Eigenschaften/WirkungenATC-Code
R03AK06
Wirkungsmechanismus/Pharmakodynamik
Seffalair Spiromax enthält die Wirkstoffe Salmeterol und Fluticason-propionat, welche unterschiedliche Wirkungsmechanismen aufweisen.
Salmeterol: Salmeterol stimuliert selektiv die adrenergen β2-Rezeptoren und bewirkt durch seine lange Seitenkette, welche mit der Exo-Site des Rezeptors eine Bindung eingeht, einen wirksamen Schutz gegen Histamin-induzierte Bronchokonstriktion und eine langandauernde Bronchodilatation von über 12 Stunden.
In vitro-Tests haben gezeigt, dass Salmeterol wie auch andere β-Adrenergika die Freisetzung der Mastzellmediatoren (wie Histamin, Leukotriene und Prostaglandin D2) in der menschlichen Lunge hemmt.
Salmeterol hemmt beim Menschen die Sofort- und Spättypreaktion inhalierter Allergene; die Hemmung der Spättypreaktion dauert nach einer Einmaldosis bis über 30 Stunden, wenn der bronchodilatatorische Effekt schon nicht mehr nachweisbar ist. Bereits eine Einmaldosierung setzt die Hyperreaktivität der Bronchien herab.
Fluticason-propionat: Fluticason-propionat weist eine anti-inflammatorische Wirkung auf. Durch die inhalative Verabreichung kommt es direkt in der Lunge zur Wirkung und vermindert die Häufigkeit des Auftretens von Symptomen und Exazerbationen des Asthmas. Inhalativ verabreichtes Fluticason-propionat hat den Vorteil, dass die Nebennierenrindenfunktion bei der üblichen Dosierung innerhalb des normalen Bereiches bleibt.
Bei einer regelmässigen Anwendung vermindert Salmeterol beim Asthma die Symptome einer Bronchokonstriktion und Fluticason-propionat verbessert die Lungenfunktion und beugt eine Exazerbation der Erkrankung vor.
Klinische Wirksamkeit
Die Sicherheit und Wirksamkeit von Seffalair Spiromax wurden bei 3004 Patienten mit Asthma untersucht.
Das Entwicklungsprogramm umfasste 2 konfirmatorische Studien mit einer Dauer von 12 Wochen, eine 26-wöchige Sicherheitsstudie und drei Dosisfindungsstudien. Die Erkenntnisse zur Wirksamkeit von Seffalair Spiromax beruhen in erster Linie auf den nachfolgend beschriebenen Dosisfindungsstudien und konfirmatorischen Studien.
Dosisfindungsstudien
In 2 randomisierten, doppelblinden, placebokontrollierten 12-wöchigen Studien wurden 6 Dosierungen von Fluticasonpropionat im Bereich von 16 µg bis 434 µg (Nominaldosis) zweimal täglich über einen Multidosen-Pulverinhalator (MDPI) verabreicht. Studie 201 wurde mit Patienten mit zu Beginn der Studie unkontrolliertem Asthma durchgeführt, die mit einem kurz wirksamen Beta-2-Agonisten alleine oder in Kombination mit nicht-corticosteroidalen Asthmamedikamenten behandelt worden waren. Patienten, die niedrig dosierte inhalative Glucocorticoide (ICS) erhalten hatten, konnten nach einer Mindestauswaschzeit von 2 Wochen in die Studie aufgenommen werden. In dieser Studie diente zweimal täglich verabreichtes Fluticasonpropionat 100 µg Pulver zur Inhalation als unverblindetes Vergleichspräparat. Studie 202 wurde mit Patienten mit zu Beginn der Studie unkontrolliertem Asthma durchgeführt, die mit hochdosiertem ICS mit oder ohne lang wirksamen Beta-2-Agonisten (LABA) behandelt worden waren.
