ZusammensetzungWirkstoffe
Zilucoplan (als Zilucoplan-Natrium)
Hilfsstoffe
Natriumdihydrogenphosphat-Monohydrat
Natriummonohydrogenphosphat
Natriumchlorid
Wasser für Injektionszwecke
Jede Fertigspritze mit 0,416 ml enthält 1,72–2,05 mg Natrium.
Jede Fertigspritze mit 0,574 ml enthält 2,39–2,85 mg Natrium.
Jede Fertigspritze mit 0,810 ml enthält 3,37–4,01 mg Natrium.
Indikationen/AnwendungsmöglichkeitenZilbrysq wird angewendet als Zusatztherapie zur Standardbehandlung der generalisierten Myasthenia gravis (gMG) bei erwachsenen Patienten, die Anti-Acetylcholinrezeptor(AChR)-Antikörper-positiv sind.
Dosierung/AnwendungZilbrysq ist für die Anwendung unter Anleitung und Aufsicht von medizinischem Fachpersonal vorgesehen, das Erfahrung in der Behandlung von Patienten mit neuromuskulären Erkrankungen hat.
Vor Beginn der Therapie mit Zilbrysq müssen die Patienten gegen Neisseria meningitidis geimpft werden. Wenn die Behandlung mit Zilbrysq weniger als 2 Wochen nach der Impfung gegen eine Meningokokken-Infektion beginnen muss, muss der Patient bis 2 Wochen nach der ersten Impfung eine geeignete prophylaktische Antibiotikabehandlung erhalten (siehe Abschnitte «Kontraindikationen» und «Warnhinweise und Vorsichtsmassnahmen»).
Übliche Dosierung
Die empfohlene Dosis ist als subkutane Injektion einmal täglich und jeden Tag ungefähr zur gleichen Uhrzeit zu verabreichen.
Tabelle 1: Tagesgesamtdosis gemäss Körpergewicht
|
Körpergewicht des Patienten
|
Dosis*
|
Anzahl der Fertigspritzen (Farbe)
| |
Weniger als 56 kg
|
16,6 mg
|
1 rubinrote Fertigspritze
| |
56 bis weniger als 77 kg
|
23 mg
|
1 orangefarbene Fertigspritze
| |
77 kg und mehr
|
32,4 mg
|
1 dunkelblaue Fertigspritze
|
* Die empfohlene Dosis entspricht etwa 0,3 mg/kg
Zilucoplan wurde bei gMG-Patienten mit Myasthenia Gravis Foundation of America (MGFA) Klasse V nicht untersucht.
Es liegen nur begrenzte Erfahrungen mit Patienten unter 43 kg und über 150 kg vor.
Versäumte Dosis
Wenn eine Dosis Zilbrysq ausgelassen wurde, soll sie noch am selben Tag verabreicht werden; danach ist am nächsten Tag mit der üblichen Anwendung fortzufahren. Es soll nicht mehr als eine Dosis pro Tag verabreicht werden.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit leichter und mittelschwerer Leberfunktionsstörung ist keine Dosisanpassung erforderlich. Für Patienten mit schwerer Leberfunktionsstörung liegen keine Daten vor (siehe Abschnitt «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit Nierenfunktionsstörung ist keine Dosisanpassung erforderlich. Es liegen keine Daten zu dialysepflichtigen Patienten vor (siehe Abschnitt «Pharmakokinetik»).
Ältere Patienten
Bei älteren Patienten ist keine Dosisanpassung erforderlich (siehe Abschnitt «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Zilbrysq bei Kindern und Jugendlichen ist nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Zilbrysq wird als subkutane Injektion angewendet.
Geeignete Injektionsstellen sind die Vorderseite der Oberschenkel, der Bauch und die Rückseite der Oberarme (siehe Abschnitt «Pharmakokinetik» in diesem Dokument und «Hinweise zur Anwendung» in der Patienteninformation).
Die Injektionsstellen sollten täglich gewechselt werden und die Injektionen sollten nicht in Bereichen verabreicht werden, in denen die Haut empfindlich, erythematös, blutunterlaufen oder verhärtet ist oder in denen die Haut Narben oder Dehnungsstreifen aufweist.
Die Verabreichung sollte durch eine Person erfolgen, die in die Injektionsmethode eingewiesen wurde. Die detaillierten Anweisungen unter «Hinweise zur Anwendung» in der Patienteninformation sind zu befolgen.
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der im Abschnitt «Zusammensetzung» genannten sonstigen Bestandteile.
Patienten, die derzeit nicht gegen Neisseria meningitidis geimpft sind (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Patienten mit nicht abgeklungener Neisseria meningitidis-Infektion.
