ZusammensetzungWirkstoffe
Talimogen laherparepvec, ein attenuiertes Herpes-simplex-Virus Typ 1 (HSV-1), das mittels rekombinanter DNA-Technologie in Vero-Zellen (Affennierenzellen) hergestellt wird und welches durch die funktionelle Deletion von 2 Genen (ICP34.5 und ICP47) und die Insertion der kodierenden DNA-Sequenz für das humane GM-CSF, verändert wurde. Talimogen laherparepvec ist ein genetisch modifizierter Organismus.
Hilfsstoffe
Dinatriumphosphatdihydrat, Natriumdihydrogenphosphatdihydrat, Natriumchlorid, myo-Inositol, Sorbitol (E420; 20 mg/ml), Wasser für Injektionszwecke. Der Natriumgehalt entspricht max. 7,7 mg pro ml.
Indikationen/AnwendungsmöglichkeitenIMLYGIC ist als Monotherapie zur Behandlung von nicht resezierbaren Melanomen mit regionalen oder entfernten Metastasen (Stadien IIIB, IIIC und IVM1a) ohne Knochen-, Hirn-, Lungen- oder andere viszerale Metastasen bei Erwachsenen indiziert.
Dosierung/AnwendungDie Behandlung mit IMLYGIC sollte durch einen qualifizierten Arzt bzw. durch eine qualifizierte Ärztin, der/die in der Behandlung von Krebserkrankungen erfahren ist, eingeleitet und überwacht werden.
Um die Rückverfolgbarkeit von IMLYGIC sicherzustellen, müssen Handelsname und Chargennummer bei jeder Behandlung in der Patientenakte dokumentiert werden.
IMLYGIC ist in 1 ml-Durchstechflaschen zum Einmalgebrauch in zwei verschiedenen Konzentrationen verfügbar:
·106 (1 Million) PFU/ml - nur für die Anfangsdosis
·108 (100 Millionen) PFU/ml - für alle nachfolgenden Dosen
Das Gesamtinjektionsvolumen pro Behandlungstermin sollte maximal 4 ml betragen. Die empfohlene Anfangsdosis beträgt maximal 4 ml IMLYGIC mit einer Konzentration von 106 (1 Million) PFU/ml. Nachfolgende Dosen sollten maximal 4 ml IMLYGIC mit einer Konzentration von 108 (100 Millionen) PFU/ml betragen.
Das empfohlene Dosierungsschema für IMLYGIC ist in Tabelle 1 dargestellt.
Tabelle 1: Empfohlenes Dosierungsschema für IMLYGIC
|
Behandlungstermin
|
Behandlungsintervall
|
Maximales Gesamtinjektionsvolumen
|
Dosiskonzentration
|
Priorisierung der zu injizierenden Läsionen
| |
Erster
|
-
|
Bis zu 4 ml
|
106
(1 Million) PFU/ml
|
·Grösste Läsion(en) zuerst behandeln.
·Übrige Läsionen anhand der Läsionsgrösse priorisieren, bis das maximale Injektionsvolumen erreicht ist.
| |
Zweiter
|
3 Wochen nach Erstbehandlung
|
Bis zu 4 ml
|
108
(100 Millionen) PFU/ml
|
·Neue Läsionen zuerst behandeln (Läsionen, die sich eventuell seit der Erstbehandlung entwickelt haben).
·Übrige Läsionen anhand der Läsionsgrösse priorisieren, bis das maximale Injektionsvolumen erreicht ist.
| |
Alle nachfolgenden Behandlungstermine (einschliesslich Wiederaufnahme)
|
2 Wochen nach vorausgegangener Behandlung
|
Bis zu 4 ml
|
108
(100 Millionen) PFU/ml
|
·Neue Läsionen zuerst behandeln (Läsionen, die sich eventuell seit der vorangegangenen Behandlung entwickelt haben).
·Übrige Läsionen anhand der Läsionsgrösse priorisieren, bis das maximale Injektionsvolumen erreicht ist.
|
Bestimmung des IMLYGIC-Dosiervolumens (pro Läsion)
Das Volumen, das in die einzelnen Läsionen injiziert werden sollte, hängt von der Läsionsgrösse ab und sollte gemäss Tabelle 2 bestimmt werden. Das Gesamtinjektionsvolumen sollte maximal 4 ml pro Behandlungstermin betragen.
Tabelle 2: Auswahl des IMLYGIC-Injektionsvolumens basierend auf der Läsionsgrösse
|
Läsionsgrösse
(längste Ausdehnung)
|
IMLYGIC Injektionsvolumen
| |
> 5 cm
|
Bis zu 4 ml
| |
> 2,5 cm bis 5 cm
|
Bis zu 2 ml
| |
> 1,5 cm bis 2,5 cm
|
Bis zu 1 ml
| |
> 0,5 cm bis 1,5 cm
|
Bis zu 0,5 ml
| |
≤ 0,5 cm
|
Bis zu 0,1 ml
|
Bei den Patienten kann es zu einer Vergrösserung der vorhandenen Läsionen oder zur Bildung neuer Läsionen kommen, bevor sie auf die Therapie ansprechen (siehe «Warnhinweise und Vorsichtsmassnahmen»). So lange noch eine injizierbare Läsion/injizierbare Läsionen vorhanden ist/sind, sollte IMLYGIC für mindestens 6 Monate fortgesetzt werden, es sei denn, der Arzt bzw. die Ärztin entscheidet, dass der Patient nicht von der Behandlung mit IMLYGIC profitiert oder dass eine andere Behandlung erforderlich ist.
Die Behandlung mit IMLYGIC kann wieder aufgenommen werden, falls nach dem vollständigen Ansprechen neue Läsionen auftreten, und der Arzt bzw. die Ärztin davon ausgeht, dass der Patient von der Behandlung profitieren wird.
Spezielle Dosierungsanweisungen
Patienten mit Leber- und Nierenfunktionsstörungen
Es wurden keine klinischen Studien zur Untersuchung der Auswirkungen einer Leber- oder Nierenfunktionsstörung auf die Pharmakokinetik von IMLYGIC durchgeführt. Es ist jedoch keine Dosisanpassung bei Patienten mit Leber- oder Nierenfunktionsstörung notwendig.
Ältere Patienten
Bei Patienten im Alter von ≥65 Jahren ist keine Dosisanpassung erforderlich (siehe «Eigenschaften/Wirkungen»).
Kinder und Jugendliche (Alter < 18)
Die Sicherheit und Wirksamkeit von IMLYGIC bei Kindern und Jugendlichen ist nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
IMLYGIC wird als intraläsionale Injektion in kutane, subkutane, und/oder nodale Läsionen, die sichtbar, tastbar oder per Ultraschallkontrolle nachweisbar sind, angewendet.
Vorsichtsmassnahmen vor der Handhabung oder vor der Anwendung des Arzneimittels
Dieses Arzneimittel enthält gentechnisch veränderte Organismen. Während der Vorbereitung oder Anwendung von IMLYGIC muss eine persönliche Schutzausrüstung getragen werden (siehe «Hinweise für die Handhabung und Anwendung, persönliche Schutzausrüstung, unbeabsichtigtes Verschütten und Abfallentsorgung» unter «Sonstige Hinweise»).
Medizinische Fachpersonen, die immungeschwächt oder schwanger sind, sollten IMLYGIC nicht verabreichen und nicht mit den IMLYGIC-Injektionsstellen oder Körperflüssigkeiten von behandelten Patienten in direkten Kontakt kommen (siehe «Warnhinweise und Vorsichtsmassnahmen» und «Kontraindikationen»).
Die folgenden Anweisungen sollten während der Vorbereitung und Verabreichung von IMLYGIC befolgt werden:
Vor der Injektion
·Tauen Sie die IMLYGIC-Durchstechflaschen bei einer Temperatur von 20°C bis 25°C auf (siehe «Besondere Lagerungshinweise»). Aufgetautes IMLYGIC kann vor der Anwendung aufbewahrt werden (siehe «Sonstige Hinweise»).
·Ziehen Sie mittels aseptischer Technik die gewünschte Menge IMLYGIC aus der Durchstechflasche in eine Spritze auf.
