ZusammensetzungWirkstoffe
Toxinum botulinicum A*.
* aus Clostridium botulinum
Hilfsstoffe
Albuminum humanum, 0,45 (50U) bzw. 0,9 (100U) mg Natrii chloridum.
Eine Durchstechflasche BOTOX 50 Allergan-Einheiten enthält 0,18 mg Natrium.
Eine Durchstechflasche BOTOX 100 Allergan-Einheiten enthält 0,35 mg Natrium.
Indikationen/AnwendungsmöglichkeitenBOTOX ist indiziert für:
Neurologische Erkrankungen:
·Symptomatische Behandlung des Blepharospasmus, des Spasmus hemifacialis und assoziierter fokaler Dystonien, ebenso zur Korrektur des Strabismus bei Patienten über 12 Jahren.
·Symptomatische Behandlung der zervikalen Dystonie (Torticollis spasticus) bei Erwachsenen.
·Symptomatische Behandlung der fokalen Spastizität der oberen und unteren Extremität bei Erwachsenen.
·Symptomatische Behandlung der fokalen Spastizität der oberen und unteren Extremität bei Jugendlichen und Kindern ab 2 Jahren.
·Prophylaxe von Kopfschmerzen bei erwachsenen Patienten mit chronischer Migräne.
Blasenfunktionsstörungen:
·Behandlung der überaktiven Blase mit den Symptomen Harninkontinenz, Harndrang und häufige Miktion bei erwachsenen Patienten, die nicht ausreichend auf Anticholinergika ansprechen oder eine Unverträglichkeit gegenüber diesen Arzneimitteln aufweisen.
·Behandlung der Harninkontinenz infolge neurogener Detrusorhyperaktivität in Zusammenhang mit einer neurologischen Erkrankung (wie z.B. Rückenmarksverletzung, Multiple Sklerose) bei Erwachsenen.
·Behandlung der neurogenen Detrusorhyperaktivität im Zusammenhang mit einer neurologischen Erkrankung (wie z.B. Spina bifida, Rückenmarksläsion) bei pädiatrischen Patienten ab 5 Jahren, bei denen die Blase durch regelmässigen Einmalkatheterismus zuverlässig entleert wird und die unzureichend auf anticholinerge Arzneimittel ansprechen oder diese nicht vertragen.
Erkrankungen der Haut und mit der Haut verbundene Erkrankungen:
·Behandlung der primären Hyperhidrosis axillae bei Erwachsenen.
Dosierung/AnwendungBotulinumtoxin-Einheiten sind nicht von einem Präparat auf andere übertragbar. Die in Allergan-Einheiten empfohlenen Dosierungen unterscheiden sich von denen anderer Botulinumtoxin-Präparate.
Hinsichtlich «Hinweise zur Handhabung» und «Entsorgung» der Durchstechflaschen siehe «Sonstige Hinweise».
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Spezielle Dosierungsanweisungen
Ältere Patienten
Es ist keine spezielle Dosisanpassung bei der Anwendung für ältere Patienten erforderlich. Initial sollte mit der niedrigsten empfohlenen Wirkdosis für die jeweilige Indikation begonnen werden. Für Wiederholungsinjektionen wird die niedrigste Wirkdosis und Einhaltung des grösstmöglichen klinisch vertretbaren Zeitraums zwischen den Injektionen empfohlen. Ältere Patienten mit umfangreicher medizinischer Vorgeschichte und Begleitmedikation sollten mit Vorsicht behandelt werden (für weitere Informationen siehe unter «Unerwünschte Wirkungen» und «Eigenschaften/Wirkungen»).
Kinder und Jugendliche
Über die Sicherheit und Wirksamkeit von BOTOX bei der Behandlung von Blepharospasmus, Spasmus hemifacialis und Strabismus liegen keine Untersuchungen bei Kindern unter 12 Jahren vor.
Die Sicherheit und Wirksamkeit von BOTOX bei der Behandlung der neurogenen Detrusorhyperaktivität wurde bei Kindern unter 5 Jahren nicht untersucht.
Die Sicherheit und Wirksamkeit von BOTOX bei der Behandlung der überaktiven Blase wurde bei Patienten unter 18 Jahren nicht nachgewiesen (für weitere Informationen siehe «Klinische Wirksamkeit»).
Die Sicherheit und Wirksamkeit von BOTOX bei der Behandlung von zervikaler Dystonie, Prophylaxe von Kopfschmerzen bei chronischer Migräne und Behandlung von primärer Hyperhidrosis axillae wurden bei Kindern und Jugendlichen (unter 18 Jahren) nicht untersucht.
Art der Anwendung
BOTOX darf nur von Ärzten mit geeigneter Qualifikation und Fachkenntnis in der Behandlung und der Anwendung der erforderlichen Ausstattung angewendet werden. Je nach Indikationsgebiet sollte die Diagnosestellung und Anwendung von BOTOX möglichst in Zusammenarbeit mit einem Neurologen, Ophthalmologen, Pädiater, Kinderorthopäden, Dermatologen oder Urologen erfolgen.
Es liegen keine ausreichenden klinischen Daten vor, die die Festlegung einer allgemeingültigen optimalen Dosis und der Anzahl an Injektionsstellen im jeweiligen Muskel ermöglichen würden. Deshalb ist die Behandlung eines Patienten vom behandelnden Facharzt individuell zu gestalten. Dabei ist zu beachten, dass die Festlegung einer optimalen Dosis durch eine Dosistitration angestrebt werden sollte. Es sollte zudem angestrebt werden, das Potenzial für die Bildung von neutralisierenden Antikörpern zu minimieren, indem die niedrigste wirksame Dosis in den längsten klinisch indizierten Abständen zwischen den Injektionen verabreicht wird (siehe «Warnhinweise und Vorsichtsmassnahmen» und «Klinische Wirksamkeit»).
NEUROLOGISCHE ERKRANKUNGEN:
Blepharospasmus/Spasmus hemifacialis
Nur bei Patienten über 12 Jahren
Empfohlene Nadel: Sterile 27-30 Gauge/0,40-0,30 mm Nadel.
Applikationshinweise: Eine elektromyographische Kontrolle (EMG) ist nicht erforderlich.
Empfohlene Dosis: Die empfohlene initiale Dosis beträgt 1,25-2,5 Einheiten. Die Injektion erfolgt in den medialen und lateralen prätarsalen M. orbicularis oculi des Oberlids und den lateralen prätarsalen M. orbicularis oculi des Unterlids. Zusätzliche Injektionsstellen in der Augenbraue, dem lateralen M. orbicularis oculi und im oberen Gesichtsbereich sind möglich, wenn Spasmen hier das Sehen beeinträchtigen.
Maximale Gesamtdosis: Die Initialdosis pro Auge sollte 25 Einheiten nicht übersteigen. Für die Behandlung des Blepharospasmus sollte die Gesamtdosis von 100 Einheiten alle 12 Wochen nicht überschritten werden.
Zusätzliche Informationen: Injektionen in die Nähe des M. levator palpebrae sup. sollten vermieden werden, um so das Auftreten einer Ptosis gering zu halten. Aufgrund der Diffusion der Botulinumtoxin Typ A Lösung in den M. obliquus inf. kann sich eine Diplopie manifestieren. Diese unerwünschte Wirkung kann geringgehalten werden, wenn auf die mediale Injektion am unteren Augenlid verzichtet wird. Abbildung 1 zeigt die möglichen Injektionsstellen:
Abbildung 1: Mögliche Injektionsstellen

In der Regel tritt die Wirkung in den ersten drei Tagen ein und erreicht ein Maximum 1-2 Wochen nach der Behandlung. Jede Behandlung hält etwa 3 Monate an, dann können die Injektionen unbegrenzt wiederholt werden. Anlässlich weiterer Behandlungen kann die Dosis maximal verdoppelt werden, wenn die Dosierung der Initialbehandlung sich als nicht ausreichend erwiesen hat.
Der Nutzen scheint gering zu sein, wenn mehr als 5,0 Einheiten pro Injektionsstelle injiziert werden. Auch kommt es zu keinem zusätzlichen Nutzen, wenn in kürzeren Abständen behandelt wird als alle drei Monate.
Patienten mit hemifaszialem Spasmus oder Störungen des VII. Hirnnervs (N. facialis) sind wie auf unilateralen Blepharospasmus zu behandeln, wobei in die übrigen betroffenen Fazialmuskeln BOTOX entsprechend dem Grad des Spasmus injiziert wird.
Strabismus
Nur bei Patienten über 12 Jahren
Empfohlene Nadel: Sterile 27 Gauge/0,40 mm Nadel.
Applikationshinweise: Eine BOTOX-Lösung mit 2,5 Einheiten pro 0,1 ml wird zubereitet (siehe «Hinweise für die Handhabung» und «Entsorgung» unter «Sonstige Hinweise»).
BOTOX ist zur Injektion in die extraokularen Muskeln mittels elektromyographischer Führung bestimmt.
Um das Auge auf die BOTOX Injektion vorzubereiten, sollten einige Tropfen eines Lokalanästhetikums und einer okularen abschwellenden Lösung einige Minuten vor der Injektion verabreicht werden.
Empfohlene Dosis: Es sollen initial die niedrigsten Dosen zur Behandlung von leichten Abweichungen und höhere Dosen für stärker ausgeprägte Abweichungen angewendet werden.
Folgende Initialdosen in Einheiten werden empfohlen (die niedrigere Dosis ist für kleinere Schielwinkel vorgesehen):
·Für vertikale Muskeln und für horizontales Schielen von weniger als 20 Prismendioptrien: 1,25 Einheiten - 2,5 Einheiten (0,05–0,10 ml) in jeden Muskel.
·Für horizontales Schielen von 20-50 Prismendioptrien: 2,5 Einheiten - 5,0 Einheiten (0,10–0,20 ml) in jeden Muskel.
·Für Lähmung des äusseren Nervus oculomotorius, die einen Monat oder länger anhält: 1,25 Einheiten - 2,5 Einheiten in den M. rectus medialis.
Maximale Gesamtdosis: Die maximale empfohlene Dosis als Einzelinjektion in einen Augenmuskel beträgt 25 Einheiten.
Das empfohlene Injektionsvolumen von BOTOX, das zur Behandlung des Strabismus verabreicht wird, beträgt zwischen 0,05 ml - 0,15 ml pro Muskel.
Zusätzliche Informationen: Die Initialdosen von verdünntem BOTOX führen üblicherweise ein bis zwei Tage nach der Injektion zu einer Paralyse der injizierten Muskeln. Die Intensität der Paralyse nimmt in der ersten Woche zu. Die Lähmung hält 2-6 Wochen an und nimmt dann schrittweise über eine etwa gleich lange Periode ab. Überkorrekturen, die länger als 6 Monate anhalten, sind selten.
Etwa die Hälfte der behandelten Patienten benötigen zusätzliche Dosen infolge einer unzureichenden Paralyse des Muskels nach der Initialdosis oder wegen mechanischen Faktoren wie ausgeprägte Abweichungen oder Einschränkung oder wegen Fehlen von motorischer binokularer Fusion, um die Anpassung zu stabilisieren.
Folgedosen bei residualem Schielen oder Rezidiven:
Es wird empfohlen, die Patienten 7-14 Tage nach jeder Injektion wieder zu untersuchen und die Wirkung der Dosis zu beurteilen. Folgedosen bei Patienten, die eine ausreichende Paralyse des Zielmuskels aufweisen, sollten gleich hoch wie die Initialdosis sein.
Folgedosen bei Patienten, die eine unvollständige Paralyse des Zielmuskels aufweisen, können bis doppelt so hoch sein wie die zuvor verabreichte Dosis. Folgeinjektionen sollten nicht verabreicht werden, bis die Wirkungen der vorangehenden Dosis abgeklungen sind, was sich in der wieder hergestellten Funktion der injizierten und benachbarten Muskeln nachweisen lässt.
Zervikale Dystonie
Nur bei Erwachsenen
Empfohlene Nadel: Geeignete sterile Nadel (in der Regel 25-30 Gauge/0,50-0,30 mm).
Applikationshinweise: In klinischen Studien wurde BOTOX zur Behandlung der zervikalen Dystonie üblicherweise in den M. sternocleidomastoideus, M. levator scapulae, M. scalenus, M. splenius capitis, M. semispinalis, M. longissimus und/oder den M. trapezius injiziert. Diese Liste ist nicht vollständig, da alle Muskeln, die für die Kontrolle der Kopfhaltung verantwortlich sind, beteiligt sein können und deshalb eventuell behandelt werden müssen.
Dosisabhängige Nebenwirkungen werden bei Frauen häufiger beobachtet, deshalb sollte bei der Wahl der Dosis die Muskelmasse und der Grad der Hypertrophie bzw. der Atrophie des zu behandelnden Muskels in Betracht gezogen werden. Das Muster der Muskelaktivierung kann sich bei der zervikalen Dystonie spontan ändern, ohne dass sich das klinische Erscheinungsbild der Dystonie ändert.
Wenn der betreffende Muskel nicht eindeutig identifiziert werden kann, sollte die Injektion unter EMG-Kontrolle erfolgen.
Empfohlene Dosis: Bei der Ersttherapie sind nicht mehr als 200 Einheiten insgesamt zu injizieren. Bei Folgeinjektionen können je nach Initialwirkung Änderungen vorgenommen werden.
In den ursprünglichen kontrollierten klinischen Studien zum Wirksamkeits- und Sicherheitsnachweis bei zervikaler Dystonie wurden Dosen von rekonstituiertem BOTOX von 140 bis 280 Einheiten eingesetzt. In aktuelleren Untersuchungen wurde eine Dosierung von 95-360 Einheiten (mit einer ungefähren mittleren Dosis von 240 Einheiten) gewählt.
Bislang unbehandelte Patienten sollten als Initialdosis die niedrigste Wirkdosis erhalten. Pro Injektionsstelle sollten nicht mehr als 50 Einheiten appliziert werden. In den M. sternocleidomastoideus sollen nicht mehr als 100 Einheiten verabreicht werden. Um das Auftreten von Dysphagie zu minimieren, ist der M. sternocleidomastoideus nicht bilateral zu injizieren.
Maximale Gesamtdosis: Eine Gesamtdosis von 300 Einheiten pro Behandlung darf nicht überschritten werden. Die optimale Anzahl der Injektionsstellen hängt von der Grösse des Muskels ab. Eine Wiederholungsbehandlung ist vor Ablauf von 10 Wochen nicht empfohlen.
Zusätzliche Informationen: Klinische Verbesserungen sind im Allgemeinen innerhalb der ersten beiden Wochen mit einem Maximum nach ca. 6 Wochen nach der Behandlung zu beobachten. Die Wirkungsdauer zeigte im Rahmen der klinischen Studien starke Schwankungsbreiten (von 2 bis 33 Wochen) bei einer mittleren Dauer von 12 Wochen.
Fokale Spastik der oberen Extremitäten
Erwachsene
Empfohlene Nadel: Sterile 25, 27 oder 30 Gauge Nadel für oberflächliche Muskeln und 22 Gauge Nadel für tiefer liegende Muskeln.
Applikationshinweise: Für die Lokalisation der involvierten Muskeln kann die EMG-Führung, Nervenstimulation oder Ultraschall hilfreich sein. Durch Anwendung von BOTOX an mehreren Injektionsstellen wird ein gleichmässigerer Kontakt mit den Innervationsbereichen der Muskeln erzielt, was bei grossen Muskeln besonders nützlich ist.
Empfohlene Dosis: Die genaue Dosierung erfolgt individuell. Dosis und Anzahl der Applikationsorte sind abhängig von Grösse, Anzahl und Lage der involvierten Muskeln, vom Vorliegen einer lokalen Muskelschwäche und dem Ansprechen des Patienten auf frühere Behandlung.
Höhere Dosen können zu einer längerdauernden Muskeltonusreduktion führen. Das Ausmass der Muskelspastik und der involvierten Muskelgruppen kann sich mit der Zeit ändern und eine Änderung in der Dosierung von BOTOX und der zu injizierenden Muskeln erwogen werden.
Folgende initiale Dosierungen und Applikationsorte sind empfohlen:
|
Muskel
|
Gesamtdosierung; Anzahl der Injektionsstellen
| |
Biceps brachii
|
50-200 Einheiten; bis zu 4 Stellen
| |
Flexor digitorum profundus
|
7,5-30 Einheiten; 1-2 Stellen
| |
Flexor digitorum sublimis
|
7,5-30 Einheiten; 1-2 Stellen
| |
Flexor carpi radialis
|
15-60 Einheiten; 1-2 Stellen
| |
Flexor carpi ulnaris
|
10-40 Einheiten; 1-2 Stellen
|
Maximale Gesamtdosis: In klinischen Studien wurde bei Erwachsenen eine Dosierung von 360 Einheiten nicht überschritten. Die Gesamtdosis wurde jeweils auf die ausgewählten Muskeln aufgeteilt (meist in den Flexormuskel des Ellbogens, des Handgelenks und der Finger). Generell sollte die Dosis 6 Einheiten/kg Körpergewicht nicht überschreiten.
Zusätzliche Informationen: In der Regel tritt eine Besserung des Muskeltonus innerhalb von 2 Wochen mit einem Maximum nach 4 bis 6 Wochen nach der Behandlung ein. Wiederholungsinjektionen sollten erst verabreicht werden, wenn der klinische Effekt der vorangegangenen Injektion abgeklungen ist, jedoch nicht häufiger als alle zwei Monate.
Jugendliche und Kinder ab 2 Jahren
Empfohlene Nadel: Geeignete sterile Nadelgrösse. Die Länge der Nadel sollte entsprechend der Lage und der Tiefe des Muskels gewählt werden.
Applikationshinweise: Für die Lokalisation der involvierten Muskeln wird die EMG-Führung, Nervenstimulation oder Ultraschall empfohlen.
Abbildung 2 zeigt die Injektionsstellen zur Behandlung der Spastik der oberen Extremitäten bei Kindern:
Abbildung 2: Injektionsstellen zur Behandlung der Spastik der oberen Extremitäten bei Kindern
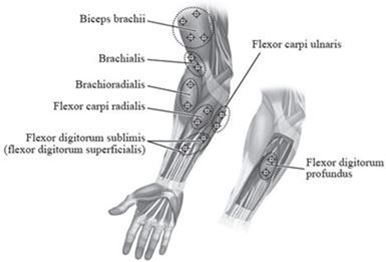
Empfohlene Dosis: Die empfohlene Dosis zur Behandlung der Spastik der oberen Extremitäten bei Kindern beträgt 3 bis 6 Einheiten/kg Körpergewicht aufgeteilt auf die betroffenen Muskeln.
Folgende Dosierung und Applikationsorte sind empfohlen:
|
Muskel
|
Gesamtdosierung; Anzahl der Injektionsstellen
| |
Biceps brachii
|
1,5 bis 3 Einheiten /kg; aufgeteilt auf 4 Stellen
| |
Brachialis
|
1 bis 2 Einheiten/kg; aufgeteilt auf 2 Stellen
| |
Brachioradialis
|
0,5 bis 1 Einheiten/kg; aufgeteilt auf 2 Stellen
| |
Flexor carpi radialis
|
1 bis 2 Einheiten/kg; aufgeteilt auf 2 Stellen
| |
Flexor carpi ulnaris
|
1 bis 2 Einheiten/kg; aufgeteilt auf 2 Stellen
| |
Flexor digitorum profundus
|
0,5 bis 1 Einheiten/kg; aufgeteilt auf 2 Stellen
| |
Flexor digitorum sublimis
|
0,5 bis 1 Einheiten/kg; aufgeteilt auf 2 Stellen
|
Maximale Gesamtdosis: Die Gesamtdosis von BOTOX pro Behandlung der oberen Extremitäten sollte 6 Einheiten/kg Körpergewicht bzw. 200 Einheiten nicht überschreiten, je nachdem, welches niedriger ist. Wenn der behandelnde Arzt es für angebracht hält, sollte beim Patienten eine Wiederholungsbehandlung in Betracht gezogen werden, wenn die klinische Wirkung der vorherigen Injektion nachgelassen hat, jedoch frühestens 12 Wochen nach der vorherigen Injektion. Bei gleichzeitiger Behandlung der oberen und unteren Extremitäten sollte die Gesamtdosis innerhalb eines 12 Wochen Intervalls den unteren Wert von 10 Einheiten/kg Körpergewicht bzw. 340 Einheiten nicht überschreiten.
Zusätzliche Informationen: In der Regel tritt eine Besserung des Muskeltonus innerhalb 7 Tage nach der Behandlung ein. Die Behandlung mit BOTOX ersetzt nicht die üblichen Standardbehandlungen bei Rehabilitationsmassnahmen.
Fokale Spastik der unteren Extremitäten
Erwachsene
Empfohlene Nadel: Sterile 25, 27 oder 30 Gauge Nadel. Die Länge der Nadel sollte entsprechend der Lage und der Tiefe des Muskels gewählt werden.
Applikationshinweise: Für die Lokalisation der involvierten Muskeln kann die EMG-Führung, Nervenstimulation oder Ultraschall hilfreich sein. Durch Anwendung von BOTOX an mehreren Injektionsstellen wird ein gleichmässigerer Kontakt mit den Innervationsbereichen der Muskeln erzielt, was bei grossen Muskeln besonders nützlich ist.
