ZusammensetzungWirkstoffe
Nemolizumab, hergestellt mittels rekombinanter DNA-Technologie in Ovarialzellen des chinesischen Hamsters.
Hilfsstoffe
Pulver: Saccharose, Trometamol, Trometalmolhydrochlorid (zur pH-Wert Einstellung), Argininhydrochlorid, Poloxamer 188
Lösungsmittel: Wasser für Injektionszwecke
Indikationen/AnwendungsmöglichkeitenAtopische Dermatitis (AD)
Nemluvio ist indiziert für die Behandlung von mittelschwerer bis schwerer atopischer Dermatitis in Kombination mit topischen Kortikosteroiden und/oder Calcineurininhibitoren bei Erwachsenen und Jugendlichen ab 12 Jahren mit einem Körpergewicht von mindestens 30 kg, wenn eine Therapie mit topischen Arzneimitteln alleine keine angemessene Krankheitskontrolle ermöglicht (siehe «Dosierung/Anwendung» und «Klinische Wirksamkeit»).
Prurigo nodularis (PN)
Nemluvio ist indiziert zur Behandlung von Erwachsenen mit mittelschwerer bis schwerer Prurigo nodularis, die für eine systemische Therapie in Frage kommen.
Dosierung/AnwendungAtopische Dermatitis (AD)
Die empfohlene Dosierung von Nemluvio bei Erwachsenen und Jugendlichen ab 12 Jahren beträgt:
§Eine Anfangsdosis von 60 mg (zwei 30-mg-Injektionen), gefolgt von 30 mg alle 4 Wochen (Q4W).
§Nach 16 Behandlungswochen beträgt die empfohlene Erhaltungsdosis von Nemluvio bei Patienten, die ein klinisches Ansprechen erreichen, 30 mg alle 8 Wochen (Q8W).
Begleitende topische Therapien:
Nemluvio wird zusammen mit topischen Kortikosteroiden mit niedriger oder mittlerer Wirkstärke und/oder topischen Calcineurininhibitoren angewendet. Topische Calcineurininhibitoren sollten auf Problembereiche wie Gesicht und Hals sowie den intertriginösen und genitalen Bereich vorbehalten bleiben. Wenn sich die Krankheit ausreichend gebessert hat, soll die Anwendung topischer Therapien ausgeschlichen und anschliessend beendet werden.
Bei Patienten, die nach 16 Wochen nicht auf die Behandlung ansprechen, ist ein Abbruch der Behandlung in Betracht zu ziehen.
Prurigo nodularis (PN)
Die empfohlene Dosis Nemluvio für Patienten mit einem Gewicht von unter 90 kg ist eine Anfangsdosis von 60 mg (zwei 30-mg-Injektionen), gefolgt von 30 mg alle 4 Wochen (Q4W).
Die empfohlene Dosis Nemluvio für Patienten mit einem Gewicht ab 90 kg ist eine Anfangsdosis von 60 mg (zwei 30-mg-Injektionen), gefolgt von 60 mg alle 4 Wochen (Q4W).
Bei Patienten, die nach 16 Wochen Behandlung der Prurigo nodularis kein Ansprechen gezeigt haben, sollte ein Abbruch der Behandlung in Betracht gezogen werden.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Versäumte Dosis (bei allen Indikationen)
Falls eine Dosis versäumt wurde, ist sie so rasch wie möglich nachzuholen und anschliessend wie vorgesehen fortzusetzen.
Spezielle Dosierungsanweisungen
Ältere Patienten (≥65 Jahre)
Bei älteren Patienten ist keine Dosisanpassung erforderlich (siehe Abschnitt «Pharmakokinetik»).
Patienten mit Leber- und Nierenfunktionsstörungen
Bei Patienten mit mit leichter oder mittelschwerer Leber- oder Nierenfunktionsstörung ist keine Dosisanpassung erforderlich. Zu Patienten mit einer schweren Leber- oder Niereninsuffizienz liegen nur sehr begrenzte Daten vor (siehe Abschnitt «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Nemluvio bei Kindern unter 12 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis sind noch nicht erwiesen.
Die Sicherheit und Wirksamkeit von Nemluvio bei Kindern und Jugendlichen unter 18 Jahren mit Prurigo nodularis sind nicht erwiesen. Es liegen keine Daten vor.
Körpergewicht
Für Patienten ab 12 Jahren mit atopischer Dermatitis wird keine Dosisanpassung bezüglich des Körpergewichts empfohlen (siehe Abschnitt «Pharmakokinetik»).
Für Patienten mit Prurigo nodularis und einem Körpergewicht ab 90 kg wird eine Dosis zu 60 mg (zwei 30-mg-Injektionen) empfohlen (siehe Abschnitt «Pharmakokinetik»).
Art der Anwendung
Die Behandlung ist durch medizinisches Fachpersonal einzuleiten, das in der Diagnose und Behandlung von Erkrankungen, bei denen Nemluvio indiziert ist (siehe Abschnitt «Indikationen/Anwendungsmöglichkeiten»), erfahren ist. Nemluvio kann durch den Patienten selbst oder durch eine Pflegeperson injiziert werden, wenn das medizinische Fachpersonal dies als angemessen erachtet. Vor der Anwendung sind der Patient und/oder die Pflegepersonen gemäss den Hinweisen zur Anwendung in der Packungsbeilage in der Vorbereitung und Verabreichung von Nemluvio zu unterweisen.
Subkutane Anwendung
Nemluvio wird subkutan in den vorderen Oberschenkel oder das Abdomen ausserhalb eines Umkreises von 5 cm um den Bauchnabel herum injiziert. Falls die Injektion durch eine andere Person erfolgt, kommt als Injektionsstelle auch der Oberarm infrage.
Die Einstichstelle muss bei jeder Injektion gewechselt werden. Nemluvio sollte nicht in Hautareale injiziert werden, die empfindlich, entzündet, geschwollen oder geschädigt sind oder blaue Flecken, Narben oder offene Wunden aufweisen.
Weitere Informationen zur Verabreichung dieses Arzneimittels sind dem Abschnitt «Hinweise für die Handhabung» zu entnehmen.
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der im Abschnitt «Zusammensetzung» genannten Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenÜberempfindlichkeitsreaktionen:
Bei einer (unmittelbaren oder verzögerten) allgemeinen systemischen Überempfindlichkeitsreaktion ist die Anwendung von Nemluvio sofort zu beenden und eine geeignete Behandlung einzuleiten.
Unkontrolliertes Asthma
Patienten mit unkontrolliertem Asthma wurden von den Studien ausgeschlossen und es liegen keine Daten zu Nemluvio in dieser Population vor.