·Die Injektionsstelle kann mit einem topischen Anästhetikum behandelt werden. Injizierbare Anästhetika können entlang der Peripherie der Läsion injiziert werden, sollten aber nicht direkt in die Läsion injiziert werden.
·Reinigen Sie die Läsion und die umgebenden Bereiche mit einem Alkoholtupfer und lassen Sie sie trocknen.
Injektion
·Spritzen Sie IMLYGIC intraläsional in kutane, subkutane und/oder nodale Läsionen, die entweder sichtbar, tastbar oder durch Ultraschallkontrolle auffindbar sind.
·Bestimmen Sie das Injektionsvolumen für jede Läsion gemäss Tabelle 2.
·Wählen Sie nur eine einzige Einstichstelle und injizieren Sie IMLYGIC fächerförmig soweit der Radius der Nadel innerhalb der Läsion reicht, um eine gleichmässige und vollständige Verteilung zu erreichen. Wenn die Läsion grösser ist als die Reichweite der Nadel, können mehrere Einstichstellen verwendet werden.
|
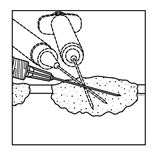
Kutane Läsionen
|
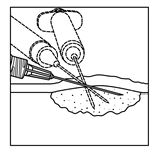
Subkutane Läsionen
|
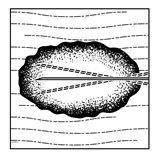
Nodale Läsionen
| |
|
|
|
|
|
| |
Abbildung 1: Injektion in kutane Läsionen
|
Abbildung 2: Injektion in subkutane Läsionen
|
Abbildung 3: Injektion in nodale Läsionen
|
·Verteilen Sie IMLYGIC gleichmässig und vollständig innerhalb der Läsion, indem Sie die Nadel zurückziehen, ohne sie ganz aus der Läsion herauszuziehen. Ändern Sie die Nadelrichtung so oft wie nötig, während Sie die restliche Dosis injizieren. Fahren Sie so lange fort, bis die Gesamtdosis gleichmässig und vollständig verteilt ist.
·Wenn Sie die Nadel entfernen, ziehen Sie diese langsam aus der Läsion heraus, um ein Auslaufen oder Zurückspritzen von IMLYGIC an der Einstichstelle zu vermeiden.
·Wiederholen Sie diese Schritte für andere Läsionen, die injiziert werden müssen. Benutzen Sie jedes Mal eine neue Nadel, wenn die Nadel vollständig aus einer Läsion entfernt wird und wenn eine andere Läsion injiziert wird.
Nach der Injektion
·Drücken Sie für mindestens 30 Sekunden mit einer sterilen Gaze auf die Injektionsstelle.
·Betupfen Sie die Injektionsstelle und den umgebenden Bereich mit Alkohol und decken Sie die injizierte Läsion mit einer absorbierenden Kompresse und einem trockenen Okklusivverband ab.
Weitere Anweisungen für den Gebrauch, die Handhabung und die Entsorgung sind unter «Sonstige Hinweise» aufgeführt.
KontraindikationenIMLYGIC ist kontraindiziert bei:
·Patienten mit einer Vorgeschichte einer Überempfindlichkeit gegenüber Talimogen laherparepvec oder einem seiner Hilfsstoffe (siehe «Zusammensetzung»).
·Stark immungeschwächten Patienten (z.B. Patienten mit schwerer angeborener oder erworbener zellulärer und/oder humoraler Immunschwäche) (siehe «Warnhinweise und Vorsichtsmassnahmen» und «Präklinische Daten»).
Warnhinweise und VorsichtsmassnahmenIMLYGIC untersteht speziellen risikominimierenden Massnahmen. Der behandelnde Arzt bzw. die behandelnde Ärztin erhält eine ärztespezifische Broschüre («Physician Education Booklet») mit Informationen zu den Risiken einer Übertragung von Herpes und Herpes-Komplikationen sowie zum sicheren Gebrauch und Umgang mit IMLYGIC. Zudem wird er bzw. sie angehalten, dem betroffenen Patienten eine Patientenbroschüre, eine Patientenkarte und eine Patienteninformation abzugeben.
Patienten mit vorhergehender Behandlung
Es liegen nur begrenzte Daten zur Wirksamkeit von IMLYGIC in der gegenwärtigen zweiten oder in späteren Behandlungslinien vor.
Disseminierte Herpesinfektionen
Bei Patienten, die mit IMLYGIC behandelt wurden, sind disseminierte Herpesinfektionen, einschliesslich schwerer Fälle von disseminierten Herpesinfektionen, berichtet worden (siehe «Unerwünschte Wirkungen»).
IMLYGIC wurde bei immungeschwächten Patienten nicht untersucht. Basierend auf Erkenntnissen aus epidemiologischen Daten kann bei immungeschwächten Patienten (z.B. Patienten mit HIV/AIDS, Leukämie, Lymphom, variablem Immundefektsyndrom oder die chronisch hochdosierte Steroide oder andere Immunsuppressiva benötigen) ein erhöhtes Risiko für eine disseminierte Herpesinfektion bestehen. Die Risiken und der Nutzen der Behandlung sollten geprüft werden, bevor IMLYGIC bei immungeschwächten Patienten angewendet wird.
Basierend auf Erkenntnissen aus Tierstudien kann bei stark immungeschwächten Patienten ein erhöhtes Risiko für eine disseminierte Herpesinfektion bestehen. Solche Patienten sollten nicht mit IMLYGIC behandelt werden (siehe «Kontraindikationen» und «Präklinische Daten»).
Unbeabsichtigte IMLYGIC-Exposition
Eine unbeabsichtigte Exposition kann zur Übertragung von IMLYGIC und einer Herpesinfektion führen. Medizinische Fachpersonen und enge Kontaktpersonen (z.B. Haushaltsmitglieder, Pflegepersonen, Sexualpartner oder Personen, die dasselbe Bett teilen) sollten den direkten Kontakt mit injizierten Läsionen oder Körperflüssigkeiten der behandelten Patienten während der gesamten Behandlungszeit und bis zu 30 Tage nach der letzten Anwendung meiden (siehe «Unbeabsichtigte Exposition» unter «Sonstige Hinweise»). Während der Vorbereitung und Verabreichung von IMLYGIC wurde von unbeabsichtigten Nadelstichverletzungen und einem Entgegenspritzen der Injektionslösung bei medizinischen Fachpersonen berichtet.
Enge Kontaktpersonen, die schwanger oder immungeschwächt sind, sollten weder die Verbände der Patienten wechseln noch deren Injektionsstelle reinigen. Schwangere Frauen, Neugeborene und immungeschwächte Personen dürfen nicht mit potenziell kontaminierten Gegenständen in Kontakt kommen.
Stellen Sie sicher, dass die Patienten dazu in der Lage sind, die Injektionsstellen mit Okklusivverbänden abzudecken (siehe «Hinweise für die Handhabung und Anwendung, persönliche Schutzausrüstung, unbeabsichtigtes Verschütten und Abfallentsorgung» unter «Sonstige Hinweise»). Die Patienten sollten zudem angewiesen werden, das Berühren oder Kratzen der Injektionsstellen zu vermeiden, weil IMLYGIC dadurch unbeabsichtigt auf andere Bereiche ihres Körpers oder auf ihre engen Kontaktpersonen übertragen werden könnte.
Obwohl nicht bekannt ist, ob IMLYGIC durch sexuellen Kontakt übertragen werden kann, ist bekannt, dass der Wildtyp HSV-1 durch Sexualkontakt übertragen werden kann. Patienten sollten angewiesen werden, während sexueller Kontakte ein Latexkondom zu benutzen, um eine mögliche Übertragung von IMLYGIC zu vermeiden. Frauen im gebärfähigen Alter sollten angewiesen werden, eine zuverlässige Verhütungsmethode anzuwenden, um eine Schwangerschaft während der Behandlung zu vermeiden (siehe «Schwangerschaft, Stillzeit»).
Pflegepersonen sollten angewiesen werden, Schutzhandschuhe zu tragen, wenn sie Patienten beim Anlegen oder Wechseln von Okklusivverbänden helfen, und die Sicherheitsmassnahmen zur Entsorgung von gebrauchten Verbänden und Reinigungsmaterial zu befolgen (siehe «Hinweise für die Handhabung und Anwendung, persönliche Schutzausrüstung, unbeabsichtigtes Verschütten und Abfallentsorgung» unter «Sonstige Hinweise»).
