ZusammensetzungWirkstoffe
Dexamethason.
Hilfsstoffe
Poly(D,L-Lactid-co-Glycolid) 50:50 mit Ester-Endgruppen, Poly(D,L-Lactid-co-Glycolid) 50:50 mit Säure-Endgruppen.
Indikationen/AnwendungsmöglichkeitenOZURDEX wird angewendet zur Behandlung von Erwachsenen mit:
·einer Sehbeeinträchtigung aufgrund eines diabetischen Makulaödems (DMÖ), die pseudophak sind oder auf eine Therapie mit Nicht-Kortikosteroiden unzureichend ansprechen oder bei denen diese als unpassend angesehen wird.
·Makulaödem nach retinalem Venenastverschluss (VAV) oder retinalem Zentralvenenverschluss (ZVV).
·Entzündung des posterioren Augensegments, die sich als nicht infektiöse Uveitis darstellt.
Dosierung/AnwendungOZURDEX muss durch einen qualifizierten Ophthalmologen verabreicht werden, der Erfahrungen mit intravitrealen Injektionen hat.
Übliche Dosierung
Erwachsene
Die empfohlene Dosierung beträgt ein OZURDEX Implantat, das intravitreal in das betroffene Auge appliziert wird. Die gleichzeitige Verabreichung in beide Augen wird nicht empfohlen.
DMÖ
Bei mit OZURDEX behandelten Patienten, die anfänglich auf die Behandlung angesprochen haben und nach Auffassung des Arztes von einer erneuten Behandlung profitieren könnten, ohne einem signifikanten Risiko ausgesetzt zu sein, sollte eine erneute Behandlung in Erwägung gezogen werden.
Wiederholungsbehandlungen können nach ungefähr sechs Monaten durchgeführt werden, wenn sich, sekundär zu einem rezidivierenden oder sich verschlechternden diabetischen Makulaödem der Visus des Patienten verschlechtert und/oder die Netzhautdicke zunimmt.
Es gibt derzeit keine Erfahrungen bezüglich der Wirksamkeit und Sicherheit wiederholter Verabreichungen bei DMÖ für mehr als sieben Implantate.
Retinaler Venenverschluss und Uveitis
Wiederholungsdosen sind in Betracht zu ziehen, wenn ein Patient auf die Behandlung anspricht, im weiteren Verlauf einen Sehverlust aufweist und nach Auffassung des Arztes bzw. der Ärztin von einer Wiederholungsbehandlung profitieren würde, ohne einem signifikanten Risiko ausgesetzt zu sein (siehe «Eigenschaften/Wirkungen»).
Patienten, die eine dauerhafte Verbesserung des Visus aufweisen, sollten nicht erneut behandelt werden. Patienten, die eine Visusverschlechterung aufweisen, die durch OZURDEX nicht verlangsamt wird, sollten nicht erneut behandelt werden.
Es liegen nur sehr begrenzte Informationen über Wiederholungsbehandlungen in einem Intervall von weniger als sechs Monaten vor (siehe «Eigenschaften/Wirkungen»). Für Informationen hinsichtlich aktueller Erfahrungen zur Sicherheit mit wiederholten Verabreichungen von mehr als zwei Implantaten bei einer nicht infektiösen Uveitis im posterioren Segment und bei retinalem Venenverschluss siehe «Unerwünschte Ereignisse».
Patienten müssen nach der Injektion überwacht werden, um im Falle einer Infektion oder eines erhöhten Augeninnendrucks eine frühzeitige Behandlung sicherzustellen.
Art der Anwendung
Intravitreales Implantat in einem Applikator zur einmaligen und ausschliesslich intravitrealen Anwendung. Jeder Applikator darf nur für die Behandlung eines einzigen Auges verwendet werden.
Die intravitreale Injektion muss unter kontrollierten aseptischen Bedingungen erfolgen. Es sind sterile Handschuhe, ein steriles Tuch und ein steriles Lidspekulum (oder ein entsprechendes anderes Instrument) zu verwenden.
Vor und nach jeder Injektion muss 3 Tage lang täglich ein topisches Breitband-Antibiotikum verabreicht werden. Vor der Injektion müssen die periokuläre Haut, das Augenlid und die Augenoberfläche desinfiziert werden (z.B. mit Tropfen einer 5%igen Povidon-Jod-Lösung auf die Konjunktiva, wie in den klinischen Studien zur Zulassung von OZURDEX), und eine adäquate Lokalanästhesie ist durchzuführen.
Nehmen Sie den Folienbeutel aus der Schachtel und kontrollieren Sie, dass der Beutel nicht beschädigt ist (siehe «Sonstige Hinweise»). Öffnen Sie den Folienbeutel in einem sterilen Bereich und legen Sie den Applikator behutsam auf eine sterile Ablage. Entfernen Sie vorsichtig die Kappe vom Applikator. Ist der Folienbeutel einmal geöffnet, muss der Applikator sofort verwendet werden.
Halten Sie den Applikator mit einer Hand fest und ziehen Sie die Sicherheitslasche gerade vom Applikator ab. Die Lasche nicht drehen oder biegen. Halten Sie die Nadel mit dem Kanülenschliff nach oben, weg von der Sklera, und führen Sie die Nadel etwa 1 mm in die Sklera ein, ändern Sie die Richtung zur Augenmitte hin und führen Sie die Nadel in den Glaskörperraum ein, bis die Silikonhülse die Konjunktiva berührt. Drücken Sie langsam den Auslöseknopf, bis ein Klicken zu hören ist. Achten Sie darauf, dass der Auslöseknopf vollständig gedrückt und bündig mit der Applikatoroberfläche arretiert ist, bevor Sie den Applikator aus dem Auge herausziehen. Ziehen Sie die Nadel in der gleichen Richtung heraus, in der sie in den Glaskörper eingeführt wurde.
Für eine bildliche Darstellung der Applikation von OZURDEX siehe Hinweise für die Handhabung unter «Sonstige Hinweise».
Unmittelbar nach der Injektion von OZURDEX sollte der Erfolg der Implantation mittels indirekter Ophthalmoskopie im Injektionsquadranten bestätigt werden. In den meisten Fällen ist die Visualisierung positiv. In seltenen Fällen, in denen das Implantat nicht visualisiert werden kann, sollte mit einem sterilen Wattestäbchen leicht auf die Injektionsstelle gedrückt werden, um das Implantat sichtbar zu machen.
Die Patienten sollten nach der intravitrealen Injektion mit einem Breitband-Antibiotikum weiterbehandelt werden.