Impfungen
Es wird empfohlen, vor der Behandlung mit Nemluvio den Impfstatus von Patienten entsprechend den aktuellen Impfempfehlungen auf den neuesten Stand zu bringen. Bei Patienten, die mit Nemluvio behandelt werden, sind Lebendimpfstoffe zu vermeiden. Es ist nicht bekannt, ob Lebendimpfstoffe während der Behandlung mit Nemluvio bezüglich ihrer Sicherheit und Wirksamkeit beeinflusst werden. Es liegen keine Daten hinsichtlich Ansprechen auf Totimpfstoffe vor.
InteraktionenInteraktionen mit Cytochrom P450
Die Auswirkungen von Nemolizumab auf die Pharmakokinetik von Midazolam (CYP3A4/5-Substrat), Warfarin (CYP2C9-Substrat), Omeprazol (CYP2C19-Substrat), Metoprolol (CYP2D6-Substrat) und Koffein ( CYP1A2-Substrat) wurden in einer Studie (SPR.201593) an 14 Probanden mit mittelschwerer bis schwerer AD untersucht, die eine erste subkutane Dosis von 60 mg erhielten, gefolgt von einer subkutanen Dosis von 30 mg alle 4 Wochen über einen Zeitraum von 12 Wochen. Es wurden keine klinisch signifikanten Veränderungen in der Exposition von CYP450-Substraten vor und nach mehreren Nemolizumab-Injektionen beobachtet, wobei die Cmax- und AUC-Verhältnisse zwischen 88,24 und 107,81 % lagen. Eine Auswirkung von Nemolizumab auf die Pharmakokinetik von gleichzeitig verabreichten Medikamenten ist nicht zu erwarten.
Schwangerschaft, StillzeitSchwangerschaft
Bisher liegen nur sehr begrenzte Erfahrungen mit der Anwendung von Nemolizumab bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe «Präklinische Daten»). Aus Vorsichtsgründen soll eine Anwendung von Nemolizumab während der Schwangerschaft vermieden werden.
Stillzeit
Es ist nicht bekannt, ob Nemluvio in die Muttermilch übergeht oder ob es Auswirkungen auf den gestillten Säugling und Auswirkungen auf die Milchproduktion hat. Es liegen keine Daten über die Ausscheidung von Nemolizumab in die Muttermilch vor. Beim Menschen erfolgt die Ausscheidung von IgG-Antikörpern in der Milch in den ersten Tagen nach der Geburt, die bald darauf auf niedrige Konzentrationen abfällt. Daher kann es in den ersten Tagen zu einer Übertragung von IgG-Antikörpern auf das Neugeborene über die Milch kommen. In diesem kurzen Zeitraum kann ein Risiko für das gestillte Kind nicht ausgeschlossen werden.
Fertilität
In tierexperimentellen Studien wurde keine Beeinträchtigung der Fertilität nachgewiesen (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDie Auswirkungen von Nemluvio auf die Fahrtüchtigkeit oder das Bedienen von Maschinen wurden nicht direkt untersucht.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die häufigsten Nebenwirkungen bei atopischer Dermatitis und Prurigo nodularis sind Typ-I-Überempfindlichkeitsreaktionen (1,1 %; umfasst Urtikaria 1,0 % und Angioödem 0,1 %) und Reaktionen an der Injektionsstelle (1,2 %). Weitere Nebenwirkungen wie Kopfschmerzen (7,0 %), atopische Dermatitis (4,6 %), Ekzem (3,8 %) und nummuläres Ekzem (3,5 %) wurden bei Prurigo nodularis berichtet
(siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Liste der unerwünschten Wirkungen
Die Anwendungssicherheit von Nemluvio wurde in einem Pool von drei randomisierten, placebokontrollierten Studien mit Patienten mit atopischer Dermatitis (1’192 Patienten, die Nemluvio erhielten, und 640 Patienten, die Placebo erhielten) und zwei randomisierten, placebokontrollierten Studien mit Patienten mit Prurigo nodularis (370 Patienten, die Nemluvio erhielten und 186 Patienten, die Placebo erhielten) ausgewertet.
In Tabelle 1 sind in klinischen Studien beobachtete unerwünschte Wirkungen aufgeführt, geordnet nach MedDRA-Systemorganklassen und Häufigkeit. Dabei wurden die folgenden Kategorien verwendet: sehr häufig (≥1/10), häufig (≥1/100 bis <1/10), gelegentlich (≥1/1000 bis <1/100), selten (≥1/10‘000 bis <1/1000) und sehr selten (<1/10‘000). Innerhalb jeder Häufigkeitsgruppe werden die unerwünschten Wirkungen nach abnehmendem Schweregrad dargestellt.
Tabelle 1: Liste der unerwünschten Wirkungen
|
MedDRA-Systemorganklasse
|
Häufigkeit
|
Unerwünschten Wirkungen
| |
Erkrankung des Nervensystems
|
Häufig
|
Kopfschmerzen (inkl. Spannungskopfschmerzen)*
| |
Erkrankungen der Haut und des Unterhautzellgewebes
|
Häufig
|
Urtikaria†
Atopische Dermatitis*, Ekzeme*, nummuläre Ekzeme*
| |
Gelegentlich
|
Angioödeme*
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Gelegentlich
|
Reaktionen an der Injektionsstelle (einschliesslich Erythem, Pruritus, Schmerzen†, Reizung†, Bluterguss*)
| |
Selten
|
Ödem an der Injektionsstelle†
|
† Aufgetreten in Studien zu atopischer Dermatitis
* Aufgetreten in Studien zu Prurigo nodularis
Bei atopischer Dermatitis stimmte das Sicherheitsprofil von Nemluvio in der offenen Studie (ARCADIA LTE) bis Woche 52 generell mit dem in Woche 16 beobachteten Sicherheitsprofil überein.
Bei Prurigo nodularis stimmte das Sicherheitsprofil von Nemluvio in der offenen Studie (OLYMPIA LTE) bis Woche 52 generell mit dem in Woche 16 und Woche 24 beobachteten Sicherheitsprofil überein.
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Überempfindlichkeit
Überempfindlichkeitsreaktionen vom Typ 1 (Ig-E-vermittelte Reaktionen) wurden bei Patienten berichtet, die mit Nemluvio bei atopischer Dermatitis und Prurigo nodularis behandelt wurden. Dazu gehörten leichte Urtikaria und ein Bericht über leichte (periokulare) Angioödeme im Gesicht (0,3 %), die nicht zum Abbruch der Behandlung führten. Es gab keine Berichte über anaphylaktischen Schock oder eine Serumkrankheit.
