführt.
Tabelle 3: Wirksamkeit von Comirnaty (Original, 30 Mikrogramm/Dosis) – Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2, nach Altersuntergruppen – Teilnehmende ohne Nachweis einer Infektion und Teilnehmende mit oder ohne Nachweis einer Infektion vor Ablauf von 7 Tagen nach Dosis 2 – auswertbare Wirksamkeitspopulation (7 Tage) während des placebokontrollierten Nachbeobachtungszeitraums
|
Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2 bei Teilnehmenden ohne Nachweis einer früheren SARS-CoV-2-Infektion* | |||
|
Untergruppe |
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
Wirksamkeit des Impfstoffs % |
|
Alle Teilnehmendenf |
77 |
850 |
91.3 |
|
16 bis 64 Jahre |
70 |
710 |
90.6 |
|
65 Jahre und älter |
7 |
124 |
94.5 |
|
65 bis 74 Jahre |
6 |
98 |
94.1 |
|
75 Jahre und älter |
1 |
26 |
96.2 |
|
Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2 bei Teilnehmenden mit oder ohne Nachweis einer früheren SARS-CoV-2-Infektion | |||
|
Untergruppe |
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
Wirksamkeit des Impfstoffs % |
|
Alle Teilnehmendenf |
81 |
873 |
91.1 |
|
16 bis 64 Jahre |
74 |
727 |
90.2 |
|
65 Jahre und älter |
7 |
128 |
94.7 |
|
65 bis 74 Jahre |
6 |
102 |
94.3 |
|
75 Jahre und älter |
1 |
26 |
96.2 |
|
Hinweis: Bestätigte Fälle wurden bestimmt durch Reverse-Transkriptase-Polymerase-Kettenreaktion (RT-PCR) und mindestens 1 auf COVID-19 hindeutendes Symptom (Symptome umfassten: Fieber, neu aufgetretener oder verstärkter Husten, neu aufgetretene oder verstärkte Atemnot, Schüttelfrost, neu aufgetretene oder verstärkte Muskelschmerzen, neu aufgetretener Verlust des Geschmacks- oder Geruchssinns, Halsschmerzen, Durchfall oder Erbrechen). | |||
Die aktualisierten Untergruppenanalysen der Wirksamkeit der primären Immunisierung mit Comirnaty (Original, 30 Mikrogramm/Dosis) nach demografischen Merkmalen sind in Tabelle 4 und 5 aufgeführt.
Tabelle 4: Wirksamkeit von Comirnaty (Original, 30 Mikrogramm/Dosis) – Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2 – Teilnehmende ohne* Nachweis einer Infektion vor Ablauf von 7 Tagen nach Dosis 2 nach demografischen Merkmalen – auswertbare Wirksamkeitspopulation (7 Tage) während des placebokontrollierten Nachbeobachtungszeitraums
|
Untergruppe |
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
Wirksamkeit des Impfstoffs % |
|
Geschlecht | |||
|
Männlich |
42 |
399 |
90.1 |
|
Weiblich |
35 |
451 |
92.4 |
|
Ethnizität | |||
|
Hispanisch oder Latino |
29 |
241 |
88.5 |
|
Nicht hispanisch oder Latino |
47 |
609 |
92.6 |
|
Hautfarbe | |||
|
Schwarz oder Afro-Amerikanisch |
4 |
48 |
91.9 |
|
Weiss |
67 |
747 |
91.3 |
|
Sonstigef |
6 |
55 |
90.0 |
|
Land | |||
|
Argentinien |
15 |
108 |
86.5 |
|
Brasilien |
12 |
80 |
86.2 |
|
Deutschland |
0 |
1 |
100.0 |
|
Südafrika |
0 |
9 |
100.0 |
|
Türkei |
0 |
5 |
100.0 |
|
USA |
50 |
647 |
92.6 |
|
Hinweis: Bestätigte Fälle wurden bestimmt durch Reverse-Transkriptase-Polymerase-Kettenreaktion (RT-PCR) und mindestens 1 auf COVID-19 hindeutendes Symptom (Symptome umfassten: Fieber, neu aufgetretener oder verstärkter Husten, neu aufgetretene oder verstärkte Atemnot, Schüttelfrost, neu aufgetretene oder verstärkte Muskelschmerzen, neu aufgetretener Verlust des Geschmacks- oder Geruchssinns, Halsschmerzen, Durchfall oder Erbrechen). | |||
Tabelle 5: Wirksamkeit von Comirnaty (Original, 30 Mikrogramm/Dosis) – Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2 – Teilnehmende mit oder ohne* Nachweis einer Infektion vor Ablauf von 7 Tagen nach Dosis 2 nach demografischen Merkmalen – auswertbare Wirksamkeitspopulation (7 Tage) während des placebokontrollierten Nachbeobachtungszeitraums
|
Untergruppe |
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
Wirksamkeit des Impfstoffs % |
|
Geschlecht | |||
|
Männlich |
44 |
411 |
89.9 |
|
Weiblich |
37 |
462 |
92.1 |
|
Ethnizität | |||
|
Hispanisch oder Latino |
32 |
245 |
87.4 |
|
Nicht hispanisch oder Latino |
48 |
628 |
92.6 |
|
Hautfarbe | |||
|
Schwarz oder Afro-Amerikanisch |
4 |
49 |
92.0 |
|
Weiss |
69 |
768 |
91.3 |
|
Sonstigef |
8 |
56 |
86.8 |
|
Land | |||
|
Argentinien |
16 |
110 |
85.7 |
|
Brasilien |
14 |
82 |
84.2 |
|
Deutschland |
0 |
1 |
100.0 |
|
Südafrika |
0 |
10 |
100.0 |
|
Türkei |
0 |
6 |
100.0 |
|
USA |
51 |
664 |
92.6 |
|
Hinweis: Bestätigte Fälle wurden bestimmt durch Reverse-Transkriptase-Polymerase-Kettenreaktion (RT-PCR) und mindestens 1 auf COVID-19 hindeutendes Symptom (Symptome umfassten: Fieber, neu aufgetretener oder verstärkter Husten, neu aufgetretene oder verstärkte Atemnot, Schüttelfrost, neu aufgetretene oder verstärkte Muskelschmerzen, neu aufgetretener Verlust des Geschmacks- oder Geruchssinns, Halsschmerzen, Durchfall oder Erbrechen). | |||
Die aktualisierten Untergruppenanalysen der Wirksamkeit des Impfstoffs nach Risikostatus der Teilnehmenden sind in Tabelle 6 und 7 aufgeführt.
