ZusammensetzungWirkstoffe
Atezolizumabum (gentechnologisch hergestellt unter Verwendung von CHO [Chinese Hamster Ovary]-Zellen]).
Hilfsstoffe
Hyaluronidasum humanum (gentechnisch hergestellt unter Verwendung von CHO [Chinese Hamster Ovary]-Zellen), L-histidinum, acidum aceticum, L-methioninum, saccharum (aus gentechnisch veränderter Zuckerrübe hergestellt), polysorbatum 20 (aus gentechnisch verändertem Mais hergestellt), aqua ad iniectabile.
Indikationen/AnwendungsmöglichkeitenNicht-kleinzelliges Lungenkarzinom (NSCLC) im Frühstadium
Tecentriq als Monotherapie ist indiziert zur adjuvanten Therapie für Patienten mit NSCLC im Stadium II und IIIA (7. Ausgabe UICC/AJCC-Klassifikationssystems), deren Tumor keine Progression nach cisplatinhaltiger Chemotherapie und eine PD-L1-Tumorexpression ≥50% aufweist (siehe «Eigenschaften/Wirkungen»).
Metastasiertes nicht-kleinzelliges Lungenkarzinom (NSCLC)
Tecentriq ist in Kombination mit Nab-Paclitaxel und Carboplatin zur Erstlinienbehandlung von Patienten mit metastasiertem, nicht-plattenepithelialem NSCLC ohne genomische EGFR- oder ALK-Tumoraberrationen indiziert.
Tecentriq ist in Kombination mit Paclitaxel und Carboplatin als Erstlinientherapie zur Behandlung des metastasierten nichtplattenepithelialen NSCLC bei Patienten indiziert, bei denen keine genomische EGFR- oder ALK-Tumoraberrationen festgestellt wurden und deren Tumoren eine PD-L1-Expression von ≥1% aufweisen.
Tecentriq ist als Monotherapie indiziert zur Behandlung von Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC, nach vorausgegangener Chemotherapie.
Kleinzelliges Lungenkarzinom (SCLC)
Tecentriq ist in Kombination mit Carboplatin und Etoposid indiziert für die Erstlinientherapie von Patienten mit fortgeschrittenem kleinzelligem Lungenkarzinom (ES-SCLC, extensive-stage small cell lung cancer).
Urothelkarzinom
Tecentriq ist als Monotherapie indiziert für die Behandlung von erwachsenen Patienten mit lokal fortgeschrittenem oder metastasiertem Urothelkarzinom nach vorangegangener Platin-basierter Chemotherapie.
Triple-negatives Mammakarzinom
Tecentriq ist in Kombination mit Nab-Paclitaxel indiziert für die Behandlung von erwachsenen Patientinnen mit nicht-resezierbarem lokal fortgeschrittenem oder metastasiertem triple-negativem Mammakarzinom (TNBC), deren Tumore eine PD-L1-Expression ≥1% aufweisen und die keine vorherige Chemotherapie oder zielgerichtete systemische Therapie wegen ihrer fortgeschrittenen Erkrankung erhalten haben (Nab-Paclitaxel Dosierung siehe «Dosierung/Anwendung»).
Tecentriq soll nicht in Kombination mit Paclitaxel für die Behandlung von erwachsenen Patientinnen mit nicht-resezierbarem, lokal fortgeschrittenem oder metastasiertem TNBC angewendet werden (siehe «Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»).
Melanom
Tecentriq ist in Kombination mit Cobimetinib und Vemurafenib indiziert zur Behandlung von erwachsenen Patienten mit einem metastasierten oder nicht resezierbaren Melanom, das positiv auf eine BRAF-V600E-Mutation getestet wurde (siehe «Klinische Wirksamkeit»).
Hepatozelluläres Karzinom
Tecentriq ist in Kombination mit Bevacizumab zur Behandlung von Patienten mit inoperablem oder metastasiertem hepatozellulärem Karzinom (HCC), die keine vorgängige systemische Therapie erhalten haben, indiziert (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen» und «Eigenschaften/Wirkungen»).
Dosierung/AnwendungAllgemein
Tecentriq muss unter der Aufsicht einer qualifizierten medizinischen Fachperson verabreicht werden. Es ist unbedingt die Produktkennzeichnung zu überprüfen, um sicherzustellen, dass Patienten verordnungsgemäss die richtige Formulierung (Tecentriq zur Anwendung i.v. oder Tecentriq zur Anwendung s.c.) erhalten.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Tecentriq in der Formulierung zur subkutanen Anwendung (Tecentriq s.c.) ist nicht für die intravenöse Gabe bestimmt. Tecentriq s.c. darf nur als subkutane Injektion gegeben werden (siehe «Sonstige Hinweise: Hinweise zur Handhabung»). Vor der Anwendung Tecentriq s.c. aus dem Kühlschrank nehmen und die Lösung Raumtemperatur annehmen lassen.
15 ml Tecentriq s.c. Lösung über ungefähr 7 Minuten subkutan in den Oberschenkel injizieren. Es wird die Verwendung eines s.c. Infusionssets (z.B. mit Flügeln/Butterfly) empfohlen. Das im Infusionsschlauch verbleibende Totvolumen NICHT verabreichen.
Die Injektionsstellen sollten nur zwischen dem linken und dem rechten Oberschenkel gewechselt werden. Neue Injektionen sollten mindestens 2,5 cm von der vorherigen Stelle entfernt in gesunde Haut und auf keinen Fall an Stellen verabreicht werden, an denen die Haut gerötet, verletzt, empfindlich oder verhärtet ist. Während der Behandlung mit Tecentriq s.c. sollten andere Medikamente zur subkutanen Verabreichung vorzugsweise an anderen Stellen injiziert werden.
Patientenauswahl
PD-L1 Test für Patienten mit TNBC, die mit Tecentriq in Kombination mit Nab-Paclitaxel behandelt werden sollen, für Patienten mit NSCLC im Frühstadium, die mit einer Monotherapie mit Tecentriq behandelt werden sollen, und für Patienten mit metastasiertem 1L NSCLC, die mit Tecentriq in Kombination mit Paclitaxel und Carboplatin behandelt werden sollen
Für die Behandlung mit Tecentriq sollten die erwachsenen Patienten eine positive PD-L1 Expression aufweisen, die durch einen für Tecentriq validierten Test bestimmt wurde (siehe Rubrik «Eigenschaften/Wirkungen»).
Test für Patienten mit Melanom
Bei Melanompatienten muss für die Behandlung mit der Kombination von Tecentriq mit Cobimetinib und Vemurafenib eine BRAF-V600E-Mutation mit einem validierten Test bestätigt worden sein.
Monotherapie mit Tecentriq
Tabelle 1: Empfohlene Dosierung für die Tecentriq Monotherapie durch subkutane Injektion
|
Indikation
|
Empfohlene Dosierung und Schema
|
Dauer der Behandlung
(siehe «Klinische Wirksamkeit»)
| |
2L Urothelkarzinom
|
1875 mg alle 3 Wochen
|
Bis zum Verlust des klinischen Nutzens oder bis zum Auftreten einer nicht behandelbaren Toxizität
| |
2L metastasiertes NSCLC*
| |
NSCLC im Frühstadium
|
1 Jahr lang, ausser bei Auftreten eines Krankheitsrezidivs oder inakzeptabler Toxizität
|
* Nur in dieser Population wurde die subkutane Formulierung von Tecentriq untersucht (siehe «Eigenschaften/Wirkungen: klinische Wirksamkeit»).
Kombinationstherapie mit Tecentriq
Zur Anwendung von Tecentriq in Kombinationstherapie ist auch die vollständige Fachinformation des Kombinationspartners zu beachten.
Tabelle 2: Empfohlene Dosierung für die Tecentriq Kombinationstherapie subkutane Injektion
|
Indikation
|
Empfohlene Dosierung und Schema
|
Dauer der Behandlung
(siehe «Klinische Wirksamkeit»)
| |
Tecentriq
|
Kombinationsarzneimittel
| |
1L metastasiertes nicht-plattenepitheliales NSCLC
Tecentriq mit Paclitaxel und Carboplatin
|
Induktionsphase:
1875 mg alle 3 Wochen
Bei Verabreichung am gleichen Tag soll Tecentriq vor der Kombinationstherapie verabreicht werden.
Erhaltungsphase (ohne Chemotherapie):
1875 mg alle 3 Wochen
|
Induktionsphase:
·Tecentriq gefolgt von Paclitaxel und danach Carboplatin als i.v. Infusion werden alle 3 Wochen verabreicht.
|
Induktionsphase:
·über vier oder sechs Zyklen
Erhaltungsphase:
·Bis zum Verlust des klinischen Nutzens oder bis zum Auftreten einer nicht behandelbaren Toxizität
| |
1L metastasiertes nicht-plattenepitheliales NSCLC
Tecentriq mit Nab-Paclitaxel und
Carboplatin
|
Induktionsphase:
·Nab-Paclitaxel und Carboplatin werden alle drei Wochen als i.v. Infusion verabreicht.
·In jedem 21-Tage-Zyklus werden Tecentriq, Nab-Paclitaxel und Carboplatin an Tag 1 gegeben.
·Nab-Paclitaxel wird ausserdem an den Tagen 8 und 15 gegeben.
| |
1L ES-SCLC
Tecentriq mit Carboplatin und Etoposid
|
Induktionsphase:
·Carboplatin und dann Etoposid werden alle drei Wochen als i.v. Infusion verabreicht.
·Tecentriq, Carboplatin und danach Etoposid werden an Tag 1 jedes Zyklus verabreicht.
·Etoposid wird zudem mittels i.v. Infusion an den Tagen 2 und 3 verabreicht.
|
Induktionsphase:
·während vier Zyklen
Erhaltungsphase:
·Bis zum Verlust des klinischen Nutzens oder bis zum Auftreten einer nicht behandelbaren Toxizität
| |
1L TNBC
Tecentriq mit Nab-Paclitaxel
|
1875 mg alle 3 Wochen
Bei Verabreichung am gleichen Tag soll Tecentriq vor der Kombinationstherapie verabreicht werden.
|
·Für jeden 28-tägigen Zyklus wird 100 mg/m2 Nab-Paclitaxel mittels i.v. Infusion an den Tagen 1, 8 und 15 verabreicht.
·Tecentriq wird in einem 3-wöchentlichem Intervall verabreicht.
·Die Substitution von Nab-Paclitaxel durch andere Paclitaxel-Formulierungen für die Behandlung des TNBC ist nicht erlaubt (siehe «Indikationen/Anwendungsmöglichkeiten» und «Warnhinweise und Vorsichtsmassnahmen»).
|
Bis zur Krankheitsprogression oder nicht akzeptabler Toxizität behandelt
| |
Melanom
Tecentriq mit Cobimetinib und Vemurafenib
|
1875 mg alle 3 Wochen
|
·Vor dem Einleiten der Behandlung mit Tecentriq erhalten die Patienten einen 28-tägigen Behandlungszyklus mit 60 mg Cobimetinib oral einmal täglich (21 Tage mit Medikationsgabe und 7 Tage Einnahmepause) und 960 mg Vemurafenib oral zweimal täglich an den Tagen 1-21 und 720 mg Vemurafenib oral zweimal täglich an den Tagen 22-28.
·Während der Behandlung mit Tecentriq wird 60 mg Cobimetinib einmal täglich (21 Tage mit Medikationsgabe und 7 Tage Einnahmepause) und 720 mg Vemurafenib oral zweimal täglich verabreicht (siehe «Klinische Wirksamkeit»).
|
Bis zur Krankheitsprogression oder nicht akzeptabler Toxizität behandelt
| |
1L HCC
Tecentriq mit Bevacizumab
|
1875 mg alle 3 Wochen
Bei Verabreichung am gleichen Tag soll Tecentriq vor der Kombinationstherapie verabreicht werden.
|
·Tecentriq gefolgt von 15 mg Bevacizumab pro kg Körpergewicht mittels i.v. Infusion werden alle 3 Wochen verabreicht.
|
Bis zum Verlust des klinischen Nutzens oder bis zum Auftreten einer nicht behandelbaren Toxizität
|
Dosisanpassung
Es werden keine Dosisreduktionen von Tecentriq empfohlen.
Dosisanpassung aufgrund immunvermittelter unerwünschter Wirkungen/Interaktionen
In Tabelle 3 sind Empfehlungen in Bezug auf bestimmte unerwünschte Arzneimittelreaktionen angegeben (siehe Rubriken «Warnhinweise und Vorsichtsmassnahmen» und «Unerwünschte Wirkungen»).
Tabelle 3: Empfehlung zur Anpassung der Dosis von Tecentriq
|
Immunvermittelte unerwünschte Reaktion
|
Schweregrad
|
Behandlungsanpassung
| |
Infektionen
|
Grad 3 oder 4
|
Dosisgabe bis zu einer Besserung bis auf Grad 1 oder bis zum vollständigen Abklingen unterbrechen
| |
Infusionsbedingte Reaktionen
|
Grad 1 oder 2
|
Geschwindigkeit der Injektion reduzieren oder Injektion unterbrechen. Die Behandlung kann wieder aufgenommen werden, wenn das Ereignis abgeklungen ist.
Bei anschliessenden Dosen kann eine Vormedikation mit Antipyretika und Antihistaminika erwogen werden.
| |
Grad 3 oder 4
|
Tecentriq dauerhaft absetzen
| |
Hämophagozytische Lymphohistiozytose
|
Verdacht auf Hämophagozytische Lymphohistiozytose (unabhängig von der Schwere)
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Pneumonitis
|
Grad 2
|
Tecentriq-Injektion unterbrechen
Bei Besserung des Ereignisses innerhalb 12 Wochen auf Grad 0 oder Grad 1 und Senkung der Kortikosteroidgaben auf ≤10 mg Prednison oder Äquivalent pro Tag kann die Behandlung wieder aufgenommen werden.
| |
Grad 3 oder 4
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Hepatitis bei Patienten ohne HCC
|
Grad 2:
(ALT oder AST > 3 bis 5 x oberer Normwert [Upper Limit of Normal, ULN]
oder
Blutbilirubin > 1,5 bis 3 x ULN)
|
Tecentriq-Injektion unterbrechen
Bei Besserung des Ereignisses innerhalb 12 Wochen auf Grad 0 oder Grad 1 und Senkung der Kortikosteroidgaben auf ≤10 mg Prednison oder Äquivalent pro Tag kann die Behandlung wieder aufgenommen werden.
| |
Grad 3 oder 4:
(ALT oder AST > 5 x ULN
oder
Blutbilirubin > 3 x ULN)
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Hepatitis bei Patienten mit HCC
|
AST/ALT-Werte im normalen Bereich zur Baseline und Anstieg auf > 3x bis ≤ 10x ULN
AST/ALT-Wert > 1 bis ≤ 3x ULN zur Baseline und Anstieg auf > 5x bis ≤ 10x ULN
AST/ALT-Wert > 3x bis ≤ 5x ULN zur Baseline und Anstieg auf > 8x bis ≤ 10x ULN
|
Tecentriq-Injektion unterbrechen
Die Behandlung kann wieder aufgenommen werden, wenn sich der Grad der Nebenwirkung innerhalb von 12 Wochen auf Grad 0 oder Grad 1 verbessert hat und die Kortikosteroid-Dosis auf ≤ 10 mg Prednison oder eines Äquivalents pro Tag verringert wurde.
| |
Anstieg des AST/ALT-Werts auf > 10x ULN oder Anstieg des Gesamtbilirubins auf > 3x ULN
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Kolitis
|
Diarrhoe Grad 2 oder 3 (Erhöhung um ≥4 Stuhlgänge/Tag gegenüber Baseline)
oder
Symptomatische Kolitis
|
Tecentriq-Injektion unterbrechen
Bei Besserung des Ereignisses innerhalb 12 Wochen auf Grad 0 oder Grad 1 und Senkung der Kortikosteroidgaben auf ≤10 mg Prednison oder Äquivalent pro Tag kann die Behandlung wieder aufgenommen werden.
| |
Diarrhoe oder Kolitis Grad 4 (lebensbedrohlich; Indikation für eine dringliche Intervention)
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Hypothyreose oder Hyperthyreose
|
Symptomatisch
|
Tecentriq-Injektion unterbrechen
Hypothyreose:
Wenn die Symptome mit einer Schilddrüsensubstitutionstherapie kontrolliert sind und die TSH-Werte fallen, kann die Behandlung wieder aufgenommen werden.
