Eigenschaften/WirkungenATC-Code
S01LA06
Wirkungsmechanismus
Die erhöhte Signalgebung über den VEGF-A-Pfad (vaskulärer endothelialer Wachstumsfaktor A) ist mit einer pathologischen okularen Angiogenese und einem Netzhautödem verbunden. Brolucizumab bindet mit hoher Affinität an VEGF-A-Isoformen (z.B. VEGF110, VEGF121 und VEGF165) und verhindert so, dass VEGF-A an seine Rezeptoren VEGFR-1 und VEGFR-2 bindet. Durch Inhibierung der Bindung an VEGF-A unterdrückt Brolucizumab die Endothelzell-Proliferation, wodurch die pathologische Neovaskularisierung reduziert und die Gefässpermeabilität verringert werden.
Pharmakodynamik
Feuchte AMD
In den Studien HAWK und HARRIER waren verwandte anatomische Parameter Teil der Beurteilungen der Krankheitsaktivität, die als Grundlage für Behandlungsentscheidungen dienten. Eine Reduktion der zentralen retinalen Netzhautdicke (central subfield thickness, CST) sowie des Vorhandenseins intraretinaler/subretinaler Flüssigkeit (IRF/SRF) oder subretinaler Pigmentepithel-(sub-RPE)-Flüssigkeit wurde bereits 4 Wochen nach Behandlungsbeginn und bis zu Woche 48 und Woche 96 bei Patienten beobachtet, die mit Beovu behandelt wurden.
In diesen Studien wurde bei mit Beovu behandelten Patienten bereits 12 Wochen nach Behandlungsbeginn sowie in Wochen 48 und 96 nach Behandlungsbeginn eine Reduktion der CNV-Läsionsgrösse beobachtet.
Diabetisches Makulaödem (DME)
In den Studien KESTREL und KITE waren verwandte anatomische Parameter Teil der Bewertung der Krankheitsaktivität, die die Behandlungsentscheidungen leitete. Eine Reduktion der zentralen retinalen Netzhautdicke (CST) und der vorhandenen intraretinalen/subretinalen Flüssigkeit (IRF/SRF) wurde bei Patienten, die mit Beovu behandelt wurden, bereits 4 Wochen nach Behandlungsbeginn und bis zu Woche 52 beobachtet. Diese Reduktionen wurden bis zu Woche 100 erhalten.
Klinische Wirksamkeit
Behandlung von feuchter AMD
Die Sicherheit und Wirksamkeit von Beovu wurde in zwei randomisierten, multizentrischen, doppelblinden, aktivkontrollierten Phase-III-Studien (HAWK und HARRIER) bei Patienten mit neovaskulärer AMD untersucht. Total wurden 1'817 Patienten im Rahmen dieser Studien zwei Jahre lang behandelt (1'088 mit Brolucizumab und 729 mit Aflibercept). Das Alter der Patienten lag zwischen 50 und 97 Jahren, mit einem Mittelwert von 76 Jahren.
In der HAWK-Studie wurden die Patienten im Verhältnis 1:1:1 randomisiert und einem der folgenden Dosierungsschemata zugewiesen:
1.Brolucizumab 3 mg, verabreicht alle 12 oder 8 Wochen («q12w/q8w») nach den ersten 3 monatlichen Dosen.
2.Brolucizumab 6 mg, verabreicht alle 12 oder 8 Wochen («q12w/q8w») nach den ersten 3 monatlichen Dosen.
3.Aflibercept 2 mg, verabreicht alle 8 Wochen («q8w») nach den ersten 3 monatlichen Dosen.
In der HARRIER-Studie wurden die Patienten im Verhältnis 1:1 randomisiert und einem der folgenden Dosierungsschemata zugewiesen:
1.Brolucizumab 6 mg, verabreicht alle 12 oder 8 Wochen («q12w/q8w») nach den ersten 3 monatlichen Dosen.
2.Aflibercept 2 mg, verabreicht alle 8 Wochen («q8w») nach den ersten 3 monatlichen Dosen.