Im Falle einer unbeabsichtigten IMLYGIC-Exposition sind die unter Sonstige Hinweise aufgeführten Anweisungen zu befolgen. Falls sich Anzeichen oder Symptome einer Herpesinfektion entwickeln, sollten die Betroffenen eine medizinische Fachperson kontaktieren. Bei Verdacht auf Herpesläsionen besteht für Patienten, enge Kontaktpersonen oder medizinisches Fachpersonal die Möglichkeit einer Nachfolgeuntersuchung zur näheren Charakterisierung der Infektion durch die Zulassungsinhaberin.
Herpesinfektion bei mit IMLYGIC behandelten Patienten
Bei Patienten, die mit IMLYGIC behandelt wurden, sind Herpesinfektionen (unter anderem Lippenherpes und herpetische Keratitis) und schwere Fälle von disseminierten Herpesinfektionen berichtet worden (siehe «Unerwünschte Wirkungen»). Es wird erwartet, dass die Symptome einer möglicherweise mit IMLYGIC in Zusammenhang stehenden lokalen oder systemischen Infektion den Symptomen von durch Wildtyp HSV-1 verursachten Infektionen ähnlich sind.
Es ist bekannt, dass Personen mit einer Wildtyp HSV-1-Infektion, bedingt durch die Reaktivierung von latentem Wildtyp HSV-1, ein lebenslanges Risiko für symptomatische Herpesinfektionen haben. Eine symptomatische Herpesinfektion durch eine mögliche Reaktivierung von IMLYGIC sollte in Betracht gezogen werden.
Patienten, welche Herpesinfektionen entwickeln, sollten angewiesen werden, Standardhygienemassnahmen zu befolgen, um eine virale Übertragung zu verhindern.
IMLYGIC ist gegenüber Aciclovir empfindlich. Die Risiken und der Nutzen einer IMLYGIC-Behandlung sollten berücksichtigt werden, bevor Aciclovir oder andere antivirale Wirkstoffe, die zur Behandlung von Herpesinfektionen indiziert sind, angewendet werden. Diese Wirkstoffe könnten die Wirksamkeit der Behandlung beeinträchtigen, wenn sie systemisch oder topisch direkt an der Injektionsstelle angewendet werden.
Cellulitis an der Injektionsstelle
Nach der Behandlung mit IMLYGIC können Nekrosen oder Ulzerationen des Tumorgewebes auftreten. Es wurden Fälle von Cellulitis und systemischen bakteriellen Infektionen berichtet. Eine sorgfältige Wundpflege und Infektionsschutzmassnahmen werden empfohlen, insbesondere wenn Gewebenekrosen zu offenen Wunden führen.
Wundheilungsstörungen an der Injektionsstelle
In klinischen Studien wurde über Wundheilungsstörungen an der Injektionsstelle berichtet. Bei Patienten mit zugrundeliegenden Risikofaktoren (z.B. vorangegangene Bestrahlung an der Injektionsstelle oder Läsionen in schlecht vaskularisierten Bereichen) kann IMLYGIC das Risiko von Wundheilungsstörungen erhöhen.
Im Falle einer persistierenden Infektion oder verzögerten Heilung sollten die Risiken und der Nutzen von IMLYGIC abgewogen werden, bevor die Behandlung fortgesetzt wird.
Immunvermittelte Ereignisse
In klinischen Studien wurden bei Patienten, die mit IMLYGIC behandelt wurden, über immunvermittelte Ereignisse berichtet, einschliesslich Glomerulonephritis, Vaskulitis, Pneumonitis, Verschlechterung einer Psoriasis und Vitiligo.
Bevor die Behandlung bei Patienten mit einer zugrunde liegenden Autoimmunerkrankung eingeleitet wird oder bevor die Behandlung bei Patienten, die immunvermittelte Ereignisse entwickeln, fortgesetzt wird, sollten die Risiken und der Nutzen von IMLYGIC abgewogen werden.
Plasmozytom an der Injektionsstelle
Nach der Verabreichung von IMLYGIC wurde über ein Plasmozytom in der Nähe der Injektionsstelle berichtet. Bei Patienten mit multiplem Myelom oder bei Patienten, die während der Behandlung ein Plasmozytom entwickeln, sollten die Risiken und der Nutzen von IMLYGIC abgewogen werden.
Obstruktive Atemwegserkrankung
Nach Anwendung von IMLYGIC wurde über obstruktive Atemwegserkrankungen berichtet. Bei der Injektion von Läsionen, die sich in der Nähe der Hauptatemwege befinden, ist Vorsicht geboten.
Leberblutungen bei transkutaner intrahepatischer Verabreichung
IMLYGIC ist nicht für die transkutane intrahepatische Verabreichung indiziert.
In klinischen Studien wurden bei Patienten, die transkutane intrahepatische IMLYGIC-Injektionen erhielten, Fälle von Leberblutungen gemeldet, die zu einer Hospitalisierung und zum Tod führten.
Pseudoprogression
Bei Patienten kann es während einer Behandlung mit IMLYGIC zunächst zu einer Vergrösserung von injizierten Läsionen und von vorhandenen nicht injizierten Läsionen kommen oder zur Bildung neuer Läsionen, bevor ein Ansprechen auf die Therapie erreicht wird.
HSV-1-seronegative Patienten
Es wurde berichtet, dass bei anfänglich HSV-1-seronegativen Patienten die Inzidenz an Pyrexie, Schüttelfrost und grippeähnlichen Erkrankungen höher war als bei jenen Patienten, die anfänglich HSV-1-seropositiv waren, insbesondere während den ersten 3 Monaten der Behandlung (siehe «Unerwünschte Wirkungen»).
Warnhinweise für sonstige Bestandteile
Dieses Arzneimittel enthält 20 mg Sorbitol (E420) pro 1-ml Durchstechflasche. Die additive Wirkung gleichzeitig angewendeter Arzneimittel, die Sorbitol (oder Fructose) enthalten, und die ernährungsbedingte Aufnahme von Sorbitol (oder Fructose) sollten berücksichtigt werden.
Bei Patienten mit der seltenen hereditären Fructoseintoleranz sollte dieses Arzneimittel nicht angewendet werden.
Dieses Arzneimittel enthält 7,7 mg Natrium pro 1 ml-Durchstechflasche, entsprechend 0,4 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
Kombinationstherapie
Es gibt noch keine Daten, welche die Sicherheit und Wirksamkeit von IMLYGIC als Kombinationstherapie belegen. Daher sollte IMLYGIC nur als Monotherapie angewendet werden.
InteraktionenEs wurden keine Interaktionsstudien mit IMLYGIC durchgeführt. Aciclovir oder andere antivirale Wirkstoffe können die Wirksamkeit der Behandlung beeinträchtigen, wenn sie systemisch oder topisch direkt an der Injektionsstelle angewendet werden. Die Risiken und der Nutzen einer IMLYGIC-Behandlung sollten berücksichtigt werden, bevor Aciclovir oder andere antivirale Wirkstoffe, die zur Behandlung von Herpesinfektionen indiziert sind, angewendet werden.
Schwangerschaft, StillzeitÜbertragung von IMLYGIC durch Sexualkontakt
Alle Patienten sollten angewiesen werden, während sexueller Kontakte ein Latexkondom zu benutzen, um eine mögliche Übertragung von IMLYGIC zu vermeiden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Verhütung
Frauen im gebärfähigen Alter sollten angewiesen werden, eine zuverlässige Verhütungsmethode anzuwenden, um während der IMLYGIC-Behandlung eine Schwangerschaft zu vermeiden.
Schwangerschaft
Es wurden keine Studien mit IMLYGIC bei Schwangeren durchgeführt. In Tierstudien wurden keine Auswirkungen auf die embryo-fetale Entwicklung beobachtet (siehe «Präklinische Daten»). Als vorsorgliche Massnahme sollte eine Anwendung von Talimogen laherparepvec während der Schwangerschaft vermieden werden.
