ZusammensetzungWirkstoffe
Letermovir.
Hilfsstoffe
Filmtablette:
Mikrokristalline Cellulose, Croscarmellose-Natrium, Povidon 25, hochdisperses Siliciumdioxid, Magnesiumstearat und folgende Hilfsstoffe im Filmüberzug: Laktosemonohydrat, Hypromellose 2910, Titandioxid, Triacetin, Eisenoxid gelb, und (nur 480-mg-Filmtabletten) Eisenoxid rot; Carnaubawachs als Poliermittel.
Jede 240 mg Filmtablette enthält 4,0 mg Lactose (als Monohydrat) und bis zu 1,90 mg Natrium.
Jede 480 mg Filmtablette enthält 6,4 mg Lactose (als Monohydrat) und bis zu 3,80 mg Natrium.
Granulat im Beutel:
Mikrokristalline Cellulose, Croscarmellose-Natrium, Povidon K-29/32, hochdisperses Siliciumdioxid, Magnesiumstearat und folgende Hilfsstoffe im Filmüberzug: Laktosemonohydrat, Hypromellose 2910, Titandioxid, Triacetin, Eisenoxid gelb und Eisenoxid rot.
Jedes 20 mg Granulat im Beutel enthält 1,7 mg Lactose (als Monohydrat) und bis zu 0,23 mg Natrium.
Jedes 120 mg Granulat im Beutel enthält 9,9 mg Lactose (als Monohydrat) und bis zu 1,35 mg Natrium.
Konzentrat zur Herstellung einer Infusionslösung:
Hydroxypropylbetadex (150 mg/ml), Natriumchlorid (3,1 mg/ml), Natriumhydroxid (1,2 mg/ml), Aqua ad iniectabilia. Die Natriumhydroxidmenge kann angepasst werden, um einen pH-Wert von ca. 7,5 zu erreichen.
Jede 240 mg Durchstechflasche enthält 23 mg Natrium und 1800 mg Hydroxypropylbetadex (Cyclodextrin).
Jede 480 mg Durchstechflasche enthält 46 mg Natrium und 3600 mg Hydroxypropylbetadex (Cyclodextrin).
Indikationen/AnwendungsmöglichkeitenCMV-Prophylaxe bei Empfängern eines hämatopoetischen Stammzelltransplantats (HSZT)
Prevymis ist indiziert zur Prophylaxe von Cytomegalovirus-(CMV)-Infektionen oder -Erkrankungen bei Erwachsenen und pädiatrischen Patienten ab dem Alter von 2 Monaten und mit einem Körpergewicht von mindestens 5 kg, die CMV-seropositive Empfänger [R+] eines allogenen hämatopoetischen Stammzelltransplantats sind.
CMV-Prophylaxe bei Nierentransplantatempfängern
Prevymis ist indiziert zur Prophylaxe der CMV-Erkrankung bei Erwachsenen und Jugendlichen ab dem Alter von 12 Jahren und mit einem Körpergewicht von mindestens 40 kg, die Nierentransplantatempfänger mit hohem Risiko (CMV-seropositiver Spender/CMV-seronegativer Empfänger [D+/R-]) sind.
Dosierung/AnwendungDie Behandlung mit Prevymis sollte von einem Arzt eingeleitet werden, der über Erfahrung in der Behandlung von Patienten verfügt, die eine allogene hämatopoetische Stammzelltransplantation oder eine Nierentransplantation erhalten haben.
Dosierungshinweise
Prevymis ist in drei Darreichungsformen erhältlich:
Prevymis Filmtabletten
Die Tablette ist ganz zu schlucken und kann mit oder ohne Nahrung eingenommen werden. Die Tablette darf nicht geteilt, zerdrückt oder zerkaut werden.
Prevymis Granulat im Beutel
Prevymis Granulat wird oral, gemischt mit weicher Nahrung oder über eine nasogastrale Sonde (NG-Sonde) oder eine Gastrostomie-Sonde (G-Sonde) verabreicht (siehe Abschnitt «Sonstige Hinweise»). Das Granulat sollte nicht zerdrückt oder zerkaut werden.
Prevymis Konzentrat zur Herstellung einer Infusionslösung
Prevymis Konzentrat zur Herstellung einer Infusionslösung, das Hydroxypropylbetadex enthält, sollte nur bei Patienten angewendet werden, die keine orale Therapie einnehmen können. Patienten sollten auf orales Prevymis umgestellt werden, sobald sie in der Lage sind, orale Arzneimittel einzunehmen. Siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen» und Abschnitt «Pharmakokinetik» von Letermovir nach i.v. und oraler Verabreichung.
Prevymis Konzentrat zur Herstellung einer Infusionslösung muss vor der Verabreichung verdünnt werden (siehe Abschnitt «Sonstige Hinweise»). Nur als intravenöse (i.v.) Infusion durch einen sterilen 0,2 μm oder 0,22 μm Inline-Filter aus Polyethersulfon (PES) verabreichen. Nicht als intravenöse Schnellinfusion oder Bolus verabreichen. Verabreichen Sie Prevymis nach der Verdünnung durch intravenöse Infusion über einen peripheren oder zentralen Venenkatheter über einen Gesamtzeitraum von ca. 60 Minuten. Verabreichen Sie den gesamten Inhalt des Infusionsbeutels.
Prevymis Filmtabletten, Granulat im Beutel und Konzentrat zur Herstellung einer Infusionslösung können abwechselnd nach Ermessen des Arztes angewendet werden. Bei pädiatrischen HSZT-Empfängern mit einem Körpergewicht von unter 30 kg kann beim Wechsel zwischen einer oralen und der intravenösen Darreichungsform eine Dosisanpassung erforderlich sein (siehe Empfohlene Dosierung und Dosierungsanpassung bei pädiatrischen Patienten mit einem Körpergewicht von mindestens 5 kg bis unter 30 kg).
HSZT
Die Gabe von Prevymis ist nach der HSZT zu beginnen. Die Gabe von Prevymis kann am Tag der Transplantation und bis spätestens 28 Tage nach HSZT gestartet werden. Die Gabe von Prevymis kann vor oder nach dem Einwachsen der Stammzellen (Engraftment) begonnen werden. Die Behandlung mit Prevymis wird bis 100 Tage nach HSZT weitergeführt.
Zusätzlich gibt es klinische Daten über die Gabe von Prevymis bis 200 Tage nach HSZT bei erwachsenen Patienten mit einem Risiko für eine späte CMV-Infektion und -Erkrankung (siehe Abschnitte «Unerwünschte Wirkungen» und «Eigenschaften/Wirkungen»).
Nierentransplantation
Prevymis sollte am Tag der Transplantation und bis spätestens 7 Tage nach der Nierentransplantation begonnen und bis 200 Tage nach der Transplantation fortgesetzt werden.
Empfohlene Dosierung bei erwachsenen HSZT- oder Nierentransplantatempfängern und pädiatrischen HSZT-Empfängern mit einem Körpergewicht von mindestens 30 kg und jugendlichen Nierentransplantatempfängern mit einem Körpergewicht von mindestens 40 kg
Die empfohlene Dosierung von Prevymis bei Erwachsenen beträgt 480 mg einmal täglich, oral oder intravenös verabreicht.
Die empfohlene Dosierung von Prevymis bei pädiatrischen HSZT-Empfängern mit einem Körpergewicht von mindestens 30 kg und jugendlichen Nierentransplantatempfängern mit einem Körpergewicht von mindestens 40 kg beträgt 480 mg einmal täglich, oral oder intravenös verabreicht.
Wenn Prevymis oral verabreicht wird, beträgt die empfohlene Dosierung eine 480-mg-Tablette einmal täglich oder zwei 240-mg-Tabletten einmal täglich. Bei Patienten, die keine Tabletten schlucken können, können vier Beutel à 120 mg Granulat angewendet werden.
Dosierungsanpassung bei erwachsenen HSZT- oder Nierentransplantatempfängern und pädiatrischen HSZT-Empfängern mit einem Körpergewicht von mindestens 30 kg und jugendlichen Nierentransplantatempfängern mit einem Körpergewicht von mindestens 40 kg
Wenn Prevymis zusammen mit Ciclosporin verabreicht wird, ist die Dosierung von Prevymis auf 240 mg einmal täglich zu reduzieren (siehe Abschnitte «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»).
·Wird nach Beginn der Gabe von Prevymis eine Ciclosporin-Therapie eingeleitet, ist die nächste Dosis Prevymis auf 240 mg einmal täglich zu reduzieren.
·Wird Ciclosporin nach Beginn der Gabe von Prevymis abgesetzt, ist die nächste Dosis Prevymis auf 480 mg einmal täglich zu erhöhen.
·Wird die Ciclosporingabe aufgrund hoher Ciclosporinspiegel vorübergehend unterbrochen, ist keine Dosisanpassung von Prevymis erforderlich.
Empfohlene Dosierung und Dosierungsanpassung bei pädiatrischen HSZT-Empfängern mit einem Körpergewicht von mindestens 5 kg bis unter 30 kg
Die empfohlenen Dosierungen von Prevymis für pädiatrische HSZT-Empfänger mit einem Körpergewicht von mindestens 5 kg bis unter 30 kg sind in Tabelle 1 (Granulat im Beutel oder Tabletten) und Tabelle 2 (Konzentrat zur Herstellung einer Infusionslösung) aufgeführt (siehe auch Abschnitt «Pharmakokinetik»). Prevymis ist einmal täglich oral oder intravenös zu verabreichen.
Wenn Prevymis zusammen mit Ciclosporin verabreicht wird, muss die Dosierung von Prevymis möglicherweise wie in Tabelle 1 und Tabelle 2 aufgeführt angepasst werden (siehe Tabelle 3 im Abschnitt «Interaktionen»).
·Wird nach Beginn der Gabe von Prevymis eine Ciclosporin-Therapie eingeleitet, sollte die nächste Dosis Prevymis der täglichen oralen Dosis bei Kombination mit Ciclosporin (Tabelle 1) bzw. der täglichen intravenösen Dosis mit Ciclosporin (Tabelle 2) entsprechen.
·Wird Ciclosporin nach Beginn der Gabe von Prevymis abgesetzt, sollte die nächste Dosis Prevymis der täglichen oralen Dosis bei Verabreichung ohne Ciclosporin (Tabelle 1) bzw. der täglichen intravenösen Dosis ohne Ciclosporin (Tabelle 2) entsprechen.
·Wird die Ciclosporingabe aufgrund hoher Ciclosporinspiegel vorübergehend unterbrochen, ist keine Dosisanpassung von Prevymis erforderlich.
Tabelle 1: Empfohlene Dosierung von Prevymis Granulat im Beutel oder Filmtabletten bei Verabreichung ohne oder mit Ciclosporin bei pädiatrischen HSZT-Empfängern mit einem Körpergewicht von mindestens 5 kg bis unter 30 kg
|
Körpergewicht
|
Verabreichung ohne Ciclosporin
|
Verabreichung in Kombination mit Ciclosporin
| |
Tägliche orale Dosis
|
Anzahl Prevymis-Beutel oder -Tabletten einmal täglich
|
Tägliche orale Dosis
|
Anzahl Prevymis-Beutel oder -Tabletten einmal täglich
| |
15 kg bis unter 30 kg
|
240 mg
|
Zwei 120-mg-Beutel
oder
Eine 240-mg-Tablette
|
120 mg
|
Ein 120-mg-Beutel
| |
7,5 kg bis unter 15 kg
|
120 mg
|
Ein 120-mg-Beutel
|
60 mg
|
Drei 20-mg-Beutel
| |
5 kg bis unter 7,5 kg
|
80 mg
|
Vier 20-mg-Beutel
|
40 mg
|
Zwei 20-mg-Beutel
|
Tabelle 2: Empfohlene Dosierung von Prevymis Konzentrat zur Herstellung einer Infusionslösung ohne oder mit Ciclosporin bei pädiatrischen HSZT-Empfängern mit einem Körpergewicht von mindestens 5 kg bis unter 30 kg
|
Körpergewicht
|
Tägliche intravenöse Dosis ohne oder mit Ciclosporin
| |
15 kg bis unter 30 kg
|
120 mg
| |
7,5 kg bis unter 15 kg
|
60 mg
| |
5 kg bis unter 7,5 kg
|
40 mg
|
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei leichter (Child-Pugh-Klasse A) bis mässiger (Child-Pugh-Klasse B) Einschränkung der Leberfunktion ist keine Dosisanpassung von Prevymis erforderlich. Prevymis wird bei Patienten mit schwerer Einschränkung der Leberfunktion (Child-Pugh-Klasse C) nicht empfohlen.
Prevymis wird bei Patienten mit mässiger Einschränkung der Leberfunktion in Kombination mit mässiger oder schwerer Einschränkung der Nierenfunktion nicht empfohlen (siehe Abschnitt «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit einer Kreatininclearance >10 ml/min ist keine Dosisanpassung von Prevymis erforderlich (siehe Abschnitt «Pharmakokinetik»).
Es liegen keine Daten bei Patienten mit Nierenerkrankung im Endstadium (KrCl unter 10 ml/min) oder Dialysepatienten vor, so dass keine Dosierungsempfehlungen möglich sind.
Prevymis Konzentrat zur Herstellung einer Infusionslösung enthält Hydroxypropylbetadex. Die erwartete klinische Exposition gegenüber Hydroxypropylbetadex bei intravenös verabreichtem Letermovir beträgt ca. 3600 mg/Tag bei einer Letermovirdosis von 480 mg (siehe auch Abschnitt «Warnhinweise und Vorsichtsmassnahmen», Hilfsstoffe, Cyclodextrin). Bei Patienten mit einer eingeschränkten Nierenfunktion (Kreatininclearance <50 ml/min) oder bei kleinen Kindern (weniger als 2 Jahre alt), die Prevymis Infusionslösung erhalten, könnte eine Akkumulation von Hydroxypropylbetadex auftreten. Bei diesen Patienten ist eine engmaschige Überprüfung der Serumkreatininspiegel erforderlich (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Ältere Patienten
Von den 373 in einer randomisierten, doppelblinden, placebokontrollierten Phase-3-Studie mit Prevymis behandelten Probanden (P001) waren 56 (15,0%) 65 Jahre und älter. Von den 144 Probanden, die in einer randomisierten, doppelblinden, placebokontrollierten Phase-3-Studie (P040) mit Prevymis behandelt wurden, waren 32 (22,2%) 65 Jahre oder älter. Von den 292 Probanden, die in einer randomisierten, doppelblinden, aktiv kontrollierten Phase-3-Studie (P002) mit Prevymis behandelt wurden, waren 48 (16,4%) 65 Jahre oder älter. Die Sicherheit und Wirksamkeit waren bei älteren und jüngeren Probanden in jeder Studie ähnlich. Eine altersentsprechende Dosisanpassung von Prevymis ist nicht erforderlich.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Prevymis bei pädiatrischen HSZT-Empfängern mit einem Körpergewicht von unter 5 kg und bei pädiatrischen Nierentransplantatempfängern mit einem Körpergewicht von unter 40 kg sind nicht erwiesen. Es liegen keine Daten vor (siehe Abschnitt «Pharmakokinetik»). Prevymis wurde bei pädiatrischen Nierentransplantatempfängern nicht untersucht.
Verspätete Dosisgabe
Weisen Sie die Patienten an, die Einnahme der vergessenen Dosis Prevymis nachzuholen, sobald sie sich daran erinnern. Falls der nächste Einnahmezeitpunkt unmittelbar bevorsteht, sollen die Patienten die Einnahme nicht nachholen, sondern mit dem gewohnten Einnahmeschema fortfahren. Weisen Sie die Patienten an, ihre nächste Dosis nicht zu verdoppeln und nicht mehr als die verordnete Dosis einzunehmen.
KontraindikationenPrevymis ist bei Patienten mit Überempfindlichkeit gegenüber Letermovir oder einem der Hilfsstoffe kontraindiziert.
Pimozid
Die gleichzeitige Gabe von Prevymis mit Pimozid kann wegen der Hemmung von Cytochrom P450 (CYP3A) durch Letermovir zu erhöhten Pimozidkonzentrationen führen, die QT-Verlängerung und Torsades de pointes zur Folge haben (siehe Abschnitte «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»).
Ergotalkaloide
Die gleichzeitige Gabe von Prevymis mit Ergotalkaloiden kann wegen der Hemmung von CYP3A durch Letermovir zu erhöhten Konzentrationen von Ergotalkaloiden (Ergotamin und Dihydroergotamin) führen, was Ergotismus zur Folge haben kann (siehe Abschnitte «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»).
Johanniskraut (Hypericum perforatum)
Die gemeinsame Anwendung mit Johanniskraut (Hypericum perforatum) ist kontraindiziert (siehe Abschnitt «Interaktionen»).
Kombination von Letermovir mit Ciclosporin
Die gemeinsame Anwendung mit Dabigatran, Atorvastatin, Simvastatin, Rosuvastatin oder Pitavastatin ist kontraindiziert (siehe Abschnitte «Warnhinweise und Vorsichtsmassnahmen» sowie «Interaktionen»).