Hyperthyreose:
Wenn die Symptome mit einem Arzneimittel zur Suppression der Schilddrüse kontrolliert sind und die Schilddrüsenfunktion sich bessert, kann die Behandlung wieder aufgenommen werden.
| |
Immunvermittelte Nebenniereninsuffizienz
|
Symptomatisch
|
Tecentriq-Injektion unterbrechen
Bei Besserung der Symptome innerhalb 12 Wochen auf Grad 0 oder Grad 1 und Senkung der Kortikosteroidgaben auf ≤10 mg Prednison oder Äquivalent pro Tag und sofern der Patient unter der Substitutionstherapie stabil eingestellt ist, kann die Behandlung wieder aufgenommen werden.
| |
Immunvermittelte Hypophysitis
|
Grad 2 oder 3
|
Tecentriq-Injektion unterbrechen
Bei Besserung der Symptome innerhalb 12 Wochen auf Grad 0 oder Grad 1 und Senkung der Kortikosteroidgaben auf ≤10 mg Prednison oder Äquivalent pro Tag und sofern der Patient unter der Substitutionstherapie stabil eingestellt ist, kann die Behandlung wieder aufgenommen werden.
| |
Grad 4
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelter Typ 1 Diabetes mellitus
|
Hyperglykämie Grad 3 oder 4 (Nüchternglukose > 250 mg/dl oder 13,9 mmol/l)
|
Tecentriq-Injektion unterbrechen
Bei Erreichen der metabolischen Kontrolle unter der Insulin-Substitutionstherapie kann die Behandlung wieder aufgenommen werden.
| |
Immunvermittelte(s) Myasthenisches Syndrom/Myasthenia gravis, Guillain-Barré Syndrom und Meningoenzephalitis
|
Alle Grade
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Myelitis
|
Alle Grade
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Fazialisparese
|
Grad 1 oder 2
|
Tecentriq-Injektion unterbrechen
Es sollte eine Behandlung mit Kortikosteroiden (1–2 mg/kg/Tag Prednison oder Äquivalent) eingeleitet werden. Bei vollständigem oder teilweisem Abklingen des Ereignisses (Grad 0 bis 1) innerhalb von 12 Wochen und nach Senkung der Kortikosteroidgabe auf ≤10 mg/Tag orales Prednison oder Äquivalent kann die Behandlung mit Tecentriq wieder aufgenommen werden.
| |
Grad 3 oder 4
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Pankreatitis
|
Auf Grad 3 oder 4 erhöhte Serumamylasewerte oder erhöhte Serumlipasewerte (> 2 x ULN)
oder Pankreatitis Grad 2 oder 3
|
Tecentriq-Injektion unterbrechen
Bei Besserung der Serumamylase- und Serumlipasewerte innerhalb 12 Wochen auf Grad 0 oder Grad 1 bzw. nach Abklingen der Symptome der Pankreatitis sowie Senkung der Kortikosteroidgaben auf ≤10 mg Prednison oder Äquivalent pro Tag kann die Behandlung wieder aufgenommen werden.
| |
Rezidivierende Pankreatitis Grad 4 oder beliebigen Grades
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Myokarditis
|
Grad 2 oder höher
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Myositis
|
Grad 2 oder 3
|
Tecentriq-Injektion unterbrechen
Bei Besserung der Symptome innerhalb 12 Wochen auf Grad 0 oder Grad 1 und Senkung der Kortikosteroidgaben auf ≤10 mg Prednison oder Äquivalent pro Tag kann die Behandlung wieder aufgenommen werden.
| |
Grad 4 oder rezidivierende Grad 3 Myositis
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Nephritis
|
Grad 2:
(Kreatininwert > 1,5 - 3,0 x Baseline oder > 1,5 - 3,0 x ULN)
|
Tecentriq-Injektion unterbrechen
Bei Besserung des Ereignisses innerhalb 12 Wochen auf Grad 0 oder Grad 1 und Senkung der Kortikosteroidgaben auf ≤10 mg Prednison oder Äquivalent pro Tag kann die Behandlung wieder aufgenommen werden.
| |
Grad 3:
(Kreatininwert > 3,0 x Baseline oder > 3,0 - 6,0 x ULN)
oder Grad 4:
(Kreatininwert > 6,0 x ULN)
|
Tecentriq dauerhaft absetzen
| |
Immunvermittelte Perikarderkrankungen
|
Grad 1 Perikarditis
|
Tecentriq-Injektion unterbrechen
Eine ausführliche kardiologische Abklärung zur Feststellung der Ätiologie durchführen und angemessen behandeln.
| |
Grad 2 oder höher
|
Tecentriq dauerhaft absetzen
| |
Hautausschlag/Schwere immunvermittelte Hautreaktionen
|
Grad 3
oder Verdacht auf Stevens-Johnson-Syndrom (SJS) oder toxische epidermale Nekrolyse (TEN)
|
Tecentriq-Injektion unterbrechen
Nach Abklingen des Hautausschlags und Senkung der Kortikosteroidgaben auf ≤10 mg Prednison oder Äquivalent pro Tag kann die Behandlung wieder aufgenommen werden.
| |
Grad 4
oder Bestätigung eines Stevens-Johnson-Syndroms (SJS) oder einer toxischen epidermalen Nekrolyse (TEN)
|
Tecentriq dauerhaft absetzen
| |
Andere immunvermittelte unerwünschte Reaktionen
|
Grad 2 oder Grad 3
|
Unterbrechen bis zu einer Besserung der unerwünschten Reaktionen innerhalb 12 Wochen auf Grad 0-1 und Senkung der Kortikosteroidgaben auf ≤10 mg Prednison oder Äquivalent pro Tag.
| |
Grad 4 oder rezidivierend Grad 3
|
Tecentriq dauerhaft absetzen (ausgenommen bei Endokrinopathien, die durch Ersatzhormone kontrolliert sind)
| |
Persistierende immunvermittelte unerwünschte Wirkung Grad 2 oder 3 (ausgenommen Endokrinopathien)
|
Immunvermittelte unerwünschte Wirkung Grad 2 oder 3, die sich nicht innerhalb von 12 Wochen nach der letzten Dosis Tecentriq bis auf Grad 0 oder 1 bessert
|
Dauerhaft absetzen
| |
Ausschleichen von Kortikosteroiden nicht möglich
|
Reduzierung auf ≤10 mg Prednison oder Äquivalent pro Tag innerhalb von 12 Wochen nach der letzten Dosis Tecentriq nicht möglich
|
Dauerhaft absetzen
| |
Rezidivierende immunvermittelte unerwünschte Wirkung Grad 3 oder 4
|
Rezidivierende immunvermittelte unerwünschte Wirkung Grad 3 oder 4 (schwer oder lebensbedrohlich)
|
Dauerhaft absetzen
|
Hinweis: Die Toxizitätsgrade entsprechen den Allgemeinen Terminologiekriterien des National Cancer Institute für unerwünschte Ereignisse (National Cancer Institute Common Terminology Criteria for Adverse Event), Version 5.0 (NCI-CTCAE v.5.).
Verspätete Dosisgabe
Wenn eine planmässige Dosis von Tecentriq versäumt wird, sollte sie so bald wie möglich nachgeholt werden. Nicht die nächste planmässige Dosis abwarten. Das Verabreichungsschema sollte angepasst werden, um einen dreiwöchigen Abstand zwischen den Dosen aufrechtzuerhalten.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Ausgehend von einer populationspharmakokinetischen Analyse sind bei Patienten mit leichter und mittelschwerer Leberfunktionsstörung keine Dosisanpassungen erforderlich. Für Patienten mit schwerer Leberfunktionsstörung liegen keine Daten vor (siehe «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Gemäss den Ergebnissen einer populationspharmakokinetischen Analyse ist bei Patienten mit leichter bis mässiger Nierenfunktionsstörung keine Dosisanpassung erforderlich (siehe Abschnitt «Pharmakokinetik»). Die Daten zu Patienten mit schwerer Nierenfunktionsstörung sind zu begrenzt, um Rückschlüsse auf diese Population zuzulassen.
Ältere Patienten
Von 3'040 Patienten mit Urothelkarzinom, Lungenkrebs, dreifach negativem Brustkrebs, hepatozellulärem Karzinom und Melanom, die in klinischen Studien mit Tecentriq behandelt wurden, waren 43% 65 Jahre und älter und 12% 75 Jahre und älter. Ausgehend von einer populationspharmakokinetischen Analyse sind bei Patienten ≥65 Jahren keine Anpassungen der Tecentriq-Dosis erforderlich (siehe «Pharmakokinetik»).
Kinder und Jugendliche
Tecentriq ist nicht für die Anwendung bei Patienten unter 18 Jahren zugelassen. Die Sicherheit und Wirksamkeit von Tecentriq in dieser Population ist bisher nicht erwiesen. Tecentriq zeigte bei Kindern und Jugendlichen keinen klinischen Nutzen in einer klinischen Studie (siehe «Eigenschaften/Wirkungen: Klinische Wirksamkeit»).
Genotyp/Genetische Polymorphismen
Asiatische Patienten
Bei asiatischen Patienten wird aufgrund von erhöhter hämatologischer Toxizität, welche in der Studie IMpower150 beobachtet wurde, eine Anfangsdosis von Paclitaxel von 175 mg/m2 alle drei Wochen empfohlen.
KontraindikationenTecentriq ist bei Patienten mit bekannter Überempfindlichkeit gegenüber Atezolizumab oder einem der Hilfsstoffe kontraindiziert.
Warnhinweise und VorsichtsmassnahmenInfektionen
Tecentriq kann schwere Infektionen verursachen, die auch tödlich verlaufen können (siehe «Unerwünschte Wirkungen, Klinische Studien»). Die Patienten sind auf Anzeichen und Symptome einer Infektion zu überwachen. Bei Infektionen vom Grad 3 oder höher sollte die Gabe von Tecentriq unterbrochen und erst nach Eintreten einer klinischen Stabilisierung wieder aufgenommen werden (siehe Dosierung/Anwendung, Tabelle 1: Empfehlung zur Anpassung der Dosis von Tecentriq).
In der Studie IMscin001 traten unerwünschte Infektionsereignisse vom Grad 5 häufiger bei Patienten auf, die Tecentriq subkutan erhielten, als bei Patienten, die Tecentriq intravenös erhielten (3,2% bzw. 1,6%).
In der Melanomstudie (CO39262) traten Infektionen bei Patienten, die Tecentriq iv in Kombination mit Cobimetinib und Vemurafenib erhielten, häufiger auf (siehe «Unerwünschte Wirkungen»).
Infusionsbedingte Reaktionen
In klinischen Studien mit Tecentriq wurden infusionsbedingte Reaktionen beobachtet (siehe «Unerwünschte Wirkungen»).
Bei Patienten mit infusionsbedingten Reaktionen vom Grad 1 oder 2 sollte die Injektionsgeschwindigkeit verringert oder die Behandlung pausiert werden. Bei Patienten mit infusionsbedingten Reaktionen vom Grad 3 oder 4 sollte Tecentriq dauerhaft abgesetzt werden. Bei Patienten mit infusionsbedingten Reaktionen vom Grad 1 oder 2 kann die Behandlung mit Tecentriq unter engmaschiger Überwachung fortgesetzt werden; eine Prämedikation mit Antipyretika und Antihistaminika kann in Betracht gezogen werden.
Die vorläufigen präklinischen Daten (siehe «Präklinische Daten») lassen erkennen, dass Atezolizumab möglicherweise primäre und sekundäre Immunglobulin G (IgG)-Antworten auf T-Zell-abhängige Antigene verringert.
Immunvermittelte Pneumonitis
In klinischen Studien mit Tecentriq wurden Fälle von Pneumonitiden, darunter auch tödlich verlaufende Fälle, beobachtet (siehe «Unerwünschte Wirkungen, Klinische Studien»). Die Patienten sollten hinsichtlich Anzeichen und Symptomen einer Pneumonitis überwacht werden.
Bei einer Pneumonitis Grad 2 sollte die Behandlung mit Tecentriq ausgesetzt und eine Behandlung mit 1-2 mg/kg Prednison oder Äquivalent pro Tag eingeleitet werden. Bei Verbesserung der Symptome auf einen Grad ≤1 sollten die Kortikosteroide während ≥1 Monat ausgeschlichen werden. Bei Verbesserung des Ereignisses innerhalb 12 Wochen auf einen Grad ≤1 und Senkung der Kortikosteroidgaben auf ≤10 mg orales Prednison oder Äquivalent pro Tag kann die Behandlung mit Tecentriq wieder aufgenommen werden. Bei einer Pneumonitis Grad 3 oder 4 sollte die Behandlung mit Tecentriq dauerhaft abgebrochen werden.
In der Melanomstudie (CO39262) trat eine immunbedingte Pneumonitis bei Patienten, die Tecentriq iv in Kombination mit Cobimetinib und Vemurafenib erhielten, häufiger auf (siehe «Unerwünschte Wirkungen»).
Immunvermittelte Hepatitis
In klinischen Studien mit Tecentriq wurden Fälle von Hepatitiden, darunter auch tödlich verlaufende Fälle, beobachtet (siehe «Unerwünschte Wirkungen, Klinische Studien»). Die Patienten sollten hinsichtlich Anzeichen und Symptomen einer Hepatitis überwacht werden. Vor und regelmässig während der Behandlung mit Tecentriq sollten die Aspartat-Aminotransferase (AST), die Alanin-Aminotransferase (ALT) und Bilirubin überwacht werden. Bei Patienten mit anomalen Ergebnissen von Leberfunktionstests (LFT) vor Beginn der Behandlung sollten geeignete Massnahmen in Betracht gezogen werden.
Halten die Symptome von Grad 2 (ALT oder AST > 3x obere Normbereichsgrenze (upper limit of normal, ULN) oder Bilirubin im Blut > 1,5x ULN) während mehr als 5-7 Tagen an sollte die Behandlung ausgesetzt und eine Behandlung mit 1-2 mg/kg Prednison oder Äquivalent pro Tag eingeleitet werden. Bei Verbesserung der LFT-Abweichungen auf einen Grad ≤1 sollten die Kortikosteroide während ≥1 Monat ausgeschlichen werden. Bei Verbesserung des Ereignisses innerhalb 12 Wochen auf einen Grad ≤1 und Senkung der Kortikosteroidgaben auf ≤10 mg orales Prednison oder Äquivalent pro Tag kann die Behandlung mit Tecentriq wieder aufgenommen werden. Bei Ereignissen vom Grad 3 oder Grad 4 (ALT oder AST > 5,0x ULN oder Bilirubin im Blut > 3x ULN) sollte die Behandlung mit Tecentriq dauerhaft abgebrochen werden.
In der Melanomstudie (CO39262) trat eine immunbedingte Hepatitis bei Patienten, die Tecentriq iv in Kombination mit Cobimetinib und Vemurafenib erhielten, häufiger auf (siehe «Unerwünschte Wirkungen»).
Immunvermittelte Kolitis
In klinischen Studien mit Tecentriq wurden Fälle von Diarrhoe, Kolitis und anderen gastrointestinalen Ereignissen wie Darmperforation beobachtet (siehe «Unerwünschte Wirkungen, Klinische Studien»). Die Patienten sollten hinsichtlich Anzeichen und Symptomen einer Kolitis überwacht werden.