In dieser Studie diente Fluticasonpropionat 250 µg Pulver zur Inhalation zweimal täglich als unverblindetes Vergleichspräparat. Bei den Studien handelte es sich um Studien, die zur Dosisfindung der Fluticasonpropionat Monokomponente dienten (nachfolgend Fluticason Spiromax genannt, welche in den USA als Armonair RespiClick Inhalator zugelassen ist).
Die Studien waren nicht für die Erhebung von Vergleichsdaten zur Wirksamkeit ausgelegt und sollten nicht als Nachweis der Überlegenheit/Unterlegenheit gegenüber Fluticasonpropionat Pulver zur Inhalation interpretiert werden. Die in den Studien 201 und 202 für den Fluticason-Multidosen-Pulverinhalator verwendeten Nominaldosen (16, 28, 59, 118, 225, 434 µg) (siehe Abbildung 1) weichen geringfügig ab von den Nominaldosen für die Vergleichspräparate (Fluticason Pulver zur Inhalation) und für die Prüfpräparate für Phase III, auf welchen die Dosisstärken und Dosierungsempfehlungen der kommerziell erhältlichen Präparate gründen (55, 113, 232 µg für Fluticason). Die Dosisstärkenänderungen zwischen Phase II und III resultierten aus der Optimierung des Herstellungsprozesses.
Abbildung 1: Baseline-adjustierte Veränderung des Least-Square-Mittelwerts der morgendlichen Tal-FEV1 (l) nach 12 Wochen (FAS)a

FAS: Full Analysis Set;
a Die Studien waren nicht für die Erhebung von Vergleichsdaten zur Wirksamkeit ausgelegt und sollten nicht als Nachweis der Überlegenheit/Unterlegenheit gegenüber Fluticasonpropionat Pulver zur Inhalation interpretiert werden
Die Wirksamkeit und Sicherheit von vier Dosierungen Salmeterol xinafoat wurden in einer doppelblinden, 6-phasigen Crossover-Studie bei Patienten mit persistierendem Asthma im Vergleich zu einer Einzeldosis eines Fluticasonpropionat-MDPI und einem Fluticasonpropionat/Salmeterol-100/50 µg-Pulverinhalator als unverblindetem Vergleichspräparat beurteilt. Ziel der Studien war die Dosisfindung der Salmeterol-Komponente des Seffalair Spiromax; sie waren nicht für die Erhebung von Vergleichsdaten zur Wirksamkeit ausgelegt und sollten nicht als Nachweis der Überlegenheit/Unterlegenheit gegenüber Fluticasonpropionat/Salmeterol Pulver zur Inhalation interpretiert werden. Salmeterol wurde in Dosierungen von 6,8 µg, 13,2 µg, 26,8 µg und 57,4 µg in Kombination mit 118 µg Fluticasonpropionat untersucht, verabreicht mittels MDPI (Nominaldosen). Die in dieser Studie verwendeten Nominaldosen für Salmeterol (6,8 µg, 13,2 µg, 26,8 µg, 57,4 µg) weichen geringfügig ab von den Nominaldosen für die Vergleichspräparate und für die Prüfpräparate für Phase III, auf welchen die Dosisstärken und Dosierungsempfehlungen der kommerziell erhältlichen Präparate gründen (55 µg, 113 µg, 232 µg für Fluticason und 14 µg für Salmeterol). Die Präparate für Phase III und die kommerziellen Produkte wurden optimiert, um die Dosisstärken besser an die Vergleichspräparate anzupassen. In jeder Dosierphase wurde Plasma für die pharmakokinetische Charakterisierung gewonnen. Der Fluticasonpropionat/Salmeterolxinafoat-MDPI 118/13,2 µg wies im Vergleich zu den 50 µg Salmeterol im Fluticasonpropionat/Salmeterol-100/50 µg-Pulverinhalator eine ähnliche klinische Wirksamkeit bei geringerer systemischer Exposition auf (Abbildung 2).