In beiden Studien wurden Brolucizumab-Patienten im Anschluss an die ersten 3 monatlichen Dosen (Wochen 0, 4 und 8) alle 12 Wochen behandelt, mit der Option, basierend auf der Krankheitsaktivität auf ein 8-wöchiges Behandlungsintervall umzustellen. Die Krankheitsaktivität wurde von einem Arzt im ersten 12-Wochen-Intervall (in Wochen 16 und 20) und bei jeder nachfolgend geplanten 12-Wochen-Behandlungsvisite beurteilt. Patienten, die bei einem dieser Besuche Krankheitsaktivität zeigten (z.B. verminderte Sehschärfe, erhöhte zentrale retinale Netzhautdicke (CST) oder Vorhandensein retinaler Flüssigkeiten (IRF/SRF, Sub-RPE), wurden auf ein 8-wöchiges Behandlungsintervall umgestellt.
Ergebnisse
Der primäre Wirksamkeitsendpunkt der Studien war die Veränderung der bestmöglich korrigierten Sehschärfe (BCVA) gegenüber Baseline in Woche 48, gemessen mithilfe der ETDRS-Buchstabentafeln, mit dem primären Ziel, die Nichtunterlegenheit von Beovu gegenüber Aflibercept nachzuweisen. In beiden Studien wurde die Nichtunterlegenheit in Bezug auf die Wirksamkeit von Beovu (verabreicht nach einem 12-/8-wöchigen Behandlungsschema) gegenüber Aflibercept 2 mg, verabreicht alle 8 Wochen, nachgewiesen.
In der Studie HAWK erreichten die Patienten in Woche 48 in der Beovu-6 mg- und Aflibercept-Gruppe eine mittlere Veränderung gegenüber Baseline von +6,6 Buchstaben bzw. +6,8 Buchstaben (p < 0,0001). Die mittlere Veränderung gegenüber der Baseline in der Beovu-3-mg-Gruppe lag bei +6,1 Buchstaben (p = 0,0003). Der Anteil der Patienten, die einen Zugewinn an Sehschärfe von mindestens 15 Buchstaben gegenüber Baseline erreichten, betrug 33,6 % in der Brolucizumab-Gruppe gegenüber 25,4 % in der Aflibercept-Gruppe. Der Anteil der Patienten, die 15 Buchstaben oder mehr Sehschärfe gegenüber dem Ausgangswert verloren, betrug 6,4 % in der 6 mg Brolucizumab-Gruppe gegenüber 5,5 % in der Aflibercept-Gruppe.
In der Studie HARRIER erreichten die Patienten in Woche 48 in der Beovu- und Aflibercept-Gruppe eine mittlere Veränderung gegenüber Baseline von +6,9 Buchstaben bzw. +7,6 Buchstaben (p < 0,0001). Der Anteil der Patienten, die einen Zugewinn an Sehschärfe von mindestens 15 Buchstaben gegenüber Baseline erreichten, lag in der Brolucizumab-Gruppe bei 29,3 % gegenüber 29,9 % in der Aflibercept-Gruppe. Der Anteil der Patienten, die gegenüber dem Ausgangswert 15 Buchstaben oder mehr Sehschärfe verloren, betrug 3,8 % in der 6 mg Brolucizumab-Gruppe gegenüber 4,8 % in der Aflibercept-Gruppe
Der im ersten Jahr beobachtete Zugewinn an Sehschärfe wurde im zweiten Jahr beibehalten.
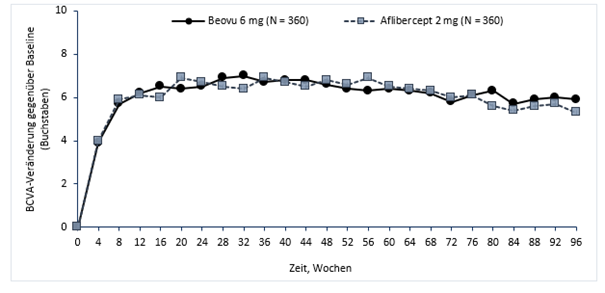
Abbildung 1: Mittlere Veränderung der Sehschärfe gegenüber Baseline bis Woche 96 in den Studien HAWK und HARRIER
HAWK
HARRIER
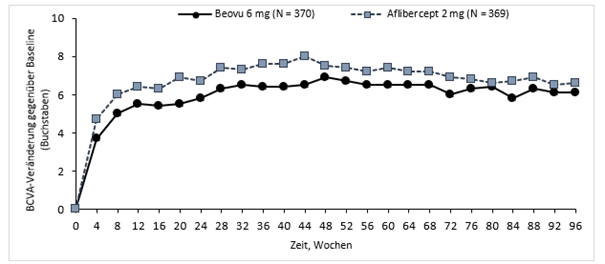
In den Studien HAWK und HARRIER erreichten 56 % bzw. 51 % der Patienten, die mit Beovu in einem 12-wöchigen Behandlungsintervall behandelt wurden, in Woche 48 diesen Zugewinn an Sehschärfe (mittlere Veränderung im Vergleich zum Ausgangswert), und 45 % bzw. 39 % der Patienten in Woche 96.