Tabelle 6: Wirksamkeit von Comirnaty (Original, 30 Mikrogramm/Dosis) – Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2 nach Risikostatus – Teilnehmende ohne* Nachweis einer Infektion vor Ablauf von 7 Tagen nach Dosis 2 – auswertbare Wirksamkeitspopulation (7 Tage) während des placebokontrollierten Nachbeobachtungszeitraums
|
Untergruppe |
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
Wirksamkeit des Impfstoffs % |
|
Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2f |
77 |
850 |
91.3 |
|
Mit Risikog | |||
|
Ja |
35 |
401 |
91.6 |
|
Nein |
42 |
449 |
91.0 |
|
Altersgruppe (Jahre) und Risikostatus | |||
|
16 bis 64 ohne Risiko |
41 |
385 |
89.8 |
|
16 bis 64 mit Risiko |
29 |
325 |
91.5 |
|
65 und älter ohne Risiko |
1 |
53 |
98.1 |
|
65 und älter mit Risiko |
6 |
71 |
91.8 |
|
Adipösh | |||
|
Ja |
27 |
314 |
91.6 |
|
Nein |
50 |
536 |
91.1 |
|
Altersgruppe (Jahre) und Adipositasstatus | |||
|
16 bis 64 und nicht adipös |
46 |
444 |
90.1 |
|
16 bis 64 und adipös |
24 |
266 |
91.3 |
|
65 und älter und nicht adipös |
4 |
79 |
95.2 |
|
65 und älter und adipös |
3 |
45 |
93.2 |
|
Hinweis: Bestätigte Fälle wurden bestimmt durch Reverse-Transkriptase-Polymerase-Kettenreaktion (RT-PCR) und mindestens 1 auf COVID-19 hindeutendes Symptom (Symptome umfassten: Fieber, neu aufgetretener oder verstärkter Husten, neu aufgetretene oder verstärkte Atemnot, Schüttelfrost, neu aufgetretene oder verstärkte Muskelschmerzen, neu aufgetretener Verlust des Geschmacks- oder Geruchssinns, Halsschmerzen, Durchfall oder Erbrechen). | |||
Tabelle 7: Wirksamkeit von Comirnaty (Original, 30 Mikrogramm/Dosis) – Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2 nach Risikostatus – Teilnehmende mit oder ohne Nachweis einer Infektion vor Ablauf von 7 Tagen nach Dosis 2 – auswertbare Wirksamkeitspopulation (7 Tage) während des placebokontrollierten Nachbeobachtungszeitraums
|
Untergruppe |
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
Wirksamkeit des Impfstoffs % |
|
Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2f |
81 |
873 |
91.1 |
|
Mit Risikog | |||
|
Ja |
36 |
410 |
91.6 |
|
Nein |
45 |
463 |
90.6 |
|
Altersgruppe (Jahre) und Risikostatus | |||
|
16 bis 64 ohne Risiko |
44 |
397 |
89.3 |
|
16 bis 64 mit Risiko |
30 |
330 |
91.3 |
|
65 und älter ohne Risiko |
1 |
55 |
98.2 |
|
65 und älter mit Risiko |
6 |
73 |
92.1 |
|
Adipösh | |||
|
Ja |
28 |
319 |
91.4 |
|
Nein |
53 |
554 |
90.8 |
|
Altersgruppe (Jahre) und Adipositasstatus | |||
|
16 bis 64 und nicht adipös |
49 |
458 |
89.8 |
|
16 bis 64 und adipös |
25 |
269 |
91.0 |
|
65 und älter und nicht adipös |
4 |
82 |
95.3 |
|
65 und älter und adipös |
3 |
46 |
93.4 |
|
Hinweis: Bestätigte Fälle wurden bestimmt durch Reverse-Transkriptase-Polymerase-Kettenreaktion (RT-PCR) und mindestens 1 auf COVID-19 hindeutendes Symptom (Symptome umfassten: Fieber, neu aufgetretener oder verstärkter Husten, neu aufgetretene oder verstärkte Atemnot, Schüttelfrost, neu aufgetretene oder verstärkte Muskelschmerzen, neu aufgetretener Verlust des Geschmacks- oder Geruchssinns, Halsschmerzen, Durchfall oder Erbrechen). | |||
Wirksamkeit gegen COVID-19 mit schwerem Verlauf
Aktualisierte Wirksamkeitsanalysen der sekundären Wirksamkeitsendpunkte unterstützten den Nutzen der primären Immunisierung mit Comirnaty (Original, 30 Mikrogramm/Dosis) hinsichtlich der Prävention von COVID-19 mit schwerem Verlauf.