Abbildung 2: Mittlere Baseline-adjustierte FEV1 (ml) nach 12 Stunden (FAS)a

FS MDPI: Fluticasonpropionat/Salmeterol-Multidosen-Pulverinhalator;
Fp MDPI: Fluticasonpropionat- Multidosen-Pulverinhalator;
FS DPI: Fluticasonpropionat/Salmeterol-Pulverinhalator;
FAS: Full Analysis Set;
FEV1: forciertes exspiratorisches Volumen in einer Sekunde;
a Die Studien waren nicht für die Erhebung von Vergleichsdaten zur Wirksamkeit ausgelegt und sollten nicht als Nachweis der Überlegenheit/Unterlegenheit gegenüber Fluticasonpropionat/ Salmeterol Pulver zur Inhalation interpretiert werden.
Studien zur Erhaltungstherapie bei Asthma
Erwachsene und jugendliche Patienten ab 12 Jahren:
Es wurden zwei klinische Studien der Phase III durchgeführt; 2 Studien zum Vergleich von Seffalair Spiromax mit Fluticason Spiromax alleine oder Placebo (Studie 1 und Studie 2).
Studien zum Vergleich von Seffalair Spiromax mit Fluticasonpropionat alleine oder Placebo
Mit Seffalair Spiromax wurden zwei doppelblinde klinische Studien im Parallelgruppen-Design durchgeführt (Studie 1 und Studie 2), an denen 1375 erwachsene und jugendliche Patienten im Alter ab 12 Jahren teilnahmen. Die Patienten hatten Asthma, das unter der bisherigen Therapie nicht optimal kontrolliert wurde und einen FEV1-Ausgangswert von 40 % bis 85 % des vorhergesagten Normwertes. Alle Behandlungen wurden als 1 Inhalation zweimal täglich mithilfe des Spiromax Inhalators verabreicht, andere Erhaltungstherapien wurden abgesetzt.
Studie 1: Diese randomisierte, doppelblinde, placebokontrollierte, 12-wöchige, weltweite Studie zur Beurteilung der Wirksamkeit und Sicherheit diente dem Vergleich des Fluticasonpropionat-Multidosen-Pulverinhalators (Fluticason Spiromax) 55 µg und 113 µg (1 Inhalation zweimal täglich) mit dem Fluticason/Salmeterol-Multidosen-Pulverinhalator (Seffalair Spiromax) 55/14 µg und 113/14 µg (1 Inhalation zweimal täglich) und Placebo bei jugendlichen und erwachsenen Patienten mit persistierendem symptomatischem Asthma, trotz Behandlung mit niedrig oder mittelhoch dosierten inhalativen Corticosteroiden oder inhalativen Corticosteroiden/LABA. Die Patienten erhielten einen einfach verblindeten Placebo-MDPI und wurden während der Run-in-Phase von ihrer ursprünglichen Therapie mit ICS auf QVAR 40 µg zweimal täglich umgestellt. Patienten, die alle Kriterien für die Randomisierung erfüllten, wurden nach dem Zufallsprinzip einer der folgenden Behandlungen zugewiesen: 130 Patienten erhielten das Placebo, 129 Patienten erhielten FLUTICASON SPIROMAX 55 µg, 130 Patienten erhielten FLUTICASON SPIROMAX 113 µg, 129 Patienten erhielten SEFFALAIR SPIROMAX 55/14 µg und 129 Patienten erhielten SEFFALAIR SPIROMAX 113/14 µg. Die Messungen des FEV1-Ausgangswertes waren über alle Behandlungsgruppen hinweg vergleichbar: FLUTICASON SPIROMAX 55 µg 2,132 l, FLUTICASON SPIROMAX 113 µg 2,166 l, SEFFALAIR SPIROMAX 55/14 µg 2,302 l, SEFFALAIR SPIROMAX 113/14 µg 2,162 l und Placebo 2,188 l. Die primären Endpunkte für diese Studie waren die Veränderung des Tal-FEV1 in Woche 12 gegenüber dem Ausgangswert für alle Patienten und die standardisierte Baseline-adjustierte Fläche unter der Plasmaeffekt-Zeitkurve über 12 Stunden (AUEC0-12h) für das FEV1 in Woche 12, die für eine Subgruppe von 312 Patienten analysiert wurde, die sich nach der Dosisverabreichung seriellen Spirometrien unterzogen.