Bei einer Diarrhoe Grad 2 oder 3 (Zunahme um ≥4 Stuhlgänge/Tag gegenüber Behandlungsbeginn) oder einer Kolitis (symptomatisch) sollte die Behandlung mit Tecentriq ausgesetzt werden. Bei einer Diarrhoe oder Kolitis Grad 2 sollte beim Persistieren der Symptome für > 5 Tage oder beim Wiederauftreten eine Behandlung mit 1-2 mg/kg Prednison oder Äquivalent pro Tag eingeleitet werden. Eine Diarrhoe oder Kolitis vom Grad 3 sollte mit i.v. Kortikosteroiden (1-2 mg/kg Methylprednisolon oder Äquivalent pro Tag) behandelt werden. Nach Besserung sollte auf orale Kortikosteroide (1-2 mg/kg Prednison oder Äquivalent pro Tag) umgestellt werden. Bei Verbesserung der Symptome auf einen Grad ≤1 sollten die Kortikosteroide während ≥1 Monat ausgeschlichen werden. Bei Verbesserung des Ereignisses innerhalb von 12 Wochen auf einen Grad ≤1 und Senkung der Kortikosteroidgaben auf ≤10 mg orales Prednison oder Äquivalent pro Tag kann die Behandlung mit Tecentriq wieder aufgenommen werden. Bei einer Diarrhoe oder Kolitis vom Grad 4 (lebensbedrohlich: dringende Intervention indiziert) sollte die Behandlung mit Tecentriq dauerhaft abgebrochen werden.
In der Melanomstudie (CO39262) trat Diarrhoe oder Kolitis bei Patienten, die Tecentriq iv in Kombination mit Cobimetinib und Vemurafenib erhielten, häufiger auf (siehe «Unerwünschte Wirkungen»).
Immunvermittelte Endokrinopathien
In klinischen Studien mit Tecentriq wurden Fälle von Hypothyreose, Hyperthyreose, Nebenniereninsuffizienz, Hypophysitis und Diabetes mellitus Typ 1 einschliesslich diabetischer Ketoazidose beobachtet (siehe «Unerwünschte Wirkungen, Klinische Studien»). Die Patienten sollten hinsichtlich klinischer Anzeichen und Symptomen von Endokrinopathien überwacht werden. Vor und regelmässig während der Behandlung mit Tecentriq sollte die Schilddrüsenfunktion überwacht werden. Bei Patienten mit anomalen Ergebnissen von Schilddrüsenfunktionstests vor Beginn der Behandlung sollten geeignete Massnahmen in Betracht gezogen werden.
Asymptomatische Patienten mit anomalen Ergebnissen von Schilddrüsenfunktionstests können mit Tecentriq behandelt werden. Bei symptomatischer Hypothyreose sollte die Behandlung mit Tecentriq ausgesetzt und bei Bedarf eine Schilddrüsenhormon-Substitutionstherapie eingeleitet werden. Eine isolierte Hypothyreose kann gegebenenfalls mit einer Substitutionstherapie ohne Kortikosteroide behandelt werden. Bei symptomatischer Hyperthyreose sollte die Behandlung mit Tecentriq ausgesetzt und bei Bedarf eine Behandlung mit einem Thyreostatikum, beispielsweise mit Methimazol oder Carbimazol, eingeleitet werden. Wenn die Symptome kontrolliert sind und die Schilddrüsenfunktion sich verbessert, kann die Behandlung mit Tecentriq wieder aufgenommen werden.
Bei symptomatischer Nebenniereninsuffizienz sollte die Behandlung mit Tecentriq ausgesetzt und eine Behandlung mit 1-2 mg/kg i.v. Methylprednisolon oder Äquivalent pro Tag eingeleitet werden. Sobald sich die Symptome verbessern, sollte sich eine Behandlung mit 1-2 mg/kg oralem Prednison oder Äquivalent pro Tag anschliessen. Bei Verbesserung der Symptome auf einen Grad ≤1 sollten die Kortikosteroide während ≥1 Monat ausgeschlichen werden. Bei Verbesserung des Ereignisses innerhalb von 12 Wochen auf einen Grad ≤1 und Senkung der Kortikosteroidgaben auf ≤10 mg orales Prednison oder Äquivalent pro Tag und sofern der Patient unter der Substitutionstherapie (falls erforderlich) stabil eingestellt ist, kann die Behandlung mit Tecentriq wieder aufgenommen werden.
Bei einer Hypophysitis Grad 2 oder 3 sollte die Behandlung mit Tecentriq ausgesetzt werden. Eine Therapie mit 1-2 mg/kg i.v. Methylprednisolon oder Äquivalent pro Tag sollte eingeleitet werden, je nach Bedarf ist zudem eine Hormonersatztherapie erforderlich. Nach Besserung sollte auf orale Kortikosteroide (1-2 mg/kg Prednison oder Äquivalent pro Tag) umgestellt werden. Bei Verbesserung der Symptome auf Grad ≤1 sollten die Kortikosteroide während ≥1 Monat ausgeschlichen werden. Bei Verbesserung des Ereignisses innerhalb von 12 Wochen auf einen Grad ≤1 und Senkung der Kortikosteroidgaben auf ≤10 mg orales Prednison oder Äquivalent pro Tag und sofern der Patient unter der Ersatztherapie (falls erforderlich) stabil eingestellt ist, kann die Behandlung mit Tecentriq wieder aufgenommen werden. Bei einer Hypophysitis Grad 4 sollte die Behandlung mit Tecentriq dauerhaft abgebrochen werden.
Bei Diabetes mellitus Typ 1 sollte eine Behandlung mit Insulin eingeleitet werden. Bei einer Hyperglykämie Grad ≥3 (Nüchternglukose > 250 mg/dl) sollte die Behandlung mit Tecentriq ausgesetzt werden. Bei Erreichen der Kontrolle des Stoffwechsels unter der Insulin-Substitutionstherapie kann die Behandlung mit Tecentriq wieder aufgenommen werden.
In der Melanomstudie (CO39262) traten Hypothyreose und Hyperthyreose bei Patienten, die Tecentriq iv in Kombination mit Cobimetinib und Vemurafenib erhielten, häufiger auf (siehe «Unerwünschte Wirkungen»).
Immunvermittelte Meningoenzephalitis
In klinischen Studien mit Tecentriq wurden Fälle von Meningoenzephalitiden beobachtet (siehe «Unerwünschte Wirkungen, Klinische Studien»). Die Patienten sollten hinsichtlich klinischer Anzeichen und Symptomen einer Meningitis oder Enzephalitis überwacht werden.
Bei Meningitiden oder Enzephalitiden jeden Grades sollte die Behandlung mit Tecentriq dauerhaft abgebrochen werden. Eine Behandlung sollte mit 1−2 mg/kg i.v. Methylprednisolon oder Äquivalent pro Tag erfolgen. Nach Besserung des Patienten sollte die Behandlung auf 1-2 mg/kg orales Prednison oder Äquivalent pro Tag umgestellt werden. Bei Verbesserung der Symptome auf einen Grad ≤1 sollten die Kortikosteroide während ≥1 Monat ausgeschlichen werden.
Immunvermittelte Neuropathien
Bei Patienten, die Tecentriq erhalten hatten, wurden myasthenisches Syndrom/Myasthenia gravis oder das unter Umständen lebensbedrohliche Guillain-Barré-Syndrom und eine Fazialisparese beobachtet (siehe «Unerwünschte Wirkungen, Klinische Studien»). Die Patienten sollten hinsichtlich Symptome einer motorischen oder sensorischen Neuropathie überwacht werden.
Bei myasthenischem Syndrom/Myasthenia gravis oder Guillain-Barré-Syndrom jeden Grades sollte die Behandlung mit Tecentriq dauerhaft abgebrochen werden. Die Einleitung einer Behandlung mit systemischen Kortikosteroiden in einer Dosis von 1-2 mg/kg oralem Prednison oder Äquivalent pro Tag sollte erwogen werden.
Immunvermittelte Myelitis
In klinischen Studien mit Tecentriq wurden Fälle von Myelitis beobachtet (siehe «Unerwünschte Wirkungen, Klinische Studien» und «Unerwünschte Wirkungen, Post-Marketing-Daten»). Die Patienten sollten hinsichtlich Anzeichen und Symptomen überwacht werden, die auf eine Myelitis hindeuten. Siehe «Dosierung/Anwendung» zu empfohlenen Dosisanpassungen.
Immunvermittelte Pankreatitis
In klinischen Studien mit Tecentriq wurden Fälle von Pankreatitis, einschliesslich erhöhter Serumamylase- und Serumlipasewerte, beobachtet (siehe «Unerwünschte Wirkungen, Klinische Studien»). Die Patienten sollten engmaschig hinsichtlich Anzeichen und Symptomen überwacht werden, die auf eine akute Pankreatitis hinweisen können.
Bei Erhöhungen der Serumamylase- oder Serumlipasewerte von einem Grad ≥3 (> 2,0 ULN) oder einer Pankreatitis vom Grad 2 oder 3 sollte die Behandlung mit Tecentriq ausgesetzt und eine Behandlung mit 1-2 mg/kg i.v. Methylprednisolon oder Äquivalent pro Tag eingeleitet werden. Sobald sich die Symptome bessern, ist eine Behandlung mit 1-2 mg/kg oralem Prednison oder Äquivalent pro Tag anzuschliessen. Bei Verbesserung der Serumamylase- und Serumlipasewerte innerhalb von 12 Wochen auf einen Grad ≤1 bzw. nach Abklingen der Symptome der Pankreatitis sowie Senkung der Kortikosteroidgaben auf ≤10 mg orales Prednison oder Äquivalent pro Tag kann die Behandlung mit Tecentriq wieder aufgenommen werden. Bei einer Pankreatitis vom Grad 4 oder bei Wiederauftreten einer Pankreatitis jeden Grades sollte die Behandlung mit Tecentriq dauerhaft abgebrochen werden.
Kardiale Erkrankungen
Immunvermittelte Myokarditis
In klinischen Studien mit Tecentriq wurde Myokarditis, einschliesslich Fälle mit tödlichem Verlauf, beobachtet (siehe «Unerwünschte Wirkungen, Klinische Studien»). Patienten sollten hinsichtlich Anzeichen und Symptomen einer Myokarditis überwacht werden. Sobald eine Myokarditis von einem Grad ≥2 festgestellt wird, soll die Behandlung mit Tecentriq dauerhaft abgesetzt werden (siehe «Dosierung/Anwendung»). Myokarditis kann auch eine klinische Manifestation einer Myositis sein und sollte entsprechend behandelt werden.
Immunvermittelte Perikarderkrankungen
Perikarderkrankungen, darunter Perikarditis, Perikarderguss und Herztamponade, einige mit tödlichem Ausgang, wurden in klinischen Studien mit Tecentriq beobachtet (siehe «Unerwünschte Wirkungen: Klinische Studien und Post-Marketing-Daten»). Die Patienten sollten hinsichtlich klinischer Anzeichen und Symptome von Perikarderkrankungen überwacht werden. Empfehlungen zur Dosisanpassungen sind im Abschnitt «Dosierung/Anwendung» enthalten.
Andere kardiale Erkrankungen
In der Studie IMscin001 traten unerwünschte kardiale Ereignisse vom Grad 5 häufiger bei Patienten auf, die Tecentriq subkutan erhielten, als bei Patienten, die Tecentriq intravenös erhielten (1,2% bzw. 0,8%).
Immunvermittelte Myositis
In klinischen Studien mit Tecentriq sind Fälle von Myositis, mitunter mit tödlichem Ausgang (auch mit Herzbeteiligung), aufgetreten (siehe «Unerwünschte Wirkungen, Klinische Studien»). Die Patienten sollten auf Anzeichen und Symptome einer Myositis überwacht werden. Wenn ein Patient Anzeichen und Symptome einer Myositis entwickelt, sollte eine engmaschige Überwachung durchgeführt werden und der Patient unverzüglich an eine spezialisierte Abteilung zur Beurteilung und Behandlung überwiesen werden. Patienten, bei denen eine Myositis vermutet wird, sollten auf Anzeichen einer Myokarditis überwacht werden. Bezüglich der empfohlenen Dosisanpassungen ist das Kapitel «Dosierung/Anwendung» zu beachten.
Immunvermittelte Nephritis
In klinischen Studien mit Tecentriq wurden Nephritiden beobachtet (siehe «Unerwünschte Wirkungen, Klinische Studien»). Patienten sollten in Hinblick auf Veränderungen ihrer Nierenfunktion überwacht werden. Siehe «Dosierung/Anwendung» zu empfohlenen Dosisanpassungen.
Sonstige immunbedingte Ereignisse
Hämophagozytische Lymphohistiozytose (HLH)
Bei Patienten unter Behandlung mit Tecentriq sind Fälle einer hämophagozytischen Lymphohistiozytose (HLH), auch mit tödlichem Verlauf, aufgetreten (siehe «Unerwünschte Wirkungen, Post-Marketing-Daten»). Eine HLH ist in Betracht zu ziehen, wenn ein Zytokinfreisetzungssyndrom vom klinischen Bild her atypisch oder verlängert ist. Die Patienten sind auf klinische Anzeichen und Symptome einer HLH zu überwachen. Siehe «Dosierung/Anwendung» zu empfohlenen Dosisanpassungen.
HLH ist ein potentiell lebensbedrohliches Syndrom mit pathologischer Aktivierung der lmmunabwehr. Falls die HLH nicht frühzeitig erkannt und behandelt wird, verläuft sie häufig letal. Die Erkrankung ist gekennzeichnet durch klinische Anzeichen und Symptome einer schweren systemischen Inflammation wie Fieber, Hautausschlag, Hepatosplenomegalie, Zytopenie (v.a. Anämie und Thrombozytopenie), Lymphadenopathie, neurologische Symptome, hohes Serum-Ferritin, Hypertriglyceridämie sowie Störungen der Leberfunktion und der Koagulation. Patienten, bei denen solche Anzeichen und Symptome auftreten, müssen unverzüglich untersucht und im Hinblick auf eine mögliche HLH-Diagnose beurteilt werden. Die Gabe von Tecentriq sollte ausgesetzt werden, solange keine alternative Ätiologie etabliert werden kann.
Unter der Behandlung mit Tecentriq und/oder anderen Immun-Checkpoint-Inhibitoren wurden Fälle von hämolytischer Anämie und aplastischer Anämie beobachtet. Die Patienten sollten hinsichtlich Anzeichen und Symptomen überwacht werden, die auf diese immunvermittelten Nebenwirkungen hindeuten.
Schwere immunvermittelte Hautreaktionen
Bei Patienten unter Behandlung mit Tecentriq wurden schwere immunvermittelte Hautreaktionen (Severe Cutaneous Adverse Reactions – SCARs) gemeldet, darunter Fälle von Stevens-Johnson-Syndrom (SJS) und toxischer epidermaler Nekrolyse (TEN; einschliesslich letaler Verläufe). Die Patienten sollten auf schwere Hautreaktionen überwacht werden. Tecentriq sollte bei Grad-3-Hautreaktionen bis zur Besserung auf Grad ≤1 unterbrochen werden und bei Grad-4-Hautreaktionen dauerhaft abgesetzt werden, und Kortikosteroide sind zu verabreichen (siehe «Dosierung/Anwendung»).
Bei Verdacht auf SCARs sind die Patienten an den Facharzt/Spezialist zur weiteren Beurteilung und Behandlung zu überweisen. Bei Patienten mit Verdacht auf SJS oder TEN sollte die Tecentriq-Behandlung unterbrochen werden. Bei Bestätigung eines SJS oder einer TEN sollte Tecentriq dauerhaft abgesetzt werden.