Ab dem 13. März 2021 wird die Wirksamkeit der primären Immunisierung mit Comirnaty (Original, 30 Mikrogramm/Dosis) gegen COVID-19 mit schwerem Verlauf nur für Teilnehmende mit oder ohne früherer SARS-CoV-2-Infektion dargestellt (Tabelle 8), da die COVID-19-Fallzahlen sowohl in der Comirnaty (Original, 30 Mikrogramm/Dosis)- als auch in der Placebo-Gruppe bei Teilnehmenden ohne frühere SARS-CoV-2-Infektion dieselben waren wie bei Teilnehmenden mit oder ohne frühere SARS-CoV-2-Infektion.
Tabelle 8: Wirksamkeit der primären Immunisierung mit Comirnaty (Original, 30 Mikrogramm/Dosis) – Erstes Auftreten von COVID-19 mit schwerem Verlauf bei Teilnehmenden mit oder ohne frühere SARS-CoV-2-Infektion gemäss Definition der Food and Drug Administration (FDA)* oder der Centers for Disease Control and Prevention (CDC)† nach Dosis 1 oder ab 7 Tage nach Dosis 2 während der placebokontrollierten Nachbeobachtung
|
Wirksamkeit des Impfstoffs – Erstes Auftreten von COVID-19 mit schwerem Verlauf gemäss Definition der FDA | |||
|
|
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
Wirksamkeit des Impfstoffs % |
|
Nach Dosis 1d |
1 |
30 |
96.7 |
|
7 Tage nach Dosis 2f |
1 |
21 |
95.3 |
|
Wirksamkeit des Impfstoffs – Erstes Auftreten von COVID-19 mit schwerem Verlauf gemäss Definition der CDC | |||
|
|
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
Wirksamkeit des Impfstoffs % |
|
Nach Dosis 1d |
1 |
45 |
97.8 |
|
7 Tage nach Dosis 2f |
0 |
32 |
100 |
|
Hinweis: Bestätigte Fälle wurden bestimmt durch Reverse-Transkriptase-Polymerase-Kettenreaktion (RT-PCR) und mindestens 1 auf COVID-19 hindeutendes Symptom (Symptome umfassten: Fieber, neu aufgetretener oder verstärkter Husten, neu aufgetretene oder verstärkte Atemnot, Schüttelfrost, neu aufgetretene oder verstärkte Muskelschmerzen, neu aufgetretener Verlust des Geschmacks- oder Geruchssinns, Halsschmerzen, Durchfall oder Erbrechen). | |||
Wirksamkeit und Immunogenität bei Jugendlichen im Alter von 12 bis 15 Jahren
In einer Analyse der Studie 2 bei Jugendlichen im Alter von 12 bis 15 Jahren ohne Nachweis einer vorherigen Infektion traten bei 1'005 Teilnehmenden, die Comirnaty (Original, 30 Mikrogramm/Dosis) erhielten, keine Fälle auf, und bei 978 Teilnehmenden, die Placebo erhielten, waren es 16 Fälle. Die Punktschätzung für die Wirksamkeit beträgt 100% (95% Konfidenzintervall 75.3 bis 100.0). Bei den Teilnehmenden mit oder ohne Nachweis einer vorherigen Infektion gab es 0 Fälle bei den 1'119 Teilnehmenden, die Comirnaty (Original, 30 Mikrogramm/Dosis) erhielten, und 18 Fälle bei den 1'110 Teilnehmenden, die Placebo erhielten. Dies zeigt ebenfalls, dass die Punktschätzung für die Wirksamkeit 100% beträgt (95% Konfidenzintervall 78.1 bis 100.0).
In Studie 2 wurde eine Analyse der SARS-CoV-2-neutralisierenden Titer einen Monat nach der zweiten Dosis mit Comirnaty (Original, 30 Mikrogramm/Dosis) bei einer zufällig ausgewählten Untergruppe von Teilnehmenden durchgeführt, die bis zu einen Monat nach der zweiten Dosis keine serologischen oder virologischen Hinweise auf eine frühere SARS-CoV-2-Infektion hatten, wobei das Ansprechen bei Jugendlichen im Alter von 12 bis 15 Jahren (n=190) mit Teilnehmenden im Alter von 16 bis 25 Jahren (n=170) verglichen wurde.
Das Verhältnis der geometrischen Mittelwerte der Titer (GMT) in der Altersgruppe der 12- bis 15-Jährigen zur Altersgruppe der 16- bis 25-Jährigen betrug 1.76 mit einem zweiseitigen 95%-Konfidenzintervall von 1.47 bis 2.10. Somit wurde das Kriterium der 1.5-fachen Nichtunterlegenheit erfüllt, da die untere Grenze des zweiseitigen 95%-Konfidenzintervalls für das geometrische Mittelverhältnis [GMR] >0.67 war.