Warnhinweise und VorsichtsmassnahmenÜberwachung der CMV-DNA bei HSZT-Empfängern
In einer Phase-3-Studie (P001), wurden bei HSZT-Empfängern mit einem negativen CMV-DNA-Testergebnis die Sicherheit und Wirksamkeit von Letermovir vor Einleitung der Prophylaxe nachgewiesen. Die CMV-DNA wurde wöchentlich bis Woche 14 nach Transplantation kontrolliert und anschliessend alle 2 Wochen bis Woche 24. Im Falle einer klinisch signifikanten CMV-DNA-ämie oder einer CMV-Erkrankung wurde die Prophylaxe mit Letermovir beendet und eine Standardversorgung mit einer präemptiven Therapie (PET) oder eine Behandlung eingeleitet. Bei den Patienten, bei denen eine Letermovir-Prophylaxe eingeleitet wurde und deren CMV-DNA-Test anschliessend positiv war, konnte die Prophylaxe fortgesetzt werden, wenn die Kriterien für eine PET nicht erfüllt waren.
Risiko im Zusammenhang mit dem Hilfsstoff Hydroxypropylbetadex in der intravenösen Formulierung
Bei Patienten mit einer eingeschränkten Nierenfunktion (Kreatininclearance <50 ml/min) oder bei kleinen Kindern (weniger als 2 Jahre alt), die Prevymis Infusionslösung erhalten, könnte eine Akkumulation von Hydroxypropylbetadex auftreten. Bei diesen Patienten ist eine engmaschige Überprüfung der Serumkreatininspiegel erforderlich (siehe Abschnitt «Dosierung/Anwendung»).
Risiko von unerwünschten Reaktionen oder einer Verminderung der therapeutischen Wirkung aufgrund von Arzneimittelinteraktionen
Die gleichzeitige Anwendung von Prevymis und bestimmten Arzneimitteln kann zu bekannten oder potenziell signifikanten Arzneimittelinteraktionen führen. Mögliche Folgen können sein:
·Klinisch signifikante unerwünschte Reaktionen durch erhöhte Exposition gegenüber Begleitmedikamenten oder Prevymis.
·Signifikante Verringerung der Plasmakonzentration von Begleitmedikamenten, die zu einer Verminderung der therapeutischen Wirkung des Begleitmedikaments führen kann.
Massnahmen zur Vermeidung bzw. Beherrschung dieser bekannten oder potenziell signifikanten Arzneimittelinteraktionen einschliesslich Dosierungsempfehlungen sind Tabelle 1 zu entnehmen (siehe Abschnitte «Kontraindikationen» und «Interaktionen»).
Prevymis sollte mit Vorsicht zusammen mit Arzneimittel, die CYP3A-Substrate mit enger therapeutischer Breite sind (z.B. Alfentanil, Fentanyl und Chinidin) verwendet werden, da die gleichzeitige Gabe zu Erhöhungen der Plasmakonzentrationen von CYP3A-Substraten führen kann. Eine engmaschige Überwachung und/oder Dosisanpassung gleichzeitig verabreichter CYP3A-Substrate wird empfohlen. Die Angaben in der Fachinformation der betroffenen CYP3A-Substrate sind zu beachten (siehe Tabelle 1 und Abschnitt «Interaktionen»).
Generell wird eine engmaschigere Überwachung der Konzentrationen von Ciclosporin, Tacrolimus und Sirolimus in den ersten beiden Wochen nach Beginn und nach Beendigung der Anwendung von Letermovir sowie auch nach Änderung der Anwendungsart empfohlen (siehe «Interaktionen»).
Letermovir ist ein moderater Induktor von Enzymen und Transportern. Diese Induktion kann die Plasmakonzentrationen von einigen metabolisierten und transportierten Arzneimitteln senken (siehe «Interaktionen»).
Ein therapeutisches Monitoring (therapeutic drug monitoring, TDM) wird daher für Voriconazol empfohlen. Die gemeinsame Anwendung von Dabigatran ist wegen des Risikos einer verminderten Wirksamkeit von Dabigatran zu vermeiden.
Letermovir kann die Plasmakonzentrationen von Arzneimitteln erhöhen, die von OATP1B1/3 transportiert werden, wie beispielsweise von vielen Statinen (siehe «Interaktionen»).
Hilfsstoffe
Laktose
Die Filmtabletten und das Granulat im Beutel enthalten Laktosemonohydrat. Patienten mit der seltenen hereditären Galaktose-Intoleranz, völligem Laktase-Mangel oder Glukose-Galaktose-Malabsorption sollten Prevymis Filmtabletten oder Granulat im Beutel nicht anwenden.
Natrium
Prevymis 240 mg Konzentrat zur Herstellung einer Infusionslösung enthält 23 mg (oder 1,0 mmol) Natrium je Durchstechflasche, entsprechend 1,15% der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
Prevymis 480 mg Konzentrat zur Herstellung einer Infusionslösung enthält 46 mg (oder 2,0 mmol) Natrium je Durchstechflasche, entsprechend 2,30% der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
Die Filmtabletten und das Granulat im Beutel enthalten weniger als 1 mmol Natrium (23 mg) pro Tablette oder Beutel, d.h. sie sind nahezu «natriumfrei».
Cyclodextrin
Konzentrat zur Herstellung einer Infusionslösung:
Dieses Arzneimittel enthält 300 mg Hydroxypropylbetadex (Cyclodextrin) pro 40-mg-Dosis.
Dieses Arzneimittel enthält 450 mg Hydroxypropylbetadex (Cyclodextrin) pro 60-mg-Dosis.
Dieses Arzneimittel enthält 900 mg Hydroxypropylbetadex (Cyclodextrin) pro 120-mg-Dosis.
Dieses Arzneimittel enthält 1800 mg Hydroxypropylbetadex (Cyclodextrin) pro 240-mg-Dosis.
Dieses Arzneimittel enthält 3600 mg Hydroxypropylbetadex (Cyclodextrin) pro 480-mg-Dosis.
InteraktionenAllgemeine Informationen über Unterschiede in der Exposition zwischen den verschiedenen Therapieregimen mit Letermovir
·Die geschätzte Letermovir-Plasmaexposition ist unterschiedlich, abhängig vom gewählten Dosierungsregime. Daher sind die klinischen Auswirkungen der Wechselwirkungen abhängig vom gewählten Letermovir-Therapieregime und davon, ob Letermovir mit Ciclosporin kombiniert wird oder nicht.
·Die Kombination von Ciclosporin und Letermovir kann zu stärker ausgeprägten oder zusätzlichen Wirkungen auf gleichzeitig angewendete Arzneimittel führen im Vergleich zur alleinigen Gabe von Letermovir.
Wirkung anderer Arzneimittel auf Letermovir
Die Elimination von Letermovir erfolgt in vivo über biliäre Ausscheidung und Glucuronidierung. Die relative Bedeutung dieser Eliminationswege ist nicht bekannt. Bei beiden Eliminationswegen erfolgt die aktive Aufnahme in Hepatozyten über den hepatischen Aufnahmetransporter OATP1B1/3. Nach der Aufnahme wird die Glucuronidierung von Letermovir über UGT1A1 und 3 vermittelt. Ausserdem scheint wohl Letermovir dem P-gp- (P-Glykoprotein) und BCRP- (Brustkrebs-Resistenz-Protein, breast cancer resistance protein) vermittelten Efflux in Leber und Darm zu unterliegen.
Induktoren von metabolisierenden Enzymen oder Transportern
Die gemeinsame Anwendung von Prevymis (mit oder ohne Ciclosporin) mit starken und moderaten Induktoren von Transportern (z.B. P-gp) und/oder Enzymen (z.B. UGTs) wird nicht empfohlen, da dies zu einem subtherapeutischen Plasmaspiegel von Letermovir führen kann.
·Beispiele für starke Induktoren sind Rifampicin, Phenytoin, Carbamazepin, Johanniskraut (Hypericum perforatum), Rifabutin und Phenobarbital.
·Beispiele für moderate Induktoren sind Thioridazin, Modafinil, Ritonavir, Lopinavir, Efavirenz und Etravirin.
Die gemeinsame Anwendung mit Rifampicin führte zu einem initialen, klinisch nicht relevanten Anstieg der Plasmakonzentrationen von Letermovir (aufgrund von OATP1B1/3- und/oder P-gp-Inhibition), gefolgt von einer klinisch relevanten Abnahme der Plasmakonzentrationen von Letermovir (aufgrund der Induktion von P-gp/UGT) bei kontinuierlicher Anwendung von Rifampicin.
Zusätzliche Wirkungen anderer Arzneimittel auf Letermovir, die bei einer Kombination mit Ciclosporin relevant sind
Inhibitoren von OATP1B1 oder 3
Die gemeinsame Anwendung von Prevymis mit Inhibitoren der OATP1B1/3-Transporter kann zu erhöhten Plasmakonzentrationen von Letermovir führen. Bei gemeinsamer Anwendung von Prevymis und Ciclosporin (einem potenten OATP1B1/3-Inhibitor) wird bei erwachsenen HSZT- und Nierentransplantatempfängern, pädiatrischen HSZT-Empfängern mit einem Körpergewicht von mindestens 30 kg und jugendlichen Nierentransplantatempfängern mit einem Körpergewicht von mindestens 40 kg eine einmal tägliche Gabe von 240 mg Prevymis empfohlen. Wenn Prevymis bei pädiatrischen HSZT-Empfängern mit einem Körpergewicht von unter 30 kg gemeinsam mit Ciclosporin verabreicht wird, ist möglicherweise eine Dosisanpassung erforderlich (siehe Abschnitte «Dosierung/Anwendung» und «Pharmakokinetik»). Vorsicht ist geboten bei der gemeinsamen Anwendung von anderen OATP1B1/3-Inhibitoren mit Letermovir und Ciclosporin.
·Beispiele für OATP1B1-Inhibitoren sind Gemfibrozil, Erythromycin, Clarithromycin sowie einige Proteaseinhibitoren (Atazanavir, Simeprevir).
Inhibitoren von P-gp/BCRP
In-vitro-Ergebnisse zeigen, dass Letermovir ein P-gp-/BCRP-Substrat ist. Die Änderungen der Letermovir-Plasmakonzentrationen aufgrund einer Inhibition von P-gp/BCRP durch Itraconazol waren in einer Interaktionsstudie klinisch nicht relevant.
Wirkung von Letermovir auf andere Arzneimittel
Arzneimittel, die hauptsächlich über den Stoffwechsel ausgeschieden oder durch aktiven Transport beeinflusst werden
Letermovir ist in aller Regel in vivo ein Induktor von Enzymen und Transportern. Im Allgemeinen kann eine Induktion erwartet werden, ausser es wird gleichzeitig ein bestimmtes Enzym oder Transporter inhibiert. Daher kann Letermovir potenziell zu niedrigeren Plasmakonzentrationen und möglicherweise verminderter Wirksamkeit von gemeinsam verabreichten Arzneimitteln führen, die hauptsächlich über den Metabolismus oder durch aktiven Transport eliminiert werden.
Das Ausmass des induzierenden Effekts hängt von der Art der Anwendung von Letermovir ab und davon, ob Letermovir gemeinsam mit Ciclosporin angewendet wird.
Der volle induzierende Effekt kann 10 bis 14 Tage nach Beginn der Anwendung von Letermovir erwartet werden. Die Zeit, die ein spezifisch betroffenes Arzneimittel jeweils braucht, den Steady-State zu erreichen, wird ebenfalls die Zeit beeinflussen, bis die volle Wirkung auf die Plasmakonzentrationen erreicht ist.
In vitro ist Letermovir ein Inhibitor von CYP3A, CYP2C8, CYP2B6, BCRP, UGT1A1, OATP2B1 und OAT3 bei in vivo relevanten Konzentrationen. In-vivo-Studien zur Untersuchung des Nettoeffektes auf CYP3A4, P-gp, OATP1B1/3 sowie auf CYP2C19 stehen zur Verfügung. Der Nettoeffekt in vivo auf die anderen genannten Enzyme und Transporter ist nicht bekannt. Detaillierte Informationen werden nachfolgend aufgeführt.
Es ist nicht bekannt, ob Letermovir die Exposition von Piperacillin/Tazobactam, Amphotericin B und Micafungin beeinflusst. Mögliche Wechselwirkungen zwischen Letermovir und diesen Arzneimitteln wurden nicht untersucht. Aufgrund der Induktion besteht ein theoretisches Risiko für eine Senkung der Plasmaspiegel, deren Ausmass und klinische Bedeutung jedoch gegenwärtig nicht bekannt sind.
Über CYP3A metabolisierte Arzneimittel
In vivo ist Letermovir ein moderater Inhibitor von CYP3A. Die gemeinsame Anwendung von Prevymis mit oralem Midazolam (ein CYP3A-Substrat) führt zu 2- bis 3-fach erhöhten Plasmakonzentrationen von Midazolam. Die Anwendung von Prevymis kann zu klinisch relevanten Erhöhungen der Plasmakonzentrationen von gemeinsam angewendeten Substraten von CYP3A führen. Siehe «Kontraindikationen» sowie «Warnhinweise und Vorsichtsmassnahmen».
·Beispiele für solche Arzneimittel umfassen bestimmte Immunsuppressiva (z.B. Ciclosporin, Tacrolimus, Sirolimus), HMG-CoA-Reduktase-Hemmer und Amiodaron. Pimozid und Mutterkornalkaloide sind kontraindiziert (siehe «Kontraindikationen»).
Das Ausmass des durch CYP3A verursachten inhibitorischen Effekts hängt von der Art der Anwendung von Letermovir ab und davon, ob Letermovir gemeinsam mit Ciclosporin angewendet wird.
Aufgrund der zeitabhängigen Inhibition und der gleichzeitigen Induktion wird der Nettoeffekt der Enzymhemmung möglicherweise erst nach 10 bis 14 Tagen erreicht. Die Zeitdauer, die ein spezifisch betroffenes Arzneimittel braucht, den Steady-State zu erreichen, wird ebenfalls die Zeit beeinflussen, bis die volle Wirkung auf die Plasmakonzentrationen erreicht ist. Nach dem Behandlungsende dauert es 10 bis 14 Tage, bis der inhibitorische Effekt verschwunden ist. Bei einer Überwachung wird empfohlen, diese in den ersten beiden Wochen nach Beginn und Absetzen der Anwendung von Letermovir durchzuführen (siehe «Warnhinweise und Vorsichtsmassnahmen») sowie bei Änderung der Anwendungsart von Letermovir.
Von OATP1B1/3 transportierte Arzneimittel
Letermovir ist ein Inhibitor von OATP1B1/3-Transportern. Die Anwendung von Prevymis kann zu einem klinisch relevanten Anstieg der Plasmakonzentrationen von gemeinsam angewendeten Substraten von OATP1B1/3 führen.
·Beispiele für solche Arzneimittel umfassen HMG-CoA-Reduktase-Hemmer, Fexofenadin, Repaglinid und Glibenclamid. Bei Vergleich der Letermovir-Regime ohne Ciclosporin ist die Wirkung nach i.v.-Gabe ausgeprägter als nach oraler Gabe von Letermovir.
Das Ausmass der OATP1B1/3-Inhibition auf gemeinsam angewendete Arzneimittel ist vermutlich grösser, wenn Prevymis zusammen mit Ciclosporin (einem potenten OATP1B1/3-Inhibitor) angewendet wird. Dies ist zu beachten, wenn das Letermovir-Regime während der Behandlung mit einem OATP1B1/3-Substrat geändert wird.
Über CYP2C9 und/oder CYP2C19 metabolisierte Arzneimittel
Die gemeinsame Anwendung von Prevymis mit Voriconazol (ein CYP2C19-Substrat) führt zu einer signifikant verminderten Voriconazol-Plasmakonzentration, was darauf hindeutet, dass Letermovir CYP2C19 induziert.
Auch CYP2C9 wird vermutlich induziert. Letermovir kann die Exposition von CYP2C9- und/oder CYP2C19-Substraten verringern, so dass möglicherweise subtherapeutische Konzentrationen erreicht werden. Zu diesen Arzneimitteln zählen u.a. Warfarin, Voriconazol, Diazepam, Lansoprazol, Omeprazol, Esomeprazol, Pantoprazol, Tilidin und Tolbutamid.
Es wird davon ausgegangen, dass die Wirkung bei oraler Anwendung von Letermovir ohne Ciclosporin nicht so ausgeprägt ist, wie unter i.v.-Anwendung von Letermovir mit oder ohne Ciclosporin oder oraler Anwendung von Letermovir mit Ciclosporin. Dies ist zu beachten, wenn das Letermovir-Regime während der Behandlung mit einem CYP2C9- oder CYP2C19-Substrat geändert wird. Bitte beachten Sie bezüglich des zeitlichen Verlaufs der Interaktion auch die obenstehenden allgemeinen Hinweise zur Induktion.
Über CYP2C8 metabolisierte Arzneimittel
In vitro hemmt Letermovir CYP2C8, kann aber aufgrund seines Induktionspotenzials CYP2C8 auch induzieren. Der Nettoeffekt in vivo ist nicht bekannt.
Zu den Arzneimitteln, die hauptsächlich über CYP2C8 eliminiert werden, zählt Repaglinid (siehe Tabelle 1). Die gemeinsame Anwendung von Repaglinid und Letermovir mit oder ohne Ciclosporin wird nicht empfohlen.
Arzneimittel, die intestinal über P-gp transportiert werden
Letermovir ist ein Induktor intestinalen P-gps. Die Anwendung von Prevymis kann die Plasmakonzentrationen gemeinsam angewendeter Arzneimittel, für die der intestinale P-gp-Transport eine wichtige Rolle spielt, wie z.B. bei Dabigatran und Sofosbuvir, in klinisch bedeutsamem Ausmass verringern.