Warnhinweise und VorsichtsmassnahmenNeisseria-Infektionen
Meningokokken-Infektion
Aufgrund seines Wirkmechanismus kann die Anwendung von Zilbrysq die Anfälligkeit des Patienten für Infektionen mit Neisseria meningitidis erhöhen. Aus Vorsichtsgründen müssen alle Patienten mindestens 2 Wochen vor Beginn der Behandlung mit Zilbrysq gegen Meningokokken-Infektionen geimpft werden.
Wenn die Behandlung mit Zilbrysq weniger als 2 Wochen nach der Impfung gegen Meningokokken-Infektionen beginnen muss, muss der Patient bis 2 Wochen nach der ersten Impfung eine geeignete prophylaktische Antibiotikabehandlung erhalten. Meningokokken-Impfstoffe reduzieren das Risiko von Meningokokken-Infektionen, schliessen sie aber nicht vollständig aus.
Impfstoffe gegen die Serogruppen A, C, Y, W und, sofern verfügbar, gegen Serogruppe B werden zur Prävention der häufig pathogenen Meningokokken-Serogruppen empfohlen. Die Impfung und die prophylaktische Antibiotikabehandlung sollen gemäss dem aktuellen Schweizerischen Impfplan erfolgen.
Während der Behandlung mit Zilbrysq sollten die Patienten auf Anzeichen und Symptome einer Meningokokken-Infektion überwacht und bei Verdacht auf eine Infektion sofort untersucht werden. Bei Verdacht auf eine Meningokokken-Infektion sind geeignete Massnahmen wie die Behandlung mit Antibiotika und das Absetzen der Behandlung mit Zilbrysq zu ergreifen, bis eine Meningokokken-Infektion ausgeschlossen werden kann. Die Patienten sollten angewiesen werden, sofort ärztlichen Rat einzuholen, wenn Anzeichen oder Symptome von Meningokokken-Infektionen auftreten.
Die verordnenden Ärzte sollten mit den Schulungsmaterialien zur Behandlung von Meningokokken-Infektionen vertraut sein und den mit Zilucoplan behandelten Patienten eine Patientenkarte («Patientenkarte zur sicheren Anwendung») und einen Leitfaden («Leitfaden für die sichere Anwendung - Patienten und Betreuungspersonen») zur Verfügung stellen.
Andere Neisseria-Infektionen
Zusätzlich zu Neisseria meningitidis können Patienten, die mit Zilbrysq behandelt werden, auch anfällig für Infektionen mit anderen Neisseria-Arten sein, wie z.B. Gonokokken-Infektionen. Die Patienten sollten über die Bedeutung der Vorbeugung und Behandlung von Gonorrhö informiert werden.
Immunisierung
Es wird empfohlen, dass sich Patienten vor Beginn der Zilucoplan-Therapie Immunisierungen gemäss den aktuellen Impfempfehlungen unterziehen.
Natriumgehalt
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Fertigspritze, d.h. es ist nahezu natriumfrei.
InteraktionenAufgrund der Ergebnisse von In-vitro-Tests sind keine klinisch relevanten Interaktionen zwischen Zilucoplan und Inhibitoren oder Induktoren der wichtigsten CYP-Enzyme und Transportern zu erwarten.
Das Potential von Zilucoplan, CYP-Enzyme (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A und 4F2) und UGTs (1A1, 1A3, 1A4, 1A6, 1A9, 2B7 und 2B15) oder Transporter (P-gp, BCRP, BSEP, MRP2, MRP3, MATE1, MATE2-k, NTCP, OCT1, OCT2, OAT1, OAT3, OATP1B1 und OATP1B3) zu hemmen, wurde in vitro untersucht. Ausserdem wurde das Potential einer CYP-Induktion von CYP1A2, 2B6 und CYP3A4 durch Zilucoplan untersucht.
Zilucoplan hemmt in therapeutischen Konzentrationen MRP3 in vitro; die klinische Bedeutung für diese Inhibition ist nicht bekannt.
Aufgrund der potenziellen Hemmwirkung von Zilucoplan auf die komplementabhängige Zytotoxizität von Rituximab kann Zilucoplan die erwarteten pharmakodynamischen Wirkungen von Rituximab mindern.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine Daten zur Anwendung von Zilbrysq bei schwangeren Frauen vor.
Tierexperimentelle Studien zeigten einen leichten Anstieg der pränatalen Verluste im Vergleich zu historischen Kontrolldaten, jedoch keine Auswirkungen auf die Geburt, die postnatale Entwicklung des Säuglings oder die postnatalen Verluste (siehe Abschnitt «Präklinische Daten»).
Die Anwendung von Zilucoplan während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen und sollte nur in Betracht gezogen werden, wenn der klinische Nutzen die Risiken überwiegt.
Stillzeit
Es ist nicht bekannt, ob Zilucoplan in die Muttermilch ausgeschieden wird oder nach der Aufnahme durch das Neugeborene/Kind systemisch resorbiert wird. Ein Risiko für das Neugeborene/Kind kann nicht ausgeschlossen werden.
Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist, oder ob die Behandlung mit Zilucoplan zu unterbrechen ist. Dabei soll sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau berücksichtigt werden.
Fertilität
Die Wirkung von Zilucoplan auf die Fertilität des Menschen wurde nicht untersucht. In einigen Studien zur Fertilität und Toxizität bei wiederholter Gabe an nicht-humane Primaten wurden an den männlichen und weiblichen Reproduktionsorganen Befunde unklarer klinischer Bedeutung beobachtet (siehe Abschnitt «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenZilbrysq hat keinen oder einen zu vernachlässigenden Einfluss auf die Fahrtüchtigkeit und die Fähigkeit, Maschinen zu bedienen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
In placebokontrollierten klinischen Studien zu gMG wurden insgesamt 115 Patienten mit Zilucoplan behandelt; dies entspricht 26,4 Patientenjahren der Exposition. Die am häufigsten berichteten unerwünschten Wirkungen waren Reaktionen an der Injektionsstelle und Infektionen der oberen Atemwege.
Liste der unerwünschten Wirkungen
Tabelle 2 stellt die Nebenwirkungen für Zilucoplan nach folgender Häufigkeitsklassifizierung dar: sehr häufig (≥1/10), häufig (≥1/100, < 1/10), gelegentlich (≥1/1 000, < 1/100), selten (≥1/10 000, < 1/1 000), sehr selten (< 1/10 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Die Häufigkeit der Nebenwirkungen in Tabelle 2 basiert auf Daten aus den gepoolten placebokontrollierten (n=115) Studien in gMG, mit Ausnahme von Morphea, welches nur in Langzeit-Open-Label-Erweiterungsstudien (n=213) in gMG berichtet wurde.
Tabelle 2: Unerwünschte Wirkungen
|
Systemorganklasse
|
Häufigkeit
|
Unerwünschte Wirkung
| |
Infektionen und parasitäre Erkrankungen
|
Sehr häufig
|
Infektionen der oberen Atemwege (13,0 %)
| |
Erkrankungen des Gastrointestinaltrakts
|
Häufig
|
Durchfall
| |
Erkrankungen der Haut und des Unterhautgewebes
|
Häufig
|
Morpheaa
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Sehr häufig
|
Reaktionen an der Injektionsstelle (25,2 %)
| |
Untersuchungen
|
Häufig
|
Erhöhte Lipase
| |
Häufig
|
Erhöhte Amylase
| |
Gelegentlich
|
Erhöhte Eosinophile im Blut
|
aMorphea wurde nur in offenen klinischen Langzeitstudien berichtet. Die maximale Expositionsdauer gegenüber Zilucoplan während der klinischen Langzeitstudien betrug mehr als 4 Jahre.
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Reaktionen an der Injektionsstelle
Die häufigsten Reaktionen waren Blutergüsse, Schmerzen, Knötchen, Pruritus und Hämatome an der Injektionsstelle. Alle Fälle waren nicht schwerwiegend, leicht oder mittelschwer, und weniger als 3 % der Reaktionen führten zum Abbruch der Behandlung. In gepoolten placebokontrollierten Studien wurden Reaktionen an der Injektionsstelle bei 25,2 % der mit Zilucoplan behandelten Patienten und bei 15,5 % der mit Placebo behandelten Patienten berichtet.
Infektionen der oberen Atemwege
Die häufigsten Infektionen waren Nasopharyngitis, Infektionen der oberen Atemwege und Sinusitis. Mehr als 95 % der Fälle waren nicht schwerwiegend, leicht oder mittelschwer, und führten nicht zum Abbruch der Behandlung. In gepoolten placebokontrollierten Studien wurden Infektionen der oberen Atemwege bei 13,0 % der mit Zilucoplan behandelten Patienten und bei 7,8 % der mit Placebo behandelten Patienten berichtet.
Erhöhte Pankreasenzyme
Es wurden Fälle von erhöhter Lipase (5.2%) und/oder Amylase (6.1%) beobachtet. Diese Erhöhungen waren vorübergehend und führten selten zum Abbruch der Behandlung. Sie traten mehrheitlich innerhalb von 2 Monaten nach Beginn der Behandlung mit Zilucoplan auf und normalisierten sich innerhalb von 2 Monaten.
Erhöhte Eosinophile im Blut
Es wurden Erhöhungen der Eosinophilen im Blut beobachtet. Diese waren vorübergehend, führten nicht zum Abbruch der Behandlung und waren nicht mit klinisch relevanten Organdysfunktionen assoziiert.
Morphea
Fälle von Morphea wurden nach Langzeitbehandlung während der offenen Verlängerungsstudie beobachtet. Bei der Mehrzahl der Fälle betrug die Zeit bis zum Auftreten mehr als ein Jahr nach Beginn der Behandlung, sie waren leicht oder mittelschwer und führten nicht zum Behandlungsabbruch.