Eine aktualisierte Wirksamkeitsanalyse der primären Immunisierung mit Comirnaty (Original, 30 Mikrogramm/Dosis) von Studie 2 wurde bei etwa 2'260 Jugendlichen im Alter von 12 bis 15 Jahren durchgeführt. Dabei wurden bestätigte COVID-19-Fälle bis zum Stichtag 2. September 2021 untersucht, entsprechend einer Nachbeobachtung bis 6 Monate nach Dosis 2 für Teilnehmende in der Wirksamkeitspopulation.
Die aktualisierten Daten zur Wirksamkeit der primären Immunisierung mit Comirnaty (Original, 30 Mikrogramm/Dosis) bei Jugendlichen im Alter von 12 bis 15 Jahren sind in Tabelle 9 dargestellt.
Tabelle 9: Wirksamkeit der primären Immunisierung mit Comirnaty (Original, 30 Mikrogramm/Dosis) – Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2: ohne Nachweis einer Infektion und mit oder ohne Nachweis einer Infektion vor Ablauf von 7 Tagen nach Dosis 2 – verblindete placebokontrollierte Nachbeobachtungsphase, Jugendliche im Alter von 12 bis 15 Jahren – auswertbare Wirksamkeitspopulation (7 Tage)
|
Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2 bei Jugendlichen ab einem Alter von 12 bis 15 Jahren ohne Nachweis einer früheren SARS-CoV-2-Infektion* | |||
|
|
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
Wirksamkeit des Impfstoffs % |
|
Jugendliche im Alter von 12 bis 15 Jahren |
0 |
28 |
100.0 |
|
Erstes Auftreten von COVID-19 ab 7 Tage nach Dosis 2 bei Jugendlichen ab einem Alter von 12 bis 15 Jahren mit oder ohne Nachweis einer früheren SARS-CoV-2-Infektion | |||
|
|
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Placebo |
Wirksamkeit des Impfstoffs % |
|
Jugendliche im Alter von 12 bis 15 Jahren |
0 |
30 |
100.0 |
|
Hinweis: Bestätigte Fälle wurden bestimmt durch Reverse-Transkriptase-Polymerase-Kettenreaktion (RT-PCR) und mindestens 1 auf COVID-19 hindeutendes Symptom (Symptome umfassten: Fieber, neu aufgetretener oder verstärkter Husten, neu aufgetretene oder verstärkte Atemnot, Schüttelfrost, neu aufgetretene oder verstärkte Muskelschmerzen, neu aufgetretener Verlust des Geschmacks- oder Geruchssinns, Halsschmerzen, Durchfall, Erbrechen). | |||
Immunogenität bei Teilnehmenden ab 18 Jahren - nach Auffrischimpfung (Boosterdosis)
In Studie 2 zeigte sich in einer Analyse der SARS-CoV-2-NT50 eine Nichtunterlegenheit der Immunantworten einen Monat nach einer Auffrischimpfung mit Comirnaty (Original, 30 Mikrogramm/Dosis) im Vergleich zu einen Monat nach der 2. Dosis bei Teilnehmenden ab einem Alter von 18 bis 55 Jahren, die bis zu einen Monat nach der Auffrischimpfung keine serologischen oder virologischen Hinweise auf eine frühere SARS-CoV-2-Infektion hatten, basierend auf vordefinierten Nichtunterlegenheitskriterien sowohl für das geometrische Mittelverhältnis [GMR] als auch für die Differenz der Seroresponse-Raten. Die Seroresponse bei einem Teilnehmenden wurde als Erreichen eines ≥4-fachen Anstiegs gegenüber dem Ausgangswert (vor Dosis 1) des NT50 definiert.
Das GMR des SARS-CoV-2 NT50 einen Monat nach der Auffrischimpfung mit Comirnaty (Original, 30 Mikrogramm/Dosis) verglichen mit einem Monat nach Dosis 2 betrug 3.26 (zweiseitiges 97.5%-Konfidenzintervall: 2.76 bis 3.86) und erfüllte somit das Nichtunterlegenheitskriterium für das GMR (untere Grenze des zweiseitigen 97.5%-Konfidenzintervalls >0.67 und Punktschätzung für das GMR ≥0.8).
Ein hoher Anteil der Teilnehmenden (99.5%) zeigte einen Monat nach Dosis 3 eine Seroresponse, im Vergleich zu 95.0% einen Monat nach Dosis 2. Der Unterschied zwischen dem Anteil der Teilnehmenden mit einer Seroresponse einen Monat nach der Auffrischdosis (Dosis 3) und dem Anteil mit einer Seroresponse einen Monat nach Dosis 2 (Dosis 3 minus Dosis 2) lag bei 4.5% (zweiseitiges 97.5%-Konfidenzintervall: 1.0% bis 7.9%) und erfüllte somit das 10%-Nichtunterlegenheitskriterium (d.h. untere Grenze des zweiseitigen 97.5%-Konfidenzintervalls >-10%).