Abbildung 3 zeigt die Injektionsstellen für Spastik der unteren Extremitäten bei Erwachsenen:
Abbildung 3: Injektionsstellen für Spastik der unteren Extremitäten bei Erwachsenen

Empfohlene Dosis: Die empfohlene Dosis zur Behandlung der unteren Extremitäten einschliesslich des Fussgelenkes und des Fusses beträgt 300 bis 400 Einheiten aufgeteilt auf die betroffenen Muskeln, welche in der nachfolgenden Tabelle aufgeführt sind. Die maximale Gesamtdosis pro Behandlung beträgt 400 Einheiten. Der Grad und das Muster der Muskelspastik zum Zeitpunkt der erneuten Injektion können Änderungen der Dosis von BOTOX und der zu injizierenden Muskeln erforderlich machen.
Folgende Dosierung und Applikationsorte sind empfohlen:
|
Muskel
|
Gesamtdosierung; Anzahl der Injektionsstellen
| |
Gastrocnemius medial head
|
75 Einheiten; 3 Stellen
| |
Gastrocnemius lateral head
|
75 Einheiten, 3 Stellen
| |
Soleus
|
75 Einheiten; 3 Stellen
| |
Tibialis posterior
|
75 Einheiten; 3 Stellen
| |
Flexor hallucis longus
|
50 Einheiten; 2 Stellen
| |
Flexor digitorum longus
|
50 Einheiten; 2 Stellen
| |
Flexor digitorum brevis
|
25 Einheiten; 1 Stelle
|
Zusätzliche Informationen: Es liegt im Ermessen des behandelnden Arztes, beim Patienten eine erneute Injektion in Betracht zu ziehen, wenn die klinische Wirkung der vorherigen Injektion nachlässt, jedoch nicht früher als 12 Wochen nach der vorherigen Injektion.
Kinder ab 2 Jahren
Empfohlene Nadel: Geeignete sterile Nadelgrösse. Die Länge der Nadel sollte entsprechend der Lage und der Tiefe des Muskels gewählt werden.
Applikationshinweise: Für die Lokalisation der involvierten Muskeln wird die EMG-Führung, Nervenstimulation oder Ultraschall empfohlen.
Abbildung 4 zeigt die Injektionsstellen zur Behandlung der Spastik der unteren Extremitäten bei Kindern:
Abbildung 4: Injektionsstellen zur Behandlung der Spastik der unteren Extremitäten bei Kindern
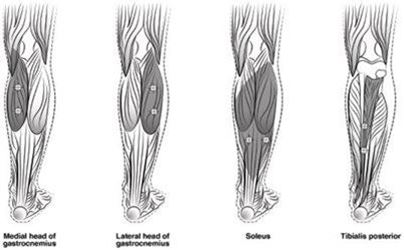
Empfohlene Dosis: Die empfohlene Dosis zur Behandlung der Spastik der unteren Extremitäten bei Kindern beträgt 4 bis 8 Einheiten/kg Körpergewicht aufgeteilt auf die betroffenen Muskeln.
Folgende Dosierung und Applikationsorte sind empfohlen:
|
Muskel
|
Gesamtdosierung; Anzahl der Injektionsstellen
| |
Gastrocnemius medial head
|
1 bis 2 Einheiten/kg; aufgeteilt auf 2 Stellen
| |
Gastrocnemius lateral head
|
1 bis 2 Einheiten/kg; aufgeteilt auf 2 Stellen
| |
Soleus
|
1 bis 2 Einheiten/kg; aufgeteilt auf 2 Stellen
| |
Tibialis posterior
|
1 bis 2 Einheiten/kg; aufgeteilt auf 2 Stellen
|
Maximale Gesamtdosis: Die Gesamtdosis von BOTOX pro Behandlung der unteren Extremitäten sollte 8 Einheiten/kg Körpergewicht bzw. 300 Einheiten nicht überschreiten, je nachdem, welcher Wert niedriger ist. Es liegt im Ermessen des behandelnden Arztes, beim Patienten eine erneute Injektion in Betracht zu ziehen, wenn die klinische Wirkung der vorherigen Injektion nachlässt, jedoch nicht früher als 12 Wochen nach der vorherigen Injektion. Bei gleichzeitiger Behandlung der oberen und unteren Extremitäten sollte die Gesamtdosis innerhalb eines 12 Wochen Intervalls den unteren Wert von 10 Einheiten/kg Körpergewicht bzw. 340 Einheiten nicht überschreiten.
Zusätzliche Informationen: In der Regel tritt eine Besserung des Muskeltonus innerhalb 7 Tage nach der Behandlung ein. Die Behandlung mit BOTOX ersetzt nicht die üblichen Standardbehandlungen bei Rehabilitationsmassnahmen.
Prophylaxe von Kopfschmerzen bei erwachsenen Patienten mit chronischer Migräne
Empfohlene Nadel: Es wird eine 30-Gauge-/13mm-Nadel verwendet.
Applikationshinweise: Die Injektionen sollten auf sieben bestimmte Muskelpartien im Kopf- und Nackenbereich aufgeteilt werden, wie in der unten stehenden Tabelle beschrieben. Mit Ausnahme des Procerus-Muskels, der an einer Stelle (mittig) injiziert werden sollte, sollten alle Muskeln bilateral mit der nachfolgend angegebenen Mindestdosis pro Muskel injiziert werden, wobei sich die Hälfte der Injektionsstellen links und die Hälfte rechts am Kopf und Nacken befinden. Wenn ein oder mehrere Schmerzpunkte vorherrschen, können zusätzliche Injektionen an einer oder beiden Seiten in bis zu drei bestimmte Muskelgruppen (Occipitalis, Temporalis und Trapezius) verabreicht werden, bis zu der in der unten stehenden Tabelle angegebenen Höchstdosis pro Muskel.
Der empfohlene Zeitplan für die erneute Behandlung beträgt 12 Wochen.
In den folgenden Abbildungen sind die Injektionsstellen dargestellt:

In den folgenden Abbildungen sind die empfohlenen Muskelgruppen für optionale zusätzliche Injektionen dargestellt:

Empfohlene Dosis: Die empfohlene Dosis beträgt 155 bis 195 Einheiten in Form von je 0,1 ml (5 Einheiten) intramuskulär (i.m.) verabreichten Injektionen aufgeteilt auf 31 bis zu 39 Stellen.
BOTOX-Dosierung je nach Muskel bei chronischer Migräne:
|
|
Empfohlene Dosis
| |
Kopf-/Nackenbereich
|
Gesamtdosierung (Anzahl der Injektionsstellena)
| |
Corrugatorb
|
10 Einheiten (2 Stellen)
| |
Procerus
|
5 Einheiten (1 Stelle)
| |
Frontalisb
|
20 Einheiten (4 Stellen)
| |
Temporalisb
|
40 Einheiten (8 Stellen) bis zu 50 Einheiten (bis zu 10 Stellen)
| |
Occipitalisb
|
30 Einheiten (6 Stellen) bis zu 40 Einheiten (bis zu 8 Stellen)
| |
Zervikale paraspinale Muskelgruppeb
|
20 Einheiten (4 Stellen)
| |
Trapeziusb
|
30 Einheiten (6 Stellen) bis zu 50 Einheiten (bis zu 10 Stellen)
| |
Gesamtdosisbereich:
|
155 Einheiten bis zu 195 Einheiten
31 bis zu 39 Stellen
|
a Jede i.m. Injektionsstelle = 0,1 ml = 5 Einheiten BOTOX
b Dosis beidseitig verteilt
Zusätzliche Informationen: Die Verbesserung tritt in der Regel innerhalb der ersten 3 Wochen nach der Injektion auf. In der Regel hält die Wirkung nach der Injektion 12 Wochen an.
BLASENFUNKTIONSSTÖRUNGEN:
Injektion in den Detrusor-Muskel - Erwachsene
Allgemeine Applikationshinweise: Zum Zeitpunkt der Behandlung sollten Patienten nicht an einer akuten Infektion der Harnwege leiden.
Antibiotika sollten prophylaktisch 1-3 Tage vor der Behandlung, am Tag der Behandlung und 1-3 Tage nach der Behandlung verabreicht werden.
Empfohlen wird, dass Patienten eine Behandlung mit Thrombozytenaggregationshemmern mindestens 3 Tage vor dem Injektionsvorgang absetzen.
Mit Antikoagulantien behandelte Patienten müssen angemessen behandelt werden, um das Blutungsrisiko zu senken.
Überaktive Blase - Erwachsene
Empfohlene Nadel: Ein flexibles oder starres Zystoskop kann verwendet werden. Die sterile Injektionsnadel sollte vor Beginn der Injektionen mit ungefähr 1 ml des rekonstituierten BOTOX (abhängig von der Nadellänge) gefüllt (vorbereitet) werden, um daraus alle Luft zu entfernen.
Applikationshinweise: Eine intravesikale Instillation mit einem verdünnten Lokalanästhetikum mit oder ohne Sedierung kann vor der Injektion, je nach üblicher Vorgehensweise vor Ort, angewendet werden. Wird eine lokale anästhetische Instillation durchgeführt, sollte die Blase entleert und mit steriler Kochsalzlösung vor den nächsten Schritten des Injektionsvorgangs gespült werden.
Das rekonstituierte BOTOX (100 Einheiten/10 ml) wird in den Detrusor-Muskel über ein flexibles oder starres Zystoskop unter Vermeidung des Trigonums injiziert. Die Blase sollte mit ausreichend Kochsalzlösung instilliert werden, um für die Injektionen eine ausreichende Visualisierung zu erreichen. Eine übermässige Dehnung sollte jedoch vermieden werden.
Die Nadel sollte ungefähr 2 mm in den Detrusor-Muskel eingeführt und 20 Injektionen von jeweils 0,5 ml (Gesamtvolumen 10 ml) in einem Abstand von ungefähr 1 cm (siehe Abbildung 5) gesetzt werden. Als Abschlussinjektion sollte ungefähr 1 ml sterile physiologische Kochsalzlösung injiziert werden, damit die vollständige Dosis verabreicht wird. Nach Verabreichung der Injektionen sollte die für die Visualisierung der Blasenwand verwendete Kochsalzlösung nicht drainiert werden, damit der Patient vor dem Verlassen der Klinik seine Fähigkeit zum Wasserlassen zeigen kann. Der Patient sollte über mindestens 30 Minuten nach der Injektion beobachtet werden und bis eine spontane Entleerung der Blase erfolgt ist.
Abbildung 5: Injektionsstellen zur Behandlung von Blasenfunktionsstörungen

Empfohlene Dosis: Die empfohlene Dosis beträgt 100 Einheiten BOTOX in Form von je 0,5 ml (5 Einheiten) Injektionen auf 20 Stellen im Detrusor-Muskel verteilt.
Zusätzliche Informationen: Eine klinische Verbesserung kann innerhalb von 2 Wochen eintreten. Eine erneute Injektion sollte für Patienten erwogen werden, bei denen der klinische Effekt der vorangegangenen Injektion nachgelassen hat (die mediane Dauer bei klinischen Phase-3-Studien betrug 166 Tage [~24 Wochen]). Die erneute Injektion sollte jedoch nicht früher als 3 Monate nach der vorangegangenen Blasen-Injektion durchgeführt werden.
Harninkontinenz infolge neurogener Detrusorhyperaktivität - Erwachsene
Empfohlene Nadel: Ein flexibles oder starres Zystoskop kann verwendet werden. Die sterile Injektionsnadel sollte vor Beginn der Injektionen mit ungefähr 1 ml (abhängig von der Nadellänge) gefüllt (vorbereitet) werden, um daraus alle Luft zu entfernen.
Applikationshinweise: Eine intravesikale Instillation mit verdünnten Anästhetika mit oder ohne Sedierung oder Allgemeinanästhesie kann vor der Injektion, je nach üblicher Vorgehensweise vor Ort, angewendet werden. Wird eine lokale anästhetische Instillation durchgeführt, sollte die Blase entleert und mit steriler Kochsalzlösung vor den nächsten Schritten des Injektionsvorgangs gespült werden.
Das rekonstituierte BOTOX (200 Einheiten/30 ml) wird in den Detrusor-Muskel über ein flexibles oder starres Zystoskop unter Vermeidung des Trigonums injiziert. Die Blase sollte mit ausreichend Kochsalzlösung instilliert werden, um für die Injektionen eine ausreichende Visualisierung zu erreichen. Eine übermässige Dehnung sollte jedoch vermieden werden.
Die Nadel sollte ungefähr 2 mm in den Detrusor-Muskel eingeführt und 30 Injektionen von jeweils 1 ml (Gesamtvolumen 30 ml) in einem Abstand von ungefähr 1 cm (siehe Abbildung 5 oben) gesetzt werden. Als Abschlussinjektion sollte ungefähr 1 ml sterile physiologische Kochsalzlösung injiziert werden, damit die vollständige Dosis verabreicht wird. Nach Verabreichung der Injektionen sollte die für die Visualisierung der Blasenwand verwendete Kochsalzlösung drainiert werden. Der Patient sollte über mindestens 30 Minuten nach der Injektion beobachtet werden.
Empfohlene Dosis: Die empfohlene Dosis beträgt 200 Einheiten BOTOX in Form von je 1 ml (~6,7 Einheiten) Injektionen auf 30 Stellen im Detrusor-Muskel verteilt.
Zusätzliche Informationen: Eine klinische Verbesserung tritt im Allgemeinen innerhalb von 2 Wochen ein. Eine erneute Injektion sollte für die Patienten erwogen werden, bei denen der klinische Effekt der vorangegangenen Injektion nachgelassen hat (die mediane Dauer bei klinischen Phase-3-Studien betrug 256-295 Tage (~36-42 Wochen) für BOTOX (200 Einheiten)). Die erneute Injektion sollte jedoch nicht früher als 3 Monate nach der vorangegangenen Blasen-Injektion durchgeführt werden.
Neurogene Detrusorhyperaktivität - Pädiatrische Patienten im Alter von 5 Jahren und älter
Allgemeine Applikationshinweise: Die Patienten dürfen zum Zeitpunkt der Behandlung nicht an einer akuten Harnwegsinfektion (urinary tract infection, UTI) leiden. Orale Antibiotika sollten prophylaktisch 1–3 Tage vor der Behandlung, am Behandlungstag und 1–3 Tage nach der Behandlung verabreicht werden, um die Wahrscheinlichkeit einer eingriffsbedingten Harnwegsinfektion zu verringern.
Alternativ kann bei Patienten, die zur Behandlung der Detrusorhyperaktivität im Zusammenhang mit einer neurologischen Erkrankung eine Vollnarkose (oder eine Sedierung bei Bewusstsein) erhalten, eine prophylaktische intravenöse Einmalgabe eines geeigneten Antibiotikums vor der Botulinumtoxin-Injektion am Behandlungstag verabreicht werden. Generell wird der Einsatz von Aminoglykosiden nicht empfohlen (siehe «Interaktionen»).
Patienten, die mit Thrombozytenaggregationshemmern behandelt werden, sollten diese mindestens 3 Tage vor der Injektion absetzen. Patienten, die eine blutgerinnungshemmende Therapie erhalten, müssen angemessen behandelt werden, um das Blutungsrisiko zu verringern.
Bei der Durchführung einer Zystoskopie ist entsprechende Vorsicht geboten.
·Bei Patienten im Alter von 5 bis unter 12 Jahren: Vor der Injektion ist eine Vollnarkose (oder eine bewusste Sedierung) in Erwägung zu ziehen, je nach der üblichen Vorgehensweise vor Ort.
·Bei Patienten im Alter von 12 Jahren oder älter: Eine intravesikale Instillation eines verdünnten Lokalanästhetikums mit oder ohne Sedierung oder eine Vollnarkose sind vor der BOTOX-Injektionsbehandlung in Erwägung zu ziehen, je nach der üblichen Vorgehensweise vor Ort.
Für alle Altersgruppen sollte zumindest eine intravesikale Instillation mit einem verdünnten Lokalanästhetikum in Erwägung gezogen werden. Wird eine lokale anästhetische Instillation durchgeführt, sollte die Blase danach entleert und mit steriler Kochsalzlösung vor den nächsten Schritten des Injektionsvorgangs gespült werden.
Empfohlene Nadel: Es kann ein flexibles oder starres Zystoskop verwendet werden. Die sterile Injektionsnadel sollte vor Beginn der Injektionen mit ca. 1 ml BOTOX Dosierlösung (abhängig von der Nadellänge) gefüllt (grundiert) werden, um vor Beginn der Injektionen die gesamte Luft aus ihr zu entfernen.
Applikationshinweise: BOTOX Dosierlösung wird über ein flexibles oder starres Zystoskop in den Detrusormuskel injiziert, wobei das Trigonum zu vermeiden ist. Die Blase sollte mit genügend Kochsalzlösung gefüllt werden, um eine ausreichende Visualisierung für die Injektionen zu erreichen, aber eine Überdehnung sollte vermieden werden.
Die Nadel sollte etwa 2 mm in den Detrusormuskel eingeführt werden, und 20 Injektionen von je 0,5 ml (Gesamtvolumen von 10 ml) sollten in einem Abstand von etwa 1 cm erfolgen (siehe Abbildung 5). Als Abschlussinjektion sollte etwa 1 ml sterile physiologische Kochsalzlösung injiziert werden, damit das restliche BOTOX aus der Nadel in den Detrusormuskel gelangt und damit die vollständige Dosis appliziert wird. Nach Verabreichung aller Injektionen sollte die für die Visualisierung der Blasenwand verwendete Kochsalzlösung abgelassen werden. Der Patient sollte nach Abschluss der Injektionen noch über mindestens 30 Minuten überwacht werden.
Empfohlene Dosis: Wenn das Körpergewicht des Patienten grösser oder gleich 34 kg ist, beträgt die empfohlene Dosierung 200 Einheiten BOTOX pro Behandlung, die nach Verdünnung als Intradetrusor-Injektionen verabreicht werden.
·Rekonstituieren Sie BOTOX so, dass sich 20 Einheiten BOTOX pro ml in der/den Durchstechflasche(n) befinden:
·BOTOX Durchstechflaschen mit 100 Einheiten: Geben Sie jeweils 5 ml konservierungsmittelfreie 0,9%-ige Natriumchlorid-Lösung zu jeder von zwei 100-Einheiten-Fläschchen von BOTOX und schütteln Sie die Durchstechflaschen vorsichtig.
·Ziehen Sie 10 ml aus den Fläschchen in eine 10-ml-Dosierspritze auf.
·Verwenden Sie die Lösung sofort nach erfolgter Rekonstitution in der Spritze. Entsorgen Sie die nicht verwendete konservierungsmittelfreie 0.9%-ige Natriumchlorid-Lösung.
Wenn das Körpergewicht des Patienten weniger als 34 kg beträgt, ist die empfohlene Dosierung 6 Einheiten pro kg Körpergewicht BOTOX pro Behandlung, die als Injektion in den Detrusor verabreicht werden:
·Rekonstituieren Sie BOTOX so, dass sich 20 Einheiten BOTOX pro ml in der/den Durchstechflasche(n) befinden:
·BOTOX Durchstechflasche(n) mit 100 Einheiten: Geben Sie 5 ml konservierungsmittelfreie 0,9%-ige Natriumchlorid-Lösung zu einer 100-Einheiten-Durchtechflasche von BOTOX (wenn die Enddosis kleiner oder gleich 100 U ist) oder zu jeder von zwei 100-Einheiten-Fläschchen von BOTOX (wenn die Enddosis grösser als 100 U ist) und schütteln Sie die Durchstechflasche(n) vorsichtig.
·In Abhängigkeit vom Körpergewicht des individuellen Patienten muss das auf eine Konzentration von 20 Einheiten pro 1ml rekonstituierte BOTOX weiter verdünnt werden. Die weiteren Verdünnungsanweisungen in Abhängigkeit vom Körpergewicht des Patienten beziehen sich auf die Menge an rekonstituiertem BOTOX (20 Einheiten pro 1ml) und an zusätzlicher konservierungsmittelfreier 0.9%-iger Natriumchlorid-Lösung, die zusammen in eine 10-ml-Dosierspritze aufgezogen werden müssen. Diese sind Tabelle 1 zu entnehmen:
Tabelle 1: BOTOX Verdünnungsanleitung und Enddosierung für Patienten mit einem Körpergewicht < 34 kg
|
Körpergewicht
(kg)
|
Volumen des rekonstituierten BOTOX (20 U/ml) und des Verdünnungsmittels* (ml) zum Aufziehen in die Dosierspritze, um ein Endvolumen von 10 ml für die Injektion in den Detrusormuskel zu erreichen
|
Enddosis BOTOX in der Dosierspritze
| |
BOTOX
(ml)
|
Verdünnungsmittel*
(ml)
| |
12 bis weniger als 14
|
3,6
|
6,4
|
72 Einheiten
| |
14 bis weniger als 16
|
4,2
|
5,8
|
84 Einheiten
| |
16 bis weniger als 18
|
4,8
|
5,2
|
96 Einheiten
| |
18 bis weniger als 20
|
5,4
|
4,6
|
108 Einheiten
| |
20 bis weniger als 22
|
6
|
4
|
120 Einheiten
| |
22 bis weniger als 24
|
6,6
|
3,4
|
132 Einheiten
| |
24 bis weniger als 26
|
7,2
|
2,8
|
144 Einheiten
| |
26 bis weniger als 28
|
7,8
|
2,2
|
156 Einheiten
| |
28 bis weniger als 30
|
8,4
|
1,6
|
168 Einheiten
| |
30 bis weniger als 32
|
9
|
1
|
180 Einheiten
| |
32 bis weniger als 34
|
9,6
|
0,4
|
192 Einheiten
|
* Konservierungsmittelfreie 0,9%-ige Natriumchlorid-Lösung
·Verwenden Sie BOTOX sofort nach Rekonstitution und Verdünnung in der Spritze. Entsorgen Sie die nicht verwendete konservierungsmittelfreie 0,9%-ige Natriumchlorid-Lösung.
