Eigenschaften/WirkungenATC-Code
M03AX01
Wirkungsmechanismus
Das Botulinumtoxin Typ A blockiert durch Spaltung von SNAP 25 («synaptosomal-associated protein of 25 kDa») die periphere Acetylcholin-Freisetzung an den präsynaptischen Nervenendigungen. SNAP 25 ist ein Protein, das wesentlich für die erfolgreiche Bindung und Freisetzung von Acetylcholin aus den Vesikeln ist, die sich in den Nervenendigungen befinden.
Pharmakodynamik
Nach der Injektion erfolgt initial eine hoch-affine Bindung des Toxins an spezifische zelluläre Oberflächenrezeptoren. Durch einen Rezeptor-vermittelten Endozytose-Prozess wird das Toxin durch die Plasmamembran transportiert und anschliessend im Cytosol freigesetzt. Dieser Prozess wird von einer progressiven Inhibition der Acetylcholin-Freisetzung begleitet. Klinische Befunde manifestieren sich innerhalb von 2-3 Tagen. Die maximale Wirkung ist 5-6 Wochen nach der Injektion zu beobachten. In sensorischen Neuronen hemmt BOTOX die Freisetzung von sensorischen Neurotransmittern (z.B. Substanz P, CGRP) und reguliert die Expression von Zelloberflächenrezeptoren (z.B. TRPV1) herunter. BOTOX verhindert auch eine Sensibilisierung nozizeptiver sensorischer Neuronen, macht diese rückgängig und hemmt somit möglicherweise auch die zentrale Sensibilisierung.
Nach intramuskulärer Injektion erfolgt innerhalb von 12 Wochen die Wiederherstellung der Impulsübertragung durch neugebildete Nervenendigungen und deren Wiederverbindung mit den motorischen Endplatten.
Bei intradermaler Injektion, wo das Ziel die ekkrinen Schweissdrüsen sind, hält der Effekt bei Patienten, die 50 Einheiten pro Axilla erhielten, ungefähr 4-7 Monate an.
Wird BOTOX zur Behandlung des Strabismus eingesetzt, dann wird angenommen, dass es auf Muskelpaare einwirkt, indem der behandelte Muskel atrophisch verlängert und der entsprechende Muskelantagonist verkürzt wird.
Nach Injektion in die Halsmuskulatur verbessert BOTOX sowohl die objektiven Anzeichen als auch die subjektiven Symptome der zervikalen Dystonie. Dies äussert sich in einer Verringerung der Drehung des Kopfes zur Seite, verringerter Anhebung der Schulter, verringerter Grösse und Kraft hypertrophischer Muskeln und Verringerung der Schmerzen.
Bei Erwachsenen mit fokalen Spastiken der oberen Extremitäten nach Schlaganfall verbessert BOTOX deutlich den Bewegungsspielraum (Ellbogen und Handgelenksstreckung).
Die Injektion von BOTOX bei Kindern mit dynamischer Spitzfussstellung (Equinus-Deformität) ohne Retraktion und ohne bedeutende Atrophie führt durch teilweise Denervierung des M. gastrocnemius zu einer veränderten Sprunggelenksposition und dadurch zu einer deutlichen Verbesserung des Gangbildes.
Nach Injektion in den Detrusor-Muskel wirkt BOTOX auf die efferenten Nervenbahnen der Detrusor-Aktivität durch Hemmung der Freisetzung von Acetylcholin. Zusätzlich hemmt BOTOX afferente Neurotransmitter und sensorische Bahnen.
Klinische Wirksamkeit
NEUROLOGISCHE ERKRANKUNGEN:
Blepharospasmus/hemifazialer Spasmus
In einer offenen Studie wurde 56 Patienten mit Spasmus hemifacialis eine Anfangsdosis von 10 bis 50 Einheiten injiziert. Die Patienten wurden anschliessend 22 Wochen beobachtet. Patienten, die in Woche 4 keine Besserung zeigten, erhielten eine erneute Injektion (5 bis 50 Einheiten). Die Patienten wurden in Woche 2, 4 (sofern eine erneute Behandlung erforderlich war), 6, 14 und 22 beobachtet. Die klinische Wirkung wurde zu jedem Kontrollzeitpunkt für die verschiedenen oberen und unteren Gesichtsmuskeln bewertet.
Bei allen 56 Patienten trat eine Besserung ein, und 62,5% (35/56) zeigten eine deutliche Besserung. Bezogen auf die oberen Gesichtsmuskeln wurde bei allen Patienten eine Besserung beobachtet. Bei der Beurteilung der unteren Gesichtsmuskeln zeigte sich, dass mit Ausnahme von 2 Patienten alle auf die Behandlung angesprochen hatten.
Bei Patienten mit gutartigem essentiellem Blepharospasmus wurde eine zusätzliche randomisierte, multizentrische Doppelblindstudie mit parallelen Gruppen zum Vergleich der Sicherheit und Wirksamkeit von 2 BOTOX-Formulierungen (der Wirkstoff stammte aus verschiedenen Master-Zellbanken - eine davon entspricht der derzeitigen BOTOX-Formulierung) durchgeführt. In die Studie wurden Patienten aufgenommen, die schon früher mit BOTOX behandelt worden waren, und die Behandlung bestand aus einem einzelnen Injektionszyklus in die M. orbicularis oculi beider Augen. Die Dosis (maximal zulässig waren 100 Einheiten) und Injektionsstellen wurden vom Prüfarzt aufgrund des Ansprechens der Patienten auf frühere BOTOX-Behandlungen festgelegt. Die Patienten wurden über einen Zeitraum von 12 Wochen nach der Behandlung beobachtet. In die Studie wurden 98 Patienten aufgenommen (48 in die Gruppe mit der derzeitigen BOTOX-Formulierung).
Die Erfolgsrate der Behandlung wurde auf einer 5-Punkte-Skala von 0 bis 4 beurteilt. Dabei stand 0 für «keinen» und 4 für «starken» behindernden Spasmus der Augenlider und möglicherweise anderer Gesichtsmuskeln. Eine Abnahme auf einen Punktwert von ≤2 an beiden Augen galt als Behandlungserfolg. Der primäre Endpunkt lag in Woche 4. Die Patienten erhielten eine durchschnittliche Dosis von 33 Einheiten pro Auge, injiziert an 3 bis 15 Injektionsstellen. Vergleichbar mit der früheren BOTOX-Formulierung lag die Erfolgsrate der Behandlung mit der derzeitigen BOTOX-Formulierung bei ca. 90% in Woche 4.
Zervikale Dystonie
Bei Patienten mit zervikaler Dystonie (CD) wurde eine randomisierte, Placebo-kontrollierte, multizentrische Doppelblindstudie durchgeführt. In diese Studie wurden erwachsene Patienten mit CD aufgenommen, die positiv auf eine frühere Behandlung mit BOTOX angesprochen hatten. Die Studie war in 2 Behandlungsphasen unterteilt, in eine offene Studienphase, in der alle Patienten mit BOTOX behandelt wurden, wobei Dosierung und Injektionsstellen individuell vom Prüfarzt festgelegt wurden, und in eine Doppelblindphase, in der Patienten, die in der offenen Phase als Responder eingestuft wurden, doppelblind entweder BOTOX oder Placebo erhielten. Zwischen diesen beiden Phasen lag ein Zeitraum von bis zu 6 Wochen. Insgesamt wurden den Patienten bis zu 360 Einheiten BOTOX verabreicht.