Falls bei einer Schwangeren eine Infektion mit Wildtyp HSV-1 (Primärinfektion oder Reaktivierung) auftritt, kann das Virus die Plazentaschranke eventuell überwinden. Es besteht ebenfalls das Risiko, dass durch die Ausscheidung von Viren (viral shedding) diese bei der Geburt übertragen werden. Infektionen mit Wildtyp HSV-1 wurden mit schwerwiegenden unerwünschten Wirkungen in Verbindung gebracht, einschliesslich Multiorganversagen und Tod, falls sich der Fetus oder das Neugeborene mit dem Wildtyp-Herpesvirus infiziert. Obwohl bisher keine klinischen Daten zu IMLYGIC-Infektionen bei Schwangeren vorliegen, könnte für den Fetus oder das Neugeborene ein Risiko bestehen, falls IMLYGIC auf die gleiche Weise wirken würde.
Transplazentare Metastasen können beim malignen Melanom vorkommen. Da IMLYGIC entwickelt wurde, um ins Tumorgewebe einzudringen und sich dort zu replizieren, könnte über Tumorgewebe, welches die Plazenta passiert hat, ein Risiko für eine fetale IMLYGIC-Exposition bestehen.
Falls IMLYGIC während der Schwangerschaft angewendet wird oder die Patientin während der Behandlung mit IMLYGIC schwanger wird, sollte die Patientin über die potenziellen Gefahren für den Fetus und/oder das Neugeborene informiert werden.
Stillzeit
Es ist nicht bekannt, ob IMLYGIC in die Muttermilch übergeht. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit IMLYGIC verzichtet werden soll bzw. die Behandlung mit IMLYGIC zu unterbrechen ist. Dabei soll sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau berücksichtigt werden.
Fertilität
Es wurden keine klinischen Studien zur Untersuchung der Auswirkungen von Talimogen laherparepvec auf die Fertilität durchgeführt.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenIMLYGIC könnte einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen haben. Aufgrund von möglichen unerwünschten Wirkungen wie Schwindel und Verwirrtheit (siehe «Unerwünschte Wirkungen»), sollten Patienten darüber informiert werden, Vorsicht beim Fahren oder beim Bedienen von Maschinen walten zu lassen, bis sie sicher sind, dass IMLYGIC keinen ungünstigen Einfluss auf sie hat.
Unerwünschte WirkungenDie Sicherheit von IMLYGIC wurde in der pivotalen Studie bei 419 Patienten (IMLYGIC: N = 292, GM-CSF: N = 127) untersucht, die mindestens eine Dosis der Studienmedikation erhalten haben (siehe «Eigenschaften/Wirkungen»). Die mediane Expositionsdauer gegenüber IMLYGIC betrug 23 Wochen (5,3 Monate). Sechsundzwanzig (26) Patienten waren IMLYGIC für mindestens ein Jahr ausgesetzt.
Die am häufigsten berichteten unerwünschten Wirkungen (≥25%) bei Patienten, die mit IMLYGIC behandelt wurden, waren Müdigkeit (50,3%), Schüttelfrost (48,6%), Fieber (42,8%), Übelkeit (35,6%), grippeähnliche Symptome (30,5%) und Schmerzen an der Injektionsstelle (27,7%). Insgesamt achtundneunzig Prozent (98%) dieser berichteten unerwünschten Wirkungen hatten einen milden bis mässigen Schweregrad. Die häufigste unerwünschte Wirkung des Grades 3 oder höher war Cellulitis (2,1%) (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Die Nebenwirkungen wurden anhand von klinischen Studien bei Melanom-Patienten, welche mit IMLYGIC im Vergleich zu GM-CSF behandelt wurden und von Erfahrungen nach Markteinführung ermittelt. Die Inzidenz der unerwünschten Wirkungen wird nach Systemorganklasse und Häufigkeit dargestellt. Häufigkeiten sind wie folgt definiert: sehr häufig (≥1/10), häufig (≥1/100 bis < 1/10) und gelegentlich (≥1/1'000 bis < 1/100). Innerhalb jeder Häufigkeitsgruppe werden die unerwünschten Wirkungen nach abnehmendem Schweregrad aufgeführt.
Infektionen und parasitäre Erkrankungen
Häufig: Cellulitis**, Herpesinfektionen***.
Gelegentlich: Infektionen an der Inzisionsstelle.
Gutartige, bösartige und unspezifische Neubildungen
Häufig: Tumorschmerzen, infiziertes Neoplasma.
Gelegentlich: Plasmozytom an der Injektionsstelle**.
Erkrankungen des Blutes und des Lymphsystems
Sehr häufig: Peripheres Ödem (12,0%).
Häufig: Anämie.
Erkrankungen des Immunsystems
Häufig: Immunvermittelte Ereignisse†**.
Gelegentlich: Überempfindlichkeit.
Stoffwechsel- und Ernährungsstörungen
Häufig: Dehydratation.
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (18,8%).
Häufig: Verwirrtheit, Angst, Depression, Schwindel, Schlaflosigkeit.
Augenerkrankungen
Gelegentlich: Herpetische Keratitis.
Erkrankungen des Ohrs und des Labyrinths
Häufig: Ohrenschmerzen.
Herzerkrankungen
Häufig: Tachykardie.
Gefässerkrankungen
Häufig: Tiefe Venenthrombose, Hypertonie, Hautrötung.
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Sehr häufig: Husten (10,6%).
Häufig: Dyspnoe, oropharyngeale Schmerzen, Infektion der oberen Atemwege.
Gelegentlich: obstruktive Atemwegserkrankung, Pneumonitis.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Erbrechen (21,2%), Diarrhö (18,8%), Verstopfung (11,6%), Übelkeit (35,6%).
Häufig: Bauchschmerzen, Bauchbeschwerden.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Vitiligo, Hautausschlag, Dermatitis.
Gelegentlich: granulomatöse Dermatitis.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Myalgie (17,5%), Arthralgie (17,1%), Schmerzen in den Extremitäten (16,4%).
Häufig: Rückenschmerzen, Schmerzen in der Leistengegend.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Grippeähnliche Symptome (30,5%)**, Fieber (42,8%), Schüttelfrost (48,6%), Müdigkeit (50,3%), Schmerzen (16,1%), Reaktionen an der Injektionsstelle*.
Häufig: Unwohlsein, Schmerzen in der Achselhöhle.
Untersuchungen
Häufig: Gewichtsverlust.
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Häufig: Wundkomplikationen, Wundsekretion, Prellungen, Schmerzen durch den Eingriff.
* Zu den Reaktionen an der Injektionsstelle zählen: sehr häufig Schmerzen (27,7%), häufig Rötungen, Blutungen, Schwellungen, Reaktionen, Entzündungen, Sekretabsonderungen, Nässen, gelegentlich Wärmegefühl.
† Immunvermittelte Ereignisse schliessen ein: gelegentlich Vaskulitis, Pneumonitis, Verschlechterung einer Psoriasis und Glomerulonephritis.
** siehe Beschreibung ausgewählter Nebenwirkungen.
*** Herpesinfektionen (unter anderem Mundherpes).
Beschreibung ausgewählter Nebenwirkungen
Immunvermittelte Ereignisse
Immunvermittelte Ereignisse, die in der pivotalen Studie berichtet wurden, umfassten je einen Fall einer sich verschlechternden Psoriasis bei einem Patienten mit einer Vorgeschichte einer Psoriasis, einen Fall einer Pneumonitis bei einem Patienten mit einer Vorgeschichte einer Autoimmunerkrankung, einen Fall einer Vaskulitis und zwei Fälle einer Glomerulonephritis, bei der in einem Fall ein akutes Nierenversagen auftrat.
Plasmozytom
In klinischen Studien wurde ein Fall eines Plasmozytoms an der Injektionsstelle bei einem Patienten beobachtet, bei dem ein Multiples Myelom festgestellt wurde.
Cellulitis
In der pivotalen Studie führten Ereignisse von Cellulitis nicht zu einem dauerhaften Abbruch der IMLYGIC-Behandlung. Eine sorgfältige Wundpflege und Infektionsschutzmassnahmen werden empfohlen, insbesondere wenn eine Gewebenekrose zu offenen Wunden führt.
