Eigenschaften/WirkungenATC-Code
L04AA32
Wirkungsmechanismus
Apremilast ist ein oraler kleinmolekularer Inhibitor der Phosphodiesterase 4 (PDE4). PDE4 ist eine für zyklisches Adenosinmonophosphat (cAMP) spezifische und in Entzündungszellen wichtige PDE. Durch PDE4-Hemmung werden intrazelluläre cAMP-Spiegel angehoben. Die spezifischen Mechanismen, über welche die Psoriasis/Psoriasis-Arthritis und der Morbus Behçet beeinflusst werden, sind nicht vollständig aufgeklärt.
Pharmakodynamik
In klinischen Studien an Patienten mit Psoriasis-Arthritis bewirkte Apremilast eine signifikante Modulation, jedoch keine vollständige Hemmung der Plasmaproteinspiegel von IL-1α, IL-6, IL-8, MCP-1, MIP-1β, MMP-3 und TNF-α. Nach 40-wöchiger Behandlung mit Apremilast fand sich eine Abnahme der Plasmaproteinspiegel von IL-17 und IL-23 sowie ein Anstieg von IL-10. In klinischen Studien an Psoriasis-Patienten verminderte Apremilast die Epidermisdicke der von Läsionen befallenen Haut, die Infiltration durch Entzündungszellen und die Expression proinflammatorischer Gene, einschliesslich derjenigen für induzierbare Stickoxidsynthase (iNOS), IL-12/IL-23p40, IL-17A, IL-22 und IL-8. Bei Patienten mit Morbus Behçet, die mit Apremilast behandelt wurden, bestand ein positiver Zusammenhang zwischen der Veränderung des Plasma-TNF-alpha und der klinischen Wirksamkeit, gemessen anhand der Anzahl von oralen Ulcera.
Bei therapeutischen Plasmakonzentrationen führte Apremilast bei gesunden Probanden zu keiner Verlängerung des QT-Intervalls.
Klinische Wirksamkeit
Psoriasis-Arthritis
Erfahrungen aus klinischen Studien an Patienten mit Psoriasis-Arthritis, welche mit kleinmolekularer DMARDs und/oder Biologika vortherapiert waren
Die Sicherheit und Wirksamkeit von Otezla wurde in 3 ähnlich aufgebauten multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studien (Studien PALACE 1, PALACE 2 und PALACE 3) an 1493 erwachsenen Patienten mit aktiver PsA (≥3 geschwollene Gelenke und ≥3 druckschmerzhafte Gelenke) trotz Vortherapie mit DMARDs, einschliesslich biologischer DMARDs (z.B. TNF-Blocker), oder bestehender kleinmolekularer DMARD-Therapie untersucht.
Bei den Patienten in diesen Studien bestand die Diagnose PsA seit mindestens 6 Monaten. In der Studie PALACE 3 war zudem eine qualifizierende Psoriasis-Hautläsion (Mindestdurchmesser 2 cm) erforderlich. Patienten, bei denen >3 bei PsA eingesetzte Substanzen (kleine Moleküle oder Biologika) oder >1 biologischer TNF-Blocker bereits versagt hatten, wurden ausgeschlossen. In die 3 Studien wurden Patienten mit allen PsA-Unterformen eingeschlossen: symmetrische Polyarthritis (62,0%), asymmetrische Oligoarthritis (26,9%), Arthritis mit Befall der distalen Interphalangealgelenke (DIP) (6,2%), Arthritis mutilans (2,7%) und prädominante Spondylitis (2,1%). Patienten mit vorbestehender Enthesitis (63%) und vorbestehender Daktylitis (42%) wurden aufgenommen.
In den 3 Studien wurden die Patienten zu Placebo (n=496), Otezla 20 mg (n=500) oder Otezla 30 mg (n=497) zweimal täglich oral randomisiert. In den Studien PALACE 1, PALACE 2 und PALACE 3 erfolgte die Behandlungszuordnung stratifiziert nach bestehender Therapie mit kleinmolekularen DMARDs in der Ausgangslage (Baseline). In PALACE 3 war ein Psoriasisbefall von ≥3% der Körperoberfläche (KOF) ein weiteres Stratifizierungskriterium. Die Patienten durften während der Studie eine Begleittherapie mit stabilen Dosen von Methotrexat (MTX) (≤25 mg/Woche; 54,5%), Sulfasalazin (SSZ) (≤2 g/Tag; 9,0%), Leflunomid (LEF) (≤20 mg/Tag; 7,4%), niedrig dosierten oralen Kortikosteroiden (entsprechend ≤10 mg Prednison täglich; 13,9%) und/oder nichtsteroidalen Antirheumatika (NSAR; 70,7%) erhalten. Die Gabe von Apremilast in Kombination mit biologischen DMARDs wurde nicht untersucht.
Eine Vortherapie nur mit kleinmolekularen DMARDs wurde bei 76,4% der Patienten und eine Vortherapie mit biologischen DMARDs bei 22,4% der Patienten angegeben, darunter 7,8% mit Versagen einer biologischen DMARD-Vortherapie. Die mediane PsA-Erkrankungsdauer betrug 5 Jahre.