Reaktionen an der Injektionsstelle
Die Inzidenz von Reaktionen an der Injektionsstelle im anfänglichen Zeitraum war bei Patienten mit atopischer Dermatitis, die entweder mit Nemluvio (1,3 % der Patienten) oder Placebo (1,1 % der Patienten) behandelt wurden, gering. Im Erhaltungszeitraum blieb die Inzidenz mit Nemluvio Q8W (0 %) und Placebo (0,5 %) niedrig.
Bei Patienten mit Prurigo nodularis war die Inzidenz von Reaktionen an der Injektionsstelle niedrig, sowohl bei Behandlung mit Nemluvio (1,1 %) als auch Placebo (1,6 %). Es traten keine schweren Reaktionen an der Injektionsstelle auf.
Bei beiden Indikationen gab es keine Reaktionen, die zum Abbruch der Behandlung führten.
Kopfschmerzen
Bei Patienten mit Prurigo nodularis wurden Kopfschmerzen häufiger bei mit Nemluvio behandelten Patienten (7,0 %) berichtet als bei Patienten, die mit Placebo behandelt wurden (3,6 %). Kopfschmerzen wurden bei Patientinnen in beiden Gruppen häufiger beobachtet. In der Nemluvio-Gruppe waren die Kopfschmerzen meist leicht oder mittelschwer und führten nicht zu einem Abbruch der Behandlung.
Ekzematöse Reaktionen
Bei Patienten mit Prurigo nodularis wurden ekzematöse Reaktionen wie atopische Dermatitis, nummuläres Ekzem oder Ekzeme häufiger bei mit Nemluvio behandelten Patienten berichtet als bei Patienten, die mit Placebo behandelt wurden: atopische Dermatitis (4,6 % der Patienten gegenüber 0,5 % der Patienten), Ekzeme (3,8 % der Patienten gegenüber 2,2 % der Patienten) und nummuläre Ekzeme (3,5 % der Patienten gegenüber 0 % der Patienten). Diese ekzematösen Reaktionen waren leicht oder mittelschwer. Atopische Dermatitis führte bei zwei Patienten (0,5 %) zum Absetzen von Nemluvio. Es gab keine Ereignisse mit nummulärem Ekzem oder Ekzemen, die zum Studienabbruch führten.
Periphere und faziale Ödeme
Bei Patienten mit atopischer Dermatitis und Prurigo nodularis wurden periphere und faziale Ödeme häufiger bei mit Nemluvio behandelten Patienten (1,6 % und 3,0%) berichtet als bei Patienten, die mit Placebo behandelt wurden (0,3 % und 1,6%).
Die derzeit verfügbaren Informationen über periphere und faziale Ödeme reichen nicht aus, um einen kausalen Zusammenhang mit Nemluvio festzustellen.
Immunogenität
Wie bei allen therapeutischen Proteinen besteht bei Nemluvio die Möglichkeit einer Immunogenität.
Die beobachtete Inzidenz von Anti-Drug-Antikörpern (ADA) hängt stark von der Sensitivität und Spezifität des Assays ab. Unterschiede in den Assay-Verfahren schliessen aussagekräftige Vergleiche der Inzidenz von Anti-Drug-Antikörpern in den unten beschriebenen Studien mit der Inzidenz von Anti-Drug-Antikörpern in anderen Studien, einschliesslich derjenigen von Nemolizumab, aus.
In den Pivotstudien der Phase III zu AD (ARCADIA 1, ARCADIA 2) und der ARCADIA LTE-Studie über bis zu 128 Wochen betrug die Inzidenz behandlungsbedingter ADA 11,2 %. Neutralisierende Antikörper wurden bei 0,5 % der Patienten beobachtet.
In den Pivotstudien der Phase III zu PN (OLYMPIA 1, OLYMPIA 2) und der OLYMPIA LTE-Studie über bis zu 116 Wochen betrug die Inzidenz behandlungsbedingter ADA 12,8 %. Neutralisierende Antikörper wurden bei 3,5 % der Patienten beobachtet.
Pädiatrische Population
Atopische Dermatitis (AD) - Jugendliche (12 bis 17 Jahre)
Die Sicherheit von Nemluvio wurde bei 176 pädiatrischen Patienten im Alter von 12 bis 17 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis untersucht, die in die Studien ARCADIA 1 und ARCADIA 2 aufgenommen wurden. Das Sicherheitsprofil von Nemluvio bei diesen Patienten bis einschliesslich Woche 16 war vergleichbar mit dem Sicherheitsprofil, das bei Erwachsenen mit atopischer Dermatitis beobachtet wurde.
Das Sicherheitsprofil von Nemluvio bei pädiatrischen Patienten, die bis Woche 48 nachkontrolliert wurden, war vergleichbar mit dem in Woche 16 beobachteten Sicherheitsprofil. Das Langzeitsicherheitsprofil von Nemluvio bei pädiatrischen Patienten im Alter von 12 bis 17 Jahren stimmte mit dem bei Erwachsenen mit atopischer Dermatitis beobachteten überein (ARCADIA LTE).
Hinweis zur Meldung unerwünschter Wirkungen
Die Meldung des Verdachts auf unerwünschte Wirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs gibt keine spezifische Behandlung für eine Überdosierung mit Nemluvio. Im Falle einer Überdosierung ist der Patient auf Anzeichen oder Symptome von unerwünschten Wirkungen zu überwachen und im Bedarfsfall ist umgehend eine geeignete symptomatische Behandlung einzuleiten.
Eigenschaften/WirkungenATC-Code
D11AH12
Wirkungsmechanismus
Nemolizumab ist ein humanisierter monoklonaler IgG2-Antikörper, der die Interleukin-31-(IL-31-)-Signaltransduktion hemmt, indem er selektiv an den Interleukin-31-Rezeptor alpha (IL-31 RA) bindet. IL-31 ist ein natürlich vorkommendes Zytokin, das an Pruritus, Entzündung, epidermaler Dysregulation und Fibrose beteiligt ist. Nemolizumab hemmt IL-31-induzierte Reaktionen, einschließlich der Freisetzung von proinflammatorischen Zytokinen und Chemokinen.