Von den Patienten, die während des ersten 12-wöchigen Behandlungsintervalls als geeignet für dieses Behandlungsintervall identifiziert worden waren, wurde bei 85 % bzw. 82 % das 12-wöchige Behandlungsintervall bis Woche 48 beibehalten. Bei 82 % bzw. 75 % der Patienten, die in Woche 48 mit dem 12-wöchigen Behandlungsintervall behandelt worden waren, wurde das 12-wöchige Behandlungsintervall von Woche 48 bis Woche 96 beibehalten.
Die Behandlungseffekte stimmten in den auswertbaren Untergruppen (z.B. Alter, Geschlecht, ethnische Zugehörigkeit, Sehschärfe bei Baseline, Netzhautdicke bei Baseline, Läsionstyp, Läsionsgrösse, Flüssigkeitsstatus) in beiden Studien weitgehend mit den Ergebnissen in der Gesamtpopulation überein.
Die Krankheitsaktivität wurde anhand von Veränderungen der Sehschärfe bzw. morphologischen Kriterien beurteilt, einschliesslich der zentralen retinalen Netzhautdicke (CST) und des Vorhandenseins retinaler Flüssigkeiten (IRF/SRF, Sub-RPE). In Woche 16, als die Krankheitsaktivität erstmalig zur Bestimmung des Behandlungsintervalls bewertet wurde, zeigten statistisch weniger mit Beovu behandelte Patienten eine Krankheitsaktivität als mit Aflibercept 2 mg behandelte Patienten (24% vs 35% in HAWK, p=0,0013; 23% vs 32% in HARRIER, p=0,0021). Die Krankheitsaktivität wurde jeweils über den Verlauf der gesamten Studien bewertet. Die morphologischen Kriterien der Krankheitsaktivität waren in Woche 48 und in Woche 96 in der Beovu-Gruppe im Vergleich zu Aflibercept geringer (Tabelle 1).
Tabelle 1: Bewertung der Krankheitsaktivität in den Studien HAWK und HARRIER bis Woche 96
|
|
|
HAWK
|
|
HARRIER
|
| |
Wirksamkeitsergebnis (präspezifizierte sekundäre Endpunkte)
|
In Woche
|
Beovu (N=360)
|
Aflibercept
2mg
(N=360)
|
Unterschied (95 %-KI) Brolucizumab und
Aflibercept
|
Beovu (N=360)
|
Aflibercept
2mg
(N=369)
|
Unterschied (95 %-KI) Brolucizumab und
Aflibercept
| |
Mittlere CST-Veränderung gegenüber Baseline (µm)
|
16 c)
|
-161,4
(SE = 6,2)
|
-133,6
(SE = 6,2)
|
-27,8
(-45,1, -10,5)
p = 0,0008 a)
|
-174,4
(SE = 6,7)
|
-134,2
(SE = 6,7)
|
-40,2
(-58,9, -21,6)
p<0,0001 a)
| |
48
|
-172,8
(SE = 6,7)
|
-143,7
(SE = 6,7)
|
-29,0
(-47,6, -10,4)
p = 0,0012 a)
|
-193,8
(SE = 6,8)
|
-143,9
(SE = 6,8)
|
-49,9
(-68,9, -30,9)
p<0,0001 a)
| |
96
|
-174,8
(SE = 7,3)
|
-148,7
(SE = 7,3)
|
-26,0
(-46,2, -5,9)
p = 0,0115 b)
|
-197,7
(SE = 7,0)
|
-155,1
(SE = 7,0)
|
-42,6
(-62,0, -23,3)
p<0,0001 b)
|
CST: zentrale retinale Netzhautdicke, IRF/SRF: intraretinale/subretinale Flüssigkeit, RPE: retinales Pigmentepithel
a) Sekundärer Endpunkt in HARRIER, bestätigende Analyse in HAWK. Einseitige p-Werte für die Überlegenheit von Brolucizumab
b) Sekundärer Endpunkt in HAWK und HARRIER; zweiseitige p-Werte
c) Bis Woche 16 war die Behandlungsexposition identisch, was einen abgestimmten Vergleich von Beovu mit Aflibercept ermöglichte.