Zusätzliche Informationen: Eine klinische Besserung tritt im Allgemeinen innerhalb von 2 Wochen ein. Die Patienten sollten für eine erneute Injektion in Betracht gezogen werden, wenn die klinische Wirkung der vorherigen Injektion nachlässt (die mediane Dauer in der klinischen Doppelblind-Parallelgruppenstudie betrug 207 Tage [~30 Wochen] für BOTOX 200 Einheiten, gewichtsangepasst, um 6 U/kg nicht zu überschreiten), jedoch nicht früher als 3 Monate nach der vorherigen Blaseninjektion.
ERKRANKUNGEN DER HAUT UND MIT DER HAUT VERBUNDENE ERKRANKUNGEN:
Primäre Hyperhidrosis axillae
Nur bei Erwachsenen
Empfohlene Nadel: Sterile 30 Gauge-Nadel.
Applikationshinweise: Der hyperhidrotische Bereich kann anhand von Anfärbungsverfahren wie z.B. mit dem Minor-Test (Jod/Stärke) ermittelt werden.
Empfohlene Dosis: 50 Einheiten BOTOX (100 Einheiten/4,0 ml) werden intradermal injiziert, und zwar gleichmässig verteilt an mehreren Stellen etwa 1-2 cm auseinander innerhalb des hyperhidrotischen Bereichs jeder betroffenen Achselhöhle.
Maximale Gesamtdosis: Da andere Dosierungen als 50 Einheiten pro Achselhöhle nicht untersucht wurden, können sie nicht empfohlen werden. Injektionen sollten nicht häufiger als alle 16 Wochen wiederholt werden.
Zusätzliche Informationen: In der Regel tritt eine Besserung des klinischen Zustandes innerhalb der ersten Woche nach der Injektion ein. Wiederholungsinjektionen sollten erst verabreicht werden, wenn die klinische Wirkung der vorangegangenen Injektion abklingt und der behandelnde Arzt bzw. Ärztin dies für erforderlich hält. Erfahrungsgemäss hält jede Behandlung 4-7 Monate an.
ALLE INDIKATIONEN:
Sollte nach durchgeführter Erstapplikation auch nach einem Monat kein therapeutischer Effekt eintreten, sind folgende Massnahmen durchzuführen:
·Klinische Verifizierung der Toxinwirkung auf den injizierten Muskel; dies kann eine elektromyographische Untersuchung in einer hierfür spezialisierten Einrichtung beinhalten.
·Analyse der Gründe für das Therapieversagen, z.B. schlechte Isolierung der Muskeln, die injiziert werden sollen, zu geringe Dosis, schlechte Injektionstechnik, fixe Kontraktur, zu schwacher Gegenmuskel, Antikörperbildung.
·Überprüfung der Behandlung mit Botulinumtoxin Typ A als angemessene Therapieform.
Sofern im Rahmen der Initialbehandlung keine unerwünschten Wirkungen aufgetreten sind, kann eine Wiederholungsbehandlung unter folgenden Voraussetzungen vorgenommen werden:
1.Dosisanpassung unter Berücksichtigung der Analyse des vorausgegangenen Therapieversagens,
2.EMG-Ableitung,
3.Einhaltung eines 3-Monatsintervalls zwischen der Initial- und der Wiederholungsbehandlung.
Beim Ausbleiben des therapeutischen Effektes oder beim Nachlassen der Wirkung bei Wiederholungsinjektionen sind alternative Behandlungsmethoden in Betracht zu ziehen.
Bei der Behandlung von Erwachsenen, einschliesslich der Behandlung mehrerer Indikationen, darf die maximale kumulative Dosis in einem Zeitraum von 12 Wochen 400 Einheiten nicht überschreiten.
Bei der Behandlung von Kindern, einschliesslich der Behandlung mehrerer Indikationen, sollte die maximale kumulative Dosis von 10 Einheiten/kg Körpergewicht oder 340 Einheiten in einem Zeitraum von 12 Wochen nicht überschritten werden, je nachdem, welcher Wert niedriger ist.
Kontraindikationen·Bekannte Überempfindlichkeit gegenüber dem Wirkstoff Botulinumtoxin Typ A oder einem der Hilfsstoffe gemäss Zusammensetzung.
·Infektion an der(n) vorgesehenen Injektionsstelle(n).
BOTOX Injektionen in den Detrusor-Muskel sind ebenfalls kontraindiziert bei:
·Patienten, die zum Zeitpunkt der Behandlung an einem Harnwegsinfekt leiden.
·Patienten, die zum Zeitpunkt der Behandlung an einer akuten Harnretention leiden und die nicht routinemässig katheterisiert werden.
·Patienten, die nicht willens und/oder fähig sind, falls notwendig nach der Behandlung einen Katheter zu verwenden.
Warnhinweise und VorsichtsmassnahmenDie empfohlenen Dosierungen und Häufigkeiten der Anwendung von BOTOX sollten nicht überschritten werden, da es zu einer Überdosierung, einer gesteigerten Muskelschwäche, der Ausbreitung des Toxins fern von der Injektionsstelle und der Bildung von neutralisierenden Antikörpern kommen kann. Als Anfangsdosis bei nicht vorbehandelten Patienten sollte die niedrigste empfohlene Dosis für die spezielle Indikation verabreicht werden.
Ärzte und Patienten müssen sich bewusst sein, dass unerwünschte Wirkungen auftreten können, obwohl vorherige Injektionen gut vertragen wurden. Daher ist bei jeder Verabreichung Vorsicht geboten.
Es wurden unerwünschte Wirkungen berichtet, die sich auf die Ausbreitung des Toxins fern von der Injektionsstelle beziehen (siehe «Unerwünschte Wirkungen»). In manchen Fällen führten diese zum Tode, die wiederum in einigen Fällen mit Dysphagie, Pneumonie und/oder Schwächezuständen assoziiert waren. Die Symptome entsprechen dem Wirkungsmechanismus von Botulinumtoxin und wurden Stunden bis Wochen nach der Injektion berichtet. Die Gefahr des Auftretens von Symptomen ist wahrscheinlich bei Patienten mit Erkrankungen und Begleiterkrankungen am grössten, die eine Prädisposition für diese Symptome darstellen. Hierzu zählen auch Kinder und Erwachsene, die aufgrund von Spastiken mit hohen Dosen behandelt werden.
Der behandelnde Arzt sollte mit der elektromyographischen Technik vertraut sein, wenn er bei Strabismus BOTOX injiziert. Die Wirkung von BOTOX bei Abweichungen über 50 Prismendioptrien bei restriktivem Strabismus, beim Duane-Syndrom und bei sekundärem Strabismus infolge einer früheren chirurgischen Überkorrektur des Muskelantagonisten ist zweifelhaft. Allenfalls sind mehrere Behandlungszyklen erforderlich.
Bei Patienten, die mit therapeutischen Dosen behandelt werden, kann auch gesteigerte Muskelschwäche auftreten.
Vor Beginn der Behandlung mit BOTOX müssen im Einzelfall Risiko und Nutzen abgewogen werden.
Über Dysphagie wurde auch nach Injektionen berichtet, die nicht in die zervikale Muskulatur erfolgten (für weitere Informationen siehe Abschnitt «Warnhinweise nach Indikation», «Zervikale Dystonie»).
BOTOX sollte nur mit äusserster Vorsicht und unter engmaschiger Überwachung bei Patienten mit subklinischen oder klinischen Anzeichen gestörter neuromuskulärer Reizleitung wie z.B. bei Myasthenia gravis oder Eaton-Lambert-Syndrom, bei Patienten mit peripheren motorisch-neuropathischen Erkrankungen (z.B. amyotropher Lateralsklerose oder motorischer Neuropathie) und bei Patienten mit neurologischen Grunderkrankungen angewendet werden. Diese Patienten können eine erhöhte Sensibilität für Wirkstoffe wie BOTOX haben, auch in therapeutischer Dosierung, was zu einer ausgeprägten Muskelschwäche und zu einem erhöhten Risiko für klinisch relevante systemische Wirkungen einschliesslich schwerer Dysphagie und respiratorischer Beeinträchtigung führen kann. Das Botulinumtoxin-Präparat soll bei diesen Patienten unter Aufsicht eines Spezialisten und nur angewendet werden, wenn der Nutzen der Behandlung das Risiko überwiegt. Patienten mit Dysphagie und Aspiration in der Anamnese sollen mit höchster Vorsicht behandelt werden.
Patienten und Pflegepersonal sollen darauf hingewiesen werden, dass sofort der Arzt zu verständigen ist, wenn Schluck-, Sprech- oder Atemstörungen auftreten.
Bislang bewegungsarme Patienten sollten darauf hingewiesen werden, körperliche Aktivitäten langsam und vorsichtig wieder zu beginnen.
Bevor der Arzt BOTOX anwendet, muss er sich mit der Anatomie des Patienten sowie irgendwelchen aufgrund chirurgischer Eingriffe entstandenen anatomischen Veränderungen vertraut machen. Injektionen in verletzliche anatomische Strukturen sind zu vermeiden.
Nach der Anwendung von BOTOX in Thoraxnähe wurde in Verbindung mit der Injektion über Pneumothorax berichtet. Vorsicht ist bei Injektionen in Nähe der Lunge (insbesondere der Apices) oder in andere verletzliche anatomische Strukturen geboten.
Es sind schwerwiegende unerwünschte Wirkungen, darunter auch mit tödlichem Verlauf, bei Patienten berichtet worden, die BOTOX nicht-indikationsgemäss (off-label) direkt in die Speicheldrüsen, in den orolingualen Rachenraum, in die Speiseröhre und in den Magen injiziert bekommen hatten. Einige Patienten hatten eine bereits bestehende Dysphagie oder ausgeprägte Schwächezustände.
Selten wurde über schwere und/oder sofortige Überempfindlichkeitsreaktionen einschliesslich Anaphylaxie, Serumkrankheit, Urtikaria, Weichteilödem und Dyspnoe berichtet. Einige dieser Reaktionen wurden nach der Anwendung von BOTOX als Monotherapie oder zusammen mit anderen Arzneimitteln berichtet, bei denen ähnliche Reaktionen bekannt sind. Bei Auftreten einer solchen Reaktion dürfen keine weiteren Injektionen mit BOTOX verabreicht werden, und es ist eine geeignete medikamentöse Therapie, z.B. Adrenalin, sofort einzuleiten. In einem Fall wurde über eine Anaphylaxie mit tödlichem Ausgang berichtet, bei einem Patienten, der eine Injektion von fälschlicherweise in 5 ml 1%iger Lidocainlösung aufgelöstes BOTOX erhielt.
Wie bei jeder Injektion kann eine mit dem Injektionsvorgang in Zusammenhang stehende Verletzung auftreten. Eine Injektion kann zu lokaler Infektion, Schmerzen, Entzündung, Parästhesie, Hypoästhesie, Druckempfindlichkeit, Schwellung, Erythem und/oder Blutung/Quetschung führen. Schmerzen und/oder Angst, die in Zusammenhang mit der Injektionsnadel stehen, können zu vasovagalen Reaktionen führen, z.B. Synkope, Hypotension, etc.
BOTOX sollte nur mit Vorsicht angewendet werden bei Entzündung der vorgesehenen Injektionsstelle(n) oder bei ausgeprägter Schwäche oder Atrophie des zu injizierenden Muskels. Bei Patienten mit peripheren motorisch-neuropathischen Erkrankungen (z.B. amyotropher Lateralsklerose oder motorischer Neuropathie) sollte BOTOX ebenfalls nur mit Vorsicht angewendet werden.
Unerwünschte Wirkungen nach Anwendung von BOTOX wurden berichtet, die das kardiovaskuläre System betrafen, wie Arrhythmie und Herzinfarkt, einige davon mit tödlichem Ausgang. Einige dieser Patienten wiesen Risikofaktoren, wie Herz-Kreislauf-Erkrankungen, auf.
Neu oder wiederholt auftretende epileptische Anfälle wurden besonders bei Patienten mit Prädisposition für solche Vorkommnisse berichtet. Der genaue Zusammenhang dieser Vorfälle mit der Botulinumtoxin Injektion ist nicht nachgewiesen. Die Berichte über Vorkommnisse bei Kindern betrafen vorwiegend Patienten mit infantiler Zerebralparese, die gegen Spastik behandelt wurden.
Die Bildung Botulinumtoxin Typ A neutralisierender Antikörper kann die Wirksamkeit einer BOTOX Behandlung durch Inaktivierung der biologischen Aktivität des Toxins vermindern. Resultate einiger Studien deuten darauf hin, dass häufigere BOTOX Injektionen oder höhere Dosen die Inzidenz einer Antikörperbildung erhöhen können. Gegebenenfalls kann eine potentielle Antikörperbildung durch Gabe der niedrigsten Wirkdosis und Einhaltung des grösstmöglichen, klinisch vertretbaren Zeitraums zwischen den Injektionen reduziert werden.
Klinische Fluktuationen, wie sie bei Folgeinjektionen mit BOTOX (und allen anderen Botulinumtoxinen) auftreten können, sind möglicherweise auf unterschiedliches Vorgehen beim Rekonstituieren, auf die gewählten Injektionsintervalle, die injizierten Muskeln und eine geringfügig variierende Aktivität des Toxins, bedingt durch die verwendete biologische Testmethode, zurückzuführen.
Nach Hautkontakt mit BOTOX sollte die betroffene Hautstelle zunächst mit verdünnter Hypochlorit-Lösung gereinigt und danach gründlich unter fliessendem Wasser abgespült werden. Im Falle einer Nadelstichverletzung sollte der betroffene Hautbereich ebenfalls sofort gereinigt und der Patient überwacht werden, wie es im Kapitel «Überdosierung» empfohlen ist. Bei Augenkontakt mit BOTOX muss das betroffene Auge gründlich unter fliessendem Wasser oder mit einer hierfür geeigneten Lösung gespült werden.
Falls eine versehentliche Kontamination vermutet wird (z.B. Hautkontakt oder Nadelstichverletzung), sollte der Hautbereich sofort gereinigt werden. Der Patient sollte medizinisch überwacht werden, wie es im Kapitel «Überdosierung» empfohlen ist.
Anwendung bei Kindern und Jugendlichen
Die Unbedenklichkeit und Wirksamkeit von BOTOX in anderen Anwendungsgebieten als denen, die für Kinder und Jugendliche im Kapitel «Indikationen/Anwendungsmöglichkeiten» beschrieben werden, sind nicht nachgewiesen. Berichte nach Markteinführung über eine mögliche Verteilung des Toxins an vom Applikationsort entfernte Stellen wurden bei Kindern und Jugendlichen mit Begleiterkrankungen hauptsächlich infantiler Zerebralparese sehr selten berichtet. Im Allgemeinen lag die Dosierung, die in diesen Fällen verwendet wurde, über der empfohlenen Dosierung (siehe «Unerwünschte Wirkungen»).
Selten wurde bei Kindern mit schwerer Zerebralparese nach einer Behandlung mit Botulinumtoxin über Todesfälle berichtet, die bisweilen mit Aspirationspneumonie im Zusammenhang stehen, darunter auch nach nicht-zugelassener (off-label) Anwendung (z.B. im Nackenbereich). Äusserste Vorsicht ist bei der Behandlung von Kindern und Jugendlichen mit ausgeprägten neurologischen Schwächezuständen, Dysphagie oder einer Vorgeschichte von Aspirationspneumonie oder Lungenerkrankung geboten. Die Behandlung bei Patienten mit schlechtem zugrundeliegendem Gesundheitszustand sollte nur erfolgen, wenn eingeschätzt wird, dass der potentielle Nutzen für den einzelnen Patienten die Risiken überwiegt.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Vial, d.h. es ist nahezu «natriumfrei».
Warnhinweise nach Indikation
NEUROLOGISCHE ERKRANKUNGEN:
Blepharospasmus
Ein verringertes Blinzeln nach der Injektion von Botulinumtoxin in den M. orbicularis oculi kann zu kornealer Belastung, anhaltendem Epitheldefekt und Hornhautulzeration führen, insbesondere bei Patienten mit Störungen des VII. Hirnnervs. Eine sorgfältige Überprüfung der Hornhautempfindlichkeit bei vorausgegangener Operation ist angezeigt. Auf Injektionen in den medialen Bereich des Unterlids sollte verzichtet werden, um Ektropium zu vermeiden, und jeglicher Epitheldefekt ist aktiv zu behandeln. Gegebenenfalls sollten «künstliche Tränen», Augensalben, therapeutische weiche Kontaktlinsen oder die Anwendung einer Augenklappe oder Ähnliches in Betracht gezogen werden.
In den Weichteilen des Augenlids ist Ekchymose möglich. Wenn nach der Injektion ein leichter Druck auf die Injektionsstelle ausgeübt wird, kann diese Reaktion minimalisiert werden.
Aufgrund der anticholinergen Wirkung des Botulinumtoxins ist Vorsicht bei der Behandlung von Patienten mit Engwinkelglaukom angeraten, einschliesslich Patienten mit anatomisch bedingt engem Kammerwinkel.
Strabismus
BOTOX ist unwirksam bei chronischem paralytischem Strabismus, ausser um die Kontraktur des Muskelantagonisten in Zusammenhang mit einer chirurgischen Korrektur zu reduzieren. Bei der Anwendung von BOTOX zur Behandlung des Strabismus können retrobulbäre Blutungen und Bulbusperforationen auftreten.
Zervikale Dystonie
Bei zervikaler Dystonie können BOTOX-Injektionen sehr milde bis schwere Dysphagien hervorrufen. Als Folge der Dysphagie können Aspiration und Dyspnoe auftreten, in ganz seltenen Fällen mit der Notwendigkeit einer künstlichen Ernährung. In seltenen Fällen wurde über das Auftreten von Dysphagie assoziiert mit Aspirationspneumonie und Tod berichtet. Die Dysphagie kann bis zu 2 bis 3 Wochen nach Injektion andauern, es wurde aber auch ein Andauern bis zu 5 Monaten nach der Injektion berichtet.
Das Auftreten von Dysphagie ist dosisabhängig und kann bei der Injektion in den M. sternocleidomastoideus durch eine Limitierung der Dosis auf < 100 Einheiten vermindert werden. Patienten mit kleineren Nackenmuskeln oder Patienten, die bilaterale Injektionen in den M. sternocleidomastoideus erhalten, haben ein grösseres Dysphagierisiko. Dysphagie ist auf die Ausbreitung des Toxins in die Ösophagusmuskulatur zurückzuführen. Patienten mit zervikaler Dystonie sollten über die mögliche unerwünschte Wirkung einer Dysphagie aufgeklärt werden.
Injektionen in den Levator scapulae können mit einem erhöhten Risiko an oberen Atemwegsinfektionen und Dysphagie assoziiert sein.
Dysphagie kann zu einer verringerten Nahrungs- und Wasseraufnahme beitragen, die zu Gewichtsverlust und Dehydration führt. Patienten mit subklinischer Dysphagie können ein erhöhtes Risiko für eine schwerere Dysphagie nach einer BOTOX Injektion haben.
Fokale Spastik
BOTOX wurde in der Behandlung fokaler Spastiken nur in Verbindung mit üblichen Standardtherapien untersucht und ist nicht als Ersatz für diese gedacht.
Es ist wenig wahrscheinlich, dass BOTOX den Bewegungsspielraum von Gelenken, die von einer fixen Kontraktur betroffen sind, verbessern kann.
BOTOX darf nicht für die Behandlung von fokaler Spastik des Fussgelenkes und des Fusses bei Erwachsenen nach einem Schlaganfall angewendet werden, wenn aufgrund der Reduzierung des Muskeltonus keine verbesserte Funktion (z.B. Verbesserung beim Gehen) oder Symptomlinderung (z.B. Schmerzlinderung) oder eine erleichterte Pflege zu erwarten ist.
Besondere Vorsicht ist geboten bei der Behandlung von Erwachsenen mit Spastik nach einem Schlaganfall, die ein erhöhtes Sturzrisiko haben könnten.
BOTOX sollte mit Vorsicht bei fokaler Spastik des Fussgelenkes und des Fusses bei älteren Schlaganfallpatienten mit ausgeprägter Komorbidität angewendet werden. Eine Behandlung sollte nur eingeleitet werden, wenn davon ausgegangen wird, dass der Nutzen der Behandlung die möglichen Risiken überwiegt.