Primärer Endpunkt war der Anteil der Patienten, die nach 16 Wochen ein American College of Rheumatology (ACR) 20-Ansprechen erreichten. Patienten, deren Anzahl an druckschmerzhaften und geschwollenen Gelenken keine mindestens 20%ige Besserung aufwiesen, wurden nach 16 Wochen als Nonresponder eingestuft. Placebo-Nonresponder wurden im Verhältnis 1:1 zu Otezla 20 mg zweimal täglich oder 30 mg zweimal täglich verblindet rerandomisiert. Otezla-Patienten erhielten weiterhin ihre initiale Behandlung. Nach 24 Wochen wurden alle verbliebenen Placebo-Patienten entweder zu Otezla 20 mg zweimal täglich oder zu Otezla 30 mg zweimal täglich rerandomisiert. Im Anschluss an die 52 Wochen Behandlung konnten die Patienten in den Langzeit-Verlängerungsstudien der PALACE 1, PALACE 2 und PALACE 3 Studien für insgesamt bis zu 5 Jahre (260 Wochen) unverblindet mit Apremilast 20 mg oder 30 mg fortfahren.
Der Anteil der Patienten, welche ein ACR-20/50/70 Ansprechen in der Studien PALACE 1/2/3 nach 16, 24 und 52 Wochen zeigten, ist in Tabelle 1 aufgelistet. Die Behandlung mit Apremilast führte zu einer signifikanten Verbesserung der Anzeichen und Symptome der PsA, welche anhand der ACR20-Ansprechkriterien bis Woche 16 im Vergleich zu Placebo erhoben wurden. Das ACR-20/50/70 Ansprechen wurden bis Woche 24 aufrechterhalten. Bei den Patienten, welche diejenige Apremilast-Behandlung durchgehend erhielten, auf die sie zu Studienbeginn randomisiert worden waren, wurde das ACR-20/50/70 Ansprechen bis einschliesslich Woche 52 aufrechterhalten. Von den 497 Patienten, welche anfänglich zu Apremilast 30 mg zweimal täglich randomisiert wurden, traten 375 (75%) in die Langzeit-Verlängerungsstudien ein und von diesen waren nach 260 Wochen noch 221 Patienten (59%) unter Behandlung. Das ACR Ansprechen wurde in den Langzeit-Verlängerungsstudien für bis zu 5 Jahre aufrechterhalten.
Tabelle 1: Anteil der Patienten mit ACR-20/50/70 Ansprechen in den Studien PALACE 1/2/3 nach 16, 24 und 52 Wochen
|
|
PALACE 1
|
PALACE 2
|
PALACE 3
| |
|
Placebo
|
Otezla 30 mg zweimal täglich
|
Placebo
|
Otezla 30 mg zweimal täglich
|
Placebo
|
Otezla 30 mg zweimal täglich
| |
Na
|
%
|
Na
|
%
|
Na
|
%
|
Na
|
%
|
Na
|
%
|
Na
|
%
| |
ACR 20
| |
Woche 16
|
168
|
19,0%
|
168
|
38,1%***
|
159
|
18,9%
|
162
|
32,1%**
|
169
|
18,3%
|
167
|
40,7%***
| |
Woche 24
|
168
|
13,1%
|
168
|
35,1%***
|
159
|
15,7%
|
162
|
24,7%*
|
169
|
15,4%
|
167
|
31,1%***
| |
Woche 52a
|
N/Ab
|
130
|
54,6%
|
N/Ab
|
116
|
52,6%
|
N/Ab
|
127
|
63,0%
| |
ACR 50
| |
Woche 16
|
168
|
6,0%
|
168
|
16,1%**
|
159
|
5,0%
|
162
|
10,5%
|
169
|
8,3%
|
167
|
15,0%
| |
Woche 24
|
168
|
4,2%
|
168
|
19,0%***
|
159
|
8,8%
|
162
|
11,7%
|
169
|
7,7%
|
167
|
16,2%*
| |
Woche 52a
|
N/Ab
|
130
|
24,6%
|
N/Ab
|
118
|
18,6%
|
N/Ab
|
126
|
30,2%
| |
ACR 70
| |
Woche 16
|
168
|
1,2%
|
168
|
4,2%
|
159
|
0,6%
|
162
|
1,2%
|
169
|
2,4%
|
167
|
3,6%
| |
Woche 24
|
168
|
0,6%
|
168
|
10,1%***
|
159
|
3,1%
|
162
|
2,5%
|
169
|
3,6%
|
167
|
5,4%
| |
Woche 52a
|
N/Ab
|
130
|
13,8%
|
N/Ab
|
118
|
6,8%
|
N/Ab
|
125
|
10,4%
|
N/A=nicht zutreffend.
* p ≤0,05 verglichen mit Placebo; ** p ≤0,01 verglichen mit Placebo; *** p ≤0,001 verglichen mit Placebo
a Die Ansprechraten über 16 und 24 Wochen bezieht sich auf N=Anzahl der randomisierten Patienten, die Ansprechrate bei Woche 52 bezieht sich auf N=Anzahl der bis zu diesem Zeitpunkt in der Studie verbliebenen Patienten.
b Kein Placebo nach Woche 24.
Die in der mit Apremilast behandelten Gruppe beobachteten Ansprechraten waren bei Patienten mit bzw. ohne Begleittherapie mit DMARDs, einschliesslich MTX, vergleichbar (Tabelle 2). Bei den Zuvor ausschliesslich mit niedermolekularen DMARDs oder Biologika vortherapierten Patienten, die Apremilast erhielten, war das ACR20-Ansprechen bis Woche 16 im Vergleich zu den mit Placebo behandelten Patienten grösser (Tabelle 3).
Ähnliche ACR20-Ansprechraten wurden bei Patienten mit verschiedenen PsA-Unterformen, einschliesslich Arthritis mit Befall der DIP, beobachtet; die Anzahl von Patienten mit den Unterformen Arthritis mutilans und prädominante Spondylitis war jedoch zu gering, um eine sinnvolle Bewertung zu erlauben.