Pharmakodynamik
Klinische Wirksamkeit
1) ) Klinische Wirksamkeit und Sicherheit bei Erwachsenen und Jugendlichen mit atopischer Dermatitis
Die Wirksamkeit und Sicherheit von Nemluvio mit begleitender topischer Hintergrundtherapie wurde in zwei randomisierten, doppelblinden, placebokontrollierten Pivotstudien (ARCADIA 1 und ARCADIA 2) beurteilt, in die total 1’728 Studienteilnehmer ab 12 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis aufgenommen wurden, die durch topische Behandlungen nicht ausreichend kontrolliert werden konnte. Der Schweregrad der Erkrankung wurde durch einen IGA (Investigator's Global Assessment)-Score von 3 (mittelschwer) und 4 (schwer) in der Gesamtbeurteilung der atopischen Dermatitis, einen EASI (Eczema Area and Severity Index)-Score von ≥ 16, eine minimale betroffene Körperoberfläche (BSA) von ≥ 10 % und einen PP-NRS (Peak Pruritus Numeric Rating Scale)-Score von ≥ 4 definiert.
Die Patienten in den Studien erhielten entweder eine erste subkutane Injektion von Nemolizumab 60 mg gefolgt von 30 mg Injektionen alle 4 Wochen (Q4W) oder entsprechendes Placebo. Begleitende TCS mit niedriger und/oder mittlerer Wirkstärke (gemäss United States classification) und/oder TCI wurden sowohl in den Nemolizumab- als auch in den Placebo-Gruppen für mindestens 14 Tage vor Baseline verabreicht und während der Studie fortgesetzt. Je nach Krankheitsaktivität konnten diese Begleittherapien nach Ermessen des Prüfarztes ausgeschlichen und/oder abgesetzt werden.
Nach 16 Wochen setzten die Patienten, die einen Erfolg nach EASI-75 oder IGA erzielten, für weitere 32 Wochen die Erhaltungsphase der Studie fort, um die Aufrechterhaltung des in Woche 16 erreichten Ansprechens zu beurteilen. Nemluvio-Responder wurden erneut randomisiert und erhielten entweder Nemluvio 30 mg alle 8 Wochen oder Placebo alle 4 Wochen (alle Gruppen setzten die Behandlung mit TCS/TCI fort). Patienten, die im anfänglichen Behandlungszeitraum zu Placebo randomisiert wurden und in Woche 16 das gleiche klinische Ansprechen erreichten, erhielten weiterhin alle 4 Wochen Placebo. Non-Responder in Woche 16, Patienten, die im Erhaltungszeitraum das klinische Ansprechen verloren, und Patienten, die den Erhaltungszeitraum absolviert hatten, hatten die Möglichkeit, in die offene Studie (ARCADIA LTE) aufgenommen zu werden und bis zu 200 Wochen lang alle 4 Wochen eine Behandlung mit Nemluvio 30 mg zu erhalten.
Endpunkte
Sowohl ARCADIA 1 als auch ARCADIA 2 bewerteten die primären Endpunkte bezüglich:
§Anteil der Patienten mit einem IGA-Erfolg (definiert als ein IGA von 0 [befallsfrei] oder 1 [nahezu befallsfrei] und einer Reduktion um ≥2 Punkte gegenüber Studienbeginn) in Woche 16
§Anteil der Patienten mit EASI-75 (≥75 % Verbesserung des EASI gegenüber Studienbeginn) in Woche 16
Sekundäre Hauptendpunkte waren PP-NRS-Verbesserung ≥4 gegenüber Baseline in den Wochen 1, 2, 4 und 16, PP-NRS <2 in Woche 4 und Woche 16, Verbesserung auf der SD-NRS (Sleep Disturbance Numeric Rating Scale) ≥4 gegenüber Baseline in Woche 16, Patienten mit einer Verbesserung sowohl des EASI-75 als auch des PP-NRS ≥4 gegenüber Baseline in Woche 16 und Patienten mit sowohl IGA-Erfolg als auch Verbesserung des PP-NRS ≥4 gegenüber Baseline in Woche 16.
Merkmale bei Studienbeginn
In diesen Studien waren bei Studienbeginn 51,0 % der Patienten männlich, 79,9 % waren weiss und 15,4 % waren 12 bis 17 Jahre alt. 70 % der Patienten hatten bei Studienbeginn einen IGA-Score von 3 (mittelschwere AD) und 30 % der Patienten einen IGA-Score von 4 (schwere AD). Der mittlere EASI-Wert bei Studienbeginn lag bei 27,5, der wöchentliche PP-NRS-Wert bei Studienbeginn betrug 7,1 (schwerer Juckreiz) und der wöchentliche SD-NRS-Wert lag bei Studienbeginn bei 5,8. Gesamthaft hatten 63,3 % der Patientinnen und Patienten andere vorgängige systemische Behandlungen für atopische Dermatitis erhalten.
Klinische Wirksamkeit - ARCADIA 1 und ARCADIA 2 – Erwachsene und Jugendliche – Induktionsphase, Woche 0 bis Woche 16
Nemluvio war im Vergleich zu Placebo hinsichtlich der hautbezogenen koprimären Endpunkte IGA-Erfolg und EASI-75 über 16 Wochen statistisch signifikant überlegen (Tabelle 2). Die Ergebnisse für beide koprimären Endpunkte waren bei der Population mit starkem Juckreiz (Baseline PP NRS ≥7) konsistent.
Tabelle 2 – Wirksamkeitsergebnisse für Nemluvio (30 mg Q4W) mit begleitenden TCS/TCI in ARCADIA 1 und ARCADIA 2 in Woche 16
|
|
ARCADIA 1
|
ARCADIA 2
| |
Nemluvio + TCS/TCI
|
Placebo +
TCS/TCI
|
Nemluvio + TCS/TCI
|
Placebo +
TCS/TCI
| |
Anzahl der randomisierten und behandelten Patienten (PP NRS bei Studienbeginn ≥4)
|
620
|
321
|
522
|
265
| |
% der Patienten mit IGA 0 oder 1a
|
35,6#
|
24,6
|
37,7#
|
26,0
| |
% der Patienten mit EASI-75a
|
43,5*
|
29,0
|
42,1#
|
30,2
|
a Patienten, die eine Bedarfsbehandlung erhielten, oder mit fehlenden Daten wurden als Non-Responder eingeordnet
* p-Wert <0,0001, #p-Wert <0,001Der schichtbereinigte p-Wert basiert auf dem CMH-Test, stratifiziert nach PP NRS und IGA-Score bei Studienbeginn
Abbildung 1 – Anteil der Patienten mit IGA-Erfolg und EASI-75 von Studienbeginn bis Woche 16 in ARCADIA 1 und ARCADIA 2
Eine signifikante Verbesserung des Pruritus bei Patienten, die in ARCADIA 1 und ARCADIA 2 mit Nemluvio behandelt wurden, im Vergleich zu Placebo, basierend auf Verbesserung der PP NRS von ≥4 gegenüber Studienbeginn wurde ab Woche 1 beobachtet und bis Woche 16 aufrechterhalten (Tabelle 3). Die Ergebnisse waren bei der Population mit schwerem Juckreiz (Ausgangswert PP-NRS ≥7) konsistent.