Grippeähnliche Symptome
Grippeähnliche Symptome traten bei 90% der mit IMLYGIC behandelten Patienten auf. Fieber, Schüttelfrost und grippeähnliche Erkrankungen, die jederzeit während der Behandlung auftreten können, klangen gewöhnlich innerhalb von 72 Stunden ab. Diese Ereignisse wurden häufiger innerhalb des Zeitraums der ersten 6 Behandlungen berichtet, insbesondere bei Patienten, die anfänglich HSV-1 negativ waren.
Meldung unerwünschter Wirkungen
Meldungen über unerwünschte Wirkungen von IMLYGIC sollen elektronisch an biovigilance@swissmedic.ch gesendet werden.
ÜberdosierungKlinische Erfahrungen zur Überdosierung von IMLYGIC liegen nicht vor. In klinischen Studien wurden alle zwei Wochen Dosen von bis zu 4 ml mit einer Konzentration von 108 PFU/ml verabreicht ohne Nachweis einer dosislimitierenden Toxizität. Die Maximaldosis, die sicher verabreicht werden kann, wurde nicht bestimmt. Im Falle einer vermuteten Überdosierung oder unbeabsichtigten intravenösen Anwendung, sollte der Patient symptomatisch behandelt werden, z.B. mit Aciclovir oder anderen antiviralen Wirkstoffen (siehe «Warnhinweise und Vorsichtsmassnahmen»), und unterstützende Massnahmen sind falls nötig einzuleiten.
Eigenschaften/WirkungenATC-Code
L01XX51
Wirkungsmechanismus
Talimogen laherparepvec ist ein onkolytisches Virus, abgeleitet von HSV-1. Talimogen laherparepvec wurde modifiziert, sodass es sich in Tumoren repliziert und das immunstimulierende Protein humanes GM-CSF produziert. Talimogen laherparepvec führt zum Absterben von Tumorzellen sowie zur Freisetzung von Antigenen, die von Tumorzellen abstammen. Es wird angenommen, dass es zusammen mit GM-CSF eine systemische Anti-Tumor-Immunantwort und eine Effektor-T-Zell-Antwort fördert. Mäuse mit einer vollständigen Rückbildung der Primärtumoren nach der Behandlung waren resistent gegenüber einer nachfolgenden Tumor-Reexposition.
Zu den Modifikationen an Talimogen laherparepvec gegenüber HSV-1 zählen die Deletion von ICP34.5 und ICP47. Während antivirale Immunantworten normale Zellen nach einer Infektion mit Talimogen laherparepvec schützen, wurde bei Tumoren gezeigt, dass diese anfällig für Schädigungen und Zelltod durch ICP34.5-defiziente HSV-1-Viren, einschliesslich Talimogen laherparepvec, sind. Die Entfernung von ICP47 verhindert die Downregulation von antigenpräsentierenden Molekülen und erhöht die Expression des HSV-US11-Gens, was wiederum die virale Replikation in Tumorzellen fördert.
Klinische Wirksamkeit und Sicherheit
Pivotale Studie
Die Sicherheit und Wirksamkeit der IMLYGIC-Monotherapie im Vergleich zu subkutan verabreichtem GM-CSF wurden in einer multinationalen, unverblindeten, randomisierten klinischen Phase-3-Studie bei Patienten mit einem Melanom im Stadium IIIB, IIIC und IV untersucht, das als nicht chirurgisch resezierbar eingestuft wurde. Eine vorangegangene systemische Melanom-Behandlung war zulässig, aber nicht erforderlich. Patienten mit aktiven Hirnmetastasen, Knochenmetastasen, extensiver viszeraler Erkrankung, primärem Augen- oder Schleimhautmelanom, Anzeichen einer Immunsuppression oder Patienten, die eine systemische antiherpetische Behandlung erhielten, wurden aus der Studie ausgeschlossen.
Die Patienten wurden im Verhältnis 2:1 randomisiert und erhielten entweder IMLYGIC oder GM-CSF (N = 436; IMLYGIC: N = 295, GM-CSF: N = 141). IMLYGIC wurde als intraläsionale Injektion mit einer Anfangskonzentration von 106 (1 Million) PFU/ml an Tag 1 verabreicht, gefolgt von 108 (100 Millionen) PFU/ml an Tag 21 und danach alle zwei Wochen mit einer Dosis von bis zu 4 ml. GM-CSF wurde in sich wiederholenden Intervallen während 14 Tagen mit einer täglichen Dosis von 125 µg/m2 subkutan verabreicht, gefolgt von einer 14tägigen behandlungsfreien Phase.
Um eine verzögerte immunvermittelte Anti-Tumor-Wirkung zu ermöglichen, wurden die Patienten für mindestens sechs Monate lang behandelt oder bis sie keine injizierbaren Läsionen mehr zeigten. Während dieses Zeitraums wurde die Behandlung auch dann fortgesetzt, wenn sich die vorhandenen Läsionen vergrösserten und/oder sich neue Läsionen bildeten, ausser der Patient entwickelte eine nicht tolerierbare Toxizität oder der klinische Zustand des Patienten machte den Beginn einer neuen Therapie erforderlich. Nach 6 Monaten der Behandlung mussten die Patienten die Behandlung bis zu einer klinisch relevanten Krankheitsprogression fortsetzen (d.h. Krankheitsprogression im Zusammenhang mit einem Rückgang des Performance-Status und/oder es waren nach Meinung des Prüfers alternative Therapien erforderlich).
Patienten, die nach zwölf Monaten auf die Behandlung ansprachen, konnten die Behandlung für bis zu weitere sechs Monate fortsetzen. Die mittlere (SD) Behandlungsdauer der ITT-Population betrug im GM-CSF-Arm 15,76 Wochen (15,79) und im IMLYGIC-Arm 26,83 (18,39) Wochen. Der primäre Endpunkt war die dauerhafte Ansprechrate (DRR) [definiert als der prozentuale Anteil an Patienten mit einem vollständigen Ansprechen (CR) oder teilweisen Ansprechen (PR), welches für mindestens 6 Monate aufrechterhalten werden konnte] gemäss verblindeter zentraler Bewertung der ansprechenden Patienten. Die sekundären Endpunkte umfassten das Gesamtüberleben (OS), die Gesamtansprechrate (ORR) [PR+CR], die Zeit bis zum Ansprechen, die Dauer des Ansprechens und die Zeit bis zum Therapieversagen (Zeit von der Randomisierung bis zur ersten Episode einer klinisch relevanten Krankheitsprogression ohne Ansprechen nach dem Progressionsereignis, oder bis zum Tod).
Das mittlere Alter betrug 63 (Bereich: 22 bis 94) Jahre, wobei 26,5% über 65 Jahre alt und 23,3% über 74 Jahre alt waren. Die Mehrheit der Patienten waren Kaukasier (98%). 57% der Studienpopulation waren männlich und 70% der Patienten hatten zu Studienbeginn einen ECOG-Performance-Status 0. Von den eingeschlossenen Patienten hatten 22% eine Erkrankung im Stadium IVM1c und 53% der Patienten hatten zuvor neben Operation, adjuvanter Therapie oder Bestrahlung eine Melanomtherapie wie Chemotherapie und Zytokin-basierte Immuntherapie erhalten. Insgesamt waren 58% aller in die Studie eingeschlossenen Patienten zu Beginn seropositiv für Wildtyp HSV-1 und 32,6% seronegativ. Der HSV-1-Serostatus für die restlichen 9,4% war unbekannt.
Durch die Behandlung mit IMLYGIC erhöhte sich in der ITT-Population die DRR statistisch signifikant (siehe Tabelle 3).