Spezielle Dosierungsanweisungen
Ältere Patienten (≥65 Jahre)
Bei älteren Patienten ist keine Dosisanpassung erforderlich.
Kinder und Jugendliche
Es gibt keinen relevanten Nutzen von OZURDEX bei Kindern und Jugendlichen mit
·diabetischem Makulaödem
·Makulaödem nach retinalem Venenastverschluss (VAV) oder retinalem Zentralvenenverschluss (ZVV).
Die Unbedenklichkeit und Wirksamkeit von OZURDEX bei Uveitis wurden bei Kindern und Jugendlichen noch nicht nachgewiesen. Es liegen keine Studien vor.
KontraindikationenOZURDEX ist in den folgenden Fällen kontraindiziert:
·Aktive oder vermutete okuläre oder periokuläre Infektion. Hierzu gehören die meisten Viruserkrankungen der Kornea und Konjunktiva, wie eine aktive epitheliale Herpes-simplex-Keratitis (dendritische Keratitis), Vaccinia, Varizellen, Mykobakterien-Infektionen und Pilzerkrankungen.
·Fortgeschrittenes Glaukom, das mit Arzneimitteln allein nicht adäquat behandelt werden kann.
·Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Zusammensetzung.
·Aphakie mit rupturierter posteriorer Linsenkapsel.
·Augen mit Vorderkammer-Intraokularlinse und rupturierter posteriorer Linsenkapsel*.
·Augen mit irisfixierter oder transskleral fixierter Intraokularlinse und rupturierter posteriorer Linsenkapsel*.
* wegen dem Zusammenhang zwischen Migration des Implantats in die Vorderkammer und konsekutivem Ödem der Kornea.
Warnhinweise und VorsichtsmassnahmenIntravitreale Injektionen, einschliesslich derjenigen mit OZURDEX, können zu konjunktivalen Hämorrhagien (Blutungen), Endophthalmitis, intraokulärer Entzündung, erhöhtem Augeninnendruck und Netzhautablösung führen. Es ist immer eine geeignete aseptische Injektionstechnik anzuwenden. Darüber hinaus müssen die Patienten nach der Injektion überwacht werden, um eine eventuelle Infektion oder Erhöhung des Augeninnendrucks frühzeitig behandeln zu können. Die Überwachung kann in einer Kontrolle der Durchblutung des Sehnervenkopfs unmittelbar nach der Injektion, einer Tonometrie innerhalb von 30 Minuten nach der Injektion und einer biomikroskopischen Untersuchung zwischen zwei und sieben Tagen nach der Injektion bestehen.
Die Unbedenklichkeit und Wirksamkeit der gleichzeitigen Anwendung von OZURDEX an beiden Augen wurde nicht untersucht. Eine Applikation an beiden Augen gleichzeitig wird deshalb nicht empfohlen.
Endophthalmitis
Die Patienten sind anzuweisen, etwaige auf eine Endophthalmitis oder eines der oben genannten Ereignisse hinweisende Symptome, z.B. Augenschmerzen, verschwommenes Sehen, usw., unverzüglich zu melden (siehe «Unerwünschte Wirkungen»).
Migration des Implantats
Bei allen Patienten mit fehlender oder gerissener posteriorer Linsenkapsel (z.B. nach einer Kataraktoperation) oder bei Patienten mit einem Irisdefekt (z.B. nach Iridektomie oder Trabekulotomie) mit oder ohne frühere Vitrektomie oder bei Patienten mit einer Dehiszenz von Zonulafasern und posteriorer Linsenkapsel besteht das Risiko einer Migration des Implantats vom Glaskörper in die Vorderkammer. Eine Migration des Implantats in die Vorderkammer kann zu einem Hornhautödem führen. Ein persistierendes schweres Hornhautödem kann so weit fortschreiten, dass eine Hornhauttransplantation erforderlich wird.
OZURDEX soll mit Vorsicht, wenn nicht kontraindiziert (siehe «Kontraindikationen»), und nur nach einer sorgfältigen Nutzen-Risiko-Abwägung bei Patienten mit fehlender oder gerissener posterioren Linsenkapsel, die für eine Therapie in Betracht gezogen werden, verwendet werden. Diese Patienten müssen eng überwacht werden, damit eine frühzeitige Diagnose und Behandlung einer Implantatmigration erfolgen kann.
Kortikosteroide
Die Anwendung von Kortikosteroiden, einschliesslich OZURDEX, kann zu Katarakten (einschliesslich posterioren subkapsulären Katarakten), einem Anstieg des intraokulären Drucks, einem Steroidglaukom und zu sekundären Augeninfektionen führen.
Wie bei einer Steroidbehandlung am Auge und intravitrealen Injektion zu erwarten, kann es zu einem Anstieg des Augeninnendrucks (IOD) kommen. Der Anstieg des IOD kann normalerweise mit IOD-senkenden Arzneimitteln behandelt werden (siehe «Unerwünschte Wirkungen»). Bei Patienten, bei welchen ein Anstieg des Augeninnendrucks von ≥10 mmHg vom Ausgangswert beobachtet wurde, trat bei der Mehrheit der Patienten dieser Anstieg 45 bis 60 Tage nach der Injektion auf. Deshalb ist eine regelmässige Überwachung des IODs unabhängig vom Basis-IOD erforderlich, und jeder Anstieg des Augeninnendrucks nach der Injektion muss entsprechend adäquat behandelt werden. Bei Patienten unter 45 Jahren mit Makulaödem nach retinalem Venenverschluss oder einer Entzündung des posterioren Segments des Auges, die sich als nicht infektiöse Uveitis darstellt, ist ein Anstieg des IOD wahrscheinlicher.
Kortikosteroide sind bei Patienten mit einer okulären Virusinfektion (z.B. Herpes simplex) in der Anamnese mit Vorsicht anzuwenden und bei einer aktiven okulären Herpes-simplex-Infektion nicht anzuwenden.
Katarakt
In den dreijährigen klinischen DMÖ-Studien unterzogen sich 59% der mit OZURDEX behandelten Patienten mit phakem behandeltem Auge einer Kataraktoperation im behandelten Auge (siehe «Unerwünschte Wirkungen»).
Nach der ersten Injektion erscheint die Inzidenz einer Katarakt bei Patienten mit nicht infektiöser Uveitis im posterioren Segment höher als bei Patienten mit VAV/ZVV. In klinischen VAV/ZVV-Studien wurde über eine Katarakt häufiger bei Patienten mit phaker Linse berichtet, die eine zweite Injektion erhalten haben (siehe «Unerwünschte Wirkungen»). Nur ein von 368 Patienten benötigte eine Kataraktoperation während der ersten Behandlung, und drei von 302 Patienten benötigten eine Kataraktoperation während der zweiten Behandlung. In der Studie zur nicht infektiösen Uveitis wurde bei einem von 62 Patienten mit phaker Linse nach einer einmaligen Injektion eine Kataraktoperation durchgeführt.
