ZusammensetzungWirkstoffe
Romiplostim (aus gentechnisch veränderten E. coli Bakterien hergestellt).
Hilfsstoffe
Pulver: Mannitol (E421), Saccharose, L-Histidin, verdünnte Salzsäure (für den Ausgleich des pH-Werts), Polysorbat 20 (Polysorbat 20 wird mit gentechnisch verändertem Mais hergestellt).
Lösungsmittel: Wasser für Injektionszwecke.
Indikationen/AnwendungsmöglichkeitenErwachsene:
Nplate ist für die Behandlung von erwachsenen Patienten mit primärer Immunthrombozytopenie (ITP) indiziert, die gegenüber anderen Therapien refraktär sind (z.B. Kortikosteroide, Immunglobuline; siehe Rubriken «Dosierung/Anwendung» und «Klinische Wirksamkeit»).
Kinder und Jugendliche:
Nplate ist für die Behandlung von pädiatrischen Patienten im Alter von 1 Jahr oder älter mit chronischer primärer Immunthrombozytopenie (ITP) indiziert, die gegenüber anderen Therapien refraktär sind (z.B. Kortikosteroide, Immunglobuline; siehe Rubriken «Dosierung/Anwendung» und «Klinische Wirksamkeit»).
Dosierung/AnwendungDie Behandlung soll durch eine erfahrene Fachperson überwacht werden.
Nplate sollte nur bei Patienten mit erhöhtem Blutungsrisiko infolge ausgeprägter Thrombozytopenie eingesetzt werden.
Die Injektionslösung von Nplate wird subkutan verabreicht.
Anweisungen zur Rekonstitution von Nplate vor der Anwendung sind unter «Sonstige Hinweise: Hinweise für die Handhabung» vorzufinden.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Medikationsfehler, einschliesslich Über- und Unterdosierung, wurden bei Patienten berichtet, die Nplate erhielten. Dies kann zu thromboembolischen Ereignissen bzw. Blutungen führen. Während der Berechnung der Dosierung, der Rekonstitution mit dem richtigen Volumen des sterilen Wassers und der Entnahme des zu verabreichenden Volumens sind die Anwendungsanweisungen strikt zu befolgen. Bei einigen pädiatrischen Patienten erfordert die genaue Dosierung einen zusätzlichen Verdünnungsschritt nach der Rekonstitution. Die Thrombozyten-Zahl ist engmaschig zu überwachen (siehe «Warnhinweise und Vorsichtsmassnahmen» und «Überdosierung»).
Die empfohlene Anfangsdosis von Nplate beträgt 1 Mikrogramm (mcg)/kg Körpergewicht, verabreicht einmal wöchentlich als subkutane Injektion. Die Dosis soll entsprechend der untenstehenden Tabelle so eingestellt werden, dass eine Thrombozytenzahl von über 50 × 109/l erreicht und aufrechterhalten wird. Die maximale Dosis soll dabei 10 Mikrogramm (mcg)/kg nicht übersteigen.
Dosisanpassungen sollen aufgrund wöchentlicher Thrombozytenbestimmungen vorgenommen werden, bis die Werte im empfohlenen Bereich liegen und stabil sind. Danach soll die Thrombozytenzahl mindestens einmal monatlich bestimmt und die Dosis entsprechend der Tabelle für die Dosisanpassung bei Erwachsenen (Tabelle 1) oder der Tabelle für die Dosisanpassung bei pädiatrischen Patienten (Tabelle 2) so angepasst werden, dass die Thrombozytenzahl im empfohlenen Bereich verbleibt. Siehe untenstehende Tabelle 1 und Tabelle 2 für Dosisanpassung und Überwachung.
Dosisbestimmung
Das anzuwendende Volumen wird basierend auf dem Körpergewicht, der erforderlichen Dosis und der Konzentration des Arzneimittels berechnet.
Tabelle 1. Richtlinien zur Dosisanpassung bei Erwachsenen gemäss Thrombozytenzahl
|
Thrombozytenzahl
(× 109/l)
|
Massnahme
|
Anpassungshäufigkeit
| |
Die Anfangsdosis beträgt 1 Mikrogramm (mcg)/kg gemäss tatsächlichem Körpergewicht
| |
< 50
|
Dosiserhöhung in Schritten von 1 mcg/kg.
|
wöchentlich
| |
> 200 bis 400
|
Dosisreduktion im Ermessen des behandelnden Arztes in Schritten von 1 mcg/kg, um eine empfohlene Thrombozytenzahl zwischen 50 und 200 × 109/l aufrechtzuerhalten.
|
Alle 2 Wochen
| |
> 400
|
Nicht verabreichen, Aussetzen der geplanten Dosis.
Die Thrombozytenzahl sollte mindestens einmal pro Woche kontrolliert werden, um eine Veränderung der Thrombozytenzahl zu ermitteln. Wiederaufnahme der Behandlung mit einer um 1 mcg/kg verminderten, einmal pro Woche anzuwendenden Dosis, mit dem Ziel eine Thrombozytenzahl von über 50 × 109/l beizubehalten. In klinischen Studien wurde die Dosierung wieder aufgenommen, wenn die Thrombozytenzahl unter 200 × 109/l fiel. Dabei soll beachtet werden, dass der Therapieeffekt von Nplate meistens mit 1-2-wöchiger Verzögerung eintritt und sich die Thrombozytenzahl rasch vermindern kann.
|
wöchentlich
| |
Falls nach einem Therapieunterbruch die Thrombozytenzahl abnimmt, ist die Therapie mit der letzten Nplate-Dosis wiederaufzunehmen.
In den placebokontrollierten Studien lagen die häufigsten wöchentlichen Dosen für splenektomierte Patienten im Bereich 2-7 mcg/kg (25.-75. Perzentil; Median 3 mcg/kg) und für nicht-splenektomierte Patienten bei 1-3 mcg/kg (25.-75. Perzentil; Median 2 mcg/kg).
| |
Siehe «Warnhinweise und Vorsichtsmassnahmen» für den Fall, dass der Patient nicht mehr auf Nplate anspricht.
|
Tabelle 2. Richtlinien zur Dosisanpassung bei pädiatrischen Patienten gemäss Thrombozytenzahl
|
Thrombozytenzahl
(× 109/l)
|
Massnahme
|
Anpassungshäufigkeit
| |
Die Anfangsdosis beträgt 1 Mikrogramm (mcg)/kg gemäss tatsächlichem Körpergewicht
| |
< 50
|
Dosiserhöhung in Schritten von 1 mcg/kg.
|
wöchentlich
| |
50 bis 200
|
Dosis bleibt konstant.
|
wöchentlich
| |
> 200 bis < 400 während 2 aufeinanderfolgender Wochen
|
Dosisreduktion in Schritten von 1 mcg/kg.
|
wöchentlich
| |
≥400
|
Nicht verabreichen, aussetzen der geplanten Dosis.
Wiederaufnahme der Therapie, wenn eine Thrombozytenzahl von < 200 × 109/l erreicht ist. Eine Reduzierung der Dosis um 1 mcg/kg zum Zeitpunkt der nächsten geplanten Dosierung ist in Betracht zu ziehen. Eine Beibehaltung der Romiplostim-Dosis ist in Betracht zu ziehen, wenn die Erhöhung der Thrombozytenzahl auf den Therapiebeginn oder eine Dosissteigerung eines gleichzeitig angewendeten ITP-Arzneimittels zurückzuführen ist.
|
wöchentlich
| |
Falls nach einem Therapieunterbruch die Thrombozytenzahl abnimmt, ist die Therapie mit der letzten Nplate-Dosis wiederaufzunehmen.
| |
Siehe «Warnhinweise und Vorsichtsmassnahmen» für den Fall, dass der Patient nicht mehr auf Nplate anspricht.
|
Tabelle 3. Richtlinien für die Berechnung der individuellen Patientendosis und des anzuwendenden Romiplostim-Volumens
|
Individuelle Patientendosis (mcg)
|
Individuelle Patientendosis (mcg) = Gewicht (kg) × Dosis in mcg/kg.
Bei der Berechnung der Anfangsdosis sollte immer das tatsächliche Körpergewicht bei Behandlungsbeginn zugrunde gelegt werden.
·Bei Erwachsenen basieren künftige Dosisanpassungen lediglich auf Veränderungen der Thrombozytenzahlen.
·Bei pädiatrischen Patienten basieren künftige Dosisanpassungen auf Veränderungen der Thrombozytenzahlen und Veränderungen des Körpergewichts. Eine erneute Überprüfung des Körpergewichts im Abstand von 12 Wochen wird empfohlen.
| |
Bei einer individuellen Patientendosis ≥23 mcg
|
Lyophilisiertes Arzneimittel rekonstituieren (wie unter «Sonstige Hinweise: Hinweise für die Handhabung» beschrieben). Dies ergibt eine Konzentration von 500 mcg/ml.
Zu verabreichendes Volumen (ml) = individuelle Patientendosis (mcg) / 500 mcg/ml
(Volumen auf hundertstel ml auf- bzw. abrunden).
| |
Bei einer individuellen Patientendosis < 23 mcg
|
Zur Sicherstellung der genauen Dosierung ist eine Verdünnung erforderlich. Lyophilisiertes Arzneimittel rekonstituieren und anschliessend verdünnen wie unter «Sonstige Hinweise: Hinweise für die Handhabung» beschrieben. Dies ergibt eine Konzentration von 125 mcg/ml.
Zu verabreichendes Volumen (ml) = individuelle Patientendosis (mcg) / 125 mcg/ml
(Volumen auf hundertstel ml auf- bzw. abrunden).
| |
Beispiel
|
Ein 10 kg schwerer Patient beginnt mit 1 mcg/kg Romiplostim.
Individuelle Patientendosis (mcg) = 10 kg × 1 mcg/kg = 10 mcg.
Da die Dosis < 23 mcg beträgt, ist die Verdünnung erforderlich, um eine genaue Dosierung sicherzustellen. Lyophilisiertes Arzneimittel rekonstituieren und anschliessend verdünnen wie unter «Sonstige Hinweise: Hinweise für die Handhabung» beschrieben. Dies ergibt eine Konzentration von 125 mcg/ml.
Zu verabreichendes Volumen (ml) = 10 mcg/125 mcg/ml = 0,08 ml.
|
Behandlungsabbruch
Nach einem Behandlungsabbruch ist mit dem Wiederauftreten der Thrombozytopenie zu rechnen (siehe «Warnhinweise und Vorsichtsmassnahmen»). Die Patienten sollen periodisch klinisch beurteilt werden und der behandelnde Arzt bzw. die behandelnde Ärztin sollte über eine Weiterführung der Behandlung individuell entscheiden. Dies sollte bei nicht-splenektomierten Patienten eine Einschätzung hinsichtlich einer Splenektomie einschliessen.