Tabelle 3 – Wirksamkeitsergebnisse zu Juckreiz bei Nemluvio mit begleitenden TCS/TCI in ARCADIA 1 und ARCADIA 2 bis Woche 16
|
|
ARCADIA 1
|
ARCADIA 2
| |
Nemluvio + TCS/TCI
|
Placebo + TCS/TCI
|
Nemluvio + TCS/TCI
|
Placebo + TCS/ TCI
| |
Anzahl der randomisierten und behandelten Patienten (PP NRS bei Studienbeginn ≥4)a
|
620
|
321
|
522
|
265
| |
% der Patienten mit PP-NRS-Verbesserung ≥4a
| |
In Woche 1
|
4,7§
|
1,2
|
6,7*
|
0,4
| |
In Woche 2
|
17,7*
|
3,1
|
16,9*
|
1,9
| |
In Woche 4
|
27,4*
|
6,5
|
26,1*
|
5,3
| |
In Woche 16
|
42,7*
|
17,8
|
41,0*
|
18,1
| |
% der Patienten mit PP NRS <2a
| |
In Woche 4
|
16,0*
|
3,7
|
15,9*
|
2,6
| |
In Woche 16
|
30,6*
|
11,2
|
28,4*
|
11,3
|
a Patienten, die eine Bedarfsbehandlung erhielten, oder mit fehlenden Daten wurden als Non-Responder eingeordnet
* p-Wert <0,0001, # p-Wert <0,001, § p-Wert <0,05 Der schichtbereinigte p-Wert basiert auf dem CMH-Test, stratifiziert nach PP NRS und IGA-Score bei Studienbeginn.
Die numerische Bewertungsskala für Schlafsörungen (SD NRS) ist eine Tagesskala, die von den Probanden verwendet wird, um den Grad ihres Schlafverlustes im Zusammenhang mit atopischer Dermatitis zu melden. In der Woche 16 wurde im Vergleich zu Placebo eine signifikante Verbesserung der Schlafstörung beobachtet (Tabelle 4). Die Ergebnisse waren bei der Population mit starken Juckreiz (Ausgangswert PP NRS ≥7) konsistent.
Tabelle 4 – Wirksamkeit bei Schlafstörungen mit Nemluvio mit begleitenden TCS/TCI in ARCADIA 1 und ARCADIA 2 in Woche 16
|
|
ARCADIA 1
|
ARCADIA 2
| |
Nemluvio + TCS/TCI
|
Placebo + TCS/TCI
|
Nemluvio + TCS/TCI
|
Placebo + TCS/ TCI
| |
Anzahl der randomisierten und behandelten Patienten (PP NRS bei Studienbeginn ≥4)a
|
620
|
321
|
522
|
265
| |
% der Patienten mit SD-NRS-Verbesserung ≥4a
Mittlere Veränderung gegenüber Studienbeginn (%)
|
37,9*
-64,6
|
19,9
-38,1
|
33,5*
-59,7
|
16,2
-35,4
|
a Teilnehmende, die eine Bedarfsbehandlung erhielten, oder mit fehlenden Daten wurden als Non-Responder eingeordnet
* p-Wert <0,0001Der schichtbereinigte p-Wert basiert auf dem CMH-Test, stratifiziert nach PP NRS und IGA-Score bei Studienbeginn
Jugendliche mit atopischer Dermatitis (12 bis 17 Jahre)
Die Ergebnisse zur Wirksamkeit der Studien ARCADIA 1 und ARCADIA 2 in Woche 16 für pädiatrische Probanden im Alter von 12 bis 17 Jahren sind in Tabelle 5 dargestellt. Die Ergebnisse in der pädiatrischen Probandengruppe stimmten im Allgemeinen mit den Ergebnissen in der erwachsenen Probandengruppe überein. Die Ergebnisse in den koprimären und wichtigsten sekundären Endpunkten waren in der Gruppe mit starkem Juckreiz (Baseline PP NRS ≥7) konsistent.
Tabelle 5 – Ergebnisse zur Wirksamkeit von Nemolizumab (30 mg Q4W) mit gleichzeitiger Gabe von TCS/TCI in ARCADIA 1 und ARCADIA 2 in Woche 16 bei pädiatrischen Probanden im Alter von 12 bis 17 Jahren
|
|
ARCADIA 1 AND ARCADIA 2
| |
Nemluvio+ TCS/TCI
|
Nemoluvio + TCS/TCI
| |
Anzahl der behandelten und randomisierten Patienten (Baseline PP NRS ≥4)
|
179
|
90
| |
% der Patienten mit IGA 0 or 1 a
|
48.9*
|
34.4
| |
% der Patienten mit EASI-75 a
|
53.4§
|
43.3
|
a Probanden, die eine Notfallbehandlung erhielten oder bei denen Daten fehlten, wurden als Non-Responder betrachtet
≠p-Wert <0,0001, #p-Wert <0,001, *p-Wert <0,05, ∞p-Wert =0,0591, §p-Wert =0,1824
Der stratifizierte p-Wert basiert auf dem CMH-Test, der nach PP-NRS- und IGA-Score zu Studienbeginn stratifiziert wurde.
Klinische Wirksamkeit - ARCADIA 1 und ARCADIA 2 – Erwachsene und Jugendliche – Erhaltungsphase, Woche 16 bis Woche 48
Das klinische Ansprechen bei Nemolizumab-Respondern (IGA 0/1 oder EASI-75 in Woche 16) wurde zwischen Woche 16 und Woche 48 in den Studien ARCADIA 1 und ARCADIA 2 ausgewertet. Für die Erhaltungsbehandlung wurden 338 Nemolizumab-Responder erneut randomisiert und erhielten entweder Nemolizumab 30 mg Q8W oder Placebo Q4W (Nemolizumab-Absetzen) mit gleichzeitiger TCS/TCI. In Woche 48 erreichten in der Nemolizumab 30 mg + TCS/TCI Q8W Gruppe 60.4% der Patienten ein IGA 0/1 und 75.7% ein EASI-75, verglichen mit 49.7% ein IGA 0/1 und 63.9% ein EASI-75 in der Placebo + TCS/TCI Q4W Gruppe.