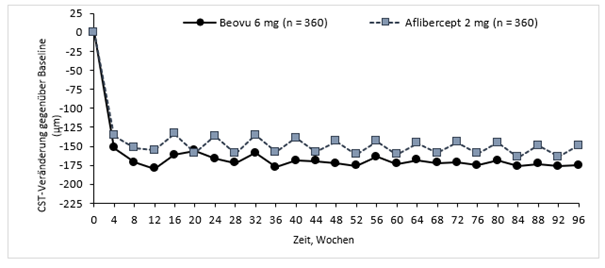
Abbildung 2: Veränderung der zentralen retinalen Netzhautdicke ab Baseline bis Woche 96 in den Studien HAWK und HARRIER
HAWK
HARRIER
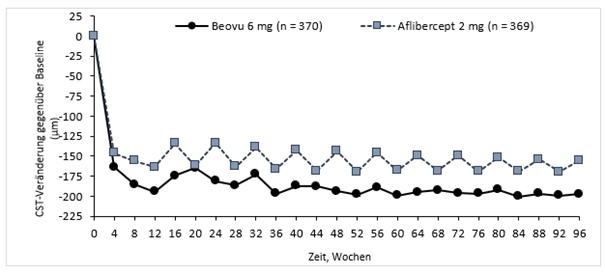
In beiden Studien führte die Behandlung mit Beovu zu klinisch relevanten Veränderungen gegenüber Baseline in Bezug auf den präspezifizierten sekundären Wirksamkeitsendpunkt «von Patienten berichtete Ergebnisse», die mithilfe des Fragebogens zur Augengesundheit des nationalen Augeninstituts der USA, NEI VFQ-25, aufgezeichnet wurden. Die Grössenordnung dieser Veränderungen war vergleichbar mit veröffentlichten Studien und entsprach einem Zugewinn von 15 Buchstaben bei der bestmöglich korrigierten Sehschärfe (BCVA). Der Nutzen der von den Patienten berichteten Ergebnisse wurde im zweiten Jahr aufrechterhalten.
Es wurden keine klinisch relevanten Unterschiede zwischen Beovu und Aflibercept hinsichtlich der Veränderungen des NEI VFQ-25-Gesamtscores und der Subskalen zwischen Baseline und Woche 48 festgestellt (generelle Sehkraft, Augenschmerzen, Nahsicht, Fernsicht, soziale Funktionsfähigkeit, psychisches Befinden, Schwierigkeiten bei der Ausübung sozialer Rollen, Abhängigkeit von anderen, Autofahren, Farbsehen und peripheres Sehen).
In-silico-Studie
Bei den Beovu-Armen der HAWK- und HARRIER-Studien wurden die ersten 3 Beovu-Dosen im Abstand von 4 Wochen (bzw. monatlich) verabreicht, gefolgt von einer Verabreichung alle 12 oder 8 Wochen («q12w/q8w»). Basierend auf den Ergebnissen einer Populations-PK/PD-Modell-Simulationsstudie sind 48 Wochen nach Behandlungsbeginn ähnliche CST und BCVA Veränderungen gegenüber Baseline zu erwarten, wenn die ersten 2 Beovu-Dosen im Abstand von 6 Wochen («q6w») verabreicht werden, gefolgt von einer Verabreichung alle 12 oder 8 Wochen («q12w/q8w»).
Behandlung von DME
Die Sicherheit und Wirksamkeit von Beovu wurde in zwei randomisierten, multizentrischen, doppelblinden, aktivkontrollierten Phase-III-Studien (KESTREL und KITE) bei Patienten mit diabetischem Makulaödem (DME) untersucht.
In diesen Studien wurden insgesamt 926 Patienten 2 Jahre lang behandelt (558 mit Brolucizumab und 368 mit Aflibercept 2 mg). Das Alter der Patienten lag zwischen 23 und 87 Jahren; mit einem Mittelwert von 63 Jahren.
In der KESTREL-Studie wurden die Patienten in einem Verhältnis von 1:1:1 randomisiert und einem der folgenden Dosierungsschemata zugewiesen:
·Brolucizumab 6 mg, verabreicht einmal alle 6 Wochen («q6w») für die ersten 5 Dosen, gefolgt von Brolucizumab 6 mg alle 12 oder 8 Wochen («q12w/q8w»).