Tabelle 3: Zusammenfassung der Ergebnisse der ITT-Population der Pivotalstudie mit IMLYGIC
|
|
Studienendpunkt
|
IMLYGIC N = 295
|
GM-CSF N = 141
| |
Dauerhafte Ansprechrate
|
primär
|
16,3% (n = 48)
(95% KI: 12,1, 20,5)
|
2,1% (n = 3)
(95% KI: 0,0, 4,5)
| |
Odds Ratio 8,9; (95% KI: 2,7, 29,2)
p < 0,0001
| |
Gesamtansprechrate
(% CR, % PR)
|
sekundär
|
26,4% (n = 78)
(95% KI: 21,4%, 31,5%)
(10,8% CR, 15,6% PR)
|
5,7% (n = 8)
(95% KI: 1,9%, 9,5%)
(0,7% CR, 5% PR)
| |
Gesamtüberleben
|
sekundär
|
Median 23,3
(95% KI: 19,5, 29,6) Monate
|
Median 18,9
(95% KI: 16,0, 23,7) Monate
| |
HR: 0,79; (95% KI: 0,62, 1,00) p = 0,051
| |
Dauer des Ansprechens (anhaltendes Ansprechen zum Zeitpunkt der letzten Tumor-Auswertung)
|
sekundär
|
Nicht erreicht
(Bereich: > 0,0 bis > 16,8 Monate)
|
Median 2,8 Monate
(Bereich: 1,2 bis > 14,9 Monate)
| |
HR: 0,46; (95% KI: 0,35, 0,60)
| |
Zeit bis zum Ansprechen (Median)
|
sekundär
|
4,1 Monate
|
3,7 Monate
| |
Zeit bis zum Therapieversagen (Median)
|
sekundär
|
8,2 Monate
(95% KI: 6,5, 9,9)
|
2,9 Monate
(95% KI: 2,8, 4,0)
| |
HR: 0,42; (95% KI: 0,32, 0,54)
|
Bei 56 der mit IMLYGIC behandelten Responder (72%) dauerte das Ansprechen zum Zeitpunkt der Primäranalyse noch an. Bei 42 der Responder (54%) nahm die Gesamtgrösse der vorhandenen Läsionen um ≥25% zu und/oder sie entwickelten eine oder mehrere neue Läsionen, bevor sie schlussendlich ein Ansprechen erreichten (Pseudoprogression).
In einer Analyse zur Untersuchung der systemischen Aktivität von IMLYGIC zeigten 27 von 79 Patienten (34,2%) eine Gesamtabnahme der nicht mit IMLYGIC injizierten nichtviszeralen Läsionen von ≥50%. Bei 8 von 71 Patienten (11,3%) lag die Gesamtabnahme der nicht mit IMLYGIC behandelten viszeralen Läsionen bei ≥50%.
Zum Zeitpunkt des medianen Follow-ups von 44,4 Monaten betrug das mediane Gesamtüberleben bei IMLYGIC 23,3 Monate (Bereich: 19,5, 29,6 Monate) und bei GM-CSF 18,9 Monate (Bereich: 16,0, 23,7 Monate) (Hazard Ratio [HR] 0,79 (95% KI: 0,62, 1.00), p = 0,051, Abbildung 4). Die Gesamtüberlebensrate war nach 1, 2, 3 und 4 Jahren bei Patienten, die dem IMLYGIC-Arm zugewiesen wurden, höher (73,7%, 49,8%, 38,6% bzw. 32,6%) als bei Patienten, die GM-CSF erhielten (69,1%, 40,3%, 30,1% bzw. 21,3%) (siehe Tabelle 4).
Es wurden insgesamt keine Unterschiede bezüglich Sicherheit oder Wirksamkeit zwischen älteren (≥65 Jahre) und jüngeren erwachsenen Patienten festgestellt.
Abbildung 4: Kaplan-Meier-Plot – Gesamtüberleben (ITT Population)
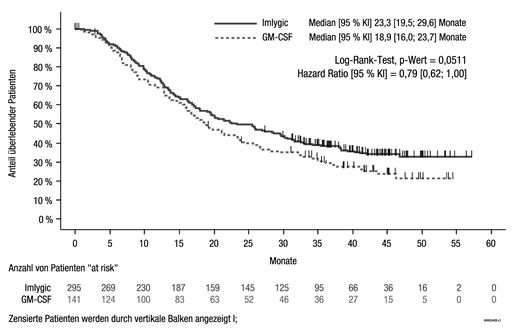
Patienten, die nicht als verstorben erfasst worden sind, sind als zensiert eingeschlossen.
Explorative Subgruppenanalysen für DRR und Gesamtüberleben nach Krankheitsstadium wurden ebenfalls durchgeführt (siehe Abbildung 5 und Tabelle 4). Obwohl die Power der Pivotalstudie nicht auf die Beurteilung der Wirksamkeit in diesen individuellen Subgruppen ausgelegt war, hatten Patienten ohne viszerale Erkrankung durch die Behandlung mit IMLYGIC einen grösseren Nutzen als jene mit einer weiter fortgeschrittenen Erkrankung.
Tabelle 4: Zusammenfassung der Ergebnisse einer explorativen Analyse der Pivotalstudie mit IMLYGIC
|
|
DRR (%)
|
ORR (%)
|
OS (Hazard Ratio)
| |
IMLYGIC
|
GM-CSF
|
IMLYGIC
|
GM-CSF
|
IMLYGIC versus GM-CSF
| |
Stadium IIIB/IIIC/ Stadium IVM1a
(IMLYGIC: n = 163; GM-CSF: n = 86)
|
25,2
|
1,2
|
40,5
|
2,3
|
0,57 (95% KI: 0,40; 0,80)
| |
Stadium IVM1b/IVM1c
(IMLYGIC: n = 131; GM-CSF: n = 55)
|
5,3
|
3,6
|
9,2
|
10,9
|
1,07 (95% KI: 0,75; 1,52)
|
Abbildung 5: Explorativer Kaplan-Meier-Plot des Gesamtüberlebens nach randomisiertem Behandlungsarm für die Krankheitsstadien IIIB, IIIC und IVM1a
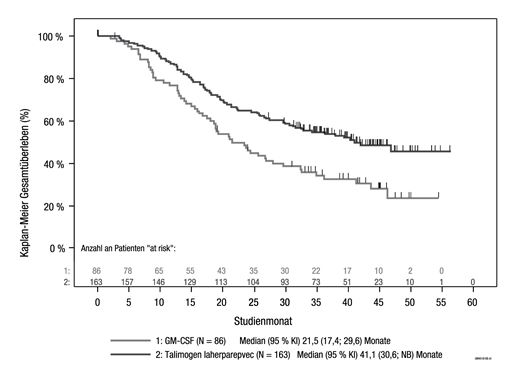
Zensierte Patienten werden durch vertikale Balken angezeigt I.
Patienten, die nicht als verstorben erfasst worden sind, sind als zensiert eingeschlossen.
Die ITT-Population umfasst alle Patienten, die zum Erhalt einer Studienbehandlung randomisiert worden sind. Die Patienten werden anhand der randomisierten Behandlung analysiert.
Ein Monat = 365,25/12 Tage. NB = nicht bestimmbar.
Aufgrund des explorativen Charakters der Analyse und basierend auf der aktuellen Datenlage ist nicht erwiesen, dass IMLYGIC mit einem Effekt auf das Gesamtüberleben assoziiert ist.
PharmakokinetikTalimogen laherparepvec ist ein genetisch modifiziertes, replikationsfähiges HSV-1-Virus. Deshalb werden dessen Pharmakokinetik und Biodistribution durch die intraläsionale Injektionsstelle, die tumor-selektive Replikation und die Freisetzung aus Tumorgewebe gesteuert.
Absorption
Die zelluläre Aufnahme von Talimogen laherparepvec erfolgt durch HSV-1-Rezeptoren auf Tumor- und nicht-tumorösen Zellen nach lokaler Injektion in Tumore. Da Talimogen laherparepvec injiziert wird und intratumoral repliziert, sind die Bioverfügbarkeit und die systemische Konzentration von Talimogen laherparepvec nicht prädiktiv für die Wirkstoffaktivität und wurden deshalb nicht berechnet.
Metabolismus und Elimination
Talimogen laherparepvec wird durch generelle Wirtsabwehrmechanismen (z.B. Autophagie, adaptive Immunantworten) eliminiert. Talimogen laherparepvec wird durch die für Proteine und DNA typischen endogenen katabolen Stoffwechselwege abgebaut. Wie bei anderen Wildtyp-HSV-1-Infektionen kann ein latenter Pool an Talimogen laherparepvec-DNA in neuronalen Zellkörpern persistieren, welche die Injektionsstellen innervieren. Das Auftreten einer latenten Infektion mit Talimogen laherparepvec ist deshalb nicht auszuschliessen.