Konjunktivale Blutung
Die Prävalenz der konjunktivalen Blutung erscheint bei Patienten mit nicht infektiöser Uveitis im posterioren Segment höher als bei Patienten mit VAV/ZVV und DMÖ. Dies könnte auf die intravitreale Injektion oder eine gleichzeitige Anwendung von topischen und/oder systemischen Kortikosteroiden oder nichtsteroidalen Antirheumatika zurückzuführen sein. Es ist keine Behandlung erforderlich, da es zu einer spontanen Auflösung kommt.
Thrombozytenaggregationshemmer, wie Clopidogrel, wurden in bestimmten Stadien während der klinischen Studien bei bis zu 56% der Patienten eingesetzt. Bei Patienten, die eine Begleitmedikation und Thrombozytenaggregationshemmer erhielten, wurden hämorrhagische unerwünschte Ereignisse bei einem etwas höheren Anteil der mit OZURDEX behandelten Patienten (bis zu 29%) im Vergleich zu den Patienten in der Scheininjektionsgruppe (bis zu 23%) berichtet, unabhängig von der Indikation oder der Anzahl Behandlungen. Das am häufigsten berichtete hämorrhagische unerwünschte Ereignis war konjunktivale Blutung (bis zu 24%).
Die Anwendung von OZURDEX sollte bei Patienten, die mit Antikoagulantien oder Thrombozytenaggregationshemmern behandelt werden, vorsichtig erfolgen.
RVO
OZURDEX wurde bei Patienten mit Makulaödem infolge eines retinalen Venenverschlusses (RVO) mit signifikanter Ischämie der Netzhaut nicht untersucht.
In klinischen Studien mit retinalem Venenverschluss wurde bei 2% der Patienten unter OZURDEX eine gerinnungshemmende Therapie angewendet, und es wurde über keine hämorrhagischen unerwünschten Wirkungen berichtet. Es liegen jedoch Berichte über Einzelfälle von hämorrhagischen unerwünschten Wirkungen während der Anwendung nach der Markteinführung (Post-Marketing) vor.
Diabetes
In den Studien der Phase III wurde eine begrenzte Anzahl Patienten mit Typ-1 Diabetes untersucht. Das Ansprechen dieser Patienten auf OZURDEX unterschied sich nicht signifikant von den Patienten mit Typ-2 Diabetes.
DMÖ
Beim DMÖ wurde bei 8% der Patienten eine gerinnungshemmende Therapie durchgeführt. Unter den Patienten, die eine gerinnungshemmende Therapie erhielten, war die Häufigkeit des Auftretens hämorrhagischer unerwünschter Ereignisse in der OZURDEX-Gruppe und der Scheininjektionsgruppe etwa gleich (29% gegenüber 32%). Unter den Patienten, die keine gerinnungshemmende Therapie erhielten, berichteten 27% der mit OZURDEX behandelten Patienten über hämorrhagische unerwünschte Ereignisse gegenüber 20% der Patienten in der Scheininjektionsgruppe. Glaskörperblutungen wurden bei einem höheren Anteil der mit OZURDEX behandelten Patienten berichtet, die eine gerinnungshemmende Therapie erhielten (11%), als bei den Patienten, die keine gerinnungshemmende Therapie erhielten (6%).
Sehstörungen
Bei der systemischen und topischen Anwendung von Kortikosteroiden können Sehstörungen auftreten. Wenn ein Patient mit Symptomen wie verschwommenem Sehen oder anderen Sehstörungen vorstellig wird, sollte eine Untersuchung auf mögliche Ursachen in Erwägung gezogen werden; diese umfassen unter anderem Katarakt, Glaukom oder seltene Erkrankungen, wie z.B. zentrale seröse Chorioretinopathie (ZSC), die nach der Anwendung systemischer oder topischer Kortikosteroide gemeldet wurden.
InteraktionenEs wurden keine Interaktionsstudien durchgeführt.
Die systemische Resorption ist minimal, und es werden keine Interaktionen erwartet.
Schwangerschaft, StillzeitIn Tierstudien haben sich nach topischer ophthalmischer Applikation teratogene Wirkungen gezeigt (siehe «Präklinische Daten»). Zur intravitrealen Anwendung von Dexamethason bei Schwangeren liegen keine hinreichenden Daten vor. Die systemischen Spiegel von Dexamethason beim Menschen haben sich als niedrig erwiesen. OZURDEX darf während der Schwangerschaft nicht angewendet werden, es sei denn, es ist klar notwendig.
Es ist nicht bekannt, ob eine intravitreale Applikation von Dexamethason zu nachweisbaren Spiegeln in der Muttermilch führt. Systemisch verabreichtes Dexamethason wird in die Muttermilch ausgeschieden. Eine Anwendung während der Stillzeit wird daher nicht empfohlen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenNach intravitrealer Injektion von OZURDEX kann die Sehfähigkeit der Patienten vorübergehend beeinträchtigt werden (siehe «Unerwünschte Wirkungen»). Der Patient soll nicht Fahrzeuge lenken oder Maschinen bedienen, bis die Sehfähigkeit wiederhergestellt ist.
Unerwünschte WirkungenDie mit der OZURDEX-Behandlung in Zusammenhang betrachteten unerwünschten Wirkungen während der klinischen Phase-III-Studien (DMÖ, VAV/ZVV und Uveitis) sowie spontane Berichte sind entsprechend den MedDRA-Systemorganklassen definiert:
Sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1000, <1/100), selten (≥1/10'000, <1/1000), sehr selten (<1/10'000). Innerhalb jeder Häufigkeitsgruppe sind die unerwünschten Wirkungen in absteigendem Schweregrad angegeben.
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen.
Gelegentlich: Migräne.
Augenerkrankungen
Sehr häufig: Erhöhter intraokulärer Druck (31%), Katarakt (38%), konjunktivale Blutung* (21%).
Häufig: Okuläre Hypertension, subkapsuläre Katarakt, Glaskörperblutung*, reduzierte Sehschärfe*, Sehbehinderung/-störung, Glaskörperabhebung*, Mouches volantes*, Glaskörpertrübungen*, Blepharitis, Augenschmerzen*, Photopsie*, konjunktivales Ödem*, konjunktivale Hyperämie*.