In der offenen Studienphase wurden 214 Patienten beurteilt, von denen 170 für den Doppelblindteil der Studie randomisiert wurden (88 Patienten wurden in die BOTOX-Gruppe, 82 in die Placebo-Gruppe aufgenommen). Anschliessend wurden die Patienten im Abstand von jeweils 2 Wochen über einen Zeitraum von bis zu 10 Wochen nach der Behandlung beobachtet. Die primären Wirksamkeitsvariablen waren eine Veränderung der Kopfhaltung (gemessen auf der Skala zur Beurteilung der Schwere der zervikalen Dystonie) und der prozentuale Anteil der Patienten mit einer Besserung des CD-Status, gemessen anhand einer ärztlich beurteilten Bewertungsskala 6 Wochen nach der Behandlung. Schmerzen als wichtiges Symptom von CD wurden von den Patienten im Hinblick auf Häufigkeit und Intensität beurteilt. Zusätzlich beurteilten Arzt und Patient die funktionale Behinderung.
BOTOX erwies sich bei beiden primären Wirksamkeitsvariablen nach 6 Wochen dem Placebo sowohl klinisch als auch statistisch signifikant überlegen. BOTOX verringerte auch die Intensität und Häufigkeit von Schmerzen statistisch signifikant besser als Placebo. Signifikante Verbesserungen wurden auch bei der Beurteilung der funktionalen Behinderung durch Arzt und Patient in Woche 6 beobachtet.
Bei Patienten mit zervikaler Dystonie wurde eine zusätzliche randomisierte Doppelblindstudie im Crossover-Design zur Untersuchung der Sicherheit und Wirksamkeit von 2 BOTOX-Formulierungen (der Wirkstoff stammte aus verschiedenen Master-Zellbanken - eine davon entspricht der derzeitigen BOTOX-Formulierung) durchgeführt. In die Studie wurden Patienten aufgenommen, die bereits früher mit zufriedenstellenden Ergebnissen behandelt worden waren; die Behandlung umfasste 2 Injektionen mit einer dazwischen liegenden Auswaschphase von 8 bis 16 Wochen. Die Patienten wurden nach jeder Behandlung über einen Zeitraum von 8 bis 16 Wochen beobachtet, wobei der primäre Endpunkt in Woche 6 nach jeder Behandlung lag.
In diese Studie wurden 135 Patienten aufgenommen. Die primäre Wirksamkeitsvariable war die Gesamtbeurteilung des Schweregrads auf der Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS). Sekundäre Wirksamkeitsvariablen waren der Behinderungs-Schmerzscore (anhand der modifizierten TWSTRS) und die Gesamtbeurteilung der zervikalen Dystonie durch Patient und Arzt. Die Ergebnisse bestätigten, dass eine maximale klinische Besserung nach Injektion von BOTOX in Woche 6 erreicht wird. Die mittlere Verringerung des Gesamtwerts auf der TWSTRS stellte eine 35%ige Besserung gegenüber den Ausgangswerten dar, und die höchste mittlere Verringerung des Behinderungs-Schmerzscores entsprach einer 50%igen Besserung gegenüber dem Ausgangswert zu diesem Zeitpunkt.
Die Gesamtbeurteilung durch Ärzte und Patienten zeigte ebenfalls den positiven Behandlungseffekt mit einem für die neue BOTOX-Formulierung berichteten Behandlungserfolg in über 85% bzw. 80% der Fälle in Woche 6.
Fokale Spastiken der oberen Extremitäten bei Erwachsenen
Bei Patienten mit Spastiken der oberen Extremitäten nach einem Schlaganfall wurde eine Placebo-kontrollierte, multizentrische Doppelblindstudie zur Untersuchung der Wirksamkeit und Sicherheit von BOTOX durchgeführt. Die Patienten waren noch nie mit BOTOX behandelte Erwachsene, deren Schlaganfall ≥6 Wochen zurücklag und die erweiterte Ashworth-Scores (EAS) von ≥3 am Handgelenk und ≥2 am Ellbogen aufwiesen. Einundneunzig Patienten wurden auf eine von vier Behandlungsgruppen randomisiert (BOTOX 90, 180 oder 360 Einheiten, oder Placebo). Der primäre Endpunkt, Spastik des Flexors im Handgelenk, wurde in Woche 6 auf der EAS gemessen. Sekundäre Wirksamkeitsvariablen waren Spastik der Flexoren in Ellbogen und Fingern, gemessen auf der EAS und nach Gesamtbeurteilung des Arztes.
In die Studie wurden 91 Patienten aufgenommen (65 Patienten erhielten BOTOX in 90, 180 oder 360 Einheiten). Die mittlere Abnahme auf der EAS für die Flexoren des Handgelenks war in allen BOTOX-Gruppen bis Woche 12 grösser als in der Placebo-Gruppe.
In allen BOTOX-Behandlungsgruppen war der Flexortonus im Handgelenk, gemessen auf der EAS, wirksamer verringert als unter Placebo. Die Flexoren der Ellbogen zeigten ähnliche Verbesserungsmuster. Bei den Fingerflexoren fand sich zwar insgesamt eine gewisse Variabilität (signifikante Verbesserungen nur in Woche 1 und 3), aber die Daten weisen auf vergleichbare Dosis/Wirkungs-Beziehungen hin. Die ärztlich beurteilte Bewertungsskala zeigte ebenfalls einen signifikanten Vorteil bei Dosen von 180 Einheiten und 360 Einheiten.
Eine weitere Placebo-kontrollierte, multizentrische Doppelblindstudie wurde bei Patienten mit Spastik im Handgelenk und den Fingern als sekundäre Folge eines Schlaganfalls durchgeführt. Die Dauer der Studie betrug 12 Wochen. Primärer Endpunkt war die Spastik des Flexors im Handgelenk in Woche 6, gemessen auf der Ashworth-Skala (AS). Weitere Variablen waren die ärztlich beurteilte Bewertungsskala, die Gesamtbeurteilung durch die Betreuungsperson des Patienten, die Skala zur Beurteilung von Behinderungen und die Grundsätzlichen Therapeutischen Interventionsziele.
126 Patienten wurden randomisiert und erhielten BOTOX (N = 64) oder Placebo (N = 62). Die injizierte Dosis von BOTOX lag zwischen 200 bis 240 Einheiten, verteilt auf die betroffenen Muskeln im Handgelenk und in den Fingern. Es zeigte sich, dass BOTOX die AS-Werte für Handgelenk und Finger zu allen Kontrollzeitpunkten nach der Injektion verbessert hatte. In Woche 6 zeigten 88% der Patienten eine Verbesserung auf der AS unter BOTOX, gegenüber 34% in der Placebo-Gruppe.
Alle anderen Variablen zeigten in Woche 6 ebenfalls einen signifikanten Vorteil gegenüber Placebo.
Fokale Spastiken der oberen Extremitäten bei Jugendlichen und Kindern ab 2 Jahren
Die Wirksamkeit und Sicherheit von BOTOX zur Behandlung der Spastiken der oberen Extremitäten wurde bei pädiatrischen Patienten ab 2 Jahren in einer randomisierten, multizentrischen, doppelblinden, Placebo-kontrollierten Studie untersucht. 234 pädiatrische Patienten (77 BOTOX 6 Einheiten/kg KG, 78 BOTOX 3 Einheiten/kg KG, 79 Placebo) mit Spastik aufgrund eines zerebralen «Palsy oder Hirnschlags (modifizierte AS Ellbogen oder Handgelenk Score von mindestens 2) wurden in die Studie aufgenommen. Die Gesamtdosis von 3 Einheiten/kg KG (maximal 100 Einheiten) oder 6 Einheiten/kg KG (maximal 200 Einheiten) oder Placebo wurde intramuskulär injiziert, verteilt auf Ellbogen oder Handgelenk und Fingermuskeln. Die Patienten wurden während 12 Wochen nach der Injektion beobachtet. Der Gebrauch einer elektromyographischen Kontrolle (EMG), Nervenstimulation oder Ultraschalltechnik wurde verlangt, um die korrekte Muskellokalisation für die Injektion zu unterstützen.
