Eigenschaften/WirkungenATC-Code
A10BJ02
Wirkungsmechanismus
Liraglutide ist ein acyliertes Analogon des humanen GLP-1 (Glucagon-like Peptid-1) mit einer 97-%-igen Aminosäurensequenz-Homologie zum endogenen humanen GLP-1. Liraglutide bindet an den GLP-1-Rezeptor (GLP-1R) und aktiviert diesen.
GLP-1 ist ein physiologischer Regulator des Appetits und der Nahrungsaufnahme, doch der genaue Wirkmechanismus ist noch nicht vollständig bekannt. In tierexperimentellen Studien führte die periphere Verabreichung zu einer Aufnahme von Liraglutide in bestimmten Hirnregionen, die mit der Appetitregulierung assoziiert sind, wo Liraglutide über die spezifische Aktivierung von GLP-1R zu einem Anstieg der wichtigsten Sättigungssignale und einer Abnahme der wichtigsten Hungersignale führte und damit zu einem geringeren Körpergewicht.
GLP-1-Rezeptoren werden an spezifischen Orten des Herzens, des Gefässsystems, des Immunsystems und der Nieren exprimiert. Human- bzw. Tierstudien belegen, dass diese Rezeptoren kardiovaskuläre Wirkungen von Liraglutide vermitteln können, einschliesslich verminderter Entzündung und der verzögerten Progression einer Atherosklerose.
Pharmakodynamik
Liraglutide reduziert das Körpergewicht beim Menschen hauptsächlich durch eine Abnahme der Fettmasse, wobei der relative Verlust an viszeralem Fett grösser ist als der Verlust an subkutanem Fett. Liraglutide reguliert den Appetit durch eine Steigerung des Völle- und Sättigungsgefühls und eine Reduzierung des Hungergefühls und des Wunsches nach Nahrungsverzehr und führt so zu einer geringeren Nahrungsaufnahme. Liraglutide erhöht im Vergleich zu Placebo nicht den Energieverbrauch.
Liraglutide stimuliert die Insulinsekretion und senkt die Glucagonsekretion in einem glucoseabhängigen Mechanismus, was zu einer Senkung des postprandialen und des Nüchternblutzuckers führt. Die blutzuckersenkende Wirkung ist bei Patienten mit Prädiabetes und Diabetes stärker ausgeprägt als bei Patienten mit Blutzuckerwerten im Normbereich. Klinische Studien legen nahe, dass Liraglutide die Betazellfunktion verbessert und unterstützt. Dabei wurden Messungen wie das HOMA-B und das Verhältnis von Proinsulin zu Insulin zugrunde gelegt.
Klinische Wirksamkeit
Die Wirksamkeit und die Sicherheit von Liraglutide für die Gewichtsregulierung in Verbindung mit einer verminderten Kalorienzufuhr und verstärkter körperlicher Aktivität wurden in vier randomisierten, doppelblinden, Placebo-kontrollierten Phase-3-Studien untersucht, an denen insgesamt 5'358 Patienten teilnahmen.
Studie 1 (SCALE Obesity & Pre-Diabetes – 1839): Insgesamt wurden 3'731 Patienten mit Adipositas (BMI ≥30 kg/m2) oder Übergewicht (BMI ≥27 kg/m2) mit Dyslipidämie und/oder Hypertonie, nach ihrem Prädiabetes-Stadium zum Zeitpunkt der Einschlussuntersuchung sowie nach ihrem BMI bei Studienbeginn (≥30 kg/m² oder < 30 kg/m²), stratifiziert. Alle 3'731 randomisierten Patienten erhielten eine 56-wöchige Behandlung, und 2'254 randomisierte Patienten, mit einem vorhandenen Prädiabetes zum Zeitpunkt der Einschlussuntersuchung, erhielten eine 160-wöchige Behandlung. Auf beide Behandlungszeiträume folgte eine 12-wöchige Nachbeobachtungszeit ohne Arzneimittel/Placebo. Hintergrundtherapie für alle Patienten war eine Lebensstilintervention in Form von einer energiereduzierten Diät sowie einer Beratung hinsichtlich körperlicher Aktivität.