Fehlendes Therapieansprechen
Patienten, die mit der höchsten wöchentlichen Dosis von Nplate 10 mcg/kg auch nach 4 Wochen eine Thrombozytenzahl von mindestens 20 × 109/l nicht erreichen, sollten die Behandlung abbrechen.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Romiplostim sollte bei Patienten mit mässig gradiger und schwerer Leberinsuffizienz (Child-Pugh, Score ≥7) nicht angewendet werden, es sei denn, der erwartete Nutzen einer Behandlung der Thrombozytopenie mit TPO-Agonisten übersteigt das bekannte Risiko einer Pfortaderthrombose bei Patienten mit einer Leberinsuffizienz (siehe «Unerwünschte Wirkungen»).
Wird die Anwendung von Romiplostim als notwendig erachtet, sollte die Thrombozytenzahl engmaschig überwacht werden, um das Risiko thromboembolischer Komplikationen zu minimieren.
Patienten mit Nierenfunktionsstörungen
Es wurden keine Studien durchgeführt.
Ältere Patienten (≥65 Jahre)
Es wurden keine signifikanten Unterschiede bezüglich Wirksamkeit oder Sicherheit zwischen Patienten < 65 und ≥65 Jahren festgestellt (siehe «Eigenschaften/Wirkungen»). Bei älteren Patienten sind keine Dosierungsanpassungen erforderlich.
Pädiatrische Population
Es gibt keine relevante Anwendung von Romiplostim bei pädiatrischen Patienten mit chronischer ITP im Alter von unter 1 Jahr.
KontraindikationenBekannte Überempfindlichkeit gegenüber dem Wirkstoff, gegenüber einem der Hilfsstoffe gemäss Zusammensetzung oder gegen Proteine, die von E. coli stammen.
Warnhinweise und VorsichtsmassnahmenDie nachfolgenden Warnungen und Vorsichtsmassnahmen betreffen identifizierte Risiken oder stellen theoretisch mögliche Klasseneffekte von TPO-Rezeptor-Stimulanzien dar.
Retikulinbildung im Knochenmark und Risiko einer Knochenmarkfibrose
Die Anwendung von Nplate erhöht das Risiko für die Entwicklung oder das Fortschreiten einer Ablagerung von Retikulinfasern im Knochenmark. In klinischen Studien wurde Nplate bei 4 von insgesamt 271 Patienten infolge von Retikulinablagerungen im Knochenmark abgesetzt. Bei 6 weiteren Patienten wurden in Knochenmarkbiopsien Retikulinablagerungen beobachtet. Alle 10 Patienten mit Retikulinablagerungen im Knochenmark hatten eine Nplate-Dosis von ≥5 mcg/kg erhalten, 6 der Patienten waren mit Dosen von ≥10 mcg/kg behandelt worden. In den kontrollierten klinischen Studien wurde keine Progression zu einer Knochenmarkfibrose mit Zytopenie beobachtet. In der Folgestudie entwickelte 1 Patient mit ITP und hämolytischer Anämie im Rahmen der Nplate-Therapie eine Knochenmarkfibrose mit gesteigerter Kollagensynthese. Das Risiko einer Knochenmarkfibrose mit Zytopenie konnte durch die klinischen Studien nicht ausgeschlossen werden.
In einer Open-Label-Studie zur Untersuchung der Veränderungen hinsichtlich des Retikulins- und Kollagens im Knochenmark entwickelten insgesamt 1,5% (2 von 132) der Patienten mit einer evaluierbaren Knochenmark Trichrom-Färbung Kollagen. Die mittlere wöchentliche Romiplostim Dosis für diese zwei Probanden betrug 7,8 und 9,7 mcg/kg. Bei dem einen Patienten, der zwölf Wochen nach Absetzen der Behandlung mit Romiplostim einem Wiederholungstest unterzogen wurde, war kein Kollagen nachweisbar. Bei 6,9% (9 von 131) der Patienten wurde ein Fortschreiten der Retikulinfaserbildung um 2 oder mehr Grade oder ein Kollagenanstieg auf Grad 4 berichtet. Der Median der mittleren wöchentlichen Romiplostim Dosis bei den 9 Patienten betrug 7,7 mcg/kg (Bereich: 3,0 – 9,7).
Vor Beginn der Nplate-Therapie muss eine genaue Analyse des peripheren Blutausstrichs vorgenommen werden, um einen Ausgangswert für Veränderungen in der Zellmorphologie zu erhalten. Nach der Bestimmung einer stabilen Nplate-Dosis sind monatlich ein peripherer Blutausstrich und ein grosses Blutbild („complete blood count“, CBC) zu erstellen und auf neue oder fortschreitende morphologische Abnormalitäten (z.B. tränenförmige und kernhaltige rote Blutkörperchen, unreife weisse Blutkörperchen) und Zytopenien zu untersuchen. Entwickelt der Patient neue oder fortschreitende morphologische Abnormalitäten oder Zytopenien, ist die Behandlung mit Nplate einzustellen und eine Knochenmarkbiopsie, inklusive Anfärbung zur Feststellung einer Fibrose, in Betracht zu ziehen.
Verstärkung der Thrombozytopenie nach Absetzen von Nplate
Das Absetzen von Nplate kann eine vor Beginn der Nplate-Therapie bestehende Thrombozytopenie verstärken (dies kann bei Erwachsenen und Kindern auftreten). Es kann ein erhöhtes Blutungsrisiko bestehen, insbesondere wenn die Behandlung mit Nplate bei gleichzeitiger Behandlung mit Antikoagulanzien oder Thrombozytenaggregationshemmern abgesetzt wird. In klinischen Studien an Patienten mit ITP, bei denen Nplate abgesetzt wurde, verschlimmerte sich die Thrombozytopenie bei 4 von 57 Patienten gegenüber dem Zustand vor der Behandlung mit Nplate. Die Verstärkung der Thrombozytopenie war innerhalb von 14 Tagen reversibel. Nach Absetzen von Nplate muss mindestens 2 Wochen lang wöchentlich das grosse Blutbild einschliesslich Thrombozytenzahl kontrolliert werden; zudem ist eine alternative Behandlung der verstärkten Thrombozytopenie gemäss den aktuellen Behandlungsrichtlinien in Betracht zu ziehen.
Thrombotische/thromboembolische Komplikationen
Thrombotische/thromboembolische Komplikationen können auf eine übermässige Erhöhung der Thrombozytenzahl zurückzuführen sein. Eine zu hohe Dosierung von Nplate oder Medikationsfehler, die zu überhöhten Nplate-Dosen führen, können die Thrombozytenzahl auf ein Mass erhöhen, das thrombotische/thromboembolische Komplikationen verursacht.
Thrombotische/thromboembolische Ereignisse, einschliesslich tiefer Beinvenenthrombose, Lungenembolie und Myokardinfarkt, wurden bei der Anwendung von Romiplostim in der ITP-Population beobachtet.
Diese Ereignisse traten unabhängig von der Thrombozytenzahl auf. Die Patienten sind auf Anzeichen und Symptome thrombotischer/thromboembolischer Ereignisse zu überwachen und unverzüglich gemäss institutionellen Richtlinien und klinischen Standardverfahren zu behandeln. Zwecks Minimierung des Risikos thrombotischer/thromboembolischer Komplikationen sind die Richtlinien zur Dosisanpassung zu befolgen, um eine Thrombozytenzahl von ≥50 × 109/l zu erreichen und aufrechtzuerhalten. Nplate darf nicht zur Normalisierung der Thrombozytenzahl verwendet werden (siehe «Dosierung/Anwendung»).
Besondere Aufmerksamkeit ist bei Patienten mit erhöhtem Thromboembolierisiko aufgrund von erblichen (z.B. Factor V Leiden) oder erworbenen Zuständen (z.B. ATIII Mangel, Antiphospholipid Syndrom) sowie bei Patienten fortgeschrittenen Alters, bei chirurgischen Eingriffen, längerer Bettlägerigkeit, malignen Tumoren, Co-Medikation mit Kontrazeptiva oder Hormon-Ersatztherapie, Obesität und Rauchen geboten.
Fälle von thromboembolischen Ereignissen, einschliesslich Pfortaderthrombose, wurden bei Patienten mit chronischer Lebererkrankung, die Romiplostim erhielten, berichtet. Romiplostim sollte in diesen Populationen mit Vorsicht angewendet werden (siehe «Dosierung/Anwendung» und «Überdosierung»).
Mangelhaftes oder Verlust des Ansprechens auf Nplate
Bei einem mangelhaften oder nur vorübergehenden Ansprechen der Thrombozyten auf Nplate sind die möglichen Ursachen zu untersuchen; unter anderem ist festzustellen, ob neutralisierende Antikörper gegen Nplate oder eine Knochenmarkfibrose vorliegen (siehe «Warnhinweise und Vorsichtsmassnahmen» sowie «Unerwünschte Wirkungen»). Die Behandlung mit Nplate sollte abgebrochen werden, wenn die Thrombozytenzahl nach vierwöchiger Behandlung mit der höchsten wöchentlichen Dosis von 10 mcg/kg nicht auf einen ausreichend hohen Wert steigt, um klinisch signifikante Blutungen zu vermeiden.
Medikationsfehler
Medikationsfehler einschliesslich Über- und Unterdosierung wurden bei Patienten, die Nplate erhielten, berichtet. Bei einigen pädiatrischen Patienten erfordert die genaue Dosierung einen zusätzlichen Verdünnungsschritt nach der Rekonstitution (siehe «Dosierung/Anwendung»). Über- oder Unterdosierung kann zu übermässig hohen oder tiefen Thrombozytenzahlen führen, welche zu thromboembolischen Ereignissen bzw. Blutungen führen können. Sollte die Thrombozytenzahl übermässig ansteigen, ist Nplate abzusetzen und die Thrombozytenzahl sollte überwacht werden. Die Wiederaufnahme der Behandlung mit Nplate sollte in Übereinstimmung mit den Dosierungs- und Anwendungsempfehlungen durchgeführt werden (siehe «Dosierung/Anwendung» und «Überdosierung»).
Maligne Erkrankungen und Progression maligner Erkrankungen
Nplate ist nicht für die Behandlung von MDS-bedingter Thrombozytopenie oder Thrombozytopenien mit anderen Ursachen als ITP indiziert. Romiplostim sollte nicht zur Behandlung von Thrombozytopenie infolge einer anderen Krankheit als ITP eingesetzt werden, weil für diese Situation kein günstiges Nutzen-Risiko-Verhältnis gezeigt worden ist.
Die durch Nplate angeregte Stimulierung des TPO-Rezeptors auf der Oberfläche von hämatopoetischen Zellen kann das Risiko für hämatologische Malignome erhöhen. In klinischen Studien mit Erwachsenen zur Behandlung von MDS-Patienten mit Romiplostim wurden Fälle von vorübergehenden Blastenanstiegen beobachtet, und es wurden Fälle einer Krankheitsprogression von MDS zu einer akuten myeloischen Leukämie (AML) berichtet. In einer randomisierten, placebokontrollierten Studie mit MDS-Patienten wurde die Behandlung mit Romiplostim aufgrund einer numerischen Häufung von Krankheitsprogression zu AML und einer Erhöhung zirkulierender Blasten von mehr als 10% bei Patienten, die mit Romiplostim behandelt wurden, vorzeitig beendet.