BOTOX sollte für die Behandlung von Spastik in den unteren Extremitäten nach einem Schlaganfall nur nach Bewertung durch Ärzte mit Erfahrung im Management der Rehabilitation von Schlaganfallpatienten angewendet werden.
Es gibt Post-Marketing-Berichte über Todesfälle (in manchen Fällen mit Aspirationspneumonie) und über mögliche Verteilung des Toxins an vom Applikationsort entfernte Stellen bei Kindern mit Begleiterkrankungen wie insbesondere Zerebralparese nach Botulinumtoxin Behandlung. Siehe Warnhinweise hinsichtlich «Anwendung bei Kindern und Jugendlichen» unter «Warnhinweise und Vorsichtsmassnahmen». Ein kausaler Zusammenhang zu BOTOX wurde in diesen Fällen nicht bewiesen.
Kopfschmerzen bei chronischer Migräne:
Die Sicherheit und Wirksamkeit bei der Prophylaxe von Kopfschmerzen bei Patienten mit episodischer Migräne (Kopfschmerzen an < 15 Tagen im Monat) oder chronischen Spannungskopfschmerzen wurden bisher nicht nachgewiesen.
BLASENFUNKTIONSSTÖRUNGEN:
Injektionen in den Detrusor-Muskel
Eine Zystoskopie sollte mit der ärztlichen Sorgfalt durchgeführt werden.
Bei nicht-katheterisierten Patienten sollte das nach der Entleerung verbleibende Harnvolumen innerhalb von 2 Wochen nach der Behandlung und danach in medizinisch angezeigten Abständen bis zu 12 Wochen beurteilt werden. Patienten sollten angewiesen werden, sich mit ihrem Arzt in Verbindung zu setzen, wenn sie beim Entleeren der Blase Beschwerden haben, da dann möglicherweise eine Katheterisierung erforderlich ist.
Überaktive Blase
BOTOX sollte mit Vorsicht bei Patienten mit Obstruktionen im Blasenhalsbereich (z.B. Obstruktion der Harnwege bei Patienten mit Prostatahyperplasie) eingesetzt werden.
Harninkontinenz infolge neurogener Detrusorhyperaktivität
Im Zusammenhang mit dem Verfahren kann eine autonome Dysreflexie auftreten. In diesem Fall ist möglicherweise eine sofortige medizinische Behandlung erforderlich.
ERKRANKUNGEN DER HAUT UND MIT DER HAUT VERBUNDENE ERKRANKUNGEN:
Primäre Hyperhidrosis axillae
Die Ursachen einer sekundären Hyperhidrosis (z.B. Hyperthyreose, Phäochromozytom) müssen vor der Behandlung ausgeschlossen werden, um eine symptomatische Behandlung der Hyperhidrosis ohne Diagnose und/oder Behandlung der Grunderkrankung zu vermeiden.
Hilfsstoffe von besonderem Interesse
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Durchstechflasche, d.h. es ist nahezu «natriumfrei».
InteraktionenTheoretisch kann die Wirkung von Botulinumtoxin bei gleichzeitiger Verabreichung von Botulinumtoxin und Aminoglykosidantibiotika, Spectinomycin oder anderen Arzneimitteln, die auf die neuromuskuläre Reizleitung wirken (wie z.B. Muskelrelaxantien) potenziert werden.
Werden unterschiedliche Botulinumneurotoxin-Serotypen gleichzeitig oder innerhalb von mehreren Monaten verabreicht, ist die Wirkung nicht bekannt. Eine stark ausgeprägte neuromuskuläre Schwäche kann sich durch die Verabreichung eines anderen Botulinumtoxins vor dem vollständigen Abklingen der Wirkungen eines zuvor verabreichten Botulinumtoxins verschlimmern.
Muskelrelaxantien sollten mit Vorsicht eingesetzt werden.
Es wurden keine Interaktionsstudien durchgeführt. Interaktionen mit klinischer Relevanz wurden nicht berichtet.
Kinder und Jugendliche
Bei Kindern und Jugendlichen wurden keine Studien zur Erfassung von Interaktionen durchgeführt. Es ist jedoch zu beachten, dass die oben genannten Interaktionen auch in der pädiatrischen Bevölkerungsgruppe auftreten können.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine ausreichenden Daten zur Anwendung von Botulinumtoxin Typ A während der Schwangerschaft vor. Tierstudien haben eine reproduktionstoxikologische Wirkung gezeigt (siehe unter «Präklinische Daten»).
Das potentielle Risiko für den Menschen ist nicht bekannt. BOTOX sollte daher während der Schwangerschaft und von Frauen im gebärfähigen Alter, die nicht verhüten, nicht angewendet werden, es sei denn, dies ist eindeutig erforderlich. Wenn eine Patientin während der Behandlung schwanger wird, sollte sie auf die potentiellen Risiken wie Fehlgeburt oder fötale Missbildungen hingewiesen werden.
Stillzeit
Es ist nicht bekannt, ob BOTOX in die Milch übergeht. Die Anwendung von BOTOX während der Stillzeit kann nicht empfohlen werden.
Fertilität
Es liegen keine klinischen Daten zur Anwendung von Botulinumtoxin Typ A vor. In tierexperimentellen Studien mit BOTOX wurden Auswirkungen auf die männliche und weibliche Fertilität festgestellt (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zu den Auswirkungen auf die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt. BOTOX kann jedoch eine Asthenie, Muskelschwäche, Schwindel und Sehstörungen hervorrufen, wodurch die Fähigkeit zur aktiven Teilnahme am Strassenverkehr und zum Bedienen von Maschinen beeinflusst werden könnte.
Unerwünschte WirkungenAllgemein
In kontrollierten klinischen Studien wurden unerwünschte Wirkungen berichtet, die nach Meinung der Prüfärzte in Zusammenhang mit BOTOX standen: bei 35% der Patienten mit Blepharospasmus, bei 28% der Patienten mit zervikaler Dystonie, bei 8% der Patienten mit infantiler Spastik, bei 11% der Patienten mit primärer Hyperhidrosis axillae, bei 16% der erwachsenen Patienten mit Spastik der oberen Extremitäten und bei 11% der erwachsenen Patienten mit Spastik der unteren Extremitäten. In klinischen Studien zur überaktiven Blase betrug die Häufigkeit 26% bei der ersten Behandlung und 22% bei einer zweiten Behandlung.
In klinischen Studien zur neurogener Detrusorhyperaktivität bei Erwachsenen betrug die Häufigkeit 32% bei der ersten Behandlung und sank auf 18% bei der zweiten Behandlung.
Bei der pädiatrischen neurogenen Detrusorhyperaktivität betrug die Inzidenz bei der ersten Behandlung 14,2 % (16/113), bei der zweiten Behandlung 16,7 % (15/90), bei der dritten Behandlung 18,2 % (10/55) und bei der vierten Behandlung 45,5 % (5/11).
In klinischen Studien zu chronischer Migräne betrug die Häufigkeit 26 % nach der ersten Behandlung und sank auf 11 % nach der zweiten Behandlung.
Generell treten Nebenwirkungen innerhalb der ersten Tage nach der Injektion auf und können, obwohl im Allgemeinen vorübergehend, mehrere Monate oder in seltenen Fällen noch länger andauern.
Lokale Muskelschwäche stellt eine erwartete pharmakologische Wirkung von Botulinumtoxin dar. Jedoch wurde über Schwäche von benachbarten Muskeln und/oder Muskeln fern von der Injektionsstelle berichtet.
Wie bei Injektionen zu erwarten, traten lokaler Schmerz, Entzündung, Parästhesie, Hypoästhesie, Druckempfindlichkeit, Schwellung/Ödem, Erythem, lokalisierte Infektion, Blutungen und/oder Blutergüsse im Bereich der Injektionsstelle auf. Mit der Injektionsnadel in Zusammenhang stehende Schmerzen und/oder Angstgefühle führten zu vasovagalen Reaktionen einschliesslich vorübergehender symptomatischer Hypotension und Synkope. Nach Injektionen mit Botulinumtoxin wurden auch Fieber und grippeähnliche Symptome berichtet.
Nebenwirkungen – Häufigkeitsangaben je Indikation
Nachfolgend sind für die einzelnen Anwendungsgebiete Angaben zur Häufigkeit von in klinischen Studien dokumentierten Nebenwirkungen gemacht.
Die Häufigkeitsangaben sind wie folgt definiert: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1000, <1/100); selten (≥1/10'000, <1/1000); sehr selten (<1/10'000), nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
NEUROLOGISCHE ERKRANKUNGEN:
Indikation Blepharospasmus/Spasmus hemifacialis
Erkrankungen des Nervensystems
Gelegentlich: Schwindel, Fazialparese, Fazialparalyse.
Augenerkrankungen
Sehr häufig: Oberlidptosis (11%).
Häufig: Keratitis punctata, Lagophthalmus, trockenes Auge, Augenirritationen, Photophobie, Zunahme der Lakrimation.
Gelegentlich: Keratitis, Ektropium, Diplopie, Entropium, visuelle Störungen, verschwommenes Sehen.
Selten: Augenlidödem.
Sehr selten: Ulzerative Keratitis, kornealer Epitheldefekt, korneale Perforation.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Ekchymose.
Gelegentlich: Exanthem, Dermatitis.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Gesichtsödem, Irritationen.
Gelegentlich: Erschöpfung.
Indikation Strabismus
Augenerkrankungen
Sehr häufig: Störungen der Augenbewegung (16,9%), Oberlidptosis (15,7%).
Gelegentlich: okulare retrobulbäre Blutungen, Augenpenetration, Holmes-Adie Pupille.
Selten: Glaskörperblutung.
Indikation Zervikale Dystonie
Infektionen und parasitäre Erkrankungen
Häufig: Rhinitis, Infektion der oberen Atemwege.
Erkrankungen des Nervensystems
Häufig: Schwindel, Kopfschmerzen, Hypoästhesie, Muskelhypertonus, Somnolenz.
Augenerkrankungen
Gelegentlich: Diplopie, Oberlidptosis.
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Gelegentlich: Dyspnoe, Dysphonie.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Dysphagie (bis zu 18,6% bei einer mittleren Dosis von 240,5 Einheiten) (siehe auch «Zusätzliche Informationen» unten).
Häufig: trockener Mund, Übelkeit.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Muskelschwäche (12,8%).
Häufig: Steifheit der Skelettmuskulatur, Muskelkater.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Schmerz (16,3%).
Häufig: Asthenie, grippeähnliche Erkrankung, Unwohlsein.
Gelegentlich: Fieber.
Indikation Fokale Spastik der oberen Extremitäten - Erwachsene
Psychiatrische Erkrankungen
Gelegentlich: Depression, Insomnie.
Erkrankungen des Nervensystems
Häufig: Muskelhypertonus.
Gelegentlich: Parästhesie, Kopfschmerzen, Hypoästhesie, Amnesie, Unkoordiniertheit.
Erkrankungen des Ohrs und des Labyrinths
Gelegentlich: Schwindel.
Gefässerkrankungen
Gelegentlich: orthostatische Hypotonie.
Erkrankungen des Gastrointestinaltrakts
Gelegentlich: Übelkeit, orale Parästhesie.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Ekchymose, Purpura.
Gelegentlich: Dermatitis, Pruritus, Exanthem.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Schmerzen in den Extremitäten, Muskelschwäche.
Gelegentlich: Arthralgie, Bursitis.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Schmerzen an der Injektionsstelle, Fieber, grippeähnliche Erkrankung, Blutung an der Einstichstelle, Irritation an der Injektionsstelle.
Gelegentlich: Schmerzen, Unwohlsein, Asthenie, Überempfindlichkeit an der Injektionsstelle, peripheres Ödem.
Einige der gelegentlich berichteten Nebenwirkungen können krankheitsbedingt sein.
Indikation Fokale Spastik der oberen Extremitäten - Jugendliche und Kinder ab 2 Jahren
Infektionen und parasitäre Erkrankung
Häufig: Infektionen der oberen Atemwege.
Erkrankungen des Gastrointestinaltrakts
Häufig: Übelkeit.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Muskelschwäche.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Schmerzen an der Injektionsstelle.
Indikation Fokale Spastik der unteren Extremitäten - Erwachsene
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Arthralgie.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: peripheres Ödem.
Bei wiederholter Verabreichung wurde insgesamt keine Veränderung des Sicherheitsprofils beobachtet.
Indikation Fokale Spastik der unteren Extremitäten - Jugendliche und Kinder ab 2 Jahren
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Schmerzen an der Injektionsstelle.
Kopfschmerzen bei chronischer Migräne
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen, Migräne, Fazialparese.
Augenerkrankungen
Häufig: Oberlidptosis.
Erkrankungen des Gastrointestinaltrakts
Gelegentlich: Dysphagie.
Erkrankungen der Haut und des Unterhautgewebes
Häufig: Pruritus, Hautausschlag.
Gelegentlich: Hautschmerzen.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Nackenschmerzen, Myalgie, Schmerzen des Bewegungsapparats, Steifigkeit des Bewegungsapparats, muskuläre Spasmen, Muskelverspannungen, Muskelschwäche
Gelegentlich: Schmerzen im Kiefer.
Nicht bekannt: Mephisto-Zeichen (seitliche Erhöhung der Augenbrauen).
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Schmerzen an der Injektionsstelle.
Migräne, einschliesslich einer Verschlimmerung der Migräne, wurde bei 3,8 % der Patienten unter BOTOX und 2,6 % der Patienten unter Placebo typischerweise innerhalb des ersten Monats nach der Behandlung berichtet.
Diese Reaktionen traten bei den nachfolgenden Behandlungszyklen nicht durchgängig erneut auf, und die Gesamthäufigkeit nahm mit wiederholten Behandlungen ab.
Die Abbruchrate aufgrund unerwünschter Ereignisse betrug in diesen Phase-3-Studien 3,8 % unter BOTOX im Vergleich zu 1,2 % unter Placebo.
BLASENFUNKTIONSSTÖRUNGEN:
Indikation Überaktive Blase - Erwachsene
Infektionen und parasitäre Erkrankungen
Sehr häufig: Infektionen der Harnwege (25,5%).
Häufig: Bakteriurie.
Erkrankungen der Nieren und Harnwege
Sehr häufig: Dysurie (10,9%).
Häufig: Harnretention, Pollakisurie.
Untersuchungen
Häufig: Restharnvolumen*.
* erhöhtes Restharnvolumen nach Blasenentleerung, das keine Katheterisierung erfordert.
Verfahrensbedingte Nebenwirkungen, die häufig auftraten, waren Dysurie und Hämaturie.
Eine saubere intermittierende Katheterisierung wurde bei 6,5% der Patienten nach Behandlung mit 100 Einheiten BOTOX gegenüber 0,4% in der Placebo-Gruppe eingeleitet.
Von 1'242 Patienten, die in den Placebo-kontrollierten klinischen Studien waren 41,4% (n = 514) ≥65 Jahre alt und 14,7% (n = 182) ≥75 Jahre alt. Es wurde in diesen Studien insgesamt kein Unterschied im Sicherheitsprofil nach der BOTOX-Behandlung zwischen Patienten im Alter von 65 Jahren und älter und Patienten unter 65 Jahren festgestellt, mit der Ausnahme, dass Harnweginfekte bei älteren Patienten sowohl in der Placebo- als auch in der BOTOX-Gruppe häufiger auftraten als bei jüngeren Patienten.
Bei wiederholter Verabreichung wurde insgesamt keine Veränderung des Sicherheitsprofils beobachtet.
Überaktive Blase bei Kindern und Jugendlichen:
Infektionen und parasitäre Erkrankungen
Häufig: Harnwegsinfektion.
Erkrankungen der Nieren und Harnwege
Häufig: Dysurie*, Schmerzen der Harnröhre*.
*verfahrensbedingte Nebenwirkung.
Erkrankungen des Gastrointestinaltrakts
Häufig: Bauchschmerzen, Schmerzen im Unterbauch.
In einer doppelblinden, randomisierten, multizentrischen Parallelgruppenstudie mit 55 Patienten im Alter von 12 bis 17 Jahren waren die Nebenwirkungen mit dem bekannten Sicherheitsprofil bei erwachsenen Patienten mit überaktiver Blase vergleichbar, jedoch wurden in dieser kleinen Studie zu pädiatrischer überaktiver Blase auch Harnröhren- und Unterleibsschmerzen festgestellt.
Indikation Harninkontinenz bei Erwachsenen infolge einer neurogenen Detrusorhyperaktivität
Unerwünschte Wirkungen aus klinischen Studien der Phase 2 und aus pivotalen klinischen Studien der Phase 3
Infektionen und parasitäre Erkrankungen
Sehr häufig: Infektion der Harnwege (49%).
Psychiatrische Erkrankungen
Häufig: Schlaflosigkeit.
Erkrankungen des Gastrointestinaltrakts
Häufig: Konstipation.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: muskuläre Schwäche, muskuläre Spasmen.
Erkrankungen der Nieren und Harnwege
Sehr häufig: Harnretention (17%).
Häufig: Hämaturie*, Dysurie*, Blasendivertikel.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Müdigkeit, Gangstörungen.
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Häufig: autonome Dysreflexie*, Sturz.
* verfahrensbedingte Nebenwirkungen
In klinischen Studien wurden bei 49,2% der Patienten, die mit 200 Einheiten BOTOX behandelt wurden, sowie bei 35,7% der mit Placebo behandelten Patienten Harnwegsinfekte berichtet (53,0% der Patienten mit Multipler Sklerose, die 200 Einheiten erhielten, gegenüber 29,3% in der Placebogruppe; 45,4% der Patienten mit Rückenmarksverletzung, die 200 Einheiten erhielten, gegenüber 41,7% mit Placebo). Bei 17,2% der Patienten, die mit 200 Einheiten BOTOX behandelt wurden, sowie bei 2,9% der mit Placebo behandelten Patienten wurde Harnverhalt berichtet (28,8% der Patienten mit Multipler Sklerose, die 200 Einheiten erhielten, gegenüber 4,5% in der Placebogruppe; 5,4% der Patienten mit Rückenmarksverletzung, die 200 Einheiten erhielten, gegenüber 1,4% mit Placebo).
Bei wiederholter Anwendung wurde keine Veränderung der Art der Nebenwirkungen beobachtet.
Bei MS-Patienten, die an den zentralen Studien teilnahmen, wurde keine Veränderung der jährlichen MS-Schubrate (d.h. Anzahl der MS-Schubereignisse pro Patientenjahr) beobachtet (BOTOX = 0,23; Placebo = 0,20).
Von den zu Studienbeginn und vor der Behandlung nicht-katheterisierten Patienten wurde bei 38,9% der Patienten nach der Behandlung mit BOTOX 200 Einheiten im Vergleich zu 17,3% der mit Placebo behandelten Patienten eine Katheterisierung eingeleitet.
Unerwünschte Wirkungen aus der Studie nach Marktzulassung mit BOTOX 100 Einheiten bei zu Studienbeginn nicht-katheterisierten MS-Patienten
Infektionen und parasitäre Erkrankungen
Sehr häufig: Infektion der Harnwege (39,4%), Bakteriurie (18,2%).
Erkrankungen der Nieren und Harnwege
Sehr häufig: Harnretention (16,7%).
Häufig: Hämaturie**, Dysurie**.
Untersuchungen
Sehr häufig: Restharnvolumen** (16,7%).
* verfahrensbedingte Nebenwirkungen.
** erhöhtes Restharnvolumen, das keine Katheterisierung erfordert
Eine Katheterisierung wurde bei 15,2% der Patienten nach der Behandlung mit BOTOX 100 Einheiten im Vergleich zu 2,6% der mit Placebo behandelten Patienten eingeleitet.
Indikation Neurogene Detrusorhyperaktivität bei katheterisierten pädiatrischen Patienten ab 5 Jahren
Unerwünschte Wirkungen (alle Dosen) aus zwei pädiatrischen Phase-3-Studien einschliesslich Behandlungszyklus 1 der Studie 191622-120 (N=113) und Wiederholungsbehandlungszyklen 2 (N=90), 3 (N=55) und 4 (N=11) der Studie 191622-121:
Infektionen und parasitäre Erkrankungen
Sehr häufig: Harnwegsinfektion (Zyklus 1=29,2%, Zyklus 2=34,4%, Zyklus 3=21,8%, Zyklus 4=18,2%), Bakteriurie (Zyklus 1=16,8%, Zyklus 2=13,3%, Zyklus 3=12,7%, Zyklus 4=18,2%).
Erkrankungen der Nieren und Harnwege
Sehr häufig: Hämaturie oder Blut im Urin nachweisbar (Zyklus 1=4.4%, Zyklus 2=10.0%, Zyklus 3=16.4%, Zyklus 4=54.5%).
Häufig: Leukozyturie.
Erkrankungen des Gastrointestinaltrakts
Häufig: Obstipation, abdominale Schmerzen*.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Suprapubische Schmerzen*.
* verfahrensbedingte Nebenwirkungen
ERKRANKUNGEN DER HAUT UND MIT DER HAUT VERBUNDENE ERKRANKUNGEN:
Indikation Primäre Hyperhidrosis axillae
Erkrankungen des Nervensystems
Häufig: Schmerzen, Kopfschmerzen, Parästhesie.