Im 56-wöchigen Teil der Studie 1 wurde der Gewichtsverlust bei allen 3'731 randomisierten Patienten bewertet (2'590 Patienten schlossen die Studie ab).
Im 160-wöchigen Teil der Studie 1 wurde die Zeit bis zum Auftreten eines Diabetes mellitus Typ 2 in den 2'254 randomisierten Patienten mit einem bereits vorhandenen Prädiabetes bewertet (1'128 Patienten schlossen die Studie ab).
Studie 2 (SCALE Diabetes – 1922): Eine 56-wöchige Studie zur Bewertung des Gewichtsverlusts als Primärendpunkt bei 846 randomisierten adipösen und übergewichtigen Patienten (628 Patienten schlossen die Studie ab) mit unzureichend kontrolliertem Diabetes mellitus Typ 2 (HbA1c-Bereich 7–10 %). Die Standardtherapie bei Studienbeginn war entweder ausschliesslich Diät und körperliche Aktivität, Metformin, ein Sulfonylharnstoff oder ein Glitazon, jeweils als Einzelwirkstoff oder in einer Kombination hiervon.
Studie 3 (SCALE Maintenance – 1923): Eine 56-wöchige Studie zur Bewertung der Erhaltung des Körpergewichts und des Gewichtsverlusts als Primärendpunkt bei 422 randomisierten adipösen und übergewichtigen Patienten (305 Patienten schlossen die Studie ab) mit Hypertonie oder Dyslipidämie nach einer vorangegangenen Gewichtsabnahme von ≥5 % infolge einer kalorienarmen Diät.
Studie 4 (SCALE Sleep Apnoe – 3970): Eine 32-wöchige Studie zur Bewertung des Schweregrads der Schlafapnoe als Primärendpunkt und des Gewichtsverlusts als Sekundärendpunkt bei 359 randomisierten adipösen Patienten (276 Patienten schlossen die Studie ab) mit mittelschwerer oder schwerer obstruktiver Schlafapnoe.
Körpergewicht
Die Daten zu Gewichtsabnahme, Therapie-Respondern, Zeitverlauf und kumulativer Verteilung der Gewichtsveränderung (%) für die Studien 1–3 sind in der Tabelle 3 und den Abbildungen 1, 2, 3 und 4 dargestellt.
Tabelle 3. Änderungen gegenüber dem Ausgangswert bei Körpergewicht in Woche 56 und 160 – Studien 1, 2 und 3
|
|
Studie 1 (Woche 56)
|
Studie 1 (Woche 160)
|
Studie 2
|
Studie 3
| |
|
Saxenda
n=2'437
|
Placebo
n=1'225
|
Saxenda (N=1'472)
|
Placebo (N=738)
|
Saxenda
n=412
|
Placebo
n=211
|
Saxenda
n=207
|
Placebo
n=206
| |
Körpergewicht
| |
Ausgangswert im Mittel, kg (SA)
|
106.3
(21.2)
|
106.3
(21.7)
|
107.6 (21.6)
|
108.0 (21.8)
|
105.6
(21.9)
|
106.7
(21.2)
|
100.7 (20.8)
|
98.9 (21.2)
| |
Änderung gegenüber Ausgangswert, %