Laborüberwachung
Vor und während der Behandlung mit Nplate sowie nach Absetzung des Präparats muss regelmässig das grosse Blutbild einschliesslich Thrombozytenzahl und peripherem Blutausstrich kontrolliert werden. Vor Behandlungsbeginn mit Nplate ist ein Differentialblutbild des peripheren Blutes zu erstellen, um einen Ausgangswert für Zellanomalien der roten und weissen Blutkörperchen zu erhalten. Während der Dosisanpassung von Nplate ist wöchentlich ein grosses Blutbild einschliesslich Thrombozytenzahl und peripherem Blutausstrich zu erstellen; nach der Ermittlung einer stabilen Dosierung muss dies einmal im Monat erfolgen. Nach Absetzung von Nplate ist mindestens 2 Wochen lang wöchentlich ein grosses Blutbild einschliesslich Thrombozytenzahl zu erstellen (siehe «Dosierung/Anwendung» sowie vorhergehende Abschnitte «Retikulinbildung im Knochenmark und Risiko einer Knochenmarkfibrose» und «Verstärkung der Thrombozytopenie nach Absetzen von Nplate»).
Auswirkungen von Romiplostim auf rote und weisse Blutzellen
Veränderungen in den Parametern roter (Abnahme) und weisser (Zunahme) Blutzellen wurden bei ITP-Patienten beobachtet. Eine gleichzeitige Anämie und Leukozytose (innerhalb eines 4-wöchigen Zeitfensters) kann bei Patienten unabhängig vom Splenektomie-Status auftreten. Diese Auswirkungen wurden allerdings häufiger bei Patienten beobachtet, die sich vorher einer Splenektomie unterzogen hatten. Eine Überwachung dieser Parameter sollte bei Patienten, die mit Romiplostim behandelt werden, erwogen werden.
InteraktionenEs wurden keine Interaktionsstudien durchgeführt.
Medikamentöse ITP-Therapien, die in klinischen Studien in Kombination mit Nplate verwendet wurden, umfassen Kortikosteroide, Danazol und/oder Azathioprin, intravenöse Immunglobuline (IVIg) und Anti-D-Immunglobulin. Bei Kombination von Nplate mit anderen medikamentösen ITP-Therapien sollen durch Überwachung der Thrombozytenzahl Werte ausserhalb des empfohlenen Bereichs verhindert werden (siehe «Dosierung/Anwendung»).
Die Verabreichung von Kortikosteroiden, Danazol und Azathioprin kann möglicherweise unter sorgfältiger Überwachung der Thrombozytenzahl reduziert oder abgebrochen werden, wenn Nplate gleichzeitig gegeben wird (siehe «Eigenschaften/Wirkungen»).
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine klinischen Studien mit schwangeren Frauen vor.
Tierexperimentelle Studien haben reproduktionstoxikologische Effekte gezeigt (siehe «Präklinische Daten»). Das potenzielle Risiko für den Menschen ist nicht bekannt.
Nplate darf während der Schwangerschaft nicht angewendet werden, es sei denn, dies ist klar notwendig.
Stillzeit
Es ist nicht bekannt, ob Romiplostim beim Menschen in die Muttermilch ausgeschieden wird. Eine Anwendung von Nplate während der Stillzeit wird nicht empfohlen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zu den Auswirkungen auf die Fahrtüchtigkeit und die Fähigkeit, Maschinen zu bedienen, durchgeführt.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Basierend auf einer Analyse aller erwachsenen ITP-Patienten, welche Romiplostim in 4 kontrollierten und 5 unkontrollierten klinischen Studien erhalten haben, betrug die Gesamtinzidenz aller unerwünschten Wirkungen bei Probanden, die mit Romiplostim behandelt wurden, 91,5% (248/271).
Die mittlere Dauer der Romiplostim-Exposition in dieser Studienpopulation war 50 Wochen.
Innerhalb jeder Systemorganklasse gemäss MedDRA und Häufigkeitsgruppe werden die unerwünschten Wirkungen nach abnehmender Inzidenz aufgeführt. Die Häufigkeiten sind wie folgt definiert: sehr häufig (≥1/10), häufig (≥1/100, < 1/10), gelegentlich (≥1/1'000, < 1/100) und nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Erkrankungen des Blut- und Lymphsystems
Häufig: Anämie, Knochenmarkstörung, Thrombozytopenie
Gelegentlich: aplastische Anämie, Knochenmarkversagen, Leukozytose, Splenomegalie, Thrombozythämie, erhöhte Thrombozytenzahl, abnorme Thrombozytenzahl, Myelofibrose
Erkrankungen des Immunsystems
Sehr häufig: Überempfindlichkeit (19,4%) (Fälle von Hautausschlägen, Urtikaria, Angioödem)
Häufig: Angioödem
Nicht bekannt: anaphylaktische Reaktion
Herzerkrankungen
Gelegentlich: Myokardinfarkt, erhöhte Herzfrequenz
Erkrankungen des Ohrs und des Labyrinths
Gelegentlich: Vertigo
Augenerkrankungen
Gelegentlich: konjunktivale Hämorrhagie, Störung der Akkommodation, Blindheit, Funktionsstörung des Auges, juckende Augen, gesteigerte Tränensekretion, Stauungspapille, Sehstörungen
Erkrankungen des Gastrointestinaltrakts
Häufig: Übelkeit, Diarrhö, Abdominalschmerzen, Konstipation, Dyspepsie
Gelegentlich: Erbrechen, rektale Hämorrhagie, Mundgeruch, Dysphagie, Gastroösophagealer Reflux, Hämatochezie, Hämorrhagie im Mund, Magenbeschwerden, Stomatitis, Zahnverfärbung
Allgemeine Beschwerden und Reaktionen an der Applikationsstelle
Häufig: Fatigue, periphere Ödeme, Influenza-ähnliche Erkrankung, Schmerzen, Asthenie, Pyrexie, Schüttelfrost, Reaktion an der Injektionsstelle
Gelegentlich: Hämorrhagie an der Injektionsstelle, Brustschmerzen, Reizbarkeit, Malaise, Gesichtsödem, Hitzegefühl, Nervosität
Leber- und Gallenerkrankungen
Gelegentlich: Pfortaderthrombose, Erhöhung der Transaminase
Infektionen und parasitäre Erkrankungen
Häufig: Sinusitis*, Bronchitis*
Gelegentlich: Influenza, örtlich begrenzte Infektion, Nasopharyngitis
* zusätzliche unerwünschte Wirkungen, beobachtet bei erwachsenen Patienten mit ITP ≤12 Monate
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Häufig: Kontusion
Untersuchungen
Gelegentlich: erhöhte Laktatdehydrogenase im Blut, erhöhte Körpertemperatur, Gewichtsabnahme, Gewichtszunahme
Stoffwechsel- und Ernährungsstörungen
Gelegentlich: Alkoholintoleranz, Anorexie, verringerter Appetit, Dehydratation, Gicht
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Arthralgie, Myalgie, Muskelkrämpfe, Schmerzen in den Extremitäten, Rückenschmerzen, Knochenschmerzen.
Gelegentlich: Anspannung der Muskeln, muskuläre Schwäche, Schulterschmerzen, Muskelzuckungen
Gutartige, bösartige und unspezifische Neubildungen (einschliesslich Zysten und Polypen)
Gelegentlich: Multiples Myelom
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (14%)
Häufig: Schwindel, Migräne, Parästhesie
Gelegentlich: Klonus, Dysgeusie, Hypästhesie, Hypogeusie, periphere Neuropathie, Sinustransversus-Thrombose
Psychiatrische Erkrankungen
Häufig: Schlaflosigkeit
Gelegentlich: Depression, abnormale Träume
Erkrankungen der Nieren und Harnwege
Gelegentlich: Proteinurie
Erkrankungen der Geschlechtsorgane und der Brustdrüse
Gelegentlich: Vaginale Hämorrhagie
Erkrankungen der Atemwege, des Brustraums und des Mediastinums
Häufig: Lungenembolie
Gelegentlich: Husten, Rhinorrhö, trockener Hals, Dyspnoe, Verstopfung der Nase, schmerzhafte Atmung
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Pruritus, Ekchymose, Hautausschlag
Gelegentlich: Alopezie, photosensible Reaktion, Akne, Kontaktdermatitis, trockene Haut, Ekzem, Erythem, exfoliativer Hautausschlag, ungewöhnliches Haarwachstum, Prurigo, Purpura, papulöser Hautausschlag, juckender Hautausschlag, Hautknötchen, ungewöhnlicher Hautgeruch, Urtikaria
Gefässerkrankungen
Häufig: Rötungen, tiefe Beinvenenthrombose
Gelegentlich: Erhöhter Blutdruck, Hypotonie, periphere Embolie, periphere Ischämie, Phlebitis, oberflächliche Thrombophlebitis, Thrombose, Erythromelalgie
Pädiatrische Population
Erkrankungen des Blut- und Lymphsystems
Häufig: Anämie, Knochenmarkstörung, Thrombozytopenie
Gelegentlich: Aplastische Anämie, Knochenmarkversagen, Leukozytose, Splenomegalie, Thrombozythämie, erhöhte Thrombozytenzahl, abnorme Thrombozytenzahl, Myelofibrose
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Schmerzen im Oberbauch (20,6%), Diarrhöe (21,3%)
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Fieber (31,6%)
Häufig: Periphere Schwellung
Infektionen und parasitäre Erkrankungen
Sehr häufig: Infektion der oberen Atemwege (26,6%), Rhinitis (11,3%)
Häufig: Pharyngitis, Konjunktivitis, Ohreninfektion, Gastroenteritis, Sinusitis
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Sehr häufig: Kontusion (28,4%)
Erkrankungen der Atemwege, des Brustraums und des Mediastinums
Sehr häufig: Husten (27,7%), Schmerzen im Oropharynx (23,0%)
Erkrankungen der Haut und des Unterhautzellgewebes
Sehr häufig: Ausschlag (14,2%)
Häufig: Purpura, Urtikaria
Immunogenität
Wie bei allen therapeutischen Proteinen besteht ein Potenzial für Immunogenität. Bei Verdacht auf die Bildung neutralisierender Antikörper, nehmen Sie bitte Kontakt mit Amgen Switzerland AG für die Antikörper-Testung auf.
Klinische Studien bei erwachsenen ITP-Patienten untersuchten Antikörper gegen Romiplostim und TPO. Während 5,7% (60/1'046) bzw. 3,2% (33/1'046) der Patienten bindende Antikörper gegen Romiplostim bzw. TPO entwickelten, waren lediglich 4 Patienten positiv hinsichtlich neutralisierender Antikörper gegen Romiplostim. Diese Antikörper zeigten jedoch keine Kreuzreaktion mit endogenem TPO. Von den 4 Patienten wurden zum jeweils letzten Testzeitpunkt 2 Patienten negativ auf neutralisierende Antikörper gegen Romiplostim getestet (vorübergehend positiv) und 2 Patienten sind weiterhin positiv (anhaltende Antikörper) geblieben. Die Inzidenz vorbestehender Antikörper gegen Romiplostim und TPO lag bei 3,3% (35/1'046) bzw. 3,0% (31/1'046).