2) Klinische Wirksamkeit und Sicherheit bei Erwachsenen mit Prurigo nodularis
Die Wirksamkeit und Sicherheit von Nemluvio als Monotherapie wurde in zwei randomisierten, doppelblinden, placebokontrollierten Pivotstudien (OLYMPIA 1 und OLYMPIA 2) untersucht, in die total 560 Teilnehmende ab 18 Jahren mit Prurigo nodularis aufgenommen wurden. Der Schweregrad der Erkrankung wurde anhand einer allgemeinen Beurteilung durch die Prüfärztin/den Prüfarzt (IGA) bei der Gesamtbeurteilung der Prurigo nodularis-Knoten auf einer Schweregradskala von 0 bis 4 definiert. Die in diese beiden Studien aufgenommenen Patienten hatten einen IGA-Score von ≥3, schweren Pruritus, definiert durch einen wöchentlichen Durchschnitt des Wertes auf der numerischen Bewertungsskala für den Pruritus-Spitzenwert (PP NRS) von ≥7 auf einer Skala von 0 bis 10 sowie mindestens 20 noduläre Läsionen. OLYMPIA 1 und OLYMPIA 2 beurteilten die Wirkung der Nemluvio-Monotherapie auf die Anzeichen und Symptome von Prurigo nodularis und zielten auf die Verbesserung der Hautläsionen und des Pruritus über 16 Wochen ab. OLYMPIA 1 hatte einen 24-wöchigen und OLYMPIA 2 einen 16-wöchigen Behandlungszeitraum.
Teilnehmende, die OLYMPIA 1 und OLYMPIA 2 abschlossen, hatten die Möglichkeit, in die offene Studie (OLYMPIA LTE) aufgenommen zu werden und bis zu 184 Wochen lang alle 4 Wochen mit Nemluvio behandelt zu werden.
Teilnehmende mit einem Körpergewicht von weniger als 90 kg in der Nemluvio-Monotherapiegruppe erhielten in Woche 0 subkutane Injektionen von Nemluvio 60 mg (2 Injektionen mit 30 mg), gefolgt von Injektionen mit 30 mg alle 4 Wochen. Teilnehmende mit einem Körpergewicht von 90 kg oder mehr in der Nemluvio-Monotherapiegruppe erhielten in Woche 0 subkutane Injektionen von Nemluvio 60 mg (2 Injektionen mit 30 mg) sowie alle 4 Wochen.
Endpunkte
Sowohl OLYMPIA 1 als auch OLYMPIA 2 bewerteten die gleichen beiden primären Endpunkte:
§Anteil der Patienten mit einer Verbesserung von ≥4 gegenüber Studienbeginn auf der numerischen Bewertungsskala für den Pruritus-Spitzenwert (NRS) in Woche 16
§Anteil der Patienten mit einem IGA-Erfolg (definiert als ein IGA von 0 [befallsfrei] oder 1 [nahezu befallsfrei] und einer Verbesserung um ≥2 Punkte gegenüber Studienbeginn) in Woche 16
Die wichtigsten sekundären Endpunkte umfassten eine PP-NRS-Verbesserung ≥4 gegenüber Studienbeginn in Woche 4, PP-NRS <2 in Woche 4 und Woche 16, eine SD-NRS-Verbesserung ≥4 gegenüber Studienbeginn in Woche 4 und 16.
Merkmale bei Studienbeginn
In diesen Studien waren zu Studienbeginn 59,6 % der Patienten weiblich, 81,4 % waren weiss und 25,4 % waren älter als 65 Jahre. Der wöchentliche durchschnittliche PP-NRS-Score bei Studienbeginn betrug im Mittel (SD) 8,4 (0,9). 58 % der Patienten hatten bei Studienbeginn einen IGA-Score von 3 (mässige PN) und 42 % der Patienten hatten bei Studienbeginn einen IGA von 4 (schwere PN).
Klinische Wirksamkeit
Monotherapiestudien (OLYMPIA 1 und OLYMPIA 2) – Woche 0 bis Woche 16
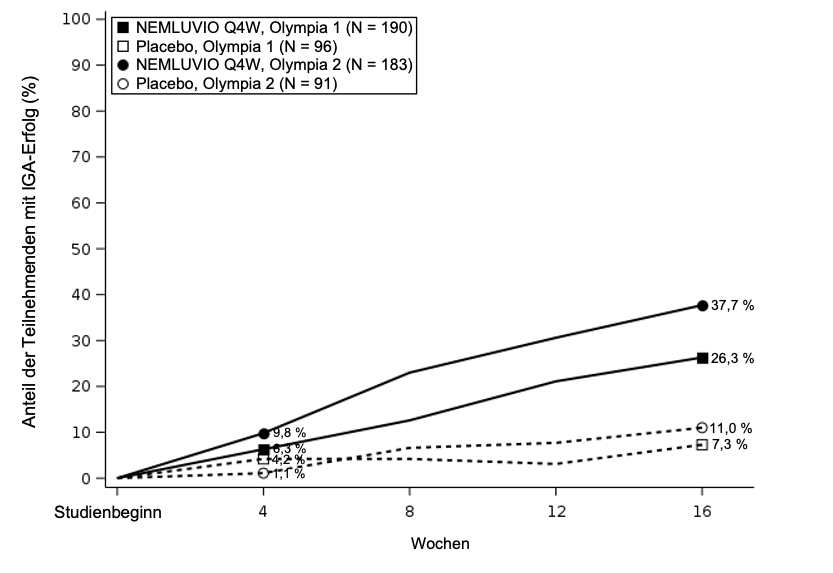
Die Ergebnisse der Pivotstudien zur Beurteilung der Behandlung von Nemluvio in OLYMPIA 1 und OLYMPIA 2 werden in Tabelle 6 dargestellt und zeigen eine signifikante Verbesserung bei mit Nemluvio behandelten Patienten im Vergleich zu Placebo sowohl für die primären Endpunkte (Abbildung 2 und Abbildung 3) als auch für die wichtigsten sekundären Endpunkte.