|
-8.0
|
-2.6
|
-6.2
|
-1.8
|
-5.9
|
-2.0
|
-6.3
|
-0.2
| |
Saxenda gegenüber
Placebo,
% (95 % KI)
|
-5.4*
(-5.8; -5.0)
|
-4.3** (-4.9; -3.7)
|
-4.0**
(-4.8; -3.1)
|
-6.1**
(-7.5; -4.6)
| |
Änderung gegenüber Ausgangswert, kg
|
-8.4
|
-2.8
|
-6.5
|
-2.0
|
-6.2
|
-2.2
|
-6.0
|
-0.2
| |
Saxenda gegenüber
Placebo,
kg (95 % KI)
|
-5.6**
(-6.0; -5.1)
|
-4.6** (-5.3; -3.9)
|
-4.1**
(-5.0; -3.1)
|
-5.9**
(-7.3; -4.4)
| |
% der Patienten mit
≥5 % Gewichtsabnahme
|
63.5
|
26.6
|
49.6
|
23.4
|
49.8
|
13.5
|
50.7
|
21.3
| |
Saxenda gegenüber
Placebo,
kg (95 % KI)
|
4.8**
(4.1; 5.6)
|
3.2** (2.6; 3.9)
|
6.4**
(4.1; 10.0)
|
3.8**
(2.4; 6.0)
| |
% der Patienten mit
> 10 % Gewichtsabnahme
|
32.8
|
10.1
|
24.4
|
9.5
|
22.9
|
4.2
|
27.4
|
6.8
| |
Saxenda gegenüber
Placebo,
kg (95 % KI)
|
4.3**
(3.5; 5.3)
|
3.1** (2.3; 4.1)
|
6.8**
(3.4; 13.8)
|
5.1**
(2.7; 9.7)
|
Gesamtgruppe (FAS=Full Analysis Set). Für Körpergewicht sind die Ausgangswerte Mittelwerte, Änderungen gegenüber den Ausgangswerten in Woche 56 und Woche 160 sind geschätzte Mittelwerte (LSMeans = kleinste Fehlerquadrate) und Behandlungsunterschiede in Woche 56 und Woche 160 sind geschätzte Behandlungsunterschiede. Für die Anteile der Patienten (%), die ≥5/> 10 % Körpergewicht verloren haben, wurden geschätzte Odds-Verhältnisse verwendet. Fehlende Werte nach Studienbeginn wurden unter Verwendung der Last Observation Carried Forward (LOCF) berechnet. * p < 0.05. ** p < 0.0001. SA=Standardabweichung. KI=Konfidenzintervall.
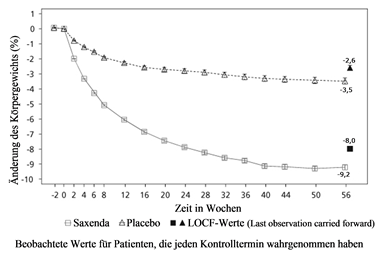
Abbildung 1. Änderung des Körpergewichts (%) im Zeitverlauf in Studie 1 (0–56 Wochen) gegenüber dem Ausgangswert
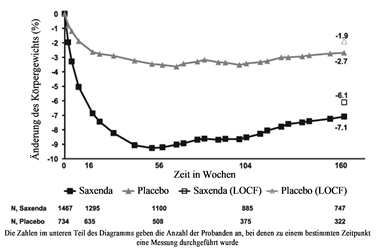
Abbildung 2. Änderung des Körpergewichts (%) im Zeitverlauf in Studie 1 (0–160 Wochen) gegenüber dem Ausgangswert
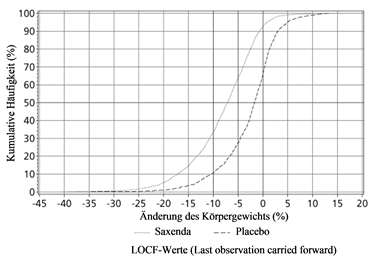
Abbildung 3. Kumulative Verteilung der Gewichtsänderung (%) nach 56 Behandlungswochen in Studie 1
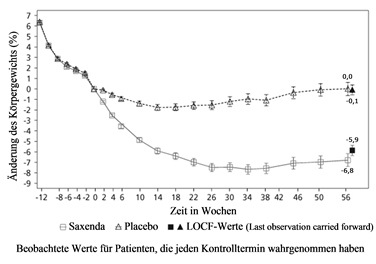
Abbildung 4. Änderung des Körpergewichts (%) im Zeitverlauf in Studie 3 gegenüber der Randomisierung (Woche 0)
Vor Woche 0 bestand die Behandlung der Patienten nur aus kalorienarmer Diät und körperlicher Aktivität. In Woche 0 wurden die Patienten randomisiert der Behandlungsgruppe mit Saxenda oder Placebo zugeteilt.