·Brolucizumab 3 mg, verabreicht einmal alle 6 Wochen («q6w») für die ersten 5 Dosen, gefolgt von Brolucizumab 3 mg alle 12 oder 8 Wochen («q12w/q8w»).
·Aflibercept 2 mg, verabreicht einmal alle 4 Wochen («q4w») für die ersten 5 Dosen, gefolgt von Aflibercept 2 mg alle 8 Wochen («q8w»).
In der KITE-Studie wurden die Patienten im Verhältnis 1:1 randomisiert und einem der folgenden Dosierungsschemata zugewiesen:
·Brolucizumab 6 mg, verabreicht einmal alle 6 Wochen («q6w») für die ersten 5 Dosen, gefolgt von Brolucizumab 6 mg alle 12 oder 8 Wochen («q12w/q8w») oder alle 16 Wochen ab Woche 72 («q16w»).
·Aflibercept 2 mg, verabreicht einmal alle 4 Wochen («q4w») für die ersten 5 Dosen, gefolgt von Aflibercept 2 mg alle 8 Wochen («q8w»).
In beiden Studien wurden Brolucizumab-Patienten im Anschluss an die ersten fünf Dosen (in den Wochen 0, 6, 12, 18 und 24) alle 12 Wochen weiterbehandelt, mit der Option, basierend auf der Krankheitsaktivität auf ein 8-wöchiges Behandlungsintervall umzustellen. Die Krankheitsaktivität wurde von einem Arzt im ersten 12-wöchigen-Behandlungsintervall (in den Wochen 32 und 36) und bei jedem nachfolgend geplanten 12-Wochen-Behandlungsbesuch beurteilt. Patienten, die bei einem dieser Besuche Krankheitsaktivität zeigten (z.B. verminderte Sehschärfe, erhöhte zentrale retinale Netzhautdicke (CST)), wurden auf ein 8-wöchiges Behandlungsintervall umgestellt. In Jahr 2 der KITE-Studie konnten Patienten, die keine Krankheitsaktivität zeigten, auf ein 16-wöchiges Behandlungsintervall umgestellt werden. Das Vergleichspräparat Aflibercept wurde nach den ersten 5 monatlichen Dosen alle 8 Wochen verabreicht.
Ergebnisse
Der primäre Wirksamkeitsendpunkt für beide Studien war die Veränderung der bestmöglich korrigierten Sehschärfe (BCVA) gegenüber Baseline in Woche 52, gemessen mithilfe der ETDRS-Buchstabentafeln (Early Treatment Diabetic Retinopathy Study, ETDRS), mit dem primären Ziel, die Nichtunterlegenheit von Beovu gegenüber Aflibercept 2 mg nachzuweisen. In beiden Studien wurde die Nichtunterlegenheit in Bezug auf die Wirksamkeit von Beovu (verabreicht nach einem 12-/8-wöchigen Behandlungsschema) gegenüber Aflibercept 2 mg, verabreicht alle 8 Wochen, nachgewiesen.
Die Ergebnisse der KESTREL-und KITE-Studien zeigten auch eine Nichtunterlegenheit von Beovu gegenüber Aflibercept 2 mg für den wichtigsten sekundären Endpunkt (durchschnittliche Veränderung der bestmöglich korrigierten Sehschärfe gegenüber Baseline über den Zeitraum Woche 40 bis Woche 52)
Die mediane Anzahl der Injektionen über 24 Monate betrug 11 bei den mit Beovu behandelten Patienten gegenüber 15 bei den mit Aflibercept 2 mg behandelten Patienten.
Detaillierte Ergebnisse der beiden Studien sind in Tabelle 2 und Abbildung 3 unten dargestellt.