Tabelle 2: Anteil der Patienten mit ACR-20 Ansprechen in den Studien PALACE 1/2/3 nach 16 Wochen mit oder ohne DMARD Begleittherapie
|
|
Placebo
|
Otezla 30 mg zweimal täglich
| |
Na
|
%
|
Na
|
%
| |
Monotherapie
| |
PALACE 1
|
58
|
10,3%
|
62
|
46,8%
| |
PALACE 2
|
46
|
15,2%
|
49
|
22,4%
| |
PALACE 3
|
68
|
13,2%
|
66
|
39,4%
| |
Mit DMARD Begleittherapie
| |
PALACE 1
|
110
|
23,6%
|
106
|
33,0%
| |
PALACE 2
|
113
|
20,4%
|
113
|
36,3%
| |
PALACE 3
|
101
|
21,8%
|
101
|
41,6%
|
a N ist die Anzahl randomisierter und behandelter Patienten innerhalb jeder Subgruppe.
Tabelle 3: Anteil der Patienten, die in PALACE 1, 2 und 3 nach vorherige Anwendung von Biologika in Woche 16 eine ACR-20-Response erreichten
|
Subgruppe
Studie
|
Placebo
|
Otezla 30 mg zweimal täglich
| |
Na
|
%
|
Na
|
%
| |
Vorherige Behandlung nur mit niedermolekularen DMARDs (Biologika-naive Patienten)
| |
PALACE 1
|
120
|
23,3%
|
124
|
41,1%
| |
PALACE 2
|
135
|
20,7%
|
134
|
34,3%
| |
PALACE 3
|
121
|
20,7%
|
124
|
42,7%
| |
Vorherige Behandlung mit Biologikab
| |
PALACE 1
|
41
|
4,9%
|
41
|
26,8%
| |
PALACE 2
|
23
|
8,7%
|
23
|
21,7%
| |
PALACE 3
|
48
|
12,5%
|
43
|
34,9%
|
DMARD=krankheitsmodifizierendes Antirheumatikum.
a N ist die Anzahl der randomisierten und behandelten Patienten innerhalb der jeweiligen Subgruppe.
b Die Teilnehmer konnten vorher mit Biologika und zusätzlich mit niedermolekularen DMARDs behandelt worden sein.
In den Studien PALACE 1, PALACE 2 und PALACE 3 waren die Verbesserungen der Krankheitsaktivitätsskala („Disease Activity Scale“, DAS) 28 mit C-reaktivem Protein (CRP) und der Anteil von Patienten, die ein modifiziertes PsA-Ansprechkriterium (PsARC) erreichten, bis Woche 16 in der Apremilast-Gruppe im Vergleich zu Placebo grösser (nominaler p <0,0004 bzw. p <0,0017). Diese Verbesserungen wurden bis Woche 24 aufrechterhalten. Bei den Patienten, welche diejenige Apremilast-Behandlung durchgehend erhielten, zu der sie zu Studienbeginn randomisiert worden waren, wurden der DAS28(CRP)-Score und das PsARC-Ansprechen bis einschliesslich Woche 52 aufrechterhalten. Die Verzögerung der Progression struktureller Schäden wurde nicht untersucht.
Bis Woche 16 und 24 wurden bei den mit Apremilast behandelten Patienten Verbesserungen bei den für die Psoriasis-Arthritis charakteristischen Parametern der peripheren Krankheitsaktivität (Anzahl geschwollener Gelenke, Anzahl (druck)schmerzempfindlicher Gelenke) und den Hautmanifestationen der Psoriasis beobachtet. Bei den Patienten, welche diejenige Apremilast-Behandlung durchgehend erhielten, zu der sie zu Studienbeginn randomisiert worden waren, wurden diese Verbesserungen bis einschliesslich Woche 52 aufrechterhalten. Das klinische Ansprechen wurde in den Langzeit-Verlängerungsstudien für dieselben Parameter der peripheren Krankheitsaktivität und der Hautmanifestationen der Psoriasis für bis zu 5 Jahre aufrechterhalten.
Körperliche Funktion und Quality of Life
Mit Apremilast 30 mg zweimal täglich behandelte Patienten wiesen eine im Vergleich zur Placebo-Gruppe signifikant grössere Verbesserung bei der mittleren Veränderung des HAQ-DI-Scores gegenüber Baseline nach 16 Wochen und nach 24 Wochen auf. Das Verhältnis der HAQ-DI-Responder (Verbesserung ≥0,3 gegenüber Baseline) nach 16 Wochen war in der 30 mg Otezla Gruppe grösser als in der Placebo-Gruppe (Tabelle 4). Bei den Patienten, welche durchgehend mit Apremilast therapiert wurden, wurde die Verbesserung im HAQ-DI-Score und im Verhältnis zu Placebo über 52 Wochen aufrechterhalten.