Der primäre Endpunkt war die durchschnittliche Änderung gegenüber dem Ausgangswert in der modifizierten AS (MAS) Score der Hauptmuskelgruppen (Ellbogen oder Handgelenk) während Woche 4 und 6. Der wichtigste sekundäre Endpunkt war der Mittelwert des «Clinical Global Impression of Overall Change by Physician (CGI)» während Woche 4 und 6. Die «goal attainment scale» (GAS) beurteilt durch den Arzt für aktive und passive Ziele wurde evaluiert als sekundärer Endpunkt während Woche 8 und 12.
Geeignete Patienten konnten an einer offenen Verlängerungsstudie teilnehmen, in welcher sie mehr als 5 Behandlungen mit Dosen bis zu 10 Einheiten/kg (maximal 340 Einheiten) erhielten, wenn mehr als eine Extremität behandelt wurde.
Eine signifikante Verbesserung im Vergleich zu Placebo für den MAS Knöchelscore gegenüber dem Ausgangswert wurde zu jedem Zeitpunkt für mit BOTOX behandelte Patienten in beiden Behandlungsgruppen beobachtet. Ähnliche Ergebnisse wurden in beiden BOTOX-Dosisgruppen mit einem statistisch signifikant höheren Anteil von MAS-Respondern (definiert als mindestens 1-Punkt-Verbesserung) im Vergleich zu Placebo zu den meisten Zeitpunkten beobachtet.
Tabelle 2: Resultate der primären und wichtigen sekundären Endpunkte
|
|
BOTOX
3 Einheiten/kg
(N=78)
|
BOTOX
6 Einheiten/kg
(N=77)
|
Placebo
(N=79)
| |
Mittlere Veränderung gegenüber dem Ausgangswert in der Hauptmuskelgruppe (Ellenbogen- oder Handgelenk auf der modifizierten Ashworth-Skala**
| |
Woche 4 und 6 Durchschnitt
|
-1,92*
|
-1,87*
|
-1,21
| |
Mittlerer Clinical Global Impression Score***
| |
Woche 4 und 6 Durchschnitt
|
1,88
|
1,87
|
1,66
|
* Signifikant unterschiedlich zu Placebo (p< 0.05)
** Der MAS ist eine 6-Punkte Skala (0 [keine Erhöhung des Muskeltonus], 1, 1+, 2, 3, and 4 [in Beugung oder Streckung starre Extremität]), die die Kraft misst, die erforderlich ist, um eine Extremität um ein Gelenk herum zu bewegen, wobei eine Verringerung der Punktzahl eine Verbesserung der Spastizität darstellt.
*** Der CGI bewertete das Ansprechen auf die Behandlung im Hinblick auf die Lebensqualität des Patienten anhand einer 9-Punkte-Skala (-4=sehr deutliche Verschlechterung bis +4=sehr deutliche Verbesserung).
Obwohl die CGI-Werte für BOTOX numerisch über Placebo lagen, war der Unterschied statistisch nicht signifikant.
Statistisch signifikante Verbesserungen im Vergleich zu Placebo wurden beim Anteil der CGI-Responder (definiert als mindestens 1-Punkt-Verbesserung) für BOTOX 3 Einheiten/kg (Woche 4 bis 12) und BOTOX 6 Einheiten/kg (Woche 6) sowie bei der funktionellen Zielerreichung (GAS durch den Arzt) für passive Ziele nur bei den BOTOX 6 Einheiten/kg (Woche 12) beobachtet.
Abbildung 6: MAS-Score (pädiatrische Spastizität obere Extremität) - Mittlere Veränderung gegenüber dem Ausgangswert

Fokale Spastik der unteren Extremitäten bei Erwachsenen
Die Wirksamkeit und Sicherheit von BOTOX zur Behandlung von Spastik der unteren Extremitäten wurde in einer randomisierten, multizentrischen, doppelblinden, Placebo-kontrollierten Studie untersucht, die 468 Patienten nach Schlaganfall (233 BOTOX und 235 Placebo) mit Spastik des Fussgelenkes (MAS Fussgelenk-Wert von mindestens 3) einschloss, deren Schlaganfall mindestens 3 Monate zurücklag. BOTOX 300 bis 400 Einheiten oder Placebo wurden intramuskulär in die für die Studie vorgeschriebenen Muskeln M. gastrocnemius, M. soleus und M. tibialis posterior sowie in optionale Muskeln, einschließlich M. flexor hallucis longus, M. flexor digitorum longus, M. flexor digitorum brevis, M. extensor hallucis und M. rectus femoris, injiziert. Der Gebrauch einer elektromyographischen Kontrolle (EMG), Nervenstimulation oder Ultraschalltechnik wurde verlangt, um die korrekte Muskellokalisation für die Injektion zu unterstützen. Die Patienten wurden während 12 Wochen nachbeobachtet.
Der primäre Endpunkt war die durchschnittliche Veränderung des MAS-Fussgelenk-Wertes ab Baseline in den Wochen 4 und 6. Ein wichtiger sekundärer Endpunkt war der durchschnittliche CGI-Wert (Bewertung des klinischen Gesamteindrucks durch den Arzt) in den Wochen 4 und 6. Statistisch und klinisch signifikante Unterschiede zwischen den Gruppen für BOTOX gegenüber Placebo wurden für den primären Wirksamkeitsparameter MAS und den sekundären Schlüsselparameter CGI gezeigt und sind in der folgenden Tabelle dargestellt. Für den primären Endpunkt des durchschnittlichen MAS-Fussgelenk-Wertes in den Wochen 4 und 6 wurde bei Patienten im Alter von 65 Jahren und älter in der BOTOX-Gruppe im Vergleich zu Placebo keine Verbesserung gegenüber dem Ausgangswert beobachtet, wahrscheinlich aufgrund der geringen Patientenzahlen.
Tabelle 3: Primäre und wichtigste sekundäre Wirksamkeitspunkte
|
|
BOTOX
300 bis 400 Einheiten (n=233)
|
Placebo
(n=235)
| |
Mittlere Veränderung bei den Plantarflexoren des Fussgelenkes ab Baseline auf der MAS-Skala
| |
Durchschnitt in Woche 4 und 6
|
-0,8*
|
-0,6
| |
Mittlerer Wert des klinischen Gesamteindruckes des Prüfarztes
| |
Durchschnitt in Woche 4 und 6
|
0,9*
|
0,7
| |
Mittlere Veränderung bei den Zehenflexoren auf der MAS-Skala
| |
FHL** Durchschnitt in Woche 4 und 6
|
-1,0*
|
-0,6
| |
FDL + FDB** Durchschnitt in Woche 4 und 6
|
-0,9
|
- 0,8
|
*Signifikant unterschiedlich zu Placebo (p<0,05)
FHL = flexor hallucis longus; FDL = flexor digitorum longus; FDB = flexor digitorum brevis
Abbildung 7: MAS-Score (Spastizität untere Extremität Erwachsene) - Mittlere Veränderung gegenüber dem Ausgangswert
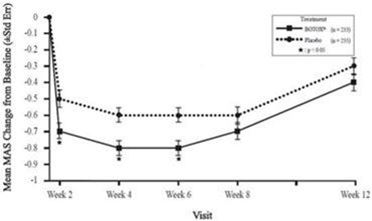
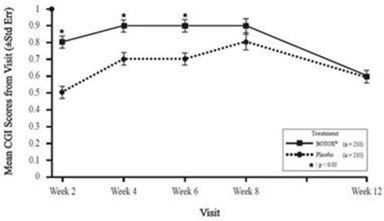
Abbildung 8: CGI des Arztes (Spastizität untere Extremitäten Erwachsene) - Durchschnittswerte pro Besuch
Fokale Spastik der unteren Extremitäten bei Jugendlichen und Kindern ab 2 Jahren
Die Wirksamkeit und Sicherheit von BOTOX zur Behandlung der Spastik der unteren Extremitäten bei pädiatrischen Patienten ab 2 Jahren wurde in einer randomisierten, multizentrischen, doppelblinden, Placebo-kontrollierten Studie untersucht. Die Studie umfasste 384 pädiatrische Patienten (128 BOTOX 8 Einheiten/kg, 126 BOTOX 4 Einheiten/kg und 128 Placebo) mit Spastik der unteren Extremitäten aufgrund einer zerebralen Lähmung (MAS Knöchelwert mind. 2). Eine Gesamtdosis von 4 Einheiten/kg (maximal 150 Einheiten) oder 8 Einheiten/kg (maximal 300 Einheiten) oder Placebo wurde intramuskulär injiziert, aufgeteilt zwischen Gastrocnemius, Soleus und Tibialis posterior. Der Gebrauch einer elektromyographischen Kontrolle (EMG), Nervenstimulation oder Ultraschalltechnik wurde verlangt, um die korrekte Muskellokalisation für die Injektion zu unterstützen. Die Patienten wurden 12 Wochen lang beobachtet. Der primäre Endpunkt war die durchschnittliche Änderung gegenüber dem Ausgangswert im MAS Knöchelscore in Woche 4 und 6, und der wichtigste sekundäre Endpunkt war der Mittelwert des CGI in Woche 4 und 6. Sekundäre Endpunkte in Wochen 8 und 12 beinhalteten GAS durch den Arzt und dem Edinburgh Visual Gait (EVG) (in einer Untergruppe von Patienten).