In pädiatrischen Studien betrug die Inzidenz von bindenden Antikörpern gegen Romiplostim zu einem beliebigen Zeitpunkt 9,6% (27/282). Von den 27 Probanden lagen bei 2 bereits zu Studienbeginn bindende, nicht-neutralisierende Antikörper gegen Romiplostim vor. Darüber hinaus entwickelten 2,8% (8/282) neutralisierende Antikörper gegen Romiplostim. Bei insgesamt 3,9% (11/282) der Probanden waren zu einem beliebigen Zeitpunkt während der Behandlung mit Romiplostim bindende Antikörper gegen TPO vorhanden. Von den 11 Probanden lagen bei 2 bereits zu Studienbeginn bindende, nicht-neutralisierende Antikörper gegen TPO vor. Ein Patient (0,35%) mit einem negativen Ergebnis zu Studienbeginn zeigte danach während der Studie ein schwach positives Ergebnis auf neutralisierende Antikörper gegen TPO (anhaltend negativ auf Anti-Romiplostim-Antikörper). Der Patient zeigte eine vorübergehende Antikörperreaktion mit neutralisierenden Antikörpern gegen TPO bei einem negativen Ergebnis zum letzten Testzeitpunkt des Patienten während des Studienzeitraums.
In der Post-Marketing-Registerstudie wurden 19 bestätigte pädiatrische Patienten aufgenommen. Nach der Behandlung betrug die Inzidenz bindender Antikörper gegen Romiplostim 16% (3/19) und 5,3% (1/19) wurden positiv auf neutralisierende Antikörper gegen Romiplostim getestet. Es wurden keine Antikörper gegen TPO nachgewiesen. In diese Studie wurden 184 bestätigte erwachsene Patienten aufgenommen. Bei ihnen betrug die Inzidenz bindender Antikörper gegen Romiplostim nach der Behandlung 3,8% (7/184), von diesen wurden 0,5% (1/184) positiv auf neutralisierende Antikörper gegen Romiplostim getestet. Insgesamt 2,2% (4/184) der erwachsenen Patienten entwickelten bindende, nicht neutralisierende Antikörper gegen TPO.
Die Ergebnisse von Immunogenitätsassays hängen stark von der Sensitivität und Spezifität des für den Nachweis verwendeten Assays ab und können durch verschiedene Faktoren beeinflusst werden, einschliesslich Probenhandhabung, Begleitmedikation und zugrunde liegende Krankheit. Daher kann der Vergleich der Inzidenz der Antikörper gegen Romiplostim mit der Inzidenz der Antikörper gegen andere Arzneimittel irreführend sein.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIn den frühen klinischen Studien betrug die maximale Dosis von Nplate 30 mcg/kg. Später wurde diese auf 10 mcg/kg reduziert, da bei höheren Dosen kein zusätzlicher klinischer Nutzen festgestellt werden konnte.
Im Falle einer Überdosierung kann die Thrombozytenzahl übermässig ansteigen und zu thrombotischen/thromboembolischen Komplikationen führen. Sollte die Thrombozytenzahl übermässig ansteigen, ist Nplate abzusetzen und die Thrombozytenzahlen sollten überwacht werden. Die Fortsetzung der Behandlung mit Nplate sollte in Übereinstimmung mit den Dosierungs- und Anwendungsempfehlungen durchgeführt werden (siehe «Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen» für die Dosisanpassung).
Eigenschaften/WirkungenATC-Code
B02BX04
Wirkungsmechanismus
Romiplostim ist ein Fc-Peptid-Fusionsprotein (Peptibody), das intrazelluläre Transkriptionsprozesse über den Signalweg des Thrombopoetin-Rezeptors (TPO-Rezeptor, auch als cMpl bezeichnet) zur Steigerung der Thrombozytenproduktion aktiviert. Das Peptibody-Molekül besteht aus einer humanen Immunoglobulin-IgG1 Fc-Domäne, bei der jede Einzelketten-Untereinheit beim C-Terminus kovalent an eine Peptidkette mit zwei Thrombopoetin-rezeptorbindenden Domänen gebunden ist. Romiplostim wird mittels rekombinanter DNA-Technologie in Escherichia coli (E. coli) hergestellt.
Romiplostim steigert die Thrombozytenbildung durch Bindung an und Aktivierung des Thrombopoetin-Rezeptors, einen dem endogenen Thrombopoetin (eTPO) analogen Mechanismus.
Klinische Wirksamkeit
Erwachsene
In klinischen Studien führte die Behandlung mit Nplate zu einer dosisabhängigen Erhöhung der Thrombozytenzahl. Nach Gabe einer subkutanen Einzeldosis von 1 bis 10 mcg/kg Nplate an ITP-Patienten erhöhte sich die Thrombozytenzahl innerhalb von 2 bis 3 Wochen auf einen individuell unterschiedlichen Spitzenwert zwischen dem 1,3- bis 14,9-fachen des Ausgangswertes; das Ansprechen der Patienten war variabel. Die Thrombozytenzahl von ITP-Patienten, denen 6 wöchentliche Dosen von 1 oder 3 mcg/kg Nplate verabreicht wurden, lag für die meisten Patienten im Bereich 50 bis 450 × 109/l; das Ansprechen war variabel.
Von den 271 Patienten, denen im Rahmen der klinischen ITP-Studien Nplate verabreicht wurde, waren 55 (20%) ≥65 Jahre alt und 27 (10%) waren ≥75. In den placebokontrollierten Studien wurden bezüglich Sicherheit und Wirksamkeit keine Unterschiede zwischen älteren und jüngeren Patienten festgestellt.
Ergebnisse der placebokontrollierten Pivotalstudien
Die Wirksamkeit und Sicherheit von Nplate wurden bei erwachsenen Patienten mit ITP, die vor Studienbeginn mindestens eine Behandlung abgeschlossen hatten und die für das Spektrum derartiger ITP-Patienten repräsentativ sind, anhand von zwei placebo-kontrollierten, doppelblinden, klinischen Studien nachgewiesen.
In der Studie S1 (20030212) wurden nicht-splenektomierte Patienten untersucht, die gegenüber vorangehenden Therapien ein ungenügendes Ansprechen oder eine Unverträglichkeit gezeigt haben. Zum Zeitpunkt des Studienbeginns lag die ITP-Diagnose der Patienten im Median 2,1 Jahre (Bereich: 0,1 bis 31,6) zurück. Die Patienten erhielten bis zum Studienbeginn 3 (Median) ITP-Behandlungen (Bereich: 1 bis 7). Zu diesen vorangehenden Behandlungen zählten Kortikosteroide (90% aller Patienten), Immunglobuline (76%), Rituximab (29%), zytotoxische Therapien (21%), Danazol (11%) und Azathioprin (5%). Bei Studienbeginn betrug die mediane Thrombozytenzahl der Patienten 19 × 109/l.
In der Studie S2 (20030105) wurden splenektomierte Patienten untersucht, bei welchen eine Thrombozytopenie bestehen blieb. Zum Zeitpunkt des Studienbeginns lag die ITP-Diagnose der Patienten im Median 8 Jahre (Bereich: 0,6 bis 44,8) zurück. Zusätzlich zur Splenektomie waren die Patienten bis zum Studienbeginn im Median 6 ITP-Behandlungen (Bereich: 3 bis 10) unterzogen worden. Zu diesen vorangehenden Behandlungen zählten Kortikosteroide (98% aller Patienten), Immunglobuline (97%), Rituximab (71%), Danazol (37%), zytotoxische Therapien (68%), und Azathioprin (24%). Bei Studienbeginn betrug die mediane Thrombozytenzahl der Patienten 14 × 109/l.
Das Studiendesign war ähnlich für beide Studien. Die Patienten (≥18 Jahre) wurden in einem Verhältnis von 2:1 randomisiert; sie erhielten eine Nplate-Anfangsdosis von 1 mcg/kg oder ein Placebo. Den Patienten wurde über 24 Wochen einmal wöchentlich eine subkutane Injektion verabreicht. Die Dosierung wurde laufend angepasst, so dass eine Thrombozytenzahl von 50 bis 200 × 109/l aufrechterhalten werden konnte. In beiden Studien wurde die Wirksamkeit durch die Zunahme des Anteils der Patienten mit einer anhaltenden Thrombozytenantwort bei den Patienten, die mit Romiplostim behandelt wurden im Vergleich zu den Patienten, die mit Placebo behandelt wurden (siehe untenstehende Tabelle) gemessen. Der Median der mittleren wöchentlichen Dosis betrug für splenektomierte Patienten 3 mcg/kg und für nicht-splenektomierte Patienten 2 mcg/kg. Die Behandlung mit Nplate ergab im Vergleich zu Placebo in beiden klinischen Studien für alle Wirksamkeits-Endpunkte signifikante Verbesserungen.
In beiden Studien erreichte ein signifikant höherer Anteil der Patienten mit Nplate eine anhaltende Antwort der Thrombozytenzahl als jener mit Placebo.
In einer Intent-To-Treat-Analyse ergab die Behandlung mit Nplate im Vergleich zum Placebo in beiden klinischen Studien für alle Wirksamkeits-Endpunkte und alle in den Studien randomisierten Patienten signifikante Verbesserungen.
In den placebokontrollierten Studien sprach die Mehrzahl der mit Nplate behandelten Patienten rasch auf die Behandlung an und erreichte nach 1 bis 3 Dosen Nplate einen Median der Thrombozytenzahl von 50 × 109/l. Während des Rests der Studie konnten sie die Thrombozytenzahl dann im therapeutischen Bereich von 50 bis 200 × 109/l halten.
Während der sechsmonatigen Behandlungsdauer konnte Nplate in den placebokontrollierten Studien bei 50% bis 70% der Patienten eine Thrombozytenzahl von ≥50 × 109/l aufrechterhalten. In der Placebo-Gruppe konnten nur zwischen 0% und 7% der Patienten während der sechsmonatigen Behandlungsdauer eine Antwort in der Thrombozytenzahl erreichen. Eine Zusammenfassung der zentralen Wirksamkeits-Endpunkte ist nachstehend gegeben.