Gelegentlich: Nekrotisierende Retinitis, Endophthalmitis*, Glaukom, Netzhautablösung*, Retinariss*, Hypotonia bulbi*, zentrale seröse Chorioretinopathie, Vorderkammerentzündung*, Vorderkammerzellen/-trübung*, Missempfindungen im Auge*, Augenlidpruritus, Hyperämie der Sklera*.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Gelegentlich: Dislokation des Implantats* (Implantatmigration) mit oder ohne Hornhautödem (siehe auch «Warnhinweise und Vorsichtsmassnahmen»), Komplikation beim Einsetzen des Implantats, die zu einer Verletzung des Augengewebes führt* (Fehlplatzierung des Implantats).
*weist darauf hin, dass die unerwünschten Wirkungen eher mit der intravitrealen Injektion in Zusammenhang gebracht werden (die Häufigkeit der unerwünschten Wirkungen ist proportional zur Anzahl der durchgeführten Behandlungen).
Diabetisches Makulaödem
Die klinische Sicherheit von OZURDEX bei Patienten mit diabetischem Makulaödem wurde in zwei randomisierten, doppelblinden, mit einer Scheininjektion kontrollierten Phase-III-Studien untersucht. In beiden Studien wurden insgesamt 347 Patienten randomisiert der Behandlung mit OZURDEX und 350 Patienten der Behandlung mit Scheininjektion zugeteilt.
Die über die gesamte Studiendauer hinweg am häufigsten berichteten unerwünschten Wirkungen im behandelten Auge der mit OZURDEX behandelten Patienten waren Katarakt und erhöhter intraokularer Druck (IOD) (siehe unten).
In den dreijährigen klinischen DMÖ-Studien wiesen 87% der mit OZURDEX behandelten Patienten mit phakem behandeltem Auge zu Studienbeginn eine gewisse Linsentrübung/frühe Katarakt auf. Die Inzidenz aller beobachteten Kataraktarten (d.h. Rindenkatarakt, diabetischer Katarakt, Kernkatarakt, subkapsulärer Katarakt, lentikulärer Katarakt, Katarakt) betrug 68% über die gesamte Studiendauer von drei Jahren bei den mit OZURDEX behandelten Patienten mit phakem behandeltem Auge. 59% der Patienten mit phakem behandeltem Auge benötigten bis zur abschliessenden Untersuchung nach drei Jahren eine Kataraktoperation, wobei die Mehrheit der Operationen im zweiten und dritten Jahr durchgeführt wurde.
Der durchschnittliche IOD im behandelten Auge war zu Studienbeginn in beiden Behandlungsgruppen gleich (15,3 mmHg). Der durchschnittliche Anstieg des IOD ab dem Ausgangswert betrug in der OZURDEX-Gruppe bei allen Untersuchungen nicht mehr als 3,2 mmHg, wobei der mittlere IOD den höchsten Wert bei der Untersuchung 1,5 Monate nach der Injektion erreichte und sechs Monate nach jeder Injektion nahezu auf die Ausgangswerte zurückging. Die Rate und das Ausmass des IOD-Anstiegs nach einer OZURDEX-Behandlung haben sich nach wiederholten Injektionen von OZURDEX nicht erhöht.
28% der mit OZURDEX behandelten Patienten verzeichneten bei einer oder mehreren Untersuchungen während der Studie einen IOD-Anstieg von ≥10 mmHg gegenüber dem Ausgangswert. Zu Studienbeginn benötigten 3% der Patienten IOD-senkende Arzneimittel. Insgesamt benötigten 42% der Patienten in irgendeiner Phase der dreijährigen Studien IOD-senkende Arzneimittel im behandelten Auge, die Mehrheit dieser Patienten benötigte mehr als eine medikamentöse Behandlung. Die häufigste Nutzung (33%) erfolgte in den ersten zwölf Monaten und blieb von Jahr zu Jahr ähnlich.
Insgesamt vier der mit OZURDEX behandelten Patienten (1%) hatten wegen eines IOD-Anstiegs am behandelten Auge einen Eingriff. Ein mit OZURDEX behandelter Patient benötigte einen operativen Eingriff (Trabekulektomie), um den durch Steroide ausgelösten IOD-Anstieg zu beheben, ein Patient hatte eine Trabekulektomie, da Fibrin in der Vorderkammer den Kammerwasserabfluss blockierte, was zu erhöhtem IOD führte. Ein Patient hatte eine Iridotomie wegen eines Engwinkelglaukoms und ein Patient hatte eine Iridektomie infolge einer Kataraktoperation. Bei keinem Patienten war eine Entfernung des Implantats durch Vitrektomie erforderlich, um den IOD zu behandeln.
VAV/ZVV
Die klinische Sicherheit von OZURDEX bei Patienten mit Makulaödem nach retinalem Zentralvenen- oder Venenastverschluss wurde in zwei randomisierten, doppelblinden, mit einer Scheininjektion kontrollierten Phase III-Studien untersucht. In den beiden Phase III-Studien wurden insgesamt 427 Patienten randomisiert der Behandlung mit OZURDEX und 426 Patienten der Behandlung mit einer Scheininjektion zugeteilt. Insgesamt 401 Patienten (94%), welche für die Behandlung mit OZURDEX randomisiert wurden, haben die initiale Behandlungsphase (bis zum Tag 180) abgeschlossen.
Bei insgesamt 47,3% der Patienten kam es zu mindestens einem unerwünschten Ereignis. Die am häufigsten bei mit OZURDEX behandelten Patienten genannten unerwünschten Ereignisse waren erhöhter intraokulärer Druck (24,0%) und konjunktivale Blutung (14,7%).
Das Profil an unerwünschten Ereignissen war bei Patienten mit Venenastverschluss und solchen mit Zentralvenenverschluss vergleichbar, obwohl die Gesamtinzidenz von unerwünschten Ereignissen in der Untergruppe der Patienten mit Zentralvenenverschluss höher war.
Der erhöhte Augeninnendruck (IOD) unter OZURDEX erreichte an Tag 60 ein Maximum und kehrte bis Tag 180 wieder auf die Ausgangswerte zurück. Die IOD-Anstiege erforderten entweder keine Behandlung oder wurden durch vorübergehende Anwendung topischer IOD-senkender Arzneimittel behandelt. In der initialen Behandlungsphase benötigten 0,7% (3/421) der mit OZURDEX behandelten Patienten gegenüber 0,2% (1/423) der mit der Scheininjektion behandelten Patienten zur Behandlung des erhöhten IOD im Studienauge eine Laser-Behandlung oder Operation.