Gewichtsabnahme nach 12-wöchiger Behandlung mit Liraglutide (3.0 mg)
Als «Early Responders» wurden die Patienten definiert, die nach 12-wöchiger Therapie mit der Behandlungsdosis von Liraglutide (4 Wochen Dosissteigerung und 12 Wochen Behandlungsdosis) eine Gewichtsabnahme von ≥5 % erzielten. In Studie 1 waren 67.5 % «Early Responders». In Studie 2 waren es 50.4 % der Patienten. Bei Fortsetzung der Behandlung mit Liraglutide erzielen voraussichtlich 86.2 % dieser «Early Responders» nach 1 Jahr Behandlung eine Gewichtsabnahme von ≥5 % und 51 % erzielen voraussichtlich eine Gewichtsabnahme von ≥10 %. Die voraussichtliche durchschnittliche Gewichtsabnahme bei den «Early Responders», die 1 Jahr Behandlung durchlaufen, beträgt 11.2 % ihres Ausgangskörpergewichts (9.7 % bei Männern und 11.6 % bei Frauen). In der Gruppe von Patienten, die nach 12-wöchiger Therapie mit 3.0 mg Liraglutide pro Tag eine Gewichtsabnahme von < 5 % erreicht haben, erreichen noch 6.6 % der Patienten eine Gewichtsabnahme von ≥10 % nach 1 Jahr.
Blutzucker und kardiometabolische Parameter
Daten zum Blutzucker und kardiometabolischen Parametern in den Studien 1 und 2 sind in der Tabelle 4 dargestellt.
Tabelle 4. Änderungen nach 56 Wochen (Studien 1 und 2) und 160 Wochen (Studie 1) gegenüber dem Ausgangswert bei Blutzucker und kardiometabolischen Parametern
|
|
Saxenda
(n=2'437)
|
Placebo
(n=1'225)
|
Saxenda gegenüber Placebo
| |
Studie 1
(Woche 56)
|
Ausgangswert
|
Änderung
|
Ausgangswert
|
Änderung
|
| |
HbA1c, %
|
5.6
|
-0.3
|
5.6
|
-0.1
|
-0.23**
(0.25; -0.21)
| |
NPG, mmol/l
|
5.3
|
-0.4
|
5.3
|
-0.01
|
-0.38**
(0.42; -0.35)
| |
Systolischer Blutdruck, mmHg
|
123.0
|
-4.3
|
123.3
|
-1.5
|
-2.8**
(-3.6; -2.1)
| |
Diastolischer Blutdruck, mmHg
|
78.7
|
-2.7
|
78.9
|
-1.8
|
-0.9*
(-1.4; - 0.4)
| |
Taillenumfang, cm
|
115.0
|
-8.2
|
114.5
|
-4.0
|
-4.2**
(-4.7; -3.7)
| |
|
Saxenda (n=1'472)
|
Placebo
(n=738)
|
Saxenda gegenüber Placebo
| |
Studie 1
(Woche 160)
|
Ausgangswert
|
Änderung
|
Ausgangswert
|
Änderung
|
| |
HbA1c, %
|
5.75
|
-0.35
|
5.74
|
-0.14
|
-0.21**
(-0.24; -0.18)
| |
NPG, mmol/l
|
5,50
|
-0,37
|
5,46
|
0,04
|
-0.41**
(-0.46; -0.36)
| |
Systolischer Blutdruck, mmHg
|
124.80
|
-3.24
|
125.01
|
-0.44
|
-2.80**
(-3.81; -1.79)
| |
Diastolischer Blutdruck, mmHg
|
79.40
|
-2.36
|
79.83
|
-1.74
|
-0.62
(-1.33; 0.09)
| |
Taillenumfang, cm
|
116.64
|
-6.88
|
116.74
|
-3.35
|
-3.53**
(-4.23; -2.83)
| |
Studie 2
|
(n=412)
|
(n=211)
|
| |
HbA1c, %
|
7.9
|
-1.3
|
7.9
|
-0.4
|
-0.9**
(-1.1; -0.8)
| |
NPG, mmol/l
|
8.8
|
-1.9
|
8.6
|
-0.1
|
-1.8**
(-2.1; -1.4)
| |
Systolischer Blutdruck, mmHg
|
128.9
|
-3.0
|
129.2
|
-0.4
|
-2.6*
(-4.6; - 0.6)
| |
Diastolischer Blutdruck, mmHg
|
79.0
|
-1.0
|
79.3
|
-0.6
|
-0.4
(1.7; 1.0)
| |
Taillenumfang, cm
|
118.1
|
-6.0
|
117.3
|
-2.8
|
-3.2**
(-4.2; -2.2)
| |
Bei der statistischen Analyse der glykämischen und kardiometabolischen Parameter wurde das multiple Testen nicht berücksichtigt, und somit müssen die Ergebnisse als statistisch nicht-konfirmatorisch betrachtet werden. Gesamtgruppe (FAS=Full Analysis Set). Für HbA1c, NPG, Blutdruck und Taillenumfang sind die Ausgangswerte Mittelwerte, Änderungen gegenüber den Ausgangswerten in Woche 56 und Woche 160 sind geschätzte Mittelwerte (LSMeans) und Behandlungsunterschiede in Woche 56 und Woche 160 sind geschätzte Behandlungsunterschiede. Fehlende Werte nach Studienbeginn wurden unter Verwendung der Last Observation Carried Forward (LOCF) berechnet. * p < 0.05. ** p < 0.0001.