Immunogenität
Wie bei allen therapeutischen Peptiden besteht auch bei Zilucoplan die Möglichkeit einer Immunogenität. Der Nachweis der Bildung von Anti-Drug-Antikörpern ist in hohem Masse abhängig von der Sensitivität und Spezifität des verwendeten Assays. Ausserdem kann die beobachtete Inzidenz des Vorhandenseins von Anti-Drug-Antikörpern durch verschiedene Faktoren beeinflusst werden, wie z.B. Methode des Assays, Handhabung der Proben, Zeitpunkt der Probennahme, Begleitmedikation und zugrundeliegende Krankheit. Aus diesen Gründen kann ein Vergleich der Inzidenz von Antikörpern gegen Zilucoplan mit der Inzidenz von Antikörpern gegen andere Wirkstoffe irreführend sein.
In MG0010 wurden die Patienten auf das Vorhandensein von Anti-Drug-Antikörpern (ADA) und Antikörpern gegen Polyethylenglycol (PEG) untersucht. Insgesamt waren jeweils 2 Patienten (2,3 %) in der Zilucoplan-Gruppe (0,3mg/kg) und in der Placebo-Gruppe ADA-positiv. Insgesamt 8 Patienten (9,3 %) in der Zilucoplan-Gruppe (0,3 mg/kg) und 6 Patienten (6,8 %) in der Placebo-Gruppe wurden positiv für PEG-Antikörper getestet. Die Antikörper-Titer waren niedrig und es gab keine Hinweise auf einen Zusammenhang zwischen positivem ADA-Status oder positivem Anti-PEG-Status und der Inzidenz unerwünschter Wirkungen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIn einer Studie mit gesunden Freiwilligen, in der 32 Teilnehmer supratherapeutische Dosen von 0,6 mg/kg erhielten, die bis 7 Tage lang subkutan verabreicht wurden, stimmten die Sicherheitsdaten mit dem Sicherheitsprofil der empfohlenen Dosis überein.
Im Falle einer Überdosierung wird empfohlen, die Patienten engmaschig auf unerwünschte Wirkungen zu überwachen und umgehend geeignete unterstützende Massnahmen einzuleiten.
Eigenschaften/WirkungenATC-Code:
L04AJ06
Wirkungsmechanismus
Zilucoplan hemmt die Wirkung von C5 durch einen dualen Wirkungsmechanismus. Es bindet spezifisch an das Komplementprotein C5 und hemmt dadurch dessen Spaltung durch die C5-Konvertase zu C5a und C5b, was zu einer Herunterregulierung der Assemblierung und zytolytischen Aktivität des Membranangriffskomplexes (MAC) führt. Darüber hinaus hindert Zilucoplan durch die Bindung an das C5b-Fragment von C5 sterisch die Bindung von C5b an C6, was die nachfolgende Assemblierung und Aktivität des MAC verhindert, sollte C5b gebildet werden.
Pharmakodynamik
Die pharmakodynamische Wirkung von Zilucoplan wurde anhand der Fähigkeit analysiert, die Ex-vivo-Lyse von Komplement-induzierten Schaf-Erythrozyten (sRBC) zu hemmen.
Daten aus den Phase-II- und Phase-III-Studien zeigen bei einer Dosierung gemäss Tabelle 1 eine schnelle, vollständige (> 95 %) und anhaltende Komplementinhibition mit Zilucoplan.
Klinische Wirksamkeit
Die Sicherheit und Wirksamkeit von Zilucoplan wurden in der 12-wöchigen multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie MG0010 (RAISE) und der offenen Verlängerungsstudie MG0011 (RAISE-XT) beurteilt.
MG0010
Insgesamt wurden 174 Patienten eingeschlossen, die mindestens 18 Jahre alt waren, eine Acetylcholin-Rezeptor-Antikörper-positive, generalisierte Myasthenia gravis, Klasse II–IV nach MGFA (leicht bis schwer), einen Myasthenia-Gravis-Score für Aktivitäten des täglichen Lebens (MG-ADL) ≥6 und einen quantitativen Myasthenia-Gravis-Score (QMG-Score) ≥12 hatten (siehe Tabelle 3).
Die Patienten wurden einmal täglich entweder mit Zilucoplan (Dosierung gemäss Tabelle 1) oder mit Placebo behandelt. Eine stabile Standardtherapie (SOC) war erlaubt.
Der primäre Endpunkt war die Veränderung des Myasthenia Gravis Activities of Daily Living Score (MG-ADL)-Gesamtscores gegenüber Baseline (change from baseline, CFB) bis Woche 12. Die wichtigsten sekundären Endpunkte waren die Veränderungen im Quantitative Myasthenia Gravis (QMG)-Gesamtscore, im Myasthenia Gravis Composite (MGC)-Gesamtscore und im Myasthenia Gravis Quality of Life (MG-QoL15r)-Gesamtscore gegenüber Baseline bis Woche 12 im.