Arzneimittel, die über CYP2B6 und UGT1A1 metabolisiert oder von BCRP oder OATP2B1 transportiert werden
Letermovir ist in vivo allgemein ein Induktor, aber wurde auch als Inhibitor von CYP2B6, UGT1A1, BCRP und OATP2B1 in vitro beobachtet. Der Nettoeffekt in vivo ist nicht bekannt. Daher können die Plasmakonzentrationen von Arzneimitteln, die Substrate dieser Enzyme oder Transporter sind, bei Kombination mit Letermovir ansteigen oder sinken. Eine zusätzliche Überwachung kann empfehlenswert sein; beachten Sie dazu die Fachinformationen solcher Arzneimittel.
·Ein Beispiel für CYP2B6-metabolisierte Arzneimittel ist Bupropion.
·Beispiele für UGT1A1-metabolisierte Arzneimittel sind Raltegravir und Dolutegravir.
·Beispiele für BCRP-transportierte Arzneimittel sind Rosuvastatin und Sulfasalazin.
·Ein Beispiel für OATP2B1-transportierte Arzneimittel ist Celiprolol.
Arzneimittel, die über den renalen Transporter OAT3 transportiert werden
In-vitro-Daten legen nahe, dass Letermovir ein Inhibitor von OAT3 ist; daher kann Letermovir auch in vivo ein OAT3-Inhibitor sein. Die Plasmaspiegel von Arzneimitteln, die über OAT3 transportiert werden, können ansteigen.
·Beispiele für OAT3-transportierte Arzneimittel sind Ciprofloxazin, Tenofovir, Imipenem und Cilastatin.
Allgemeine Informationen
Falls aufgrund einer Behandlung mit Prevymis Dosisanpassungen von gleichzeitig angewendeten Arzneimitteln durchgeführt werden, sollten nach Beendigung der Behandlung mit Prevymis die Dosen wieder neu angepasst werden. Eine Dosisanpassung kann auch bei einem Wechsel der Art der Anwendung oder des Immunsuppressivums erforderlich sein. Siehe «Kontraindikationen» sowie «Warnhinweise und Vorsichtsmassnahmen».
Kinder und Jugendliche
Studien zur Erfassung von Wechselwirkungen wurden nur bei Erwachsenen durchgeführt.
Bei gesunden erwachsenen Probanden wurden Studien zu Arzneimittelinteraktionen mit Prevymis und Arzneimitteln, die wahrscheinlich gleichzeitig verabreicht werden, oder Arzneimitteln, die häufig als Prüfmedikamente für pharmakokinetische Interaktionen verwendet werden, durchgeführt (siehe Tabelle 3).
Tabelle 3 enthält eine Liste der erwiesenermassen oder potenziell klinisch signifikanten Arzneimittelinteraktionen. Die beschriebenen Arzneimittelinteraktionen beruhen auf Studien zu Prevymis bei Erwachsenen oder es handelt sich um vorhergesagte Arzneimittelinteraktionen, die mit Prevymis auftreten können (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen» und «Kontraindikationen»).
Tabelle 3: Interaktionen mit anderen Arzneimitteln und Dosierungsempfehlungen: Eine Dosisanpassung kann aufgrund der Ergebnisse von Arzneimittel-Interaktionsstudien bei Erwachsenen oder vorhergesagter Interaktionen empfohlen werden*
|
Klasse und/oder Ausscheidungsweg der begleitend verabreichten Arzneimittel: Name des Wirkstoffs
|
Effekt auf die Konzentration†
Mittelwerte (90% Konfidenzintervall) für AUC, Cmax (wahrscheinlicher Wirkungsmechanismus)
|
Empfehlungen zur gleichzeitigen Anwendung mit Prevymis
| |
Antiarrhythmika
| |
Amiodaron
|
Interaktion nicht untersucht.
Erwartet:
↑ Amiodaron
(hauptsächlich CYP3A-Inhibition und CYP2C8-Inhibition oder -Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Amiodaron erhöht die Plasmakonzentration von Amiodaron. Eine enge klinische Überwachung auf unerwünschte Ereignisse im Zusammenhang mit Amiodaron wird während der gleichzeitigen Verabreichung empfohlen. Überwachen Sie häufig die Amiodaron-Konzentrationen.
| |
Chinidin
|
Interaktion nicht untersucht.
Erwartet:
↑ Chinidin
(CYP3A-Inhibition)
|
Letermovir kann die Plasmakonzentration von Chinidin erhöhen. Eine engmaschige klinische Überwachung sollte während der Anwendung von Prevymis mit Chinidin erfolgen. Beachten Sie dazu die entsprechende Fachinformation (siehe «Warnhinweise und Vorsichtsmassnahmen»).
| |
Antibiotika
| |
Nafcillin
|
Interaktion nicht untersucht.
Erwartet:
↓ Letermovir
(P-gp-/UGT-Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Nafcillin kann die Plasmakonzentration von Letermovir verringern.
Die gleichzeitige Verabreichung von Prevymis und Nafcillin wird nicht empfohlen.
| |
Antikoagulanzien
| |
Warfarin
|
Interaktion nicht untersucht.
Erwartet:
↓ Warfarin
(CYP2C9-Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Warfarin kann die Plasmakonzentration von Warfarin verringern.
Bei gleichzeitiger Anwendung von Warfarin mit Prevymis sollte eine häufige Überwachung der INR erfolgen#.
Die Überwachung wird während der ersten beiden Wochen nach Beginn oder Beendigung der Anwendung mit Letermovir, sowie bei Änderung der Anwendungsart von Letermovir oder des Immunsuppressivums empfohlen.
| |
Dabigatran
|
Interaktion nicht untersucht.
Erwartet:
↓ Dabigatran
(intestinale P-gp-Induktion)
|
Letermovir kann die Plasmakonzentration von Dabigatran reduzieren und seine Wirksamkeit vermindern. Aufgrund des Risikos einer verminderten Wirksamkeit von Dabigatran ist die gemeinsame Anwendung zu vermeiden. Bei Kombination von Prevymis mit Ciclosporin ist Dabigatran kontraindiziert.
| |
Antikonvulsiva
| |
Carbamazepin
|
Interaktion nicht untersucht.
Erwartet:
↓ Letermovir
(P-gp-/UGT-Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Carbamazepin kann die Plasmakonzentration von Letermovir verringern.
Die gleichzeitige Verabreichung von Prevymis mit Carbamazepin wird nicht empfohlen.
| |
Phenobarbital
|
Interaktion nicht untersucht.
Erwartet:
↓ Letermovir
(P-gp-/UGT-Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Phenobarbital kann die Plasmakonzentration von Letermovir verringern. Die gleichzeitige Verabreichung von Prevymis mit Phenobarbital wird nicht empfohlen.
| |
Phenytoin
|
Interaktion nicht untersucht.
Erwartet:
↓ Letermovir
(P-gp-/UGT-Induktion)
↓ Phenytoin
(CYP2C9/19-Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Phenytoin kann die Plasmakonzentration von Letermovir verringern. Prevymis kann die Plasmakonzentration von Phenytoin verringern.
Die gleichzeitige Verabreichung von Prevymis mit Phenytoin wird nicht empfohlen.
| |
Antidiabetika
| |
Glibenclamid
|
Interaktion nicht untersucht.
Erwartet:
↑ Glibenclamid
(OATP1B1/3-Inhibition, CYP3A-Inhibition, CYP2C9-Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Glibenclamid kann die Plasmakonzentration von Glibenclamid erhöhen. Eine häufige Überwachung der Glukosekonzentrationen wird empfohlen#.
Bei gemeinsamer Anwendung von Prevymis mit Ciclosporin ist auch die Fachinformation von Glibenclamid zu spezifischen Dosierungsempfehlungen zu beachten.
| |
Repaglinid
|
Interaktion nicht untersucht.
Erwartet:
↑ oder ↓ Repaglinid
(CYP2C8-Induktion, CYP2C8- und OATP1B-Inhibition)
|
Letermovir kann die Plasmakonzentration von Repaglinid erhöhen oder reduzieren. (Der Nettoeffekt ist nicht bekannt).
Eine gemeinsame Anwendung wird nicht empfohlen.
Falls Prevymis auch gemeinsam mit Ciclosporin angewendet wird, ist aufgrund der zusätzlichen OATP1B-Inhibition durch Ciclosporin zu erwarten, dass die Plasmakonzentrationen von Repaglinid ansteigen. Eine gemeinsame Anwendung wird nicht empfohlen#.
| |
Antimykotika
| |
Fluconazol‡
(400 mg Einzeldosis PO)/ Letermovir (480 mg Einzeldosis PO)
|
↔ Letermovir
AUC 1,11 (1,01; 1,23)
Cmax 1,06 (0,93; 1,21)
↔ Fluconazol
AUC 1,03 (0,99; 1,08)
Cmax 0,95 (0,92; 0,99)
Interaktion im Steady-State nicht untersucht.
Erwartet:
↔ Fluconazol
↔ Letermovir
|
Keine Dosisanpassung erforderlich.
| |
Posaconazol‡
(300 mg Einzeldosis)/ Letermovir (480 mg täglich)
|
↔ Posaconazol
AUC 0,98 (0,82; 1,17)
Cmax 1,11 (0,95; 1,29)
|
Keine Dosisanpassung erforderlich.
| |
Voriconazol‡
(200 mg zweimal täglich/ Letermovir (480 mg täglich)
|
↓ Voriconazol
AUC 0,56 (0,51; 0,62)
Cmax 0,61 (0,53; 0,71)
(CYP2C9/19-Induktion)
|
Wenn die gleichzeitige Verabreichung erforderlich ist, wird eine engmaschige Überwachung auf verminderte Wirksamkeit und ein therapeutisches Arzneimittel-Monitoring (TDM) von Voriconazol empfohlen (in den ersten beiden Wochen nach Beginn oder Beendigung der Anwendung von Letermovir sowie bei Änderung der Anwendungsart von Letermovir oder des Immunsuppressivums)#.
| |
Itraconazol‡
(200 mg einmal täglich PO)/ Letermovir (480 mg einmal täglich PO)
|
↔ Letermovir
AUC 1,33 (1,17; 1,51)
Cmax 1,21 (1,05; 1,39)
C24 1,90 (1,58; 2,28)
↔ Itraconazol
AUC 0,76 (0,71; 0,81)
Cmax 0,84 (0,76; 0,92)
C24 0,67 (0,61; 0,73)
|
Keine Dosisanpassung erforderlich.
| |
Antimykobakterielle Medikamente
| |
Rifabutin
|
Interaktion nicht untersucht.
Erwartet:
↓ Letermovir
(P-gp-/UGT-Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Rifabutin kann die Plasmakonzentration von Letermovir verringern.
Die gleichzeitige Verabreichung von Prevymis mit Rifabutin wird nicht empfohlen.
| |
Rifampicin‡
| |
(600 mg Einzeldosis PO)/ Letermovir (480 mg Einzeldosis PO)
|
↔ Letermovir
AUC 2,03 (1,84; 2,26)
Cmax 1,59 (1,46; 1,74)
C24 2,01 (1,59; 2,54)
(OATP1B1/3-Inhibition und/oder P-gp-Inhibition)
|
Mehrfache Verabreichung von Rifampicin verringert die Plasmakonzentration von Letermovir.
Die gleichzeitige Verabreichung von Prevymis und Rifampicin wird nicht empfohlen.
| |
(600 mg Einzeldosis IV)/ Letermovir (480 mg Einzeldosis PO)
|
↔ Letermovir
AUC 1,58 (1,38; 1,81)
Cmax 1,37 (1,16; 1,61)
C24 0,78 (0,65; 0,93)
(OATP1B1/3- und/oder P-gp-Inhibition)
| |
(600 mg einmal täglich PO)¶/ Letermovir (480 mg einmal täglich PO)
|
↓ Letermovir
AUC 0,81 (0,67; 0,98)
Cmax 1,01 (0,79; 1,28)
C24 0,14 (0,11; 0,19)
(Summe der OATP1B1/3- und/oder P-gp-Inhibition und P-gp-/UGT-Induktion)
| |
(600 mg einmal täglich PO (24 Stunden nach Rifampicin))/ Letermovir (480 mg einmal täglich PO)††
|
↓ Letermovir
AUC 0,15 (0,13; 0,17)
Cmax 0,27 (0,22; 0,31)
C24 0,09 (0,06; 0,12)
(P-gp-/UGT-Induktion)
| |
Virostatika
| |
Aciclovir‡
(400 mg Einzeldosis)/ Letermovir (480 mg täglich)
|
↔ Aciclovir
AUC 1,02 (0,87; 1,2)
Cmax 0,82 (0,71; 0,93)
|
Keine Dosisanpassung erforderlich.
| |
Valaciclovir
|
Interaktion nicht untersucht.
Erwartet:
↔ Valaciclovir
|
Keine Dosisanpassung erforderlich.
| |
Endothelin-Antagonisten
| |
Bosentan
|
Interaktion nicht untersucht.
Erwartet:
↓ Letermovir
(P-gp-/UGT-Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Bosentan kann die Plasmakonzentration von Letermovir verringern.
Die gleichzeitige Verabreichung von Prevymis mit Bosentan wird nicht empfohlen.
| |
Antipsychotika
| |
Thioridazin
|
Interaktion nicht untersucht.
Erwartet:
↓ Letermovir
(P-gp-/UGT-Induktion)
|
Thioridazin kann die Plasmakonzentration von Letermovir reduzieren.
Eine gemeinsame Anwendung von Prevymis und Thioridazin wird nicht empfohlen.
| |
Pflanzliche Präparate
| |
Johanniskraut (Hypericum perforatum)
|
Interaktion nicht untersucht.
Erwartet:
↓ Letermovir
(P-gp-/UGT-Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Johanniskraut kann die Plasmakonzentration von Letermovir verringern.
Die gleichzeitige Verabreichung von Prevymis mit Johanniskraut ist kontraindiziert.
| |
HIV-Medikamente
| |
Efavirenz
|
Interaktion nicht untersucht.
Erwartet:
↓ Letermovir
(P-gp-/UGT-Induktion)
↑ oder ↓ Efavirenz
(CYP2B6-Inhibition oder -Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Efavirenz kann die Plasmakonzentration von Letermovir verringern.
Die gleichzeitige Verabreichung von Prevymis mit Efavirenz wird nicht empfohlen.
| |
Etravirin, Nevirapin, Ritonavir, Lopinavir
|
Interaktion nicht untersucht.
Erwartet:
↓ Letermovir
(P-gp-/UGT-Induktion)
|
Diese Virostatika können die Plasmakonzentrationen von Letermovir verringern.
Die gleichzeitige Verabreichung von Prevymis mit diesen Virostatika wird nicht empfohlen.
| |
HMG-CoA-Reduktase-Inhibitoren
| |
Pitavastatin, Simvastatin, Rosuvastatin
|
Interaktion nicht untersucht.
Erwartet:
↑Pitavastatin
↑Simvastatin
↑ Rosuvastatin
(CYP3A- und/oder OATP1B1/3-, und potentielle intestinale BCRP-Inhibition)
|
Die Behandlung mit HMG-CoA-Reduktase-Inhibitoren sollte während der Behandlung mit Prevymis ausgesetzt werden#.
Wenn Prevymis zusammen mit Ciclosporin verabreicht wird, ist die Verwendung von Pitavastatin, Rosuvastatin oder Simvastatin kontraindiziert (siehe Abschnitt «Kontraindikationen»).
| |
Atorvastatin‡
(20 mg Einzeldosis)/ Letermovir (480 mg täglich)
|
↑ Atorvastatin
AUC 3,29 (2,84; 3,82)
Cmax 2,17 (1,76; 2,67)
(CYP3A-, OATP1B1/3-Inhibition)
|
Die Behandlung mit HMG-CoA-Reduktase-Inhibitoren sollte während der Behandlung mit Prevymis ausgesetzt werden#.
Obwohl nicht untersucht, wird erwartet, dass bei gemeinsamer Anwendung von Prevymis mit Ciclosporin das Ausmass des Anstiegs der Plasmakonzentrationen von Atorvastatin grösser ist als bei alleiniger Gabe von Prevymis. Wenn Prevymis zusammen mit Ciclosporin verabreicht wird, ist Atorvastatin kontraindiziert (siehe Abschnitt «Kontraindikationen»)
| |
Sonstige HMG-CoA-Reduktase-Inhibitoren
Beispiele: Fluvastatin, Lovastatin, Pravastatin
|
↑ Konzentrationen von HMG-CoA-Reduktase-Inhibitoren (nicht untersucht)
(CYP3A- und/oder OATP1B1/3- und potenziell intestinale BCRP-Inhibition)
|
Die Behandlung mit HMG-CoA-Reduktase-Inhibitoren sollte während der Behandlung mit Prevymis ausgesetzt werden#.
| |
Immunsuppressiva
| |
Ciclosporin‡
(50 mg Einzeldosis)/ Letermovir (240 mg täglich)
|
↑ Ciclosporin
AUC 1,66 (1,51; 1,82)
Cmax 1,08 (0,97; 1,19)
(CYP3A-Inhibition)
|
Wenn Prevymis zusammen mit Ciclosporin verabreicht wird, sollte die Dosis von Prevymis bei erwachsenen HSZT- und Nierentransplantatempfängern, pädiatrischen HSZT-Empfängern mit einem Körpergewicht von mindestens 30 kg und jugendlichen Nierentransplantatempfängern mit einem Körpergewicht von mindestens 40 kg auf 240 mg einmal täglich reduziert werden. Wenn Prevymis bei pädiatrischen HSZT-Empfängern mit einem Körpergewicht von unter 30 kg zusammen mit Ciclosporin verabreicht wird, ist möglicherweise eine Dosisanpassung erforderlich (siehe Abschnitte «Dosierung/Anwendung» und «Pharmakokinetik»).