SA=Standardabweichung. KI=Konfidenzintervall.
|
Immunogenität
Mit Saxenda behandelte Patienten können Anti-Liraglutide-Antikörper entwickeln. Bei einer Untersuchung nach Behandlungsbeginn wurden bei 42 (2.8 %) von 1'505 mit Saxenda behandelten Patienten Anti-Liraglutide-Antikörper nachgewiesen. Antikörper, die in einem in-vitro-Test eine neutralisierende Wirkung hatten, traten bei 18 (1.2 %) von 1'505 mit Saxenda behandelten Patienten auf. Das Vorhandensein von Antikörpern könnte im Zusammenhang mit einem vermehrten Auftreten von Reaktionen an der Injektionsstelle und Berichten von niedrigen Blutzuckerspiegeln stehen. In klinischen Studien wurden diese Ereignisse üblicherweise als leicht eingestuft und verschwanden im weiteren Behandlungsverlauf wieder.
Der Nachweis der Antikörperbildung ist stark abhängig von der Empfindlichkeit und Spezifität des Tests. Ausserdem kann die beobachtete Inzidenz von nachgewiesenen Antikörpern (einschliesslich neutralisierender Antikörper) in einem Test durch verschiedene Faktoren beeinflusst werden, zu denen die Testmethodik, die Probenhandhabung, der zeitliche Ablauf der Probenentnahme, die Begleitmedikation sowie Grunderkrankungen zählen. Aus diesen Gründen kann das Auftreten von Antikörpern gegen Saxenda nicht direkt mit dem Auftreten von Antikörpern anderer Produkte verglichen werden.
Kardiovaskuläre Bewertung
Schwere, unerwünschte kardiovaskuläre Ereignisse (MACE) wurden von einer externen unabhängigen Expertengruppe beurteilt und als nicht-tödlicher Myokardinfarkt, nicht-tödlicher Schlaganfall und kardiovaskulärer Tod klassiert. In allen Studien mit Saxenda traten 6 MACE bei Patienten, die mit Liraglutide behandelt wurden, und 10 MACE bei mit Placebo behandelten Patienten auf. Die Hazard Ratio und 95 % KI ist 0.33 [0.12–0.90] für Liraglutide gegenüber Placebo. In klinischen Phase-3-Studien wurde bei Behandlung mit Liraglutide eine mittlere Erhöhung der Herzfrequenz gegenüber dem Ausgangswert in Höhe von 2.5 Schlägen pro Minute beobachtet (in allen Studien zwischen 1.6 und 3.6 Schläge pro Minute), welche nach etwa 6 Wochen ihr Maximum erreichte und nach Absetzen von Liraglutide reversibel war (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Die Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results (LEADER) Studie untersuchte die Häufigkeit schwerer kardiovaskulärer Ereignisse (MACE: kardiovaskulärer Tod, nicht-tödlicher Myokardinfarkt, nicht-tödlicher Schlaganfall) in 9'340 Patienten mit erhöhten Diabetes mellitus Typ 2 Diabetes und einem erhöhten kardiovaskulären Risiko. Randomisiert (1:1) wurden die Patienten zusätzlich zur Standardtherapie entweder mit bis zu 1.8 mg Liraglutide täglich (4'668) oder Placebo (4'672) behandelt (mediane Behandlungsdauer zirka 3.5 Jahre). Primärer Endpunkt war die Zeit bis zum ersten Auftreten eines MACE. Dieser wurde durch Liraglutide in der untersuchten Dosierung signifikant reduziert (Hazard Ratio 0.87 [0.78; 0.97] 95 % KI).