Gefässerkrankungen
Häufig: Hitzewallungen.
Erkrankungen des Gastrointestinaltrakts
Gelegentlich: Übelkeit.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Hyperhidrosis (nicht-axillares Schwitzen), anormaler Hautgeruch, Pruritus, subkutaner Knoten, Alopezie.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Schmerzen in den Extremitäten.
Gelegentlich: Schwäche in den Armen, Myalgie, Arthropathie.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Schmerzen an der Injektionsstelle (11,5%).
Häufig: Schmerzen, Ödem/Blutung/Überempfindlichkeit/Reizung an der Injektionsstelle, Asthenie, Reaktionen an der Injektionsstelle.
Bei der Behandlung der primären axillaren Hyperhidrosis wurde für 4,5% der Patienten über einen Anstieg der Schweissbildung ausserhalb der Achselhöhlen innerhalb eines Monats nach Injektion berichtet. Hierbei ergab sich kein Muster in Bezug auf die betroffenen anatomischen Stellen. Bei etwa 30% der Patienten konnte ein Rückgang innerhalb von 4 Monaten festgestellt werden.
Gelegentlich (0,7%) wurde über eine leichte, vorübergehende, nicht behandlungsbedürftige Schwäche in den Armen berichtet, die sich ohne Folgeerscheinungen besserte. Diese Nebenwirkung könnte mit der Behandlung, der Injektionstechnik oder beidem zusammenhängen. Bei der gelegentlich berichteten Muskelschwäche kann eine neurologische Untersuchung in Betracht gezogen werden. Zusätzlich ist eine Überprüfung der Injektionstechnik vor erneuten Injektionen ratsam, um die intradermale Platzierung der Injektionen sicherzustellen.
Unerwünschte Wirkungen nach Markteinführung
Die folgende Aufstellung enthält unerwünschte Arzneimittelwirkungen oder andere medizinisch relevante unerwünschte Wirkungen, über die unabhängig von der Indikation seit der Markteinführung des Arzneimittels berichtet wurde und zusätzlich zu den unerwünschten Wirkungen auftreten können, die unter «Warnhinweise und Vorsichtsmassnahmen» und unter «Unerwünschte Wirkungen» genannt werden.
Erkrankungen des Immunsystems
Anaphylaxie, Angioödem, Serumkrankheit und Urtikaria.
Stoffwechsel- und Ernährungsstörungen
Anorexie.
Erkrankungen des Nervensystems
Brachialplexopathie, Dysphonie, Dysarthrie, Fazialparese, Hypoästhesie, Muskelschwäche, Myasthenia gravis, periphere Neuropathie, Parästhesie, Radikulopathie, Krampfanfälle, Synkope und Fazialparalyse.
Augenerkrankungen
Engwinkelglaukom (bei der Behandlung von Blepharospasmus), Strabismus, verschwommenes Sehen, Sehstörungen, trockene Augen und Augenlidödem.
Erkrankungen des Ohrs und des Labyrinths
Hypoakusis, Tinnitus und Vertigo.
Herzerkrankungen
Arrhythmie, Herzinfarkt.
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Aspirationspneumonie (manchmal mit tödlichem Ausgang), Dyspnoe, Atemdepression und respiratorische Insuffizienz.
Erkrankungen des Gastrointestinaltrakts
Abdominale Schmerzen, Diarrhö, Obstipation, trockener Mund, Dysphagie, Nausea und Erbrechen.
Erkrankungen der Haut und des Unterhautzellgewebes
Alopezie, psoriasisartige Dermatitis, Erythema multiforme, Hyperhidrose, Madarosis, Pruritus und Hautausschlag.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Muskelathropie, Myalgie und lokalisierte Muskelzuckungen/unfreiwillige Muskelkontraktionen.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Denervationsatrophie, Malaise und Fieber.
Bei der Behandlung des Strabismus ist in einem Fall eine Blutung in den Glaskörper bekannt geworden, die sich später wieder zurückbildete. Es sind einige Fälle von retrobulbärer Hämorrhagie bekannt geworden, ein Visusverlust wurde jedoch nie festgestellt. 5 Augen wiesen Pupillenveränderungen mit einem Ziliarganglionschaden (Adies-Pupille) auf.
Eine Patientin entwickelte zwei Tage nach der Injektion von 120 Einheiten BOTOX zur Behandlung der zervikalen Dystonie eine Erkrankung des Plexus brachialis, die etwa fünf Monate andauerte.
In einer Studie bei Patienten mit Spastiken der oberen Extremitäten nach Schlaganfall wurden pulmonale Spirometrie Messungen durchgeführt, welche eine 15% verminderte Lungenfunktion aufzeigten. Die Häufigkeit dieser unerwünschten Wirkung wurde vermehrt in der BOTOX-Patienten-Gruppe berichtet als in der Placebo-Gruppe, wobei die Unterschiede weder klinische noch statistische Signifikanz erreichten.
Bei der Behandlung der primären axillaren Hyperhidrosis wurde innerhalb von 1 Monat nach Injektion bei 4,5% der Patienten eine Zunahme der nicht-axillaren Schweissabsonderung wahrgenommen. Hierbei ergab sich kein Muster hinsichtlich der betroffenen anatomischen Stellen. Ein Rückgang wurde bei etwa 30% der Patienten innerhalb von 4 Monaten festgestellt.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungÜberdosierung von BOTOX ist ein relativer Begriff und abhängig von Dosierung, Injektionsstelle und den zugrundeliegenden Gewebeeigenschaften. Es wurden keine Fälle systemischer Intoxikation aufgrund versehentlicher Injektion von BOTOX-Lösung beobachtet. Überhöhte Dosierungen können lokale oder entfernt liegende, allgemeine und profunde neuromuskuläre Paralysen hervorrufen. Es wurden keine Fälle einer Ingestion von BOTOX berichtet.
Anzeichen und Symptome einer Überdosierung treten nicht unmittelbar nach einer Injektion auf. Nach versehentlicher Injektion oder Ingestion oder vermuteter Überdosierung muss der Patient mehrere Wochen hinsichtlich fortschreitender Anzeichen und Symptome einer lokalen oder vom Applikationsort entfernten Muskelschwäche wie Ptosis, Diplopie, Dysphagie, Dysarthrie, generalisierte Schwäche oder respiratorische Insuffizienz medizinisch überwacht werden. Bei diesen Patienten soll eine weitere medizinische Abklärung erwogen werden, und eine geeignete medizinische Behandlung ist sofort einzuleiten, was Hospitalisation einschliessen kann.
Wenn die Muskulatur von Oropharynx und Ösophagus betroffen ist, manifestiert sich evtl. eine Aspirationspneumonie. Wenn es zur Paralyse oder zu ausgeprägter Schwächung der Atemmuskulatur kommt, werden Intubation und assistierte Beatmung erforderlich, bis Besserung eintritt, und es können eine Tracheostomie und längere künstliche Beatmung zusätzlich zu anderen allgemeinen unterstützenden Massnahmen erforderlich sein.
Eigenschaften/WirkungenATC-Code
M03AX01
Wirkungsmechanismus
Das Botulinumtoxin Typ A blockiert durch Spaltung von SNAP 25 («synaptosomal-associated protein of 25 kDa») die periphere Acetylcholin-Freisetzung an den präsynaptischen Nervenendigungen. SNAP 25 ist ein Protein, das wesentlich für die erfolgreiche Bindung und Freisetzung von Acetylcholin aus den Vesikeln ist, die sich in den Nervenendigungen befinden.
Pharmakodynamik
Nach der Injektion erfolgt initial eine hoch-affine Bindung des Toxins an spezifische zelluläre Oberflächenrezeptoren. Durch einen Rezeptor-vermittelten Endozytose-Prozess wird das Toxin durch die Plasmamembran transportiert und anschliessend im Cytosol freigesetzt. Dieser Prozess wird von einer progressiven Inhibition der Acetylcholin-Freisetzung begleitet. Klinische Befunde manifestieren sich innerhalb von 2-3 Tagen. Die maximale Wirkung ist 5-6 Wochen nach der Injektion zu beobachten. In sensorischen Neuronen hemmt BOTOX die Freisetzung von sensorischen Neurotransmittern (z.B. Substanz P, CGRP) und reguliert die Expression von Zelloberflächenrezeptoren (z.B. TRPV1) herunter. BOTOX verhindert auch eine Sensibilisierung nozizeptiver sensorischer Neuronen, macht diese rückgängig und hemmt somit möglicherweise auch die zentrale Sensibilisierung.
Nach intramuskulärer Injektion erfolgt innerhalb von 12 Wochen die Wiederherstellung der Impulsübertragung durch neugebildete Nervenendigungen und deren Wiederverbindung mit den motorischen Endplatten.
Bei intradermaler Injektion, wo das Ziel die ekkrinen Schweissdrüsen sind, hält der Effekt bei Patienten, die 50 Einheiten pro Axilla erhielten, ungefähr 4-7 Monate an.
Wird BOTOX zur Behandlung des Strabismus eingesetzt, dann wird angenommen, dass es auf Muskelpaare einwirkt, indem der behandelte Muskel atrophisch verlängert und der entsprechende Muskelantagonist verkürzt wird.
Nach Injektion in die Halsmuskulatur verbessert BOTOX sowohl die objektiven Anzeichen als auch die subjektiven Symptome der zervikalen Dystonie. Dies äussert sich in einer Verringerung der Drehung des Kopfes zur Seite, verringerter Anhebung der Schulter, verringerter Grösse und Kraft hypertrophischer Muskeln und Verringerung der Schmerzen.
Bei Erwachsenen mit fokalen Spastiken der oberen Extremitäten nach Schlaganfall verbessert BOTOX deutlich den Bewegungsspielraum (Ellbogen und Handgelenksstreckung).
Die Injektion von BOTOX bei Kindern mit dynamischer Spitzfussstellung (Equinus-Deformität) ohne Retraktion und ohne bedeutende Atrophie führt durch teilweise Denervierung des M. gastrocnemius zu einer veränderten Sprunggelenksposition und dadurch zu einer deutlichen Verbesserung des Gangbildes.
Nach Injektion in den Detrusor-Muskel wirkt BOTOX auf die efferenten Nervenbahnen der Detrusor-Aktivität durch Hemmung der Freisetzung von Acetylcholin. Zusätzlich hemmt BOTOX afferente Neurotransmitter und sensorische Bahnen.
Klinische Wirksamkeit
NEUROLOGISCHE ERKRANKUNGEN:
Blepharospasmus/hemifazialer Spasmus
In einer offenen Studie wurde 56 Patienten mit Spasmus hemifacialis eine Anfangsdosis von 10 bis 50 Einheiten injiziert. Die Patienten wurden anschliessend 22 Wochen beobachtet. Patienten, die in Woche 4 keine Besserung zeigten, erhielten eine erneute Injektion (5 bis 50 Einheiten). Die Patienten wurden in Woche 2, 4 (sofern eine erneute Behandlung erforderlich war), 6, 14 und 22 beobachtet. Die klinische Wirkung wurde zu jedem Kontrollzeitpunkt für die verschiedenen oberen und unteren Gesichtsmuskeln bewertet.
Bei allen 56 Patienten trat eine Besserung ein, und 62,5% (35/56) zeigten eine deutliche Besserung. Bezogen auf die oberen Gesichtsmuskeln wurde bei allen Patienten eine Besserung beobachtet. Bei der Beurteilung der unteren Gesichtsmuskeln zeigte sich, dass mit Ausnahme von 2 Patienten alle auf die Behandlung angesprochen hatten.
Bei Patienten mit gutartigem essentiellem Blepharospasmus wurde eine zusätzliche randomisierte, multizentrische Doppelblindstudie mit parallelen Gruppen zum Vergleich der Sicherheit und Wirksamkeit von 2 BOTOX-Formulierungen (der Wirkstoff stammte aus verschiedenen Master-Zellbanken - eine davon entspricht der derzeitigen BOTOX-Formulierung) durchgeführt. In die Studie wurden Patienten aufgenommen, die schon früher mit BOTOX behandelt worden waren, und die Behandlung bestand aus einem einzelnen Injektionszyklus in die M. orbicularis oculi beider Augen. Die Dosis (maximal zulässig waren 100 Einheiten) und Injektionsstellen wurden vom Prüfarzt aufgrund des Ansprechens der Patienten auf frühere BOTOX-Behandlungen festgelegt. Die Patienten wurden über einen Zeitraum von 12 Wochen nach der Behandlung beobachtet. In die Studie wurden 98 Patienten aufgenommen (48 in die Gruppe mit der derzeitigen BOTOX-Formulierung).
Die Erfolgsrate der Behandlung wurde auf einer 5-Punkte-Skala von 0 bis 4 beurteilt. Dabei stand 0 für «keinen» und 4 für «starken» behindernden Spasmus der Augenlider und möglicherweise anderer Gesichtsmuskeln. Eine Abnahme auf einen Punktwert von ≤2 an beiden Augen galt als Behandlungserfolg. Der primäre Endpunkt lag in Woche 4. Die Patienten erhielten eine durchschnittliche Dosis von 33 Einheiten pro Auge, injiziert an 3 bis 15 Injektionsstellen. Vergleichbar mit der früheren BOTOX-Formulierung lag die Erfolgsrate der Behandlung mit der derzeitigen BOTOX-Formulierung bei ca. 90% in Woche 4.
Zervikale Dystonie
Bei Patienten mit zervikaler Dystonie (CD) wurde eine randomisierte, Placebo-kontrollierte, multizentrische Doppelblindstudie durchgeführt. In diese Studie wurden erwachsene Patienten mit CD aufgenommen, die positiv auf eine frühere Behandlung mit BOTOX angesprochen hatten. Die Studie war in 2 Behandlungsphasen unterteilt, in eine offene Studienphase, in der alle Patienten mit BOTOX behandelt wurden, wobei Dosierung und Injektionsstellen individuell vom Prüfarzt festgelegt wurden, und in eine Doppelblindphase, in der Patienten, die in der offenen Phase als Responder eingestuft wurden, doppelblind entweder BOTOX oder Placebo erhielten. Zwischen diesen beiden Phasen lag ein Zeitraum von bis zu 6 Wochen. Insgesamt wurden den Patienten bis zu 360 Einheiten BOTOX verabreicht.
In der offenen Studienphase wurden 214 Patienten beurteilt, von denen 170 für den Doppelblindteil der Studie randomisiert wurden (88 Patienten wurden in die BOTOX-Gruppe, 82 in die Placebo-Gruppe aufgenommen). Anschliessend wurden die Patienten im Abstand von jeweils 2 Wochen über einen Zeitraum von bis zu 10 Wochen nach der Behandlung beobachtet. Die primären Wirksamkeitsvariablen waren eine Veränderung der Kopfhaltung (gemessen auf der Skala zur Beurteilung der Schwere der zervikalen Dystonie) und der prozentuale Anteil der Patienten mit einer Besserung des CD-Status, gemessen anhand einer ärztlich beurteilten Bewertungsskala 6 Wochen nach der Behandlung. Schmerzen als wichtiges Symptom von CD wurden von den Patienten im Hinblick auf Häufigkeit und Intensität beurteilt. Zusätzlich beurteilten Arzt und Patient die funktionale Behinderung.
BOTOX erwies sich bei beiden primären Wirksamkeitsvariablen nach 6 Wochen dem Placebo sowohl klinisch als auch statistisch signifikant überlegen. BOTOX verringerte auch die Intensität und Häufigkeit von Schmerzen statistisch signifikant besser als Placebo. Signifikante Verbesserungen wurden auch bei der Beurteilung der funktionalen Behinderung durch Arzt und Patient in Woche 6 beobachtet.
Bei Patienten mit zervikaler Dystonie wurde eine zusätzliche randomisierte Doppelblindstudie im Crossover-Design zur Untersuchung der Sicherheit und Wirksamkeit von 2 BOTOX-Formulierungen (der Wirkstoff stammte aus verschiedenen Master-Zellbanken - eine davon entspricht der derzeitigen BOTOX-Formulierung) durchgeführt. In die Studie wurden Patienten aufgenommen, die bereits früher mit zufriedenstellenden Ergebnissen behandelt worden waren; die Behandlung umfasste 2 Injektionen mit einer dazwischen liegenden Auswaschphase von 8 bis 16 Wochen. Die Patienten wurden nach jeder Behandlung über einen Zeitraum von 8 bis 16 Wochen beobachtet, wobei der primäre Endpunkt in Woche 6 nach jeder Behandlung lag.
In diese Studie wurden 135 Patienten aufgenommen. Die primäre Wirksamkeitsvariable war die Gesamtbeurteilung des Schweregrads auf der Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS). Sekundäre Wirksamkeitsvariablen waren der Behinderungs-Schmerzscore (anhand der modifizierten TWSTRS) und die Gesamtbeurteilung der zervikalen Dystonie durch Patient und Arzt. Die Ergebnisse bestätigten, dass eine maximale klinische Besserung nach Injektion von BOTOX in Woche 6 erreicht wird. Die mittlere Verringerung des Gesamtwerts auf der TWSTRS stellte eine 35%ige Besserung gegenüber den Ausgangswerten dar, und die höchste mittlere Verringerung des Behinderungs-Schmerzscores entsprach einer 50%igen Besserung gegenüber dem Ausgangswert zu diesem Zeitpunkt.
Die Gesamtbeurteilung durch Ärzte und Patienten zeigte ebenfalls den positiven Behandlungseffekt mit einem für die neue BOTOX-Formulierung berichteten Behandlungserfolg in über 85% bzw. 80% der Fälle in Woche 6.
Fokale Spastiken der oberen Extremitäten bei Erwachsenen
Bei Patienten mit Spastiken der oberen Extremitäten nach einem Schlaganfall wurde eine Placebo-kontrollierte, multizentrische Doppelblindstudie zur Untersuchung der Wirksamkeit und Sicherheit von BOTOX durchgeführt. Die Patienten waren noch nie mit BOTOX behandelte Erwachsene, deren Schlaganfall ≥6 Wochen zurücklag und die erweiterte Ashworth-Scores (EAS) von ≥3 am Handgelenk und ≥2 am Ellbogen aufwiesen. Einundneunzig Patienten wurden auf eine von vier Behandlungsgruppen randomisiert (BOTOX 90, 180 oder 360 Einheiten, oder Placebo). Der primäre Endpunkt, Spastik des Flexors im Handgelenk, wurde in Woche 6 auf der EAS gemessen. Sekundäre Wirksamkeitsvariablen waren Spastik der Flexoren in Ellbogen und Fingern, gemessen auf der EAS und nach Gesamtbeurteilung des Arztes.
In die Studie wurden 91 Patienten aufgenommen (65 Patienten erhielten BOTOX in 90, 180 oder 360 Einheiten). Die mittlere Abnahme auf der EAS für die Flexoren des Handgelenks war in allen BOTOX-Gruppen bis Woche 12 grösser als in der Placebo-Gruppe.
In allen BOTOX-Behandlungsgruppen war der Flexortonus im Handgelenk, gemessen auf der EAS, wirksamer verringert als unter Placebo. Die Flexoren der Ellbogen zeigten ähnliche Verbesserungsmuster. Bei den Fingerflexoren fand sich zwar insgesamt eine gewisse Variabilität (signifikante Verbesserungen nur in Woche 1 und 3), aber die Daten weisen auf vergleichbare Dosis/Wirkungs-Beziehungen hin. Die ärztlich beurteilte Bewertungsskala zeigte ebenfalls einen signifikanten Vorteil bei Dosen von 180 Einheiten und 360 Einheiten.
Eine weitere Placebo-kontrollierte, multizentrische Doppelblindstudie wurde bei Patienten mit Spastik im Handgelenk und den Fingern als sekundäre Folge eines Schlaganfalls durchgeführt. Die Dauer der Studie betrug 12 Wochen. Primärer Endpunkt war die Spastik des Flexors im Handgelenk in Woche 6, gemessen auf der Ashworth-Skala (AS). Weitere Variablen waren die ärztlich beurteilte Bewertungsskala, die Gesamtbeurteilung durch die Betreuungsperson des Patienten, die Skala zur Beurteilung von Behinderungen und die Grundsätzlichen Therapeutischen Interventionsziele.
126 Patienten wurden randomisiert und erhielten BOTOX (N = 64) oder Placebo (N = 62). Die injizierte Dosis von BOTOX lag zwischen 200 bis 240 Einheiten, verteilt auf die betroffenen Muskeln im Handgelenk und in den Fingern. Es zeigte sich, dass BOTOX die AS-Werte für Handgelenk und Finger zu allen Kontrollzeitpunkten nach der Injektion verbessert hatte. In Woche 6 zeigten 88% der Patienten eine Verbesserung auf der AS unter BOTOX, gegenüber 34% in der Placebo-Gruppe.
Alle anderen Variablen zeigten in Woche 6 ebenfalls einen signifikanten Vorteil gegenüber Placebo.