Vorsicht ist geboten, wenn die Anwendung von Tecentriq bei einem Patienten erwogen wird, bei dem im Rahmen einer früheren Therapie mit anderen immunstimulierenden Wirkstoffen gegen eine Krebserkrankung eine schwere oder lebensbedrohliche unerwünschte Hautreaktion auftrat.
Wirkungen auf die Klasse der Immuncheckpoint-Inhibitoren
Während der Behandlung mit anderen Immuncheckpoint-Inhibitoren wurde über Fälle von exokriner Pankreasinsuffizienz berichtet, die auch unter Behandlung mit Atezolizumab vorkommen kann.
Zöliakie
Es wurde über Fälle von Zöliakie während der Behandlung mit Tecentriq berichtet.
Immunogenität
Explorative, nicht-adjustierte Metaanalysen zur Wirksamkeit (ohne Anpassung von Ungleichgewichten hinsichtlich Baseline Charakteristika) weisen auf eine Verminderung der Wirksamkeit von Tecentriq, hinsichtlich Gesamtüberleben bei ADA-positiven Patienten im Vergleich zu ADA-negativen Patienten hin (siehe «Eigenschaften/Wirkungen: Klinische Wirksamkeit» und «Pharmakokinetik»).
Patienten mit vorbestehender Autoimmunerkrankung
Bei Patienten mit vorbestehender Autoimmunerkrankung (AIE) deuten Daten aus Beobachtungsstudien auf ein erhöhtes Risiko für immunvermittelte unerwünschte Wirkungen nach Therapie mit einem Immun-Checkpoint-Inhibitor im Vergleich zu Patienten ohne vorbestehende AIE hin. Darüber hinaus traten häufig Schübe der zugrundeliegenden AIE auf, die aber überwiegend leicht und gut behandelbar waren.
Krankheitsspezifische Vorsichtsmassnahmen
Anwendung von Atezolizumab in Kombination mit Bevacizumab bei hepatozellulärem Karzinom
Bei Patienten, die mit Bevacizumab behandelt werden, besteht ein erhöhtes Blutungsrisiko. Bei Patienten mit hepatozellulärem Karzinom (HCC) unter Behandlung mit Atezolizumab in Kombination mit Bevacizumab wurden Fälle schwerer gastrointestinaler Blutungen, einschliesslich tödlich verlaufender Ereignisse, berichtet. Bei Patienten mit HCC sollten vor Beginn der Behandlung mit einer Kombination aus Atezolizumab und Bevacizumab ein Screening auf Ösophagusvarizen und gegebenenfalls eine anschliessende Behandlung der klinischen Praxis entsprechend durchgeführt werden. Bevacizumab sollte bei Patienten mit Blutung vom Grad 3 oder 4 während der Kombinationsbehandlung dauerhaft abgesetzt werden. Es ist die Fachinformation für Bevacizumab zu beachten.
Erhöhtes Mortalitätsrisiko für Patientinnen mit metastasiertem TNBC bei Anwendung von Tecentriq mit Paclitaxel
In einer randomisierten Studie mit Patientinnen mit metastasiertem TNBC wurde in der PD-L1-positiven Population bei Patientinnen, die mit Tecentriq iv plus Paclitaxel behandelt wurden, im Vergleich zu jenen, die mit Placebo und Paclitaxel behandelt wurden, ein erhöhtes Mortalitätsrisiko beobachtet. Das Gesamtüberleben in der Population mit PD-L1-Expression, in der die Mortalität 42% betrug, ergab eine HR von 1,11 (95-%-KI: 0,76; 1,64). Das mediane Überleben betrug 22,1 Monate (95-%-KI: 19,2; 30,5) bei Patientinnen, die Tecentriq in Kombination mit Paclitaxel erhielten, und 28,3 Monate (95-%-KI: 19,1; NE) bei Patientinnen, die ein Placebo mit Paclitaxel erhielten (siehe «Indikationen/Anwendungsmöglichkeiten» und «Dosierung/Anwendung»). Die häufigste Todesursache war eine Krankheitsprogression und zwischen den Armen wurde kein Ungleichgewicht der toxizitätsassoziierten Mortalität beobachtet.
Die Wirksamkeit von Tecentriq in Kombination mit Paclitaxel in Patienten mit lokal fortgeschrittenem oder metastasiertem TNBC wurde nicht gezeigt. Zur Behandlung von metastasiertem TNBC in der klinischen Praxis ausserhalb kontrollierter Studien sollte proteingebundenes Paclitaxel nicht durch Paclitaxel in Kombination mit Tecentriq ersetzt werden.
Spezielle Patientengruppen
Aus klinischen Studien ausgeschlossene Patienten
Patienten mit den folgenden Erkrankungen oder Merkmalen waren von der Teilnahme an klinischen Studien ausgeschlossen:
·Vorgeschichte mit Pneumonitis,
·aktiven Hirnmetastasen,
·HIV- oder Hepatitis-B- oder Hepatitis-C-Infektion,
·Patienten, denen innerhalb von 28 Tagen vor der Aufnahme in die Studie ein lebend-attenuierter Impfstoff verabreicht wurde,
·Patienten, die innerhalb von 4 Wochen vor der Aufnahme in die Studie systemische Immunstimulanzien oder innerhalb von 2 Wochen vor der Aufnahme in die Studie systemische Immunsuppressiva erhielten,
·Autoimmunerkrankung in der Vorgeschichte,
·Patienten mit erheblichen kardiovaskulären Erkrankungen und Patienten mit unzureichender hämatologischer Funktion bzw. unzureichenden Funktionen von Endorganen.
Patienten mit einem ECOG Performancestatus ≥2 waren von der Teilnahme an den klinischen Studien bei NSCLC und UC mit Zweitlinientherapie ausgeschlossen.
Embryofötale Toxizität
Auf der Grundlage des Wirkmechanismus kann die Anwendung von Tecentriq schädliche Wirkungen auf den Fötus haben. Tierexperimentelle Studien haben gezeigt, dass die Hemmung des PD-L1/PD-1-Signalwegs zu einem erhöhten Risiko für eine immunvermittelte Abstossung des sich entwickelnden Fötus und daraus resultierenden fötalen Tod führen kann.
Schwangere Frauen sollten auf das potenzielle Risiko für den Fötus hingewiesen werden. Frauen im gebärfähigen Alter sollten angewiesen werden, während der Behandlung mit Tecentriq und während 5 Monaten nach der letzten Dosis hochwirksame Methoden der Schwangerschaftsverhütung anzuwenden (siehe «Schwangerschaft» und «Präklinische Daten: Teratogenität»).
Unerwünschte Wirkungen bei Transplantationspatienten
Bei mit PD-1/PD-L1-Inhibitoren behandelten Patienten wurde im Postmarketing-Umfeld eine Abstossung von soliden Organtransplantaten beobachtet. Die Behandlung mit Tecentriq kann das Abstossungsrisiko bei Empfängern solider Organtransplantate erhöhen. Bei diesen Patienten sollte der Nutzen der Behandlung mit Atezolizumab gegen das Risiko einer möglichen Organabstossung abgewogen werden.
InteraktionenPharmakokinetische Interaktionen
Es wurden keine formalen pharmakokinetischen Interaktionsstudien mit Atezolizumab durchgeführt. Da Atezolizumab durch katabolischen Abbau aus dem Blutkreislauf eliminiert wird, sind keine metabolischen Arzneimittelwechselwirkungen zu erwarten.
Pharmakodynamische Interaktionen
Eine Verwendung systemischer Kortikosteroide oder Immunsuppressiva vor Behandlungsbeginn mit Atezolizumab sollte aufgrund möglicher Beeinträchtigungen der pharmakodynamischen Aktivität und der Wirksamkeit von Atezolizumab vermieden werden. Systemische Kortikosteroide oder andere Immunsuppressiva können jedoch nach Beginn der Therapie mit Atezolizumab zur Behandlung von immunvermittelten Nebenwirkungen angewendet werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Schwangerschaft, StillzeitSchwangerschaft
Aufgrund seines Wirkungsmechanismus kann Tecentriq den Fötus schädigen, wenn es an Schwangere verabreicht wird. Es liegen keine klinischen Studien zur Anwendung von Tecentriq bei Schwangeren vor. Tierexperimente haben gezeigt, dass die Hemmung des PD-L1/PD1-Signalweges zu einem erhöhten Risiko für eine immunologische Abstossung des sich entwickelnden Fötus mit nachfolgendem fötalem Tod führen kann. Tecentriq darf nicht während der Schwangerschaft und nicht an Frauen im gebärfähigen Alter, die keine wirksame Methode der Schwangerschaftsverhütung anwenden, verabreicht werden, sofern dies nicht unbedingt erforderlich ist. Wenn dieses Arzneimittel während einer Schwangerschaft angewendet wird oder eine Patientin während der Behandlung schwanger wird, muss die Patientin über das potenzielle Risiko für den Fötus aufgeklärt werden.
Patientinnen im gebärfähigen Alter sollten während der Behandlung mit Tecentriq und während mindestens 5 Monaten nach der letzten Dosis hochwirksame Methoden der Schwangerschaftsverhütung anwenden und aktive Massnahmen zur Vermeidung einer Schwangerschaft ergreifen (siehe «Warnhinweise und Vorsichtsmassnahmen: Embryofötale Toxizität» und «Präklinische Daten: Teratogenität»).
Wehen und Entbindung
Die Anwendung von Tecentriq während Wehen und Entbindung wurde nicht untersucht.
Stillzeit
Es ist nicht bekannt, ob Tecentriq in der menschlichen Muttermilch ausgeschieden wird. Es wurden keine Studien zur Untersuchung der Auswirkungen von Tecentriq auf die Milchbildung oder seiner Anwesenheit in der Muttermilch durchgeführt. Da viele Medikamente, darunter Antikörper, in die Muttermilch ausgeschieden werden, kann ein Risiko für das Neugeborene/Kleinkind nicht ausgeschlossen werden. Wegen möglicher Schäden für den gestillten Säugling wird es empfohlen während der Behandlung mit Tecentriq und während mindestens 5 Monaten nach der letzten Dosis nicht zu stillen.
Fertilität
Tierexperimente lassen darauf schliessen, dass Tecentriq eine Auswirkung auf den Menstruationszyklus von Frauen im gebärfähigen Alter haben kann (siehe «Präklinische Daten»). Patientinnen im gebärfähigen Alter müssen während der Behandlung mit Tecentriq und während mindestens 5 Monaten nach der letzten Dosis hochwirksame Methoden der Schwangerschaftsverhütung anwenden und aktive Vorsichtsmassnahmen zur Vermeidung einer Schwangerschaft ergreifen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenTecentriq hat einen geringen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit Maschinen zu bedienen. Patienten, bei denen eine Fatigue auftritt, sind anzuweisen, bis zum Abklingen der Symptome kein Fahrzeug zu führen und keine Maschinen zu bedienen.
Unerwünschte WirkungenKlinische Studien
Die Beurteilung der Sicherheit einer Monotherapie mit Tecentriq basiert auf gepoolten Daten von 5464 Patienten mit verschiedenen Tumorarten sowie unterstützenden Daten aus kumulativer Exposition von >13'000 Patienten in allen klinischen Studien (siehe Tabelle 4). Die häufigsten in klinischen Studien identifizierten Nebenwirkungen im Zusammenhang mit einer Tecentriq-Monotherapie (> 10%) waren Fatigue (27,9%), verminderter Appetit (19,3%), Hautausschlag (18,8%), Nausea (17,9%), Diarrhoe (17,1%), Pyrexie (17,0 %), Husten (16,8%), Obstipation (16,2%), Dyspnoe (15,6%), Arthralgie (15,5%), Anämie (14,5%), Pruritus (13,0%), Asthenie (12,6%), Rückenschmerzen (12,0%), Harnwegsinfektion (11,5%), Hepatitis [anomale Laborwerte] (11,2%) und Erbrechen (11,1%).
Die in klinischen Studien im Zusammenhang mit der Anwendung von Tecentriq iv im Rahmen einer Kombinationstherapie (n=5'196) festgestellten unerwünschten Wirkungen (UAW) sind ebenfalls in Tabelle 4 zusammengefasst. Die häufigsten unerwünschten Wirkungen (≥10%) waren Anämie (35,3%), Neutropenie (35,0%), Nausea (34,5%), Fatigue (32,4%), Hautausschlag (30,4%), Alopezie (29,8%), Diarrhoe (28,6%), periphere Neuropathie (24,8%), Verstopfung (24,7%), Thrombozytopenie (24,2%), verminderter Appetit (23,4%), Arthralgie (20,9%), Pyrexie (19,2%), Erbrechen (18,9%), Asthenie (18,6%), Hepatitis-abnormale Laborwerte (18,4%), Husten (17,9%), Dyspnoe (15,0%), muskuloskelettale Schmerzen (15,6%), Kopfschmerz (14,5%), Pruritus (13,9%), Leukopenie (13,9%), Hypothyreose (13,7%), Hypertonie (13,2%), Lungeninfektion (12,3%), Rückenschmerzen (11,4%), erhöhter ALT-Wert (11,4%), erhöhter AST-Wert (11,0%), peripheres Ödem (10,8%) und Nasopharyngitis (10,3%).
Tecentriq als Kombinationstherapie bei Melanom
In der Studie CO39262 (IMspire150) hatten die Patienten, welche Tecentriq iv in Kombination mit Cobimetinib und Vemurafenib erhielten, eine höhere Frequenz von hepatischen Laboranomalitäten (114/230, 49,6%), Pankreatitis (89/230, 38,7%), Hypothyreose (60/230, 26,1%), Hyperthyreose (43/230, 18,7%), Pneumonitis (29/230, 12,6%), Meningoencephalitis (6/230, 2,6%), Diabetes Mellitus (4/230, 1,7%), Myositis (3/230, 1,3%), Nephritis (3/230, 1,3%) und Hypophysitis (2/230, 0,9%). Die häufigsten (≥2%) schwerwiegenden unerwünschten Reaktionen waren Pyrexie (5,7%) und erhöhte Alaninaminotransferase (2,2%).
Weitere Informationen zu möglicherweise schwerwiegenden Nebenwirkungen sind im Abschnitt «Beschreibung ausgewählter Nebenwirkungen» beschrieben.
Die folgenden Häufigkeitskategorien wurden verwendet: «sehr häufig» (≥1/10), «häufig» (≥1/100, <1/10), «gelegentlich» (≥1/1000, <1/100), «selten» (≥1/10'000, <1/1000), «sehr selten» (<1/10'000).
Tabelle 4 fasst alle relevanten unerwünschten Wirkungen unter Tecentriq zusammen, welche auf gepoolten Daten aus Mono- und Kombinationstherapien verschiedener Tumorarten basieren.