Tabelle 10: Zusammenfassung des geometrischen Mittelverhältnisses für 50% neutralisierende Titer – Vergleich von 1 Monat nach Auffrischungsimpfung (Boosterdosis) mit Comirnaty (Original, 30 Mikrogramm/Dosis) zu 1 Monat nach Dosis 2 – Teilnehmende ohne Anzeichen einer Infektion bis zu 1 Monat nach der Auffrischungsimpfung (Boosterdosis)* – Population mit auswertbarer Immunogenität nach Auffrischungsimpfung (Boosterdosis)±
|
Test |
na |
Comirnaty (Original, 30 Mikrogramm/Dosis) |
| ||
|
1 Monat nach Auffrischungsimpfung (Boosterdosis) |
1 Monat nach Dosis 2 |
1 Monat nach Auffrischungsimpfung (Boosterdosis) - 1 Monat nach Dosis 2 |
Ziel der Nichtunterlegenheit erreichtd | ||
|
GMTb |
GMTb |
GMRc | |||
|
SARS-CoV-2-Neutralisationstest - Referenzstamm - NT50 (Titer)e |
212 |
2466.0 |
755.7 |
3.26 |
J |
|
Abkürzungen: KI = Konfidenzintervall, GMR = geometrisches Mittelverhältnis (geometric mean ratio), GMT = geometrischer Mittelwert der Titer (geometric mean titre), LLOQ = untere Bestimmungsgrenze (lower limit of quantitation), N-Bindung = SARS-CoV-2 Nukleoprotein-Bindung, NAAT = Nukleinsäure-Amplifikationstest (Nucleic Acid Amplification Test), NT50 = 50% neutralisierender Titer, SARS-CoV-2 = schweres akutes respiratorisches Syndrom Coronavirus 2, J/N = ja/nein. | |||||
Tabelle 11: Differenz der Prozentsätze der Teilnehmenden mit Seroresponse – Vergleich von 1 Monat nach Auffrischungsimpfung (Boosterdosis) mit Comirnaty (Original, 30 Mikrogramm/Dosis) zu 1 Monat nach Dosis 2 – Phase 3 – Teilnehmende ohne Anzeichen einer Infektion bis zu 1 Monat nach der Auffrischungsimpfung (Boosterdosis)* – Population mit auswertbarer Immunogenität nach Auffrischungsimpfung (Boosterdosis)±
|
Test |
Na |
Comirnaty (Original, 30 Mikrogramm/Dosis) |
Differenz |
Ziel der Nichtunterlegenheit erreichtf | |
|
1 Monat nach Auffrischungsimpfung (Boosterdosis) |
1 Monat nach Dosis 2 | ||||
|
nb |
nb |
%d | |||
|
SARS-CoV-2-Neutralisationstest - Referenzstamm - NT50 (Titer)g |
200 |
199 |
190 |
4.5 |
J |
|
Abkürzungen: KI = Konfidenzintervall, LLOQ = untere Bestimmungsgrenze (lower limit of quantitation), N-Bindung = SARS-CoV-2 Nukleoprotein-Bindung, NAAT = Nukleinsäure-Amplifikationstest (Nucleic Acid Amplification Test), NT50 = 50% neutralisierender Titer, SARS-CoV-2 = schweres akutes respiratorisches Syndrom Coronavirus 2, J/N = ja/nein. | |||||
Relative Wirksamkeit von Comirnaty (Original, 30 Mikrogramm/Dosis) bei Teilnehmenden ab 16 Jahren nach Auffrischimpfung (Boosterdosis)
Eine Interim-Wirksamkeitsanalyse von Studie 4, einer placebokontrollierten Studie zur Auffrischimpfung mit Comirnaty (Original, 30 Mikrogramm/Dosis), wurde bei rund 10'000 Teilnehmenden ab 16 Jahren durchgeführt, die aus Studie 2 rekrutiert wurden. Dabei wurden bestätigte COVID-19-Fälle untersucht, die im Zeitraum von mindestens 7 Tagen nach der Auffrischimpfung bis zum Datenstichtag am 8. Februar 2022 (einer Periode, in der Delta und dann Omicron die vorherrschenden Varianten waren) angefallen waren, was einer medianen Nachbeobachtungszeit von 2.8 Monaten (Spanne von 0.3 bis 7.5 Monaten) entspricht. Untersucht wurde die Wirksamkeit einer Auffrischimpfung (Boosterdosis) mit Comirnaty nach der primären Serie im Vergleich zur Placebo-Booster-Gruppe, die nur die Dosen der primären Serie erhielt. Die relative Wirksamkeit von Comirnaty (Original, 30 Mikrogramm/Dosis) bei Teilnehmern mit oder ohne Hinweise auf eine vorherige SARS-CoV-2-Infektion betrug 62.4% (95%-Konfidenzintervall von 49.5% bis 72.2%), ähnlich wie bei den Teilnehmern ohne Nachweis einer früheren Infektion. Primäre COVID-19-Fälle, die 7 Tage nach der Auffrischungsdosis beobachtet wurden, waren 67 primäre Fälle in der Comirnaty-Gruppe und 150 primäre Fälle in der Placebogruppe.
Dritte Dosis in Patienten mit geschwächtem Immunsystem
Literaturdaten zeigen, dass bei Patienten mit eingeschränkter Funktion des Immunsystems nach zwei Impfungen mit einer mRNA-Impfung gegen SARS-CoV-2 teilweise nur eine geringe oder gar keine Immunantwort gegen SARS-CoV-2, gemäss der Titer neutralisierender anti-RBD-Antikörper, ausgelöst wurde. In einer Publikation konnte gezeigt werden, dass in Patienten deren Immunsystem infolge einer Organtransplantation mutmasslich beeinträchtigt war, eine dritte Dosis mit einem mRNA-Impfstoff gegen SARS-CoV-2 (mRNA1273) etwa 28 Tage nach der zweiten Impfung verabreicht, einen signifikanten Anstieg neutralisierender anti-RBD-Antikörper auszulösen vermochte.