Geeignete Patienten konnten an einer offenen Verlängerungsstudie teilnehmen, in der sie bis zu fünf Behandlungen mit Dosen bis zu 10 Einheiten/kg (maximal 340 Einheiten) erhielten bei Behandlung von mehr als einer Extremität.
Eine statistisch signifikante Verbesserung im primären Endpunkt gegenüber Placebo wurde bei den mit 4 und mit 8 Einheiten/kg BOTOX behandelten Patienten bis Woche 12 beobachtet (siehe Abbildung 9). Ähnliche Ergebnisse wurden in beiden BOTOX-Dosisgruppen mit einem höheren Anteil von MAS-Respondern (definiert als mindestens 1-Punkt-Verbesserung) im Vergleich zu Placebo zu allen Zeitpunkten beobachtet.
Tabelle 4: Primäre und wichtige sekundäre Wirksamkeitsendpunkt-Ergebnisse
|
|
BOTOX 4 Units/kg
(n = 125)
|
BOTOX 8 Units/kg
(n=127)
|
Placebo
(n=129)
| |
Mittlere Änderung im MAS-Ausgangswert der Plantarflexoren
| |
Durchschnitt Woche 4 und 6
|
-1,01*
|
-1,06*
|
-0,80
| |
Mittlerer CGI Wert
| |
Durchschnitt Woche 4 und 6
|
1,49
|
1,65*
|
1,36
|
* Signifikanter Unterschied zu Placebo (p< 0,05)
Statistisch signifikante Verbesserung im Vergleich zu Placebo wurde mit 4 und 8 Einheiten/kg BOTOX für den primären Endpunkt mittlerer MAS Knöchelscore in Wochen 4 und 6 als Veränderung gegenüber dem Ausgangswert und mit 8 Einheiten/kg BOTOX für den wichtigsten sekundären Endpunkt mittlerer CGI in den Wochen 4 und 6 nachgewiesen. Eine statistisch signifikante Verbesserung im Vergleich zu Placebo wurde für CGI vom Arzt im Verlauf von durchschnittlich 4 und 6 Wochen für BOTOX 8 Einheiten/kg beobachtet. Statistisch signifikante Verbesserungen der funktionellen aktiven und passiven Zielerreichung (GAS) wurden in Woche 8 und 12 für BOTOX 8 Einheiten/kg und in der Woche 8 aber nicht in Woche 12 für BOTOX 4 Einheiten/kg beobachtet. Die Gangverbesserung war dosisabhängig, mit statistisch signifikanten Verbesserungen des EVG in Woche 8 für BOTOX 8 Einheiten/kg.
Abbildung 9: MAS-Score (pädiatrische Spastizität untere Extremität) - Mittlere Veränderung gegenüber dem Ausgangswert

Bei pädiatrischen Patienten mit Spastik der unteren Extremitäten, deren Proben aus einer Phase-3-Studie und der Open-Label-Extension-Studie analysiert wurden, entwickelten 2 von 264 Patienten (0,8%) neutralisierende Antikörper, die mit BOTOX für bis zu 5 Behandlungszyklen behandelt wurden. Beide Patienten hatten anschliessend weiterhin einen klinischen Nutzen von der BOTOX-Behandlung.
Prophylaxe von Kopfschmerzen bei erwachsenen Patienten mit chronischer Migräne
BOTOX wurde in zwei 56wöchigen multinationalen, multizentrischen Studien untersucht, die Folgendes umfassten: 24wöchige, doppelblinde Phase mit 2 Injektionszyklen zum Vergleich von BOTOX mit Placebo, gefolgt von einer 32wöchigen offenen Phase mit 3 Injektionszyklen mit BOTOX. Insgesamt 1'384 Erwachsene mit chronischer Migräne, die entweder noch nie eine begleitende Kopfschmerzprophylaxe erhalten hatten oder diese bei Studienbeginn für 28 Tage nicht angewendet hatten, mit Kopfschmerzen an ≥15 Tagen, davon 50 % Migräne/wahrscheinliche Migräne, und ≥4 Kopfschmerzepisoden, wurden in zwei klinischen Phase-3-Studien untersucht. Diese Patienten wurden randomisiert und erhielten alle 12 Wochen Injektionen mit Placebo oder 155–195 Einheiten BOTOX für die doppelblinde Phase mit 2 Injektionszyklen. Sie konnten 3 weitere offene BOTOX-Zyklen erhalten, bis zu maximal 5 Injektionszyklen.
Die Akutbehandlung von Kopfschmerzen war den Patienten erlaubt (bei 65,5 % lag eine übermässige Akutbehandlung während der Studienbeginn-Phase vor).
Die Behandlung mit BOTOX zeigte im Vergleich zu Placebo statistisch signifikante (p < 0,001) und klinisch bedeutsame Verbesserungen gegenüber dem Ausgangswert in Bezug auf die Häufigkeit von Kopfschmerztagen, die Häufigkeit von Migräne/ wahrscheinlichen Migränetagen, die Häufigkeit von mittelschweren/schweren Kopfschmerztagen und die kumulativen Gesamtstunden an Kopfschmerztagen.
Die Ergebnisse des Headache Impact Test (HIT-6) und des Migraine-Specific Quality of Life Questionnaire (MSQ) wiesen auf eine anhaltende Wirkungsdauer von BOTOX sowie eine Verbesserung der Funktionsfähigkeit, Vitalität, psychischen Belastung und Lebensqualität insgesamt hin.