Patienten, die SEFFALAIR SPIROMAX 55/14 µg und SEFFALAIR SPIROMAX 113/14 µg erhielten, zeigten im Vergleich zu FLUTICASON SPIROMAX 55 µg (Veränderung des LS-Mittelwertes um 0,172 l nach 12 Wochen), FLUTICASON SPIROMAX 113 µg (Veränderung des LS-Mittelwertes um 0,204 l nach 12 Wochen) und Placebo (Veränderung des LS-Mittelwertes um 0,053 l nach 12 Wochen) eine signifikant deutlichere Verbesserung des Tal-FEV1 (SEFFALAIR SPIROMAX 55/14 µg: Veränderung des LS-Mittelwertes um 0,319 l nach 12 Wochen und SEFFALAIR SPIROMAX 113/14 µg: Veränderung des LS-Mittelwertes um 0,315 l nach 12 Wochen). Die geschätzten Mittelwertsunterschiede zwischen SEFFALAIR SPIROMAX 55/14 µg und SEFFALAIR SPIROMAX 113/14 µg im Vergleich zu Placebo betrugen 0,266 l (95%-KI: 0,172, 0,360) bzw. 0,262 l (95%-KI: 0,168, 0,356).
Die geschätzten Mittelwertsunterschiede zwischen FLUTICASON SPIROMAX 55 µg und FLUTICASON SPIROMAX 113 µg im Vergleich zu Placebo betrugen 0,119 l (95%-KI: 0,025, 0,212) bzw. 0,151 l (95%-KI: 0,057, 0,244). Der geschätzte Mittelwertsunterschied zwischen SEFFALAIR SPIROMAX 113/14 µg und FLUTICASON SPIROMAX 113 µg betrug 0,111 l (95%-KI: 0,017, 0,206). Der geschätzte Mittelwertsunterschied zwischen SEFFALAIR SPIROMAX 55/14 µg und FLUTICASON SPIROMAX 55 µg betrug 0,147 l (95%-KI: 0,053, 0,242). Zusätzlich sind in Abbildung 3 die Ergebnisse für den FEV1-Mittelwert an den einzelnen Besuchsterminen angegeben.
Abbildung 3: Veränderung des Tal-FEV1-Mittelwertes gegenüber dem Ausgangswert an den jeweiligen Besuchsterminen nach Behandlungsgruppe, Studie 1 (FAS)
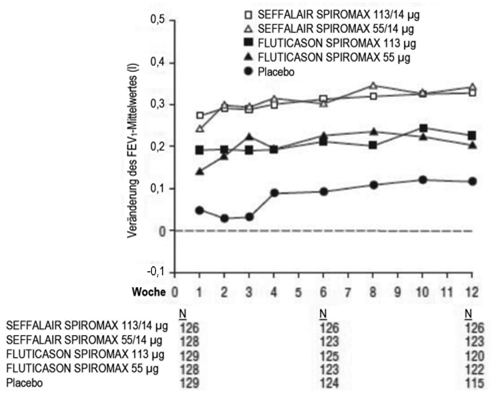
FAS: Full Analysis Set; FEV1: forciertes exspiratorisches Volumen in einer Sekunde
Im Hinblick auf die sekundären Endpunkte wurden unterstützende Wirksamkeitsnachweise für SEFFALAIR SPIROMAX im Vergleich zu Placebo erbracht, beispielsweise in Bezug auf den Wochendurchschnitt des täglichen morgendlichen Talwertes des exspiratorischen Spitzenflusses und die gesamte tägliche Anwendung von Bedarfsmedikamenten. In Studie 1 wurde der AQLQ (Asthma Quality of Life Questionnaire) für Patienten ab 18 Jahren bzw. der pädiatrische AQLQ (PAQLQ) für Patienten von 12‒17 Jahren bewertet. Die Responder-Rate wurde für beide Messinstrumente als Verbesserung des Punktwertes um 0,5 oder mehr als Grenzwert definiert. In Studie 1 lag die Responder-Rate unter den Patienten, die SEFFALAIR SPIROMAX 55/14 µg oder SEFFALAIR SPIROMAX 113/14 µg erhielten, bei 51 % bzw. 57 %, verglichen mit 40 % unter den Patienten, die das Placebo erhielten. Das Odds Ratio betrug 1,53 (95%-KI: 0,93, 2,55) bzw. 2,04 (95%-KI: 1,23, 3,41).