Tabelle 4: Zusammenfassung der Nebenwirkungen bei Patienten, die in klinischen Studien mit Tecentriq i.v. oder s.c. behandelt wurden
|
|
Monotherapie mit Tecentriq
n=5464
|
Kombinationstherapie mit Tecentriq
n=5196
| |
Infektionen und parasitäre Erkrankungen
| |
Sehr häufig
|
Harnwegsinfektion a (alle Grade 11,5%, Grad 3-4 3,1%)
|
Lungeninfektion b (alle Grade 12,3%, Grad 3-4 4,7%, Grad 5 0,6%)
| |
Erkrankungen des Blutes und des Lymphsystems
| |
Sehr häufig
|
Anämie (alle Grade 14,5%, Grad 3-4 3,8%)
|
Anämie (alle Grade 35,3%, Grad 3-4 12,8%), Thrombozytopenie c (alle Grade 24,2%, Grad 3-4 9,4%, Grad 5<0,1%), Neutropenie d (alle Grade 35,1%, Grad 3-4 23,1%, Grad 5 0,1%), Leukopenie e (alle Grade 13,9%, Grad 3-4 5,6%)
| |
Häufig
|
Thrombozytopenie c
|
Lymphopenie f
| |
Selten
|
Hämophagozytische Lymphohistiozytose **
|
Hämophagozytische Lymphohistiozytose **
| |
Erkrankungen des Immunsystems
| |
Häufig
|
Überempfindlichkeit
|
| |
Endokrine Erkrankungen
| |
Sehr häufig
|
|
Hypothyreose g (alle Grade 14,0%, Grad 3-4 0,2%)
| |
Häufig
|
Hypothyreose g, Hyperthyreose h
|
Hyperthyreose h
| |
Gelegentlich
|
Nebenniereninsuffizienz i, Diabetes Mellitus j, Hypophysitis k
|
Nebenniereninsuffizienz i, Hypophysitis k
| |
Stoffwechsel- und Ernährungsstörungen
| |
Sehr häufig
|
Verminderter Appetit (alle Grade 19,3%, Grad 3-4 0,9%)
|
Verminderter Appetit (alle Grade 23,4%, Grad 3-4 1,5%, Grad 5<0,1%)
| |
Häufig
|
Hyperglykämie, Hypokaliämie m, Hyponatriämie n
|
Hypokaliämie m, Hyponatriämie n, Hypomagnesiämie l
| |
Erkrankungen des Nervensystems
| |
Sehr häufig
|
Kopfschmerzen (alle Grade 10,2%, Grad 3-4 0,3%)
|
Kopfschmerzen (alle Grade 14,7%, Grad 3-4 0,3%), periphere Neuropathie o (alle Grade 24,9%, Grad 3-4 3,0%)
| |
Häufig
|
|
Schwindel, Dysgeusie, Synkope
| |
Gelegentlich
|
Guillain-Barré-Syndrom p, Meningoencephalitis q
|
| |
Selten
|
Myasthenisches Syndrom r, Fazialisparese **, Myelitis **
|
Fazialisparese **, Myelitis **
| |
Herzerkrankungen
| |
Häufig
|
Perikarderkrankungen s
|
| |
Gelegentlich
|
|
Perikarderkrankungen s
| |
Selten
|
Myokarditis t
|
| |
Gefässerkrankungen
| |
Sehr häufig
|
|
Hypertonie u (alle Grade 13,2%, Grad 3-4 5,8%)
| |
Häufig
|
Hypotonie
|
| |
Erkrankungen der Atemwege, des Brustraums und Mediastinums
| |
Sehr häufig
|
Husten (alle Grade 16,8%, Grad 3-4 0,2%), Dyspnoe (alle Grade 15,6%, Grad 3-4 2,4%)
|
Husten (alle Grade 18,0%, Grad 3-4 0,2%), Dyspnoe (alle Grade 15,1%, Grad 3-4 1,7%, Grad 5<0,1%), Nasopharyngitis v (alle Grade 10,6%, Grad 3-4<0,1%)
| |
Häufig
|
Hypoxie w, Nasopharyngitis v, Pneumonitis x
|
Pneumonitis x, Dysphonie
| |
Gelegentlich
|
|
Hypoxie w
| |
Erkrankungen des Gastrointestinaltrakts
| |
Sehr häufig
|
Diarrhoe y (alle Grade 17,1%, Grad 3-4 1,0%), Nausea (alle Grade 17,9%, Grad 3-4 0,7%), Erbrechen (alle Grade 11,1%, Grad 3-4 0,6%)
|
Verstopfung (alle Grade 24,7%, Grad 3-4 0,5%), Diarrhoe y (alle Grade 28,7%, Grad 3-4 2,6%), Nausea (alle Grade 34,5%, Grad 3-4 1,5%), Erbrechen (alle Grade 19,0%, Grad 3-4 1,4%)
| |
Häufig
|
Bauchschmerzen, Kolitis z, Dysphagie, oropharyngeale Schmerzen aa, trockener Mund
|
Stomatitis, Pankreatitis bb, trockener Mund
| |
Gelegentlich
|
Pankreatitis bb
|
| |
Selten
|
Zöliakie **
|
Zöliakie **
| |
Leber- und Gallenerkrankungen
| |
Sehr häufig
|
Hepatitis- abnormale Laborwerte cc (Alle Grade 11,2%, Grad 3-4 3,2%)
|
Hepatitis-abnormale Laborwerte cc (Alle Grade 18,5%, Grad 3-4 5,6%, Grad 5<0,1%), erhöhter ALT-Wert (Alle Grade 11,4%, Grad 3-4 2,5%), erhöhter AST-Wert (Alle Grade 11,0%, Grad 3-4 2,3%)
| |
Häufig
|
Erhöhter ALT-Wert, erhöhter AST-Wert, Hepatitis dd
|
| |
Erkrankungen der Haut und des Unterhautgewebes
| |
Sehr häufig
|
Pruritus (alle Grade 13,0%, Grad 3-4 0,2%), Hautausschlag ee (alle Grade 18,8%, Grad 3-4 1,1%)
|
Pruritus (alle Grade 14,0%, Grad 3-4 0,2%), Hautausschlag ee (alle Grade 30,6%, Grad 3-4 2,7%), Alopezie ff (alle Grade 29,8%, Grad 3-4 <0,1%)
| |
Häufig
|
Trockene Haut
|
| |
Gelegentlich
|
Schwere unerwünschte Hautreaktionen (Toxische epidermale Nekrolyse, Stevens-Johnson-Syndrom) gg, Psoriasis hh
|
Schwere unerwünschte Hautreaktionen (Toxische epidermale Nekrolyse, Stevens-Johnson-Syndrom) gg, Psoriasis hh
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
| |
Sehr häufig
|
Arthralgie (alle Grade 15,5%, Grad 3-4 0,8%), Rückenschmerzen (alle Grade 12,0%, Grad 3-4 1,1%)
|
Arthralgie (alle Grade 21,8%, Grad 3-4 1,1%), Rückenschmerzen (alle Grade 11,5%, Grad 3-4 0,8%), muskuloskelettale Schmerzen ii (alle Grade 15,1%, Grad 3-4 0,7%)
| |
Häufig
|
Muskuloskelettale Schmerzen ii
|
| |
Gelegentlich
|
Myositis jj, kk
|
| |
Erkrankungen der Nieren und Harnwege
| |
Häufig
|
Kreatinin im Blut erhöht ll
|
Proteinurie mm, Kreatinin im Blut erhöht ll
| |
Gelegentlich
|
Nephritis nn
|
Nephritis nn
| |
Laborwerte
| |
Häufig
|
|
Alkalische Phosphatase im Blut erhöht
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
| |
Sehr häufig
|
Asthenie (alle Grade 12,6%, Grad 3-4 1,5%), Fatigue (alle Grade 27,9%, Grad 3-4 2,3%), Pyrexie (alle Grade 17,0%, Grad 3-4 0,5%)
|
Asthenie (alle Grade 18,7%, Grad 3-4 2,8%), Fatigue (alle Grade 32,5%, Grad 3-4 3,5%), Pyrexie (alle Grade 19,3%, Grad 3-4 0,9%), Peripheres Ödem (alle Grade 10,9%, Grad 3-4 0,2%)
| |
Häufig
|
Schüttelfrost, grippeartige Erkrankung, Infusionsbedingte Reaktionen oo, Reaktion an der Injektionsstelle pp, peripheres Ödem
|
|
a Umfasst Meldungen von Harnwegsinfektion, Zystitis, Pyelonephritis, Harnwegsinfektion mit Escherichia, bakterieller Harnwegsinfektion, Niereninfektion, akuter Pyelonephritis, Pilzinfektion der Harnwege, pseudomonaler Harnwegsinfektion, Harnwegsinfektion mit Enterokokken, chronischer Pyelonephritis, Harnwegsinfektion mit Staphylokokken, renalem Abzess, Pyelitis und Urethritis, Harnwegsinfekt durch Streptokokken
b Umfasst Meldungen von Pneumonie, Bronchitis, Infektion der unteren Atemwege, infektbedingter Exazerbation einer chronischen obstruktiven Lungenerkrankung, infektiösem Pleuraerguss, atypischer Pneumonie, Lungenabszess, Pleurainfektion, postprozeduraler Pneumonie, Tracheobronchitis, parakanzeröser Pneumonie, Pyopneumothorax
c Umfasst Meldungen von Thrombozytopenie und verringerter Thrombozytenzahl
d Umfasst Meldungen von Neutropenie, verringerter Neutrophilenzahl, febriler Neutropenie, neutropenischer Sepsis, Agranulozytose und Granulozytopenie
e Umfasst Meldungen von erniedrigter Zahl weisser Blutzellen und Leukopenie
f Umfasst Meldungen von Lymphozytopenie und verringerter Lymphozytenzahl
g Umfasst Meldungen von Hypothyreose, erhöhten und verminderten Blutspiegeln des Thyreoidea-stimulierenden Hormons, Autoimmunthyreoiditis, Kropf, Thyreoiditis, verminderten Blutspiegeln von freiem Thyroxin oder von Triiodthyronin, Schilddrüsenstörungen, erhöhtem Blutspiegel des freien Thyroxins oder von Gesamt-Thyroxin, vermindertem Blutspiegel des Gesamt-Triiodthyronins, erhöhtem Blutspiegel von freiem Triiodthyronin, anomalen Blutwerten des Thyroidea-stimulierenden Hormons, Euthyroid-Sick-Syndrom, Myxödemkoma, anomalem Schilddrüsenfunktionstest, verringertem Thyroxin, anomalem Triiodthyronin, Positivität auf Anti-Thyreoidea-Antikörper, stummer Thyreoiditis, chronischer Thyreoiditis, immunvermittelter Thyreoiditis, Myxödem, akuter Thyreoiditis, anomalem freiem Triiodthyronin, erhöhtem Triiodthyronin, autoimmunbedingter Hypothyreose, primärer Hypothyreose, hypothyreoter Struma, immunvermittelter Hypothyreose
h Umfasst Meldungen von Hyperthyreose, Basedow-Krankheit, endokriner Ophthalmopathie, Exophthalmus und primärer Hyperthyreose
i Umfasst Meldungen von Nebenniereninsuffizienz, Glukokortikoidmangel, primärer Nebenniereninsuffizienz, erniedrigtem Kortisol, akuter Nebennierenrindeninsuffizienz, abnormalem ACTH-Stimulationstest, Morbus Addison, Adrenalitis, Mangel an adrenocorticotropem Hormon (ACTH), abnormalen Blutwerten von Corticotropin, erhöhten Blutwerten von Corticotropin, sekundärer Nebennierenrindeninsuffizienz, erniedrigtem Corticotropin im Blut
j Umfasst Meldungen von Diabetes mellitus, Diabetes mellitus Typ 1, diabetischer Ketoazidose und Ketoazidose
k Umfasst Meldungen von Hypophysitis, Hypopituitarismus, gestörter Temperaturregulierung, sekundärer Insuffizienz der Nebennierenrinde
l Umfasst Meldungen von Hypomagnesiämie und erniedrigtem Magnesium im Blut
m Umfasst Meldungen von Hypokaliämie und erniedrigtem Kalium im Blut
n Umfasst Meldungen von Hyponatriämie und erniedrigtem Natrium im Blut
o Umfasst Meldungen von peripherer Neuropathie, immunvermittelter Neuropathie, peripherer sensorischer Neuropathie, Polyneuropathie, Herpes zoster, peripherer motorischer Neuropathie, autoimmuner Neuropathie, neuralgischer Amyotrophie, peripherer sensorimotorischer Neuropathie, axonaler Neuropathie, lumbosakraler Plexopathie, neuropathischer Arthropathie, toxischer Neuropathie, peripherer Nerveninfektion, Neuritis
p Umfasst Meldungen von Guillain-Barré-Syndrom, demyelinisierender Polyneuropathie, aufsteigender schlaffer Paralyse
q Umfasst Meldungen von Enzephalitis, Meningitis, Photophobie, autoimmuner Enzephalitis. aseptischer Meningitis
r Umfasst Meldungen von Myasthenia gravis
s Umfasst Meldungen von Perikarditis, Perikarderguss, Herztamponade und Pericarditis constrictiva
t Umfasst Meldungen von Myokarditis, autoimmunbedingte Myokarditis, immunvermittelte Myokarditis (Fälle von Myokarditis wurden in Studien ausserhalb des gepoolten Datensatzes berichtet)
u Umfasst Meldungen von Hypertonie, erhöhtem Blutdruck, hypertensiver Krise, erhöhtem systolischem Blutdruck, diastolischer Hypertonie, unzureichend eingestelltem Blutdruck und hypertensiver Retinopathie, essentieller Hypertonie, hypertensiver Nephropathie, orthostatischer Hypertonie
v Umfasst Meldungen von Nasopharyngitis, verstopfter Nase und Rhinorrhoe
w Umfasst Meldungen von Hypoxie, erniedrigter Sauerstoffsättigung und erniedrigtem PO2
x Umfasst Meldungen von Pneumonitis, Alveolitis, pulmonaler Toxizität, Lungeninfiltration, Bronchiolitis, interstitieller Lungenkrankheit, Bestrahlungspneumonitis, Lungentrübung, alveolärer Lungenkrankheit, eosinophiler Pneumonie, immunvermittelter Pneumonitis, Lungenfibrose, pulmonalen Strahlungsschäden, immunvermittelter Lungenerkrankung
y Umfasst Meldungen von Diarrhoe, häufigem Stuhlgang, gastrointestinaler Hypermotilität, Stuhldrang, hämorrhagischer Diarrhoe
z Umfasst Meldungen von Kolitis, Diversionskolitis, autoimmuner Kolitis, ischämischer Kolitis, mikroskopischer Kolitis, Colitis ulcerosa, immunvermittelter Enterokolitis, eosinophiler Kolitis
aa Umfasst Meldungen von oropharyngealen Schmerzen, oropharyngealen Beschwerden und Rachenreizung
bb Umfasst Meldungen von Pankreatitis, autoimmuner Pankreatitis, akuter Pankreatitis, erhöhter Lipase, erhöhter Amylase, erhöhten Bauchspeicheldrüsenenzymen, abnormaler Amylase
cc Umfasst Meldungen von erhöhten ALT-Werten, erhöhten AST-Werten, erhöhtem Bilirubin im Blut, erhöhter Gamma-Glutamyltransferase, Aszites, erhöhten Transaminasen, hepatischen Schmerzen, Hyperbilirubinämie, erhöhte Werte im Leberfunktionstest, erhöhte Leberenzyme, gestörte Leberfunktion, Hypertransaminasämie, erhöhten Blutwerten von unkonjugiertem Bilirubin, Hyperammonämie, erhöhtem Gallensäure-Gesamtspiegel, erhöhten Ammoniakwerten, kongestiver Hepatopathie, erhöhten Bilirubinwerten im Urin, erhöhten Blutwerten von konjugiertem Bilirubin, Hepatomegalie, abnormalen Leberenzymen, abnormalen Werten im Leberfunktionstest, anomalem Blut-Bilirubin
dd Umfasst Meldungen von Aszites, Autoimmunhepatitis, Leberzellschädigung, Hepatitis, akuter Hepatitis, Hepatotoxizität, Lebererkrankung, arzneimittelinduzierter Leberschädigung, Leberversagen, hepatischer Steatose, hepatischer Läsion, Ösophagusvarizen-Hämorrhagie, Ösophagusvarizen, Hepatomegalie und toxischer Hepatitis, hepatischer Zytolyse, immunvermittelter Hepatitis, Leberschädigung, spontaner bakterieller Peritonitis
ee Umfasst Meldungen von Hautausschlag, makulo-papulösem Hautausschlag, Erythem, juckendem Hautausschlag, akneartiger Dermatitis, Ekzem, Dermatitis, erythematösem Hautausschlag, Hautulzeration, papulösem Hautausschlag, Follikulitis, makulösem Hautausschlag, Hautabschuppung, Handdermatitis, infiziertem Ekzem, Skrotaldermatitis, nodulärem Hautausschlag, follikulärem Hautausschlag, morbilliformem Hautausschlag, pustulösem Hautausschlag, Furunkel, Akne, Arzneimittelexanthem, palmar-plantarem Erythrodysästhesie-Syndrom, seborrhoischer Dermatitis, allergischer Dermatitis, Erythem des Augenlids, Hauttoxizität, Hautausschlag am Augenlid, fixem Exanthem, papulosquamösem Hautausschlag, vesikulärem Hautausschlag, Hautblase, Lippenbläschen, Pemphigoid, Blutblase im Mund, Exanthem an der Gefässzugangstelle, immunvermittelte Dermatitis
ff Umfasst Meldungen von Alopezie, Madarosis, Alopecia areata, Alopecia totalis und Hypotrichose
gg Umfasst Meldungen von bullöser Dermatitis, exfoliativem Exanthem, Erythema multiforme, generalisierter exfoliativer Dermatitis, toxischem Exanthem, Stevens-Johnson-Syndrome (SJS), exfoliativer Dermatitis, Arzneimittelreaktion mit Eosinophilie und systemischen Symptomen (DRESS), toxischer epidermaler Nekrolyse (TEN) und kutaner Vaskulitis (es wurde über Fälle von SJS und DRESS in Studien ausserhalb des kombinierten Datensatzes berichtet)
hh Umfasst Meldungen von psoriasiformer Dermatitis, Psoriasis guttata und Psoriasis
ii Umfasst Meldungen von Schmerzen im Bewegungsapparat, Myalgie und Knochenschmerzen
jj Umfasst Meldungen von Myositis, Rhabdomyolyse, Polymyalgia rheumatica, Dermatomyositis, Muskelabszess, Vorhandensein von Myoglobin im Urin, Myopathie, Polymyositis
kk Es liegen Berichte über tödliche Fälle in Studien ausserhalb des kombinierten Datensatzes vor
ll Umfasst Meldungen von erhöhtem Kreatinin im Blut und Hyperkreatininämie
mm Umfasst Meldungen von Proteinurie, Mikroalbuminurie, Vorhandensein von Proteinen im Urin, Hämoglobinurie, nephrotischem Syndrom, Urinanomalie und Albuminurie
nn Umfasst Meldungen von Nephritis, paraneoplastischer Glomerulonephritis, chronischer Glomerulonephritis, Purpura Schönlein-Henoch-Nephritis, tubulo-interstitieller Nephritis, autoimmunbedingter Nephritis, allergischer Nephritis, Glomerulonephritis, nephrotischem Syndrom und mesangioproliferativer Glomerulonephritis
oo Umfasst Meldungen von infusionsbedingter Reaktion Zytokin-Freisetzungssyndrom; Hypersensitivität, anaphylaktische Reaktion
pp Berichtet in Studie IMscin001 (bezogen auf subkutane Gabe). Die Häufigkeit basiert auf der Exposition gegenüber Tecentriq s.c. in der Studie IMscin001 und umfasst Berichte über Reaktionen an der Injektionsstelle, Schmerzen an der Injektionsstelle, Erythem an der Injektionsstelle und Hautausschlag an der Injektionsstelle
** Nach Markteinführung ausserhalb des gepoolten Datensatzes berichtet. Die Häufigkeitsangabe basiert auf der Exposition innerhalb des gesamten Programms
Post-Marketing-Daten
Die folgenden unerwünschten Arzneimittelreaktionen wurden im Rahmen der Post-Marketing-Überwachung mit Tecentriq identifiziert. Unerwünschte Arzneimittelreaktionen im Rahmen der Post-Marketing-Überwachung sind nach MedDRA-Systemorganklasse gelistet.