Während der Behandlung, sowie bei Anwendungsänderung und Abbruch der Anwendung mit Prevymis sollte eine häufige Überwachung der Ciclosporinkonzentrationen im Vollblut erfolgen und die Ciclosporindosis entsprechend angepasst werden#.
| |
Ciclosporin‡
(200 mg Einzeldosis)/ Letermovir (240 mg täglich)
|
↑ Letermovir
AUC 2,11 (1,97; 2,26)
Cmax 1,48 (1,33; 1,65)
(OATP1B1/3-Inhibition)
| |
Mycophenolat-Mofetil‡
(1 g Einzeldosis)/ Letermovir (480 mg täglich)
|
↔ Mycophenolsäure
AUC 1,08 (0,97; 1,20)
Cmax 0,96 (0,82; 1,12)
↔ Letermovir
AUC 1,18 (1,04; 1,32)
Cmax 1,11 (0;92; 1,34)
|
Keine Dosisanpassung für Mycophenolat-Mofetil erforderlich.
| |
Sirolimus‡
(2 mg Einzeldosis)/ Letermovir (480 mg täglich)
|
↑ Sirolimus
AUC 3,40 (3,01; 3,85)
Cmax 2,76 (2,48; 3,06)
(CYP3A-Inhibition)
Interaktion nicht untersucht.
Erwartet:
↔ Letermovir
|
Während der Behandlung, sowie bei Anwendungsänderung und Abbruch der Anwendung mit Prevymis sollte eine häufige Überwachung der Sirolimuskonzentrationen im Vollblut erfolgen und die Sirolimusdosis entsprechend angepasst werden#.
Es wird empfohlen, die Sirolimuskonzentrationen zu Beginn und nach Beendigung der gemeinsamen Verabreichung von Ciclosporin mit Prevymis häufig zu überwachen.
Wenn Prevymis zusammen mit Ciclosporin verabreicht wird, beziehen Sie sich bitte auf die Fachinformation von Sirolimus, um spezifische Dosierungsempfehlungen für die Verwendung von Sirolimus mit Ciclosporin zu erhalten#.
Bei Kombination von Prevymis mit Ciclosporin kann der Anstieg der Plasmakonzentration von Sirolimus ausgeprägter sein als unter Prevymis allein.
| |
Tacrolimus‡
(5 mg Einzeldosis)/ Letermovir (480 mg täglich)
|
↑ Tacrolimus
AUC 2,42 (2,04; 2,88)
Cmax 1,57 (1,32; 1,86)
(CYP3A-Inhibition)
|
Während der Behandlung, sowie bei Anwendungsänderung und Abbruch der Anwendung mit Prevymis sollte eine häufige Überwachung der Tacrolimuskonzentrationen im Vollblut erfolgen und die Tacrolimusdosis entsprechend angepasst werden#.
| |
Tacrolimus
(5 mg Einzeldosis)/ Letermovir (80 mg zweimal täglich)
|
↔ Letermovir
AUC 1,02 (0,97; 1,07)
Cmax 0,92 (0,84; 1,00)
| |
Orale Kontrazeptiva
| |
Ethinylestradiol (EE) (0.03 mg)/ Levonorgestrel (LNG)‡ (0.15 mg) Einzeldosis/ Letermovir (480 mg täglich)
|
↔ EE
AUC 1,42 (1,32; 1,52)
Cmax 0,89 (0,83; 0,96)
↔ LNG
AUC 1,36 (1,30; 1,43)
Cmax 0,95 (0,86; 1,04)
|
Prevymis kann mit hormonellen Verhütungsmitteln verwendet werden. Die klinische Relevanz der von Prevymis erwarteten Erhöhung der Ethinylestradiol- und Levonorgestrelspiegel bei mehrfacher Verabreichung dieser Wirkstoffe ist nicht bekannt.
| |
Andere kontrazeptive Steroide mit systemischer Wirkung
|
Risiko für ↓ kontrazeptiver Steroide
|
Letermovir kann die Plasmakonzentrationen anderer oraler kontrazeptiver Steroide reduzieren und dadurch deren Wirksamkeit beeinträchtigen.
| |
Protonenpumpeninhibitoren
| |
Omeprazol, Pantoprazol
|
Interaktion nicht untersucht.
Erwartet:
↓ Omeprazol
(CYP2C19-Induktion)
↓ Pantoprazol
(wahrscheinlich aufgrund von CYP2C19-Induktion)
Interaktion nicht untersucht.
Erwartet:
↔ Letermovir
|
Die gleichzeitige Verabreichung von Prevymis mit diesen Protonenpumpeninhibitoren (PPI) kann die Plasmakonzentration der PPIs verringern. Klinische Überwachung und Dosisanpassung können erforderlich sein, wenn sie gemeinsam mit Prevymis verabreicht werden#.
| |
Wachhaltende Medikamente
| |
Modafinil
|
Interaktion nicht untersucht.
Erwartet:
↓ Letermovir
(P-gp-/UGT-Induktion)
|
Die gleichzeitige Verabreichung von Prevymis mit Modafinil kann die Plasmakonzentration von Letermovir verringern.
Die gleichzeitige Verabreichung von Prevymis mit Modafinil wird nicht empfohlen.
| |
CYP3A-Substrate
| |
Sedativa
Midazolam‡
(1 mg Einzeldosis IV)/ Letermovir (240 mg einmal täglich PO)
Midazolam‡ (2 mg Einzeldosis PO) / Letermovir (240 mg einmal täglich PO)
|
↑ Midazolam
IV:
AUC 1,47 (1,37; 1,58)
Cmax 1,05 (0,94; 1,17)
PO:
AUC 2,25 (2,04; 2,49)
Cmax 1,72 (1,55; 1,92)
(CYP3A-Inhibition)
|
Bei gemeinsamer Anwendung von Prevymis mit Midazolam sollte eine engmaschige klinische Überwachung hinsichtlich einer Atemdepression und/oder einer verlängerten Sedierung vorgenommen werden. Eine Dosisanpassung von Midazolam sollte in Betracht gezogen werden#. Der Anstieg der Plasmakonzentrationen von Midazolam kann grösser sein, wenn Midazolam oral gemeinsam mit Letermovir in klinischer Dosis gegeben wird, als bei der untersuchten Dosis.
| |
Opioidagonisten
Andere Beispiele: Alfentanil, Fentanyl
|
Interaktion nicht untersucht.
Erwartet:
↑ Konzentrationen von über CYP3A metabolisierte Opioide
(CYP3A-Inhibition)
|
Bei gleichzeitiger Anwendung wird eine häufige Überwachung auf unerwünschte Reaktionen im Zusammenhang mit diesen Arzneimitteln empfohlen. Eventuell ist eine Dosisanpassung von über CYP3A metabolisierte Opioide erforderlich# (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Eine Überwachung ist auch bei Änderung der Anwendungsart empfohlen. Bei gemeinsamer Anwendung von Prevymis mit Ciclosporin kann das Ausmass der Erhöhung der Plasmakonzentrationen der über CYP3A metabolisierten Opioide noch grösser sein. Eine engmaschige klinische Überwachung hinsichtlich einer Atemdepression und/oder einer länger anhaltenden Sedierung sollte während der gemeinsamen Anwendung von Prevymis mit Ciclosporin und Alfentanil oder Fentanyl erfolgen. Beachten Sie dazu die jeweilige Fachinformation (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
| |
P-gp-Substrate
| |
Digoxin‡
(0.5 mg Einzeldosis)/ Letermovir (240 mg zweimal täglich)
|
↔ Digoxin
AUC 0,88 (0,80; 0,96)
Cmax 0,75 (0,63; 0,89)
(P-gp-Induktion)
|
Keine Dosisanpassung erforderlich.
| |
Abkürzungen: PO= oral
* Diese Tabelle ist nicht allumfassend.
† ↓ =verringert, ↑=erhöht, ↔ =keine klinisch relevante Veränderung
‡ Diese Interaktionen wurden untersucht.
# Siehe die jeweilige Fachinformation.
¶ C24 GMR [90%] ist 0,14 (0,11; 0,19)
†† Diese Daten zeigen die Wirkung von Rifampicin auf Letermovir 24 Stunden nach der letzten Rifampicindosis. C24 GMR [90%] ist 0,09 (0,06; 0,12).
|
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine klinischen Daten zur Anwendung von Prevymis bei Schwangeren vor. In tierexperimentellen Studien wurde Reproduktionstoxizität bei maternal-toxischen Dosen beobachtet (siehe Abschnitt «Präklinische Daten»).
Prevymis wird während einer Schwangerschaft und bei Frauen im gebärfähigen Alter, die keine Empfängnisverhütung verwenden, nicht empfohlen.
Stillzeit
Es ist nicht bekannt, ob Letermovir beim Menschen in die Muttermilch übergeht.
Bei Verabreichung an laktierende Ratten war Letermovir in der Milch nachweisbar.
Ein Risiko für Neugeborene/Säuglinge kann nicht ausgeschlossen werden. Der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau sind bei einer Entscheidung zu berücksichtigen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenPrevymis kann einen Einfluss auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen haben. Bei einigen Patienten wurde während der Behandlung mit Prevymis Ermüdung, Vertigo und Kopfschmerzen berichtet, was die Fahrtüchtigkeit und das Bedienen von Maschinen beeinträchtigen kann (siehe «Unerwünschte Wirkungen»).
Unerwünschte WirkungenErfahrung aus klinischen Studien
Prophylaxe bis Woche 14 (~100 Tage) nach HSZT
Die Sicherheit von Prevymis wurde in einer randomisierten, doppelblinden, placebokontrollierten Phase-3-Studie (P001) beurteilt, in der 565 erwachsene Probanden randomisiert und bis Woche 14 nach HSZT mit Prevymis (N=373) oder Placebo (N=192) behandelt und bis Woche 24 nach HSZT einer Nachbeobachtung der Sicherheit unterzogen wurden (siehe Abschnitt «Pharmakokinetik»).
Das Studienmedikament wurde in 4,8% der Prevymis-Probanden aufgrund einer unerwünschten Wirkung abgesetzt. Die am häufigsten gemeldeten unerwünschten Wirkungen, die zum Absetzen des Studienmedikaments führten, waren Übelkeit (1,6%), Erbrechen (0,8%) und Abdominalschmerzen (0,5%).
Prophylaxe von Woche 14 (~100 Tage) bis Woche 28 (~200 Tage) nach HSZT
Die Sicherheit von Prevymis wurde in einer randomisierten, doppelblinden, placebokontrollierten Phase-3-Studie (P040) bewertet, in der 218 erwachsene Probanden, die eine Prevymis-Prophylaxe bis ~100 Tage nach HSZT abgeschlossen hatten, randomisiert einer Behandlung mit Prevymis (N=144) oder Placebo (N=74) bis Woche 28 (~200 Tage) nach HSZT zugeteilt und hinsichtlich der Sicherheit bis Woche 48 nach HSZT nachbeobachtet wurden. Die gemeldeten unerwünschten Wirkungen stimmten mit dem Sicherheitsprofil von Prevymis, wie in Studie P001 beschrieben, überein.
Erwachsene Nierentransplantatempfänger [D+/R-]
Die Sicherheit von Prevymis wurde in einer randomisierten, doppelblinden, aktiv kontrollierten Phase-3-Studie (P002) bewertet, in der 589 erwachsene Probanden bis Woche 28 nach Transplantation mit Prevymis (N = 292) oder Valganciclovir (N = 297) behandelt wurden.
Unerwünschte Wirkungen wurden während der Behandlungsphase und in den zwei Wochen nach Abschluss/Abbruch der Behandlung gesammelt. Die folgenden unerwünschten Wirkungen wurden bei erwachsenen Patienten unter Prevymis in den klinischen Studien identifiziert. Die unerwünschten Wirkungen sind in Tabelle 4 nach Systemorganklasse und Häufigkeit aufgeführt. Die Häufigkeiten sind wie folgt definiert: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1'000, <1/100); selten (≥1/10'000, <1/1'000); sehr selten (<1/10'000).
Tabelle 4: Mit Prevymis identifizierte unerwünschte Wirkungen (Studie P001, P040 und P002).
|
Häufigkeit
|
Unerwünschte Wirkungen
| |
Erkrankungen des Immunsystems
| |
Gelegentlich
|
Überempfindlichkeit
| |
Stoffwechsel- und Ernährungsstörungen
| |
Gelegentlich
|
Verminderter Appetit
| |
Erkrankungen des Nervensystems
| |
Gelegentlich
|
Dysgeusie, Kopfschmerzen
| |
Störungen des Ohrs und Labyrinths
| |
Gelegentlich
|
Vertigo
| |
Erkrankungen des Gastrointestinaltrakts
| |
Häufig
|
Übelkeit, Diarrhö, Erbrechen
| |
Gelegentlich
|
Abdominalschmerzen
| |
Leber- und Gallenerkrankungen
| |
Gelegentlich
|
Erhöhte Alaninaminotransferase, erhöhte Aspartataminotransferase
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
| |
Gelegentlich
|
Muskelspasmen
| |
Erkrankungen der Nieren und Harnwege
| |
Gelegentlich
|
Erhöhtes Kreatinin im Blut
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
| |
Gelegentlich
|
Ermüdung, peripheres Ödem
|
Kardiale Ereignisse
In der Studie P001 war die Rate der kardialen Ereignisse (unabhängig von der vom Prüfarzt bewerteten Kausalität) bei den Probanden, die Prevymis erhielten, höher (13%) als bei den Probanden, die Placebo erhielten (6%). Die häufigsten kardialen Ereignisse waren Tachykardie (berichtet für 4% der Prevymis-Probanden und für 2% der Placebo-Probanden) und Vorhofflimmern (berichtet für 3% der Prevymis-Probanden und für 1% der Placebo-Probanden). Unter den 6 Probanden, die ein oder mehrere kardiale Ereignisse hatten, wiesen 85% der Prevymis- und 92% der Placebo-Probanden Ereignisse auf, die als leicht oder mittelgradig eingestuft wurden. In Studie P040 betrug die Rate kardialer unerwünschter Ereignisse (unabhängig von der vom Prüfarzt beurteilten Kausalität) 4% in der Prevymis- und der Placebo-Gruppe; in beiden Gruppen wurde kein kardiales unerwünschtes Ereignis mehr als einmal berichtet.
Rezidivierende akute myeloische Leukämie (AML)
In Studie P040 wurde rezidivierende AML von Woche 14 bis Woche 28 nach HSZT für 6,3% der Probanden in der Prevymis-Gruppe berichtet, im Vergleich zu 1,4% der Probanden in der Placebo-Gruppe. Von Woche 14 bis Woche 48 nach HSZT wurde rezidivierende AML für 7,6% der Probanden in der Prevymis-Gruppe berichtet, im Vergleich zu 4,1% der Probanden in der Placebo-Gruppe. Keines der Ereignisse wurde vom Prüfarzt als arzneimittelbedingt eingestuft oder führte zum Absetzen von Prevymis. In Studie P001 wurde rezidivierende AML vom Start der Studienmedikation bis Woche 48 nach HSZT für 7,0% der Probanden in der Prevymis-Gruppe berichtet, im Vergleich zu 9,9% der Probanden in der Placebo-Gruppe.
Abnorme Laborwerte
Insgesamt war der Anteil der Patienten mit potenziell klinisch signifikanten Veränderungen der Laborwerte (z.B. Hämatologie, Chemie, Nieren- und Leberfunktion) in der Prevymis- und der Placebo-Gruppe in Studie P001 ähnlich. Es gab keine Unterschiede in der Inzidenz oder der Zeit bis zum Einwachsen der Stammzellen (Engraftment) zwischen der Prevymis- und der Placebo-Gruppe.
In P001 wurden bei männlichen Patienten Biomarker der testikulären Toxizität bewertet (siehe Abschnitt «Präklinische Daten»). Veränderungen der männlichen Geschlechtshormone (Serum-Inhibin B, luteinisierendes Hormon (LH), follikelstimulierendes Hormon (FSH) und Testosteron) gegenüber dem Ausgangswert waren in der Prevymis- und der Placebo-Gruppe ähnlich.
Die Rate hämatologischer Laboranomalien war in der Prevymis- und der Placebo-Gruppe in Studie P040 vergleichbar. Eine Verschlechterung der Serumkreatininwerte auf >1,8 mg/dl (Grad 3 oder 4) trat bei 4% der Prevymis- und 0% der Placebo-Probanden auf.
Ausgewählte abnorme Laborwerte bei Patienten mit Nierentransplantation der Studie P002, die bis Woche 28 nach der Transplantation gemeldet wurden, sind in der nachstehenden Tabelle aufgeführt.
Tabelle 5: Ausgewählte abnorme Laborwerte in der Studie P002
|
|
Prevymis
N=292
|
Valganciclovir
N=297
| |
Absolute Neutrophilenzahl (Zellen/µl)
| |
<500
|
2%
|
7%
| |
500 – <750
|
1%
|
2%
| |
750 – <1000
|
1%
|
7%
| |
Hämoglobin (g/dl)
| |
<6,5
|
1%
|
0%
| |
6,5 – <8,0
|
4%
|
4%
| |
8,0 – <9,5
|
30%
|
32%
| |
Thrombozyten (Zellen/µl)
| |
<25000
|
0%
|
0%
| |
25000 – <50000
|
0%
|
0%
| |
50000 – <100000
|
1%
|
3%
| |
Leukozyten (Zellen/µl)
| |
<1000
|
1%
|
2%
| |
1000 – <2000
|
5%
|
16%
| |
2000 – <3500
|
16%
|
36%
| |
Serumkreatinin (mg/dl)
| |
>2,5
|
22%
|
21%
| |
>1,5 – 2,5
|
51%
|
52%
|
Pädiatrische HSZT-Population
Die Sicherheit von Prevymis wurde in einer offenen, einarmigen Phase-2b-Studie (P030) bewertet, in der 63 Probanden im Alter von 2 Monaten bis 18 Jahren bis Woche 14 nach HSZT mit Prevymis behandelt wurden.