Gepoolte Ergebnisse der wichtigsten Wirksamkeitsendpunkte in den beiden Zulassungsstudien in Woche 24 (primärer Zeitpunkt)
|
Wirksamkeit über 28 Tagen
|
BOTOX
(N = 688)
|
Placebo
(N = 696)
|
p-Wert
| |
Veränderung der Anzahl an Tagen mit Kopfschmerzen gegenüber dem Ausgangswert
|
-8,4
|
-6,6
|
< 0,001
| |
Veränderung der Anzahl an Tagen mit Migräne/wahrscheinlicher Migräne gegenüber dem Ausgangswert
|
-8,2
|
-6,2
|
< 0,001
| |
Veränderung der Anzahl an Tagen mit mittelstarken/starken Kopfschmerzen gegenüber dem Ausgangswert
|
-7,7
|
-5,8
|
< 0,001
| |
Veränderung der Gesamtzahl an Kopfschmerzstunden an den Tagen mit Kopfschmerzen gegenüber dem Ausgangswert
|
-119,7
|
-80,5
|
< 0,001
| |
Veränderung der Anzahl an Kopfschmerzepisoden gegenüber dem Ausgangswert
|
-5,2
|
-4,9
|
0,009
| |
Prozentsatz der Patienten mit 50 % weniger Kopfschmerztagen
|
47%
|
35%
|
< 0,001
| |
Anteil der Patienten mit hohen Scores bei HIT-6-Kategorien
|
67,6 %
|
78,2 %
|
< 0,001
| |
Veränderung des HIT-6-Gesamtscores gegenüber dem Ausgangswert
|
-4,8
|
-2,4
|
< 0,001
|
Obwohl die Studien nicht darauf ausgerichtet waren, Unterschiede bei Untergruppen aufzuweisen, schien der Behandlungseffekt in der Untergruppe der männlichen Studienteilnehmer (n = 188) und der Studienteilnehmer nicht kaukasischer Herkunft (n = 137) geringer zu sein als in der gesamten Studienpopulation.
BLASENFUNKTIONSSTÖRUNGEN:
Überaktive Blase bei Erwachsenen
Zwei doppel-blinde, Placebo-kontrollierte, randomisierte, multizentrische 24-wöchige klinische Phase-3-Studien wurden bei Patienten mit überaktiver Blase mit den Symptomen Harninkontinenz, Harndrang und häufige Miktion durchgeführt. Insgesamt wurden1'105 Patienten, deren Symptome durch Anticholinergika nicht adäquat kontrolliert werden konnten (unzureichendes Ansprechen oder inakzeptable Nebenwirkungen), entweder auf 100 Einheiten BOTOX (n = 557) oder Placebo (n = 548) randomisiert.
In beiden Studien wurden für BOTOX (100 Einheiten) signifikante Verbesserungen im Vergleich zu Placebo bei der Änderung der täglichen Häufigkeit von Harninkontinenzepisoden verglichen mit dem Ausgangswert, einschliesslich des Anteils trockener Patienten, zum primären Zeitpunkt in Woche 12 festgestellt. Gemessen anhand der Skala für den Behandlungsnutzen war der Anteil der Patienten, die über ein positives Behandlungsansprechen berichteten (ihr Zustand hatte sich «stark verbessert» oder «verbessert»), in beiden Studien in der BOTOX-Gruppe signifikant grösser als in der Placebo-Gruppe. Signifikante Verbesserungen im Vergleich zu Placebo wurden auch bei der täglichen Miktionshäufigkeit und der Häufigkeit der Episoden von Harndrang und Nykturie beobachtet. Die pro Miktion ausgeschiedene Urinmenge war ebenfalls signifikant grösser. Signifikante Verbesserungen wurden ab Woche 2 bei allen Symptomen der überaktiven Blase festgestellt.
Die BOTOX-Behandlung war gegenüber Placebo mit signifikanten Verbesserungen der gesundheitsbezogenen Lebensqualität verbunden, gemessen anhand des I-QOL-Fragebogens (Incontinence Quality of Life) (mit den Faktoren Vermeidungsverhalten und Einschränkungen, psychosoziale Folgen und soziale Peinlichkeit) und des KHQ-Fragebogens (King's Health Questionnaire) (mit den Faktoren Folgen der Inkontinenz, Einschränkungen bei Alltagsaktivitäten, soziale Einschränkungen, körperliche Einschränkungen, persönliche Beziehungen, Gefühlszustand, Schlaf/Energie und Schweregrad/Gegenmassnahmen).
Zwischen Patienten im Alter von 65 Jahren und älter und Patienten unter 65 Jahren wurde insgesamt nach der BOTOX-Behandlung kein Unterschied in der Wirkung festgestellt.
Die Ergebnisse der gepoolten zentralen Studien sind unten dargestellt:
Tabelle 5: Primäre und sekundäre Wirksamkeitsendpunkte zu Studienbeginn und Veränderung gegenüber Studienbeginn in den gepoolten zentralen Studien
|
|
BOTOX
100 Einheiten
(n = 557)
|
Placebo
(n = 548)
|
p-Wert
| |
Tägliche Häufigkeit von Harninkontinenzepisoden*
| |
Mittelwert zu Studienbeginn
|
5,49
|
5,39
|
| |
Mittlere Veränderung in Woche 2
|
-2,85
|
-1,21
|
<0,001
| |
Mittlere Veränderung in Woche 6
|
-3,11
|
-1,22
|
<0,001
| |
Mittlere Veränderung in Woche 12
|
-2,80
|
-0,95
|
<0,001
| |
Anteil der Patienten mit positivem Behandlungsansprechen auf der Skala des Behandlungsnutzens (%)
| |
Woche 2
|
64,4
|
34,7
|
<0,001
| |
Woche 6
|
68,1
|
32,8
|
<0,001
| |
Woche 12
|
61,8
|
28,0
|
<0,001
| |
Tägliche Häufigkeit von Miktionsepisoden
| |
Mittelwert zu Studienbeginn
|
11,99
|
11,48
|
| |
Mittlere Veränderung in Woche 2
|
-1,53
|
-0,78
|
<0,001
| |
Mittlere Veränderung in Woche 6
|
-2,18
|
-0,97
|
<0,001
| |
Mittlere Veränderung in Woche 12
|
-2,35
|
-0,87
|
<0,001
| |
Tägliche Häufigkeit der Harndrangepisoden
| |
Mittelwert zu Studienbeginn
|
8,82
|
8,31
|
| |
Mittlere Veränderung in Woche 2
|
-2,89
|
-1,35
|
<0,001
| |
Mittlere Veränderung in Woche 6
|
-3,56
|
-1,40
|
<0,001
| |
Mittlere Veränderung in Woche 12b
|
-3,30
|
-1,23
|
<0,001
| |
I-QOL-Gesamtscore
| |
Mittelwert zu Studienbeginn
|
34,1
|
34,7
|
| |
Mittlere Veränderung nach Woche 12bc
|
+22,5
|
+6,6
|
<0,001
| |
KHQ-Fragebogen: Rolleneinschränkungen
| |
Mittelwert zu Studienbeginn
|
65,4
|
61,2
|
| |
Mittlere Veränderung nach Woche 12bc
|
-25,4
|
-3,7
|
<0,001
| |
KHQ-Fragebogen: Soziale Einschränkungen
| |
Mittelwert zu Studienbeginn
|
44,8
|
42,4
|
<0,001
| |
Mittlere Veränderung nach Woche 12bc
|
-16,8
|
-2,5
|
<0,001
|
* Der prozentuale Anteil der Patienten, die in Woche 12 trocken (frei von Inkontinenz) waren, betrug 27,1% in der BOTOX-Gruppe und 8,4% in der Placebo-Gruppe. Die prozentualen Anteile der Patienten, die eine Verringerung der Harninkontinenzepisoden im Vergleich zum Ausgangswert von mindestens 75% bzw. 50% erzielten, waren 46,0% bzw. 60,5% in der BOTOX-Gruppe und 17,7% bzw. 31,0% in der Placebo-Gruppe.
a Koprimäre Endpunkte
b Sekundäre Endpunkte
c Die vorab definierte bedeutsame Mindeständerung im Vergleich zur Baseline war +10 Punkte für I-QOL und 5 Punkte für KHQ.
Die mediane Dauer des Ansprechens nach der BOTOX-Behandlung, basierend auf dem Verlangen der Patienten nach einer erneuten Behandlung, betrug 166 Tage (~24 Wochen).
Bei Patienten, die auch noch an der offenen Verlängerungsstudie teilnahmen und nur mit BOTOX 100 Einheiten behandelt wurden (N = 438), betrug die mediane Dauer des Ansprechens 212 Tage (~30 Wochen).