Erkrankung des Blutes und des Lymphsystems
Selten: hämophagozytische Lymphohistiozytose a.
Erkrankungen des Nervensystems
Selten: Fazialisparese a.
Selten: Myelitis a.
Augenerkrankungen
Gelegentlich: Uveitis a.
a Gemeldet im Rahmen der Post-Marketing-Erfahrung ausserhalb des kombinierten Datensatzes. Die Häufigkeit basiert auf der programmübergreifenden Exposition.
Beschreibung ausgewählter Nebenwirkungen
Die nachstehenden Daten enthalten Angaben zu wesentlichen unerwünschten Reaktionen unter Monotherapie mit Tecentriq. Details zu den wesentlichen unerwünschten Reaktionen auf Tecentriq bei Anwendung im Rahmen einer Kombinationstherapie sind angegeben, wenn klinisch relevante Unterschiede im Vergleich zu einer Monotherapie mit Tecentriq festzustellen waren. Zum Umgang mit folgenden Ereignissen siehe «Warnhinweise und Vorsichtsmassnahmen, Allgemein»:
Infektionen
In klinischen Studien bei 5464 Patienten mit verschiedenen Arten von Krebs, die Tecentriq erhielten, traten bei 41,1% der Patienten Infektionen auf, darunter Infektionen vom Grad 3 (8,1%), Grad 4 (1,6%) und Grad 5 (1,0%). Harnwegsinfektionen und Pneumonie waren die häufigsten Infektionen vom Grad 3 oder höher und traten bei 2,5 resp. 2,4% der Patienten auf.
In der Melanomstudie (CO39262) traten Infektionen bei Patienten, die Tecentriq iv in Kombination mit Cobimetinib und Vemurafenib erhielten, häufiger auf. Infektionen traten bei 60,4% (139/230) der Patienten auf; einschliesslich solcher vom Grad 3–4 bei 8,7% (20/230) und solcher vom Grad 5 bei 1,7% (4/230) der Patienten. Die häufigste Infektion war eine Harnwegsinfektion, die bei 7,8% (18/230) der Patienten auftrat.
Hämophagozytische Lymphohistiozytose
Eine hämophagozytische Lymphohistiozytose (HLH) trat bei < 0,1 % (2/5464) der Patienten unter Tecentriq-Monotherapie auf. Der Zeitraum bis zum Auftreten betrug 2,7 Monate (Bereich: 1,6 bis 3,8 Monate). Die Dauer betrug 1,1 Monate (Bereich: 0,9 bis 1,4 Monate). Bei 1 Patienten (< 0,1 %) führte die HLH zum Absetzen von Tecentriq. Beide Patienten benötigten keine Kortikosteroide.
Immunvermittelte Pneumonitis
Eine Pneumonitis trat bei 2,9% (156/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Bei drei der 156 Patienten verlief dieses Ereignis tödlich. Die mediane Zeit bis zum Auftreten betrug 4,0 Monate (Bereich: 0,1 bis 29,8 Monate). Die mediane Dauer betrug 1,8 Monate (Bereich: 0 bis 27,8+ Monate; + kennzeichnet einen zensierten Wert). Bei 36 (0,7%) Patienten hatte die Pneumonitis einen Abbruch der Behandlung mit Tecentriq zur Folge. Eine Pneumonitis, die die Anwendung von Kortikosteroiden erforderlich machte, trat bei 1,6% (88/5464) der Patienten unter Tecentriq auf.
In der Studie IMscin001 trat bei 2,0% (5/247) der mit Tecentriq s.c. behandelten Patienten eine Pneumonitis auf.
In der Melanomstudie (CO39262) trat eine immunbedingte Pneumonitis bei Patienten, die Tecentriq iv in Kombination mit Cobimetinib und Vemurafenib erhielten, häufiger auf. Eine immunbedingte Pneumonitis trat bei 12,6% (29/230) der Patienten auf und war bei 1,3% (3/230) der Patienten vom Grad 3–4.
Immunvermittelte Hepatitis
Eine Hepatitis trat bei 1,7% (90/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Bei drei der 90 Patienten verlief dieses Ereignis tödlich. Die mediane Zeit bis zum Auftreten betrug 1,9 Monate (Bereich: 0,20 bis 18,8 Monate). Die mediane Dauer betrug 1,1 Monate (Bereich: 0 bis 32,4+ Monate; + kennzeichnet einen zensierten Wert). Hepatitis führte bei 18 (0,3%) Patienten zum Absetzen von Tecentriq. Bei 0,6% (32/5464) der Patienten, die Tecentriq erhielten, erforderte eine Hepatitis den Einsatz von Kortikosteroiden.
In der Studie IMscin001 trat bei 1,2% (3/247) der mit Tecentriq s.c. behandelten Patienten eine immunvermittelte Hepatitis auf.
In der Melanomstudie (CO39262) trat eine immunbedingte Hepatitis bei Patienten, die Tecentriq iv in Kombination mit Cobimetinib und Vemurafenib erhielten, häufiger auf. Eine immunbedingte Hepatitis trat bei 53,0% (122/230) der Patienten auf und war bei 22,2% (51/230) der Patienten vom Grad 3–4 und bei 0,9% (2/230) der Patienten vom Grad 5. Bei den meisten Ereignissen im Zusammenhang mit der immunbedingten Hepatitis, die bei Patienten unter Behandlung mit Tecentriq in Kombination mit Cobimetinib und Vemurafenib berichtet wurden, handelte es sich um abnormale Laborwerte (114 von 122 Patienten [93,4%]).
Immunvermittelte Kolitis
Eine Kolitis trat bei 1,1% (62/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 4,9 Monate (Bereich: 0,5 bis 17,2 Monate). Die mediane Dauer betrug 1,4 Monate (Bereich: 0,1 bis 50,2+ Monate; + kennzeichnet einen zensierten Wert). Bei 23 (0,4%) Patienten hatte die Kolitis einen Abbruch der Behandlung mit Tecentriq zur Folge. Bei 0,5% (29/5464) der Patienten unter Behandlung mit Tecentriq trat eine Kolitis auf, welche die Anwendung von Kortikosteroiden erforderte.
In der Melanomstudie (CO39262) trat Diarrhoe oder Kolitis bei Patienten, die Tecentriq iv in Kombination mit Cobimetinib und Vemurafenib erhielten, häufiger auf. Diarrhoe oder Kolitis trat bei 50,4% (116/230) der Patienten auf und war bei 3,0% (7/230) der Patienten vom Grad 3–4.
Immunvermittelte Endokrinopathien
Schilddrüsenerkrankungen
Eine Hypothyreose trat bei 8,6% (471/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 4,2 Monate (Bereich: 0 bis 34,5 Monate). Die Hypothyreose führte bei 10 (0,2%) der Patienten zum Abbruch der Behandlung mit Tecentriq. Die mittlere Dauer betrug 51,1 Monate (Bereich: 0,1+ bis 56,2+ Monate; + bezeichnet einen zensierten Wert).
Eine Hyperthyreose trat bei 2,5% (138/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 2,7 Monate (Bereich: 0 bis 24,3 Monate). Die mediane Dauer betrug 2,6 Monate (Bereich 0+ bis 47,7+; + kennzeichnet einen zensierten Wert). Die Hyperthyreose führte bei 5 (<0,1%) der Patienten zum Absetzen von Tecentriq.
Eine Hyperthyreose trat bei 4,9% (23/473) der Patienten auf, die Tecentriq in Kombination mit Carboplatin und Nab-Paclitaxel erhielten. Ein Patient (0,2%) brach die Behandlung aufgrund einer Hyperthyreose ab.
In der Studie IMscin001 trat eine Hypothyreose bei 10,5% (26/247) und eine Hyperthyreose bei 2,0% (5/247) der mit Tecentriq s.c. behandelten Patienten auf.
In der Melanomstudie (CO39262) traten Hypothyreose und Hyperthyreose bei Patienten, die Tecentriq iv in Kombination mit Cobimetinib und Vemurafenib erhielten, häufiger auf. Hypothyreose trat bei 26,1% (60/230) der Patienten auf; es gab keine Ereignisse vom Grad 3–4. Hyperthyreose trat bei 18,7% (43/230) der Patienten auf und war bei 0,9% (2/230) der Patienten vom Grad 3–4.
Nebenniereninsuffizienz
Eine Nebenniereninsuffizienz trat bei 0,5% (26/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 6,7 Monate (Bereich: 0,1 bis 21,4 Monate). Eine Nebenniereninsuffizienz führte bei 5 (<0,1%) Patienten zu einem Absetzen der Behandlung. Bei 0,4% (22/5464) der Patienten unter Behandlung mit Tecentriq trat eine Nebenniereninsuffizienz auf, welche die Anwendung von Kortikosteroiden erforderte.
In der Studie IMscin001 trat bei 0,4% (1/247) der mit Tecentriq s.c. behandelten Patienten eine Nebenniereninsuffizienz auf.
Eine Nebenniereninsuffizienz trat bei 1,5% (7/473) der Patienten auf, die Tecentriq iv in Kombination mit Carboplatin und Nab-Paclitaxel erhielten. Bei 0,8% (4/473) der Patienten, die Tecentriq in Kombination mit Carboplatin und Nab-Paclitaxel erhielten, trat eine Nebenniereninsuffizienz auf, welche die Anwendung von Kortikosteroiden erforderte.
Hypophysitis
Hypophysitis trat bei 0,1% (6/5464) der Patienten auf die Tecentriq als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug bei diesen Patienten 6,1 Monate (Bereich: 0,8 bis 13,7 Monate). Hypophysitis führte zu einem Absetzen von Tecentriq bei 1 (<0,1%) Patienten und machte den Einsatz von Kortikosteroiden bei <0,1% (5/5464) der Patienten unter Behandlung mit Tecentriq erforderlich. Hypophysitis trat bei 0,8% (3/393) der Patienten auf, die Tecentriq iv in einer laufenden Studie zusammen mit Bevacizumab, Paclitaxel und Carboplatin erhielten. Die mediane Zeit bis zum Auftreten betrug 7,7 Monate (Bereich: 5,0 bis 8,8 Monate). Alle drei Patienten benötigten eine Behandlung mit Kortikosteroiden.
Diabetes Mellitus
Diabetes mellitus trat bei 0,5% (26/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 5,4 Monate (Bereich: 0,1 bis 29,0 Monate). Die mediane Dauer betrug 9,2 Monate (Bereich 0,1 bis 51,2+ Monate; + kennzeichnet einen zensierten Wert). Bei 3 (<0,1%) Patienten hatte Diabetes mellitus einen Abbruch der Behandlung mit Tecentriq zur Folge und vier Patienten benötigten eine Behandlung mit Kortikosteroiden.
Ein Diabetes mellitus trat bei 2,0% (10/493) der HCC Patienten auf, die Tecentriq iv in Kombination mit Bevacizumab erhielten. Die mediane Zeit bis zum Auftreten betrug 4,4 Monate (Bereich: 1,2 bis 8,3 Monate). Ein Absetzen von Tecentriq war in keinem der Fälle von Diabetes mellitus erforderlich.
Immunvermittelte Meningoenzephalitis
Meningoenzephalitis trat bei 0,5% (24/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Die Zeitspanne bis zum Ausbruch betrug 0,5 Monate (Bereich 0 bis 12,5 Monate). Die mediane Dauer betrug 0,7 Monate (Bereich 0,1+ bis 14,5+ Monate; + kennzeichnet einen zensierten Wert). Eine Meningoenzephalitis führte zu einem Absetzen von Tecentriq bei 9 (0,2%) Patienten und machte den Einsatz von Kortikosteroiden bei 0,3% (14/5464) der Patienten unter Behandlung mit Tecentriq erforderlich.
Immunvermittelte Neuropathien
Guillain-Barré-Syndrom und demyelinisierende Polyneuropathie
Guillain-Barré-Syndrom und demyelinisierende Polyneuropathie traten bei 0,1% (6/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten dieses Ereignisses betrug 4,1 Monate (Bereich: 0,6 bis 8,1 Monate). Die mediane Dauer betrug 8,0 Monate (Bereich: 0,6 bis 24,5+ Monate; + kennzeichnet einen zensierten Wert). Guillain-Barré-Syndrom führte bei 3 (<0,1%) Patienten zum Absetzen von Tecentriq. Bei <0,1% (3/5464) der Patienten erforderte ein Guillain-Barré-Syndrom den Einsatz von Kortikosteroiden.