Die Nebenwirkungen stimmten mit den bei Erwachsenen beobachteten überein. Die am häufigsten gemeldete unerwünschte Wirkung war Erbrechen (17,5%). Es kam zu 2 Behandlungsabbrüchen wegen unerwünschter Wirkungen (Vorhofflimmern und Begleitmedikation unter der therapeutischen Konzentration).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIn klinischen Studien der Phase 1 erhielten 86 gesunde erwachsene Probanden bis zu 14 Tage Prevymis-Dosen von 720 mg/Tag bis 1'440 mg/Tag. Das Profil der unerwünschten Reaktionen entsprach etwa dem Profil unter der klinischen Dosis von 480 mg/Tag. Es steht kein spezifisches Antidot für den Fall einer Überdosierung von Prevymis zur Verfügung. Bei einer Überdosierung wird empfohlen, den betroffenen Patienten engmaschig auf unerwünschte Reaktionen zu überwachen. Gegebenenfalls muss eine geeignete symptomatische Therapie eingeleitet werden.
Es ist nicht bekannt, ob Prevymis durch Dialyse in bedeutendem Umfang aus dem systemischen Kreislauf entfernt wird.
Eigenschaften/WirkungenATC-Code
J05AX18
Wirkungsmechanismus
Prevymis (Letermovir) ist ein antivirales Arzneimittel gegen CMV.
Letermovir hemmt den Terminase-Komplex der CMV-DNA, der für die virale Replikation erforderlich ist. Biochemische Charakterisierung und Elektronenmikroskopie haben gezeigt, dass Letermovir die Bildung von Genomen mit geeigneter Einheitslänge beeinflusst und die Virion-Reifung beeinträchtigt.
Pharmakodynamik
Antivirale Aktivität
Der mittlere EC50-Wert von Letermovir gegen eine Reihe von klinischen CMV-Isolaten in einem Zellkultur-Infektionsmodell betrug 2,1 nM (Spanne 0,7 nM bis 6,1 nM n=74).
Virale Resistenz
In Zellkulturen
Die CMV-Gene UL51, UL56 und UL89 kodieren Untereinheiten der CMV-DNA-Terminase. CMV-Mutanten mit reduzierter Empfindlichkeit gegenüber Letermovir sind in Zellkultur selektiert worden, und die Mutationen korrespondieren mit pUL51 (P91S, A95V), pUL56 (C25F, S229F, V231A, V231L, N232Y, V236A, V236L, V236M, E237D, L241P, T244K, T244R, L254F, L257F, L257I, K258E, F261C, F261L, F261S, Y321C, C325F, C325R, C325W, C325Y, L328V, M329T, A365S, N368D, R369G, R369M, R369S) und pUL89 (N320H, D344E). Die EC50-Werte für die rekombinanten CMV-Mutanten, die diese Substitutionen exprimieren, sind um das 1,6- bis 9'300-Fache höher als beim Wildtyp-Referenzvirus.
In klinischen Studien
In einer Phase-2b-Studie zur Beurteilung von Letermovir-Dosen von 60, 120 oder 240 mg/Tag oder Placebo über einen Zeitraum von bis zu 84 Tagen bei 131 erwachsenen HSZT-Empfängern wurde eine DNA-Sequenzanalyse einer ausgewählten UL56-Region (Aminosäuren 231 bis 369) an Proben von 12 mit Letermovir behandelten Probanden durchgeführt, bei denen es zu Prophylaxeversagen kam und von denen Proben zur Analyse verfügbar waren. Bei einem Probanden (der 60 mg/Tag erhielt) lag eine Letermovir-resistente Genotyp-Variante (GV) (V236M) vor.
In einer Phase-3-Studie (P001) wurde eine DNA-Sequenzanalyse der gesamten codierenden Regionen von UL56 und UL89 an Proben von 40 mit Letermovir behandelten erwachsenen Probanden der FAS-Population durchgeführt, bei denen es zu Prophylaxeversagen kam und von denen Proben zur Analyse verfügbar waren. Bei zwei Probanden wurden insgesamt 2 mit Letermovir-Resistenz assoziierte Substitutionen festgestellt, die beide mit pUL56 korrespondieren. Ein Proband hatte die Substitution V236M, der andere E237G. Ein weiterer Proband, der nachweisbare CMV-DNA zu Studienbeginn hatte (und daher nicht in der FAS-Population enthalten war) hatte pUL56 Substitutionen, C325W und R369T, die nach dem Absetzen von Letermovir nachgewiesen wurden.
In einer Phase-3-Studie (P040) wurde eine DNA-Sequenzanalyse der gesamten codierenden Regionen von UL51, UL56 und UL89 an Proben von 32 erwachsenen Probanden (unabhängig von der Behandlungsgruppe) durchgeführt, bei denen die Prophylaxe versagte, oder die aufgrund einer CMV-Virämie vorzeitig abbrachen. Es wurden keine mit Letermovir-Resistenz assoziierten Substitutionen oberhalb der validierten Assay-Grenze von 5% nachgewiesen.
In einer Phase-3-Studie (P002) wurde eine DNA-Sequenzanalyse der gesamten codierenden Regionen von UL51, UL56 und UL89 an Proben von 52 mit Letermovir behandelten erwachsenen Probanden durchgeführt, bei denen eine CMV-Erkrankung auftrat oder die aufgrund einer CMV-Virämie vorzeitig abbrachen. Es wurden keine mit Letermovir-Resistenz assoziierten Substitutionen oberhalb der validierten Assay-Grenze von 5% nachgewiesen.
In einer Phase-2b-Studie (P030) wurde eine DNA-Sequenzanalyse der gesamten codierenden Regionen von UL51, UL56 und UL89 an Proben von 10 mit Letermovir behandelten pädiatrischen Probanden, die bei einer Kontrolle zur Beurteilung einer CMV-Infektion entnommen wurden, durchgeführt. Bei 2 Probanden wurden insgesamt 2 mit Letermovir-Resistenz assoziierte Substitutionen festgestellt, die beide mit pUL56 korrespondieren. Ein Proband hatte die Substitution C325W (9300-fache Reduktion) und der andere R369S (38-fache Reduktion).
Kreuzresistenz
Eine Kreuzresistenz mit Arzneimitteln ausserhalb dieser Klasse ist nicht wahrscheinlich. Letermovir ist voll wirksam gegen Viruspopulationen mit Substitutionen, die Resistenz gegen CMV-DNA-Polymerase-Hemmer (Ganciclovir, Cidofovir und Foscarnet) verleihen. Eine Gruppe von rekombinanten CMV-Stämmen mit Substitutionen, die eine Resistenz gegen Letermovir verleihen, sprach vollständig auf Cidofovir, Foscarnet und Ganciclovir an – mit Ausnahme eines rekombinanten Stammes mit der pUL56 E237G-Substitution, die eine 2,1-fache Reduktion der Ganciclovir-Empfindlichkeit gegenüber dem Wildtyp bewirkt.
Pharmakogenomik
Die Auswirkung genetischer Varianten im OATP1B1-Gen SLCO1B1 (rs4149056, rs2306283, rs4149032) und UGT1A1 (rs4148323 und Promoter-TA-Repeat-Varianten) auf die Pharmakokinetik von Letermovir wurde bei 299 erwachsenen Studienteilnehmern beurteilt. Klinisch relevante Auswirkungen dieser Varianten auf die Letermovir-Expositionen lagen nicht vor.
Kardiale Elektrophysiologie
Die Auswirkung von Letermovir-Dosen bis zu 960 mg i.v. auf das QTc-Intervall wurde in einer randomisierten, sowohl Placebo- als auch aktiv (Moxifloxacin 400 mg oral) kontrollierten, vierphasigen Einzeldosis-Crossover-Studie zur QT-Zeit an 38 gesunden erwachsenen Probanden untersucht. Letermovir verlängert das QTc-Intervall nicht in klinisch relevantem Umfang nach der i.v.-Dosis von 960 mg mit Plasmakonzentrationen, die etwa doppelt so hoch sind wie bei der i.v.-Dosis von 480 mg.
Klinische Wirksamkeit
Erwachsene CMV-seropositive Empfänger [R+] eines allogenen hämatopoetischen Stammzelltransplantats
Prophylaxe bis Woche 14 (~100 Tage) nach HSZT
Zur Beurteilung der Letermovir-Prophylaxe als Präventionsstrategie gegen eine CMV-Infektion oder -Erkrankung wurde die Wirksamkeit von Letermovir in einer multizentrischen, doppelblinden, placebokontrollierten Phase-3-Studie (P001) mit erwachsenen CMV-seropositiven Empfängern [R+] eines allogenen HSZT untersucht. Die Patienten erhielten per Randomisierung (2:1) entweder Letermovir oder Placebo. Die Randomisierung war nach Studienzentrum und Risiko einer CMV-Reaktivierung (hoch gegenüber niedrig) bei Eintritt in die Studie stratifiziert. Die Letermovir-Therapie wurde nach HSZT (Tag 0-28 nach HSZT) eingeleitet und bis Woche 14 nach HSZT fortgesetzt. Letermovir wurde entweder oral oder i.v. verabreicht. Bis Woche 24 nach HSZT wurden die Patienten im Hinblick auf den primären Wirksamkeitsendpunkt überwacht; bis Woche 48 nach HSZT wurde eine kontinuierliche Nachbeobachtung durchgeführt.
Von den 565 behandelten Patienten erhielten 373 Letermovir (einschliesslich 99 Patienten, die mindestens eine i.v.-Dosis erhielten) und 192 Placebo (einschliesslich 48 Patienten, die mindestens eine i.v.-Dosis erhielten). Die mediane Zeit bis zum Beginn der Letermovir-Behandlung betrug 9 Tage nach der Transplantation. Bei 37% der Patienten war das Einwachsen der Stammzellen (Engraftment) zu Studienbeginn erfolgt. Das mediane Alter betrug 54 Jahre (Spanne 18 bis 78 Jahre). Zu Studienbeginn erhielten 50% der Patienten eine myeloablative Therapie, 52% bekamen Ciclosporin und 42% Tacrolimus. Die häufigsten primären Gründe für die Transplantation waren akute myeloische Leukämie (38%), myeloblastisches Syndrom (15%) und Lymphom (13%). Zwölf Prozent (12%) der Patienten waren zu Studienbeginn positiv auf CMV-DNA getestet.
Zu Studienbeginn bestand bei 31% der Patienten ein hohes Risiko einer Reaktivierung, definiert durch mindestens eines der folgenden Kriterien: Humanes-Leukozytenantigen-(HLA-)verwandter Spender (Bruder oder Schwester) mit mindestens einem Mismatch an einem der folgenden drei HLA-Genloci: HLA-A, -B oder -DR, haploidentischer Spender; nicht verwandter Spender mit mindestens einem Mismatch an einem der folgenden vier HLA-Genloci: HLA-A, -B, -C und -DRB1; Verwendung von Nabelschnurblut als Stammzellquelle; Verwendung von Ex-vivo-T-Zellerschöpften Transplantaten; Graft-versus-Host-Krankheit (GVHD) Grad 2 oder höher, die systemische Kortikosteroide erforderlich machte.
Klinisch signifikante CMV-Infektion
Der primäre Wirksamkeitsendpunkt von P001 war das Auftreten einer klinisch signifikanten CMV-Infektion bis Woche 24 nach HSZT. Als klinisch signifikante CMV-Infektion wurde entweder das Auftreten einer CMV-Endorgan-Erkrankung oder die Einleitung einer präemptiven Therapie (PET) aufgrund einer dokumentierten CMV-Virämie (unter Verwendung des Roche COBAS AmpliPrep/COBAS TaqMan Assay) definiert.
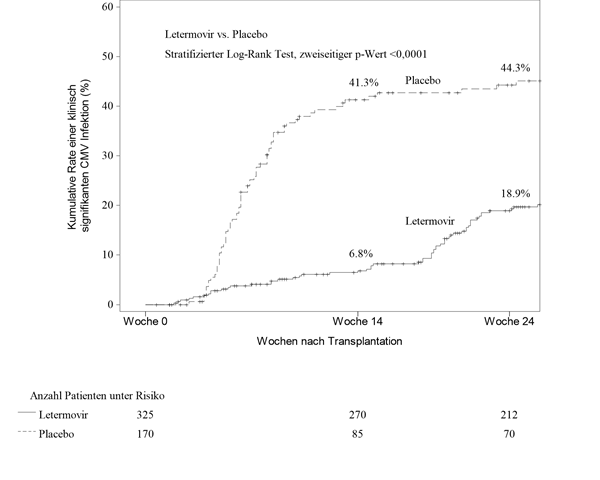
Letermovir zeigte in der Analyse des primären Endpunkts eine überlegene Wirksamkeit gegenüber Placebo, wie in Tabelle 6 dargestellt. Der geschätzte Behandlungsunterschied von -23,5% war statistisch signifikant (einseitiger p-Wert <0,0001).
Tabelle 6: P001: Wirksamkeitsergebnisse bei HSZT-Empfängern (NC=F-Ansatz, FAS-Population)
|
Parameter
|
Letermovir
(N=325)
n (%)
|
Placebo
(N=170)
n (%)
| |
Primärer Endpunkt
(Anteil der Patienten mit Prophylaxeversagen)
|
122 (37,5)
|
103 (60,6)
| |
Gründe für Versagen†
|
|
| |
Klinisch signifikante CMV-Infektion bis Woche 24‡
|
57 (17,5)
|
71 (41,8)
| |
Einleitung einer PET basierend auf dokumentierter CMV-Virämie
|
52 (16,0)
|
68 (40,0)
| |
CMV-Endorganerkrankung
|
5 (1,5)
|
3 (1,8)
| |
Vor Woche 24 aus Studie ausgeschieden
|
56 (17,2)
|
27 (15,9)
| |
Fehlendes Outcome in Besuchsfenster Woche 24
|
9 (2,8)
|
5 (2,9)
| |
Stratum-adjustierter Behandlungsunterschied (Letermovir-Placebo)§
|
|
| |
Unterschied (95%-KI)
|
-23,5 (-32,5; -14,6)
| |
p-Wert
|
<0,0001
| |
†
Die Kategorien für das Versagen schliessen sich gegenseitig aus und beruhen auf der Hierarchie der Kategorien in der aufgeführten Reihenfolge.
‡ Eine klinisch signifikante CMV-Infektion wurde definiert als CMV-Endorganerkrankung oder Einleitung einer PET auf Basis einer dokumentierten CMV-Virämie und des klinischen Zustands des Patienten.
§ 95%-KIs und p-Wert für die Behandlungsunterschiede im prozentualen Ansprechen wurden unter Verwendung der Stratum-adjustierten Mantel-Haenszel-Methode berechnet, wobei der Unterschied nach dem harmonischen Mittelwert der Stichprobengrösse je Arm für jede Schicht (hohes oder niedriges Risiko) gewichtet wurde. Zur Erklärung der statistischen Signifikanz wurde ein einseitiger p-Wert von ≤0,0249 verwendet.
Hinweis: FAS= komplettes Analyse-Set; FAS umfasst die randomisierten Patienten, die mindestens eine Dosis des Studienmedikaments erhalten haben; ausgeschlossen sind Patienten mit nachweisbarer CMV-DNA zu Studienbeginn. Ansatz zum Umgang mit fehlenden Werten: Ansatz Abbruch=Versagen (Non-Completer=Failure - NC=F). Bei dem NC=F-Ansatz wurde Versagen definiert als alle Patienten, die eine klinisch signifikante CMV-Infektion entwickelten oder vorzeitig aus der Studie ausschieden oder bei denen bis zum Besuchsfenster in Woche 24 nach HSZT kein Outcome vorlag.
N = Anzahl der Patienten in jeder Behandlungsgruppe.
n (%) = Anzahl (Prozent) der Patienten in jeder Unterkategorie.
|
In Woche 24 nach HSZT betrug die Kaplan-Meier-(KM)-Ereignisrate für klinisch signifikante CMV-Infektion 18,9% in der Letermovir-Gruppe im Vergleich zu 44,3% in der Placebo-Gruppe (nomineller zweiseitiger stratifizierter Log-Rank-p-Wert <0,0001) (siehe Abb. 1). Die folgenden Faktoren standen bei den mit Letermovir behandelten Patienten mit einer klinisch signifikanten CMV-Infektion zwischen Woche 14 und Woche 24 nach HSZT in Zusammenhang:
·hohes Risiko einer CMV-Reaktivierung zu Studienbeginn,
·bestehende GVHD, und
·Anwendung eines Steroids zu irgendeinem Zeitpunkt nach der Randomisierung.
Abbildung 1: P001: Kaplan-Meier-Plot der Zeit bis zum Beginn einer klinisch signifikanten CMV-Infektion bis Woche 24 nach der Transplantation bei HSZT-Empfängern (FAS-Population)
Im Hinblick auf die Wirksamkeit war Letermovir in allen Untergruppen, einschliesslich der folgenden, deutlich vorteilhafter:
·geringes oder hohes Risiko einer CMV-Reaktivierung,
·Konditionierungstherapien, und
·Begleittherapien mit Immunsuppressiva.