Biodistribution (im Körper) und Virusausscheidung (Exkretion/Sekretion)
Die DNA von Talimogen laherparepvec wurde mit einem hoch sensitiven und spezifischen qPCR-Assay quantifiziert, der nicht mit dem viralen Infektionsrisiko korrelieren muss. In klinischen Studien wurde Talimogen laherparepvec bei ausgewählten Patienten auch mittels Assays zur viralen Infektiosität an der Injektionsstelle und in einigen Fällen an potenziellen Herpesläsionen quantifiziert.
Klinische Biodistribution, Elimination und Ausscheidung
Die Biodistribution und Ausscheidung von intraläsional verabreichtem Talimogen laherparepvec wurden in einer klinischen Studie untersucht, bei der die Konzentration von Talimogen laherparepvec-DNA im Blut, im Urin, an der Injektionsstelle, an der Aussenseite der Okklusivverbände, in der Mundschleimhaut und Anogenitalregion und an den mutmasslichen Herpesläsionen gemessen wurde. 60 Patienten mit Melanom erhielten IMLYGIC als intraläsionale Injektion mit derselben Dosierung und demselben Behandlungsschema wie in der pivotalen Studie (siehe «Klinische Wirksamkeit und Sicherheit»). Während der Behandlung wurden Proben der Okklusivverbände gesammelt. Blut- und Urinproben wurden während der Behandlung und bis zu 30 Tage nach Ende der Behandlung gesammelt. Proben von Injektionsstelle, Mundschleimhaut und Anogenitalregion wurden während der Behandlung und bis zu 60 Tage nach Ende der Behandlung entnommen. Proben von mutmasslichen Herpesläsionen wurden immer dann entnommen, wenn beim Patienten Läsionen auftraten, die möglicherweise durch Herpes verursacht wurden. Wenn die qPCR Untersuchung für Talimogen laherparepvec positiv war, wurde ein TCID50-Assay durchgeführt, um die virale Infektiosität zu messen. Bei den 60 behandelten Patienten zeigten die Daten, dass die Talimogen laherparepvec-DNA während der Studie an allen Stellen vorhanden war (siehe Tabelle 5).
Tabelle 5: Patienten mit nachweisbarer DNA während der Behandlung
|
Körperflüssigkeit/Stelle
|
Patienten mit nachweisbarer DNA während der Behandlung
(n = 60)
| |
Blut
|
59 (98%)
| |
Urin
|
19 (32%)
| |
Injektionsstelle
|
60 (100%)
| |
Aussenseite des Okklusivverbandes
|
48 (80%)
| |
Mundschleimhaut
|
8 (13%)
| |
Anogenitalregion
|
5 (19%)a
|
aFür die Anogenitalregion wurden 26 Patienten auf IMLYGIC-DNA getestet.
Der Anteil der Proben und Probanden mit Talimogen laherparepvec-DNA war im Behandlungszyklus 2 am höchsten für Blut, Urin, Injektionsstelle und Okklusivverbände, im Behandlungszyklus 1 am höchsten für die Mundschleimhaut und in den Zyklen 1 und 2 am höchsten für die Anogenitalregion. Bei den Patienten mit nachweisbarer Talimogen laherparepvec-DNA in Blut, Urin, Mundschleimhaut und Anogenitalregion enthielt 30 Tage nach Ende der Behandlung keine der Proben nachweisbare Talimogen laherparepvec-DNA. Bei Patienten mit nachweisbarer DNA in den behandelten Läsionen war 60 Tage nach Ende der Behandlung in keiner Probe Talimogen laherparepvec-DNA nachweisbar.
Insgesamt wiesen 3 von 19 Patienten mit Läsionen, die möglicherweise durch Herpes verursacht wurden, zu jeder Zeit während der Studie Talimogen laherparepvec-DNA auf.
Bei Talimogen laherparepvec-DNA-positiven Proben von Injektionsstelle, Okklusivverbänden, Mundschleimhaut, Anogenitalregion und mutmasslichen Herpesläsionen wurde die Virenaktivität gemessen. In keiner der Proben von Okklusivverbänden, Mundschleimhaut, Anogenitalregion und mutmasslichen Herpesläsionen war eine Virenaktivität nachweisbar. Ein infektiöses Talimogen laherparepvec-Virus wurde bei 7 (11% der) Patienten zu mehreren Zeitpunkten der Studie an der Injektionsstelle festgestellt; nach dem Behandlungszyklus 2 oder nach Ende der Behandlung war keine der Proben positiv für eine virale Infektiosität.
Kinetik spezieller Patientengruppen
Es wurden keine pharmakokinetischen Studien zur Anwendung von Talimogen laherparepvec in speziellen Patientengruppen durchgeführt.
Präklinische DatenTalimogen laherparepvec wurde in Dosen von bis zu 4 × 108 PFU/kg oder 107 PFU/Dosis (60fach über der höchsten empfohlenen klinischen Dosis) bei einmaliger oder wiederholter Verabreichung als subkutane, intravenöse oder intratumorale Injektion von immunkompetenten Mäusen, Ratten und Hunden gut vertragen. Die Wirkungen, die nach der wiederholten Injektion von Talimogen laherparepvec bei Mäusen beobachtet wurden, waren im Allgemeinen mild, auf lokale Gewebereaktionen an der Injektionsstelle beschränkt und entsprachen den bei viralen Infektionen erwarteten Entzündungsreaktionen (z.B. vorübergehende Veränderungen der Leukozytenpopulationen, lymphoide Hyperplasie in der Milz und erhöhte Hämatopoese). Sie besserten sich bei fortgesetzter Dosierung oder nach Ende der Behandlung. Es wurden keine Neuropathologie oder neurologische Nebenwirkungen beobachtet. In einer in vivo-Studie mit intrazerebraler Injektion war Talimogen laherparepvec im Vergleich zu einer Wildtyp HSV-1-Dosis, die bei Mäusen in 50% der Fälle zum Tod führte, 10'000fach weniger neurovirulent.
Talimogen Laherparepvec wurde bei immundefizienten Mäusen (Nacktmäuse und SCID) in Dosen von bis zu 2 × 108 PFU/kg (30fach über der höchsten empfohlenen klinischen Dosis) in verschiedene Xenograft-Tumoren injiziert. Bei bis zu 20% der Nacktmäuse (hauptsächlich ungenügende Funktion der T-Lymphozyten) und bei 100% der SCID-Mäuse (ohne T- und B-Lymphozyten) wurde eine letale systemische Virusinfektion beobachtet. In allen Studien wurde bei 14% der Nacktmäuse nach der Behandlung mit Talimogen laherparepvec-Dosen, die 10- bis 100fach höher waren, als diejenigen, die mit Wildtyp HSV-1 zu einer 100%igen Letalität führen, eine fatale disseminierte Virusinfektion beobachtet.
Mutagenität
Das genotoxische Potenzial von Talimogen laherparepvec wurde nicht in Langzeitstudien am Tier oder Menschen untersucht. Da sich Wildtyp HSV-1 nicht ins Wirtsgenom integriert, ist das Risiko einer Insertionsmutagenese mit Talimogen laherparepvec vernachlässigbar.
Karzinogenität
Das karzinogene Potenzial von Talimogen laherparepvec wurde nicht in Langzeitstudien am Tier oder am Menschen untersucht. Verfügbare Daten zu Talimogen laherparepvec und Wildtyp HSV-1 weisen auf kein karzinogenes Risiko beim Menschen hin.
Reproduktions- und Entwicklungstoxizität
Es gab keine Auswirkungen auf männliches oder weibliches Reproduktionsgewebe nach einer Behandlung ausgewachsener Mäuse mit Dosen von bis zu 4 × 108 PFU/kg (basierend auf PFU/kg, 60fach höher im Vergleich zur maximalen klinischen Dosis). Bei trächtigen Mäusen, denen während der Organogenese Talimogen laherparepvec-Dosen von bis zu 4 × 108 (400 Millionen) PFU/kg (60fach höher als die maximale klinische Dosis, auf PFU/kg-Basis) verabreicht wurden, wurden keine Auswirkungen auf die embryo-fetale Entwicklung beobachtet. Im fetalen Blut wurden vernachlässigbare Mengen an Talimogen laherparepvec-DNA (< 0,001% der mütterlichen Blutspiegel) gefunden.