Fokale Spastiken der oberen Extremitäten bei Jugendlichen und Kindern ab 2 Jahren
Die Wirksamkeit und Sicherheit von BOTOX zur Behandlung der Spastiken der oberen Extremitäten wurde bei pädiatrischen Patienten ab 2 Jahren in einer randomisierten, multizentrischen, doppelblinden, Placebo-kontrollierten Studie untersucht. 234 pädiatrische Patienten (77 BOTOX 6 Einheiten/kg KG, 78 BOTOX 3 Einheiten/kg KG, 79 Placebo) mit Spastik aufgrund eines zerebralen «Palsy oder Hirnschlags (modifizierte AS Ellbogen oder Handgelenk Score von mindestens 2) wurden in die Studie aufgenommen. Die Gesamtdosis von 3 Einheiten/kg KG (maximal 100 Einheiten) oder 6 Einheiten/kg KG (maximal 200 Einheiten) oder Placebo wurde intramuskulär injiziert, verteilt auf Ellbogen oder Handgelenk und Fingermuskeln. Die Patienten wurden während 12 Wochen nach der Injektion beobachtet. Der Gebrauch einer elektromyographischen Kontrolle (EMG), Nervenstimulation oder Ultraschalltechnik wurde verlangt, um die korrekte Muskellokalisation für die Injektion zu unterstützen.
Der primäre Endpunkt war die durchschnittliche Änderung gegenüber dem Ausgangswert in der modifizierten AS (MAS) Score der Hauptmuskelgruppen (Ellbogen oder Handgelenk) während Woche 4 und 6. Der wichtigste sekundäre Endpunkt war der Mittelwert des «Clinical Global Impression of Overall Change by Physician (CGI)» während Woche 4 und 6. Die «goal attainment scale» (GAS) beurteilt durch den Arzt für aktive und passive Ziele wurde evaluiert als sekundärer Endpunkt während Woche 8 und 12.
Geeignete Patienten konnten an einer offenen Verlängerungsstudie teilnehmen, in welcher sie mehr als 5 Behandlungen mit Dosen bis zu 10 Einheiten/kg (maximal 340 Einheiten) erhielten, wenn mehr als eine Extremität behandelt wurde.
Eine signifikante Verbesserung im Vergleich zu Placebo für den MAS Knöchelscore gegenüber dem Ausgangswert wurde zu jedem Zeitpunkt für mit BOTOX behandelte Patienten in beiden Behandlungsgruppen beobachtet. Ähnliche Ergebnisse wurden in beiden BOTOX-Dosisgruppen mit einem statistisch signifikant höheren Anteil von MAS-Respondern (definiert als mindestens 1-Punkt-Verbesserung) im Vergleich zu Placebo zu den meisten Zeitpunkten beobachtet.
Tabelle 2: Resultate der primären und wichtigen sekundären Endpunkte
|
|
BOTOX
3 Einheiten/kg
(N=78)
|
BOTOX
6 Einheiten/kg
(N=77)
|
Placebo
(N=79)
| |
Mittlere Veränderung gegenüber dem Ausgangswert in der Hauptmuskelgruppe (Ellenbogen- oder Handgelenk auf der modifizierten Ashworth-Skala**
| |
Woche 4 und 6 Durchschnitt
|
-1,92*
|
-1,87*
|
-1,21
| |
Mittlerer Clinical Global Impression Score***
| |
Woche 4 und 6 Durchschnitt
|
1,88
|
1,87
|
1,66
|
* Signifikant unterschiedlich zu Placebo (p< 0.05)
** Der MAS ist eine 6-Punkte Skala (0 [keine Erhöhung des Muskeltonus], 1, 1+, 2, 3, and 4 [in Beugung oder Streckung starre Extremität]), die die Kraft misst, die erforderlich ist, um eine Extremität um ein Gelenk herum zu bewegen, wobei eine Verringerung der Punktzahl eine Verbesserung der Spastizität darstellt.
*** Der CGI bewertete das Ansprechen auf die Behandlung im Hinblick auf die Lebensqualität des Patienten anhand einer 9-Punkte-Skala (-4=sehr deutliche Verschlechterung bis +4=sehr deutliche Verbesserung).
Obwohl die CGI-Werte für BOTOX numerisch über Placebo lagen, war der Unterschied statistisch nicht signifikant.
Statistisch signifikante Verbesserungen im Vergleich zu Placebo wurden beim Anteil der CGI-Responder (definiert als mindestens 1-Punkt-Verbesserung) für BOTOX 3 Einheiten/kg (Woche 4 bis 12) und BOTOX 6 Einheiten/kg (Woche 6) sowie bei der funktionellen Zielerreichung (GAS durch den Arzt) für passive Ziele nur bei den BOTOX 6 Einheiten/kg (Woche 12) beobachtet.
Abbildung 6: MAS-Score (pädiatrische Spastizität obere Extremität) - Mittlere Veränderung gegenüber dem Ausgangswert

Fokale Spastik der unteren Extremitäten bei Erwachsenen
Die Wirksamkeit und Sicherheit von BOTOX zur Behandlung von Spastik der unteren Extremitäten wurde in einer randomisierten, multizentrischen, doppelblinden, Placebo-kontrollierten Studie untersucht, die 468 Patienten nach Schlaganfall (233 BOTOX und 235 Placebo) mit Spastik des Fussgelenkes (MAS Fussgelenk-Wert von mindestens 3) einschloss, deren Schlaganfall mindestens 3 Monate zurücklag. BOTOX 300 bis 400 Einheiten oder Placebo wurden intramuskulär in die für die Studie vorgeschriebenen Muskeln M. gastrocnemius, M. soleus und M. tibialis posterior sowie in optionale Muskeln, einschließlich M. flexor hallucis longus, M. flexor digitorum longus, M. flexor digitorum brevis, M. extensor hallucis und M. rectus femoris, injiziert. Der Gebrauch einer elektromyographischen Kontrolle (EMG), Nervenstimulation oder Ultraschalltechnik wurde verlangt, um die korrekte Muskellokalisation für die Injektion zu unterstützen. Die Patienten wurden während 12 Wochen nachbeobachtet.
Der primäre Endpunkt war die durchschnittliche Veränderung des MAS-Fussgelenk-Wertes ab Baseline in den Wochen 4 und 6. Ein wichtiger sekundärer Endpunkt war der durchschnittliche CGI-Wert (Bewertung des klinischen Gesamteindrucks durch den Arzt) in den Wochen 4 und 6. Statistisch und klinisch signifikante Unterschiede zwischen den Gruppen für BOTOX gegenüber Placebo wurden für den primären Wirksamkeitsparameter MAS und den sekundären Schlüsselparameter CGI gezeigt und sind in der folgenden Tabelle dargestellt. Für den primären Endpunkt des durchschnittlichen MAS-Fussgelenk-Wertes in den Wochen 4 und 6 wurde bei Patienten im Alter von 65 Jahren und älter in der BOTOX-Gruppe im Vergleich zu Placebo keine Verbesserung gegenüber dem Ausgangswert beobachtet, wahrscheinlich aufgrund der geringen Patientenzahlen.
Tabelle 3: Primäre und wichtigste sekundäre Wirksamkeitspunkte
|
|
BOTOX
300 bis 400 Einheiten (n=233)
|
Placebo
(n=235)
| |
Mittlere Veränderung bei den Plantarflexoren des Fussgelenkes ab Baseline auf der MAS-Skala
| |
Durchschnitt in Woche 4 und 6
|
-0,8*
|
-0,6
| |
Mittlerer Wert des klinischen Gesamteindruckes des Prüfarztes
| |
Durchschnitt in Woche 4 und 6
|
0,9*
|
0,7
| |
Mittlere Veränderung bei den Zehenflexoren auf der MAS-Skala
| |
FHL** Durchschnitt in Woche 4 und 6
|
-1,0*
|
-0,6
| |
FDL + FDB** Durchschnitt in Woche 4 und 6
|
-0,9
|
- 0,8
|
*Signifikant unterschiedlich zu Placebo (p<0,05)
FHL = flexor hallucis longus; FDL = flexor digitorum longus; FDB = flexor digitorum brevis
Abbildung 7: MAS-Score (Spastizität untere Extremität Erwachsene) - Mittlere Veränderung gegenüber dem Ausgangswert
Abbildung 8: CGI des Arztes (Spastizität untere Extremitäten Erwachsene) - Durchschnittswerte pro Besuch
Fokale Spastik der unteren Extremitäten bei Jugendlichen und Kindern ab 2 Jahren
Die Wirksamkeit und Sicherheit von BOTOX zur Behandlung der Spastik der unteren Extremitäten bei pädiatrischen Patienten ab 2 Jahren wurde in einer randomisierten, multizentrischen, doppelblinden, Placebo-kontrollierten Studie untersucht. Die Studie umfasste 384 pädiatrische Patienten (128 BOTOX 8 Einheiten/kg, 126 BOTOX 4 Einheiten/kg und 128 Placebo) mit Spastik der unteren Extremitäten aufgrund einer zerebralen Lähmung (MAS Knöchelwert mind. 2). Eine Gesamtdosis von 4 Einheiten/kg (maximal 150 Einheiten) oder 8 Einheiten/kg (maximal 300 Einheiten) oder Placebo wurde intramuskulär injiziert, aufgeteilt zwischen Gastrocnemius, Soleus und Tibialis posterior. Der Gebrauch einer elektromyographischen Kontrolle (EMG), Nervenstimulation oder Ultraschalltechnik wurde verlangt, um die korrekte Muskellokalisation für die Injektion zu unterstützen. Die Patienten wurden 12 Wochen lang beobachtet. Der primäre Endpunkt war die durchschnittliche Änderung gegenüber dem Ausgangswert im MAS Knöchelscore in Woche 4 und 6, und der wichtigste sekundäre Endpunkt war der Mittelwert des CGI in Woche 4 und 6. Sekundäre Endpunkte in Wochen 8 und 12 beinhalteten GAS durch den Arzt und dem Edinburgh Visual Gait (EVG) (in einer Untergruppe von Patienten).
Geeignete Patienten konnten an einer offenen Verlängerungsstudie teilnehmen, in der sie bis zu fünf Behandlungen mit Dosen bis zu 10 Einheiten/kg (maximal 340 Einheiten) erhielten bei Behandlung von mehr als einer Extremität.
Eine statistisch signifikante Verbesserung im primären Endpunkt gegenüber Placebo wurde bei den mit 4 und mit 8 Einheiten/kg BOTOX behandelten Patienten bis Woche 12 beobachtet (siehe Abbildung 9). Ähnliche Ergebnisse wurden in beiden BOTOX-Dosisgruppen mit einem höheren Anteil von MAS-Respondern (definiert als mindestens 1-Punkt-Verbesserung) im Vergleich zu Placebo zu allen Zeitpunkten beobachtet.
Tabelle 4: Primäre und wichtige sekundäre Wirksamkeitsendpunkt-Ergebnisse
|
|
BOTOX 4 Units/kg
(n = 125)
|
BOTOX 8 Units/kg
(n=127)
|
Placebo
(n=129)
| |
Mittlere Änderung im MAS-Ausgangswert der Plantarflexoren
| |
Durchschnitt Woche 4 und 6
|
-1,01*
|
-1,06*
|
-0,80
| |
Mittlerer CGI Wert
| |
Durchschnitt Woche 4 und 6
|
1,49
|
1,65*
|
1,36
|
* Signifikanter Unterschied zu Placebo (p< 0,05)
Statistisch signifikante Verbesserung im Vergleich zu Placebo wurde mit 4 und 8 Einheiten/kg BOTOX für den primären Endpunkt mittlerer MAS Knöchelscore in Wochen 4 und 6 als Veränderung gegenüber dem Ausgangswert und mit 8 Einheiten/kg BOTOX für den wichtigsten sekundären Endpunkt mittlerer CGI in den Wochen 4 und 6 nachgewiesen. Eine statistisch signifikante Verbesserung im Vergleich zu Placebo wurde für CGI vom Arzt im Verlauf von durchschnittlich 4 und 6 Wochen für BOTOX 8 Einheiten/kg beobachtet. Statistisch signifikante Verbesserungen der funktionellen aktiven und passiven Zielerreichung (GAS) wurden in Woche 8 und 12 für BOTOX 8 Einheiten/kg und in der Woche 8 aber nicht in Woche 12 für BOTOX 4 Einheiten/kg beobachtet. Die Gangverbesserung war dosisabhängig, mit statistisch signifikanten Verbesserungen des EVG in Woche 8 für BOTOX 8 Einheiten/kg.
Abbildung 9: MAS-Score (pädiatrische Spastizität untere Extremität) - Mittlere Veränderung gegenüber dem Ausgangswert

Bei pädiatrischen Patienten mit Spastik der unteren Extremitäten, deren Proben aus einer Phase-3-Studie und der Open-Label-Extension-Studie analysiert wurden, entwickelten 2 von 264 Patienten (0,8%) neutralisierende Antikörper, die mit BOTOX für bis zu 5 Behandlungszyklen behandelt wurden. Beide Patienten hatten anschliessend weiterhin einen klinischen Nutzen von der BOTOX-Behandlung.
Prophylaxe von Kopfschmerzen bei erwachsenen Patienten mit chronischer Migräne
BOTOX wurde in zwei 56wöchigen multinationalen, multizentrischen Studien untersucht, die Folgendes umfassten: 24wöchige, doppelblinde Phase mit 2 Injektionszyklen zum Vergleich von BOTOX mit Placebo, gefolgt von einer 32wöchigen offenen Phase mit 3 Injektionszyklen mit BOTOX. Insgesamt 1'384 Erwachsene mit chronischer Migräne, die entweder noch nie eine begleitende Kopfschmerzprophylaxe erhalten hatten oder diese bei Studienbeginn für 28 Tage nicht angewendet hatten, mit Kopfschmerzen an ≥15 Tagen, davon 50 % Migräne/wahrscheinliche Migräne, und ≥4 Kopfschmerzepisoden, wurden in zwei klinischen Phase-3-Studien untersucht. Diese Patienten wurden randomisiert und erhielten alle 12 Wochen Injektionen mit Placebo oder 155–195 Einheiten BOTOX für die doppelblinde Phase mit 2 Injektionszyklen. Sie konnten 3 weitere offene BOTOX-Zyklen erhalten, bis zu maximal 5 Injektionszyklen.
Die Akutbehandlung von Kopfschmerzen war den Patienten erlaubt (bei 65,5 % lag eine übermässige Akutbehandlung während der Studienbeginn-Phase vor).
Die Behandlung mit BOTOX zeigte im Vergleich zu Placebo statistisch signifikante (p < 0,001) und klinisch bedeutsame Verbesserungen gegenüber dem Ausgangswert in Bezug auf die Häufigkeit von Kopfschmerztagen, die Häufigkeit von Migräne/ wahrscheinlichen Migränetagen, die Häufigkeit von mittelschweren/schweren Kopfschmerztagen und die kumulativen Gesamtstunden an Kopfschmerztagen.
Die Ergebnisse des Headache Impact Test (HIT-6) und des Migraine-Specific Quality of Life Questionnaire (MSQ) wiesen auf eine anhaltende Wirkungsdauer von BOTOX sowie eine Verbesserung der Funktionsfähigkeit, Vitalität, psychischen Belastung und Lebensqualität insgesamt hin.
Gepoolte Ergebnisse der wichtigsten Wirksamkeitsendpunkte in den beiden Zulassungsstudien in Woche 24 (primärer Zeitpunkt)
|
Wirksamkeit über 28 Tagen
|
BOTOX
(N = 688)
|
Placebo
(N = 696)
|
p-Wert
| |
Veränderung der Anzahl an Tagen mit Kopfschmerzen gegenüber dem Ausgangswert
|
-8,4
|
-6,6
|
< 0,001
| |
Veränderung der Anzahl an Tagen mit Migräne/wahrscheinlicher Migräne gegenüber dem Ausgangswert
|
-8,2
|
-6,2
|
< 0,001
| |
Veränderung der Anzahl an Tagen mit mittelstarken/starken Kopfschmerzen gegenüber dem Ausgangswert
|
-7,7
|
-5,8
|
< 0,001
| |
Veränderung der Gesamtzahl an Kopfschmerzstunden an den Tagen mit Kopfschmerzen gegenüber dem Ausgangswert
|
-119,7
|
-80,5
|
< 0,001
| |
Veränderung der Anzahl an Kopfschmerzepisoden gegenüber dem Ausgangswert
|
-5,2
|
-4,9
|
0,009
| |
Prozentsatz der Patienten mit 50 % weniger Kopfschmerztagen
|
47%
|
35%
|
< 0,001
| |
Anteil der Patienten mit hohen Scores bei HIT-6-Kategorien
|
67,6 %
|
78,2 %
|
< 0,001
| |
Veränderung des HIT-6-Gesamtscores gegenüber dem Ausgangswert
|
-4,8
|
-2,4
|
< 0,001
|
Obwohl die Studien nicht darauf ausgerichtet waren, Unterschiede bei Untergruppen aufzuweisen, schien der Behandlungseffekt in der Untergruppe der männlichen Studienteilnehmer (n = 188) und der Studienteilnehmer nicht kaukasischer Herkunft (n = 137) geringer zu sein als in der gesamten Studienpopulation.
BLASENFUNKTIONSSTÖRUNGEN:
Überaktive Blase bei Erwachsenen
Zwei doppel-blinde, Placebo-kontrollierte, randomisierte, multizentrische 24-wöchige klinische Phase-3-Studien wurden bei Patienten mit überaktiver Blase mit den Symptomen Harninkontinenz, Harndrang und häufige Miktion durchgeführt. Insgesamt wurden1'105 Patienten, deren Symptome durch Anticholinergika nicht adäquat kontrolliert werden konnten (unzureichendes Ansprechen oder inakzeptable Nebenwirkungen), entweder auf 100 Einheiten BOTOX (n = 557) oder Placebo (n = 548) randomisiert.
In beiden Studien wurden für BOTOX (100 Einheiten) signifikante Verbesserungen im Vergleich zu Placebo bei der Änderung der täglichen Häufigkeit von Harninkontinenzepisoden verglichen mit dem Ausgangswert, einschliesslich des Anteils trockener Patienten, zum primären Zeitpunkt in Woche 12 festgestellt. Gemessen anhand der Skala für den Behandlungsnutzen war der Anteil der Patienten, die über ein positives Behandlungsansprechen berichteten (ihr Zustand hatte sich «stark verbessert» oder «verbessert»), in beiden Studien in der BOTOX-Gruppe signifikant grösser als in der Placebo-Gruppe. Signifikante Verbesserungen im Vergleich zu Placebo wurden auch bei der täglichen Miktionshäufigkeit und der Häufigkeit der Episoden von Harndrang und Nykturie beobachtet. Die pro Miktion ausgeschiedene Urinmenge war ebenfalls signifikant grösser. Signifikante Verbesserungen wurden ab Woche 2 bei allen Symptomen der überaktiven Blase festgestellt.
Die BOTOX-Behandlung war gegenüber Placebo mit signifikanten Verbesserungen der gesundheitsbezogenen Lebensqualität verbunden, gemessen anhand des I-QOL-Fragebogens (Incontinence Quality of Life) (mit den Faktoren Vermeidungsverhalten und Einschränkungen, psychosoziale Folgen und soziale Peinlichkeit) und des KHQ-Fragebogens (King's Health Questionnaire) (mit den Faktoren Folgen der Inkontinenz, Einschränkungen bei Alltagsaktivitäten, soziale Einschränkungen, körperliche Einschränkungen, persönliche Beziehungen, Gefühlszustand, Schlaf/Energie und Schweregrad/Gegenmassnahmen).
Zwischen Patienten im Alter von 65 Jahren und älter und Patienten unter 65 Jahren wurde insgesamt nach der BOTOX-Behandlung kein Unterschied in der Wirkung festgestellt.