Das bei 341 Patienten nach einer zweiten OZURDEX-Injektion untersuchte Profil an unerwünschten Ereignissen war dem nach der ersten Injektion vergleichbar. Insgesamt ist es bei 54% der Patienten zu mindestens einer unerwünschten Reaktion gekommen. Die Inzidenz eines erhöhten IOD (24,9%) glich der nach der ersten Injektion beobachteten, und es kam genauso wie nach der ersten Injektion bis zum nicht verblindeten Tag 180 zu einer Normalisierung. Die Gesamtinzidenz an Katarakten war nach 1 Jahr höher als nach den ersten 6 Monaten.
Die klinische Sicherheit wurde zusätzlich in einer multizentrischen, 24-monatigen, post-marketing Beobachtungsstudie bei Patienten während der Behandlung von Makulaödem nach retinalem Venenverschluss und bei Patienten mit einer nicht infektiösen Uveitits des posterioren Segmentes des Auges, untersucht. Die häufigsten unerwünschten Wirkungen, welche während dieser Studie beobachtet wurden, waren konsistent mit den häufigsten unerwünschten Wirkungen aus den klinischen Studien. Gewichtet nach Häufigkeit der Injektionen ergab sich eine erhöhte Inzidenz unerwünschter Wirkungen bei Patienten, welche mehr als 2 Injektionen erhalten haben, im Vergleich zu Patienten, welche ≤2 Injektionen erhalten haben. Bei Patienten, welche mehr als 2 Injektionen erhalten haben, waren insbesondere Katarakt (Kataraktbildung bei 24,7% [44/178] und Kataraktprogression bei 32,0% [57/178] der Patienten mit einem phaken Linsenstatus bei Therapiebeginn), Glaskörperblutung bei 6,0% [17/283] und erhöhter intraokulärer Druck bei 24,0% [68/283] häufiger als bei Patienten mit ≤2 Injektionen.
Uveitis
Die klinische Sicherheit von OZURDEX bei Patienten mit einer Entzündung des posterioren Segments des Auges, die sich als nicht infektiöse Uveitis darstellt, wurde in einer einzelnen, multizentrischen, verblindeten, randomisierten Studie ermittelt.
Insgesamt 77 Patienten wurden randomisiert der Behandlung mit OZURDEX und 76 der Behandlung mit der Scheininjektion zugeteilt. Insgesamt 73 der randomisierten und mit OZURDEX behandelten Patienten (95%) haben die 26 Wochen dauernde Studie abgeschlossen.
Die am häufigsten berichteten unerwünschten Wirkungen im behandelten Auge von Patienten, die OZURDEX erhielten, waren konjunktivale Blutung (30,3%), erhöhter intraokulärer Druck (25,0%) und Katarakt (11,8%).
Die klinische Sicherheit wurde zusätzlich in einer multizentrischen, 24-monatigen, post-marketing Beobachtungsstudie bei Patienten während der Behandlung von Makulaödem nach retinalem Venenverschluss und bei Patienten mit einer nicht infektiösen Uveitis des posterioren Segmentes des Auges untersucht (siehe Abschnitt «VAV/ZVV»).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch
ÜberdosierungEs wurden keine Fälle von Überdosierung berichtet
Eigenschaften/WirkungenATC-Code
S01BA01
Wirkungsmechanismus
Dexamethason, ein starkes Kortikosteroid, unterdrückt Entzündungen, indem es die im Zuge der entzündlichen Reaktion auftretenden Ödeme, Fibrinablagerungen, Flüssigkeitsaustritte aus Kapillaren und Phagozytenmigration hemmt. Der Vascular Endothelial Growth Factor (VEGF) ist ein Zytokin, das im Zusammenhang mit Makulaödemen in erhöhten Konzentrationen exprimiert wird. Es ist ein starker Promotor der Gefässpermeabilität. Es wurde gezeigt, dass Kortikosteroide die VEGF-Expression hemmen. Darüber hinaus verhindern Kortikosteroide die Freisetzung von Prostaglandinen, von denen einige als Mediatoren von zystoiden Makulaödemen identifiziert wurden.
Pharmakodynamik
Keine Angaben.
Klinische Wirksamkeit
Diabetisches Makulaödem
Die Wirksamkeit von OZURDEX wurde in zwei dreijährigen, multizentrischen, doppelblinden, randomisierten, mit Scheininjektion kontrollierten, parallelen Studien identischen Designs ermittelt, die zusammen 1'048 Patienten eingeschlossen haben (Studien 206207 010 und 206207 011). Insgesamt wurden 351 Patienten randomisiert der Behandlung mit OZURDEX, 347 der Behandlung mit Dexamethason 350 µg und 350 der Behandlung mit Scheininjektion zugeteilt.
Patienten kamen für eine erneute Behandlung in Frage, wenn die Netzhautdicke im Zentralbereich gemäss OCT-Messung (optische Kohärenztomografie) über 175 Mikrometer betrug oder Prüfärzte die OCT-Werte als Beleg für ein verbleibendes Netzhautödem bewerteten, bestehend aus intraretinalen Zysten oder irgendwelcher Bereiche erhöhter Netzhautdicke innerhalb oder ausserhalb des Zentralbereichs. Patienten erhielten bis zu sieben Behandlungen in Intervallen von nicht häufiger als ca. alle sechs Monate.
Ausweichtherapien waren im Ermessen der Prüfärzte in jeder Phase zulässig, führten jedoch zum Abbruch der Teilnahme an den Studien.
Aus unterschiedlichen Gründen beendeten insgesamt 36% der mit OZURDEX behandelten Patienten gegenüber 57% der mit einer Scheininjektion behandelten Patienten die Teilnahme an der Studie vorzeitig. Die Abbruchraten aufgrund von unerwünschten Ereignissen waren bei den Behandlungs- und Scheininjektionsgruppen ähnlich (13% gegenüber 11%). Die Abbruchraten aufgrund fehlender Wirksamkeit waren in der OZURDEX-Gruppe geringer als in der Scheininjektionsgruppe (7% gegenüber 24%).
Die primären und wichtigen sekundären Endpunkte der Studien 206207-010 und -011 werden in der nachfolgenden Tabelle dargestellt. Die Verbesserung der Sehfähigkeit in der DEX700-Gruppe wurde durch Kataraktbildung beeinträchtigt – die verbesserte Sehfähigkeit wurde nach Beseitigung der Katarakt wiederhergestellt.