Mortalität
Die KM-Ereignisrate für die Gesamtmortalität in der Letermovir- gegenüber der Placebo-Gruppe betrug 12,1% gegenüber 17,2% in Woche 24 nach HSZT (nomineller zweiseitiger stratifizierter Log-Rank-p-Wert = 0,0401) bzw. 23,8% gegenüber 27,6% in Woche 48 nach HSZT (nomineller zweiseitiger stratifizierter Log-Rank-p-Wert = 0,2117). Die K-M-Ereignisrate für die Gesamtmortalität nach Geschlecht (Männer vs. Frauen) in der Woche 24 nach HSZT betrug 16,4% und 6,6% in der Letermovir-Gruppe und lag bei 14,2% und 25,4% in der Placebo-Gruppe; diese nach Geschlecht aufgeschlüsselten Ereignisraten sind mit Vorsicht zu interpretieren, da die Randomisierung nicht nach Geschlecht stratifiziert wurde, was bei Baseline zu Ungleichgewichten beim geschlechtsspezifischen Mortalitätsrisiko zwischen den Behandlungsgruppen führte.
Prophylaxe von Woche 14 (~100 Tage) bis Woche 28 (~200 Tage) nach HSZT
Die Wirksamkeit der Verlängerung der Letermovir-Prophylaxe von Woche 14 (~100 Tage) bis Woche 28 (~200 Tage) nach HSZT bei Patienten mit einem Risiko einer späten CMV-Infektion und -Erkrankung wurde in einer multizentrischen, doppelblinden, placebokontrollierten Phase-3-Studie (P040) bei erwachsenen CMV-seropositiven Empfängern [R+] einer allogenen HSZT bewertet. Geeignete Probanden, die die Letermovir-Prophylaxe bis etwa 100 Tage nach HSZT abgeschlossen hatten, wurden randomisiert (2:1) und erhielten Letermovir oder Placebo von Woche 14 bis Woche 28 nach HSZT. Die Probanden erhielten eine Tagesdosis von 480 mg Letermovir, die bei gleichzeitiger Verabreichung mit Ciclosporin auf 240 mg angepasst wurde. Das Studienmedikament wurde entweder oral oder i.v. verabreicht. Ein Proband erhielt Letermovir i.v. für 2 Tage. Die Probanden wurden bis Woche 28 nach HSZT auf den primären Wirksamkeitsendpunkt mit fortgesetzter Nachbeobachtung ausserhalb der Behandlung bis Woche 48 nach HSZT überwacht.
Von den 218 behandelten Probanden erhielten 144 Probanden Letermovir und 74 Placebo. Das Durchschnittsalter betrug 55 Jahre (Bereich: 20 bis 74 Jahre). Die häufigsten Gründe für eine Transplantation waren akute myeloische Leukämie (42%), akute lymphatische Leukämie (15%) und myelodysplastisches Syndrom (11%).
Bei Studieneintritt hatten alle Probanden Risikofaktoren für eine späte CMV-Infektion und -Erkrankung, wobei 64% zwei oder mehr Risikofaktoren aufwiesen. Zu den Risikofaktoren gehörten: HLA-verwandter (Geschwister-)Spender mit mindestens einem Mismatch an einem der folgenden drei HLA-Genloci: HLA-A, -B oder -DR; haploidentischer Spender; nicht verwandter Spender mit mindestens einem Mismatch an einem der folgenden vier HLA-Genloci: HLA-A, -B, -C und -DRB1; Verwendung von Nabelschnurblut als Stammzellenquelle; Verwendung von ex-vivo T-Zell-depletierten Transplantaten; Erhalt von Anti-Thymozyten-Globulin; Erhalt von Alemtuzumab; Anwendung von systemischem Prednison (oder Äquivalent) in einer Dosis von ≥1 mg/kg Körpergewicht pro Tag.
Klinisch signifikante CMV-Infektion
Der primäre Wirksamkeitsendpunkt von P040 war das Auftreten einer klinisch signifikanten CMV-Infektion bis Woche 28 nach HSZT. Als klinisch signifikante CMV-Infektion wurde entweder das Auftreten einer CMV-Endorganerkrankung oder die Einleitung einer PET aufgrund einer dokumentierten CMV-Virämie und des klinischen Zustands des Patienten definiert. Es wurde der Observed Failure (OF)-Ansatz verwendet, bei dem Probanden, die die Studie aus irgendeinem Grund ohne Virämie vorzeitig abbrachen oder zu denen zu diesem Zeitpunkt Daten fehlten, nicht als Versagen galten.
Die Anzahl der Probanden, die aus der Studie vor Woche 28 ohne Virämie ausschieden, betrug 14 (9,7%) in der Prevymis-Gruppe und 0 in der Placebo-Gruppe. Die Anzahl der Probanden, bei denen in dem Besuchsfenster der Woche 28 kein Outcome vorlag, betrug 3 (2,1%) in der Prevymis-Gruppe und 4 (5,4%) in der Placebo-Gruppe, keiner hatte zuvor eine Virämie.
Die Wirksamkeitsergebnisse der Studie P040 sind in Tabelle 7 aufgeführt. Die Wirksamkeit favorisierte Letermovir gegenüber Placebo in allen Untergruppen basierend auf Patientenmerkmalen (Alter, Geschlecht, ethnische Zugehörigkeit) und Risikofaktoren für späte CMV-Infektion und -Erkrankung bei Verwendung des OF-Ansatzes.
Tabelle 7: P040-Wirksamkeitsergebnisse bei HSZT-Empfängern mit einem Risiko für eine späte CMV-Infektion und -Erkrankung (OF-Ansatz, FAS-Population)
|
Parameter
|
Letermovir
(~200 Tage Letermovir)
(N=144)
n (%)
|
Placebo
(~100 Tage Letermovir)
(N=74)
n (%)
| |
Versagen†
|
4 (2,8)
|
14 (18,9)
| |
Klinisch signifikante CMV-Infektion bis Woche 28‡
|
2 (1,4)
|
13 (17,6)
| |
Einleitung einer PET basierend auf dokumentierter CMV-Virämie
|
1 (0,7)
|
11 (14,9)
| |
CMV-Endorganerkrankung
|
1 (0,7)
|
2 (2,7)
| |
Vor Woche 28 mit CMV-Virämie aus Studie ausgeschieden
|
2 (1,4)
|
1 (1,4)
| |
Stratum-adjustierter Behandlungsunterschied (Letermovir (~200 Tage Letermovir)-Placebo (~100 Tage Letermovir)) §
| |
Differenz (95%-KI)
|
-16,1 (-25,8; -6,5)
| |
p-Wert
|
0,0005
| |
†
Die Kategorien für das Versagen schliessen sich gegenseitig aus und beruhen auf der Hierarchie der Kategorien in der aufgeführten Reihenfolge.
‡ Eine klinisch signifikante CMV-Infektion wurde definiert als CMV-Endorganerkrankung (bewiesen oder wahrscheinlich) oder Einleitung einer PET, basierend auf einer dokumentierten CMV-Virämie und dem klinischen Zustand des Patienten.
§ 95%-KIs und p-Wert für die Behandlungsunterschiede im prozentualen Ansprechen wurden unter Verwendung der Stratum-adjustierten Mantel-Haenszel-Methode berechnet, wobei die Differenz nach dem harmonischen Mittel der Stichprobengrösse pro Arm für jede Schicht gewichtet wurde (haploidentischer Spender ja oder nein). Zur Feststellung einer statistischen Signifikanz wurde ein einseitiger p-Wert ≤0,0249 verwendet.
Ansatz zum Umgang mit fehlenden Werten: Observed Failure (OF)-Ansatz. Beim OF-Ansatz wurde Versagen definiert als alle Probanden, die eine klinisch signifikante CMV-Infektion entwickelten oder die Studie vorzeitig mit CMV-Virämie von Woche 14 (~100 Tage) bis Woche 28 (~200 Tage) nach HSZT abbrachen.
N = Anzahl der Probanden in jeder Behandlungsgruppe.
n (%) = Anzahl (Prozent) der Probanden in jeder Unterkategorie.
|
Die Zeit bis zur klinisch signifikanten CMV-Infektion ist in Abbildung 2 gezeigt.
Abbildung 2: P040 Kaplan-Meier-Plot der Zeit bis zum Einsetzen einer klinisch signifikanten CMV-Infektion von Woche 14 bis Woche 48 nach Transplantation bei HSZT-Empfängern mit Risiko für späte CMV-Infektion und -Erkrankung (FAS-Population)
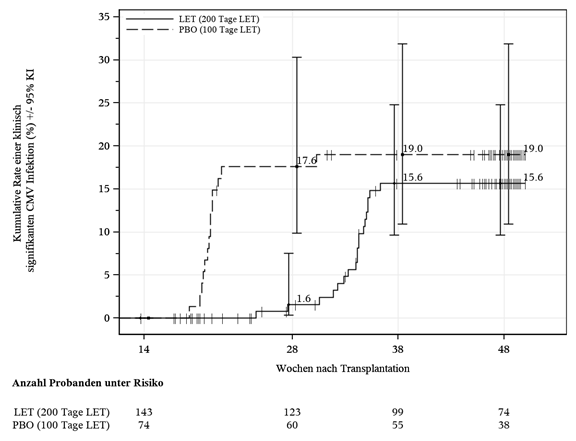
LET = Letermovir; PBO = Placebo
Mortalität
Die Gesamtmortalitätsrate im Letermovir- resp. Placebo-Arm war 2,1% resp. 1,4% in Woche 28 nach HSZT (nomineller einseitiger p-Wert=0,6244), und 8,3% resp. 8,1% in Woche 48 nach HSZT (nomineller einseitiger p-Wert=0,5264).
Erwachsene CMV-seronegative Empfänger eines Nierentransplantats von einem CMV-seropositiven Spender [D+/R-]
Um die Letermovir-Prophylaxe als präventive Strategie für CMV-Erkrankungen bei Nierentransplantatempfängern zu bewerten, wurde die Wirksamkeit von Letermovir in einer multizentrischen, doppelblinden, aktiv kontrollierten Phase-3-Nichtunterlegenheitsstudie (P002) bei erwachsenen Nierentransplantatempfängern mit hohem Risiko [D+/R-] bewertet. Die Probanden wurden randomisiert (1:1) und erhielten entweder Letermovir oder Valganciclovir. Letermovir wurde in einer Dosis von 480 mg einmal täglich verabreicht (angepasst auf 240 mg bei gleichzeitiger Anwendung mit Ciclosporin). Letermovir wurde gleichzeitig mit Aciclovir verabreicht. Valganciclovir wurde gleichzeitig mit einem Placebo zu Aciclovir verabreicht. Die Randomisierung wurde nach Anwendung oder Nichtanwendung einer stark zytolytischen Anti-Lymphozyten-Immuntherapie während der Induktion stratifiziert. Das Studienmedikament wurde zwischen Tag 0 und Tag 7 nach Nierentransplantation begonnen und bis Woche 28 (~200 Tage) nach Transplantation fortgesetzt. Das Studienmedikament wurde entweder oral oder i.v. verabreicht. Drei Probanden erhielten Letermovir i.v. für eine mittlere Dauer von 1,7 Tagen. Die Probanden wurden bis Woche 52 nach der Transplantation überwacht.
Von den 589 behandelten Probanden erhielten 292 Letermovir und 297 Valganciclovir. Das mediane Alter betrug 51 Jahre (Bereich: 18 bis 82 Jahre); 72% waren männlich; 84% Weiss; 2% Asiaten; 9% Schwarz; 17% Hispano- oder Lateinamerikaner; und 60% erhielten eine Niere von einem verstorbenen Spender. Die häufigsten primären Gründe für eine Transplantation waren angeborene zystische Nierenerkrankung (17%), Bluthochdruck (16%) und Diabetes/diabetische Nephropathie (14%).
CMV-Erkrankung
Der primäre Wirksamkeitsendpunkt von P002 war das Auftreten einer CMV-Erkrankung (definiert als CMV-Endorganerkrankung oder CMV-Syndrom, bestätigt von einem unabhängigen Beurteilungsausschuss) bis Woche 52 nach der Transplantation. Es wurde der Observed Failure (OF)-Ansatz verwendet, bei dem Probanden, die die Studie aus irgendeinem Grund vorzeitig abbrachen oder zu denen zu diesem Zeitpunkt Daten fehlten, nicht als Versagen galten.
Die Anzahl der Probanden, die aus der Studie vor Woche 52 ausschieden, betrug 32 (11%) in der Prevymis-Gruppe und 28 (9%) in der Valganciclovir-Gruppe. Die Anzahl der Probanden, bei denen in dem Besuchsfenster der Woche 52 kein Outcome vorlag, betrug 24 (8%) in der Prevymis-Gruppe und 25 (8%) in der Valganciclovir-Gruppe.
Die Wirksamkeitsergebnisse der Studie P002 sind in Tabelle 8 aufgeführt.
Tabelle 8: P002-Wirksamkeitsergebnisse bei Nierentransplantatempfängern (OF-Ansatz, FAS-Population)
|
Parameter
|
Letermovir
(N=289)
n (%)
|
Valganciclovir
(N=297)
n (%)
| |
CMV-Erkrankung† bis Woche 52
CMV-Syndrom‡
CMV-Endorganerkrankung
|
30 (10,4)
24 (8,3)
6 (2,1)
|
35 (11,8)
34 (11,4)
1 (0,3)
| |
Stratum-adjustierter Behandlungsunterschied (Letermovir-Valganciclovir) §
| |
Differenz (95% KI)
|
-1,4 (-6,5, 3,8) ¶
| |
†
Fälle von CMV-Erkrankungen, die von einem unabhängigen Bewertungsausschuss bestätigt wurden.
‡ Definiert als Nachweis von CMV im Blut durch Virusisolierung, Schnellkultur, Antigenämie oder Nukleinsäuretesten und zwei oder mehr der folgenden Symptome: 1) Fieber ≥38 °C für mindestens 2 Tage, 2) neues oder verstärktes Unwohlsein/Ermüdung, 3) Leukopenie oder Neutropenie bei zwei separaten Messungen im Abstand von min. 24 h, 4) ≥5% atypische Lymphozyten, 5) Thrombozytopenie, 6) ALT- oder AST-Erhöhung auf das Zweifache der oberen Normgrenze.
§ Die 95%-KIs für die Behandlungsunterschiede im prozentualen Ansprechen wurden unter Verwendung der Stratum-adjustierten Mantel-Haenszel-Methode berechnet, wobei die Differenz nach dem harmonischen Mittel der Stichprobengrösse pro Arm für jedes Stratum gewichtet wurde (Anwendung/Nichtanwendung einer stark zytolytischen Anti-Lymphozyten-Immuntherapie während Induktion).
¶ Basierend auf einer Nichtunterlegenheitsspanne von 10%.
Ansatz zum Umgang mit fehlenden Werten: Observed Failure (OF)-Ansatz.
N = Anzahl der Probanden in jeder Behandlungsgruppe.
n (%) = Anzahl (Prozent) der Probanden in jeder Unterkategorie.
|
Die Wirksamkeit war in allen Untergruppen vergleichbar, einschliesslich Geschlecht, Alter, Rasse, Region, und der Anwendung/Nichtanwendung einer stark zytolytischen Anti-Lymphozyten-Immuntherapie während der Induktion.
Pädiatrische Population
Pädiatrische Empfänger einer allogenen hämatopoetischen Stammzelltransplantation
Die Wirksamkeit von Letermovir wurde in einer multizentrischen, offenen, einarmigen Phase-2b-Studie (P030) bei pädiatrischen Empfängern eines allogenen HSZT bewertet. Das Studienmedikament wurde nach der HSZT (Tag 0–28 nach HSZT) eingeleitet und bis Woche 14 nach HSZT fortgesetzt. Das Studienmedikament wurde entweder oral oder i.v. verabreicht; die Dosis von Letermovir war abhängig von Alter, Körpergewicht und Darreichungsform.
Von den 63 behandelten Probanden waren 8 im Alter von 0 bis unter 2 Jahren, 27 im Alter von 2 bis unter 12 Jahren und 28 im Alter von 12 bis unter 18 Jahren. Das mediane Alter betrug 11 Jahre (Spanne: 2 Monate bis 17 Jahre), das mediane Gewicht betrug 32 kg (Spanne: 5 bis 95 kg), 70% waren männlich, 70% weiss, 14% Asiaten, 5% schwarz, 22% Hispano- oder Lateinamerikaner. Zu Studienbeginn erhielten 87% der Probanden eine myeloablative Therapie, 67% erhielten Ciclosporin und 27% erhielten Tacrolimus. Die häufigsten primären Gründe für die Transplantation waren akute myeloische Leukämie (18%) und aplastische Anämie (10%). 11% der Probanden waren positiv für CMV-DNA zu Studienbeginn.
Klinisch signifikante CMV-Infektion
Die Wirksamkeitsendpunkte in P030 waren sekundär und beinhalteten das Auftreten einer klinisch signifikanten CMV-Infektion bis Woche 24 nach HSZT. Als klinisch signifikante CMV-Infektion wurde das Auftreten einer CMV-Endorganerkrankung oder die Einleitung einer PET aufgrund einer dokumentierten CMV-Virämie und des klinischen Zustands des Probanden definiert. Die Wirksamkeitsanalysen basierten auf einer Population, die 56 Probanden umfassten, welche mindestens 1 Dosis des Studienmedikaments erhielten und keine detektierbare virale CMV-DNA bei Studienbeginn hatten. Der Anteil Probanden, bei denen die CMV-Prophylaxe bis Woche 24 nach HSZT versagte, war 25% (14 der 56 Probanden). Bei 6 Probanden wurde eine PET aufgrund einer CMV-Virämie initiiert, und 8 Probanden brachen die Studie vor Woche 24 ab. Keiner der Probanden hatte eine CMV-Endorganerkrankung.