Tabelle 4: Veränderung des HAQ-DI-Scores gegenüber der Baseline in den Studien PALACE 1/2/3 in Woche 16, 24 und 52
|
|
Placebo
|
Otezla 30 mg zweimal täglich
| |
Na
|
Änderung
|
Na
|
Änderung
| |
PALACE 1
| |
Woche 16
|
168
|
-0,086
|
168
|
-0,244**
| |
Woche 24
|
168
|
-0,076
|
168
|
-0,258***
| |
Woche 52
|
N/Ab
|
132
|
-0,318
| |
PALACE 2
| |
Woche 16
|
159
|
-0,053
|
162
|
-0,193**
| |
Woche 24
|
159
|
-0,085
|
162
|
-0,206*
| |
Woche 52
|
N/Ab
|
117
|
-0,330
| |
PALACE 3
| |
Woche 16
|
169
|
-0,065
|
167
|
-0,192**
| |
Woche 24
|
169
|
-0,053
|
167
|
-0,192**
| |
Woche 52
|
N/Ab
|
127
|
-0,350
|
* p ≤0,05 für Apremilast vs. Placebo; **p ≤0,01 für Apremilast vs. Placebo; ***p ≤0,001 für Apremilast vs. Placebo.
a Die Ansprechraten über 16 und 24 Wochen bezieht sich auf N=Anzahl der randomisierten Patienten, die Ansprechrate bei Woche 52 bezieht sich auf N=Anzahl der bis zu diesem Zeitpunkt in der Studie verbliebenen Patienten.
b Kein Placebo nach Woche 24.
HAQ-DI=Health Assessment Questionnaire-Disability Index; 0=beste Bewertung; 3=schlechteste Bewertung; mit diesem Fragebogen wird die Fähigkeit des Patienten, folgende Tätigkeiten zu verrichten, ermittelt: Ankleiden/Körperpflege, Aufstehen, Essen, Gehen, Erreichen von Gegenständen, Greifen, Hygiene und Verrichtung von Aktivitäten des täglichen Lebens.
In den Studien PALACE 1, PALACE 2 und PALACE 3 wurden bei den mit Apremilast behandelten Patienten im Vergleich zu Placebo signifikante Verbesserungen der gesundheitsbezogenen Lebensqualität nachgewiesen, erfasst anhand der gegenüber Baseline erhobenen Veränderungen in Gesundheitsfragebogen «Short Form Health Survey» Version 2 (SF-36v2) und im Score des Instruments Funktionelle Beurteilung der Therapie chronischer Erkrankung – Fatigue (FACIT-Fatigue). Eine verbesserte physische Funktion, bewertet gemäss HAQ-DI und SF-36v2PF, sowie der FACIT Fatigue Score wurden in den Langzeit-Verlängerungsstudien für bis zu 5 Jahre aufrechterhalten.
Psoriasis
Die Sicherheit und Wirksamkeit von Otezla wurden in zwei multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studien (Studien ESTEEM 1 und ESTEEM 2) beurteilt, in welche insgesamt 1257 Patienten ab 18 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis eingeschlossen wurden, bei denen ≥10% der Körperoberfläche (KOF) befallen war, ein PASI (Psoriasis Area and Severity Index)-Score ≥12 und ein sPGA (static Physician Global Assessment)-Score ≥3 (mittelschwer oder schwer) vorlagen und die für eine Phototherapie oder systemische Therapie in Frage kamen.
Diese Studien waren bis zur Woche 32 ähnlich aufgebaut. In beiden Studien wurden die Patienten für eine 16-wöchige Behandlung im Verhältnis 2:1 zu Otezla 30 mg zweimal täglich oder Placebo randomisiert (placebokontrollierte Phase), und von Woche 16 bis 32 erhielten alle Patienten Otezla 30 mg zweimal täglich (Erhaltungsphase). Während der randomisierten Therapie-Absetzphase (Woche 32 bis 52) wurden diejenigen Patienten, die ursprünglich zu Otezla randomisiert worden waren und eine mindestens 75%ige Reduktion ihres PASI-Scores (PASI-75) (ESTEEM 1) bzw. eine mindestens 50%ige Reduktion ihres PASI-Scores (PASI-50) (ESTEEM 2) erreichten, nach 32 Wochen entweder zu Placebo oder zu Otezla 30 mg zweimal täglich rerandomisiert. Patienten, die zu Placebo rerandomisiert wurden und nach 32 Wochen ihr PASI-75-Ansprechen (ESTEEM 1) bzw. 50% ihrer gegenüber Baseline verzeichneten PASI-Verbesserung (ESTEEM 2) einbüssten, wurden erneut mit Otezla 30 mg zweimal täglich behandelt. Patienten, welche das vorgegebene PASI-Ansprechen bis Woche 32 nicht erreichten oder die anfangs zu Placebo randomisiert worden waren, erhielten Otezla bis Woche 52. Die Gabe von Apremilast in Kombination mit biologischen DMARDs wurde nicht untersucht.
Im Anschluss an die 52 Wochen Behandlung konnten die Patienten in den Langzeit-Verlängerungsstudien der ESTEEM 1 und ESTEEM 2 Studien für insgesamt bis zu 5 Jahre (260 Wochen) unverblindet mit Apremilast 30 mg fortfahren.
Primärer Endpunkt war in beiden Studien der Anteil von Patienten, die nach 16 Wochen ein PASI-75-Ansprechen erreichten. Der wichtigste sekundäre Endpunkt war der Anteil von Patienten, die nach 16 Wochen einen sPGA-Score von befallsfrei (0) oder nahezu befallsfrei (1) erreichten. Zu den weiteren Endpunkten gehörten befallene KOF, Pruritus-VAS, Nagelbefall (NAPSI), Kopfhautbefall (ScPGA) und Resultate aus Fragebögen zur Lebensqualität (DLQI und SF-36 MCS).