Tabelle 2: Ergebnisse zur Wirksamkeit in den Wochen 52 und 100 in den Phase-III-Studien KESTREL und KITE
|
|
|
KESTREL
|
KITE
| |
Ergebnis der Wirksamkeit
|
In Woche
|
Beovu (n=189)
|
Aflibercept
2 mg
(n=187)
|
Unterschied
(95 %-KI) Beovu - Aflibercept
|
Beovu (n=179)
|
Aflibercept
2 mg
(n=181)
|
Unterschied
(95 %-KI) Beovu - Aflibercept
| |
Veränderung der BCVA gegenüber Baseline, gemessen mithilfe der ETDRS-Buchstabentafeln, - LS-Mittelwert (SE)
|
52
|
9,2
(0,57)
|
10,5
(0,57)
|
-1,3
(-2,9, 0,3)
P < 0,001a
|
10,6
(0,66)
|
9,4
(0,66)
|
1,2
(-0,6; 3,1)
P < 0,001a
| |
40-52
|
9,0
(0,53)
|
10,5
(0,53)
|
-1,5
(-3,0; 0,0)
P < 0,001a
|
10,3
(0,62)
|
9,4
(0,62)
|
0,9
(-0,9; 2,6)
P < 0,001a
| |
100
|
8,8 (0,75)
|
10,6
(0,75)
|
-1,7
(-3,8, 0,4)
|
10,9
(0,85)
|
8,4
(0,85)
|
2,6
(0,2, 4,9)
| |
Verbesserung der BCVA um mindestens 15 Buchstaben gegenüber Baseline oder BCVA von mindestens 84 Buchstaben (%)
|
52
|
36,0
|
40,1
|
-4,1
(-13,3; 5,9)
|
46,8
|
37,2
|
9,6
(-0,4; 20,2)
| |
100
|
39,2
|
42,2
|
-3,0
(-12,5, 6,3)
|
50,4
|
36,9
|
13,6
(3,3, 23,5)
| |
Durchschnittliche Veränderung der CST gegenüber Baseline (Mikrometer) - LS-Mittelwert (SE)
|
40-52
|
-159,5 (5,88)
|
-158,1 (5,91)
|
-1,4
(-17,9; 15,0)
|
-187,1
(6,91)
|
-157,7
(6,89)
|
-29,4
(-48,6;-10,2)
P =0,001b
| |
88-100
|
-171,9 (6,18)
|
-168,5 (6,22)
|
-3,5
(-20,7, 13,8)
|
-196,6 (7,28)
|
-173,4 (7,26)
|
-23,2
(-43,5, -3,0)
| |
Vorhandene IRF und/oder SRF (%)
|
52
|
60,4
|
73,5
|
-13,2
(-23,2; -3,8)
|
54,5
|
72,9
|
-18,4
(-28,5;-8,3)
| |
100
|
41,8
|
54,2
|
-12,4
(-22,8, -2,1)
|
40,7
|
56,9
|
-16,2
(-26,4, -5,9)
| |
BCVA: Bestmöglich korrigierte Sehschärfe; BCVA-Beurteilungen nach Beginn der alternativen DME-Behandlung im Studienauge wurden zensiert und durch den letzten Wert vor Beginn dieser alternativen Behandlung ersetzt
CST: Zentrale retinale Netzhautdicke
IRF: Intraretinale Flüssigkeit; SRF: Subretinale Flüssigkeit
Beurteilung der CST und des Flüssigkeitsstatus nach Beginn einer alternativen DME-Behandlung im Studienauge wurde zensiert und durch den letzten Wert vor Beginn dieser alternativen Behandlung ersetzt
a p-Wert bezieht sich auf Nichtunterlegenheits-Hypothese mit einer Nichtunterlegenheitsspanne von 4 Buchstaben
b p-Wert bezieht sich auf den Überlegenheitstest bei einseitigem Typ-I-Fehler von 0,025
|
Abbildung 3: Mittlere Veränderung der Sehschärfe gegenüber Baseline bis Woche 100 in den Studien KESTREL und KITE
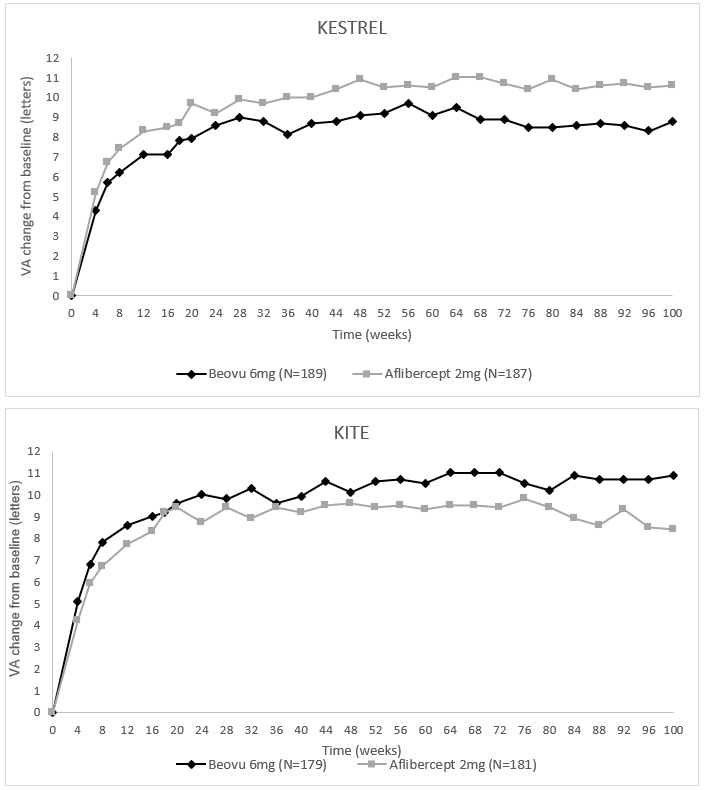