Pharmakokinetik
Absorption
Nicht zutreffend.
Distribution
Nicht zutreffend.
Metabolismus
Nicht zutreffend.
Elimination
Nicht zutreffend.
Präklinische Daten
Basierend auf konventionellen Studien zur Toxizität bei wiederholter Gabe sowie zur Reproduktions- und Entwicklungstoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Allgemeine Toxizität
Ratten, denen intramuskulär Comirnaty verabreicht wurde (einmal wöchentlich 3 volle Humandosen, die bei Ratten aufgrund von Körpergewichtsunterschieden relativ höhere Werte erzeugen), zeigten an der Injektionsstelle leichte Ödeme und Erytheme, Vergrösserungen der lokalen Lymphknoten und der Milz und einen Anstieg der Leukozyten (einschliesslich Basophile und Eosinophile), was auf eine Entzündungsreaktion hindeutet, sowie eine Vakuolisierung der portalen Hepatozyten ohne Nachweis einer Leberschädigung. Alle Erscheinungen waren reversibel.
Genotoxizität/Karzinogenität
Es wurden weder Genotoxizitäts- noch Karzinogenitätsstudien durchgeführt. Es ist nicht damit zu rechnen, dass die Bestandteile des Impfstoffs (Lipide und mRNA) ein genotoxisches Potential besitzen.
Reproduktions- und Entwicklungstoxizität
Die Reproduktions- und Entwicklungstoxizität wurde an Ratten in einer kombinierten Fertilitäts- und Entwicklungstoxizitätsstudie untersucht, bei der weiblichen Ratten Comirnaty vor der Paarung und während der Trächtigkeit intramuskulär verabreicht wurde (Gabe von 4 vollen Humandosen, die bei Ratten aufgrund von Körpergewichtsunterschieden relativ höhere Dosen erzeugen, und sich zwischen dem Tag 21 vor der Paarung und dem Tag 20 der Gravidität erstreckten). SARS-CoV-2 neutralisierende Antikörperreaktionen waren bei den mütterlichen Tieren von vor der Paarung bis zum Ende der Studie am postnatalen Tag 21 sowie bei den Föten und Nachkommen vorhanden. Es wurden keine impfstoffbedingten Wirkungen auf die weibliche Fertilität, die Trächtigkeit oder die embryofötale Entwicklung oder auf die Entwicklung der Nachkommen festgestellt. Es liegen keine Daten zu Comirnaty zum Plazentatransfer des Impfstoffs oder zur Ausscheidung in der Milch vor.
Sonstige Hinweise
Alle Angaben in dieser Rubrik beziehen sich ausschliesslich auf Comirnaty 30 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion für Personen ab 12 Jahren (Durchstechflasche mit VIOLETTER Kunststoffkappe).
Für Angaben zu Comirnaty 10 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion für Kinder im Alter von 5 bis <12 Jahren (Durchstechflasche mit ORANGER Kunststoffkappe), Comirnaty 30 Mikrogramm/Dosis gebrauchsfertige Injektionsdispersion für Personen ab 12 Jahren (Durchstechflasche mit GRAUER Kunststoffkappe), Comirnaty Original/Omicron BA.1 15/15 Mikrogramm pro Dosis gebrauchsfertige Injektionsdispersion für Personen ab 18 Jahren (Durchstechflasche mit GRAUER Kunststoffkappe) oder Comirnaty Original/Omicron BA.4-5 15/15 Mikrogramm pro Dosis gebrauchsfertige Injektionsdispersion für Personen ab 12 Jahren (Durchstechflasche mit GRAUER Kunststoffkappe) beachten Sie bitte die jeweiligen separaten Fachinformationen!
Inkompatibilitäten
Das Arzneimittel darf, ausser mit den unten unter «Hinweise für die Handhabung – Comirnaty 30 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion für Personen ab 12 Jahren (VIOLETTE Kappe)» aufgeführten, nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit der ungeöffneten Durchstechflasche: 2 Jahre bei -90 °C bis -60 °C.
Alternativ können ungeöffnete Durchstechflaschen für insgesamt 2 Wochen bei -25 °C bis -15 °C gelagert und transportiert werden und können einmal auf die empfohlene Lagerungstemperatur von -90 °C bis -60 °C zurückgebracht werden. Dabei darf das aufgedruckte Verfallsdatum («EXP») nicht überschritten werden. Die gesamte Lagerungszeit bei -25 °C bis -15 °C sollte 2 Wochen nicht überschreiten.
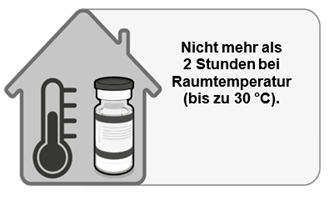
Nach dem Herausnehmen aus dem Gefrierschrank kann die ungeöffnete Durchstechflasche vor der Verwendung bis zu 1 Monat bei 2 °C bis 8 °C gelagert werden. Dabei darf das aufgedruckte Verfallsdatum («EXP») nicht überschritten werden. Innerhalb der Haltbarkeitsdauer von 1 Monat bei 2 °C bis 8 °C können bis zu 48 Stunden für den Transport genutzt werden. Vor der Verwendung kann die ungeöffnete Durchstechflasche bis zu 2 Stunden bei Temperaturen bis 30 °C gelagert werden.