Bioverteilung/Ausscheidung
Bei Mäusen wurde nach der intraläsionalen Verabreichung in etwa 40% der Tumorproben und in ≤20% der Blut- und Organgewebeproben (z.B. Milz, Lymphknoten, Leber, Herz und Nieren) Talimogen laherparepvec-DNA nachgewiesen. In ≤2% der Gehirn-, Ovarial- und Speicheldrüsenproben wurde Talimogen laherparepvec-DNA nachgewiesen. Im Knochenmark, in den Augen, in Sekret absondernden Geweben (Tränendrüsen, Nasenschleimhaut) oder im Kot war sie nicht nachweisbar. Die höchste Konzentration von Talimogen laherparepvec-DNA wurde in den Läsionen festgestellt. Alle anderen Gewebe wiesen eine deutlich geringere Talimogen laherparepvec-DNA-Konzentration auf (< 0,5% der höchsten in Tumoren nachgewiesenen Konzentration). In behandelten Tumoren konnte bis 84 Tage nach der letzten Dosis Talimogen laherparepvec-DNA nachgewiesen werden. In den meisten Blutproben (94%) war sie jedoch innerhalb von sieben Tagen nach der letzten Dosis abgebaut.
Bei Mäusen wurde nach der intravenösen Verabreichung in etwa 8% der Proben von peripheren Nerven Talimogen laherparepvec-DNA nachgewiesen.
Sonstige HinweiseInkompatibilitäten
Da keine Verträglichkeitsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Vorbereitung und Aufbewahrung vor der Anwendung
IMLYGIC soll nach dem Auftauen sobald wie möglich angewendet werden.
Aufgetautes IMLYGIC ist stabil, wenn es bei einer Temperatur von 2°C bis 25°C und vor Licht geschützt in der Original-Durchstechflasche, in einer Spritze, oder in der Original-Durchstechflasche gefolgt von einer Spritze gelagert wird. Die Lagerungszeiten gemäss Tabelle 6 und Tabelle 7 dürfen nicht überschritten werden.
·Falls aufgetautes IMLYGIC in der Original-Durchstechflasche und anschliessend in einer Spritze aufbewahrt wird:
·Sollte derselbe Temperaturbereich während der gesamten Aufbewahrung bis zur Anwendung eingehalten werden.
·Darf die Aufbewahrungszeit in einer Spritze bei einer Temperatur von bis zu 25°C nicht länger sein als 2 Stunden für 106 (1 Million) PFU/ml sowie 4 Stunden für 108 (100 Millionen) PFU/ml (siehe Tabelle 6).
·Darf die maximale kumulative Aufbewahrungszeit (Aufbewahrungszeit in der Durchstechflasche zuzüglich Aufbewahrungszeit in einer Spritze) die in Tabelle 7 angegebene Dauer nicht überschreiten.
Nach dem Auftauen darf IMLYGIC nicht wieder eingefroren werden. Entsorgen Sie alle aufgetauten IMLYGIC-Durchstechflaschen oder Spritzen, die über die unten vorgegebene Zeit hinaus aufbewahrt wurden.
Tabelle 6: Maximale Aufbewahrungszeit für aufgetautes IMLYGIC in einer Spritze
|
|
106 (1 Million) PFU/ml
|
108 (100 Millionen) PFU/ml
| |
2°C bis 8°C
|
8 Stunden
|
8 Stunden
| |
Bis 25°C
|
2 Stunden
|
4 Stunden
|
Tabelle 7: Maximale kumulative Aufbewahrungszeit für aufgetautes IMLYGIC (Aufbewahrungszeit in der Durchstechflasche zuzüglich Aufbewahrungszeit in einer Spritze)
|
|
106 (1 Million) PFU/ml
|
108 (100 Millionen) PFU/ml
| |
2°C bis 8°C
|
24 Stunden
|
1 Woche [7 Tage]
| |
Bis 25°C
|
12 Stunden
|
24 Stunden
|
Besondere Lagerungshinweise
Bei -90°C bis -70°C tiefgekühlt lagern und transportieren.
Im Originalkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Auftauen der IMLYGIC-Durchstechflaschen
·Tauen Sie die gefrorenen IMLYGIC-Durchstechflaschen vor der Anwendung bei einer Temperatur von 20°C bis 25°C auf, bis IMLYGIC flüssig ist. Die Zeit zum Erreichen des vollständigen Auftauens der Durchstechflaschen beträgt voraussichtlich 30 bis 70 Minuten, abhängig von der Raumtemperatur. Behutsam schwenken. NICHT schütteln.
·Die IMLYGIC-Durchstechflaschen sollten bis zur Anwendung im Originalkarton aufgetaut und gelagert werden, um den Inhalt vor Licht zu schützen.
Hinweise für die Handhabung und Anwendung, persönliche Schutzausrüstung, unbeabsichtigtes Verschütten und Abfallentsorgung
Befolgen Sie die lokalen Richtlinien betreffend Handhabung und Anwendung, persönlicher Schutzausrüstung, versehentlichem Verschütten und Abfallentsorgung.
·Tragen Sie während der Vorbereitung und Verabreichung von IMLYGIC Schutzkleidung oder einen Labormantel, eine Schutzbrille oder einen Gesichtsschutz und Handschuhe. Decken Sie exponierte Wunden vor der Verabreichung ab. Vermeiden Sie den Kontakt mit der Haut, den Augen oder den Schleimhäuten.
·Wechseln Sie die Handschuhe nach der Anwendung bevor Sie die injizierten Läsionen mit Okklusivverbänden abdecken. Wischen Sie die Aussenseite der Okklusivverbände mit einem Alkoholtupfer ab. Es wird empfohlen, die Injektionsstellen immer, wenn möglich, mit luft- und wasserdichten Verbänden abzudecken. Um das Risiko einer viralen Übertragung zu minimieren, sollten Patienten ihre Injektionsstelle für mindestens 8 Tage nach der letzten Behandlung oder, falls die Injektionsstelle nässt oder Flüssigkeit absondert, für längere Zeit abdecken. Weisen Sie die Patienten an, die Verbände so anzulegen, wie es das medizinische Fachpersonal gezeigt hat, und den Verband zu ersetzen, falls er sich löst.
·Entsorgen Sie alle Materialien, die mit IMLYGIC in Kontakt gekommen sind (z.B. Durchstechflasche, Spritze, Nadel, Verbandsmaterial) gemäss den lokalen Richtlinien.
Unbeabsichtigte Exposition
·Im Falle eines versehentlichen, berufsbedingten Kontakts mit IMLYGIC (durch einen Spritzer in die Augen oder auf Schleimhäute) während der Vorbereitung oder Verabreichung, spülen Sie für mindestens 15 Minuten mit klarem Wasser. Im Falle einer Exposition verletzter Haut oder durch einen Nadelstich, reinigen Sie die betroffene Stelle gründlich mit Seife und Wasser und/oder Desinfektionsmittel.
·Entfernen Sie jegliches verschüttetes IMLYGIC mit einem viruziden Mittel und saugfähigen Materialen.
·Weisen Sie Patienten an, benutzte Verbände und Reinigungsmaterial in einem verschliessbaren Plastikbeutel unterzubringen, da diese potenziell kontaminiert sind, und den Beutel im Haushaltsabfall zu entsorgen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu entsorgen.
Zulassungsnummer65812 (Swissmedic)
PackungenIMLYGIC wird in zwei verschiedenen Aufmachungen angeboten:
Durchstechflasche zum Einmalgebrauch dauerhaft eingeführt in einer klaren Kunststoffhülse
ODER
Durchstechflasche zum Einmalgebrauch ohne klare Kunststoffhülse
IMLYGIC 106 Plaque-bildende Einheiten (PFU)/ml: 1 Durchstechflasche (mit hellgrünem Deckel). [A]
IMLYGIC 108 Plaque-bildende Einheiten (PFU)/ml: 1 Durchstechflasche (mit königsblauem Deckel). [A]
ZulassungsinhaberinAmgen Switzerland AG, Risch
Domizil: 6343 Rotkreuz
Stand der InformationNovember 2022.
Version #200622
|