Die Besserung der Lungenfunktion trat innerhalb von 15 Minuten nach Verabreichung der ersten Dosis ein (15 Minuten nach Dosisgabe lag der Unterschied bezüglich der Veränderung des LS-Mittelwertes für das FEV1 gegenüber dem Ausgangswert für SEFFALAIR SPIROMAX 55/14 µg und 113/14 µg bei 0,216 l bzw. 0,164 l im Vergleich zu Placebo; der nicht adjustierte p-Wert betrug < 0,0001 für beide Dosen im Vergleich zu Placebo. Siehe Abbildung 4).
Die maximale Verbesserung des FEV1 trat im Allgemeinen innerhalb von 3 Stunden für SEFFALAIR SPIROMAX 55/14 µg und innerhalb von 6 Stunden für SEFFALAIR SPIROMAX 113/14 µg ein. Die Verbesserungen hielten in Woche 1 und 12 über die Prüfzeit von 12 Stunden an (Abbildung 4 und Abbildung 5). Nach der ersten Dosis verbesserte sich der vor der Dosisgabe bestimmte FEV1 über die erste Behandlungswoche hinweg gegenüber dem an Tag 1 gemessenen Ausgangswert deutlich und die Verbesserung hielt während der 12 Behandlungswochen der Studie an. Für keine der SEFFALAIR SPIROMAX Dosen wurde nach 12 Behandlungswochen eine Beeinträchtigung der anhand des FEV1-Wertes beurteilten bronchienerweiternden Wirkung über 12 Stunden beobachtet.
Abbildung 4: Serielle Spirometrie: Veränderung des FEV1-Mittelwertes gegenüber dem Ausgangswert (l) an Tag 1 nach Zeitpunkt und Behandlungsgruppe, Studie 1 (FAS; Subgruppe mit serieller Spirometrie)
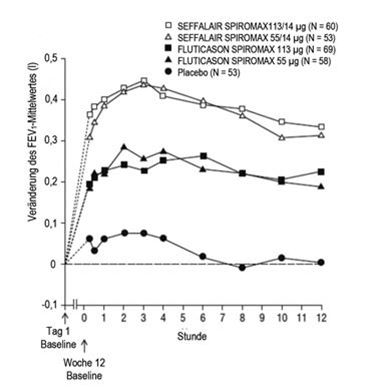
FAS: Full Analysis Set; FEV1: forciertes exspiratorisches Volumen in einer Sekunde
Abbildung 5: Serielle Spirometrie: Veränderung des FEV1-Mittelwertes gegenüber dem Ausgangswert (l) in Woche 12 nach Zeitpunkt und Behandlungsgruppe, Studie 1 (FAS; Subgruppe mit serieller Spirometrie)
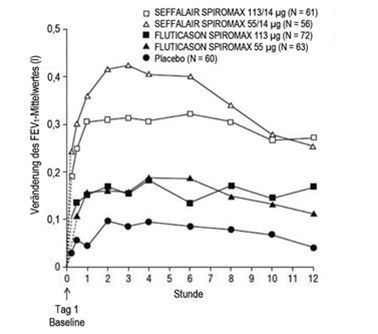
FAS: Full Analysis Set; FEV1: forciertes exspiratorisches Volumen in einer Sekunde
Studie 2: Diese randomisierte, doppelblinde, placebokontrollierte, 12-wöchige, weltweite Studie zur Beurteilung der Wirksamkeit und Sicherheit diente dem Vergleich des Fluticasonpropionat-Multidosen-Pulverinhalators (FLUTICASON SPIROMAX) 113 µg und 232 µg (1 Inhalation zweimal täglich) mit dem Fluticason/Salmeterol-Multidosen-Pulverinhalator (SEFFALAIR SPIROMAX) 113/14 µg und 232/14 µg (1 Inhalation zweimal täglich) und Placebo bei jugendlichen und erwachsenen Patienten mit persistierendem