Zusammenfassung der zentralen Ergebnisse der placebokontrollierten Studien zur Wirksamkeit
|
|
Studie 1
Nicht-splenektomierte Patienten
|
Studie 2
Splenektomierte Patienten
|
Studien 1 & 2
gepoolt
| |
|
Nplate
(n = 41)
|
Placebo
(n = 21)
|
Nplate
(n = 42)
|
Placebo
(n = 21)
|
Nplate
(n = 83)
|
Placebo
(n = 42)
| |
Anzahl (%) Patienten mit anhaltender Thrombozytenantworta
|
25 (61%)
|
1 (5%)
|
16 (38%)
|
0 (0%)
|
41 (50%)
|
1 (2%)
| |
(95% KI)
|
(45%, 76%)
|
(0%, 24%)
|
(24%, 54%)
|
(0%, 16%)
|
(38%, 61%)
|
(0%, 13%)
| |
p-Wert
|
< 0,0001
|
0,0013
|
< 0,0001
| |
Anzahl (%) Patienten mit Gesamt-Thrombozytenantwortb
|
36 (88%)
|
3 (14%)
|
33 (79%)
|
0 (0%)
|
69 (83%)
|
3 (7%)
| |
(95% KI)
|
(74%, 96%)
|
(3%, 36%)
|
(63%, 90%)
|
(0%, 16%)
|
(73%, 91%)
|
(2%, 20%)
| |
p-Wert
|
< 0,0001
|
< 0,0001
|
< 0,0001
| |
Mittlere Anzahl Wochen mit Thrombozytenantwortc
|
15
|
1
|
12
|
0
|
14
|
1
| |
(Standardabweichung)
|
3,5
|
7,5
|
7,9
|
0,5
|
7,8
|
2,5
| |
p-Wert
|
< 0,0001
|
< 0,0001
|
< 0,0001
| |
Anzahl (%) Patienten, welche Notfall-Medikationen benötigtend
|
8 (20%)
|
13 (62%)
|
11 (26%)
|
12 (57%)
|
19 (23%)
|
25 (60%)
| |
(95% KI)
|
(8,8%, 35%)
|
(38%, 82%)
|
(14%, 42%)
|
(34%, 78%)
|
(14%, 34%)
|
(43%, 74%)
| |
p-Wert
|
0,0010
|
0,0175
|
< 0,0001
| |
Anzahl (%) Patienten mit anhaltender Thrombozytenantwort bei stabiler Dosise
|
21 (51%)
|
0 (0%)
|
13 (31%)
|
0 (0%)
|
34 (41%)
|
0 (0%)
| |
(95% KI)
|
(35%, 67%)
|
(0%, 16%)
|
(18%, 47%)
|
(0%, 16%)
|
(30%, 52%)
|
(0%, 8%)
| |
p-Wert
|
0,0001
|
0,0046
|
< 0,0001
| |
a
Anhaltende Thrombozytenantwort war definiert als wöchentliche Thrombozytenzahl von ≥50 × 109/l mindestens sechsmal während der Studienwochen 18-25 in Abwesenheit von Notfall-Medikation zu irgendeinem Zeitpunkt während der Behandlungsdauer.
b Gesamt-Thrombozytenantwort ist definiert als das Erreichen von anhaltender oder transienter Thrombozytenantwort. Transiente Thrombozytenantwort war definiert als wöchentliche Thrombozytenzahl von ≥50 × 109/l mindestens viermal während der Studienwochen 2-25, ohne aber eine anhaltende Thrombozytenantwort zu erreichen. Eine Antwort innerhalb von 8 Wochen nach Gebrauch einer Notfall-Medikation zählte nicht als wöchentliche Thrombozytenantwort.
c Die Anzahl Wochen mit Thrombozytenantwort ist definiert als Anzahl Wochen mit Thrombozytenzahl von ≥50 × 109/l während der Studienwochen 2-25. Eine Antwort innerhalb von 8 Wochen nach Gebrauch einer Notfall-Medikation zählte nicht als wöchentliche Thrombozytenantwort.
d Notfall-Therapien umfassten alle Therapien, die zur Steigerung der Thrombozytenzahl verabreicht wurden. Patienten, die Notfall-Medikationen benötigten, wurden für anhaltende Thrombozytenantwort nicht in Betracht gezogen. In den Studien erlaubte Notfall-Medikationen umfassten IVIg, Thrombozytentransfusionen, Anti-D-Immmunglobulin und Kortikosteroide.
e Stabile Dosis war definiert als Dosis, die während der letzten 8 Behandlungswochen auf ± 1 mcg/kg konstant gehalten wurde.
|
Studienergebnisse bei erwachsenen Patienten mit neu diagnostizierter und persistierender ITP
Studie S3 (20080435) war eine einarmige, offene Studie bei erwachsenen Patienten, die ein unzureichendes Ansprechen (Thrombozytenzahl ≤30 × 109/l) auf die Erstlinientherapie zeigten. An der Studie nahmen 75 Patienten mit einem medianen Durchschnittsalter von 39 Jahren (Bereich: 19 bis 85) teil, darunter 59% Frauen.
Die mediane Zeit von der ITP-Diagnose bis zur Aufnahme in die Studie betrug 2,2 Monate (Bereich: 0,1 bis 6,6). Bei 60% der Patienten (n = 45) bestand die ITP < 3 Monate und bei 40% (n = 30) ≥3 Monate. Beim Screening lag die mediane Thrombozytenzahl bei 20 × 109/l. Zu den Vorbehandlungen zählten Kortikosteroide, Immunglobuline und Anti-D-Immunglobuline. Patienten, die bereits medikamentöse ITP-Therapien mit einem konstanten Dosierungsschema erhielten, durften diese medikamentösen Behandlungen während der Studiendauer fortsetzen. Notfall-Therapien (wie Kortikosteroide, IVIg, Thrombozytentransfusionen, Anti-D-Immunglobulin, Dapson, Danazol und Azathioprin) waren erlaubt.
Die Patienten erhielten eine 12monatige Behandlung mit Romiplostim einmal wöchentlich als subkutane Injektion. Die Dosis wurde individuell angepasst, um Thrombozytenwerte zwischen 50 × 109/l und 200 × 109/l beizubehalten. Während der Studie betrug die mediane wöchentliche Romiplostim-Dosis 3 mcg/kg (jeweils 25.-75. Perzentile: 2 -4 mcg/kg).
Von den 75 Patienten, die in die Studie 20080435 eingeschlossen wurden, zeigten 70 (93%) ein Thrombozytenansprechen von ≥50 × 109/l während der 12monatigen Behandlungsdauer. Die mittlere Anzahl Monate mit Thrombozytenantwort lag während der 12monatigen Behandlungsdauer bei 9,2 (95% KI: 8,3; 10,1) Monate; der Median betrug 11 (95% KI: 10; 11) Monate. Die Kaplan-Meier-Schätzung der medianen Zeit bis zur ersten Thrombozytenantwort betrug 2,1 Wochen (95% KI: 1,1; 3,0). 24 (32 %) Patienten zeigten eine dauerhafte behandlungsfreie Remission, definiert als das Erreichen einer anhaltenden Thrombozytenzahl von ≥50 × 109/l über mindestens 6 Monate ohne Romiplostim bzw. eine andere medikamentöse ITP-Behandlung (begleitende oder Notfall-Therapie). Die mediane Zeit bis zum Erreichen von Thrombozytenwerten ≥50 × 109/l über mindestens 6 Monate betrug 27 Wochen (Bereich: 6 bis 57).
In einer integrierten Analyse zur Wirksamkeit wurden 277 erwachsene Patienten mit einer ITP-Dauer von ≤12 Monaten einbezogen, wobei diese Patienten aus 9 ITP-Studien (einschliesslich der Studie S3) stammten und mindestens eine Romiplostim-Dosis erhalten hatten. Von den 277 mit Romiplostim behandelten Patienten wiesen 140 Patienten eine neu diagnostizierte ITP (ITP-Dauer < 3 Monate) und 137 Patienten eine persistierende ITP (ITP-Dauer ≥3 bis ≤12 Monate) auf. Der prozentuale Anteil der Patienten, die ein dauerhaftes Thrombozytenansprechen erreichten (definiert als mindestens 6 wöchentliche Thrombozytenzahlen ≥50 × 109/l in den Behandlungswochen 18 bis einschliesslich 25), lag bei den 140 Patienten mit neu diagnostizierter ITP bei 50% (95% KI: 41,4% bis 58,6%) und bei den 137 Patienten mit persistierender ITP bei 55% (95 % KI: 46,7% bis 64,0%). Die mediane (Q1, Q3) Zeit (Prozent) mit einem Thrombozytenansprechen ≥50 × 109/l betrug 100% (70,3%; 100,0%) bei Patienten mit neu diagnostizierter ITP und 93,5% (72,2%; 100,0%) bei Patienten mit persistierender ITP. Zudem betrug der prozentuale Anteil der Patienten, die eine Rescue-Medikation benötigten, 47,4% bei Patienten mit neu diagnostizierter ITP und 44,9% bei Patienten mit persistierender ITP.
Studienergebnisse bei nicht-splenektomierten Patienten im Vergleich zur Standardtherapie (standard of care, SOC)
Studie S4 (20060131) war eine offene, randomisierte 52-wöchige Studie bei Patienten, die Romiplostim oder eine medizinische Standardbehandlung (SOC) erhielten. Zum Zeitpunkt des Studieneintritts betrug die diagnostizierte mediane ITP-Dauer bei den Patienten 2 Jahre (Bereich: 0,01 bis 44,2). Diese Studie untersuchte nicht-splenektomierte Patienten mit ITP und Thrombozytenzahlen von < 50 × 109/l. Romiplostim wurde bei 157 Patienten einmal wöchentlich als subkutane (s.c.) Injektion angewendet, beginnend mit einer Dosis von 3 mcg/kg, die während der Studie innerhalb eines Bereiches von 1-10 mcg/kg angepasst wurde, um Thrombozytenwerte zwischen 50 und 200 × 109/l beizubehalten. 77 Patienten wurden entsprechend den institutionellen Standardverfahren oder therapeutischen Richtlinien mit SOC behandelt.
Die Gesamtinzidenz von Patienten mit einem Therapieversagen lag in der Romiplostim-Gruppe bei 11,5% (18 von 157 Patienten) im Vergleich zu 29,9% (23 von 77 Patienten) in der SOC-Gruppe mit einer Odds-Ratio (Romiplostim versus SOC) von 0,31 (95% KI: 0,15; 0,61).
Unter den 154 Patienten, die Romiplostim erhielten, lag die mediane Gesamtexposition gegenüber Romiplostim bei 52 Wochen. Die am häufigsten angewendete wöchentliche Dosis lag zwischen 3-5 mcg/kg (jeweils 25.-75. Perzentile; Median 3 mcg/kg).
Unter den 75 Patienten, die mindestens eine Dosis der SOC erhielten, lag die mediane Gesamtexposition gegenüber SOC bei 51 Wochen.
Pädiatrie
Die Sicherheit und Wirksamkeit von Nplate wurde in zwei placebokontrollierten, doppelblinden Studien untersucht. Bei Studie S5 (20080279) handelte es sich um eine Phase-III-Studie mit einer 24wöchigen Behandlung mit Nplate und bei Studie S6 (20060195) um eine Phase-I/II-Studie mit einer 12-wöchigen Behandlung mit Nplate (bis zu 16 Wochen bei geeigneten Respondern, die in eine 4-wöchige pharmakokinetische Beurteilung eingeschlossen wurden).
Bei beiden Studien wurden pädiatrische Probanden (≥1 Jahr bis < 18 Jahre alt) mit Thrombozytopenie (in beiden Studien definiert durch den Mittelwert aus 2 Thrombozytenzahlen von ≤30 × 109/l, ohne dass eine der Thrombozytenzahlen > 35 × 109/l betrug) mit ITP, unabhängig vom Splenektomie-Status, aufgenommen.
In Studie S5 wurden 62 Probanden in einem Verhältnis von 2:1 randomisiert und erhielten entweder Romiplostim (n = 42) oder Placebo (n = 20), stratifiziert nach 1 von 3 Altersgruppen. Die Anfangsdosis von 1 mcg/kg Nplate und die nachfolgenden Dosen wurden angepasst, um eine Thrombozytenzahl von 50 – 200 × 109/l aufrechtzuerhalten. Die häufigste wöchentliche Dosis lag im Bereich von 3 – 10 mcg/kg und die in der Studie zulässige Maximaldosis betrug 10 mcg/kg. Die Patienten erhielten über 24 Wochen einmal wöchentlich eine subkutane Injektion.