In den beiden pivotalen Studien nahm nur eine begrenzte Anzahl männlicher Patienten (ca. 12.2% der gesamten Studienpopulation) teil. Eine statistisch signifikante Wirksamkeit konnte in dieser kleinen Subgruppe nicht gezeigt werden.
Insgesamt wurden 834 Patienten in einer Langzeit-Verlängerungsstudie untersucht. Bei erneuten Behandlungen zeigten die Patienten für alle Wirksamkeitsendpunkte ein gleichbleibendes Ansprechen.
In den zentralen Studien entwickelte keiner der 615 Patienten, bei denen Proben analysiert wurden, neutralisierende Antikörper. Bei Patienten, bei denen Proben aus der pivotalen Phase-3-Studie und den offenen Verlängerungsstudien analysiert wurden, entwickelten sich in den folgenden Fällen neutralisierende Antikörper: bei 0 von 954 Patienten (0,0%) während der Behandlung mit 100 Einheiten BOTOX und bei 3 von 260 Patienten (1,2%) nach anschliessender Behandlung mit mindestens einer Dosis von 150 Einheiten. Einer dieser drei Patienten zeigte einen anhaltenden klinischen Nutzen.
Überaktive Blase bei Kindern und Jugendlichen
Aus einer doppelblinden, randomisierten, multizentrischen Parallelgruppenstudie (191622 137) mit Patienten im Alter von 12 bis 17 Jahren mit überaktiver Blase und Symptomen einer Harninkontinenz liegen begrenzte Daten zur Wirksamkeit vor. Eingeschlossen wurden Patienten, die auf mindestens ein anticholinerges Arzneimittel unzureichend angesprochen oder dieses nicht vertragen hatten. Aufgrund von Rekrutierungsproblemen wurden insgesamt 55 von den geplanten 108 Patienten eingeschlossen, so dass die Fallzahl nicht ausreichte, um eine Aussage über die Wirksamkeit in dieser Population treffen zu können. Diese Patienten wurden randomisiert und erhielten 25 Einheiten, 50 Einheiten oder 100 Einheiten, wobei 6 Einheiten/kg Körpergewicht nicht überschritten werden durften; BOTOX 25 Einheiten N = 18, BOTOX 50 Einheiten N = 17 und BOTOX 100 Einheiten N = 20. Vor der Anwendung erhielten die Patienten eine Anästhesie gemäss der lokalen Praxis. Alle Patienten erhielten eine Allgemeinanästhesie oder eine leichte Sedierung.Im primären Endpunkt in Woche 12 «Tägliche Häufigkeit von Tages-Harninkontinenzepisoden» sowie in weiteren Endpunkten wie Häufigkeit von Tages-Miktionsepisoden, Häufigkeit von Tages-Harndrangepisoden oder positivem Behandlungsansprechen konnten teils numerische, aber keine statistisch signifikanten Differenzen zwischen den Behandlungsgruppen festgestellt werden. Von den 55 pädiatrischen Patienten, die einen negativen Ausgangswert für bindende Antikörper oder neutralisierende Antikörper und nach Studienbeginn mindestens einen auswertbaren Wert nach dem Ausgangswert aus einer randomisierten doppelblinden Studie hatten, entwickelte kein Patient nach der Behandlung mit 25–100 Einheiten BOTOX neutralisierende Antikörper.
Harninkontinenz bei Erwachsenen infolge neurogener Detrusorhyperaktivität
Zwei doppel-blinde, Placebo-kontrollierte, randomisierte, multizentrische klinische Studien der Phase 3 wurden bei Patienten mit Harninkontinenz aufgrund neurogener Überaktivität des Detrusor-Muskels und entweder mit spontaner Harnentleerung oder Katheterisierung durchgeführt. Insgesamt wurden 691 Patienten mit Rückenmarksverletzung oder Multipler Sklerose aufgenommen, die mit mindestens einem anticholinergen Wirkstoff nicht angemessen behandelt wurden. Diese Patienten wurden entweder auf 200 Einheiten BOTOX (n = 227), 300 Einheiten BOTOX (n = 223) oder Placebo (n = 241) randomisiert.
Im Vergleich zu Placebo wurden in beiden Phase-3-Studien signifikante Verbesserungen des primären Wirksamkeitsparameters, Veränderung der wöchentlichen Häufigkeit von Inkontinenz-Episoden gegenüber Studienbeginn, beobachtet, wobei BOTOX (200 Einheiten und 300 Einheiten) zum primären Wirksamkeitszeitpunkt in Woche 6 einschliesslich des Anteils bei Patienten, bei denen keine Harninkontinenz mehr auftrat, am besten abschnitt. Es wurden signifikante Verbesserungen der urodynamischen Parameter einschliesslich eines Anstiegs der maximalen zystometrischen Kapazität und einem verringerten Maximaldruck auf den Detrusor-Muskel bei der ersten unbeabsichtigten Detrusor-Kontraktion beobachtet. Ebenso wurden im Vergleich zu Placebo signifikante Verbesserungen in den vom Patienten beurteilten Inkontinenz-spezifischen, gesundheitsbezogenen Wert zur Lebensqualität, die mit dem I-QOL–Fragebogen erfasst wurden, beobachtet (einschliesslich einschränkender Vermeidungstaktiken, psychosozialer Auswirkungen und Schamgefühle). Ein zusätzlicher Nutzen von BOTOX 300 Einheiten gegenüber 200 Einheiten konnte nicht gezeigt werden.
Die Ergebnisse der gepoolten zentralen Studien werden nachfolgend vorgestellt:
Tabelle 6: Primäre und sekundäre Endpunkte zu Studienbeginn und Veränderung gegenüber Studienbeginn in den gepoolten zentralen Studien
|
|
BOTOX
200 Einheiten
(n = 227)
|
Placebo
(n = 241)
|
p-Wert
| |
Wöchentliche Häufigkeit der Harninkontinenz*
| |
Mittelwert zu Studienbeginn
|
32,4
|
31,5
|
| |
Mittlere Veränderung in Woche 2
|
-17,7
|
-9,0
|
<0,001
| |
Mittlere Veränderung in Woche 6a
|
-21,3
|
-10,5
|
<0,001
| |
Mittlere Veränderung in Woche 12
|
-20,6
|
-9,9
|
<0,001
| |
Maximale zystometrische Kapazität (ml)
| |
Mittelwert zu Studienbeginn
|
250,2
|
253,5
|
| |
Mittlere Veränderung in Woche 6b
|
+153,6
|
+11,9
|
<0,001
| |
Maximaler Detrusor-Druck während der 1. unbeabsichtigten Detrusor-Kontraktion (cmH2O):
| |
Mittelwert zu Studienbeginn
|
51,5
|
47,3
|
| |
Mittlere Veränderung in Woche 6b
|
-32,4
|
+1,1
|
<0,001
| |
Gesamtscore Inkontinenz-spezifische Lebensqualitätc,d
| |
Mittelwert zu Studienbeginn
|
35,37
|
35,32
|
| |
Mittlere Veränderung in Woche 6b
|
+25,89
|
+11,15
|
<0,001
| |
Mittlere Veränderung in Woche 12
|
+28,89
|
+8,86
|
<0,001
|
* Der Prozentsatz der Patienten, bei denen eine Inkontinenz nicht mehr auftrat, lag während der gesamten Woche 6 bei 37% in der mit 200 BOTOX Einheiten behandelten Gruppe und bei 9% in der Placebo-Gruppe. Der Anteil, der eine um mindestens 75% reduzierte Häufigkeit von Inkontinenz-Episoden gegenüber Studienbeginn erreichte, lag bei 63% bzw. 24%. Der Anteil, der eine um mindestens 50% reduzierte Häufigkeit gegenüber Studienbeginn erreichte, lag bei 76% bzw. 39%.
a Primärer Endpunkt
b Sekundärer Endpunkt
c Die Skala des I-QOL-Gesamtscores reicht von 0 (grösstes Problem) bis 100 (überhaupt kein Problem).
d In den zentralen Studien lag die vordefinierte minimal-bedeutsame Veränderung (MID – Minimally Important Difference) des I-QOL-Gesamtscores bei 8 Punkten, ausgehend von einem geschätzten MID-Wert von 4-11 Punkten, der von Patienten mit neurogener Überaktivität des Detrusor-Muskels angegeben wurde.