Tabelle 3: Demografische Charakteristika und Krankheitsmerkmale von Patienten, die in die Studie MG0010 aufgenommen wurden, bei Baseline
|
|
Zilucoplan (n= 86)
|
Placebo (n = 88)
| |
Alter, Jahre, Mittelwert (SD)
|
52,6 (14,6)
|
53,3 (15,7)
| |
Alter bei Krankheitsbeginn, Jahre, Mittelwert (SD)
|
43,5 (17,4)
|
44,0 (18,7)
| |
Geschlecht, männlich, n (%)
|
34 (39,5)
|
41 (46,6)
| |
Baseline-MG-ADL-Score, Mittelwert (SD)
|
10,3 (2,5)
|
10,9 (3,4)
| |
Baseline-QMG-Score, Mittelwert (SD)
|
18,7 (3,6)
|
19,4 (4,5)
| |
Baseline-MGC-Score, Mittelwert (SD)
|
20,1 (6,0)
|
21,6 (7,2)
| |
Baseline-MG-QoL 15r-Score, Mittelwert (SD)
|
18,6 (6,6)
|
18,9 (6,8)
| |
Refraktär gegenüber der Behandlung, ja, n (%)
|
44 (51,2)
|
44 (50,0)
| |
Krankheitsdauer, Jahre, Mittelwert (SD)
|
9,3 (9,5)
|
9,0 (10,4)
|
Die Mehrzahl der Studienteilnehmer waren zu Studienbeginn bereits mit Parasympathomimetika (84,5 %), systemischen Kortikosteroide (63,2 %) und Immunsuppressiva (51,1 %) behandelt.
Der Behandlungseffekt in der Zilucoplan-Gruppe für alle 4 Endpunkte begann schnell in Woche 1, stieg bis Woche 4 weiter an und blieb bis Woche 12 erhalten.
In Woche 12 wurde eine klinisch bedeutsame und statistisch hochsignifikante Verbesserung des MG-ADL-Gesamtscores (Abbildung 1) und des QMG-Gesamtscores für Zilucoplan gegenüber Placebo beobachtet.
Abbildung 1: Veränderungen gegenüber Baseline (CFB) beim MG-ADL-Gesamtscore
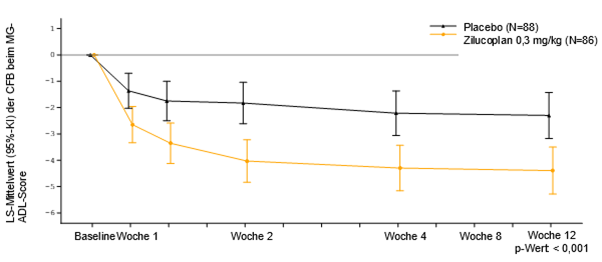
Analyse basierend auf MMRM-ANCOVA-Modell
Klinisch bedeutsame Veränderung = Veränderung des MG-ADL-Scores um 2 Punkte
Tabelle 4 zeigt die Veränderungen gegenüber Baseline in Woche 12 bei den Gesamtscores für MG-ADL, QMG, MGC und MG-QoL15r.
Tabelle 4: Veränderungen gegenüber Baseline in Woche 12 bei den Gesamtscores für MG-ADL, QMG, MGC und MG-QoL15r
|
Endpunkte: Veränderungen gegenüber Baseline beim Gesamtscore in Woche 12: LS-Mittelwert (95%-KI)
|
Zilucoplan (n = 86)
|
Placebo (n = 88)
|
Veränderung, LS-Mittelwert-Differenz zwischen Zilucoplan und Placebo (95%-KI)
|
p-Wert*
| |
MG-ADL
|
-4,39 (-5,28, -3,50)
|
-2,30 (-3,17, -1,43)
|
-2,09 (-3,24, -0,95)
|
< 0,001
| |
QMG
|
-6,19 (-7,29, -5,08)
|
-3,25 (-4,32, -2,17)
|
-2,94 (-4,39, -1,49)
|
< 0,001
| |
MGC
|
-8,62 (-10,22, -7,01)
|
-5,42 (-6,98, -3,86)
|
-3,20 (-5,24, -1,16)
|
0,0023
| |
MG-QoL15r
|
-5,65 (-7,17, -4,12)
|
-3,16 (-4,65, -1,67)
|
-2,49 (-4,45, -0,54)
|
0,0128
|
*Analyse basierend auf MMRM-ANCOVA-Modell
In Woche 12 war der kumulierte Anteil der Patienten, die eine Rescue-Therapie (intravenöses Immunglobulin G oder Plasmaaustausch) benötigten, in der Zilucoplan-Gruppe (5 %) niedriger als in der Placebo-Gruppe (12 %).