symptomatischem Asthma, trotz Behandlung mit niedrig oder mittelhoch dosierten inhalativen Corticosteroiden oder inhalativen Corticosteroiden/LABA. Die Patienten erhielten einen einfach verblindeten Placebo-MDPI und wurden während der Run-in-Phase von ihrer ursprünglichen Therapie mit ICS auf FLUTICASON SPIROMAX 55 µg zweimal täglich umgestellt. Patienten, die alle Kriterien für die Randomisierung erfüllten, wurden nach dem Zufallsprinzip einer der folgenden Behandlungen zugewiesen: 145 Patienten erhielten das Placebo, 146 Patienten erhielten FLUTICASON SPIROMAX 113 µg, 146 Patienten erhielten FLUTICASON SPIROMAX 232 µg, 145 Patienten erhielten SEFFALAIR SPIROMAX 113/14 µg und 146 Patienten erhielten SEFFALAIR SPIROMAX 232/14 µg. Die Messungen des FEV1-Ausgangswertes waren über alle Behandlungsgruppen hinweg vergleichbar: FLUTICASON SPIROMAX 113 µg 2,069 l, FLUTICASON SPIROMAX 232 µg 2,075 l, SEFFALAIR SPIROMAX 113/14 µg 2,157 l, SEFFALAIR SPIROMAX 232/14 µg 2,083 l und Placebo 2,141 l. Die primären Endpunkte für diese Studie waren die Veränderung des Tal-FEV1 in Woche 12 gegenüber dem Ausgangswert für alle Patienten und die standardisierte Baseline-adjustierte Fläche unter der Plasmaeffekt-Zeitkurve über 12 Stunden (AUEC0-12h) für das FEV1 in Woche 12, die für eine Subgruppe von 312 Patienten analysiert wurde, die sich nach der Dosisverabreichung seriellen Spirometrien unterzogen.
In dieser Studie wurden ähnliche Ergebnisse zur Wirksamkeit beobachtet, wie in Studie 1. Patienten, die SEFFALAIR SPIROMAX 113/14 µg und SEFFALAIR SPIROMAX 232/14 µg erhielten, zeigten im Vergleich zu FLUTICASON SPIROMAX 113 µg (Veränderung des LS-Mittelwertes von 0,119 l nach 12 Wochen), FLUTICASON SPIROMAX 232 µg (Veränderung des LS-Mittelwertes von 0,179 l nach 12 Wochen) und Placebo (Veränderung des LS-Mittelwertes von -0,004 l nach 12 Wochen) eine signifikant deutlichere Verbesserung des Tal-FEV1 (SEFFALAIR SPIROMAX 113/14 µg: Veränderung des LS-Mittelwertes von 0,271 l nach 12 Wochen und SEFFALAIR SPIROMAX 232/14 µg: Veränderung des LS-Mittelwertes von 0,272 l nach 12 Wochen). Die geschätzten Mittelwertsunterschiede zwischen SEFFALAIR SPIROMAX 113/14 µg und SEFFALAIR SPIROMAX 232/14 µg im Vergleich zu Placebo betrugen 0,274 l (95%-KI: 0,189, 0,360) bzw. 0,276 l (95%-KI: 0,191, 0,361).