In den beiden zentralen Studien wurde die mediale Dauer des Ansprechens auf die Therapie, ausgehend von der Bitte der Patienten nach erneuter Behandlung, ermittelt und lag bei 256-295 Tagen (36-42 Wochen) in der mit 200 Einheiten behandelten Gruppe im Vergleich zu 92 Tagen (13 Wochen) bei der Gruppe unter Placebo. Bei Patienten, die auch noch an der offenen Verlängerungsstudie teilnahmen und nur mit BOTOX 200 Einheiten behandelt wurden (N = 174), betrug die mediane Dauer des Ansprechens 253 Tage (~36 Wochen).
Bei erneuter Behandlung wiesen die Patienten ein gleichbleibendes Ansprechen auf die Therapie hinsichtlich aller Endpunkte zur Wirksamkeit auf.
In den zentralen Studien entwickelte keiner der 475 Patienten mit neurogener Überaktivität des Detrusor-Muskels, bei denen Proben untersucht wurden, neutralisierende Antikörper.
Bei Patienten, bei denen Proben aus dem Programm zur Entwicklung neuer Medikamente (einschliesslich offene Verlängerungsstudie) analysiert wurden, entwickelten sich in den folgenden Fällen neutralisierende Antikörper: bei 3 von 300 Patienten (1,0%), die nur mit BOTOX 200 Einheiten behandelt wurden, und bei 5 von 258 Patienten (1,9%) nach der Behandlung mit mindestens einer Dosis von 300 Einheiten. Vier dieser acht Patienten zeigten einen anhaltenden klinischen Nutzen.
Nach Marktzulassung wurde eine Placebo-kontrollierte, doppel-blinde Studie bei Patienten mit multipler Sklerose (MS) durchgeführt, die aufgrund einer neurogenen Detrusorhyperaktivität eine Harninkontinenz zeigten und die mit mindestens einem anticholinergen Wirkstoff nicht angemessen behandelt wurden und zu Studienbeginn nicht katheterisiert waren. Diese Patienten wurden entweder auf BOTOX 100 Einheiten (n = 66) oder Placebo (n = 78) randomisiert.
Signifikante Verbesserungen des primären Wirksamkeitsparameters, Veränderung der täglichen Häufigkeit von Inkontinenz-Episoden, gegenüber Studienbeginn wurden im Vergleich zu Placebo für BOTOX (100 Einheiten) zum primären Wirksamkeitszeitpunkt in Woche 6 beobachtet. Dazu zählte auch der Anteil der Patienten, bei denen eine Harninkontinenz nicht mehr auftrat. Es wurden signifikante Verbesserungen der urodynamischen Parameter und des vom Patienten beurteilten I-QOL–Fragebogens beobachtet. Hierzu gehören auch einschränkende Vermeidungstaktiken, psychosoziale Auswirkungen und Schamgefühle im Beisein anderer.
Die Ergebnisse der Studie nach Markteinführung werden nachfolgend vorgestellt:
Tabelle 7: Primäre und sekundäre Endpunkte zu Studienbeginn und Veränderung gegenüber Studienbeginn in der Studie nach Markteinführung mit BOTOX 100 Einheiten und bei zu Studienbeginn nicht-katheterisierten MS-Patienten
|
|
BOTOX
100 Einheiten
(n=66)
|
Placebo
(n=78)
|
p-Wert
| |
Tägliche Häufigkeit von Harninkontinenzepisoden*
| |
Mittelwert zu Studienbeginn
|
4,2
|
4,3
|
| |
Mittlere Veränderung in Woche 2
|
-2,9
|
-1,2
|
p<0,001
| |
Mittlere Veränderung in Woche 6a
|
-3,3
|
-1,1
|
p<0,001
| |
Mittlere Veränderung in Woche 12
|
-2,8
|
-1,1
|
p<0,001
| |
Maximale zystometrische Kapazität (ml)
| |
Mittelwert zu Studienbeginn
|
246,4
|
245,7
|
| |
Mittlere Veränderung in Woche 6b
|
+127,2
|
-1,8
|
p<0,001
| |
Maximaler Detrusor-Druck während der 1. unbeabsichtigten Detrusor-Kontraktion (cmH2O):
| |
Mittelwert zu Studienbeginn
|
35,9
|
36,1
|
| |
Mittlere Veränderung in Woche 6b
|
-19,6
|
+3,7
|
p=0,007
| |
Gesamtscore Inkontinenz-spezifische Lebensqualitätc, d
| |
Mittelwert zu Studienbeginn
|
32,4
|
34,2
|
| |
Mittlere Veränderung in Woche 6b
|
+40,4
|
+9,9
|
p<0,001
| |
Mittlere Veränderung in Woche 12
|
+38,8
|
+7,6
|
p<0,001
|
* Der Prozentsatz der Patienten, bei denen eine Inkontinenz nicht mehr auftrat, lag während der gesamten Woche 6 bei 53,0% in der mit 100 BOTOX Einheiten behandelten Gruppe und bei 10,3% in der Placebo-Gruppe.
a Primärer Endpunkt
b Sekundäre Endpunkte
c Die Skala des I-QOL-Gesamtscores reicht von 0 (grösstes Problem) bis 100 (überhaupt kein Problem).
d Die vordefinierte minimal-bedeutsame Veränderung (MID – Minimally Important Difference) des I-QOL-Gesamtscores lag bei 11 Punkten, ausgehend von einem geschätzten MID-Wert von 4–11 Punkten, der von Patienten mit neurogener Überaktivität des Detrusor-Muskels angegeben wurde.
Die mediane Dauer des Ansprechens in dieser Studie, basierend auf dem Verlangen der Patienten nach einer erneuten Behandlung, betrug 362 Tage (~52 Wochen) in der mit BOTOX 100 Einheiten behandelten Gruppe im Vergleich zu 88 Tagen (~13 Wochen) bei der Gruppe unter Placebo.
Neurogene Detrusorhyperaktivität bei pädiatrischen Patienten ab 5 Jahren
Eine randomisierte, multizentrische klinische Doppelblindstudie mit parallelen Dosierungsgruppen, aber ohne Placebo-Arm [aufgrund der untersuchten pädiatrischen Population] (191622-120) wurde bei Patienten zwischen 5 und 17 Jahren mit Harninkontinenz infolge einer Detrusorhyperaktivität in Verbindung mit einer neurologischen Erkrankung durchgeführt, die mit sauberer intermittierender Katheterisierung behandelt wurden und die auf eine Therapie mit Anticholinergika unzureichend ansprachen oder diese nicht tolerierten. Insgesamt wurden 113 Patienten (darunter 99 mit Neuralrohrdefekten wie Spina bifida oder Tethered Cord-Syndrom, 13 mit Rückenmarksverletzungen [T1 oder darunter] und 1 mit transverser Myelitis) mit BOTOX behandelt. Diese Patienten erhielten nach dem Zufallsprinzip 50 Einheiten, 100 Einheiten oder 200 Einheiten, wobei 6 Einheiten/kg Körpergewicht nicht überschritten werden durften. Patienten, die aufgrund der Höchstmenge von 6 Einheiten/kg weniger als die randomisierte Dosis erhielten, wurden für die Analyse der nächstgelegenen Dosisgruppe zugeordnet. Insgesamt wurden 38 Patienten der Gruppe mit 50 Einheiten, 45 Patienten der Gruppe mit 100 Einheiten und 30 Patienten der Gruppe mit 200 Einheiten BOTOX zugewiesen. Vor der Behandlung erhielten die Patienten eine Narkose entsprechend ihres Alter und der örtlichen Praxis. 109 Patienten (97,3 %) erhielten eine Vollnarkose oder eine bewusste Sedierung und 3 Patienten (2,7 %) eine Lokalanästhesie.