Tabelle 6 – Wirksamkeitsergebnisse für Nemluvio-Monotherapie (Q4W) in OLYMPIA 1 und OLYMPIA 2
|
|
OLYMPIA 1
|
OLYMPIA 2
| |
Nemluvio
|
Placebo
|
Nemluvio
|
Placebo
| |
Anzahl der randomisierten Patienten
|
190
|
96
|
183
|
91
| |
% der Patienten mit einer PP-NRS-Verbesserung ≥4 gegenüber Studienbeginna
| |
Woche 4
|
41,1*
|
6,3
|
41,0*
|
7,7
| |
Woche 16
|
58,4*
|
16,7
|
56,3*
|
20,9
| |
% der Patienten mit IGA 0 oder 1 in Woche 16a
|
26,3#
|
7,3
|
37,7*
|
11
| |
% der Patienten mit PP NRS <2a
| |
Woche 4
|
21,6*
|
1,0
|
19,7*
|
2,2
| |
Woche 16
|
34,2*
|
4,2
|
35,0*
|
7,7
| |
% der Patienten mit einer SD-NRS-Verbesserung ≥4 gegenüber Studienbeginna
| |
Woche 4
|
31,1*
|
5,2
|
37,2*
|
9,9
| |
Woche 16
|
50,0*
|
11,5
|
51,9*
|
20,9
|
a Wenn Teilnehmende eine Bedarfstherapie erhalten haben, wird eine Strategie mit zusammengesetzten Variablen angewandt: Die zugrunde liegenden Daten bei/nach Erhalt der Bedarfstherapie werden als schlechtest möglicher Wert festgelegt, und das Ansprechen wird aus dem zugrunde liegenden Datenwert abgeleitet. Teilnehmende mit fehlenden Ergebnissen gelten als Non-Responder.
b Nicht angepasst für Multiplizität
* p-Wert <0,0001, # p-Wert = 0,0025 Strata angepasst anhand der randomisierten Stratifizierungsvariablen (Analysenzentrum und Körpergewicht bei Studienbeginn (<90 kg, ≥90 kg)
§ p-Wert <0,0001 Strata-bereinigt vs. Placebo (ANCOVA MI-MAR)
|
Abbildung 2 – Anteil der Patienten mit PP-NRS-Verbesserung von ≥4 gegenüber Studienbeginn bis Woche 16
| |
| |
Abbildung 3 – Anteil der IGA-Responder von Studienbeginn bis Woche 16
| |
|
|
PharmakokinetikAbsorption
Nach einer anfänglichen subkutanen Dosis von 60 mg in einer Phase-I-Studie (96 Patienten pro Arm) erreichte Nemolizumab die mittlere (SD) Spitzenkonzentration (Cmax) von 7,5 (2,31) µg/ml etwa 6 Tage nach der Verabreichung.
Nach mehreren Behandlungen mit Nemluvio bei Patienten mit atopischer Dermatitis betrug die geschätzte mittlere (SD) Steady-State-Talkonzentration von Nemolizumab 2,63 (1,27) µg/ml bei 30 mg Q4W und 0,74 (0,44) µg/ml bei 30 mg Q8W.
Nach mehreren Behandlungen mit Nemluvio bei Patienten mit Prurigo nodularis betrug die geschätzte mittlere (SD) Steady-State-Talkonzentrationen der von Nemolizumab 3,04 (1,23) µg/ml bei Patienten mit einem Körpergewicht <90 kg bei 30 mg Q4W und 3,66 (1,63) µg/ml bei Patienten mit einem Körpergewicht ≥90 kg bei 60 mg Q4W.
Sowohl bei der Population mit atopischer Dermatitis als auch bei der Population mit Prurigo nodularis wurden Steady-State-Konzentrationen von Nemolizumab in Woche 4 nach einer Aufsättigungsdosis von 60 mg und in Woche 12 ohne Aufsättigungsdosis erreicht.
Distribution
Basierend auf einer populationspharmakokinetischen Analyse betrug das Verteilungsvolumen 7,67 l.
Metabolismus
Da es sich bei Nemolizumab um ein Protein handelt, wurden keine spezifischen Metabolisierungsstudien durchgeführt. Es ist zu erwarten, dass Nemolizumab durch katabole Signalwege in kleine Peptide metabolisiert wird.
Elimination
Es ist zu erwarten, dass Nemolizumab auf die gleiche Weise abgebaut wird wie endogenes IgG. In der populationspharmakokinetischen Analyse wurde die terminale Eliminationshalbwertszeit (SD) von Nemolizumab auf 18,9 (4,96) Tage und die systemische Clearance auf 0,263 l/Tag geschätzt.
Linearität/Nichtlinearität
Nach einer Einzelbehandlung zeigte Nemolizumab eine lineare Pharmakokinetik, wobei die Exposition dosisproportional zwischen 0,03 und 3 mg/kg anstieg.
Nach mehreren Behandlungen stieg die systemische Exposition von Nemolizumab im ganzen subkutanen Dosisbereich bis 30 mg ungefähr dosisproportional an. Es gab eine leichte Abnahme der Bioverfügbarkeit um 9 % bei der subkutanen Dosis von 60 mg und um 15 % bei der subkutanen Dosis von 90 mg.
Kinetik spezieller Patientengruppen
Geschlecht, Alter und ethnische Herkunft
Basierend auf einer populationspharmakokinetischen Analyse hatten Geschlecht, Alter (12 bis 85 Jahre für atopische Dermatitis und 18 bis 84 Jahre für Prurigo nodularis) und ethnische Herkunft keinen signifikanten Einfluss auf die Pharmakokinetik von Nemoluzimab.
Leberfunktionsstörungen
Es wird nicht erwartet, dass Nemolizumab als monoklonaler Antikörper eine signifikante hepatische Elimination durchläuft. Es wurden keine klinischen Studien durchgeführt, um die Wirkung einer Leberfunktionsstörung auf die Pharmakokinetik von Nemolizumab zu beurteilen. Es wurde festgestellt, dass eine leichte bis mittelschwere Leberfunktionsstörung keinen Einfluss auf die durch die populationspharmakokinetische Analyse ermittelte Pharmakokinetik von Nemolizumab hat. Für Patienten mit schwerer Leberfunktionsstörung liegen keine Daten vor.
Nierenfunktionsstörungen
Es wird nicht erwartet, dass Nemolizumab als monoklonaler Antikörper eine signifikante renale Elimination durchläuft. Es wurden keine klinischen Studien durchgeführt, um die Wirkung einer Nierenfunktionsstörung auf die Pharmakokinetik von Nemolizumab zu beurteilen. Die populationspharmakokinetische Analyse ergab keinen klinisch signifikanten Einfluss einer leichten oder mittelschweren Nierenfunktionsstörung auf die systemische Exposition von Nemolizumab. Für Patienten mit schwerer Nierenfunktionsstörung liegen nur sehr begrenzte Daten vor.
Körpergewicht
Bei Patienten mit höherem Körpergewicht war die Nemolizumab-Exposition geringer.