Der primäre Endpunkt war die Inzidenz eines dauerhaften Ansprechens, definiert als das Erreichen von mindestens 6 wöchentlichen Thrombozytenzahlen ≥50 × 109/l während der Behandlungswochen 18 – 25. Insgesamt erreichte ein signifikant grösserer Anteil der Probanden in der Nplate-Gruppe den primären Endpunkt als in der Placebo-Gruppe (p = 0,0018). In der Nplate-Gruppe zeigten insgesamt 22 Probanden (52%) ein dauerhaftes Thrombozytenansprechen gegenüber 2 Probanden (10%) in der Placebo-Gruppe: ≥1 und < 6 Jahre 38% vs. 25%; ≥6 und < 12 Jahre 56% vs. 11%; ≥12 und < 18 Jahre 56% vs. 0%.
Die kombinierte Blutungsepisode wurde definiert als klinisch signifikante Blutungsereignisse oder Anwendung einer Notfallmedikation zur Prävention eines klinisch signifikanten Blutungsereignisses während der Behandlungswochen 2 – 25. Ein klinisch signifikantes Blutungsereignis wurde definiert als Blutungsereignis mit einem Schweregrad von ≥2 gemäss den «Allgemeinen Terminologiekriterien für unerwünschte Ereignisse» (Common Terminology Criteria for Adverse Events, CTCAE), Version 3.0. Die mittlere Anzahl (SD) der kombinierten Blutungsepisoden lag in der Nplate-Gruppe bei 1,9 (4,2) und in der Placebo-Gruppe bei 4,0 (6,9) mit einer medianen Anzahl (Q1; Q3) der Blutungsereignisse von 0,0 (0; 2) in der Nplate-Gruppe und von 0,5 (0; 4,5) in der Placebo-Gruppe.
In Studie S6, wurden 22 Probanden in einem Verhältnis von 3:1 randomisiert und erhielten entweder Nplate (n = 17) oder Placebo (n = 5). Die Dosen wurden alle 2 Wochen in Schritten von 2 mcg/kg erhöht und die Zielthrombozytenzahl lag bei ≥50 × 109/l. Die Behandlung mit Nplate führte zu einer statistisch signifikant höheren Inzidenz des Thrombozytenansprechens als unter Placebo (p = 0,0008).
Von den 17 Probanden, die Nplate erhielten, erreichten 15 während 2 aufeinanderfolgender Wochen (unter Ausschluss der Thrombozytenzahlen innerhalb von 4 Wochen nach der Anwendung der Notfallmedikation) des Behandlungszeitraums eine Thrombozytenzahl von ≥50 × 109/l (88,2%, 95% KI: 63,6%, 98,5%). Die gleichen 15 Probanden erreichten zudem während 2 aufeinanderfolgender Wochen (unter Ausschluss der Thrombozytenzahlen innerhalb von 4 Wochen nach der Anwendung der Notfallmedikation) des Behandlungszeitraums eine Erhöhung der Thrombozytenzahl von ≥20 × 109/l gegenüber den Ausgangswerten (88,2%, 95% KI: 63,6%, 98,5%). Keiner der mit Placebo behandelten Probanden erreichte einen der Endpunkte.
Reduktion der zulässigen gleichzeitig verabreichten ITP-Medikationen
In beiden placebokontrollierten doppelblinden Studien mit Erwachsenen durften Patienten, denen bereits vorher ITP-Medikationen nach einem konstanten Dosierungsschema verabreicht wurden, diese Therapien während der Studie weiter erhalten (Kortikosteroide, Danazol und/oder Azathioprin). Bei Studienbeginn erhielten 21 nicht-splenektomierte und 18 splenektomierte Patienten derartige ITP-Therapien (zumeist Kortikosteroide). Bis zum Ende der Studie konnten alle (100%) der splenektomierten und mit Nplate behandelten Patienten die Dosis dieser gleichzeitig verabreichten ITP-Medikationen um mehr als 25% reduzieren oder sie ganz absetzen, verglichen mit 17% bei den mit Placebo behandelten Patienten. Bis zum Ende der Studie konnten 73% der nichtsplenektomierten und mit Nplate behandelten Patienten die Dosis dieser gleichzeitig verabreichten ITP-Medikationen um mehr als 25% reduzieren oder sie ganz absetzen, verglichen mit 50% bei den mit Placebo behandelten Patienten (siehe «Interaktionen»).
In pädiatrischen Studien betrug die Prävalenz der gleichzeitigen Anwendung einer ITP-Therapie zu irgendeinem Zeitpunkt während der Behandlung 42,6% (120 von 282 Probanden) und es bestand ein Trend zur Reduktion der angewendeten Begleitmedikation im Zeitverlauf.
Verwendung der Notfall-Therapien
Erwachsene
Notfall-Therapien (wie Kortikosteroide, IVIg, Thrombozytentransfusionen, Anti-D-Immunglobulin) waren bei Blutungen, feuchter Purpura oder bei akuter Gefahr für den Patienten in beiden placebokontrollierten doppelblinden Studien erlaubt. Wie aus der obenstehenden Tabelle ersichtlich ist, war die Häufigkeit des Einsatzes von Notfall-Therapien bei mit Placebo behandelten Patienten deutlich höher als bei mit Nplate behandelten Patienten.
Pädiatrische Patienten
Die Inzidenz der Anwendung einer Notfall-Medikation bei den Probanden betrug zu einem beliebigen Zeitpunkt während der Behandlung 33,7% (95 von 282 Probanden), und es bestand ein Trend zur Reduktion der angewendeten Notfall-Medikation im Zeitverlauf.
Langzeitwirksamkeit
Erwachsene
Patienten, die eine vorgängige Studie mit Nplate abgeschlossen hatten (einschliesslich S1 und S2), wurde die Teilnahme an einer offenen Langzeit-Folgestudie (S7; Studie 20030213) angeboten. Patienten in der Langzeit-Folgestudie fuhren mit der wöchentlichen Dosierung und individuellen Dosisanpassungen von Nplate auf Basis der Thrombozytenzahl fort. Patienten, die in den placebokontrollierten Studien Placebo erhalten hatten, erhielten in der Folgestudie eine Anfangsdosis von 1 mcg/kg Nplate. Patienten, die in den placebokontrollierten Studien mit Nplate behandelt worden waren, setzten die Therapie mit ihrer vorherigen Dosis von Nplate fort, wenn die Nplate-freie Phase kürzer als 24 Wochen war. Betrug die Nplate-freie Phase mehr als 24 Wochen, so erhielten die Patienten eine Anfangsdosis von 1 mcg/kg Nplate.
Die Ergebnisse einer integrierten Analyse von Patienten, die eine vorgängige Studie mit Romiplostim beendeten (einschliesslich S1 und S2), die sich an der Folgestudie beteiligten, unterstützen die Eignung von Nplate zur Langzeitbehandlung (der Median der Behandlungsdauer für 291 erwachsene Patienten betrug 78 Wochen, mit Behandlung bis 277 Wochen).
Nach der ersten Dosisanpassungsphase war die Mehrheit (> 75%) der erwachsenen Patienten in der Lage, ihre Dosis im Bereich von 2 mcg/kg aufrechtzuerhalten, was darauf schliessen lässt, dass der klinische Effekt im Laufe der Zeit ohne eine signifikante Erhöhung der Dosierung von Nplate erhalten bleibt. Die Gesamtinzidenz der Anwendung einer Notfall-Medikation bei erwachsenen Patienten betrug 33,3%. Etwa 13% (37/291) der erwachsenen Patienten erhielten eine begleitende ITP-Therapie zum Zeitpunkt des Studieneintritts. Davon setzten 20 Patienten (54,1%) die begleitende ITP-Therapie bis zum Ende der Studie ab. Bei Patienten, bei denen Knochenmarkbiopsien (n = 38) durchgeführt wurden, waren keine Anzeichen von Kollagen Typ I zu erkennen. Die Trichrom-Färbung für Kollagen Typ I wurde jedoch uneinheitlich durchgeführt.
Die Daten von Patienten, die zuvor in einer der placebokontrollierten Studien mit Nplate behandelt worden waren, bestätigen, dass Nplate bei der Mehrheit der Patienten ein anhaltendes Ansprechen über einen längeren Zeitraum vermitteln kann. Darüber hinaus zeigen diese Daten, dass Nplate die Thrombozytenzahlen bei Patienten erhöhen kann, die in den Studien vorher Placebo erhalten hatten. Patienten, die vorgängig mit Placebo behandelt worden waren und während der Folgestudie Nplate erhielten, zeigten bei der Erhöhung der Thrombozytenzahl ein ähnliches Entwicklungsmuster wie Patienten, die während der Pivotalstudien mit Nplate behandelt worden waren.
Pädiatrie
Bei 20 pädiatrischen Patienten wurde eine Verlängerung der Studie S6 durchgeführt, in der diese einmal pro Woche Romiplostim erhielten (Studie S7). Die Dosierung begann entweder mit der gleichen Dosis, die in der vorangegangenen Studie am Ende der Behandlung verabreicht wurde, oder mit einer Dosis von 1 mcg/kg (bei Probanden, die in der vorangegangenen Studie Placebo erhalten hatten).
Das Thrombozytenansprechen (≥50 × 109/l zu irgendeinem Zeitpunkt während der Studie) wurde bei 100,0% der pädiatrischen Probanden erreicht (95% KI: 83,2%, 100,0%). Eine Thrombozytenzahl von ≥100 × 109/l wurde bei 90,0% der pädiatrischen Probanden erreicht, der Thrombozyten-Spitzenwert von ≥150 × 109/l wurde bei 85,0% der pädiatrischen Probanden erreicht. Die Gesamtinzidenz der Anwendung einer Notfallmedikation bei der pädiatrischen Population betrug 20,0% (4 Probanden).
Pädiatrische Probanden, die eine vorgängige Studie mit Romiplostim abgeschlossen hatten (einschliesslich Studie S5), wurden in die Studie S8 (20090340) eingeschlossen, einer offenen Folgestudie zur Beurteilung der Sicherheit und Wirksamkeit der Langzeit-Anwendung von Romiplostim bei thrombozytopenischen Kindern und Jugendlichen mit ITP.
In diese Studie wurden insgesamt 66 Probanden aufgenommen, von denen 54 (82%) zuvor die Studie S5 abgeschlossen hatten. 65 dieser Probanden (98,5%) erhielten mindestens eine Dosis Romiplostim. Der Median (Q1, Q3) der Behandlungsdauer betrug 135,0 Wochen (95,0 Wochen, 184,0 Wochen). Der Median (Q1, Q3) der durchschnittlichen Wochendosis betrug 4,82 mcg/kg (1,88 mcg/kg, 8,79 mcg/kg). Der Median (Q1, Q3) der im Behandlungszeitraum am häufigsten angewendeten Dosis betrug 5,0 mcg/kg (1,0 mcg/kg, 10,0 mcg/kg).