Wirksamkeit in den Studien 206207-010 und 206207-011 (ITT-Population)
|
Endpunkt
|
Studie
206207-010
|
Studie
206207-011
|
«Pooled» Studien 206207-010 und 206207-011
| |
DEX 700
N = 163
|
Schein-injektion
N = 165
|
DEX 700
N = 188
|
Schein-injektion
N = 185
|
DEX 700
N = 351
|
Schein-injektion
N = 350
| |
Änderung des mittleren BCVA-Werts über 3 Jahre, AUC-Methode (Buchstaben)
|
4,1
|
1,9
|
2,9
|
2,0
|
3,5
|
2,0
| |
p-Wert
|
0,016
|
0,366
|
0,023
| |
BCVA ≥15 Buchstaben Verbesserung gegenüber dem Ausgangswert in Jahr 3/ Abschlussuntersuchung (%)
|
22,1
|
13,3
|
22,3
|
10,8
|
22,2
|
12,0
| |
p-Wert
|
0,038
|
0,003
|
< 0,001
| |
Änderung des mittleren BCVA-Werts gegenüber dem Ausgangswert in Jahr 3/Abschlussuntersuchung (Buchstaben)
|
4,1
|
0,8
|
1,3
|
-0,0
|
2,6
|
0,4
| |
p-Wert
|
0,020
|
0,505
|
0,054
| |
Änderung der mittleren OCT-Netzhautdicke im Zentralbereich über 3 Jahre, AUC-Methode (µm)
|
-101,1
|
-37,8
|
-120,7
|
-45,8
|
-111,6
|
-41,9
| |
p-Wert
|
< 0,001
|
< 0,001
|
< 0,001
|
Die primären und wichtigen sekundären Endpunkte der zusammengefassten Analyse für pseudophake Patienten werden in der nachfolgenden Tabelle dargestellt.
Wirksamkeit in pseudophaken Patienten (zusammengefasste Studien 206207-010 und 206207-011)
|
Endpunkt
|
DEX 700
N = 86
|
Schein-injektion
N = 101
|
p-Wert
| |
Änderung des mittleren BCVA-Werts über 3 Jahre, AUC-Methode (Buchstaben)
|
6,5
|
1,7
|
< 0,001
| |
BCVA ≥15 Buchstaben Verbesserung gegenüber dem Ausgangswert in Jahr 3/Abschlussuntersuchung (%)
|
23,3
|
10,9
|
0,024
| |
Änderung des mittleren BCVA-Werts gegenüber dem Ausgangswert in Jahr 3/Abschlussuntersuchung
|
6,1
|
1,1
|
0,004
| |
Änderung der mittleren OCT-Netzhautdicke im Zentralbereich über 3 Jahre, AUC-Methode (µm)
|
-131,8
|
-50,8
|
< 0,001
|
Die primären und wichtigen sekundären Endpunkte der zusammengefassten Analyse für Patienten, die zuvor irgendeine Behandlung erhalten haben, sind in der nachfolgenden Tabelle dargestellt.
Wirksamkeit in Patienten, die zuvor irgendeine Behandlung erhalten haben (zusammengefasste Studien 206207-010 und 206207-011)
|
Endpunkt
|
DEX 700
N = 247
|
Schein-injektion
N = 261
|
p-Wert
| |
Änderung des mittleren BCVA-Werts über 3 Jahre, AUC-Methode (Buchstaben)
|
3,2
|
1,5
|
0,024
| |
BCVA ≥15 Buchstaben Verbesserung gegenüber dem Ausgangswert in Jahr 3/Abschlussuntersuchung (%)
|
21,5
|
11,1
|
0,002
| |
Änderung des mittleren BCVA-Werts gegenüber dem Ausgangswert in Jahr 3/Abschlussuntersuchung
|
2,7
|
0,1
|
0,055
| |
Änderung der mittleren OCT-Netzhautdicke im Zentralbereich über 3 Jahre, AUC-Methode (µm)
|
-126,1
|
-39,0
|
< 0,001
|
VAV/ZVV
Die Wirksamkeit von OZURDEX wurde im Rahmen von zwei multizentrischen, doppelblinden, randomisierten, mit Scheininjektion kontrollierten, parallelen Studien mit identischem Design untersucht, die zusammen 1'267 Patienten umfassten, die randomisiert mit 350 µg oder 700 µg Dexamethason-Implantaten oder mit einer Scheininjektion behandelt wurden (Studien 206207 008 und 206207 009). Insgesamt 427 Patienten wurden randomisiert der Behandlung mit OZURDEX (700 µg), 414 Patienten der Behandlung mit Dexamethason 350 µg und 426 Patienten der Behandlung mit der Scheininjektion zugeteilt.
Auf der Grundlage der zusammengefassten Analysenergebnisse war der Anteil der Responder, definiert als Besserung der BCVA (beste korrigierte Sehschärfe) 90 Tage nach Injektion eines einzelnen Implantats um ≥15 Buchstaben, unter OZURDEX-Implantaten signifikant höher als nach einer Behandlung mit der Scheininjektion (p < 0,001).
Die nachfolgende Tabelle zeigt den Anteil der Patienten, die den primären Endpunkt der Wirksamkeitsbewertung (Besserung der BCVA um ≥15 Buchstaben gegenüber dem Ausgangswert nach Injektion eines einzelnen Implantats) erreichten. Ein Behandlungseffekt wurde am ersten Messpunkt an Tag 30 beobachtet. Der maximale Behandlungseffekt war an Tag 60 festzustellen, und die Differenz im Anteil der Responder fiel an allen Messpunkten bis Tag 90 nach der Injektion statistisch signifikant zugunsten von OZURDEX gegenüber der Scheininjektion aus. An Tag 180 war bei den mit OZURDEX behandelten Patienten weiterhin ein numerisch höherer Anteil von Respondern mit Verbesserung um ≥15 Buchstaben gegenüber dem BCVA-Ausgangswert zu beobachten als bei den Patienten mit der Scheininjektion.
Anteil der Patienten mit ≥15 Buchstaben Verbesserung gegenüber dem Ausgangswert des bestkorrigierten Visus (BCVA) im behandelten Auge (zusammengefasst, ITT Population)
|
Visite
|
OZURDEX
N = 427
|
Scheininjektion
N = 426
| |
Tag 30
|
21,3%a
|
7,5%
| |
Tag 60
|
29,3%a
|
11,3%
| |
Tag 90
|
21,8%a
|
13,1%
| |
Tag 180
|
21,5%
|
17,6%
|
a Proportion signifikant höher bei OZURDEX im Vergleich zur Scheininjektion (p < 0,001)
Die mittlere Änderung der BCVA gegenüber dem Ausgangswert war zu allen Zeitpunkten unter OZURDEX signifikant grösser als unter der Behandlung mit der Scheininjektion.
