PharmakokinetikNach oraler Gabe wird Fosamprenavir vor Erreichen der systemischen Zirkulation rasch und nahezu vollständig zu Amprenavir und anorganischem Phosphat hydrolysiert. Die Umwandlung von Fosamprenavir zu Amprenavir scheint hauptsächlich in Darmepithel zu erfolgen.
Die pharmakokinetischen Eigenschaften von Amprenavir nach gemeinsamer Anwendung von Telzir mit Ritonavir wurden bei gesunden Freiwilligen und mit HIV infizierten Patienten untersucht, wesentliche Unterschiede zwischen diesen Gruppen wurden nicht gefunden.
Absorption
Nach Einmalgabe von Fosamprenavir werden maximale Plasmakonzentrationen ungefähr 2 Stunden nach der Einnahme beobachtet. Die AUC-Werte von Fosamprenavir liegen im Allgemeinen bei weniger als 1 % der für Amprenavir beobachteten Werte. Die absolute Bioverfügbarkeit von Fosamprenavir beim Menschen ist nicht bekannt.
Nach mehrfacher oraler Gabe äquivalenter Fosamprenavir- und Amprenavir-Dosen wurden ähnliche AUC-Werte beobachtet; nach Gabe von Fosamprenavir lagen jedoch die Cmax-Werte um ungefähr 30 % niedriger und die Cmin-Werte um ungefähr 28 % höher.
Nach mehrfacher oraler Gabe von 700 mg Fosamprenavir mit 100 mg Ritonavir 2x täglich wurde Amprenavir rasch resorbiert, die maximale Plasmakonzentration (Cmax) von Amprenavir im Steady-State betrug 6,08 (5,38 bis 6,86) µg/ml im geometrischen Mittel (95 % KI) und wurde ungefähr 1,5 (0,75 bis 5,0) Stunden (tmax) nach Gabe erreicht. Im Steady-State betrug die Trough-Konzentration (Cmin) von Amprenavir im Plasma im geometrischen Mittel 2,12 (1,77 bis 2,54) µg/ml, die AUC0-τ betrug 39,6 (34,5 bis 45,3) h•µg/ml.
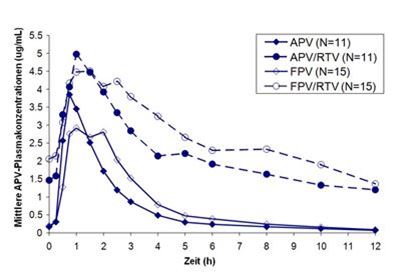
In einer vergleichenden Studie an Probanden wurden über 14 Tage 2 x 700 mg Fosamprenavir (FPV) jeweils mit und ohne Ritonavir (RTV) mit 2 x 600 mg Amprenavir (APV) jeweils mit und ohne 2 x 100 mg Ritonavir verglichen. Dabei konnten folgende Plasmaspiegelkurven für Amprenavir ermittelt werden, die sowohl das Verhältnis zwischen Amprenavir und Fosamprenavir als auch den Effekt der zusätzlichen Gabe von Ritonavir darstellen:
APV10022: Mittlere Konzentrationen von Amprenavir im Plasma
Die Gabe von Fosamprenavir in der Tablettenformulierung mit einer fettreichen Mahlzeit (standardisierte fettreiche Mahlzeit: 967 kcal, 67 Gramm Fett, 33 Gramm Protein, 58 Gramm Kohlenhydrate) war mit keinen Veränderungen in der Pharmakokinetik (Cmax oder AUC) von Amprenavir im Vergleich zur Gabe dieser Formulierung im Nüchternzustand verbunden. Durch Einnahme der Tabletten mit einer fettreichen Mahlzeit nahm die t½ (+29,6%) und die tmax (32,6%) signifikant zu. Die Telzir Filmtabletten können unabhängig von den Mahlzeiten eingenommen werden.
Die Einnahme von Amprenavir zusammen mit Grapefruitsaft war nicht mit signifikanten Veränderungen in der Pharmakokinetik von Amprenavir verbunden.
Distribution
Das scheinbare Verteilungsvolumen von Amprenavir nach Gabe von Telzir beträgt ungefähr 430 l (6 l/kg bei einem Körpergewicht von 70 kg) und lässt auf ein grosses Verteilungsvolumen sowie eine ungehinderte Penetration von Amprenavir aus dem Blutkreislauf in das Gewebe schliessen. Dieser Wert wird, wahrscheinlich auf Grund einer Erhöhung der Bioverfügbarkeit von Amprenavir, um ungefähr 40 % verringert, wenn Telzir mit Ritonavir gegeben wird.