MG0011
Zweihundert Patienten, die an der placebokontrollierten Phase-II-Studie (MG0009) oder Phase-III-Studie (MG0010) teilgenommen hatten, fuhren mit der offenen Verlängerungsstudie MG0011 fort, in der alle Patienten Zilucoplan (Dosierung gemäss Tabelle 1) erhielten. Primäres Ziel war die Gewinnung von langfristigen Sicherheitsinformationen und sekundär die Änderung der Wirksamkeitsendpunkte gegenüber Baseline beim MG-ADL-, QMG-, MGC- and MG-QoL15r-Score in Woche 12 der offenen Verlängerungsphase (entsprechend Woche 24 der gesamten Studienteilnahme).
Abbildung 2: Mittlere Veränderung des MG-ADL-Gesamtscores von der Baseline der doppelblinden Studie bis Woche 60
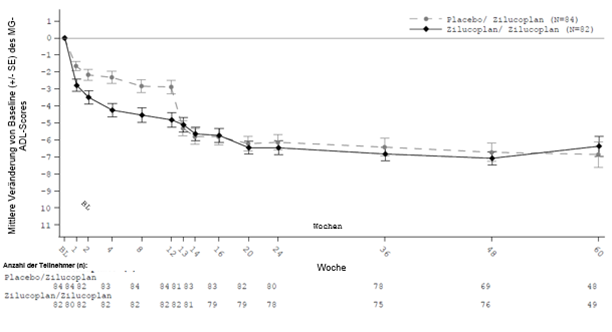
Tabelle 5: Mittlere Veränderung gegenüber der Baseline (MG0010) der doppelblinden Studie bei den Gesamtscores für MG-ADL, QMG, MGC und MG-QoL15r bis Woche 24 (Woche 12 in MG0011) und Woche 60 (Woche 48 in MG0011)
|
Endpunkte: Veränderungen von Baseline-Gesamtscore in Woche 24 und Woche 60: LS-Mittelwert (95%-KI)
|
Zilucoplan (n=82)
|
Placebo/zilucoplan (n=84)
| |
MG-ADL
|
| |
Woche 24
|
-5,46 (0,59)
|
-5,20 (0,52)
| |
Woche 60
|
-5,16 (0,61)
|
-4,37 (0,54)
| |
QMG
|
| |
Woche 24
|
-7,10 (0,80)
|
-7,19 (0,69)
| |
Woche 60
|
-6,44 (0,83)
|
-6,15 (0,71)
| |
MGC
|
| |
Woche 24
|
-10,37 (1,15)
|
-11,12 (1,00)
| |
Woche 60
|
-8,89 (1,20)
|
-9,01 (1,04)
| |
MG-QoL15r
|
| |
Woche 24
|
-8,09 (0,96)
|
-7,96 (0,89)
| |
Woche 60
|
-7,22 (0,99)
|
-6,09 (0,91)
|
Analyse basierend auf einem MMRM-ANCOVA-Modell mit Rescue-Therapie und Absetzen imputiert als Behandlungsversagen. Tod wird mit dem schlechtesten Score (z.B. Score 24 für MG-ADL) imputiert.
SE = Standardfehler
PharmakokinetikDie pharmakokinetischen Eigenschaften von Zilucoplan und den wichtigsten zirkulierenden Metaboliten (RA102758 and RA103488) wurden bei erwachsenen gesunden Probanden und bei Patienten mit gMG untersucht.
Absorption
Nach einmaliger und mehrfacher täglicher subkutaner Verabreichung von Zilucoplan in der empfohlenen Dosis (Tabelle 1) bei gesunden Studienteilnehmern erreichte Zilucoplan die maximale Plasmakonzentration im Allgemeinen zwischen 3 und 6 Stunden nach der Dosisgabe.
In der Studie MG0010 bei Patienten mit gMG waren nach täglicher wiederholter subkutaner Verabreichung der empfohlenen Zilucoplan Dosis die Plasmakonzentrationen von Zilucoplan konsistent, wobei die Steady-State-Talkonzentrationen bis Woche 4 der Zilucoplan-Behandlung erreicht und bis Woche 12 aufrechterhalten wurden.
Die Expositionen nach subkutaner Verabreichung einzelner Zilucoplan-Dosen in Bauch, Oberschenkel oder Oberarm waren vergleichbar.
Distribution
Zilucoplan und seine zwei wichtigsten Metaboliten sind stark an Plasmaproteine gebunden (> 99 %). Das mittlere Verteilungsvolumen für Zilucoplan (Vc/F) unter Verwendung einer populationspharmako-kinetischen Analyse beträgt 3,51 l.
Zilucoplan ist kein Substrat für Transporter (P-gp, BCRP, OATP1B1 und OATP1B3).