Über beide Studien hinweg betrachtet, lag das mediane Alter bei 45,8 Jahren (18-83 Jahre). Bei Baseline betrug der mittlere KOF-Befall 25,19% (Median 21,0%), der mittlere PASI-Score 19,07 (Median 16,80) und der Anteil von Patienten mit einem sPGA-Score von 3 (mittelschwer) und 4 (schwer) 70,0% resp. 29,8%. Etwa 30% aller Patienten hatten zur Behandlung der Psoriasis bereits eine Phototherapie und 54% eine konventionelle systemische und/oder biologische Vortherapie erhalten (einschliesslich Therapieversagern), davon 37% eine herkömmliche systemische Vorbehandlung und 30% eine Vortherapie mit Biologika. Etwa ein Drittel der Patienten war weder mit einer Phototherapie noch mit einer konventionellen systemischen oder biologischen Therapie vorbehandelt worden. Bei insgesamt 18% der Patienten war eine Psoriasis-Arthritis in der Vorgeschichte bekannt.
Die Anteile von Patienten mit einem PASI-50-, PASI-75- und PASI-90-Ansprechen und einem sPGA-Score von befallsfrei (0) oder nahezu befallsfrei (1) sind in der nachfolgenden Tabelle (Tabelle 5) dargestellt. Otezla bewirkte eine im Vergleich zu Placebo signifikante Besserung der mittelschweren bis schweren Plaque-Psoriasis, erhoben anhand des Anteils von Patienten mit einem PASI-75-Ansprechen nach 16 Wochen. Auch für das sPGA-, PASI-50- und PASI-90-Ansprechen konnten nach 16 Wochen klinische Verbesserungen belegt werden.
Tabelle 5: Klinisches Ansprechen nach 16 Wochen in den Studien ESTEEM 1 und ESTEEM 2 (FASc; LOCF)
|
|
ESTEEM 1
|
ESTEEM 2
| |
|
Placebo
|
Otezla 30 mg zweimal täglich*
|
Placebo
|
Otezla 30 mg zweimal täglich*
| |
N
|
282
|
562
|
137
|
274
| |
PASIa 75, n (%)
|
15 (5,3)
|
186 (33,1)
|
8 (5,8)
|
79 (28,8)
| |
sPGAb «befallsfrei» oder
«nahezu befallsfrei», n (%)
|
11 (3,9)
|
122 (21,7)
|
6 (4,4)
|
56 (20,4)
| |
PASI 50, n (%)
|
48 (17,0)
|
330 (58,7)
|
27 (19,7)
|
152 (55,5)
| |
PASI 90, n (%)
|
1 (0,4)
|
55 (9,8)
|
2 (1,5)
|
24 (8,8)
|
* p <0,0001 für alle Vergleiche versus Placebo ausser für PASI-90-Ansprechen in der Studie ESTEEM 2 – dort p=0,0042.
a PASI=Psoriasis Area and Severity Index.
b sPGA=Static Physician Global Assessment.
c FAS=Full Analysis Set.
Der klinische Nutzen von Otezla wurde für verschiedene Subgruppen nachgewiesen, welche anhand von demographischen Charakteristika bei Baseline, klinischen Charakteristika der Erkrankung im Ausgangsbefund (einschliesslich Dauer der Psoriasis-Erkrankung und Patienten mit Psoriasis-Arthritis in der Vorgeschichte), Vortherapie mit Psoriasismitteln und Ansprechen auf Psoriasis-Vortherapien definiert wurden. Über alle nach dem Körpergewicht definierten Subgruppen hinweg wurden vergleichbare Ansprechraten beobachtet. Das Ansprechen auf Otezla setzte rasch ein, wobei bereits innert 2 Wochen im Vergleich zu Placebo signifikant grössere Verbesserungen der Anzeichen und Symptome der Psoriasis, einschliesslich PASI, Hautbeschwerden/-schmerzen und Pruritus, verzeichnet wurden. Das PASI-Ansprechen wurde generell innert 16 Wochen erreicht und bis Woche 32 aufrechterhalten.
Während der randomisierten Therapie-Absetzphase (Woche 32-52) in der Studie ESTEEM 1, blieb die mittlere prozentuale Besserung im PASI bei den Pateinten, welche in Woche 32 zu Otezla rerandomisiert wurden, gegenüber Baseline stabil (81–88%). Annähernd 61% dieser Patienten hatten eine PASI-75-Antwort in Woche 52. Von den Patienten welche in Woche 32 zu Placebo rerandomisiert wurden hatten 11,7% eine PASI-75-Antwort in Woche 52. Patienten welche zu Placebo rerandomisiert wurden verloren die PASI-75-Antwort schneller als die Patienten welche zu Otezla rerandomisiert wurden. Die mittlere Zeitdauer bis zum Verlust der PASI-75-Antwort war bei Patienten, welche zu Placebo bzw. zu Otezla rerandomisiert wurden 5,1 bzw. 17,7 Wochen.
In der Studie ESTEEM 2 betrug die mediane Zeit bis zum ersten Verlust der PASI-50-Antwort für Patienten, welche in Woche 32 zu Placebo bzw. zu Otezla rerandomisiert wurden, 12,4 Wochen bzw. 21,9 Wochen.
In der Studie ESTEEM 1 fanden sich nach 16 Wochen bei den Patienten unter Otezla im Vergleich zur Placebo-Gruppe signifikante Verbesserungen (Rückgänge) der Nagelpsoriasis, erhoben anhand der mittleren prozentualen Veränderung des NAPSI (Nail Psoriasis Severity Index) gegenüber Baseline (Otezla 30 mg zweimal täglich: −22,5%; Placebo: +6,5%; p <0,0001). Ähnliche Verbesserungen wurden auch in der Studie ESTEEM 2 beobachtet (Otezla 30 mg zweimal täglich: −29,0%; Placebo: −7,1%, p=0,0052). Weitere Besserungen der Nagelpsoriasis wurden bei Patienten beobachtet, die fortlaufend mit Otezla behandelt wurden, wobei die mittlere prozentuale Veränderung des NAPSI nach 32 Wochen gegenüber Baseline in ESTEEM 1 bei −43,6% und in ESTEEM 2 bei −60,0% lag.