In Invitro-Studien beträgt die Proteinbindung von Amprenavir ca. 90 %. Amprenavir wird an das alpha-1-Säure-Glykoprotein (AAG) sowie an Albumin gebunden, zeigt aber eine höhere Affinität zu AAG. Es wurde gezeigt, dass die AAG-Konzentrationen im Verlauf der antiretroviralen Therapie abnehmen. Diese Änderung führt zu einer Abnahme der Gesamtkonzentration des Wirkstoffs im Plasma, wobei die Menge von ungebundenem Amprenavir, das den aktiven Anteil darstellt, wahrscheinlich unverändert bleibt.
Die Penetration von Amprenavir in die zerebrospinale Flüssigkeit ist beim Menschen vernachlässigbar. Amprenavir scheint in die Samenflüssigkeit zu penetrieren, allerdings sind die Konzentrationen in der Samenflüssigkeit geringer als im Plasma.
Metabolismus
Nach oraler Gabe wird Fosamprenavir während der Resorption durch das Darmepithel rasch und beinahe vollständig zu Amprenavir und anorganischem Phosphat hydrolysiert. Amprenavir wird hauptsächlich über die Leber metabolisiert, nur ca. 1% der eingenommenen Dosis wird in unveränderter Form mit dem Urin ausgeschieden. Der Abbau über das Enzym Cytochrom-P450 CYP3A4 stellt den Hauptstoffwechselweg dar. Die Verstoffwechselung von Amprenavir wird durch Ritonavir über die Inhibierung von CYP3A4 gehemmt mit der Folge erhöhter Plasmakonzentrationen von Amprenavir. Zusätzlich ist Amprenavir auch ein Hemmstoff des CYP3A4-Enzyms, allerdings in geringerem Ausmass als Ritonavir. Daher müssen Arzneimittel, die CYP3A4 induzieren, inhibieren oder ein Substrat für CYP3A4 darstellen, bei gleichzeitiger Gabe von Telzir mit Ritonavir mit Vorsicht angewendet werden (vgl. «Kontraindikationen» und «Interaktionen»).
Elimination
Nach Einnahme von Telzir beträgt die Eliminationshalbwertszeit von Amprenavir 7,7 Stunden. Wenn Telzir zusammen mit Ritonavir gegeben wird, wird die Eliminationshalbwertszeit von Amprenavir auf 15 bis 23 Stunden verlängert.
Die Elimination von Amprenavir erfolgt hauptsächlich über den Leberstoffwechsel, weniger als 1% werden unverändert im Urin ausgeschieden, in den Faeces ist Amprenavir nicht nachweisbar. Ca. 14% der gegebenen Amprenavir-Dosis werden in Form von Metaboliten mit dem Urin und ca. 75% mit den Faeces ausgeschieden.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Fosamprenavir ist ein Prodrug, das beim Menschen zum aktiven Amprenavir umgewandelt wird. Der Hauptweg der Elimination von Amprenavir und Ritonavir führt über den Leberstoffwechsel.
Die Pharmakokinetik von Amprenavir im Plasma wurde in einer Studie an HIV-infizierten erwachsenen Patienten mit Leberfunktionsstörung (Child Pugh A, B und C) untersucht und mit jener bei HIV-Patienten mit normaler Leberfunktion als Kontrolle verglichen. Die Patienten erhielten über 2 Wochen eine Kombinationstherapie aus Fosamprenavir und Ritonavir, wobei die Ritonavir-Dosis bei Patienten mit Leberfunktionsstörung auf einmal täglich 100mg reduziert war.
Dabei wiesen Patienten mit leichter Leberfunktionsstörung gegenüber Lebergesunden eine um 17% höhere Cmax, eine um 22% höhere AUC0-t sowie vergleichbare trough-Konzentrationen auf.
Patienten mit mittelschwerer Leberfunktionsstörung wiesen gegenüber Patienten mit normaler Leberfunktion eine um ca. 70% höhere AUC bei ebenfalls vergleichbaren trough-Konzentrationen auf.
Patienten mit schwerer Leberfunktionsstörung (Child Pugh Score 10-13) wiesen mit 2x täglich 300 mg Fosamprenavir und 1x täglich 100 mg Ritonavir gegenüber Patienten mit normaler Leberfunktion unter 2x täglich 700 mg Fosamprenavir und 2x täglich 100 mg Ritonavir eine um 19% tiefere Plasma Amprenavir Cmax, eine um 23% tiefere AUC (0-τ) und um 38% tiefere Cτ-Werte bei vergleichbaren ungebundenen Plasma Amprenavir Cτ-Werten auf. Trotz Reduktion der Dosierungsfrequenz von Ritonavir wiesen die Patienten mit schwerer Leberfunktionsstörung eine um 64% höhere Ritonavir Cmax, eine um 40% höhere mittlere Ritonavir Plasmakonzentration (Cavg) und eine um 38% höhere Ritonavir Cτ auf als Patienten mit normaler Leberfunktion unter Fosamprenavir 700 mg / Ritonavir 100 mg 2x täglich.