Die geschätzten Mittelwertsunterschiede zwischen FLUTICASON SPIROMAX 113 µg und FLUTICASON SPIROMAX 232 µg im Vergleich zu Placebo betrugen 0,123 l (95%-KI: 0,038, 0,208) bzw. 0,183 l (95%-KI: 0,098, 0,268). Der geschätzte Mittelwertsunterschied zwischen SEFFALAIR SPIROMAX 232/14 µg und FLUTICASON SPIROMAX 232 µg betrug 0,093 l (95%-KI: 0,009, 0,178). Der geschätzte Mittelwertsunterschied zwischen SEFFALAIR SPIROMAX 113/14 µg und FLUTICASON SPIROMAX 113 µg betrug 0,152 l (95%-KI: 0,066, 0,237). Zusätzlich sind in Abbildung 6 die Ergebnisse für den FEV1-Mittelwert an den einzelnen Besuchsterminen angegeben.
Abbildung 6: Veränderung des Tal-FEV1-Mittelwertes gegenüber dem Ausgangswert an den jeweiligen Besuchsterminen nach Behandlungsgruppe, Studie 2 (FAS)
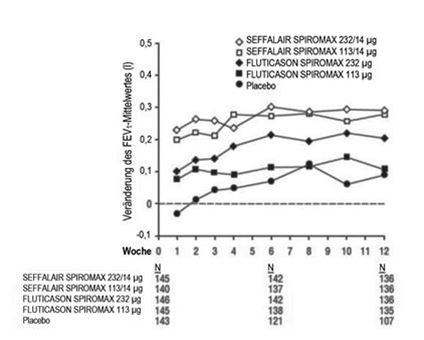
FAS: Full Analysis Set; FEV1: forciertes exspiratorisches Volumen in einer Sekunde
Im Hinblick auf die sekundären Endpunkte wurden unterstützende Wirksamkeitsnachweise für SEFFALAIR SPIROMAX im Vergleich zu Placebo erbracht, beispielsweise in Bezug auf den Wochendurchschnitt des täglichen morgendlichen Talwertes des exspiratorischen Spitzenflusses und die gesamte tägliche Anwendung von Bedarfsmedikamenten. Unter den mit SEFFALAIR SPIROMAX behandelten Patienten brach ein geringerer Anteil die Studie aufgrund einer Verschlimmerung des Asthmas ab, als unter den Patienten, die das Placebo erhielten. In Studie 2 wurde der AQLQ (Asthma Quality of Life Questionnaire) für Patienten ab 18 Jahren bzw. der pädiatrische AQLQ (PAQLQ) für Patienten von 12‒17 Jahren bewertet. Die Responder-Rate wurde für beide Messinstrumente als Verbesserung des Punktwertes um 0,5 oder mehr als Grenzwert definiert. In Studie 2 lag die Responder-Rate unter den Patienten, die SEFFALAIR SPIROMAX 113/14 µg oder SEFFALAIR SPIROMAX 232/14 µg erhielten, bei 48 % bzw. 41 %, verglichen mit 27 % unter den Patienten, die das Placebo erhielten. Das Odds Ratio betrug 2,59 (95%-KI: 1,56, 4,31) bzw. 1,94 (95%-KI: 1,16, 3,23).
Die Besserung der Lungenfunktion trat innerhalb von 15 Minuten nach Verabreichung der ersten Dosis ein (15 Minuten nach Dosisgabe lag der Unterschied bezüglich der Veränderung des LS-Mittelwertes für das FEV1 gegenüber dem Ausgangswert für SEFFALAIR SPIROMAX 113/14 µg und 232/14 µg bei 0,160 l bzw. 0,187 l im Vergleich zu Placebo; der nicht adjustierte p-Wert betrug < 0,0001 für beide Dosen im Vergleich zu Placebo). Die maximale Verbesserung des FEV1 trat in beiden SEFFALAIR SPIROMAX Dosisgruppen im Allgemeinen innerhalb von 3 Stunden ein. Die Verbesserungen hielten in Woche 1 und 12 über die Prüfzeit von 12 Stunden an (Abbildung 7 und Abbildung 8). Nach der ersten Dosis verbesserte sich der vor der Dosisgabe bestimmte FEV1 über die erste Behandlungswoche hinweg gegenüber dem an Tag 1 gemessenen Ausgangswert deutlich und die Verbesserung hielt während der 12 Behandlungswochen der Studie an. Für keine der SEFFALAIR SPIROMAX Dosen wurde nach 12 Behandlungswochen eine Beeinträchtigung der anhand des FEV1-Wertes beurteilten bronchienerweiternden Wirkung über 12 Stunden beobachtet.