In beiden Phase-III-Studien und in der gepoolten Analyse unterschieden sich die kumulativen Ansprechkurven in Bezug auf die Zeit bis zum Erreichen einer Besserung der BCVA um ≥15 Buchstaben (3 Zeilen) unter OZURDEX und der Behandlung mit der Scheininjektion signifikant (p < 0,001), wobei die mit OZURDEX behandelten Patienten die Verbesserung der BCVA um 3 Zeilen signifikant früher erzielten als die Patienten mit der Scheininjektion.
OZURDEX war der Behandlung mit der Scheininjektion in Bezug auf die Prävention eines Sehverlusts zahlenmässig überlegen, was sich in einem geringeren Anteil an Patienten in der OZURDEX-Gruppe äusserte, bei denen es über den Beurteilungszeitraum von 6 Monaten zu einer Verschlechterung des Sehvermögens um ≥15 Buchstaben kam.
In beiden Phase III-Studien und in der gepoolten Analyse war die mittlere Retinadicke unter OZURDEX an Tag 90 signifikant geringer und die mittlere Reduktion gegenüber dem Ausgangswert signifikant grösser (207,9 Mikron) als unter der Behandlung mit der Scheininjektion (95,0 Mikron) (p < 0,001, zusammengefasste Daten). Der über die BCVA an Tag 90 untersuchte Behandlungseffekt wurde durch diesen anatomischen Befund unterstützt. An Tag 180 fiel die mittlere Verminderung der Retinadicke (119,3 Mikron) gegenüber der Behandlung mit der Scheininjektion nicht signifikant aus.
Patienten, die einen BCVA-Score von < 84 oder eine Retinadicke > 250 Mikron in der optischen Kohärenztomographie (OCT) aufwiesen und nach Meinung des Prüfarztes ohne Risiko behandelt werden konnten, waren für eine Behandlung mit OZURDEX innerhalb einer offenen Fortsetzungsphase geeignet. Von den innerhalb der offenen Fortsetzungsphase behandelten Patienten erhielten 98% der Patienten zwischen 5 und 7 Monaten nach der Initialbehandlung eine OZURDEX-Injektion.
Wie auch während der Initialbehandlung wurde innerhalb der offenen Phase an Tag 60 das maximale Ansprechen verzeichnet. Die kumulierten Ansprechraten fielen während der gesamten offenen Phase bei Patienten, die zwei aufeinanderfolgende OZURDEX-Injektionen erhalten hatten, höher aus als bei Patienten, die in der Initialphase keine OZURDEX-Injektion erhalten hatten.
Der Anteil von Respondern war zu jedem Zeitpunkt nach der zweiten Behandlung grundsätzlich höher als nach der ersten Behandlung. Hingegen führte ein Aufschub der Behandlung um 6 Monate zu allen Zeitpunkten in der offenen Phase zu einem geringeren Anteil von Respondern, verglichen mit Patienten, die eine zweite OZURDEX-Injektion erhalten hatten.
Uveitis
Die klinische Wirksamkeit von OZURDEX wurde in einer einzelnen, multizentrischen, verblindeten, randomisierten Studie zur Behandlung einer nicht infektiösen Entzündung des posterioren Segments des Auges bei Patienten mit Uveitis ermittelt.
Insgesamt 229 Patienten wurden randomisiert und der Behandlung mit Dexamethason-Implantaten (350 µg oder 700 µg) oder mit Scheininjektion zugeteilt. Davon wurden insgesamt 77 Patienten randomisiert der Behandlung mit OZURDEX, 76 Patienten der Behandlung mit Dexamethason 350 µg und 76 der Behandlung mit der Scheininjektion zugeteilt. Insgesamt 95% der Patienten haben die 26 Wochen dauernde Studie abgeschlossen.
Der Anteil der Patienten mit einem Glaskörpertrübungswert von 0 im behandelten Auge in Woche 8 (primärer Endpunkt) war bei OZURDEX viermal so hoch (46,8%) wie bei der Scheininjektion (11,8%), p < 0,001. Die statistische Überlegenheit blieb bis einschliesslich Woche 26 bestehen (p ≤0,014), wie in der nachfolgenden Tabelle dargestellt.
Die kumulativen Ansprechkurven (Zeit bis zum Glaskörpertrübungswert von 0) der OZURDEX-Gruppe unterschieden sich signifikant von denen der Scheininjektionsgruppe (p < 0,001), wobei Patienten, die Dexamethason erhielten, ein früheres und besseres Ansprechen auf die Behandlung zeigten.
Die Reduzierung der Glaskörpertrübung ging mit einer Verbesserung des Visus einher. Der Anteil der Patienten mit einer Verbesserung von mindestens 15 Buchstaben gegenüber dem BCVA-Ausgangswert im behandelten Auge betrug in Woche 8 bei OZURDEX mehr als das Sechsfache (42,9%) im Vergleich zur Scheininjektion (6,6%), p < 0,001. Die statistische Überlegenheit wurde in Woche 3 erzielt und blieb bis einschliesslich Woche 26 bestehen (p ≤0,001), wie in der nachfolgenden Tabelle dargestellt.
Der prozentuale Anteil der Patienten, die vom Ausgangspunkt bis Woche 8 zusätzliche Medikation benötigten, betrug bei der OZURDEX-Gruppe nur rund ein Drittel (7,8%) gegenüber der Scheininjektionsgruppe (22,4%), p = 0,012.
Anteil der Patienten mit einem Glaskörpertrübungswert von Null und einer Verbesserung von ≥15 Buchstaben ab dem Ausgangswert des bestkorrigierten Visus im behandelten Auge (ITT-Population)
|
Visite
|
Glaskörpertrübungswert von Null
|
BCVA-Verbesserung von ≥15 Buchstaben ab dem Ausgangswert
| |
|
DEX 700
N = 77
|
Scheininjektion
N = 76
|
DEX 700
N = 77
|
Scheininjektion
N = 76
| |
Woche 3
|
23,4%
|
11,8%
|
32,5%a
|
3,9%
| |
Woche 6
|
42,9%a
|
9,2%
|
41,6%a
|
7,9%
| |
Woche 8
|
46,8%a
|
11,8%
|
42,9%a
|
6,6%
| |
Woche 12
|
45,5%a
|
13,2%
|
41,6%a
|
13,2%
| |
Woche 16
|
40,3%b
|
21,1%
|
39,0%a
|
13,2%
| |
Woche 20
|
39,0%c
|
19,7%
|
40,3%a
|
13,2%
| |
Woche 26
|
31,2%d
|
14,5%
|
37,7%a
|
13,2%
|
a p < 0,001; b p = 0,010; c p = 0,009; d p = 0,014
PharmakokinetikAbsorption
An einer Untergruppe von 21 Patienten der beiden 6-monatigen Wirksamkeitsstudien wurden vor der Applikation und an Tag 7, 30, 60 und 90 nach Applikation eines einzelnen intravitrealen Implantats mit 350 µg oder 700 µg Dexamethason die Plasmakonzentrationen bestimmt. 95% der in der 350 µg-Dosisgruppe und 86% der in der 700 µg-Dosisgruppe gemessenen Dexamethason-Plasmakonzentrationen lagen unterhalb der Nachweisgrenze (0,05 ng/ml). Die höchste Plasmakonzentration betrug 0,094 ng/ml bei einem Patienten der 700 µg-Gruppe. Die Dexamethason-Plasmakonzentration schien keine Beziehung zu Alter, Körpergewicht oder Geschlecht des Patienten zu haben.