Die Ergebnisse der gepoolten zentralen Studien sind unten dargestellt:
Tabelle 5: Primäre und sekundäre Wirksamkeitsendpunkte zu Studienbeginn und Veränderung gegenüber Studienbeginn in den gepoolten zentralen Studien
|
|
BOTOX
100 Einheiten
(n = 557)
|
Placebo
(n = 548)
|
p-Wert
| |
Tägliche Häufigkeit von Harninkontinenzepisoden*
| |
Mittelwert zu Studienbeginn
|
5,49
|
5,39
|
| |
Mittlere Veränderung in Woche 2
|
-2,85
|
-1,21
|
<0,001
| |
Mittlere Veränderung in Woche 6
|
-3,11
|
-1,22
|
<0,001
| |
Mittlere Veränderung in Woche 12
|
-2,80
|
-0,95
|
<0,001
| |
Anteil der Patienten mit positivem Behandlungsansprechen auf der Skala des Behandlungsnutzens (%)
| |
Woche 2
|
64,4
|
34,7
|
<0,001
| |
Woche 6
|
68,1
|
32,8
|
<0,001
| |
Woche 12
|
61,8
|
28,0
|
<0,001
| |
Tägliche Häufigkeit von Miktionsepisoden
| |
Mittelwert zu Studienbeginn
|
11,99
|
11,48
|
| |
Mittlere Veränderung in Woche 2
|
-1,53
|
-0,78
|
<0,001
| |
Mittlere Veränderung in Woche 6
|
-2,18
|
-0,97
|
<0,001
| |
Mittlere Veränderung in Woche 12
|
-2,35
|
-0,87
|
<0,001
| |
Tägliche Häufigkeit der Harndrangepisoden
| |
Mittelwert zu Studienbeginn
|
8,82
|
8,31
|
| |
Mittlere Veränderung in Woche 2
|
-2,89
|
-1,35
|
<0,001
| |
Mittlere Veränderung in Woche 6
|
-3,56
|
-1,40
|
<0,001
| |
Mittlere Veränderung in Woche 12b
|
-3,30
|
-1,23
|
<0,001
| |
I-QOL-Gesamtscore
| |
Mittelwert zu Studienbeginn
|
34,1
|
34,7
|
| |
Mittlere Veränderung nach Woche 12bc
|
+22,5
|
+6,6
|
<0,001
| |
KHQ-Fragebogen: Rolleneinschränkungen
| |
Mittelwert zu Studienbeginn
|
65,4
|
61,2
|
| |
Mittlere Veränderung nach Woche 12bc
|
-25,4
|
-3,7
|
<0,001
| |
KHQ-Fragebogen: Soziale Einschränkungen
| |
Mittelwert zu Studienbeginn
|
44,8
|
42,4
|
<0,001
| |
Mittlere Veränderung nach Woche 12bc
|
-16,8
|
-2,5
|
<0,001
|
* Der prozentuale Anteil der Patienten, die in Woche 12 trocken (frei von Inkontinenz) waren, betrug 27,1% in der BOTOX-Gruppe und 8,4% in der Placebo-Gruppe. Die prozentualen Anteile der Patienten, die eine Verringerung der Harninkontinenzepisoden im Vergleich zum Ausgangswert von mindestens 75% bzw. 50% erzielten, waren 46,0% bzw. 60,5% in der BOTOX-Gruppe und 17,7% bzw. 31,0% in der Placebo-Gruppe.
a Koprimäre Endpunkte
b Sekundäre Endpunkte
c Die vorab definierte bedeutsame Mindeständerung im Vergleich zur Baseline war +10 Punkte für I-QOL und 5 Punkte für KHQ.
Die mediane Dauer des Ansprechens nach der BOTOX-Behandlung, basierend auf dem Verlangen der Patienten nach einer erneuten Behandlung, betrug 166 Tage (~24 Wochen).
Bei Patienten, die auch noch an der offenen Verlängerungsstudie teilnahmen und nur mit BOTOX 100 Einheiten behandelt wurden (N = 438), betrug die mediane Dauer des Ansprechens 212 Tage (~30 Wochen).
In den beiden pivotalen Studien nahm nur eine begrenzte Anzahl männlicher Patienten (ca. 12.2% der gesamten Studienpopulation) teil. Eine statistisch signifikante Wirksamkeit konnte in dieser kleinen Subgruppe nicht gezeigt werden.
Insgesamt wurden 834 Patienten in einer Langzeit-Verlängerungsstudie untersucht. Bei erneuten Behandlungen zeigten die Patienten für alle Wirksamkeitsendpunkte ein gleichbleibendes Ansprechen.
In den zentralen Studien entwickelte keiner der 615 Patienten, bei denen Proben analysiert wurden, neutralisierende Antikörper. Bei Patienten, bei denen Proben aus der pivotalen Phase-3-Studie und den offenen Verlängerungsstudien analysiert wurden, entwickelten sich in den folgenden Fällen neutralisierende Antikörper: bei 0 von 954 Patienten (0,0%) während der Behandlung mit 100 Einheiten BOTOX und bei 3 von 260 Patienten (1,2%) nach anschliessender Behandlung mit mindestens einer Dosis von 150 Einheiten. Einer dieser drei Patienten zeigte einen anhaltenden klinischen Nutzen.
Überaktive Blase bei Kindern und Jugendlichen
Aus einer doppelblinden, randomisierten, multizentrischen Parallelgruppenstudie (191622 137) mit Patienten im Alter von 12 bis 17 Jahren mit überaktiver Blase und Symptomen einer Harninkontinenz liegen begrenzte Daten zur Wirksamkeit vor. Eingeschlossen wurden Patienten, die auf mindestens ein anticholinerges Arzneimittel unzureichend angesprochen oder dieses nicht vertragen hatten. Aufgrund von Rekrutierungsproblemen wurden insgesamt 55 von den geplanten 108 Patienten eingeschlossen, so dass die Fallzahl nicht ausreichte, um eine Aussage über die Wirksamkeit in dieser Population treffen zu können. Diese Patienten wurden randomisiert und erhielten 25 Einheiten, 50 Einheiten oder 100 Einheiten, wobei 6 Einheiten/kg Körpergewicht nicht überschritten werden durften; BOTOX 25 Einheiten N = 18, BOTOX 50 Einheiten N = 17 und BOTOX 100 Einheiten N = 20. Vor der Anwendung erhielten die Patienten eine Anästhesie gemäss der lokalen Praxis. Alle Patienten erhielten eine Allgemeinanästhesie oder eine leichte Sedierung.Im primären Endpunkt in Woche 12 «Tägliche Häufigkeit von Tages-Harninkontinenzepisoden» sowie in weiteren Endpunkten wie Häufigkeit von Tages-Miktionsepisoden, Häufigkeit von Tages-Harndrangepisoden oder positivem Behandlungsansprechen konnten teils numerische, aber keine statistisch signifikanten Differenzen zwischen den Behandlungsgruppen festgestellt werden. Von den 55 pädiatrischen Patienten, die einen negativen Ausgangswert für bindende Antikörper oder neutralisierende Antikörper und nach Studienbeginn mindestens einen auswertbaren Wert nach dem Ausgangswert aus einer randomisierten doppelblinden Studie hatten, entwickelte kein Patient nach der Behandlung mit 25–100 Einheiten BOTOX neutralisierende Antikörper.
Harninkontinenz bei Erwachsenen infolge neurogener Detrusorhyperaktivität
Zwei doppel-blinde, Placebo-kontrollierte, randomisierte, multizentrische klinische Studien der Phase 3 wurden bei Patienten mit Harninkontinenz aufgrund neurogener Überaktivität des Detrusor-Muskels und entweder mit spontaner Harnentleerung oder Katheterisierung durchgeführt. Insgesamt wurden 691 Patienten mit Rückenmarksverletzung oder Multipler Sklerose aufgenommen, die mit mindestens einem anticholinergen Wirkstoff nicht angemessen behandelt wurden. Diese Patienten wurden entweder auf 200 Einheiten BOTOX (n = 227), 300 Einheiten BOTOX (n = 223) oder Placebo (n = 241) randomisiert.
Im Vergleich zu Placebo wurden in beiden Phase-3-Studien signifikante Verbesserungen des primären Wirksamkeitsparameters, Veränderung der wöchentlichen Häufigkeit von Inkontinenz-Episoden gegenüber Studienbeginn, beobachtet, wobei BOTOX (200 Einheiten und 300 Einheiten) zum primären Wirksamkeitszeitpunkt in Woche 6 einschliesslich des Anteils bei Patienten, bei denen keine Harninkontinenz mehr auftrat, am besten abschnitt. Es wurden signifikante Verbesserungen der urodynamischen Parameter einschliesslich eines Anstiegs der maximalen zystometrischen Kapazität und einem verringerten Maximaldruck auf den Detrusor-Muskel bei der ersten unbeabsichtigten Detrusor-Kontraktion beobachtet. Ebenso wurden im Vergleich zu Placebo signifikante Verbesserungen in den vom Patienten beurteilten Inkontinenz-spezifischen, gesundheitsbezogenen Wert zur Lebensqualität, die mit dem I-QOL–Fragebogen erfasst wurden, beobachtet (einschliesslich einschränkender Vermeidungstaktiken, psychosozialer Auswirkungen und Schamgefühle). Ein zusätzlicher Nutzen von BOTOX 300 Einheiten gegenüber 200 Einheiten konnte nicht gezeigt werden.
Die Ergebnisse der gepoolten zentralen Studien werden nachfolgend vorgestellt:
Tabelle 6: Primäre und sekundäre Endpunkte zu Studienbeginn und Veränderung gegenüber Studienbeginn in den gepoolten zentralen Studien
|
|
BOTOX
200 Einheiten
(n = 227)
|
Placebo
(n = 241)
|
p-Wert
| |
Wöchentliche Häufigkeit der Harninkontinenz*
| |
Mittelwert zu Studienbeginn
|
32,4
|
31,5
|
| |
Mittlere Veränderung in Woche 2
|
-17,7
|
-9,0
|
<0,001
| |
Mittlere Veränderung in Woche 6a
|
-21,3
|
-10,5
|
<0,001
| |
Mittlere Veränderung in Woche 12
|
-20,6
|
-9,9
|
<0,001
| |
Maximale zystometrische Kapazität (ml)
| |
Mittelwert zu Studienbeginn
|
250,2
|
253,5
|
| |
Mittlere Veränderung in Woche 6b
|
+153,6
|
+11,9
|
<0,001
| |
Maximaler Detrusor-Druck während der 1. unbeabsichtigten Detrusor-Kontraktion (cmH2O):
| |
Mittelwert zu Studienbeginn
|
51,5
|
47,3
|
| |
Mittlere Veränderung in Woche 6b
|
-32,4
|
+1,1
|
<0,001
| |
Gesamtscore Inkontinenz-spezifische Lebensqualitätc,d
| |
Mittelwert zu Studienbeginn
|
35,37
|
35,32
|
| |
Mittlere Veränderung in Woche 6b
|
+25,89
|
+11,15
|
<0,001
| |
Mittlere Veränderung in Woche 12
|
+28,89
|
+8,86
|
<0,001
|
* Der Prozentsatz der Patienten, bei denen eine Inkontinenz nicht mehr auftrat, lag während der gesamten Woche 6 bei 37% in der mit 200 BOTOX Einheiten behandelten Gruppe und bei 9% in der Placebo-Gruppe. Der Anteil, der eine um mindestens 75% reduzierte Häufigkeit von Inkontinenz-Episoden gegenüber Studienbeginn erreichte, lag bei 63% bzw. 24%. Der Anteil, der eine um mindestens 50% reduzierte Häufigkeit gegenüber Studienbeginn erreichte, lag bei 76% bzw. 39%.
a Primärer Endpunkt
b Sekundärer Endpunkt
c Die Skala des I-QOL-Gesamtscores reicht von 0 (grösstes Problem) bis 100 (überhaupt kein Problem).
d In den zentralen Studien lag die vordefinierte minimal-bedeutsame Veränderung (MID – Minimally Important Difference) des I-QOL-Gesamtscores bei 8 Punkten, ausgehend von einem geschätzten MID-Wert von 4-11 Punkten, der von Patienten mit neurogener Überaktivität des Detrusor-Muskels angegeben wurde.
In den beiden zentralen Studien wurde die mediale Dauer des Ansprechens auf die Therapie, ausgehend von der Bitte der Patienten nach erneuter Behandlung, ermittelt und lag bei 256-295 Tagen (36-42 Wochen) in der mit 200 Einheiten behandelten Gruppe im Vergleich zu 92 Tagen (13 Wochen) bei der Gruppe unter Placebo. Bei Patienten, die auch noch an der offenen Verlängerungsstudie teilnahmen und nur mit BOTOX 200 Einheiten behandelt wurden (N = 174), betrug die mediane Dauer des Ansprechens 253 Tage (~36 Wochen).
Bei erneuter Behandlung wiesen die Patienten ein gleichbleibendes Ansprechen auf die Therapie hinsichtlich aller Endpunkte zur Wirksamkeit auf.
In den zentralen Studien entwickelte keiner der 475 Patienten mit neurogener Überaktivität des Detrusor-Muskels, bei denen Proben untersucht wurden, neutralisierende Antikörper.
Bei Patienten, bei denen Proben aus dem Programm zur Entwicklung neuer Medikamente (einschliesslich offene Verlängerungsstudie) analysiert wurden, entwickelten sich in den folgenden Fällen neutralisierende Antikörper: bei 3 von 300 Patienten (1,0%), die nur mit BOTOX 200 Einheiten behandelt wurden, und bei 5 von 258 Patienten (1,9%) nach der Behandlung mit mindestens einer Dosis von 300 Einheiten. Vier dieser acht Patienten zeigten einen anhaltenden klinischen Nutzen.
Nach Marktzulassung wurde eine Placebo-kontrollierte, doppel-blinde Studie bei Patienten mit multipler Sklerose (MS) durchgeführt, die aufgrund einer neurogenen Detrusorhyperaktivität eine Harninkontinenz zeigten und die mit mindestens einem anticholinergen Wirkstoff nicht angemessen behandelt wurden und zu Studienbeginn nicht katheterisiert waren. Diese Patienten wurden entweder auf BOTOX 100 Einheiten (n = 66) oder Placebo (n = 78) randomisiert.
Signifikante Verbesserungen des primären Wirksamkeitsparameters, Veränderung der täglichen Häufigkeit von Inkontinenz-Episoden, gegenüber Studienbeginn wurden im Vergleich zu Placebo für BOTOX (100 Einheiten) zum primären Wirksamkeitszeitpunkt in Woche 6 beobachtet. Dazu zählte auch der Anteil der Patienten, bei denen eine Harninkontinenz nicht mehr auftrat. Es wurden signifikante Verbesserungen der urodynamischen Parameter und des vom Patienten beurteilten I-QOL–Fragebogens beobachtet. Hierzu gehören auch einschränkende Vermeidungstaktiken, psychosoziale Auswirkungen und Schamgefühle im Beisein anderer.
Die Ergebnisse der Studie nach Markteinführung werden nachfolgend vorgestellt:
Tabelle 7: Primäre und sekundäre Endpunkte zu Studienbeginn und Veränderung gegenüber Studienbeginn in der Studie nach Markteinführung mit BOTOX 100 Einheiten und bei zu Studienbeginn nicht-katheterisierten MS-Patienten
|
|
BOTOX
100 Einheiten
(n=66)
|
Placebo
(n=78)
|
p-Wert
| |
Tägliche Häufigkeit von Harninkontinenzepisoden*
| |
Mittelwert zu Studienbeginn
|
4,2
|
4,3
|
| |
Mittlere Veränderung in Woche 2
|
-2,9
|
-1,2
|
p<0,001
| |
Mittlere Veränderung in Woche 6a
|
-3,3
|
-1,1
|
p<0,001
| |
Mittlere Veränderung in Woche 12
|
-2,8
|
-1,1
|
p<0,001
| |
Maximale zystometrische Kapazität (ml)
| |
Mittelwert zu Studienbeginn
|
246,4
|
245,7
|
| |
Mittlere Veränderung in Woche 6b
|
+127,2
|
-1,8
|
p<0,001
| |
Maximaler Detrusor-Druck während der 1. unbeabsichtigten Detrusor-Kontraktion (cmH2O):
| |
Mittelwert zu Studienbeginn
|
35,9
|
36,1
|
| |
Mittlere Veränderung in Woche 6b
|
-19,6
|
+3,7
|
p=0,007
| |
Gesamtscore Inkontinenz-spezifische Lebensqualitätc, d
| |
Mittelwert zu Studienbeginn
|
32,4
|
34,2
|
| |
Mittlere Veränderung in Woche 6b
|
+40,4
|
+9,9
|
p<0,001
| |
Mittlere Veränderung in Woche 12
|
+38,8
|
+7,6
|
p<0,001
|
* Der Prozentsatz der Patienten, bei denen eine Inkontinenz nicht mehr auftrat, lag während der gesamten Woche 6 bei 53,0% in der mit 100 BOTOX Einheiten behandelten Gruppe und bei 10,3% in der Placebo-Gruppe.
a Primärer Endpunkt
b Sekundäre Endpunkte
c Die Skala des I-QOL-Gesamtscores reicht von 0 (grösstes Problem) bis 100 (überhaupt kein Problem).
d Die vordefinierte minimal-bedeutsame Veränderung (MID – Minimally Important Difference) des I-QOL-Gesamtscores lag bei 11 Punkten, ausgehend von einem geschätzten MID-Wert von 4–11 Punkten, der von Patienten mit neurogener Überaktivität des Detrusor-Muskels angegeben wurde.
Die mediane Dauer des Ansprechens in dieser Studie, basierend auf dem Verlangen der Patienten nach einer erneuten Behandlung, betrug 362 Tage (~52 Wochen) in der mit BOTOX 100 Einheiten behandelten Gruppe im Vergleich zu 88 Tagen (~13 Wochen) bei der Gruppe unter Placebo.
Neurogene Detrusorhyperaktivität bei pädiatrischen Patienten ab 5 Jahren
Eine randomisierte, multizentrische klinische Doppelblindstudie mit parallelen Dosierungsgruppen, aber ohne Placebo-Arm [aufgrund der untersuchten pädiatrischen Population] (191622-120) wurde bei Patienten zwischen 5 und 17 Jahren mit Harninkontinenz infolge einer Detrusorhyperaktivität in Verbindung mit einer neurologischen Erkrankung durchgeführt, die mit sauberer intermittierender Katheterisierung behandelt wurden und die auf eine Therapie mit Anticholinergika unzureichend ansprachen oder diese nicht tolerierten. Insgesamt wurden 113 Patienten (darunter 99 mit Neuralrohrdefekten wie Spina bifida oder Tethered Cord-Syndrom, 13 mit Rückenmarksverletzungen [T1 oder darunter] und 1 mit transverser Myelitis) mit BOTOX behandelt. Diese Patienten erhielten nach dem Zufallsprinzip 50 Einheiten, 100 Einheiten oder 200 Einheiten, wobei 6 Einheiten/kg Körpergewicht nicht überschritten werden durften. Patienten, die aufgrund der Höchstmenge von 6 Einheiten/kg weniger als die randomisierte Dosis erhielten, wurden für die Analyse der nächstgelegenen Dosisgruppe zugeordnet. Insgesamt wurden 38 Patienten der Gruppe mit 50 Einheiten, 45 Patienten der Gruppe mit 100 Einheiten und 30 Patienten der Gruppe mit 200 Einheiten BOTOX zugewiesen. Vor der Behandlung erhielten die Patienten eine Narkose entsprechend ihres Alter und der örtlichen Praxis. 109 Patienten (97,3 %) erhielten eine Vollnarkose oder eine bewusste Sedierung und 3 Patienten (2,7 %) eine Lokalanästhesie.
Die Studienergebnisse zeigten in Woche 6 (primärer Zeitpunkt) keine statistisch signifikanten Unterschiede in der primären Wirksamkeitsvariable [vordefinierter statistischer Ansatz] der Änderung der durchschnittlichen täglichen Häufigkeit von Harninkontinenz-Episoden gegenüber dem Ausgangswert zwischen der 100 Einheiten und 50 Einheiten BOTOX-Gruppe (p=0,9949) oder zwischen der 200 Einheiten und 50 Einheiten BOTOX-Gruppe (p=0,9123). Beim Vergleich innerhalb einer jeden Gruppe wurden in allen drei Dosierungsgruppen Verbesserungen der Anzahl täglicher Harninkontinenz-Episoden (normalisiert auf 12 Stunden) von etwa 30 % im Vergleich zum Ausgangswert festgestellt.
Hinsichtlich der sekundären Wirksamkeitsvariable Urinvolumen nach der ersten morgendlichen Katheterisierung ergaben sich in Woche 6 signifikante Gruppenunterschiede zwischen der mit 200 U gegenüber der mit 50 U BOTOX behandelten Dosierungsgruppe (p= 0,0055), während der Anstieg in der 100 Einheiten BOTOX Gruppe im Vergleich zur 50 Einheiten Gruppe statistisch nicht signifikant war (p=0,5117).
Im Hinblick auf Massnahmen zur Verringerung des maximalen Blasendrucks in Woche 6 erwies sich die Verabreichung von 200 Einheiten BOTOX im Vergleich zu 50 Einheiten BOTOX als signifikant wirksamer (p=0,0157), die 100 U-Dosis hingegen nicht (p=0,1737).