Metabolismus
Zilucoplan ist kein Substrat wichtiger CYP-Enzyme (1A2, 2B6, 2C8, 2C9, 2C19, 2D6 und 3A). Im Plasma wurden zwei Hauptmetaboliten, RA103488 und RA102758, nachgewiesen. Die Bildung von RA103488 erfolgt hauptsächlich über Cytochrom CYP450 4F2. RA103488 hat eine ähnliche pharmakologische Aktivität wie Zilucoplan, ist aber im Vergleich zu Zilucoplan in einer viel geringeren Konzentration vorhanden. Der Beitrag von RA103488 zur pharmakologischen Aktivität ist gering. RA102758 ist pharmakologisch inaktiv.
Elimination
Es ist zu erwarten, dass Zilucoplan als Peptid über katabole Wege in kleinere Peptide und Aminosäuren abgebaut wird. Die mittlere terminale Eliminationshalbwertszeit im Plasma betrug etwa 172 Stunden (7–8 Tage). Die Halbwertszeit betrug 220 Stunden beim aktiven (RA103488) bzw. 96 Stunden beim wichtigsten inaktiven Metaboliten (RA102758). Die Ausscheidung von Zilucoplan und seinen Metaboliten, die sowohl im Urin als auch im Stuhl gemessen wurde, war vernachlässigbar.
Linearität/Nicht-Linearität.
In der populationspharmakokinetischen Analyse (bei Dosen von 0,05 bis 0,6 mg/kg) ist die Pharmakokinetik von Zilucoplan durch eine Target-abhängige Wirkstoffverteilung gekennzeichnet, wobei es bei zunehmenden Dosen zu einem weniger als dosisproportionalen Anstieg der Exposition kommt, auch nach dem Vergleich der Gabe von Mehrfachdosen mit Einzeldosen.
Antikörper
Die Inzidenz von Anti-Drug- (ADA) und Anti-PEG-Antikörpern in der Phase-III-Studie bei Patienten mit gMG war in der Zilucoplan-Behandlungsgruppe und der Placebo-Behandlungsgruppe vergleichbar (siehe Abschnitt «Unerwünschte Wirkungen»).
Es konnte kein Einfluss auf die Zilucoplan Konzentration in den ADA- und Anti-PEG-Antikörper-positiven Patienten beobachtet werden.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Die Auswirkungen einer mittelschweren Leberfunktionsstörung auf die Pharmakokinetik von Zilucoplan und seine Metaboliten wurden in einer offenen Phase-I-Studie untersucht, in der gesunden Studienteilnehmern und solchen mit mittelschwerer Leberfunktionsstörung eine Einzeldosis Zilucoplan 0,3 mg/kg verabreicht wurde.
Die systemische Exposition gegenüber Zilucoplan war bei Studienteilnehmern mit mittelschwerer Leberfunktionsstörung im Vergleich zu gesunden Studienteilnehmern um 24 % niedriger, was in Einklang mit einer höheren systemischen und Spitzenexposition beider Metaboliten bei Studienteilnehmern mit Leberfunktionsstörungen im Vergleich zu gesunden Studienteilnehmern steht. Die maximale Exposition gegenüber Zilucoplan sowie die terminale Halbwertszeit waren in beiden Gruppen vergleichbar. Eine pharmakodynamische Analyse ergab keine bedeutsamen Unterschiede in den Komplementspiegeln oder der Hemmung der Komplementaktivität zwischen beiden Gruppen. Basierend auf diesen Ergebnissen ist keine Dosisanpassung für Patienten mit leichter und mittelschwerer Leberfunktionsstörung erforderlich.
Nierenfunktionsstörungen
Die Wirkung einer Nierenfunktionsstörung auf die Pharmakokinetik von Zilucoplan und seinen Metaboliten wurde in einer offenen Phase-I-Studie untersucht, in der gesunden Studienteilnehmern und solchen mit schwerer Nierenfunktionsstörung eine Einzeldosis Zilucoplan 0,3 mg/kg verabreicht wurde.
Basierend auf den pharmakokinetischen Ergebnissen ist bei Patienten mit Nierenfunktionsstörung keine Dosisanpassung erforderlich.
Ältere Patienten
Basierend auf der populationspharmakokinetischen Analyse hatte das Alter keinen Einfluss auf die Pharmakokinetik von Zilucoplan. Eine Dosisanpassung ist nicht erforderlich.
Ethnische Zugehörigkeit
In einer klinischen Phase-I-Studie mit gesunden kaukasischen und japanischen Studienteilnehmern wurde das pharmakokinetische Profil von Zilucoplan und seinen beiden wichtigsten Metaboliten nach einer Einzeldosis von 0,3 mg/kg und nach mehrfacher Gabe von 0,3 mg/kg über 14 Tage verglichen. Die Ergebnisse waren in beiden Gruppen im Allgemeinen vergleichbar.