Die Studienergebnisse zeigten in Woche 6 (primärer Zeitpunkt) keine statistisch signifikanten Unterschiede in der primären Wirksamkeitsvariable [vordefinierter statistischer Ansatz] der Änderung der durchschnittlichen täglichen Häufigkeit von Harninkontinenz-Episoden gegenüber dem Ausgangswert zwischen der 100 Einheiten und 50 Einheiten BOTOX-Gruppe (p=0,9949) oder zwischen der 200 Einheiten und 50 Einheiten BOTOX-Gruppe (p=0,9123). Beim Vergleich innerhalb einer jeden Gruppe wurden in allen drei Dosierungsgruppen Verbesserungen der Anzahl täglicher Harninkontinenz-Episoden (normalisiert auf 12 Stunden) von etwa 30 % im Vergleich zum Ausgangswert festgestellt.
Hinsichtlich der sekundären Wirksamkeitsvariable Urinvolumen nach der ersten morgendlichen Katheterisierung ergaben sich in Woche 6 signifikante Gruppenunterschiede zwischen der mit 200 U gegenüber der mit 50 U BOTOX behandelten Dosierungsgruppe (p= 0,0055), während der Anstieg in der 100 Einheiten BOTOX Gruppe im Vergleich zur 50 Einheiten Gruppe statistisch nicht signifikant war (p=0,5117).
Im Hinblick auf Massnahmen zur Verringerung des maximalen Blasendrucks in Woche 6 erwies sich die Verabreichung von 200 Einheiten BOTOX im Vergleich zu 50 Einheiten BOTOX als signifikant wirksamer (p=0,0157), die 100 U-Dosis hingegen nicht (p=0,1737).
Tabelle 8: Ausgangswert und Veränderung gegenüber dem Ausgangswert in Bezug auf die tägliche Häufigkeit von Harninkontinenz-Episoden, das Urinvolumen beim ersten morgendlichen Katheterisieren und den maximalen Detrusor-Druck während der Speicherphase (cmH2O) und die maximale zystometrische Kapazität (mL) in der Studie 191622-120
|
|
BOTOX 200 Einheiten
N=30
|
p-Wert*
| |
Tägliche durchschnittliche Häufigkeit von Harninkontinenz-Episoden während des Tagesa
| |
Mittelwert zu Studienbeginn
|
3,7
|
| |
Mittlere Veränderung* in Woche 2 (95% CI)
|
–1,1 (–1,7, –0,6)
|
<0.0001
| |
Mittlere Veränderung* in Woche 6** (95% CI)
|
–1,3 (–1,8, –0,9)
|
<0.0001
| |
Mittlere Veränderung* in Woche 12 (95% CI)
|
–0,9 (–1,5, –0,4)
|
0.0009
| |
Urinvolumen beim ersten morgendlichen Katheterisieren (mL)b
| |
Mittelwert zu Studienbeginn
|
187,7
|
| |
Mittlere Veränderung* in Woche 2 (95% CI)
|
63,2 (27,9, 98,6)
|
0.0006
| |
Mittlere Veränderung* in Woche 6** (95% CI)
|
87,5 (52,1, 122,8)
|
<0.0001
| |
Mittlere Veränderung* in Woche 12 (95% CI)
|
45,2 (10,0, 80,5)
|
0.0125
| |
Maximaler Detrusordruck (Pdetmax) während der Speicherphase (cm H2O)b
| |
Mittelwert zu Studienbeginn
|
56,7
|
| |
Mittlere Veränderung* in Woche 6** (95% CI)
|
–27,3 (–36,4, –18,2)
|
<0.0001
| |
Maximale zystometrische Kapazität (mL) (MCC)b
| |
Mittelwert zu Studienbeginn
|
202,3
|
| |
Mittlere Veränderung* in Woche 6** (95% CI)
|
63,6 (29,0, 98,1)
|
| |
Unbeabsichtigten Detrusor-Kontraktion
|
|
| |
- Studienbeginn
|
25 (92.6)
|
| |
- Woche 6**
|
13 (46.4)
|
0.0004
|
KI = Konfidenzintervall
* Die Veränderung des Least-Square-Mittelwerts, und der p-Wert basieren auf dem ANCOVA-Modell mit dem Ausgangswert als Kovariable und der Behandlungsgruppe, dem Alter (< 12 Jahre oder ≥12 Jahre), den Tages- Harninkontinenz-Episoden zu Beginn (≤6 oder > 6) und der anticholinergen Therapie (ja/nein) zu Beginn als Faktoren.
** Primärer Zeitpunkt
a Primärer Endpunkt
b Sekundärer Endpunkt
Insgesamt zeigten die Daten zur Wiederholungsbehandlung einen anhaltenden Nutzen.
Keiner von 99 pädiatrischen Patienten, die einen negativen Ausgangswert für bindende oder neutralisierende Antikörper zu Studienbeginn aufwiesen und für die mindestens ein weiterer Wert im Verlauf einer randomisierten Doppelblindstudie und einer Doppelblind-Verlängerungsstudie ausgewertet werden konnte, entwickelte neutralisierende Antikörper nach bis zu vier Behandlungszyklen mit 50 bis 200 Einheiten BOTOX.
ERKRANKUNGEN DER HAUT UND MIT DER HAUT VERBUNDENE ERKRANKUNGEN:
Primäre Hyperhidrosis axillaris
Es wurde eine doppel-blinde, multizentrische klinische Studie bei Patienten mit persistierender bilateraler primärer Hyperhidrosis axillaris durchgeführt. Eine primäre Hyperhidrosis axillaris lag vor, wenn bei einer Ausgangsmessung in Ruhe und bei Raumtemperatur pro Achsel über 5 Minuten mindestens 50 mg Schweiss spontan produziert wurden. 320 Patienten wurden randomisiert und erhielten entweder 50 Einheiten BOTOX (N = 242) oder Placebo (N = 78). Patienten wurden als auf die Therapie ansprechend definiert, wenn sie zumindest eine Reduzierung der Achselschweissproduktion um 50% von der Ausgangsmessung zeigten. Am primären Endpunkt 4 Wochen nach den Injektionen sprachen in der BOTOX-Gruppe 93,8% der Patienten im Vergleich zu 35,9% der Patienten in der Placebo-Gruppe (p < 0,001) auf die Behandlung an. Die Rate der auf die Therapie ansprechenden Patienten war in der BOTOX-Gruppe kontinuierlich an allen nachfolgenden Messpunkten bis zu 16 Wochen nach den Injektionen signifikant grösser (p < 0,001) als in der Placebo-Gruppe.
In einer offenen Folgestudie wurden 207 geeignete Patienten eingeschlossen, die bis zu 3 BOTOX-Behandlungen erhielten. Insgesamt schlossen 174 Patienten die volle Dauer von 16 Monaten der zwei kombinierten Studien ab (4 Monate doppel-blind und 12 Monate offene Nachfolgestudie). Die Ansprechrate 16 Wochen nach der ersten (n = 287), der zweiten (n = 123) und der dritten (n = 30) Behandlung war jeweils 85,0%, 86,2% und 80%. Die mittlere Dauer der Wirkung basierend auf der Einzelbehandlung kombiniert mit der offenen Nachfolgestudie war 7,5 Monate nach der ersten Behandlung, jedoch hielt der Effekt bei 27,5% der Patienten über ein Jahr oder länger an.
|