Abbildung 7: Serielle Spirometrie: Veränderung des FEV1-Mittelwertes gegenüber dem Ausgangswert (l) an Tag 1 nach Zeitpunkt und Behandlungsgruppe, Studie 2 (FAS; Subgruppe mit serieller Spirometrie)
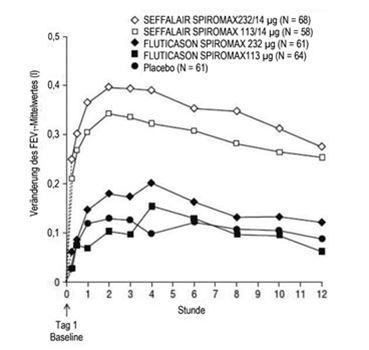
FAS: Full Analysis Set; FEV1: forciertes exspiratorisches Volumen in einer Sekunde
Abbildung 8: Serielle Spirometrie: Veränderung des FEV1-Mittelwertes gegenüber dem Ausgangswert (l) in Woche 12 nach Zeitpunkt und Behandlungsgruppe, Studie 2 (FAS; Subgruppe mit serieller Spirometrie)
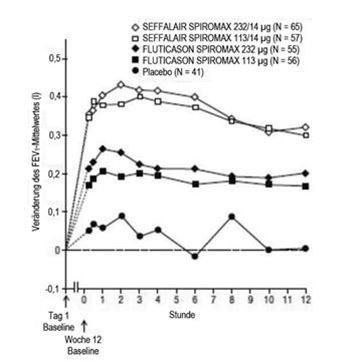
FAS: Full Analysis Set; FEV1: forciertes exspiratorisches Volumen in einer Sekunde
Fluticasonpropionat (FP)-haltige Arzneimittel zur Behandlung von Asthma während der Schwangerschaft
In einer retrospektiven, epidemiologischen Kohortenbeobachtungsstudie wurde das Risiko erheblicher angeborener Missbildungen (EAM) nach einer Exposition gegenüber inhalativem FP allein und Salmeterol-FP im ersten Trimenon im Vergleich zu nicht-FP-haltigen ICS (Inhaled Corticosteroids)-Präparaten anhand elektronischer Patientenakten aus UK ausgewertet. Bei dieser Studie wurde kein Placebovergleich eingeschlossen.
In der Asthmakohorte der 5362 Schwangerschaften mit ICS-Exposition im ersten Trimenon wurden 131 diagnostizierte EAM identifiziert. Bei 1612 (30%) hatte eine Exposition gegenüber FP oder Salmeterol-FP stattgefunden, mit 42 diagnostizierten Fällen von EAM. Die adjustierte Odds-Ratio für eine innerhalb eines Jahres diagnostizierte EAM unter FP-Exposition vs nicht-FP ICS-Exposition betrug 1,1 (95%-KI: 0,5–2,3) bei Frauen mit Asthmabehandlung analog GINA-Stufe 2, und 1,2 (95%-KI: 0,7–2,0) bei Frauen mit Asthmabehandlung analog GINA-Stufe 3 oder höher. Es wurde kein Unterschied bezüglich des EAM-Risikos bei FP-Exposition allein im Vergleich zu Salmeterol-FP-Exposition im ersten Trimenon festgestellt. Das absolute Risiko für EAM über alle Schweregrade der Asthmaerkrankung lag bei 2,0 bis 2,9 pro 100 Schwangerschaften mit FP-Exposition, was vergleichbar ist mit den Resultaten einer Studie bei 15'840 Schwangerschaften ohne Exposition gegenüber Asthmamedikamenten aus der «General Practice Research Database» (2,8 EAM pro 100 Schwangerschaften).
|