In der gesamten Studie betrug die Inzidenz der Thrombozytenantwort (mindestens einmalige Messung einer Thrombozytenzahl ≥ 50 × 109/l ohne Notfall-Medikation) 93,8% (n = 61) und war in allen Altersgruppen vergleichbar. In der gesamten Studienpopulation betrug der Median (Q1, Q3) der Anzahl der Monate mit einer Thrombozytenantwort 30,0 Monate (13,0 Monate, 43,0 Monate), und der Median (Q1, Q3) der Dauer der Studienteilnahme betrug 34,0 Monate (24,0 Monate, 46,0 Monate). In der gesamten Studienpopulation betrug der Median (Q1; Q3) des prozentualen Anteils der Monate mit einer Thrombozytenantwort 93,33% (67,57%; 100,00%) und war in allen Altersgruppen vergleichbar.
Insgesamt 31 Probanden (47,7%) wendeten während der Studie eine begleitende ITP-Therapie an, einschliesslich der 23 Probanden (35,4%) mit Anwendung einer Notfall-Medikation und der 5 Probanden (7,7%) mit begleitender Anwendung einer ITP-Medikation bei Studienbeginn. Die Prävalenz der begleitenden ITP-Medikation zeigte bei den Probanden im Verlauf der Studie einen rückläufigen Trend: von 30,8% (Woche 1-12) auf < 20,0% (Woche 13-240) und dann auf 0% von Woche 240 bis zum Ende der Studie.
Die Prävalenz der Anwendung von Notfall-Medikation zeigte bei den Probanden im Verlauf der Studie einen rückläufigen Trend: von 24,6% (Woche 1-12) auf < 13,0% (Woche 13-216) und dann auf 0% von Woche 216 bis zum Ende der Studie.
Studie S9 (20101221) war eine einarmige, offene, multizentrische Langzeitstudie der Phase III an 203 pädiatrischen Patienten mit seit mindestens 6 Monaten diagnostizierter ITP, die zuvor mindestens 1 ITP-Behandlung (ohne Romiplostim) erhalten hatten oder für andere ITP-Therapien nicht infrage kamen. Romiplostim wurde wöchentlich als subkutane Injektion verabreicht, mit einer Anfangsdosis von 1 mcg/kg und einer schrittweisen wöchentlichen Dosiserhöhung auf maximal 10 mcg/kg, um eine Zielthrombozytenzahl zwischen 50 × 109/l und 200 × 109/l zu erreichen. Das mediane Alter der Patienten lag bei 10 Jahren (Bereich: 1 bis 17 Jahre), und die mediane Behandlungsdauer betrug 155,9 Wochen (Bereich: 8,0 bis 163,0).
Der Mittelwert (SD) und der mediane prozentuale Anteil der Zeit mit einer Thrombozytenantwort (Thrombozytenzahl ≥50 × 109/l) innerhalb der ersten 6 Monate nach Einleitung der Romiplostim-Therapie ohne Notfall-Medikation während der letzten 4 Wochen betrug 50,57% (37,01) bzw. 50,0%. Insgesamt erhielten 60 (29,6%) Patienten Notfall-Medikationen. Notfall-Medikationen (wie Kortikosteroide, Thrombozytentransfusionen, IVIg, Azathioprin, Anti-D-Immunglobulin und Danazol) waren erlaubt.
Diese Open-Label-Studie untersuchte das Knochenmark auf die Bildung von Retikulin und Kollagen sowie auf Veränderungen bei pädiatrischen Patienten mit ITP, die mit Romiplostim behandelt wurden. Für die Retikulin und KollagenAuswertungen wurde die modifizierte Graduierung nach Bauermeister-Skala angewendet, wohingegen Zytogenetik und Fluoreszenzin-situ-Hybridisierung (FISH) verwendet wurden, um Veränderungen im Knochenmark nachzuweisen. Basierend auf der Kohortenzuweisung zum Zeitpunkt der Aufnahme in die Studie wurden die Patienten auf Retikulin und Kollagen im Knochenmark nach Jahr 1 (Kohorte 1) oder Jahr 2 (Kohorte 2) im Vergleich zum Ausgangszustand des Knochenmarks bei Studienbeginn untersucht. Von den insgesamt 79 Patienten in den beiden Kohorten wurden 27 von 30 (90%) Patienten aus Kohorte 1 und 36 von 49 (73,5%) Patienten aus Kohorte 2 während der Studie einer bewertbaren Knochenmarkbiopsie unterzogen. Bei 18,5% (5 von 27) Patienten aus Kohorte 1 und 47,2% (17 von 36) Patienten aus Kohorte 2 wurde eine erhöhte Retikulinfaserbildung beobachtet. In keiner der beiden Kohorten entwickelte ein Patient eine Fibrose mit gesteigerter Kollagenbildung oder eine Knochenmarkveränderung, die im Widerspruch zu einer zugrundeliegenden ITP-Diagnose stand.
PharmakokinetikAbsorption
Erwachsene
In der Langzeit-Folgestudie, in der ITP-Patienten wöchentlich Nplate subkutan verabreicht erhielten, ergaben pharmakokinetische Untersuchungen, dass im Dosisbereich von 3 bis 15 mcg/kg die maximale Serumkonzentration von Romiplostim 7 bis 50 Stunden nach Verabreichung (Median 14 Stunden) erreicht wurde. Die Serumkonzentrationen variierten zwischen Patienten und korrelierten nicht mit der verabreichten Dosis.
Distribution
Keine Angaben.
Metabolismus
Keine Angaben.
Elimination
Die Eliminations-Halbwertszeit betrug 1 bis 34 Tage (Median 3,5 Tage). Die Elimination von Serum-Romiplostim hängt teilweise vom TPO-Rezeptor auf den Thrombozyten ab. Entsprechend weisen, für eine bestimmte Dosis, Patienten mit hohen Thrombozytenzahlen tiefe Serumkonzentrationen auf und umgekehrt. In einer weiteren klinischen ITP-Studie konnte nach 6 wöchentlichen Dosen von Nplate (3 mcg/kg) keine Akkumulation bzgl. der Serumkonzentration festgestellt werden (siehe «Dosierung/Anwendung»).
Kinetik spezieller Patientengruppen
Pädiatrie
Pharmakokinetische Daten für Nplate wurden in zwei Studien bei 21 pädiatrischen Probanden mit ITP erhoben. In Studie S6 (20060195) lag die Romiplostim-Konzentration von 17 Probanden bei Dosierungen von 1 bis 10 mcg/kg vor. In Studie S8 (20090340) lagen die intensiven Romiplostim-Konzentrationen von 4 Probanden (2 bei 7 mcg/kg und 2 bei 9 mcg/kg) vor. Die Serumkonzentrationen von Nplate bei pädiatrischen Probanden mit ITP lagen innerhalb des Bereichs, der bei erwachsenen ITP-Patienten beobachtet wurde, die den gleichen Dosisbereich von Nplate erhielten. Ähnlich wie bei Erwachsenen mit ITP ist die Pharmakokinetik von Nplate bei pädiatrischen Probanden mit ITP sehr variabel und nicht zuverlässig und prädiktiv. Die Daten sind jedoch unzureichend, um eine fundierte Schlussfolgerung bezüglich der Auswirkungen von Dosis und Alter auf die Pharmakokinetik von Nplate zu ziehen.
Präklinische DatenMutagenität und Karzinogenität
Das mutagene und karzinogene Potenzial von Romiplostim wurde nicht untersucht.
Reproduktionstoxikologie
In allen Entwicklungsstudien wurden neutralisierende Antikörper gebildet, die möglicherweise die Wirkung von Romiplostim hemmen. In Embryo-fetalen Entwicklungsstudien mit Mäusen und Ratten trat nur bei Mäusen ein verringertes Gewicht der Muttertiere auf. Bei Mäusen gab es Hinweise auf erhöhte Post-Implantationsverluste. In einer pränatalen und postnatalen Entwicklungsstudie bei Ratten traten eine Verlängerung der Gestationsdauer sowie eine geringe Erhöhung der perinatalen Sterblichkeit des Nachwuchses auf. Bei Ratten überwindet Romiplostim bekannterweise die Plazentaschranke und kann von der Mutter auf den sich entwickelnden Fötus übergehen und die fetale Thrombozytenproduktion anregen. Romiplostim zeigte keine Auswirkungen auf die Fertilität von Ratten.
Sonstige HinweiseInkompatibilitäten
Mangels Kompatibilitätsstudien darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt oder zu einer Infusion hinzugegeben werden, ausser mit denen unter «Sonstige Hinweise: Hinweise für die Handhabung».
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Nach Rekonstitution mit sterilem Wasser für Injektionszwecke:
Die chemische und physikalische in-use Stabilität wurde für 24 Stunden bei 25°C und für 24 Stunden bei 2-8°C gezeigt, sofern das Arzneimittel vor Licht geschützt und in der Original-Durchstechflasche gelagert wird.
Nplate enthält keine antimikrobiellen oder bakteriostatischen Konservierungsmittel. Aus mikrobiologischer Sicht sollte das Arzneimittel sofort verwendet werden. Sofern es nicht sofort verabreicht wird, liegen die Aufbewahrungsdauer und die Lagerbedingungen in der Verantwortung des Anwenders und sollten normalerweise nicht länger als 24 Stunden bei Raumtemperatur (25°C) oder 24 Stunden im Kühlschrank (2-8°C), vor Licht geschützt, betragen.
Nach Verdünnung der rekonstituierten Lösung mit konservierungsmittelfreier, steriler Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke:
Die chemische und physikalische Stabilität wurde bei 25°C für 4 Stunden nachgewiesen, wenn das verdünnte Arzneimittel in einer Einwegspritze aufbewahrt wurde, oder für 4 Stunden im Kühlschrank (2–8°C), wenn das verdünnte Arzneimittel in der Original-Durchstechflasche aufbewahrt wurde.
Aus mikrobiologischer Sicht sollte das verdünnte Arzneimittel sofort verwendet werden. Sofern es nicht sofort verabreicht wird, liegen Aufbewahrungsdauer und Lagerbedingungen vor dem Gebrauch in der Verantwortung des Anwenders und sollten normalerweise nicht länger als 4 Stunden bei 25°C in Einwegspritzen oder 4 Stunden im Kühlschrank (2–8°C) in den Original-Durchstechflaschen, vor Licht geschützt, betragen.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern.
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Das Nplate Lyophilisat kann für einen Zeitraum von 30 Tagen bei Raumtemperatur (bis zu 25°C) aus dem Kühlschrank entnommen werden, sofern es in der Originalverpackung aufbewahrt wird. Falls nicht innerhalb von 30 Tagen verwendet, ist Nplate zu verwerfen.
Für die Lagerungsbedingungen des rekonstituierten Arzneimittels und des verdünnten rekonstituierten Arzneimittels siehe «Sonstige Hinweise: Haltbarkeit».
Hinweise für die Handhabung
Rekonstitution:
Nplate ist ein steriles Arzneimittel ohne Konservierungsmittel und ist zum einmaligen Gebrauch bestimmt.