Kinder und Jugendliche
In einer doppelblinden Studie, in der die Wirksamkeit und Sicherheit von Saxenda gegenüber Placebo hinsichtlich des Gewichtsverlusts bei Jugendlichen ab 12 Jahren mit Adipositas untersucht wurde, war Saxenda gegenüber Placebo in der Reduktion des Körpergewichtes nach 56 Wochen überlegen (gemessen anhand des BMI-SDS; siehe Tabelle 5).
Ein grösserer Anteil der Patienten erreichten eine ≥5 % und ≥10 % BMI-Reduktion mit Saxenda als mit Placebo sowie eine grössere mittlere BMI-Abnahme und Körpergewichtsabnahme (siehe Tabelle 5).
Die Veränderung der Körperzusammensetzung wurde nicht untersucht.
Alle Patienten sollten engmaschig überwacht werden (siehe auch Abbruchregel unter «Indikation»).
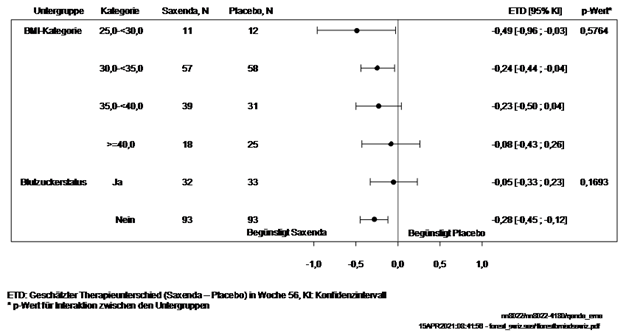
Abbildung 5.
Der Forest-Plot zeigt die geschätzten Unterschiede (Saxenda vs. Placebo) in der Änderung des BMI-SDS von Studienbeginn bis Woche 56 in Abhängigkeit vom BMI Ausgangswert ([kg/m2]: ≥25 -<30, ≥30 - <35, ≥35 - <40 und ≥40) und vom glykämischen Status zu Beginn der Studie (Normoglykämie vs. Prädiabetes oder Diabetes Typ 2) inklusive der der p-Werte für die Interaktionstests.
Nach einer 26-wöchigen Nachbeobachtungszeitraum ohne die Studienmedikation bzw. Placebo wurde in der Saxenda Gruppe gegenüber der Placebo-Gruppe eine erneute Gewichtszunahme beobachtet (siehe Tabelle 5):
Tabelle 5. NN8022-4180 Änderung des Körpergewichts und des BMI in Woche 56 und Änderung des BMI-SDS von Woche 56 bis Woche 82
|
|
Saxenda (N=125)
|
Placebo (N=126)
|
Saxenda vs.