Immunvermittelte Fazialisparese
Eine Fazialisparese trat bei <0,1% (1/5464) der Patienten unter Tecentriq-Monotherapie auf. Der Zeitraum bis zum Auftreten betrug 1,0 Monat. Die Dauer betrug 1,1 Monate. Das Ereignis erforderte keine Gabe von Kortikosteroiden und führte nicht zum Absetzen von Tecentriq.
Immunvermittelte Myelitis
Eine immunvermittelte Myelitis trat bei <0,1% (1/5464) der Patienten unter Tecentriq-Monotherapie auf. Der Zeitraum bis zum Auftreten betrug 0,76 Monate. Das Ereignis erforderte die Gabe von Kortikosteroiden, führte aber nicht zum Absetzen von Tecentriq.
Immunvermittelte Pankreatitis
Eine Pankreatitis, einschliesslich erhöhter Serumamylase- und Serumlipasewerte, trat bei 0,8% (43/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 6,2 Monate (Bereich: 0 bis 40,4 Monate). Die mediane Dauer betrug 0,9 Monate (Bereich: 0 bis 40,4+ Monate; + kennzeichnet einen zensierten Wert). Pankreatitis führte bei 3 (<0,1%) Patienten zu einem Absetzen von Tecentriq. Bei <0,1% (7/5464) der Patienten, die Tecentriq erhielten, erforderte eine Pankreatitis den Einsatz von Kortikosteroiden.
Immunvermittelte Myositis
Myositis trat bei 0,6% (33/5464) der Patienten auf, die eine Tecentriq-Monotherapie erhalten hatten. Die mediane Zeit bis zum Auftreten betrug 5,0 Monate (Bereich: 0,4 bis 11,5 Monate). Die mediane Dauer betrug 3,2 Monate (Bereich 0,0 bis 51,1+ Monate, + bezeichnet einen zensierten Wert). Bei 2 Patienten (0,1%) führte die Myositis dazu, dass Tecentriq abgesetzt wurde. Bei 0,2% (11/5464) der Patienten unter Behandlung mit Tecentriq erforderte die Myositis die Anwendung von Kortikosteroiden.
Immunvermittelte Nephritis
Nephritis trat bei 0,2% (12/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 5,1 Monate (Bereich: 0,1 bis 20,4 Monate). Nephritis führte bei 6 (0,1%) Patienten zu einem Absetzen von Tecentriq. Sechs (0,1%) Patienten bedurften einer Behandlung mit Kortikosteroiden.
Schwere immunvermittelte Hautreaktionen
Schwere unerwünschte Hautreaktionen (SCARs) traten bei 0,6% (33/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Bei zwei der 33 Patienten verlief dieses Ereignis tödlich. Die mediane Zeit bis zum Auftreten betrug 4,9 Monate (Bereich: 0,1 bis 15,5 Monate). Die mediane Dauer betrug 2,8 Monate (Bereich: 0 bis 37,5+ Monate; + kennzeichnet einen zensierten Wert). In 3 Patienten (<0,1%) führten SCARs zum Abbruch der Behandlung mit Tecentriq. SCARs, welche die systemische Anwendung von Kortikosteroiden erforderten, traten bei 0,2% (11/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten.
In der Studie IMscin001 traten SCARs bei 0,8% (2/247) der mit Tecentriq s.c. behandelten Patienten auf.
Immunvermittelte Perikarderkrankungen
Perikarderkrankungen traten bei 0,9% (48/5464) der Patienten auf, die Tecentriq als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 1,4 Monate (Bereich: 0,2 bis 17,5 Monate). Die mediane Dauer betrug 1,4 Monate (Bereich: 0 bis 51,5+ Monate; + kennzeichnet einen zensierten Wert). Bei 3 Patienten (<0,1 %) führten Perikarderkrankungen zum Abbruch der Behandlung mit Tecentriq. Perikarderkrankungen, welche die Anwendung von Kortikosteroiden erforderten, traten bei 0,1% (7/5464) der Patienten auf.
Immunogenität
Wie bei allen therapeutischen Proteinen kann auch Tecentriq potentiell eine Immunantwort auslösen. In verschiedenen Phase-III-Studien entwickelten 13,1 - 38,5% der Patienten im Zusammenhang mit der Behandlung Antikörper gegen den Arzneistoff (Anti-Drug-Antikörper – ADA), 4,3% bis 19,9% der Patienten entwickelten neutralisierende Antikörper (NAb) (siehe «Warnhinweise und Vorsichtsmassnahmen», «Eigenschaften und Wirkungen», «Pharmakokinetik»).
In gepoolten Datensätzen von Patienten, die Atezolizumab als Monotherapie (n=3'460) beziehungsweise als Kombinationstherapie (n=2'285) erhalten haben, wurden für die ADA-positive Population im Vergleich zur ADA-negativen Population die folgenden Raten unerwünschter Ereignisse beobachtet: unerwünschte Ereignisse mit Grad 3–4: 45,9% bzw. 39,1%; schwerwiegende unerwünschte Ereignisse 39,4% bzw. 33,0%; unerwünschte Ereignisse, die zum Abbruch der Behandlung führten: 8,4% bzw. 7,7% (bei Monotherapie); unerwünschte Ereignisse mit Grad 3–4: 63,9% bzw. 60,9%, schwerwiegende unerwünschte Ereignisse: 43,9% bzw. 35,6%, unerwünschte Ereignisse, die zum Abbruch der Behandlung führten: 22,8% gegenüber 18,4% (bei Kombinationstherapie).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurden keine Fälle von Überdosierung berichtet.
Eigenschaften/WirkungenATC-Code
L01FF05
Wirkungsmechanismus
Tecentriq s.c. enthält Atezolizumab und rekombinante humane Hyaluronidase (rHuPH20).
Durch die Bindung von PD-L1 an PD-1- und B7.1-Rezeptoren auf T-Zellen wird die zytotoxische T-Zell-Aktivität über eine Hemmung der Proliferation und Zytokinproduktion der T-Zellen supprimiert. PD-L1 kann auf Tumorzellen (TC) und Tumor-infiltrierenden Immunzellen (IC) exprimiert werden und zur Hemmung der antitumoralen Immunantwort in der Mikroumgebung beitragen.
Atezolizumab ist ein humanisierter monoklonaler Immunglobulin G1 (IgG1)-Antikörper mit modifiziertem Fc-Teil, der direkt an PD-L1 bindet und die Interaktionen mit den PD-1- und B7.1-Rezeptoren blockiert. Dies führt zu einer Aufhebung der über den PD-L1/PD-1-Signalweg vermittelten Hemmung der Immunantwort einschliesslich Reaktivierung der antitumoralen Immunantwort. Atezolizumab lässt die PD-L2/PD-1-Interaktion intakt. In syngenen murinen Tumormodellen führte die Blockade der PD-L1-Aktivität zu vermindertem Tumorwachstum.
Die duale Hemmung der PD-1/PD-L1- und MAPK- sowie der BRAF- und MEK-Signalwege unterdrückt in Mauskrebsmodellen das Tumorwachstum und verbessert die Tumorimmunogenität durch eine im Vergleich zur alleinigen zielgerichteten Therapie erhöhte Antigenpräsentation und T-Zell-Infiltration und -Aktivierung.
Rekombinante humane Hyaluronidase (rHuPH20) baut lokal und transient Hyaluronan (HA), ein Glykosaminoglykan, das von Natur aus überall im Körper vorkommt, in der extrazellulären Matrix des subkutanen Raums ab, indem es die Bindung zwischen den beiden Zuckern (N-Acetylglucosamin und Glucuronsäure), aus denen HA besteht, spaltet.
Klinische Wirksamkeit
Intravenöse Darreichungsform
Für detaillierte Angaben zu klinischen Studien, die mit Tecentriq i.v. durchgeführt wurden, bitte die Fachinformation von Tecentriq i.v konsultieren.
Nicht-kleinzelliges Lungenkarzinom
Chemotherapie-vorbehandeltes NSCLC
IMscin001
Es wurde eine offene, multizentrische, internationale, randomisierte Studie der Phase Ib/III, BP40657 (IMscin001), durchgeführt, um die Pharmakokinetik, Wirksamkeit und Sicherheit von Tecentriq s.c. im Vergleich zu Tecentriq i.v. bei Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC zu bewerten, die noch keine Immuntherapie gegen Krebs (CIT) erhalten hatten und bei Patienten, bei denen es nach einer vorgängigen platinbasierten Therapie zu einer Progression oder zu einem Rezidiv kam. Die Studie IMscin001 war dazu ausgelegt, die Nichtunterlegenheit des Serumtalspiegels (CTal) und des per Modell vorausberechneten AUC-Werts von Tag 0 bis 21 im Atezolizumab-Zyklus 1 (vor der Dosisgabe in Zyklus 2) von Atezolizumab s.c. im Vergleich zu Atezolizumab i.v. aufzuzeigen (Ko-primärer Endpunkt). Zu den deskriptiven sekundären Endpunkten gehörten progressionsfreies Überleben (PFS), die objektive Ansprechrate (ORR) und das Gesamtüberleben (OS).
In Teil 2 (Phase III) wurden insgesamt 371 Patienten aufgenommen und randomisiert, um entweder 1875 mg Tecentriq s.c. q3w oder 1200 mg Tecentriq i.v. q3w zu erhalten. Eine Dosisreduktion war nicht zulässig.
Patienten mit anamnestisch bekannter Autoimmunerkrankung, aktiven oder kortikosteroid-abhängigen Hirnmetastasen, Anwendung einer attenuierten Lebendvakzine in den 4 Wochen vor der Randomisierung, sowie Anwendung von systemischen immunstimulierenden Wirkstoffen in den 4 Wochen oder von systemischen immunsuppressiven Arzneimitteln in den 2 Wochen vor der Randomisierung, wurden von der Teilnahme an der Studie ausgeschlossen.
Das mediane Alter der Intention-to-treat-Population betrug 64 Jahre (Bereich: 27 bis 85 Jahre), und 69 % der Patienten waren männlich. Die meisten Patienten waren Weisse (67 %). Ungefähr zwei Drittel der Patienten (65 %) hatten eine nicht-plattenepitheliale Erkrankung, 5 % hatten eine bekannte EGFR-Mutation, 2 % hatten bekannte ALK-Translokationen, 40 % waren PD-L1-positiv (TC ≥1 % und/oder IC ≥1 %), bei 16 % lagen zur Baseline nichtaktive ZNS-Metastasen vor, 26 % hatten einen ECOG PS von 0 und 74 % einen ECOG PS von 1, und die meisten Patienten waren aktuell oder vorgängig Raucher (70 %).
Die Nichtunterlegenheit der Exposition gegenüber Atezolizumab in Tecentriq s.c. im Vergleich zu Atezolizumab i.v. wurde aufgezeigt (siehe «Pharmakokinetik»). Die Ergebnisse einer post-hoc aktualisierten Wirksamkeitsanalyse mit einer medianen Überlebensdauer von 9,5 Monaten sind in Tabelle 5 zusammengefasst.
Tabelle 5: Zusammenfassung der aktualisierten Wirksamkeitsanalysen (IMscin001)
|
Wirksamkeitsendpunkt
|
Tecentriq s.c.
|
Tecentriq i.v.
| |
Vom Prüfarzt beurteilte, bestätigte ORR (RECIST v1.1)*
|
n=245
|
n=124
| |
Anzahl Responder (%)
|
27 (11,0%)
|
13 (10,5%)
| |
Vom Prüfarzt beurteiltes PFS (RECIST v1.1)*
|
n=247
|
n=124
| |
Anzahl Ereignisse (%)
|
219 (88,7%)
|
107 (86,3%)
| |
Median (Monate)
|
2,8
|
2,9
| |
OS*
|
n=247
|
n=124
| |
Anzahl Ereignisse (%)
|
144 (58,3%)
|
79 (63,7)
| |
Median (Monate)
|
10,7
|
10,1
|
ORR = objektive Ansprechrate; OS = Gesamtüberleben; PFS = progressionsfreies Überleben; RECIST = Beurteilungskriterien des Ansprechens solider Tumoren Version 1.1
* Deskriptive Analysen
Wirksamkeit von Atezolizumab bei Vorliegen von Anti-Drug-Antikörpern
Explorative, nicht-adjustierte Metaanalysen zur Wirksamkeit (ohne Anpassungen von Ungleichgewichten der Baseline Charakteristika) wurden mit Tecentriq i.v. durchgeführt. Gemäss den Metaanalysen betrug der Schätzwert der HR für das Gesamtüberleben (OS) zwischen der ADA-positiven Subgruppe und dem Kontrollarm 0,815 (95-%-KI: 0,732; 0,908), sowie der Schätzwert der HR für das OS zwischen der ADA-negativen Subgruppe und dem Kontrollarm 0,672 (95-%-KI: 0,617; 0,732), siehe Graphiken 1 und 2.
Graphik 1: Meta-Analyse OS (unadjustiert) ADA – Positive Patienten im Vergleich zur Kontrolle
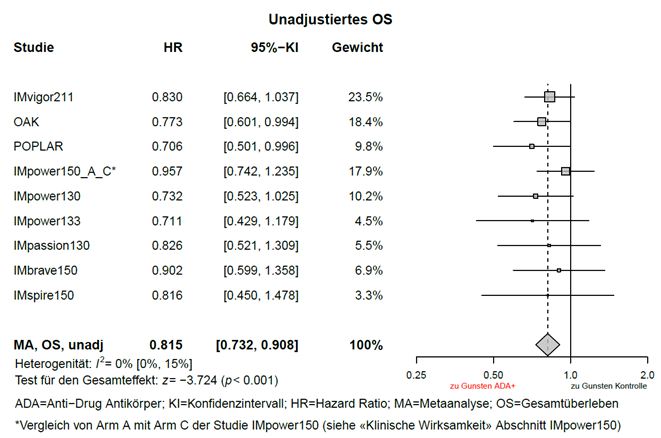
Graphik 2: Meta-Analyse OS (unadjustiert) ADA – Negative Patienten im Vergleich zur Kontrolle
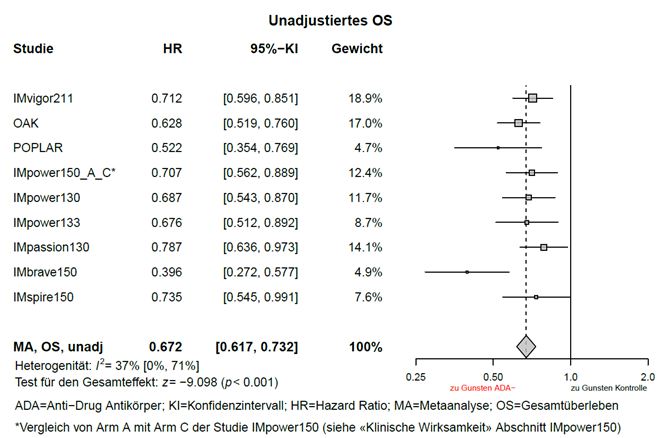
In der Studie IMscin001 war die Inzidenz behandlungsbedingter Anti-Atezolizumab-Antikörper bei Patienten, die mit Tecentriq s.c. bzw. i.v. behandelt wurden 19,5 % [43/221] bzw. 13,9 % [15/108]). Die Inzidenz behandlungsbedingter Anti-rHuPH20-Antikörper bei mit Tecentriq s.c. behandelten Patienten betrug 5,4 % (12/224).
Das mediane OS betrug bei mit Tecentriq s.c. behandelten Patienten, die ADA-negativ waren (n=178), 12,6 Monate (95% KI:10,6, 16,5) und bei Patienten, die zu einem beliebigen Zeitpunkt ADA-positiv waren (n=43), 6,9 Monate (95% KI: 5,0, nicht abschätzbar).