PharmakokinetikAllgemeine Einleitung
Die Pharmakokinetik von Letermovir wurde nach oraler und intravenöser Verabreichung an gesunde erwachsene Probanden sowie erwachsene und pädiatrische HSZT-Empfänger und nach oraler Verabreichung bei erwachsenen Nierentransplantatempfängern charakterisiert.
Gesunde erwachsene Probanden
Die Letermovir-Exposition stieg bei beiden Verabreichungswegen (oral oder i.v.) nach Einzel- und Mehrfachdosen von 240 mg und 480 mg überproportional zur Dosis an. Letermovir wurde mit einer medianen Zeit bis zur maximalen Plasmakonzentration (Tmax) von 1.5 bis 3 Stunden schnell absorbiert, und die Plasmakonzentrationen fielen biphasisch ab. Die geometrischen Mittelwerte von AUC und Cmax im Steady-State betrugen bei gesunden Probanden bei 480 mg Prevymis einmal täglich oral 71'500 ng•h/ml bzw. 13'000 ng/ml. Das Plasmakonzentrations-Zeit-Profil nach Absorption von Letermovir nach oraler Gabe ähnelte dem nach intravenöser Gabe. Letermovir erreichte in 9 bis 10 Tagen einen Gleichgewichtszustand (Steady-State) mit einem Akkumulationsverhältnis von 1,23 für AUC und 1,09 für Cmax.
Erwachsene HSZT-Empfänger
Die Letermovir-AUC wurde anhand von populationspharmakokinetischen Analysen unter Verwendung von Phase-3-Daten geschätzt (siehe Tabelle 9). Die unterschiedliche Exposition in den einzelnen Therapieschemen ist klinisch nicht relevant; die Wirksamkeit war über den gesamten in P001 beobachteten Expositionsbereich konsistent.
Tabelle 9: AUC-Werte von Letermovir (ng•h/ml) bei erwachsenen HSZT-Empfängern
|
Therapieschema
|
Median (90% Prognoseintervall)*
| |
480 mg oral, kein Ciclosporin
|
34'400 (16'900, 73'700)
| |
480 mg i.v., kein Ciclosporin
|
100'000 (65'300, 148'000)
| |
240 mg oral, mit Ciclosporin
|
60'800 (28'700, 122'000)
| |
240 mg i.v., mit Ciclosporin
|
70'300 (46'200, 106'000)
| |
* Mediane und 90% Prognoseintervall basieren auf Simulationen unter Verwendung des Phase-3-Populations-PK-Modells mit interindividueller Variabilität.
|
Erwachsene Nierentransplantatempfänger
Die Letermovir-AUC wurde anhand einer populationspharmakokinetischen Analyse unter Verwendung von Phase-3-Daten geschätzt (siehe Tabelle 10). Die Wirksamkeit war über den gesamten in P002 beobachteten Expositionsbereich konsistent.
Tabelle 10: AUC-Werte (ng•h/ml) von Letermovir bei erwachsenen Nierentransplantatempfängern
|
Therapieschema
|
Median (90% Prognoseintervall)*
| |
480 mg oral, kein Ciclosporin
|
62'200 (28'900, 145'000)
| |
240 mg oral, mit Ciclosporin
|
57'700 (26'900, 135'000)
| |
* Mediane und 90%-Prognoseintervalle basieren auf Simulationen unter Verwendung des Phase-3-Populations-PK-Modells mit interindividueller Variabilität.
|
Absorption
Auf der Basis von populationspharmakokinetischen Analysen wurde die absolute Bioverfügbarkeit von Letermovir bei gesunden erwachsenen Probanden über den Dosisbereich von 240 mg bis 480 mg auf ca. 94% geschätzt. Bei erwachsenen HSZT-Empfängern wurde die Bioverfügbarkeit von Letermovir auf ca. 35% geschätzt, wenn 480 mg Prevymis einmal täglich oral ohne Ciclosporin verabreicht wurden. Die interindividuelle Variabilität für die Bioverfügbarkeit wurde auf ca. 37% geschätzt. Bei erwachsenen Nierentransplantatempfängern wurde die Bioverfügbarkeit von Letermovir auf etwa 60% geschätzt, wenn 480 mg Prevymis einmal täglich oral ohne Ciclosporin verabreicht wurde.
Effekt von Ciclosporin
Bei erwachsenen HSZT-Empfängern erhöhte die gleichzeitige Gabe von Ciclosporin die Plasmakonzentrationen von Letermovir. Die Bioverfügbarkeit von Letermovir wurde auf ca. 85% geschätzt, wenn 240 mg Prevymis einmal täglich oral zusammen mit Ciclosporin verabreicht wurden. Wenn Prevymis gleichzeitig mit Ciclosporin verabreicht wird, beträgt die empfohlene Dosis Prevymis bei erwachsenen HSZT- und Nierentransplantatempfängern, pädiatrischen HSZT-Empfängern mit einem Körpergewicht von mindestens 30 kg und jugendlichen Nierentransplantatempfängern mit einem Körpergewicht von mindestens 40 kg 240 mg einmal täglich. Wenn Prevymis bei pädiatrischen HSZT-Empfängern mit einem Körpergewicht von unter 30 kg gleichzeitig mit Ciclosporin verabreicht wird, ist möglicherweise eine Dosisanpassung erforderlich (siehe Abschnitt «Dosierung/Anwendung»).
Einfluss von Nahrungsmitteln
Im Vergleich zum Nüchternzustand hatte die orale Verabreichung einer Einzeldosis von 480 mg Prevymis zusammen mit einer fett- und kalorienreichen Standardmahlzeit bei gesunden erwachsenen Probanden keinerlei Auswirkungen auf die Gesamtexposition (AUC) und führte zu einer Erhöhung der Spitzenkonzentrationen (Cmax) von Letermovir um ca. 30 %. Prevymis kann oral mit oder ohne Nahrung eingenommen werden (siehe Abschnitt «Dosierung/Anwendung»).
Distribution
Auf der Grundlage von populationspharmakokinetischen Analysen wird das mittlere Verteilungsvolumen im Gleichgewichtszustand (Steady-State) nach intravenöser Verabreichung bei erwachsenen HSZT-Empfängern auf 45,5 l geschätzt.
Letermovir wird in vitro stark an humane Plasmaproteine gebunden (98,7%). Bei In-vitro-Beurteilung beträgt die Blut/Plasma-Verteilung von Letermovir 0,56 und ist unabhängig vom Konzentrationsbereich (0,1 bis 10 mg/l).
In präklinischen Studien zur Distribution wurde Letermovir in Organen und Geweben verteilt, wobei die höchsten Konzentrationen im Gastrointestinaltrakt, im Gallengang und in der Leber und niedrige Konzentrationen im Gehirn beobachtet wurden.
Metabolismus
Der Grossteil der arzneimittelbezogenen Komponenten im Plasma ist unveränderte Muttersubstanz (96,6%). Im Plasma wurden keine wesentlichen Metaboliten nachgewiesen.
Die wichtigsten Ausscheidungswege von Letermovir sind die biliäre Ausscheidung und die direkte Glucuronidierung. Der Prozess beinhaltet die hepatischen Aufnahme-Transporter OATP1B1 und -3, gefolgt von UGT1A1/3-katalysierter Glucuronidierung.
Elimination
Die mittlere scheinbare terminale Halbwertszeit von Letermovir beträgt nach einer i.v.-Dosis von 480 mg Prevymis bei gesunden erwachsenen Probanden ca. 12 Stunden.
Auf Grundlage von populationspharmakokinetischen Analysen beträgt die Steady-State-CL von Letermovir nach intravenöser Verabreichung bei erwachsenen HSZT-Empfängern schätzungsweise 4,84 l/h. Die interindividuelle Variabilität für die CL wird auf 24,6% geschätzt.
Ausscheidung
Nach oraler Verabreichung von radioaktiv markiertem Letermovir wurden 93,3% der Radioaktivität im Stuhl nachgewiesen. Der Grossteil des Arzneimittels wurde als unveränderte Muttersubstanz mit einer geringen Menge (6% der Dosis) als Acylglucuronid-Metabolit im Stuhl ausgeschieden. Die Ausscheidung von Letermovir im Urin war vernachlässigbar (<2% der Dosis).
Kinetik spezieller Patientengruppen
Geschlecht
Populationspharmakokinetische Analysen zeigen keinen Unterschied der Pharmakokinetik von Letermovir bei erwachsenen Frauen im Vergleich zu Männern.
Gewicht
Auf Grundlage von populationspharmakokinetischen Phase-1-Analysen bei Erwachsenen ist die AUC von Letermovir bei Probanden, die 80-100 kg wiegen, im Vergleich zu Probanden mit einem Gewicht von 67 kg schätzungsweise 18,7% niedriger. Auf Grundlage von populationspharmakokinetischer Analyse bei erwachsenen Nierentransplantatempfängern wird geschätzt, dass die AUC von Letermovir bei Probanden mit einem Körpergewicht über 80 kg um 26% niedriger ist als bei Probanden mit einem Körpergewicht von 80 kg oder weniger. Diese Veränderungen sind klinisch nicht relevant.
Ethnische Zugehörigkeit
Auf Grundlage von populationspharmakokinetischen Analysen bei Erwachsenen ist die AUC von Letermovir bei Asiaten schätzungsweise 33,2% höher als bei Weissen. Diese Veränderung ist klinisch nicht relevant.
Leberfunktionsstörungen
Die AUC von Letermovir war bei erwachsenen Probanden mit mässiger (Child-Pugh-Klasse B [CP-B], Score von 7-9) und schwerer (Child-Pugh-Klasse C [CP-C], Score von 10-15) Einschränkung der Leberfunktion ca. 1,6- bzw. 3,8-fach höher als bei gesunden erwachsenen Probanden. Die Veränderungen der Letermovir-Exposition bei erwachsenen Probanden mit mässiger Einschränkung der Leberfunktion sind klinisch nicht relevant.
Klinisch relevante Erhöhungen der Letermovir-Exposition werden bei Patienten mit schwerer Einschränkung der Leberfunktion oder bei Patienten mit mässiger Einschränkung der Leberfunktion in Kombination mit mässiger bis schwerer Einschränkung der Nierenfunktion erwartet.
Nierenfunktionsstörungen
Klinische Studie in einer Population mit eingeschränkter Nierenfunktion
Die AUC von Letermovir war bei erwachsenen Probanden mit mässiger (eGFR 30 bis 59 ml/min/1,73 m2) und schwerer (eGFR kleiner als 30 ml/min/1,73 m2) Einschränkung der Nierenfunktion ca. 1,9- bzw. 1,4-fach höher als bei gesunden erwachsenen Probanden. Die Veränderungen der Letermovir-Exposition aufgrund einer Einschränkung der Nierenfunktion sind klinisch nicht relevant. Für Patienten mit eGFR unter 10 ml/min/1,73 m2 oder für Dialysepatienten liegen keine Daten vor.
Post-Nierentransplantation
Basierend auf einer populationspharmakokinetischen Analyse war die AUC von Letermovir etwa 1,1-, 1,3- und 1,4-fach höher bei erwachsenen Probanden mit leichter (60ml/min≤CrCl<90ml/min), mittelschwerer (30ml/min≤CrCl<60ml/min) bzw. schwerer (15ml/min≤CrCl<30ml/min) Nierenfunktionsstörung als bei erwachsenen Probanden mit einer CrCl grösser als oder gleich 90 ml/min. Diese Veränderungen sind klinisch nicht relevant.
Ältere Patienten
Populationspharmakokinetische Analysen ergaben keinen Effekt des Alters auf die Pharmakokinetik von Letermovir. Eine altersentsprechende Dosisanpassung ist nicht erforderlich.
Kinder und Jugendliche
Die Letermovir-AUC bei pädiatrischen HSZT-Empfängern wurde anhand einer populationspharmakokinetischen Analyse unter Verwendung von Phase-2b-Daten geschätzt (siehe Tabelle 11 und Tabelle 12). Die Expositionen für pädiatrische HSZT-Empfänger in den einzelnen Körpergewichtsgruppen liegen innerhalb der Expositionsbereiche, die mit den Referenzexpositionen erwachsener HSZT-Empfänger erzielt wurden (siehe Tabelle 9).
Tabelle 11: Letermovir-AUC-Werte (ng•h/ml) nach oraler Verabreichung bei pädiatrischen HSZT-Empfängern
|
Körpergewicht
|
Orale Dosis, kein Ciclosporin
|
Median
(90%-Prognoseintervall)*
|
Orale Dosis, mit Ciclosporin
|
Median
(90%-Prognoseintervall)*
| |
Ab 30 kg
|
480 mg
|
39'100
(18'700, 81'300)
|
240 mg
|
49'100
(23'200, 104'000)
| |
15 kg bis unter 30 kg
|
240 mg
|
38'900
(20'200, 74'300)
|
120 mg
|
51'000
(26'600, 98'200)
| |
7,5 kg bis unter 15 kg
|
120 mg
|
32'000
(16'700, 59'300)
|
60 mg
|
41'600
(22'300, 81'100)
| |
5 kg bis unter 7,5 kg
|
80 mg
|
30'600
(16'200, 55'000)
|
40 mg
|
39'000
(20'600, 72'200)
| |
* Mediane und 90%-Prognoseintervalle basieren auf Simulationen unter Verwendung des pädiatrischen HSZT-Populations-PK-Modells mit interindividueller Variabilität.
|
Tabelle 12: Letermovir-AUC-Werte (ng•h/ml) nach i.v. Verabreichung bei pädiatrischen HSZT-Empfängern
|
Körpergewicht
|
i.v. Dosis, kein Ciclosporin
|
Median
(90%-Prognoseintervall)*
|
i.v. Dosis, mit Ciclosporin
|
Median
(90%-Prognoseintervall)*
| |
Ab 30 kg
|
480 mg
|
111'000
(55'700, 218'000)
|
240 mg
|
59'800
(28'400, 120'000)
| |
15 kg bis unter 30 kg
|
120 mg
|
57'200
(29'700, 113'000)
|
120 mg
|
61'100
(29'900, 121'000)
| |
7,5 kg bis unter 15 kg
|
60 mg
|
46'000
(24'300, 83'900)
|
60 mg
|
49'200
(25'800, 93'800)
| |
5 kg bis unter 7,5 kg
|
40 mg
|
43'400
(24'300, 81'000)
|
40 mg
|
45'900
(24'900, 82'200)
| |
* Mediane und 90%-Prognoseintervalle basieren auf Simulationen unter Verwendung des pädiatrischen HSZT-Populations-PK-Modells mit interindividueller Variabilität.
|
Präklinische DatenLangzeittoxizität
Testikuläre Toxizität wurde bei Ratten bei systemischen Expositionen (AUC) festgestellt, die der ≥3-fachen Exposition beim Menschen unter der RHD entsprachen. Diese Toxizität war durch Degeneration der Tubuli seminiferi, Oligospermie und Zellfragmente in den Epididymiden mit verminderten Gewichten von Testikeln und Epididymiden gekennzeichnet. Der NOAEL (No Observed Adverse Effect Level) für testikuläre Toxizität bei Ratten wurde bei ähnlichen Expositionen (AUC) wie bei Menschen unter der RHD beobachtet. Diese testikuläre Toxizität scheint artspezifisch zu sein; testikuläre Toxizität wurde bei Mäusen und Affen in den höchsten getesteten Dosen bei Expositionen bis zum 4-Fachen bzw. 2-Fachen der AUC unter der RHD nicht beobachtet. Die Relevanz für Menschen ist nicht bekannt.
Mit Ausnahme der Vakuolenbildung, die in den Nieren von Ratten festgestellt wurde, denen mit 1'500 mg/kg/Tag des Cyclodextrin-Hilfsstoffs Hydroxypropylbetadex formuliertes Letermovir i. v. verabreicht wurde, war das Toxizitätsprofil von Letermovir in Studien an Ratten und Affen mit oraler und intravenöser Applikation generell ähnlich. Es ist bekannt, dass Hydroxypropylbetadex bei Ratten Vakuolenbildung in den Nieren verursachen kann, wenn es in Dosen von mehr als 50 mg/kg/Tag verabreicht wird.
Genotoxizität
In einer Reihe von In-vitro- und In-vivo-Tests, einschliesslich Tests zur mikrobiellen Mutagenese, zu Chromosomenabberationen in CHO-Zellen sowie eines In-vivo-Mikrokerntests an Mäusen, zeigte Letermovir keine genotoxischen Eigenschaften.
Kanzerogenität
Eine 6-monatige orale Kanzerogenitätsstudie an RasH2-transgenen (Tg.RasH2)-Mäusen zeigte keine Hinweise auf eine für den Menschen relevante Tumorentstehung bis zu den höchsten getesteten Dosen, 150 mg/kg/Tag bzw. 300 mg/kg/Tag bei Männchen bzw. Weibchen (Expositionen, die der menschlichen Exposition bei therapeutischer Dosis entsprechen oder diese übersteigen).
Reproduktionstoxizität
In den Studien zur Fertilität und frühen Embryonalentwicklung bei Ratten wurden in der höchsten untersuchten Dosis von 240 mg/kg/Tag (ca. das 5-Fache der AUC beim Menschen unter der RHD) keine Effekte von Letermovir auf die weibliche Fertilität festgestellt. Bei männlichen Ratten wurden reduzierte Spermienkonzentration, reduzierte Spermienmotilität und verminderte Fertilität bei systemischen Expositionen von dem ≥3-Fachen der AUC beim Menschen unter der RHD beobachtet (siehe Langzeittoxizität).