Placebo
| |
BMI-SDS
|
|
|
| |
Ausgangswert, BMI-SDS (SD)
|
3.14 (0.65)
|
3.20 (0.77)
|
| |
Mittlere Änderung in Woche 56 (95%-KI)
|
-0.23
|
-0.00
|
-0.22*
(-0.37; -0.08)
| |
Woche 56, BMI-SDS (SD)
|
2.88 (0.94)
|
3.14 (0.98)
|
| |
Mittlere Änderung von Woche 56 bis Woche 82, BMI-SDS (95%-KI)
|
0.22
|
0.07
|
0.15**
(0.07; 0.23)
| |
Körpergewicht
|
|
|
| |
Ausgangswert, kg (SD)
|
99.3 (19.7)
|
102.2 (21,6)
|
-
| |
Mittlere Änderung in Woche 56, % (95%-KI)
|
-2.65
|
2.37
|
-5.01**
(-7.63; -2.39)
| |
Mittlere Änderung in Woche 56, kg (95%-KI)
|
-2.,26
|
2.25
|
-4.50**
(-7.17; -1.84)
| |
BMI
|
|
|
| |
Ausgangswert, kg/m2 (SD)
|
35.3 (5.1)
|
35.8 (5.7)
|
-
| |
Mittlere Änderung in Woche 56, kg/m2 (95%-KI)
|
-1.39
|
0.19
|
-1.58**
(-2.47; -0.69)
| |
Anteil der Patienten mit einer Reduzierung des BMI-Ausgangswertes von ≥5% in Woche 56, % (95%-KI)
|
43.25
|
18.73
|
3.31**
(1.78; 6.16)
| |
Anteil der Patienten mit einer Reduzierung des BMI-Ausgangswertes von ≥10% in Woche 56, % (95%-KI)
|
26.08
|
8.11
|
4.00**
(1.81; 8.83)
| |
Gesamtgruppe (FAS=Full Analysis Set). Für BMI-SDS, Körpergewicht und BMI sind die Ausgangswerte Mittelwerte, die Änderungen gegenüber den Ausgangswerten in Woche 56 sind geschätzte Mittelwerte (LSMeans) und die Behandlungskontraste in Woche 56 sind geschätzte Behandlungsunterschiede. Die BMI-SDS-Werte in Woche 56 sind Mittelwerte, die Änderungen von Woche 56 bis Woche 82 sind geschätzte Mittelwerte (LSMeans) und die Behandlungskontraste in Woche 82 sind geschätzte Behandlungsunterschiede. Für den Anteil der Patienten, bei denen sich der BMI-Ausgangswert um ≥5%/≥10% reduziert hat, wird die geschätzte Odds Ratio angegeben. Fehlende Beobachtungen wurden mittels multipler Imputation (Jump-to-Reference-Ansatz, x100) basierend auf der Placebo-Gruppe vervollständigt.
*p<0.01, **p<0.001. KI=Konfidenzintervall. SD=Standardabweichung.
|
Basierend auf der Verträglichkeit wurde die Dosis bei 103 Patienten (82.4 %) bis auf 3.0 mg, bei 11 Patienten (8.8 %) bis auf 2.4 mg, bei 4 Patienten (3.2 %) bis auf 1.8 mg, bei 4 Patienten (3.2 %) bis auf 1.2 mg erhöht und 3 Patienten (2.4 %) blieben auf 0.6 mg.
Die Wirksamkeit und Sicherheit von Saxenda bei pädiatrischen Patienten mit Prader-Willi-Syndrom und Adipositas wurde in einer 16-wöchigen doppelblinden Studie (Teil A) mit anschliessender 36-wöchiger unverblindeter (open label) Phase untersucht. Im Teil A wurden insgesamt 32 Jugendliche im Alter von ≥12 bis <18 Jahren (Tanner-Stadium 2-5) randomisiert mit Liraglutid 3 mg (n=20) bzw. Placebo (n=12) behandelt, von denen 18 bzw. 12 die doppelblinde Phase bis Woche 16 sowie 17 bzw. 12 die unverblindete Extensionsphase bis Woche 52 abschlossen. Im Teil B wurden insgesamt 24 Kinder zwischen ≥6 bis <12 Jahren (Tanner-Stadium unter 2) randomisiert mit Liraglutid 3 mg (n=17) bzw. Placebo (n=7) behandelt, von denen 16 bzw. 7 die doppelblinde Phase bis Woche 16 sowie 14 bzw. 7 die unverblindete Extensionsphase bis Woche 52 abschlossen.
Patienten mit einem Körpergewicht von weniger als 45 kg starteten die Dosiseskalation mit einer niedrigeren Dosis von 0.3 mg anstelle von 0.6 mg und erhöhten die maximale Dosis auf 2.4 mg.
Die geschätzten Änderungen des mittleren BMI-SDS in den beiden Behandlungsarmen waren sowohl in Woche 16 (Differenz Liraglutid 3 mg vs. Placebo [95% KI]: -0,07 [-0,23, 0,09] im Teil A und -0,06 [-1.06, 0.93] im Teil B) als auch in Woche 52 (Differenz Liraglutid 3 mg vs. Placebo [95% KI]: -0,14 [-0.62, 0.34] im Teil A und -0.07 [-0.89, 0.76] im Teil B) vergleichbar.
|