In der Studie ESTEEM 1 fanden sich bei den Patienten unter Otezla im Vergleich zur Placebo-Gruppe signifikante Verbesserungen der Kopfhautpsoriasis mit mindestens mittelschwerer Ausprägung (≥3), erhoben anhand des prozentualen Anteils von Patienten, die nach 16 Wochen einen ScPGA (Scalp Psoriasis Physician's Global Assessment)-Score von befallsfrei (0) oder minimal (1) erreichten (46,5% vs. 17,5%; p <0,0001). Ähnliche Resultate wurden in der Studie ESTEEM 2 beobachtet (Otezla 30 mg zweimal täglich 40,9%; Placebo 17,2%; p <0,0001).
In den Studien ESTEEM 1 und 2 wurden bei den Patienten unter Otezla im Vergleich zur Placebo-Gruppe signifikante Verbesserungen der Lebensqualität, erhoben anhand der Fragebögen DLQI (Dermatology Life Quality Index) und SF-36v2MCS, nachgewiesen.
Von den 832 Patienten, welche anfänglich zu Apremilast 30 mg zweimal täglich randomisiert wurden, traten 443 (53%) in die Langzeit-Verlängerungsstudien von ESTEEM 1 und ESTEEM 2 ein, und von diesen waren nach 260 Wochen noch 115 Patienten (26%) unter Behandlung. Bei Patienten, die in der unverblindeten Verlängerung der ESTEEM 1 und ESTEEM 2 Studien auf Behandlung mit Apremilast blieben, konnten Verbesserungen des PASI-Scores, der befallenen KOF, des Pruritus, des Nagelbefalls sowie der Lebensqualität für bis zu 5 Jahre generell aufrechterhalten werden.
LIBERATE-Studie
Die Sicherheit und Wirksamkeit von Otezla und Etanercept wurden im Rahmen einer multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie (LIBERATE) untersucht, in welche insgesamt 250 Patienten ab 18 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis aufgenommen wurden, bei denen ein KOF-Befall von ≥10%, ein PASI-Score ≥12 und ein sPGA-Score ≥3 (mittelschwer oder schwer) vorlagen und die für eine Phototherapie oder systemische Therapie in Frage kamen. Ausserdem musste bei den eingeschlossenen Patienten mindestens eine herkömmliche systemische Therapie ein unzureichendes Ansprechen bewirkt haben oder eine Unverträglichkeit gegenüber resp. eine Kontraindikation für eine solche Therapie bestanden haben, und die Studienteilnehmenden durften zudem keine Vortherapie mit Biologika erhalten haben. Die Patienten wurden im Verhältnis 1:1:1 zu Otezla 30 mg oral zweimal täglich, Etanercept 50 mg subkutan einmal wöchentlich oder Placebo für 16 Wochen randomisiert; anschliessend erhielten alle Patienten Otezla 30 mg zweimal täglich. Primärer Endpunkt war das PASI-75-Ansprechen in Woche 16 bei den mit Otezla behandelten Patienten im Vergleich zu Placebo. Ein sekundärer Endpunkt war das PASI-75-Ansprechen bei den mit Etanercept behandelten Patienten im Vergleich zu Placebo. Die Studie war nicht auf die Durchführung statistischer Vergleiche zwischen Otezla und Etanercept ausgelegt, sondern vielmehr auf einen Vergleich jeder Verumbehandlung mit Placebo.
Signifikante Verbesserungen des Anteils von Patienten, welche ein PASI-50, -75 und -90 Ansprechen und einen sPGA-Score von befallsfrei (0) oder nahezu befallsfrei (1) erreichten, fanden sich bei den mit Otezla resp. Etanercept behandelten Patienten jeweils im Vergleich zu Placebo, wie aus untenstehender Tabelle hervorgeht.
Tabelle 6: Klinisches Ansprechen in Woche 16 in der LIBERATE-Studie (mITTa; LOCF)
|
|
Placebo
|
Otezla 30 mg zweimal täglich
|
Etanercept 50 mg einmal wöchentlich
| |
N
|
84
|
83
|
83
| |
PASIb-75, n (%)
|
10 (11,9)
|
33 (39,8)
|
40 (48,2)
| |
[zweiseitiges 95%-KI]c
|
|
[14,9; 40,1]e
|
[23,3; 48,5]e
| |
PASI-50, n (%)
|
28 (33,3)
|
52 (62,7)
|
69 (83,1)
| |
[zweiseitiges 95%-KI]c
|
|
[14,9; 43,9]f
|
[36,9; 62,7]f
| |
PASI-90, n (%)
|
3 (3,6)
|
12 (14,5)
|
17 (20,5)
| |
[zweiseitiges 95%-KI]c
|
|
[2,0; 19,2]g
|
[7,2; 26,1]g
| |
sPGAdScore von befallsfrei (0) oder nahezu befallsfrei (1), n (%)
|
3 (3,6)
|
18 (21,7)
|
24 (28,9)
| |
[zweiseitiges 95%-KI]c
|
|
[8,4; 27,7]h
|
[14,8; 35,5]h
|
a mITT = modifiziertes Intent-to-Treat-Kollektiv.
b PASI = Psoriasis Area and Severity Index.
c Das zweiseitige 95%-Konfidenzintervall (KI) wurde mithilfe des nach BMI stratifizierten CMH-Tests für den Behandlungsunterschied versus Placebo berechnet.
d sPGA=Static Physician Global Assessment.
e Für PASI-75: p <0,0001 für Vergleiche Otezla vs. Placebo und Etanercept vs. Placebo.
f Für PASI-50: p=0,0002 für Otezla vs. Placebo und p <0,0001 für Etanercept vs. Placebo.
g Für PASI-90: p=0,0169 für Otezla vs. Placebo und p=0,0009 für Etanercept vs. Placebo.
h Für sPGA-Score von befallsfrei oder nahezu befallsfrei: p=0,0005 für Otezla vs. Placebo und p <0,0001 für Etanercept vs. Placebo.