Nach dem Auftauen darf der Impfstoff nicht erneut eingefroren werden.
Handhabung von Temperaturabweichungen nach Entnahme aus dem Gefrierschrank
Die Stabilitätsdaten zeigen, dass die ungeöffnete Durchstechflasche haltbar ist bis zu:
·24 Stunden bei Aufbewahrung bei Temperaturen von -3 °C bis 2 °C.
·insgesamt 4 Stunden bei Aufbewahrung bei Temperaturen von 8 °C bis 30 °C; dies schliesst die oben beschriebenen 2 Stunden bei bis zu 30 °C ein.
Diese Angaben dienen nur als Orientierungshilfe für das medizinische Fachpersonal im Falle einer versehentlichen Temperaturabweichung.
Transfer von gefrorenen Durchstechflaschen, die bei Ultratiefkühlung (<-60 °C) gelagert wurden
·Flaschenträger mit geschlossenem Deckel und 195 Durchstechflaschen, die gefroren aus der Ultratiefkühlung (<-60 °C) entnommen wurden, können bis zu 5 Minuten bei Temperaturen bis zu 25 °C bleiben.
·Flaschenträger mit geöffnetem Deckel, oder Flaschenträger mit weniger als 195 Durchstechflaschen, die gefroren aus der Ultratiefkühlung (<-60 °C) entnommen wurden, können bis zu 3 Minuten bei Temperaturen bis zu 25 °C bleiben.
·Nachdem die Flaschenträger nach der Exposition bei Temperaturen bis zu 25 °C wieder in die Tiefkühlung gebracht wurden, müssen sie mindestens 2 Stunden in der Tiefkühlung verbleiben, bevor sie wieder entnommen werden können.
Transfer von gefrorenen Durchstechflaschen, die bei -25 °C bis -15 °C gelagert wurden
·Flaschenträger mit geschlossenem Deckel und 195 Durchstechflaschen, die aus gefrorener Lagerung (-25 °C bis -15 °C) entnommen wurden, können bis zu 3 Minuten bei Temperaturen bis zu 25 °C bleiben.
·Flaschenträger mit geöffnetem Deckel, oder Flaschenträger mit weniger als 195 Durchstechflaschen, die aus gefrorener Lagerung (-25 °C bis -15 °C) entnommen wurden, können bis zu 1 Minute bei Temperaturen bis zu 25 °C bleiben.
Sobald eine Durchstechflasche aus dem Flaschenträger entnommen wurde, sollte sie zur Verwendung aufgetaut werden.
Haltbarkeit nach Anbruch
Verdünntes Arzneimittel: Die chemische und physikalische Gebrauchsstabilität, einschliesslich des Transports, ist für einen Zeitraum von 6 Stunden bei 2 °C bis 30 °C nach Verdünnung mit Natriumchlorid-Injektionslösung 9 mg/ml (0.9%) belegt. Aus mikrobiologischen Gründen sollte das Produkt sofort verwendet werden, es sei denn, die Methode des Verdünnens schliesst das Risiko einer mikrobiellen Kontamination aus. Erfolgt die Anwendung nicht sofort, liegen die Aufbewahrungszeiten und -bedingungen für die Verwendung in der Verantwortung des Anwenders.
Besondere Lagerungshinweise
Tiefgekühlt bei -90 °C bis -60 °C lagern.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Während der Lagerung ist die Exposition gegenüber Raumlicht so gering wie möglich zu halten, die Exposition gegenüber direktem Sonnenlicht und ultraviolettem Licht ist zu vermeiden.
Aufgetaute Durchstechflaschen können bei Raumlicht gehandhabt werden.
Für detaillierte Angaben zur kurzfristigen Lagerung der ungeöffneten gefrorenen Durchstechflasche bei -25 °C bis -15 °C und zu den Aufbewahrungsbedingungen nach Auftauen und Verdünnung des Arzneimittels, siehe oben unter «Haltbarkeit» und «Haltbarkeit nach Anbruch».
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung – Comirnaty 30 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion für Personen ab 12 Jahren (VIOLETTE Kappe)
Comirnaty 30 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion ist von einer medizinischen Fachperson unter Verwendung aseptischer Techniken zuzubereiten, um die Sterilität der zubereiteten Dispersion sicherzustellen.