Tabelle 8: Ausgangswert und Veränderung gegenüber dem Ausgangswert in Bezug auf die tägliche Häufigkeit von Harninkontinenz-Episoden, das Urinvolumen beim ersten morgendlichen Katheterisieren und den maximalen Detrusor-Druck während der Speicherphase (cmH2O) und die maximale zystometrische Kapazität (mL) in der Studie 191622-120
|
|
BOTOX 200 Einheiten
N=30
|
p-Wert*
| |
Tägliche durchschnittliche Häufigkeit von Harninkontinenz-Episoden während des Tagesa
| |
Mittelwert zu Studienbeginn
|
3,7
|
| |
Mittlere Veränderung* in Woche 2 (95% CI)
|
–1,1 (–1,7, –0,6)
|
<0.0001
| |
Mittlere Veränderung* in Woche 6** (95% CI)
|
–1,3 (–1,8, –0,9)
|
<0.0001
| |
Mittlere Veränderung* in Woche 12 (95% CI)
|
–0,9 (–1,5, –0,4)
|
0.0009
| |
Urinvolumen beim ersten morgendlichen Katheterisieren (mL)b
| |
Mittelwert zu Studienbeginn
|
187,7
|
| |
Mittlere Veränderung* in Woche 2 (95% CI)
|
63,2 (27,9, 98,6)
|
0.0006
| |
Mittlere Veränderung* in Woche 6** (95% CI)
|
87,5 (52,1, 122,8)
|
<0.0001
| |
Mittlere Veränderung* in Woche 12 (95% CI)
|
45,2 (10,0, 80,5)
|
0.0125
| |
Maximaler Detrusordruck (Pdetmax) während der Speicherphase (cm H2O)b
| |
Mittelwert zu Studienbeginn
|
56,7
|
| |
Mittlere Veränderung* in Woche 6** (95% CI)
|
–27,3 (–36,4, –18,2)
|
<0.0001
| |
Maximale zystometrische Kapazität (mL) (MCC)b
| |
Mittelwert zu Studienbeginn
|
202,3
|
| |
Mittlere Veränderung* in Woche 6** (95% CI)
|
63,6 (29,0, 98,1)
|
| |
Unbeabsichtigten Detrusor-Kontraktion
|
|
| |
- Studienbeginn
|
25 (92.6)
|
| |
- Woche 6**
|
13 (46.4)
|
0.0004
|
KI = Konfidenzintervall
* Die Veränderung des Least-Square-Mittelwerts, und der p-Wert basieren auf dem ANCOVA-Modell mit dem Ausgangswert als Kovariable und der Behandlungsgruppe, dem Alter (< 12 Jahre oder ≥12 Jahre), den Tages- Harninkontinenz-Episoden zu Beginn (≤6 oder > 6) und der anticholinergen Therapie (ja/nein) zu Beginn als Faktoren.
** Primärer Zeitpunkt
a Primärer Endpunkt
b Sekundärer Endpunkt
Insgesamt zeigten die Daten zur Wiederholungsbehandlung einen anhaltenden Nutzen.
Keiner von 99 pädiatrischen Patienten, die einen negativen Ausgangswert für bindende oder neutralisierende Antikörper zu Studienbeginn aufwiesen und für die mindestens ein weiterer Wert im Verlauf einer randomisierten Doppelblindstudie und einer Doppelblind-Verlängerungsstudie ausgewertet werden konnte, entwickelte neutralisierende Antikörper nach bis zu vier Behandlungszyklen mit 50 bis 200 Einheiten BOTOX.
ERKRANKUNGEN DER HAUT UND MIT DER HAUT VERBUNDENE ERKRANKUNGEN:
Primäre Hyperhidrosis axillaris
Es wurde eine doppel-blinde, multizentrische klinische Studie bei Patienten mit persistierender bilateraler primärer Hyperhidrosis axillaris durchgeführt. Eine primäre Hyperhidrosis axillaris lag vor, wenn bei einer Ausgangsmessung in Ruhe und bei Raumtemperatur pro Achsel über 5 Minuten mindestens 50 mg Schweiss spontan produziert wurden. 320 Patienten wurden randomisiert und erhielten entweder 50 Einheiten BOTOX (N = 242) oder Placebo (N = 78). Patienten wurden als auf die Therapie ansprechend definiert, wenn sie zumindest eine Reduzierung der Achselschweissproduktion um 50% von der Ausgangsmessung zeigten. Am primären Endpunkt 4 Wochen nach den Injektionen sprachen in der BOTOX-Gruppe 93,8% der Patienten im Vergleich zu 35,9% der Patienten in der Placebo-Gruppe (p < 0,001) auf die Behandlung an. Die Rate der auf die Therapie ansprechenden Patienten war in der BOTOX-Gruppe kontinuierlich an allen nachfolgenden Messpunkten bis zu 16 Wochen nach den Injektionen signifikant grösser (p < 0,001) als in der Placebo-Gruppe.
In einer offenen Folgestudie wurden 207 geeignete Patienten eingeschlossen, die bis zu 3 BOTOX-Behandlungen erhielten. Insgesamt schlossen 174 Patienten die volle Dauer von 16 Monaten der zwei kombinierten Studien ab (4 Monate doppel-blind und 12 Monate offene Nachfolgestudie). Die Ansprechrate 16 Wochen nach der ersten (n = 287), der zweiten (n = 123) und der dritten (n = 30) Behandlung war jeweils 85,0%, 86,2% und 80%. Die mittlere Dauer der Wirkung basierend auf der Einzelbehandlung kombiniert mit der offenen Nachfolgestudie war 7,5 Monate nach der ersten Behandlung, jedoch hielt der Effekt bei 27,5% der Patienten über ein Jahr oder länger an.
PharmakokinetikAufgrund der Beschaffenheit von Botulinumtoxin Typ A wurden keine klassischen Resorptions-, Verteilungs-, Biotransformations- und Eliminationsuntersuchungen mit dem Wirkstoff durchgeführt.
Allgemeine Merkmale der aktiven Substanz
Distribution/Metabolismus/Elimination
Verteilungsstudien bei Ratten zeigen eine langsame Diffusion des radioaktiv markierten Botulinumtoxin Typ A im M. gastrocnemius, gefolgt von einer raschen systemischen Metabolisierung und Ausscheidung im Urin.
Die Halbwertszeit des markierten Materials betrug etwa 10 Stunden. An der Injektionsstelle wurde Radioaktivität an grosse Protein-Moleküle gebunden. Im Plasma erfolgte die Bindung an kleine Moleküle, was auf einen schnellen systemischen Metabolismus schliessen lässt. Innerhalb von 24 Stunden nach Applikation wurden 60% der Radioaktivität über den Urin ausgeschieden. Das Toxin wird vermutlich von Proteasen metabolisiert, und die Molekülkomponenten werden auf dem normalen Stoffwechselweg wiederverwertet.
Distribution
Verteilung des arzneilich wirksamen Bestandteils im Patienten:
Es wird angenommen, dass therapeutische BOTOX-Dosen wenig systemisch verteilt werden. In klinischen Studien konnte mit Hilfe der Einzelfaser-EMG-Technik für Muskeln, die entfernt von der Injektionsstelle lagen, eine erhöhte elektrophysiologische neuromuskuläre Aktivität gezeigt werden. Gleichzeitige klinische Befunde oder Symptome wurden nicht beobachtet. Der Umfang der Diffusion und damit von unerwünschten Wirkungen ist von den anatomischen Gegebenheiten, vom Injektionsvolumen und von der Dosis abhängig.
Präklinische DatenPräklinische Daten zur Sicherheit
Zusätzlich zur Reproduktionstoxikologie wurden die folgenden präklinischen Studien zur Sicherheit von BOTOX durchgeführt: akute Toxizität, chronische Toxizität, lokale Verträglichkeit, Mutagenität, Antigenität, sowie Verträglichkeit mit menschlichem Blut. Für klinisch relevante Dosen konnte in diesen Studien kein spezielles Risiko für den Menschen nachgewiesen werden.
Nach einer einzelnen Injektion von < 50 Einheiten/kg BOTOX in den Detrusor-Muskel von Ratten wurde keine systemische Toxizität beobachtet. Zur Simulation einer versehentlichen Injektion wurde eine Einzeldosis BOTOX (~7 Einheiten/kg) in die prostatische Harnröhre und das proximale Rektum, die Samenblase und Harnblasenwand oder den Uterus von Affen (~3 Einheiten/kg) injiziert, ohne dass unerwünschte klinische Wirkungen auftraten.
In einer Studie über 9 Monate in Affen, in der wiederholte Injektionen in den Detrusor-Muskel untersucht wurden (4 Injektionen), wurde bei Dosierungen von 24 Einheiten/kg Ptosis beobachtet. Mortalität trat bei Dosierungen von 36 Einheiten/kg nach der ersten von vier Injektionen auf und bei einem Tier bei einer Dosierung von 24 Einheiten/kg nach der zweiten Injektion. Verminderte motorische Aktivität und erschwerte Atmung traten bei moribunden Tieren nach Dosierungen ≥24 Einheiten/kg auf. Bei einer Dosierung von 12 Einheiten/kg bei Affen wurden keine unerwünschten Wirkungen beobachtet, was einer um das 3-fache erhöhten Dosierung mit BOTOX gegenüber der empfohlenen klinischen Dosis von 200 Einheiten bei Harninkontinenz infolge neurogener Detrusorhyperaktivität (bei einem 50 kg schweren Patienten) entspricht.
Mutagenität
BOTOX war in bakteriellen Rückmutationstests, in Mikrokerntests in Säugetier-Zellkulturen und Chromosomenanalyse negativ. BOTOX war in einem In-vivo-Mikrokerntest bei Nagern negativ.
Karzinogenität
Es wurden keine Karzinogenitätsstudien mit BOTOX durchgeführt.
Reproduktionstoxizität
In Fertilitätsstudien war der NOEL nach intramuskulärer Injektion von BOTOX 4 Einheiten/kg bei männlichen Ratten und 8 Einheiten/kg bei weiblichen Ratten und entspricht der maximal empfohlenen Dosierung von 400 Einheiten auf der Basis des Körpergewichts (kg). Höhere Dosierungen waren mit dosisabhängigen Verringerungen der Fertilität verbunden. Sofern eine Befruchtung eintrat, gab es keine nachteiligen Auswirkungen auf die Anzahl oder Lebensfähigkeit der Embryonen, die von behandelten männlichen oder weiblichen Ratten gezeugt oder empfangen wurden.
Trächtige Mäuse, Ratten und Kaninchen erhielten während der Organogenese intramuskuläre Injektionen von BOTOX. Der NOAEL-Wert (No Observed Adverse Effect Level) in der Entwicklungsphase lag bei 4 bzw. 1 bzw. 0,125 Einheiten/kg Körpergewicht. Höhere Dosen führten zu verminderten fetalen Körpergewichten und/oder verzögerter Skelett-Ossifikation. Bei Kaninchen wurden Aborte beobachtet.
Weitere Daten
Juvenile Tiere
In einer Studie, in der juvenilen Ratten jede zweite Woche ab dem 21. postnatalen Tag während 3 Monaten BOTOX als Dosis von 0, 8, 16 oder 24 Einheiten/kg Körpergewicht intramuskulär injiziert wurde, wurden Änderungen der Knochengrösse/-geometrie verbunden mit einer verringerten Knochendichte und Knochenmasse sekundär zur Gliedmassenverletzung, fehlender Muskelkontraktion und Abnahme der Körpergewichtszunahme beobachtet. Die Veränderungen waren bei der niedrigsten getesteten Dosis weniger schwerwiegend und zeigten bei allen Dosierungen Anzeichen von Reversibilität. Jugendliche Tiere erscheinen empfindlicher als Erwachsene. In einer Studie, in der juvenilen Ratten jede zweite Woche während 12 Wochen BOTOX intramuskulär injiziert wurde, lag der NOAEL-Wert (No Observed Adverse Effect Level) bei 8 Einheiten/kg, was mit der vorgeschlagenen Höchstdosis für pädiatrische Patienten von 10 Einheiten/kg vergleichbar ist. Die kumulative Dosis bei juvenilen Ratten von 56 Einheiten/kg (8 Einheiten/kg jede zweite Woche während 12 Wochen verabreicht) ist 5,6-mal höher als die maximale kumulative Dosis in der pädiatrischen Spastikpopulation über den gleichen Zeitraum. Die nicht-wirksame Dosis für nachteilige Auswirkungen auf die Entwicklung bei Jungtieren (8 Einheiten/kg Körpergewicht) entspricht der maximalen kumulativen Dosis bei Erwachsenen (400 Einheiten) und liegt unter der maximalen pädiatrischen Dosis (340 Einheiten) auf der Basis des Körpergewichts (kg).
Sonstige HinweiseInkompatibilitäten
Da keine Verträglichkeitsstudien zu möglichen Inkompatibilitäten durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Ungeöffnete Durchstechflaschen: Im Kühlschrank bei 2-8°C oder Gefrierschrank bei –5°C oder darunter lagern.
Potenzstudien haben gezeigt, dass das Arzneimittel nach Rekonstitution bis zu 3 Tage bei 2-8°C aufbewahrt werden kann. Da das Arzneimittel kein Konservierungsmittel enthält, ist die gebrauchsfertige Zubereitung aus mikrobiologischen Gründen unmittelbar nach Verdünnung zu verwenden, es sei denn, diese hat unter kontrollierten und validierten aseptischen Bedingungen stattgefunden. Das Arzneimittel sollte üblicherweise nicht länger als 24 Stunden bei 2-8°C gelagert werden.
Hinweise für die Handhabung
Zubereitung der BOTOX-Injektionslösung
Es wird empfohlen, die Rekonstitution der Lösung und die Vorbereitung der Spritze auf einem plastiküberzogenen Papiertuch vorzunehmen, um versehentlich vergossene BOTOX-Lösung sofort wegwischen zu können. Vor Gebrauch muss BOTOX mit steriler physiologischer Kochsalzlösung (ohne Konservierungsmittel) aufbereitet werden.
Die gewünschte Menge Lösungsmittel (siehe dazu die nachstehende Tabelle) wird in die Spritze aufgezogen. Die Oberfläche des Gummistopfens der Durchstechflasche soll mit einem Alkoholtupfer gereinigt werden, bevor man die Injektionsnadel durchsticht.
Tabelle 9: Verdünnungstabelle für Durchstechflaschen mit BOTOX 50 und 100 Einheiten für alle Indikationen (ohne Injektionen in den Detrusor-Muskel)
|
|
50 Einheiten
Durchstechflaschen
|
100 Einheiten
Durchstechflaschen
| |
Resultierende Dosis (Einheiten pro 0,1 ml)
|
Menge an Lösungsmittel (Natriumchlorid 9 mg/ml (0,9%)), welches einer Durchstechflasche mit 50 Einheiten zugesetzt wird:
|
Menge an Lösungsmittel (Natriumchlorid 9 mg/ml (0,9%)), welches einer Durchstechflasche mit 100Einheiten zugesetzt wird:
| |
20,0 E
|
0,25 ml
|
0,5 ml
| |
10,0 E
|
0,5 ml
|
1,0 ml
| |
5,0 E
|
1 ml
|
2,0 ml
| |
2,5 E
|
2 ml
|
4,0 ml
| |
1,25 E
|
4 ml
|
8,0 ml
|
Überaktive Blase
Verdünnungsanleitung für zwei Durchstechflaschen mit BOTOX 50 Einheiten:
·Zwei Durchstechflaschen mit BOTOX 50 Einheiten mit je 5 ml 0,9%iger Kochsalzlösung (ohne Konservierungsmittel) rekonstituieren und den Inhalt der Durchstechflaschen behutsam mischen.
·Aus jeder Durchstechflasche je 5 ml in einer 10 ml Spritze aufziehen.
Dies ergibt nun eine 10 ml Spritze mit insgesamt 100 Einheiten rekonstituiertem BOTOX. Nach Rekonstitution in der Spritze sofort verwenden. Nicht verwendete Kochsalzlösung verwerfen.
Verdünnungsanleitung für eine Durchstechflasche mit BOTOX 100 Einheiten:
·Eine Durchstechflasche mit BOTOX 100 Einheiten mit 10 ml 0,9%iger Kochsalzlösung (ohne Konservierungsmittel) rekonstituieren und den Inhalt der Durchstechflasche behutsam mischen.
·Aus der Durchstechflasche 10 ml in einer 10 ml Spritze aufziehen.
Dies ergibt nun eine 10 ml Spritze mit insgesamt 100 Einheiten rekonstituiertem BOTOX. Nach Rekonstitution in der Spritze sofort verwenden. Nicht verwendete Kochsalzlösung verwerfen.
Harninkontinenz infolge neurogener Detrusorhyperaktivität
Verdünnungsanleitung für vier Durchstechflaschen mit BOTOX 50 Einheiten:
·Vier Durchstechflaschen mit BOTOX 50 Einheiten mit je 3 ml 0,9%iger Kochsalzlösung (ohne Konservierungsmittel) rekonstituieren und den Inhalt der Durchstechflaschen behutsam mischen.
·Aus der ersten Durchstechflasche 3 ml und 1 ml aus der zweiten Durchstechflasche in eine 10 ml Spritze aufziehen.
·Aus der dritten Durchstechflasche 3 ml und aus der vierten Durchstechflasche 1 ml in eine zweite 10 ml Spritze aufziehen.
·Die verbleibenden 2 ml aus der zweiten und vierten Durchstechflasche in eine dritte 10 ml Spritze aufziehen.
·Die Rekonstitution durch Zugabe von 6 ml 0,9%iger Kochsalzlösung in jede der drei 10 ml Spritzen abschliessen und deren Inhalt behutsam mischen.
Dies ergibt nun drei 10 ml Spritzen mit insgesamt 200 Einheiten rekonstituiertem BOTOX. Nach Rekonstitution in der Spritze sofort verwenden. Nicht verwendete Kochsalzlösung verwerfen.
Verdünnungsanleitung für zwei Durchstechflaschen mit BOTOX 100 Einheiten:
·Zwei Durchstechflaschen mit BOTOX 100 Einheiten mit je 6 ml 0,9%iger Kochsalzlösung (ohne Konservierungsmittel) rekonstituieren und den Inhalt der Durchstechflaschen behutsam mischen.
·Aus jeder Durchstechflasche je 4 ml in zwei 10 ml Spritzen aufziehen.
·Die jeweils verbleibenden 2 ml aus den beiden Durchstechflaschen in eine dritte 10 ml Spritze aufziehen.
·Die Rekonstitution durch Zugabe von je 6 ml 0,9%iger Kochsalzlösung in jede der drei 10 ml Spritzen abschliessen und deren Inhalt behutsam mischen.
Dies ergibt nun drei 10 ml Spritzen mit insgesamt 200 Einheiten rekonstituiertem BOTOX.
Nach Rekonstitution in der Spritze sofort verwenden. Nicht verwendete Kochsalzlösung verwerfen.
Eine niedrigere oder höhere BOTOX-Konzentration kann durch Zugabe eines kleineren oder grösseren Lösungsmittelvolumens erreicht werden.
Da BOTOX durch Blasenwerfen oder starkes Schütteln denaturiert werden kann, soll das Lösungsmittel vorsichtig in die Durchstechflasche eingegeben werden. Die Durchstechflasche darf nicht verwendet werden, wenn das Lösungsmittel nicht durch einen leichten Unterdruck eingezogen wird. Das aufbereitete BOTOX ist eine klare farblose bis leicht gelbliche Lösung, die frei von sichtbaren Teilchen ist. Die zubereitete Lösung sollte auf Klarheit und Schwebeteilchen hin mit dem Auge überprüft werden.
Die rekonstituierte Lösung ist zum einmaligen Gebrauch bestimmt. Wenn BOTOX zur Injektion in den Detrusor-Muskel in einer Spritze weiter verdünnt wird, sollte es sofort verbraucht werden. Verbleibende Restmengen der Toxinlösung sind fachgerecht zu verwerfen.
Entsorgung
Verschüttetes BOTOX-Pulver sollte mit einem saugfähigen Tuch aufgewischt werden, welches mit verdünnter Hypochlorit-Lösung (0,5%) getränkt wurde. Verschüttete BOTOX-Injektionslösung sollte mit einem trockenen, saugfähigen Tuch aufgewischt werden. Die verunreinigten Oberflächen sollten mit einem saugfähigen Tuch, welches mit verdünnter Hypochlorit-Lösung getränkt wurde, gereinigt und anschliessend trockengerieben werden.
Falls eine Durchstechflasche zerbrochen ist, müssen Glasscherben vorsichtig gesammelt werden, um Hautverletzungen zu vermeiden.
Ungebrauchte Durchstechflaschen sollten mit etwas Wasser aufbereitet und autoklaviert werden. Alle gebrauchten Durchstechflaschen, Spritzen, Kanülen, etc. sollten ebenfalls autoklaviert werden. Rückstände von Botulinumtoxin können auch mit verdünnter Hypochlorit-Lösung inaktiviert werden. Die Lösung sollte mindestens 5 Minuten einwirken. Andere kontaminierte Materialien wie Glasscherben, Handschuhe, etc. sollten in einem für spitze Gegenstände geeigneten Behälter entsorgt und der Müllverbrennung zugeführt werden.
Identifizierung des Arzneimittels
Wird ein authentisches BOTOX-Produkt verwendet, werden horizontale Linien in Regenbogenfarben und der Name «abbvie» auf der Etikette sichtbar (Hologramm), und zwar beim Hin- und Herdrehen der Durchstechflasche in der Hand unter einer Schreibtischlampe oder Leuchtstoffröhre. Ist dies nicht der Fall und/oder ist die Perforation nicht intakt, soll das Produkt nicht verwendet werden, und AbbVie ist zu kontaktieren.
Zusätzlich können Aufkleber mit Chargennummer und Verfalldatum von der Etikette der BOTOX-Durchstechflasche abgezogen und in die Patientenakte geklebt werden, um die Rückverfolgbarkeit der Charge zu erleichtern. Nach dem Abziehen des Aufklebers von der Etikette der Durchstechflasche wird «Verwendet/Utilisé» erscheinen.
Zulassungsnummer52433 (Swissmedic)
PackungenBOTOX
Durchstechflasche mit 50 Allergan-Einheiten (roter Aluminiumring): 2 x 1 [A]
Durchstechflasche mit 100 Allergan-Einheiten (violetter Aluminiumring): 1 und 2 x 1 [A]
ZulassungsinhaberinAbbVie AG, Cham
Stand der InformationMai 2025
|