Letermovir wurde trächtigen Ratten in Dosen von 0, 10, 50 oder 250 mg/kg/Tag an den Trächtigkeitstagen 6 bis 17 oral verabreicht. Maternale Toxizität (einschliesslich verringerter Zunahme des Körpergewichts) wurde unter 250 mg/kg/Tag beobachtet (ca. 11-fache AUC unter der RHD); bei den Nachkommen wurden reduziertes Fetalgewicht mit verzögerter Ossifikation, leicht ödematöse Feten und eine erhöhte Inzidenz von Verkürzung der Nabelschnur und Variationen sowie Fehlbildungen von Wirbeln, Rippen und Becken beobachtet. Bei Dosen von bis zu 50 mg/kg/Tag (ca. 2,5-fache AUC unter der RHD) wurden keine maternalen oder Entwicklungseffekte festgestellt.
Letermovir wurde trächtigen Kaninchen in Dosen von 0, 25, 75 oder 225 mg/kg/Tag an den Trächtigkeitstagen 6 bis 20 oral verabreicht. Bei einer Dosis von 225 mg/kg/Tag (ca. 2-fache AUC unter der RHD) wurde maternale Toxizität (einschliesslich Mortalität und Aborte) festgestellt; in der Nachkommenschaft war eine erhöhte Inzidenz von Fehlbildungen und Variationen von Wirbeln und Rippen zu beobachten. Unter der Dosis von 75 mg/kg/Tag (weniger als die AUC unter der RHD) wurden keine maternalen oder Entwicklungseffekte festgestellt.
In der Studie zur prä- und postnatalen Entwicklung in trächtigen Ratten mit Dosen von 0, 10, 45 oder 180 mg/kg Letermovir (oral) pro Tag (bis etwa zum 2-fachen der AUC unter der RHD) wurde keine Entwicklungstoxizität beobachtet.
Sonstige HinweiseInkompatibilitäten
Inkompatible Arzneimittel
Prevymis Konzentrat zur Herstellung einer Infusionslösung ist physikalisch inkompatibel mit Amiodaronhydrochlorid, Amphotericin B (liposomal), Aztreonam, Cefepimhydrochlorid, Ciprofloxacin, Ciclosporin, Diltiazemhydrochlorid, Filgrastim, Gentamicinsulfat, Levofloxacin, Linezolid, Lorazepam, Midazolam-HCl, Mycophenolat-Mofetilhydrochlorid, Ondansetron, Palonosetron.
Inkompatible Materialien für Infusionsbeutel und Infusionssets
Prevymis ist inkompatibel mit Weichmachern aus Diethylhexylphthalat (DEHP) und polyurethanhaltigen Schläuchen von Infusionssets.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Kompatible Verdünnungsmittel
Prevymis Konzentrat zur Herstellung einer Infusionslösung ist mit 0,9%igen Natriumchlorid- und 5%igen Dextroselösungen kompatibel.
Kompatible Arzneimittel
Es wurde eine Studie zur Bewertung der physikalischen Kompatibilität von Prevymis Konzentrat zur Herstellung einer Infusionslösung mit injizierbaren Arzneimitteln durchgeführt. Die Kompatibilität wurde durch visuelle Beobachtungen, Trübung und Partikelmessung bestimmt. Kompatible Arzneimittel sind unten aufgeführt.
Mit Ausnahme der unten genannten Präparate darf Prevymis nicht durch dieselbe intravenöse Leitung (oder Kanüle) gleichzeitig mit anderen Arzneimitteln oder Verdünnungsmittelkombinationen verabreicht werden.
Die folgenden kompatiblen Arzneimittel† können zusammen mit Prevymis Konzentrat zur Herstellung einer Infusionslösung verabreicht werden, nur wenn beide Arzneimittel in 0,9% Natriumchlorid zubereitet werden und über Y-Verbinder gemäss den genehmigten Anweisungen der jeweiligen Fachinformation verabreicht werden.
Antithymozytenglobulin, Caspofungin, Daptomycin, Fentanylcitrat, Fluconazol, Furosemid, Humaninsulin, Magnesiumsulfat, Methotrexat, Micafungin.
† Diese Injektionspräparate sind in der Schweiz erhältlich.
Die folgenden kompatiblen Arzneimittel† können zusammen mit Prevymis Konzentrat zur Herstellung einer Infusionslösung verabreicht werden, nur wenn beide Arzneimittel in 5%iger Dextrose zubereitet werden und über Y-Verbinder gemäss den genehmigten Anweisungen der jeweiligen Fachinformation verabreicht werden.
Amphotericin B (Lipidkomplex)*, Anidulafungin, Cefazolin-Natrium, Ceftarolin, Ceftriaxon-Natrium, Ganciclovir-Natrium, Morphinsulfat, Noradrenalin-Bitartrat, Pantoprazol-Natrium, Kaliumchlorid, Kaliumphosphat, Tacrolimus, Telavancin, Tigecyclin.
† Diese Injektionspräparate sind in der Schweiz erhältlich.
* Amphotericin B (Lipidkomplex) ist mit Prevymis kompatibel. Allerdings ist Amphotericin B (liposomal) inkompatibel (siehe Inkompatible Arzneimittel).
Kompatible Materialien für Infusionsbeutel und Infusionssets
Prevymis ist mit den folgenden Materialien für Infusionsbeutel und Infusionssets kompatibel. Unten nicht genannte Materialien für Infusionsbeutel oder Infusionssets dürfen nicht verwendet werden.
Materialien für Infusionsbeutel
Polyvinylchlorid (PVC), Ethylenvinylacetat (EVA) und Polyolefin (Polypropylen und Polyethylen).
Materialien für Infusionssets
PVC, Polyethylen (PE), Polybutadien (PBD), Silikonkautschuk (SR), Styrol-Butadien-Copolymer (SBC), Styrol-Butadien-Styrol-Copolymer (SBS), Polystyrol (PS).
Weichmacher
Tris(2-ethylhexyl)trimellitat (TOTM), Butylbenzylphthalat (BBP).
Katheter
Radioopakes Polyurethan.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Prevymis Durchstechflaschen sind nur zur einmaligen Anwendung bestimmt. Nicht verwendetes Arzneimittel ist zu entsorgen.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Nicht über 30 °C lagern.
Filmtabletten in der Originalverpackung (Blister) aufbewahren, um sie vor Feuchtigkeit zu schützen.
Prevymis Granulat bis zur Verwendung im Originalbeutel aufbewahren.
Durchstechflaschen mit Prevymis Konzentrat zur Herstellung einer Infusionslösung im Originalkarton aufbewahren, um sie vor Licht zu schützen.
Hinweise für die Handhabung
Zubereitung und Verabreichung des Granulats
Prevymis Granulat wird oral, gemischt mit weicher Nahrung, oder über eine nasogastrale Sonde (NG-Sonde) oder Gastrostomiesonde (G-Sonde) verabreicht.
Zubereitung und Verabreichung gemischt mit weicher Nahrung
Für Näheres zur Zubereitung und Verabreichung von Prevymis Granulat gemischt mit weicher Nahrung siehe Gebrauchsanweisung.
·Prevymis Granulat nicht zerstossen oder zerkauen.
·Prevymis Granulat auf 1 bis 3 Teelöffel weiche Nahrung streuen, deren Temperatur bei Raumtemperatur oder darunter liegt. Keine heisse Nahrung verwenden. Beispiele für weiche Nahrung sind Apfelmus oder Joghurt.
·Prevymis Granulat mit der weichen Nahrung mischen.
·Gesamtes Gemisch innerhalb von 10 Minuten nach dem Mischen des Prevymis Granulats mit der weichen Nahrung verabreichen.
Zubereitung und Verabreichung über eine NG-Sonde oder G-Sonde
Für Näheres zur Zubereitung und Verabreichung von Prevymis Granulat über eine NG-Sonde oder G-Sonde siehe Gebrauchsanweisung sowie Tabelle 13 (NG-Sonde) und Tabelle 14 (G-Sonde).
·Startvolumen an Flüssigkeit bei Raumtemperatur (Milch, Apfelsaft, Säuglingsmilch oder Wasser) mithilfe der Spritze in einen Arzneimittelbecher geben. Prevymis Granulat nicht mit Wasser mischen, wenn es über eine G-Sonde verabreicht wird. Prevymis Granulat nicht mit heisser oder gekühlter Flüssigkeit mischen.
·Prevymis Granulat in die Flüssigkeit im Arzneimittelbecher geben.
·10 Minuten warten. Den Arzneimittelbecher nicht schütteln oder schwenken. Prevymis Granulat löst sich nicht auf, sondern bricht auf oder zerfällt.
·Gemisch mit der Spritze umrühren. Gesamtes Gemisch mithilfe der Spritze über die NG- oder G-Sonde verabreichen.
·Spülvolumen an Flüssigkeit bei Raumtemperatur (Milch, Apfelsaft, Säuglingsmilch oder Wasser) mithilfe der Spritze in den Arzneimittelbecher geben. Arzneimittelbecher nicht mit Wasser nachspülen, wenn Prevymis über eine G-Sonde verabreicht wird.
·Gemisch mit der Spritze umrühren. Gesamtes Spülgemisch mithilfe der Spritze über die NG- oder G-Sonde verabreichen.
·NG- oder G-Sonde mit dem vom Hersteller empfohlenen Volumen an Wasser ausspülen.
Tabelle 13: Empfehlungen zur Verabreichung von Prevymis Granulat per NG-Sonde
|
Dosierung
|
NG-Sonde*
|
Art der Flüssigkeit
|
Art der Spritze†
|
Misch-behälter
|
Start-volumen (ml)
|
Spül-volumen (ml)
| |
120 mg bis
480 mg
|
Jede NG-Sonde ≥8 Ch
|
Milch, Apfelsaft, Säuglingsmilch oder Wasser
|
Spritze mit ENFit- oder Katheter-Ansatz in geeigneter Grösse
|
Arzneimittel-becher
|
15
|
15
| |
40 mg bis 80 mg
|
PUR-NG-Sonde 5 Ch
oder
Jede NG-Sonde ≥6 Ch
|
3
|
2
| |
* Ch = Charrière; PUR = Polyurethan.
† Bei Nutzung einer ENFit-Spritze ist für das Aufziehen des Gemischs aus dem Arzneimittelbecher ein Entnahmehalm (mit grossem Innendurchmesser) erforderlich.
|
Tabelle 14: Empfehlungen zur Verabreichung von Prevymis Granulat über G-Sonde
|
Dosierung
|
G-Sonde*
|
Art der Flüssigkeit
|
Art der Spritze†
|
Misch-behälter
|
Start-volumen (ml)
|
Spül-volumen (ml)
| |
120 mg bis
480 mg
|
Jede G-Sonde
|
Milch, Apfelsaft oder Säuglingsmilch
Kein Wasser verwenden
|
Spritze mit ENFit- oder Katheter-Ansatz in geeigneter Grösse
|
Arzneimittel-becher
|
15
|
15
| |
40 mg bis 80 mg
|
Jede G-Sonde 12 Ch
|
3
|
2
| |
* Ch = Charrière.
† Bei Nutzung einer ENFit-Spritze ist für das Aufziehen des Gemischs aus dem Arzneimittelbecher ein Entnahmehalm (mit grossem Innendurchmesser) erforderlich.
|
Zubereitung und Verabreichung der intravenösen Lösung
Prevymis Konzentrat zur Herstellung einer Infusionslösung wird in Einzeldosis-Durchstechflaschen mit 30 ml geliefert, die entweder 240 mg (12 ml je Durchstechflasche) oder 480 mg (24 ml je Durchstechflasche) enthalten.
Zubereitung
·Prevymis ist vor der intravenösen (i.v.) Anwendung zu verdünnen.
·Den Inhalt der Durchstechflasche vor der Verdünnung auf Verfärbung und vorhandene Partikel prüfen. Prevymis Konzentrat zur Herstellung einer Infusionslösung ist eine klare, farblose Lösung und kann wenige produktbezogene, kleine, durchsichtige oder weisse Partikel enthalten.
·Die Durchstechflasche nicht verwenden, wenn die Lösung trüb oder verfärbt ist oder anderes Material als wenige kleine, durchsichtige oder weisse Partikel enthält.
·Prevymis Konzentrat zur Herstellung einer Infusionslösung nicht mit Infusionsbeutel oder setmaterialien verwenden, welche Polyurethan oder den Weichmacher Diethylhexylphthalat (DEHP) enthalten. Materialien, die phthalatfrei sind, sind auch DEHP-frei.
·Die Prevymis Durchstechflasche nicht schütteln.
·Für die 480-mg- oder 240-mg-Dosis eine Einzeldosis-Durchstechflasche Prevymis Konzentrat zur Herstellung einer Infusionslösung in einen vorgefüllten 250-ml-Infusionsbeutel geben, der entweder 0,9% Natriumchlorid oder 5% Dextrose enthält, und den Beutel vorsichtig mischen. Nicht schütteln.
Für die 120-mg- oder 60-mg-Dosis das Prevymis Konzentrat zur Herstellung einer Infusionslösung gemäss den Angaben in der nachfolgenden Tabelle 15 mit entweder 0,9% Natriumchlorid oder 5% Dextrose zubereiten und den Beutel vorsichtig mischen. Nicht schütteln.
Für die 40-mg-Dosis das Prevymis Konzentrat zur Herstellung einer Infusionslösung gemäss den Angaben in der nachfolgenden Tabelle 16 mit entweder 0,9% Natriumchlorid oder 5% Dextrose zubereiten und den Beutel vorsichtig mischen. Nicht schütteln.
Tabelle 15: Zubereitung der intravenösen Prevymis-Lösung für die 120-mg- oder 60-mg-Dosis
|
Intravenöse Dosis
|
Volumen von Prevymis Konzentrat zur Herstellung einer Infusionslösung 20 mg/ml
|
Endgültiges Infusionsvolumen
| |
120 mg
|
6 ml von 20 mg/ml
|
75 ml
| |
60 mg
|
3 ml von 20 mg/ml
|
50 ml
|
Tabelle 16: Zubereitung der intravenösen Prevymis-Lösung für die 40-mg-Dosis
|
Intravenöse Dosis
|
Volumen der Prevymis-Verdünnung mit 2 mg/ml (1:10)*
|
Endgültiges Infusionsvolumen
| |
40 mg
|
20 ml von 2 mg/ml
|
20 ml
| |
* Herstellung der Prevymis-Verdünnung 2 mg/ml: 5 ml von Prevymis Konzentrat zur Herstellung einer Infusionslösung 20 mg/ml aus der Durchstechflasche in 45 ml Verdünnungsmittel (0,9% Natriumchlorid oder 5% Dextrose) geben und vorsichtig mischen.
|
·Nach der Verdünnung ist die Prevymis Lösung klar und die Färbung reicht von farblos bis gelb. Farbliche Schwankungen innerhalb dieses Bereichs beeinträchtigen die Produktqualität nicht. Die verdünnte Lösung sollte vor der Verabreichung einer Sichtkontrolle auf Partikel und Verfärbung unterzogen werden.
·Die verdünnte Lösung ist zu verwerfen, wenn sie trüb oder verfärbt ist oder anderes Material als wenige kleine, durchsichtige oder weisse Partikel enthält.
Aufbewahrung der verdünnten Lösung
·Aus mikrobiologischen Gründen sollte die verdünnte Lösung, die keine Konservierungsstoffe enthält, sofort verwendet werden.
·Die chemische und physikalische Stabilität im Gebrauch wurde für 48 Stunden bei 25 °C und 48 Stunden bei 2 bis 8 °C nachgewiesen.
·Dieser Zeitraum umfasst die Aufbewahrung der verdünnten Lösung im Infusionsbeutel und die Infusionsdauer.
Verabreichung
·Die verdünnte Lösung muss durch einen sterilen 0,2 oder 0,22 μm PES-Inline-Filter verabreicht werden.
·Die verdünnte Lösung nicht durch einen anderen Filter als einen sterilen 0,2 oder 0,22 μm PES-Inline-Filter verabreichen.
·Nur als intravenöse Infusion verabreichen. Nicht als intravenöse Schnellinfusion oder Bolus geben.
·Prevymis nach der Verdünnung durch intravenöse Infusion über einen peripheren oder zentralen Venenkatheter über einen Gesamtzeitraum von ca. 60 Minuten verabreichen. Den gesamten Inhalt des Infusionsbeutels verabreichen.
Zulassungsnummer66652, 66653, 69867 (Swissmedic).
PackungenPrevymis 240 mg Filmtablette: 28 Filmtabletten in perforierten Aluminium-Blisterpackungen. (A)
Prevymis 480 mg Filmtablette: 28 Filmtabletten in perforierten Aluminium-Blisterpackungen. (A)
Prevymis 20 mg Granulat im Beutel: Jeder Karton enthält 30 Beutel. (A)
Prevymis 120 mg Granulat im Beutel: Jeder Karton enthält 30 Beutel. (A)
Prevymis 12 ml (240 mg) Konzentrat zur Herstellung einer Infusionslösung: Jeder Karton enthält 1 Durchstechflasche. (A)
Prevymis 24 ml (480 mg) Konzentrat zur Herstellung einer Infusionslösung: Jeder Karton enthält 1 Durchstechflasche. (A)
ZulassungsinhaberinMSD MERCK SHARP & DOHME AG
Luzern
Stand der InformationFebruar 2025
CCDS-MK8228-MF-032024/RCN000026329-CH
|