|
ÜBERPRÜFUNG DER DURCHSTECHFLASCHE - COMIRNATY 30 MIKROGRAMM/DOSIS KONZENTRAT ZUR HERSTELLUNG EINER INJEKTIONSDISPERSION FÜR PERSONEN AB 12 JAHREN (VIOLETTE KAPPE) | ||
|
|
|
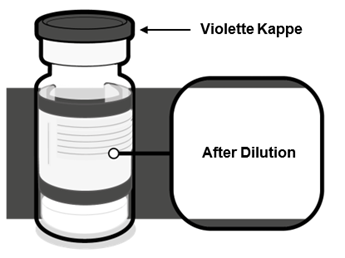
·Stellen Sie sicher, dass die Durchstechflasche mit einer violetten Kunststoffkappe versehen ist. |
|
AUFTAUEN VOR DEM VERDÜNNEN - COMIRNATY 30 MIKROGRAMM/DOSIS KONZENTRAT ZUR HERSTELLUNG EINER INJEKTIONSDISPERSION FÜR PERSONEN AB 12 JAHREN (VIOLETTE KAPPE) | ||
|
|
|
·Die Mehrfachdosis-Durchstechflasche wird gefroren gelagert und muss vor der Verdünnung aufgetaut werden. Die gefrorenen Durchstechflaschen sollten zum Auftauen in eine Umgebung von 2 °C bis 8 °C gebracht werden. Das Auftauen eines Flaschenträgers mit 195 Durchstechflaschen kann 3 Stunden dauern. Alternativ können gefrorene Durchstechflaschen zur sofortigen Verwendung auch 30 Minuten lang bei Temperaturen bis zu 30 °C aufgetaut werden. |
|
VERDÜNNUNG - COMIRNATY 30 MIKROGRAMM/DOSIS KONZENTRAT ZUR HERSTELLUNG EINER INJEKTIONSDISPERSION FÜR PERSONEN AB 12 JAHREN (VIOLETTE KAPPE) | ||
|
|
|
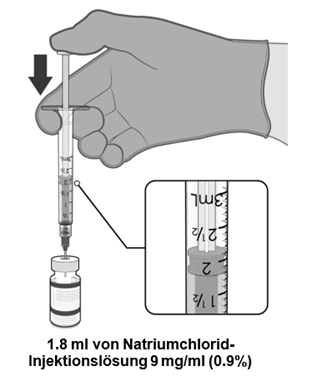
·Der aufgetaute Impfstoff muss in seiner ursprünglichen Durchstechflasche mit 1.8 ml Natriumchlorid-Injektionslösung 9 mg/ml (0.9%) unter Verwendung einer 21-Gauge- oder dünneren Nadel unter aseptischen Techniken verdünnt werden. |
|
|
|
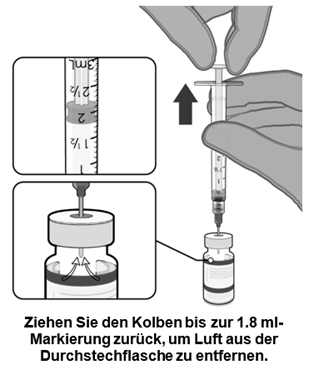
·Gleichen Sie den Druck in der Durchstechflasche aus, bevor Sie die Nadel aus der Durchstechflasche entfernen, indem Sie 1.8 ml Luft in die leere Verdünnungsmittelspritze ziehen. |
|
|
|
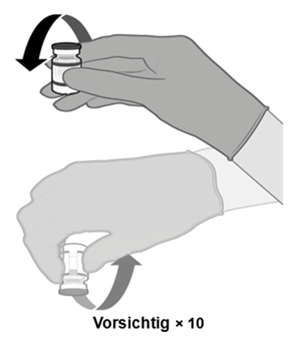
·Drehen Sie die verdünnte Dispersion 10-mal vorsichtig um. Nicht schütteln. |
|
|
|
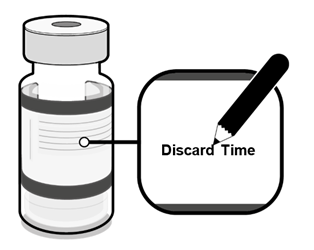
·Die Durchstechflaschen mit der verdünnten Dispersion sind mit dem entsprechenden Datum und Uhrzeit der Verdünnung zu kennzeichnen. |
|
ZUBEREITUNG VON INDIVIDUELLEN 0.3 ml DOSEN - COMIRNATY 30 MIKROGRAMM/DOSIS KONZENTRAT ZUR HERSTELLUNG EINER INJEKTIONSDISPERSION FÜR PERSONEN AB 12 JAHREN (VIOLETTE KAPPE) | ||
|
|
|
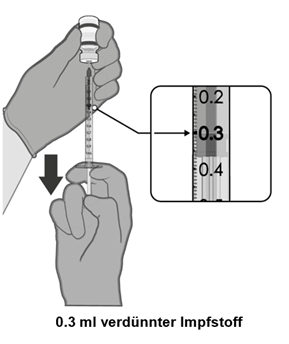
·Nach der Verdünnung enthält die Durchstechflasche 2.25 ml, aus der 6 Dosen zu 0.3 ml entnommen werden können. |
Entsorgung
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Zulassungsnummer
68225 (Swissmedic).
Packungen
Comirnaty 30 Mikrogramm/Dosis Konzentrat zur Herstellung einer Injektionsdispersion für Personen ab 12 Jahren (VIOLETTE Kappe)
1 Packung mit 195 2 ml Mehrfachdosis-Durchstechflaschen (Typ I-Glas) mit einem Stopfen (synthetischer Brombutylkautschuk) und einer violetten Flip-off-Kunststoffkappe mit einem Verschluss aus Aluminium (mit je 6 Dosen à 0.3 ml) [B].
Zulassungsinhaberin
Pfizer AG, Zürich.
Stand der Information
Mai 2024.
LLD V053