PharmakokinetikDer Wert für die gemessene CTal von Atezolizumab im Serum in Zyklus 1 (d.h. vor der Dosisgabe in Zyklus 2) deutete nicht auf eine Unterlegenheit von Atezolizumab in Tecentriq s.c. gegenüber intravenösem Atezolizumab hin, bei einem Quotienten der geometrischen Mittelwerte (GMR) von 1,05 (90 %-KI: 0,88–1,24). Der GMR für den per Modell berechneten AUC-Wert von Tag 0 bis 21 in Zyklus 1 (AUC0-21T) betrug 0,87 (90 %-KI: 0,83–0,92). Der Quotient des Mittelwerts der systemischen Akkumulation nach Gabe von 1875 mg Tecentriq s.c. q3w betrug 2,2. Die per Modell berechneten Werte von CTal und AUC für Tecentriq s.c. und intravenöses Atezolizumab im Steady-State waren vergleichbar (siehe Tabelle 6).
Tabelle 6: Atezolizumab-Exposition im Steady-State (geometrischer Mittelwert mit 5tem bis 95tem Perzentil) nach subkutaner oder intravenöser Gabe von Atezolizumab
|
Parameter
|
Atezolizumab in Tecentriq s.c.
|
Intravenöses Atezolizumab
| |
CTal im Steady-State a
(µg/ml)
|
205
(70,3–427)
|
179
(98,4–313)
| |
AUC im Steady-State a
(µg/ml•Tag)
|
6163
(2561–11340)
|
6107
(3890–9334)
|
a Die per Modell berechnete Exposition basierte auf der populationspharmakokinetischen Analyse
Absorption
Basierend auf einer populationspharmakokinetischen Analyse betrug die absolute Bioverfügbarkeit 72 % und die Absorptionsrate erster Ordnung (Ka) 0,3 (1/Tag). Der geometrische Mittelwert der maximalen Serumkonzentration (Cmax) von Atezolizumab im Zyklus 1 betrug 189 µg/ml und die mediane Zeit bis zur maximalen Serumkonzentration (Tmax) betrug 4,5 Tage (Median; 2,2–9,0 Tage, Min.-Max.).
Distribution
Eine populationspharmakokinetische Analyse ergab, dass bei einem typischen Patienten das Verteilungsvolumen des zentralen Kompartiments (V1) 3,28 l und das Verteilungsvolumen im Steady-State (Vss) 6,91 l beträgt.
Metabolismus
Der Metabolismus von Tecentriq wurde nicht direkt untersucht. Antikörper werden prinzipiell durch Katabolismus eliminiert.
Elimination
Eine populationspharmakokinetische Analyse ergab, dass die Clearance von Tecentriq 0,200 l/Tag und die typische Eliminationshalbwertszeit (t1/2) 27 Tage beträgt. In klinischen Studien mit intravenös verabreichtem Tecentriq war die mediane Atezolizumab Clearance in Patienten, welche positiv waren für behandlungsbedingte ADA, um 22% (Bereich zwischen 18% und 49%) erhöht im Vergleich zu Patienten, welche negativ waren für behandlungsbedingte ADA.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Es wurden keine spezifischen Studien mit Tecentriq bei Patienten mit Leberfunktionsstörungen durchgeführt. Die populationspharmakokinetische Analyse ergab keine klinisch bedeutsamen Unterschiede hinsichtlich der Clearance von intravenös oder subkutan angewendetem Tecentriq bei Patienten mit leichter Leberfunktionsstörung (Bilirubin ≤ ULN und AST > ULN oder Bilirubin < 1,0 bis 1,5 × ULN und beliebiger AST-Wert) oder mittelschwerer Beeinträchtigung der Leberfunktion (Bilirubin > 1,5 bis 3x ULN und beliebiger AST-Wert) im Vergleich zu Patienten mit normaler Leberfunktion (Bilirubin und AST ≤ ULN). Nur einer der mit Tecentriq behandelten Patienten mit mässiger Leberinsuffizienz erhielt Tecentriq s.c.. Von Patienten mit schwerer (Bilirubin > 3,0x ULN, beliebiger AST-Wert) Leberfunktionsstörung sind keine Daten verfügbar. Leberfunktionsstörungen wurden entsprechend den Kriterien für Leberfunktionsstörungen des National Cancer Institute (NCI) definiert (siehe «Dosierung/Anwendung: Spezielle Dosierungsanweisungen»).
Nierenfunktionsstörungen
Es wurden keine spezifischen Studien mit Tecentriq bei Patienten mit Nierenfunktionsstörungen durchgeführt. Bei Patienten, die Tecentriq entweder subkutan oder intravenös erhielten, ergab die populationspharmakokinetische Analyse bei Vorliegen einer leichten (eGFR 60 bis 89 ml/min/1,73 m2; n=111 mit Behandlung s.c. oder n=208 mit Behandlung i.v.) oder mittelschweren (eGFR 30 bis 59 ml/min/1,73 m2; n=32 mit Behandlung s.c. oder n=116 mit Behandlung i.v.) Nierenfunktionsstörung keine klinisch bedeutsamen Unterschiede hinsichtlich der Clearance im Vergleich zu Patienten mit normaler Nierenfunktion (eGFR grösser oder gleich 90 ml/min/1,73 m2; n=103 mit Behandlung s.c. oder n=140 mit Behandlung i.v.). Nur wenige, mit intravenösem Tecentriq behandelte Patienten hatten eine schwere Nierenfunktionsstörung (eGFR 15 bis 29 ml/min/1,73 m2; n=8) (siehe «Dosierung/Anwendung: Spezielle Dosierungsanweisungen»).
Ältere Patienten
Es wurden keine spezifischen Studien mit Tecentriq bei älteren Patienten durchgeführt. Der Einfluss des Alters auf die Pharmakokinetik von Tecentriq wurde in einer populationspharmakokinetischen Analyse untersucht. Basierend auf Patienten im Altersbereich 21-89 Jahre (n=472) bei einem medianen Alter von 62 Jahren wurde das Alter nicht als eine signifikante Kovariate mit Einfluss auf die Pharmakokinetik von intravenös angewendetem Tecentriq identifiziert. Es wurden keine klinisch bedeutsamen Unterschiede hinsichtlich der Pharmakokinetik von subkutan oder intravenös angewendetem Tecentriq bei Patienten < 65 Jahre (n=138 mit Behandlung s.c. oder n=274 mit Behandlung i.v.), Patienten im Alter von 65−75 Jahre (n=89 mit Behandlung s.c. oder n=152 mit Behandlung i.v.) und Patienten > 75 Jahre (n=19 mit Behandlung s.c. oder n=46 mit Behandlung i.v.) beobachtet (siehe «Dosierung/Anwendung: Spezielle Dosierungsanweisungen»).
Präklinische DatenSicherheitspharmakologie
Sicherheitspharmakologische Endpunkte von intravenös verabreichtem Tecentriq wurden in einer achtwöchigen und 26-wöchigen Studie zur chronischen Toxizität an Cynomolgusaffen untersucht ohne besondere Gefahren für den Menschen.
Toxikologische Eigenschaften bei Tieren
Bei einem spezifischen Mausstamm wurden unter intravenös verabreichten Dosierungen ≥10 mg/kg geringfügige Neuropathien des Ischiasnervs beobachtet. Geringfügige bis leichte Multiorgan-Arteriitiden/Periarteriitiden traten bei Cynomolgus-Affen unter Dosierungen ≥15 mg/kg nach subkutaner Verabreichung und 50 mg/kg nach intravenöser Verabreichung auf. Bei weiblichen Affen wurden unter intravenös verabreichten Dosierungen von 50 mg/kg unregelmässige Sexualzyklen beobachtet. Bei Affen wurden verstärkte subkutane Entzündungen (leichte Reizungen) an der Injektionsstelle beobachtet.
Eine gesteigerte Immunantwort, die zu erhöhter Mortalität führte, wurde im LCMV CL-13-, nicht jedoch im LCMV Armstrong-Virusinfektionsmodell bei Mäusen beobachtet.
In einer Studie an Cynomolgus-Affen mit Exposition gegenüber Hämocyanin der Schlüsselloch-Napfschnecke (Keyhole Limpet Hemocyanin, KLH) als T-Zell-abhängiges Antigen wurde 50 mg Atezolizumab pro kg per i.v. Injektion für bis zu 26 Wochen gut vertragen. Bei Cynomolgus-Affen, die KLH verabreicht bekamen, traten keine unerwarteten oder neuartigen Toxizitäten auf. Die langfristige Verabreichung von Atezolizumab führte zu einer nicht-signifikanten Verringerung der primären IgM- und der primären und sekundären IgG-Antworten auf KLH. Wenngleich die toxikologische Bedeutung aufgrund der geringen Anzahl der in dieser Studie verwendeten Tiere und der hohen Variabilität unter den Tieren ungewiss ist, ist daraus zu schliessen, dass Atezolizumab möglicherweise primäre und sekundäre IgG-Antworten auf T-Zell-abhängige Antigene verringert.
Mutagenität
Mit Tecentriq wurden keine Mutagenitätsstudien durchgeführt.
Karzinogenität
Mit Tecentriq wurden keine Karzinogenitätsstudien durchgeführt.
Reproduktionstoxizität
Mit Tecentriq wurden keine Reproduktions- oder Teratogenitätsstudien bei Tieren durchgeführt. Die Bedeutung des PD-L1/PD-1-Signalwegs als wesentlich für die maternofötale Toleranz und das embryofötale Überleben während der Trächtigkeit ist gut belegt. Es ist zu erwarten, dass die Gabe von Tecentriq sich ungünstig auf die Schwangerschaft auswirkt und den menschlichen Föten einem Risiko, unter anderem für embryonalen Tod, aussetzt.
Fertilität
Mit Tecentriq wurden keine Fertilitätsstudien durchgeführt. Die Studie zur chronischen Toxizität beinhaltete jedoch eine Beurteilung der Reproduktionsorgane männlicher und weiblicher Cynomolgus-Affen. Intravenös verabreichtes Tecentriq zeigte bei allen weiblichen Affen in der Gruppe mit der Dosierung 50 mg/kg eine Wirkung auf den Menstruationszyklus, welcher durch ein unregelmässiges Zyklusmuster während der Dosierungsphase verbunden mit dem Fehlen frischer Corpora lutea in den Ovarien bei der Sektion charakterisiert war. Dieser Effekt trat bei einem geschätzten AUC-Wert auf, der ungefähr 6 Mal höher war als der AUC-Wert bei Patienten nach Verabreichung der empfohlenen Dosis, und war in der verabreichungsfreien Erholungsphase reversibel. Es wurde keine Auswirkung auf die männlichen Reproduktionsorgane festgestellt.
Sonstige Angaben
Es wurden keine Karzinogenitäts-, Genotoxizitäts- oder Fertilitätsstudien mit rHuPH20 durchgeführt. In allgemeinen Studien zur Toxizität an Cynomolgus-Affen wurden keine schädlichen Wirkungen auf die Reproduktionsorgane von männlichen oder weiblichen Tieren beobachtet. Zusätzlich wurde keine Wirkung von rHuPH20 auf die Qualität der Spermien festgestellt. Reproduktionstoxikologische Studien mit rHuPH20 bei Mäusen liessen nach hoher systemischer Exposition eine embryofetale Toxizität, jedoch kein teratogenes Potenzial erkennen.
Sonstige HinweiseInkompatibilitäten
Es wurden keine Inkompatibilitäten zwischen Tecentriq s.c. und Polypropylen (PP), Polycarbonat (PC), Edelstahl (SS), Polyvinylchlorid (PVC) und Polyurethanen (PU) festgestellt.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Aus hygienischen Gründen sollte das Arzneimittel sofort verwendet werden, nachdem es von der Durchstechflasche in die Spritze überführt wurde, da es kein antimikrobielles Konservierungsmittel enthält. Wenn es nicht sofort verwendet wird, liegen die Dauer und die Bedingungen der Aufbewahrung vor der Anwendung in der Verantwortung des Benutzers und betragen normalerweise maximal 24 Stunden bei 2 °C bis 8 °C, es sei denn, die Zubereitung fand unter kontrollierten und validierten aseptischen Bedingungen statt.
Die mit der Spritzenkappe versehene Spritze kann bei ≤30 °C bis zu 8 Stunden bei diffusem Tageslicht und im Kühlschrank (2 °C bis 8 °C) bis zu 30 Tage aufbewahrt werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8 °C) lagern.
Nicht schütteln. Nicht einfrieren.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Tecentriq s.c. ist eine gebrauchsfertige Lösung zur ausschliesslichen subkutanen Injektion und sollte nicht verdünnt oder mit anderen Medikamenten vermischt werden.
Tecentriq s.c. ist vor der Anwendung einer Sichtprüfung zu unterziehen, um sicherzustellen, dass keine Partikel oder Verfärbungen vorhanden sind. Tecentriq Injektionslösung ist nur für den einmaligen Gebrauch bestimmt und sollte von medizinischem Fachpersonal vorbereitet werden.
Vorbereitung der Spritze
Tecentriq s.c. enthält keine antimikrobiellen Konservierungsmittel. Wenn die Dosis dem Patienten nicht sofort gegeben wird, sind die Angaben im Abschnitt «Aufbewahrung der Spritze» unten zu beachten.
Die Durchstechflasche vor der Anwendung aus dem Kühlschrank nehmen und die Lösung Raumtemperatur annehmen lassen.
Den gesamten Inhalt der Durchstechflasche mit Tecentriq s.c.-Lösung mit einer Spritze und einer Transfernadel (18G wird empfohlen) aufziehen. Die Transfernadel entfernen und ein Infusionsset für die subkutane Gabe (z.B. mit Flügeln/Butterfly) mit einer Edelstahlkanüle der Grösse 23–25 G für die Injektion aufsetzen. Für die Gabe an den Patienten ein SC Infusionsset mit einem verbleibenden Totvolumen von MAXIMAL 0,5 ml verwenden. Den Schlauch für die subkutane Injektion mit der Arzneimittellösung füllen, um die Luft aus dem Infusionsschlauch zu entfernen, aber den Zulauf stoppen, bevor die Flüssigkeit die Kanüle erreicht hat. Darauf achten, dass die Spritze genau 15 ml Arzneimittellösung enthält, nachdem sie gefüllt und überschüssiges Volumen aus der Spritze abgegeben worden ist. Zur Vermeidung eines Verstopfens der Kanüle, die Dosisgabe unverzüglich durchführen. Die vorbereitete Spritze mit angebrachtem und bereits gefülltem Infusionsset NICHT aufbewahren.
Aufbewahrung der Spritze
Wenn die Dosis nicht sofort gegeben wird, den gesamten Inhalt der Tecentriq s.c.-Lösung aseptisch aus der Durchstechflasche in die Spritze aufziehen, um das Dosisvolumen (15 ml) plus das Füllvolumen des SC Infusionssets aufzunehmen. Die Transfernadel durch eine Spritzenverschlusskappe ersetzen. Zur Aufbewahrung der Spritze KEIN SC Infusionsset anschliessen.
Wenn die Spritze im Kühlschrank aufbewahrt wurde, die Spritze vor der Dosisgabe Raumtemperatur annehmen lassen.
Entsorgung nicht benötigter/verfallener Arzneimittel
Die Freisetzung von Arzneimitteln in die Umwelt sollte minimiert werden. Arzneimittel sollten nicht über das Abwasser entsorgt werden. Eine Entsorgung mit dem Haushaltsabfall sollte vermieden werden. Verwenden Sie die an Ihrem Standort verfügbaren etablierten Sammelsysteme.
Zulassungsnummer69262 (Swissmedic).
PackungenDurchstechflasche mit 15 ml Injektionslösung zur subkutanen Anwendung (125 mg/ml) [A]
ZulassungsinhaberinRoche Pharma (Schweiz) AG, Basel.
Stand der InformationSeptember 2024.
|