In den Studien KESTREL und KITE erreichten 55 % bzw. 50 % der Patienten, die mit Beovu 6 mg in einem 12-wöchigen Behandlungsintervall behandelt wurden, in Woche 52 diesen Zugewinn an Sehschärfe und 44 % bzw. 37 % der Patienten, die mit Beovu in einem 12-wöchigen bzw. 12-/16-wöchigen Behandlungsintervall behandelt wurden, in Woche 100. Von den Patienten, die während dieses ersten 12-wöchigen Intervalls als geeignet für ein 12-wöchiges Behandlungsintervall identifiziert worden waren, wurde in beiden Studien bei etwa 70 % mindestens das 12-wöchige Behandlungsintervall bis Woche 100 beibehalten. In der KITE-Studie wurden 25 % der Patienten in Woche 100 mit Beovu in einem 16-wöchigen Behandlungsintervall behandelt.
Die Behandlungseffekte stimmten in den auswertbaren Untergruppen (z.B. Alter, Geschlecht, HbA1c bei Baseline, Sehschärfe bei Baseline, Netzhautdicke bei Baseline, DME-Läsionstyp, Dauer des DME seit der Diagnose, retinaler Flüssigkeitsstatus) in beiden Studien weitgehend mit den Ergebnissen in der Gesamtpopulation überein.
In beiden Studien führte die Behandlung mit Beovu zu Verbesserungen gegenüber Baseline in Bezug auf den präspezifizierten sekundären Wirksamkeitsendpunkt «von Patienten berichtete Ergebnisse», die mithilfe des Fragebogens zur Augengesundheit des nationalen Augeninstituts der USA, NEI VFQ-25, aufgezeichnet wurden. Die Grössenordnung dieser Veränderungen war vergleichbar mit veröffentlichten Studien und entsprach einem Zugewinn von 15 Buchstaben bei der bestmöglich korrigierten Sehschärfe (BCVA).
Für andere Subskalen dieses Fragebogens konnten keine Behandlungsunterschiede zwischen Beovu und Aflibercept 2 mg gefunden werden.
Sowohl in der KESTREL- als auch in der KITE-Studie wurde der Schweregrad auf der Diabetes-Retinopathie-Schweregrad-Skala (Diabetic Retinopathy Severity Score, DRSS) ermittelt. Bei Baseline hatten 98,1 % der Patienten sowohl in der KESTREL- als auch in der KITE-Studie einen bestimmbaren Schweregrad der diabetischen Retinopathie. Basierend auf der gepoolten Analyse erfuhren 28,9 % der mit Beovu behandelten Patienten bis Woche 52 eine mindestens 2-stufige Verbesserung des DRSS-Werts gegenüber Baseline. Im Vergleich dazu waren es bei den mit Aflibercept 2 mg behandelten Patienten 24,9 %. Der geschätzte Unterschied zwischen Beovu und Aflibercept 2 mg betrug 4,0 % (95 %-KI: [-0,6, 8,6]). In Woche 100 betrug der Anteil der Patienten mit einer mindestens 2-stufigen Verbesserung des DRSS-Werts bis Woche 100 gegenüber Baseline in der KESTREL-Studie bei mit Beovu behandelten Patienten 32,8 % und bei mit Aflibercept 2 mg behandelten Patienten 29,3 % und in der KITE-Studie bei mit Beovu behandelten Patienten 35,8 % und bei mit Aflibercept 2 mg behandelten Patienten 31,1 %.
|