Atopische Dermatitis
Der Unterschied in der systemischen Exposition aufgrund des Körpergewichts hatte keine klinisch bedeutsamen Auswirkungen auf die Wirksamkeit. Eine Dosisanpassung basierend auf dem Körpergewicht ist nicht erforderlich (siehe «Dosierung/ Anwendung»).
Prurigo nodularis
Die Variabilität der systemischen Exposition aufgrund des Körpergewichts hatte eine klinisch bedeutsame Auswirkung auf die Wirksamkeit bezüglich der Hautläsionen, beurteilt anhand des IGA-Ansprechens, jedoch nicht auf die Verbesserung des Pruritus, und erfordert eine Dosisanpassung bei Patienten mit Prurigo nodularis (siehe «Dosierung/Anwendung»).
Pädiatrische Population
Atopische Dermatitis
In der Populations-PK-Analyse wurde kein klinisch signifikanter Unterschied in der Pharmakokinetik von Nemolizumab bei pädiatrischen Patienten im Alter von 12–17 Jahren im Vergleich zu Erwachsenen geschätzt. Eine Dosisanpassung in dieser Population wird nicht empfohlen.
Präklinische DatenMutagenität und Kanzerogenität
Das mutagene Potenzial von Nemolizumab wurde nicht untersucht. Es ist jedoch nicht zu erwarten, dass monoklonale Antikörper DNA oder Chromosomen verändern.
Es wurden keine Kanzerogenitätsstudien mit Nemolizumab durchgeführt. Die Ergebnisse der tierexperimentellen Untersuchungen wie auch die Kenntnise in Bezug auf die Hemmung von IL-31 deuten nicht darauf hin, dass Nemolizumab kanzerogen ist.
Reproduktionstoxizität
Bei geschlechtsreifen Javaneraffen wurden nach einer langfristigen subkutanen Behandlung mit Nemolizumab keine Auswirkungen auf die Fruchtbarkeitsparameter beobachtet. Während der subkutanen Nemolizumab-Behandlung bei Muttertieren von der frühen Organogenese bis zur Entbindung sowie während der direkten Verabreichung an die Nachkommen über einen Zeitraum von 26 Wochen ab dem 35. Tag nach der Geburt wurden keine direkten oder indirekten schädlichen Auswirkungen auf die Schwangerschaft, die embryonale und fetale sowie die postnatale Entwicklung beobachtet. Allerdings wurde bei hoher Exposition (43- oder 34-mal höher als die Exposition des Menschen bei der maximal empfohlenen Dosis für AD- bzw. PN-Patienten) in der Gruppe der mit 25 mg/kg Nemolizumab alle zwei Wochen behandelten Weibchen eine leicht erhöhte Inzidenz des Todes von Nachkommen in der frühen postnatalen Phase beobachtet. Obwohl es unwahrscheinlich ist, kann ein Zusammenhang zwischen diesem Befund und Nemolizumab nicht vollständig ausgeschlossen werden).
Sonstige HinweiseDie folgenden Hinweise gelten für alle erhältlichen Darreichungsformen.
Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Beeinflussung diagnostischer Methoden
Es liegen keine Daten vor.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit „EXP“ bezeichneten Datum verwendet werden. Falls erforderlich, kann der Umkarton mit dem Fertigpen oder der Fertigspritze einmal bis zu 90 Tage lang bei Raumtemperatur (bis zu 25°C) ausserhalb des Kühlschranks aufbewahrt werden. Dabei ist das Datum der erstmaligen Entnahme aus dem Kühlschrank im dafür vorgesehenen Feld auf dem Umkarton des Fertigpens oder der Fertigspritze zu notieren. Nemluvio darf nicht nach Ablauf des Verfallsdatums oder 90 Tage nach dem Datum der ersten Entnahme aus dem Kühlschrank (je nachdem, was früher eintritt) verwendet werden.
Besondere Lagerungshinweise
Ausser Sicht- und Reichweite von Kindern aufbewahren.
Fertigspritze oder Fertigpen in der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Im Kühlschrank (2–8 °C) lagern. Arzneimittel nicht einfrieren oder erhitzen.
Lagerungsbedingungen nach Rekonstitution des Arzneimittels siehe Abschnitt Hinweise für die Handhabung.
Hinweise für die Handhabung
Umfassende Anweisungen für die Verabreichung von Nemluvio in einem Fertigpen oder in einer Fertigspritze, siehe Ende der Packungsbeilage.
Nemluvio muss 30–45 Minuten vor der Rekonstitution aus dem Kühlschrank genommen werden. Nach Abschluss der Schritte zur Rekonstitution muss Nemluvio innerhalb von 4 Stunden verwendet oder entsorgt werden.
Nemluvio ist vor der Rekonstitution visuell zu überprüfen. Nemluvio besteht aus einem weissen Pulver und einer klaren Flüssigkeit. Nemluvio darf nicht verwendet werden, wenn das Pulver nicht weiss ist oder wenn die Flüssigkeit trüb ist oder Partikel sichtbar sind. Vor der Verabreichung ist zu kontrollieren, ob Nemluvio klar und farblos bis leicht gelblich ist und keine Partikel enthält.
Der Fertigpen oder die Fertigspritze darf weder Hitze noch direktem Sonnenlicht ausgesetzt werden und darf nicht geschüttelt werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
ZulassungsnummerNemluvio, Pulver und Lösungsmittel zur Herstellung einer Injektionslösung im Fertigpen: 69707 (Swissmedic)
Nemluvio, Pulver und Lösungsmittel zur Herstellung einer Injektionslösung in einer Fertigspritze: 69818 (Swissmedic)
PackungenNemluvio, Pulver und Lösungsmittel zur Herstellung einer Injektionslösung im Fertigpen
Einweg-Kartusche mit zwei Kammern aus Borosilikatglas Typ 1 in einem Autoinjektor mit integrierter Edelstahlnadel.
Packungsgrössen:
• 1 Fertigpen [B]
• Multipack mit 2 (2 Packungen mit je 1) Fertigpens [B]
Nemluvio, Pulver und Lösungsmittel zur Herstellung einer Injektionslösung in einer Fertigspritze
Einweg-Fertigspritze mit zwei Kammern aus Borosilikatglas Typ 1, mit einer beigepackten 27G-Nadel (Edelstahl) mit Sicherheitsabdeckung.
Packungsgrösse:
• 1 Fertigspritze [B]
ZulassungsinhaberinGalderma SA
Zählerweg 10
CH – 6300 Zug
Stand der InformationDezember 2024
|