In den beiden konfirmatorischen DMÖ-Studien wurden Plasmakonzentrationen von einer Untergruppe von Patienten vor der intravitrealen Injektion sowie an Tag 1, 7 und 21 sowie 1,5 und 3 Monate nach der intravitrealen Injektion eines einzelnen Implantats mit 350 µg oder 700 µg Dexamethason bestimmt. 100% der Plasma-Dexamethason-Konzentrationswerte der 350-µg-Dosisgruppe und 90% der 700-µg-Dosisgruppe lagen unterhalb der Bestimmungsgrenze (0,05 ng/ml). Der höchste Plasmakonzentrationswert von 0,102 ng/ml wurde bei einem Patienten in der 700-µg-Gruppe gemessen. Es gab keine Anzeichen dafür, dass die Plasma-Dexamethason-Konzentration mit dem Alter, dem Körpergewicht oder dem Geschlecht der Patienten zusammenhängt.
Distribution
In einer 6-monatigen Studie an Affen lag die Cmax von Dexamethason im Glaskörper an Tag 42 nach einer einzelnen intravitrealen Injektion von OZURDEX bei 100 ng/ml und an Tag 91 bei 5,57 ng/ml. Dexamethason blieb über 6 Monate nach der Injektion im Glaskörper nachweisbar. Die Dexmethason-Konzentration nahm in der folgenden Rangfolge ab: Retina > Iris > Ziliarkörper > Glaskörper > Kammerwasser > Plasma. Dexamethason wurde bei Affen über 6 Monate in den Glaskörper abgegeben.
Metabolismus
In einer In-vitro-Metabolisierungsstudie waren nach 18-stündiger Inkubation von [14C] -Dexamethason mit Kornea-, Iris-, Ziliarkörper, Choroidea-, Retina-, Glaskörper- und Sklera-Gewebe des Menschen keine Metaboliten nachweisbar. Diese Beobachtung steht im Einklang mit den Ergebnissen einer Studie zum okulären Metabolismus bei Kaninchen und Affen.
Dexamethason wird schliesslich in lipid- und wasserlösliche Metaboliten metabolisiert, die über die Galle und im Urin ausgeschieden werden können.
Elimination
Die OZURDEX-Matrix wird über eine einfache Hydrolyse langsam in Milchsäure und Glykolsäure und dann weiter in Kohlendioxid und Wasser abgebaut.
Kinetik spezieller Patientengruppen
Keine Angaben.
Präklinische DatenEs liegen keine Daten zur Mutagenität und Kanzerogenität von OZURDEX vor. Dexamethason ruft in Tierexperimenten bei Mäusen, Ratten, Hamstern, Kaninchen und Hunden Gaumenspalten und in einem viel geringeren Umfang andere Fehlbildung hervor.
Sonstige HinweiseInkompatibilitäten
Keine Angaben.
Beeinflussung diagnostischer Methoden
Keine Angaben.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «Exp» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Nicht über 30°C lagern. Nicht einfrieren.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
OZURDEX ist nur zur einmaligen Anwendung bestimmt.
Jeder Applikator darf nur für die Behandlung eines einzigen Auges verwendet werden.
Der Applikator darf nicht verwendet werden, wenn der Siegelverschluss des Folienbeutels, welcher den Applikator enthält, beschädigt ist.
|
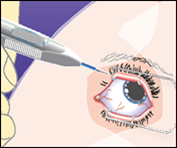
1.Halten Sie den Applikator der Länge nach parallel zum Limbus.
|
|
| |
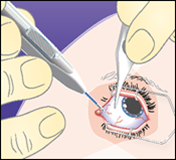
2.Richten Sie ihn in schrägem Winkel mit dem Kanülenschliff der Nadel nach oben (also von der Sklera abgewandt) auf die Sklera. Schieben Sie die Spitze der Nadel in der Sklera parallel zum Limbus etwa 1 mm vor.
|
|
| |
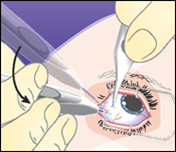
3.Ändern Sie die Richtung und richten Sie den Applikator senkrecht zum Zentrum des Auges aus, um einen geführten skleralen Zugang zu schaffen. Schieben Sie die Nadel vor, bis sie in den Glaskörperraum eingedrungen ist.
Die Nadel darf nur bis zu dem Punkt vorgeschoben werden, an dem die Hülse um die Nadel die Konjunktiva berührt.
|
|
| |
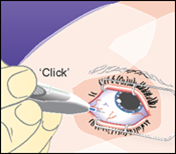
4.Drücken Sie die Auslösetaste des Applikators langsam herunter, bis Sie einen hörbaren Klick vernehmen. Bevor Sie den Applikator aus dem Auge zurückziehen, stellen Sie bitte sicher, dass die Auslösetaste vollständig heruntergedrückt ist und mit der Oberfläche des Applikators bündig abschliesst.
|
|
| |
5.Ziehen Sie den Applikator in der gleichen Richtung heraus, in der Sie ihn in den Glaskörper vorgeschoben haben.
|
|
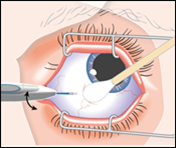
| |
6.Entsorgen Sie den Applikator sofort nach erfolgter Behandlung in einem geeigneten Sammelgefäss.
|
Jedes ungebrauchte Produkt oder Abfallmaterial muss gemäss den lokalen Vorschriften vernichtet werden.
Zulassungsnummer60324 (Swissmedic)
PackungenOZURDEX enthält: 1 steriles Implantat zu 0,7 mg in einem Applikator. [A]
ZulassungsinhaberinAbbVie AG, Cham
Stand der InformationNovember 2023
|