Nierenfunktionsstörungen
Patienten mit Nierenfunktionsstörungen wurden nicht speziell untersucht. Weniger als 1% der therapeutischen Dosis von Amprenavir wird unverändert im Urin ausgeschieden. Auch ist die renale Clearance von Ritonavir vernachlässigbar, daher dürfte die Auswirkung einer Nierenfunktionsstörung auf die Elimination von Amprenavir und Ritonavir gering sein.
Ältere Patienten
Die Pharmakokinetik von Fosamprenavir in Kombination mit Ritonavir wurde bei Patienten über 65 Jahren nicht untersucht.
Kinder und Jugendliche
In einer klinischen Studie zur Pharmakokinetik von Fosamprenavir bei pädiatrischen Patienten erhielten 8 Testpersonen im Alter von 12 bis 18 Jahren die bei Erwachsenen übliche Fosamprenavir-Tablettendosis von 700 mg zweimal täglich (mit 100 mg Ritonavir zweimal täglich). Im historischen Vergleich zu erwachsenen Patienten, die Fosamprenavir / Ritonavir 700 mg / 100 mg zweimal täglich erhielten, lagen bei den Testpersonen im Alter von 12 bis 18 Jahren die AUC(0-24)-Werte von APV im Plasma um 20 %, die Cmax- um 23 % und die Cmin-Werte um 20 % niedriger. Bei Kindern im Alter von 6 bis 11 Jahren (n=9), die Fosamprenavir / Ritonavir 18 / 3 mg/kg zweimal täglich erhielten, lagen die AUC(0-24)-Werte um 26 % höher bei ähnlichen Werten für Cmax- und Cmin im historischen Vergleich zu erwachsenen Patienten, die Fosamprenavir / Ritonavir 700 mg / 100 mg zweimal täglich erhielten.
APV20002 ist eine offene Phase-II-Studie über 48 Wochen zur Untersuchung der Pharmakokinetik, Sicherheit, Verträglichkeit und antiviralen Aktivität von Fosamprenavir mit und ohne Ritonavir bei
pädiatrischen Testpersonen im Alter von 4 Wochen bis zu < 2 Jahren. Im historischen Vergleich zu erwachsenen Patienten, die Fosamprenavir / Ritonavir 700 mg / 100 mg zweimal täglich erhielten, wurde bei einer Untergruppe von 5 Testpersonen im Alter von 6 bis < 24 Monaten, die Fosamprenavir / Ritonavir 45 / 7 mg/kg zweimal täglich erhielten, gezeigt, dass trotz eines ungefähr fünffachen Anstiegs der Dosen von Fosamprenavir / Ritonavir, bezogen auf mg/kg-Basis, die AUC(0-τ) von
Fosamprenavir im Plasma um ungefähr 48 %, Cmax um 26 % und Cτ um 29 % bei den pädiatrischen Testpersonen niedriger lagen. Daher können für sehr kleine Kinder (Kinder < 2 Jahre) keine Dosisempfehlungen gegeben werden, auch wird Telzir mit Ritonavir nicht für diese Patientengruppe empfohlen (siehe «Dosierung / Anwendung»).
Schwangerschaft
Die Pharmakokinetik von Amprenavir (APV) wurde an schwangeren Frauen untersucht, die während des zweiten Trimenons (n=6) oder dritten Trimenons (n=9) und während der postpartalen Phase mit FPV/RTV 700/100 mg zweimal täglich behandelt wurden. Die Exposition gegenüber APV fiel während der Schwangerschaft um 25-35% niedriger aus. Die geometrischen Mittelwerte von APV (95%-KI) und Ctau-Werte lagen für das zweite Trimenon bei 1,31 (0,97; 1,77), für das dritte Trimenon bei 1,34 (0,95; 1,89) und für die postpartale Phase bei 2,03 (1,46; 2,83) μg/ml und befanden sich innerhalb des Wertebereichs bei nicht-schwangeren Patienten unter verschiedenen FPV/RTV-haltigen Therapieregimen. Eine geringe bis mittelgradige Plazentagängigkeit wurde beobachtet. Diese Schlussfolgerung basierte auf einem Verhältnis der Kleinstquadratmittelwerte von APV (95%-KI) hinsichtlich der Konzentration im fetalen Nabelschnurblut zur Konzentration im maternalen peripheren Plasma von 0,27 (0,24; 0,30).
|