Morbus Behçet
Die Sicherheit und Wirksamkeit von Apremilast wurden in einer multizentrischen, randomisierten, placebokontrollierten Phase-3-Studie (RELIEF) an erwachsenen Patienten mit aktivem Morbus Behçet und oralen Ulcera untersucht. Die Patienten, die für eine systemische Therapie infrage kamen, hatten mindestens ein nicht-biologisches Morbus Behçet-Medikament gegen orale Ulcera erhalten: Colchicin (53%), NSAR (33%), andere Analgetika oder Anästhetika (18%), topische oder orale Kortikosteroide (14% bzw. 16%), Immunsuppressiva (14%). Die Patienten erfüllten die Kriterien der International Study Group (ISG) für Morbus Behçet. Die Patienten wiesen sowohl beim Screening als auch bei der Randomisierung mindestens zwei orale Ulcera auf. Patienten mit Morbus Behçet und aktiver Beteiligung wichtiger Organe (z.B. Manifestationen im Bereich der Augen, des kardiovaskulären Systems, des Gastrointestinaltrakts und des Zentralnervensystems), bei denen deshalb im Jahr vor dem Screening eine immunsuppressive Therapie erforderlich war, wurden aus der Studie ausgeschlossen. Eine Begleitbehandlung für Morbus Behçet war nicht erlaubt. Bei 37,7% der Patienten wurde eine begleitende Anwendung von Paracetamol oder NSAR angegeben.
Insgesamt wurden 207 Patienten mit Morbus Behçet im Verhältnis 1:1 auf eine Behandlung mit entweder Apremilast 30 mg zweimal täglich (n=104) oder Placebo (n=103) über 12 Wochen (placebokontrollierte Phase) randomisiert. Von Woche 12 bis 64 erhielten alle Patienten Apremilast 30 mg zweimal täglich (aktive Behandlungsphase).
Der primäre Endpunkt war die Fläche unter der Kurve (AUC) für die Anzahl der oralen Ulcera von der Baseline bis einschliesslich Woche 12. Die sekundären Endpunkte umfassten andere Parameter für orale Ulcera (Schmerzen durch orale Ulcera auf einer visuellen Analogskala (VAS)), Anteil von ulkusfreien Patienten (vollständiges Ansprechen), Zeit bis zum Beginn der Rückbildung oraler Ulcera und Anteil derPatienten, die eine Rückbildung von oralen Ulcera bis Woche 6 erreichten und über mindestens 6 weitere Wochen während der 12-wöchigen placebokontrollierten Behandlungsphase bei jedem Besuchstermin ulkusfrei blieben.
Der Altersbereich der Patienten reichte von 19 bis 72 Jahren, wobei das Durchschnittsalter bei 40 Jahren lag. Die durchschnittliche Dauer des Morbus Behçet betrug 6,84 Jahre. Alle Patienten hatten eine Vorgeschichte mit rezidivierenden oralen Ulcera, die aktuell aktiv waren. Ferner wiesen die Patienten in der Anamnese Hautläsionen (98,6%), genitale Ulcera (90,3%), muskuloskeletale Manifestationen (72,5%), Manifestationen im Bereich der Augen (17,4%), des Zentralnervensystems (9,7%) und des Gastrointestinatrakts (9,2%), Epididymitis (2,4%) und Gefässbeteiligung (1,4%) auf. Die mittlere Anzahl der oralen Ulcera bei Baseline lag bei 4,2 und 3,9 in der Apremilast- bzw. der Placebo-Gruppe.
Parameter der oralen Ulcera
Apremilast 30 mg zweimal täglich führte zu einer signifikanten Besserung oraler Ulcera, wie es durch die AUC für die Anzahl der oralen Ulcera von der Baseline bis einschliesslich Woche 12 (p<0,0001) im Vergleich zu Placebo gezeigt wurde.
In Woche 12 wurden signifikante Verbesserungen bei weiteren Parametern für orale Ulcera nachgewiesen.
Tabelle 7: Klinisches Ansprechen von oralen Ulcera in Woche 12 in der RELIEF-Studiea (ITT Population)
|
Endpunkt
|
Placebo
N=103
|
Apremilast
30 mg 2x tgl.
N=104
|
Absoluter angepasster Behandlungsunterschiedd
| |
AUCb für die Anzahl von oralen Ulcera von Baseline bis einschliesslich Woche 12 (ITT, MI)
|
LS-Mittelwert
222,14
|
LS-Mittelwert
129,54
|
92,60e
| |
Veränderung gegenüber Baseline bei den Schmerzen von oralen Ulcera gemessen anhand der VASc in Woche 12 (ITT, MMRM)
|
LS-Mittelwert
-18,7
|
LS-Mittelwert
-42,7
|
24,1e
| |
Anteil von Patienten mit Rückbildung von oralen Ulcera (ulkusfreier Mund) bis Woche 6, die über mindestens 6 weitere Wochen bei jedem Besuch während des 12-wöchigen placebokontrollierten Behandlungszeitraums ulkusfrei sind (ITT)
|
4,9%
|
29,8%
|
25,1%e
| |
Anteil der Patienten mit vollständigem Ansprechen der oralen Ulcera in der Woche 12 (ITT; NRI)
|
22,3%
|
52,9%
|
30,6%
|
a HR=Hazard Ratio; ITT=Intent-to-Treat; LS=kleinste Quadrate; MI=multiple Imputation; MMRM=Mischeffekte-Modell für wiederholte Messungen; NRI=Non-Responder Imputation.