Die populationspharmakokinetische Analyse zeigte, dass es keine Unterschiede zwischen den verschiedenen ethnischen Gruppen gibt (Schwarze/Afro-Amerikaner, Asiaten/Japaner und Kaukasier). Eine Dosisanpassung ist nicht erforderlich.
Geschlecht
In der populationspharmakokinetischen Analyse wurde kein Unterschied in der Pharmakokinetik zwischen den Geschlechtern beobachtet. Eine Dosisanpassung ist nicht erforderlich.
Körpergewicht
Die populationspharmakokinetische Analyse zeigte, dass das Körpergewicht die Pharmakokinetik von Zilucoplan signifikant beeinflusst. Die Dosierung von Zilucoplan basiert auf Körpergewichtskategorien (siehe Abschnitt «Dosierung/Anwendung»). Eine weitere Dosisanpassung ist nicht erforderlich.
Präklinische DatenToxizität bei wiederholter Gabe
In Studien zur Toxizität bei wiederholter Gabe bei nicht-humanen Primaten traten bei klinisch relevanter Exposition vesikuläre Degeneration/Hyperplasie von Epithelzellen und mononukleäre Zellinfiltrate in verschiedenen Geweben auf. Im Pankreas manifestierte sich dies gelegentlich als Degeneration von Pankreas-Azinuszellen, einige mit Fibrose und duktaler Degeneration/Regeneration, und war begleitet von erhöhten Amylase- und Lipase-Plasmakonzentrationen. Die Befunde bei nicht-humanen Primaten sind von unklarer klinischer Relevanz, wobei einige möglicherweise in Verbindung mit Infektionen stehen, die sekundär zur pharmakologischen Wirkung von Zilucoplan sind, andere Mechanismen können jedoch nicht ausgeschlossen werden.
Genotoxizität
Zilucoplan zeigte negative Ergebnisse im In-vitro-Mutagenitätstest (Ames-Test) und In-vitro-Chromosomenaberrationstest sowie im In-vivo-Mikrokerntest an Knochenmarkzellen in Ratten.
Kanzerogenität
Es wurden keine Kanzerogenitätsstudien mit Zilucoplan durchgeführt.
Reproduktionstoxizität
In einer Fertilitätsstudie mit männlichen Affen wurde eine minimale bis geringfügige Degeneration/Depletion der Keimbahn bei klinisch relevanter Exposition beobachtet. Jedoch steigerte sich der Schweregrad nicht mit der Dosis. Es liessen sich keine Auswirkungen auf die Spermatogenese feststellen. An weiblichen Reproduktionsorganen (Vagina, Zervix, Uterus) wurden mononukleäre Zellinfiltrate mit Degeneration des Epithels und Plattenepithelmetaplasie der Zervix uteri beobachtet.
Die subkutane Verabreichung von Zilucoplan (0, 1, 2 oder 4 mg/kg/Tag) an trächtige Affen während der gesamten Tragzeit führte bei allen Dosen zu einem leichten Anstieg der pränatalen Verluste, ohne dass eine mütterliche Toxizität vorlag. Die niedrigste getestete Dosis wurde mit einer mütterlichen Exposition (AUC) assoziiert, die derjenigen beim Menschen bei der empfohlenen Höchstdosis von 32,4 mg/Tag entspricht. Bei nichtmenschlichen Primaten wurden keine Auswirkungen auf die Geburt, die postnatale Entwicklung des Säuglings und die postnatalen Verluste festgestellt.
Sonstige HinweiseInkompatibilitäten
Nicht zutreffend.
Haltbarkeit
36 Monate
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2–8°C) lagern.
Nicht einfrieren.
Die Fertigspritze im Originalkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Die Patienten können die Zilbrysq-Fertigspritze im Originalumkarton bei Raumtemperatur bis zu 30°C über einen einmaligen Zeitraum von maximal 3 Monaten lichtgeschützt aufbewahren. Nachdem Zilbrysq bei Raumtemperatur gelagert wurde, darf es nicht wieder in den Kühlschrank gelegt, sondern muss entsorgt werden, wenn es nicht innerhalb von 3 Monaten bzw. bis zum Verfalldatum, je nachdem, was zuerst eintritt, verwendet wird.
Zulassungsnummer69066 (Swissmedic)
PackungenZILBRYSQ 16,6 mg Injektionslösung in einer Fertigspritze (A)
0,416 ml Injektionslösung in einer Fertigspritze mit rubinrotem Kolben
7 Fertigspritzen
ZILBRYSQ 23 mg Injektionslösung in einer Fertigspritze (A)
0,574 ml Injektionslösung in einer Fertigspritze mit orangefarbigem Kolben
7 Fertigspritzen
ZILBRYSQ 32,4 mg Injektionslösung in einer Fertigspritze (A)
0,810 ml Injektionslösung in einer Fertigspritze mit dunkelblauem Kolben
7 Fertigspritzen
ZulassungsinhaberinUCB-Pharma AG
Stand der InformationJuni 2024
|