b AUC=Fläche unter der Kurve.
c VAS=visuelle Analogskala; 0=keine Schmerzen; 100=die schlimmsten Schmerzen, die man sich vorstellen kann.
d Der angepasste Unterschied bei den Anteilen ist der gewichtete Durchschnitt der Behandlungsunterschiede über die 4 Strata der kombinierten Geschlechts- und Regionsfaktoren hinweg mit den Cochran-Mantel-Haenszel-Gewichtung.
e p-Wert <0,0001 für alle Apremilast vs. Placebo.
Von den 104 Patienten, die ursprünglich zu Apremilast 30 mg zweimal täglich randomisiert worden waren, behielten 75 Patienten (etwa 72%) diese Behandlung bis Woche 64 bei. Bei diesen Patienten wurde in der Behandlungsgruppe mit 30 mg Apremilast zweimal täglich bei jedem Besuch, bereits ab Woche 1, bis einschliesslich Woche 12, eine signifikante Reduktion der durchschnittlichen Anzahl von oralen Ulcera (p≤0,0015) sowie der Schmerzen durch orale Ulcera (p≤0,0035) im Vergleich zur Placebo-Gruppe beobachtet. Bei den Patienten, die durchgehend mit Apremilast behandelt wurden und in der Studie verblieben sind, blieb die Besserung der oralen Ulcera bis einschliesslich Woche 64 erhalten (Abbildung 1).
Bei den Patienten, die ursprünglich zu Apremilast 30 mg zweimal täglich randomisiert worden waren und in der Studie verblieben sind, blieb der Anteil der Patienten mit vollständigem Ansprechen der oralen Ulcera bis einschliesslich Woche 64 erhalten (53,3%).
Der klinische Nutzen von Apremilast 30 mg zweimal täglich wurde in verschiedenen Subgruppen nachgewiesen, die nach demographischen Ausgangsdaten, klinischen Ausgangsmerkmalen der Krankheit (einschliesslich Krankheitsdauer und Anzahl oraler Ulcera bei Baseline) und früherer Anwendung von Arzneimitteln zur Behandlung von Morbus Behçet definiert waren.
Abbildung 1: Mittlere Anzahl von oralen Ulcera nach Zeitpunkt bis einschliesslich Woche 64 (ITT-Population; DAO)
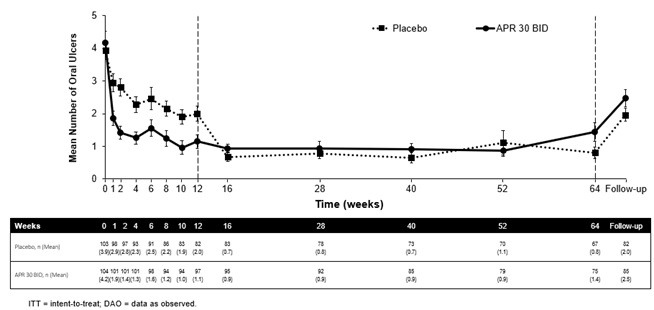
APR 30 BID=Apremilast 30 mg zweimal täglich;
Hinweis: Placebo bzw. APR 30 mg BID steht für die Behandlungsgruppe, in die die Patienten randomisiert wurden. Die Patienten der Placebo-Gruppe wurden in Woche 12 auf Apremilast 30 mg 2x tgl. umgestellt.
Der Follow-up-Zeitpunkt lag entweder 4 Wochen nach Abschluss der Studie in Woche 64 oder 4 Wochen nach vorzeitiger Beendigung der Behandlung vor Woche 64.
Gesamtverbesserungen der Krankheitsaktivität von Morbus Behçet
Apremilast 30 mg zweimal täglich führte zu einer signifikanten Abnahme der gesamthaften Krankheitsaktivität im Vergleich zu Placebo, wie die mittlere Veränderung gegenüber Baseline in Woche 12 beim Aktivitätsscore für das Behçet-Syndrom (BSAS) (p <0,0001), bei dem Formular für die aktuelle Morbus Behçet-Aktivität (BDCAF), dem aktuellen Morbus Behçet-Aktivitätsindex (BDCAI), der Wahrnehmung der Krankheitsaktivität durch den Patienten und der Gesamtwahrnehmung der Krankheitsaktivität durch den Kliniker gezeigt haben.
Bei den ursprünglich zu Apremilast 30 mg zweimal täglich randomisierten Patienten, die in der Studie verblieben sind, blieben die Verbesserungen (mittlere Veränderung gegenüber Baseline) beim BSAS und beim BDCAF bis Woche 64 erhalten.
Verbesserung der Lebensqualität
Apremilast 30 mg zweimal täglich führte im Vergleich zu Placebo zu einer signifikant grösseren Verbesserung der Lebensqualität (QoL), wie es in Woche 12 anhand des Fragebogens zur Lebensqualität bei Morbus Behçet nachgewiesen wurde.
Bei den ursprünglich zu Apremilast 30 mg zweimal täglich randomisierten Patienten, die in der Studie verblieben sind, blieb die Verbesserung der Lebensqualität bei Morbus Behçet bis Woche 64 erhalten.
|