Nplate 250 Mikrogramm (mcg) Pulver zur Herstellung einer Injektionslösung muss mit 0,72 ml sterilem Wasser für Injektionszwecke rekonstituiert werden, um ein entnehmbares Volumen von 0,5 ml (entspricht 250 mcg) zu erzielen. Jede Durchstechflasche enthält eine zusätzliche Überfüllung, um sicherzustellen, dass 250 mcg Romiplostim entnommen werden kann.
Nplate 500 Mikrogramm (mcg) Pulver zur Herstellung einer Injektionslösung muss mit 1,2 ml sterilem Wasser für Injektionszwecke rekonstituiert werden, um ein entnehmbares Volumen von 1 ml (entspricht 500 mcg) zu erzielen. Jede Durchstechflasche enthält eine zusätzliche Überfüllung, um sicherzustellen, dass 500 mcg Romiplostim entnommen werden kann.
Die Konzentration der Injektionslösung nach Rekonstitution von Nplate 250 Mikrogramm (mcg) oder Nplate 500 Mikrogramm (mcg) entspricht 500 mcg/ml. Diese Konzentration wird zur Berechnung des zu verabreichenden Volumens angewendet (siehe «Dosierung/Anwendung»).
|
Nplate Durchstechflasche zum Einmalgebrauch
|
Gesamtmenge Nplate pro Durchstechflasche
|
|
Rekonstitutionsvolumen des sterilen Wassers für Injektionszwecke
|
|
Entnehmbare Menge und Volumen
|
Endgültige Konzentration
| |
250 mcg
|
375 mcg
|
+
|
0,72 ml
|
=
|
250 mcg in 0,5 ml
|
500 mcg/ml
| |
500 mcg
|
625 mcg
|
+
|
1,2 ml
|
=
|
500 mcg in 1 ml
|
500 mcg/ml
|
Zur Rekonstitution des Arzneimittels sollte nur steriles Wasser für Injektionszwecke verwendet werden. Die Verwendung von Kochsalzlösung oder «bakteriostatischem Wasser» zur Rekonstitution ist nicht zulässig. Das Wasser für Injektionszwecke wird in die Nplate-Durchstechflasche injiziert. Der Inhalt der Durchstechflasche kann zur Auflösung vorsichtig geschwenkt und gewendet werden. Durchstechflasche nicht schütteln oder heftig bewegen. Nplate löst sich normalerweise in weniger als 2 Minuten auf. Führen Sie vor der Verabreichung eine visuelle Prüfung der Lösung durch, und achten Sie dabei speziell auf Partikel und Verfärbungen. Rekonstituiertes Nplate sollte klar und farblos sein. Falls Partikel und/oder Verfärbungen festgestellt werden, darf Nplate nicht verabreicht werden.
Das rekonstituierte Arzneimittel sollte innerhalb von 24 Stunden verabreicht werden, da es keine Konservierungsmittel enthält. Das rekonstituierte Arzneimittel kann bei Raumtemperatur (25°C) oder im Kühlschrank bei 2-8°C für bis zu 24 Stunden vor der Verabreichung aufbewahrt werden. Das rekonstituierte Arzneimittel muss vor Licht geschützt werden.
Nicht verwendetes Arzneimittel sowie Abfallmaterial sollten entsprechend den geltenden Vorschriften entsorgt werden.
Verdünnung (erforderlich, wenn die berechnete individuelle Patientendosis weniger als 23 mcg beträgt):
Die initiale Rekonstitution von Nplate mit den vorgesehenen Volumina von sterilem Wasser für Injektionszwecke ergibt bei allen Durchstechflaschen-Grössen eine Konzentration von 500 mcg/ml. Wenn die berechnete individuelle Patientendosis weniger als 23 mcg beträgt (siehe «Dosierung/Anwendung»), ist ein zusätzlicher Verdünnungsschritt auf 125 mcg/ml mit konservierungsmittelfreier, steriler Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke erforderlich, um ein genaues Volumen zu erzielen (siehe untenstehende Tabelle).
Verdünnungsrichtlinien:
|
Nplate Durchstechflasche zum Einmalgebrauch
|
Fügen Sie dieses Volumen an konservierungsmittelfreier, steriler Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke der rekonstituierten Durchstechflasche hinzu
|
Konzentration nach Verdünnung
| |
250 mcg
|
2,25 ml
|
125 mcg/ml
| |
500 mcg
|
3,75 ml
|
125 mcg/ml
|
Zur Verdünnung darf nur konservierungsmittelfreie, sterile Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke verwendet werden. Zur Verdünnung darf keine Dextrose (5%) in Wasser oder steriles Wasser für Injektionszwecke verwendet werden. Es wurden keine anderen Verdünnungsmittel getestet.
Für die Zugabe der konservierungsmittelfreien, sterilen Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke ist eine Luer-Lock-Spritze zu verwenden; der Inhalt wird nach der Rekonstitution durch den Adapter in die Durchstechflasche injiziert.
Es sollte beachtet werden, dass die benötigte konservierungsmittelfreie, sterile Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke für den zusätzlichen Verdünnungsschritt und die Luer-Lock-Spritze nicht in den Packungen enthalten sind, sondern separat bezogen werden müssen.
Zu den Aufbewahrungsbedingungen nach der Verdünnung des rekonstituierten Arzneimittels siehe «Sonstige Hinweise: Haltbarkeit».
Handhabung
|
1.Entfernen Sie den Schnappdeckel aus Plastik der Nplate Pulver-Durchstechflasche und reinigen Sie den Gummistopfen mit einem der mitgelieferten Alkoholtupfer.
|
| |
2.Setzen Sie den Adapter für die Durchstechflasche auf die Nplate-Durchstechflasche, indem Sie die Papierabdeckung der Verpackung des Adapters für die Durchstechflasche abziehen; belassen Sie dabei den Adapter für die Durchstechflasche in seiner Verpackung. Stellen Sie die Durchstechflasche auf eine stabile Oberfläche und drücken Sie den Adapter für die Durchstechflasche so lange senkrecht auf die Mitte der Durchstechflasche bis er fest sitzt.
Hinweis: Berühren Sie nicht den Dorn des Adapters für die Durchstechflasche oder den Luer-Lock-Ansatz, um eine Kontamination des Arzneimittels zu vermeiden.
|
|

| |
3.Entfernen und entsorgen Sie die Verpackung des Adapters für die Durchstechflasche.
|
| |
4.Stecken Sie die Kolbenstange in die Fertigspritze mit Wasser für Injektionszwecke und drehen Sie die Kolbenstange im Uhrzeigersinn in den Gummistopfen in der Spritze, bis Sie einen leichten Widerstand spüren.
|
| |
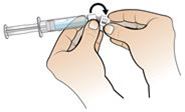
5.Halten Sie die Fertigspritze mit Wasser für Injektionszwecke in einer Hand, während Sie die Spitze der weissen Plastikabdeckung mit der anderen Hand nach unten biegen. Damit wird das Siegel der weissen Plastikabdeckung aufgebrochen. Sobald der Verschluss aufgebrochen ist, entfernen Sie die Abdeckung und den grauen Gummiaufsatz von der durchsichtigen Plastikspitze der Spritze.
|
|
| |
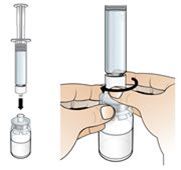
6.Stellen Sie die Durchstechflasche auf eine stabile Oberfläche und befestigen Sie die Fertigspritze mit Wasser für Injektionszwecke am vorbereiteten Adapter für die Durchstechflasche: Halten Sie das äussere Ende des Adapters für die Durchstechflasche mit einer Hand fest und drehen Sie die Spritze am unteren Ende mit der anderen Hand im Uhrzeigersinn in den Adapter für die Durchstechflasche, bis Sie einen leichten Widerstand spüren.
|
|
| |
7.Entleeren Sie das gesamte Wasser sehr langsam und vorsichtig in die Durchstechflasche, damit es langsam auf das Pulver fliesst. Schwenken Sie die Durchstechflasche VORSICHTIG, bis sich das gesamte Pulver gelöst hat und die Flüssigkeit in der Durchstechflasche klar und farblos ist.
Die Durchstechflasche nicht schütteln oder heftig bewegen.
Hinweis: Aus mikrobiologischer Sicht sollte das Arzneimittel sofort nach der Rekonstitution verwendet werden.
Falls das rekonstituierte Arzneimittel nicht sofort verwendet wird, sollte die Spritze nicht vom Adapter für die Durchstechflasche entfernt werden, um die mikrobiologische Reinheit zu erhalten.
|
|

HINWEIS: Die vollständige Auflösung des Pulvers kann bis zu 2 Minuten dauern.
| |
Bevor Sie fortfahren:
Prüfen Sie die rekonstituierte Lösung optisch auf Partikel und/oder Verfärbungen. Die rekonstituierte Lösung sollte klar und farblos sein und sollte nicht angewendet werden, wenn Partikel und/oder Verfärbungen zu sehen sind.
Stellen Sie sicher, dass die Auflösung vollständig ist, bevor Sie die Spritze entfernen.
| |
8.Entfernen Sie die leere Fertigspritze von dem Adapter für die Durchstechflasche.
|
| |
Wenn die berechnete Patientendosis 23 mcg oder mehr beträgt, fahren Sie mit Schritt 9 fort. Verdünnen Sie nicht.
Wenn die berechnete Patientendosis weniger als 23 mcg beträgt, gehen Sie wie folgt vor:
Ein zusätzlicher Verdünnungsschritt auf 125 mcg/ml mit konservierungsmittelfreier, steriler Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke ist erforderlich, um das genaue Volumen sicherzustellen (siehe «Sonstige Hinweise: Verdünnungsrichtlinien»). Verwenden Sie eine Luer-Lock-Spritze um die konservierungsmittelfreie, sterile Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke in die rekonstituierte Durchstechflasche zu geben. Welches Volumen an konservierungsmittelfreier, steriler Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke in die rekonstituierte Durchstechflasche hinzuzufügen ist, ist der nachstehenden Tabelle zu entnehmen:
|
|
Nplate-Durchstechflasche zum Einmalgebrauch
|
Fügen Sie dieses Volumen an konservierungsmittelfreier, steriler Natriumchloridlösung 9 mg/ml (0,9%) für Injektionszwecke der rekonstituierten Durchstechflasche hinzu
|
Konzentration nach Verdünnung
| |
250 mcg
|
2,25 ml
|
125 mcg/ml
| |
500 mcg
|
3,75 ml
|
125 mcg/ml
|
Stellen Sie die rekonstituierte Durchstechflasche auf eine stabile Oberfläche und befestigen Sie die mit Natriumchloridlösung gefüllte Luer-Lock-Spritze mit dem Adapter für die Durchstechflasche: Halten Sie dazu den äusseren Rand des Adapters für die Durchstechflasche mit einer Hand fest und drehen Sie mit der anderen Hand die Spritze so lange im Uhrzeigersinn auf den Adapter für die Durchstechflasche, bis Sie einen leichten Widerstand spüren. Injizieren Sie nun langsam den gesamten Inhalt in die Durchstechflasche der rekonstituierten Lösung und achten Sie darauf, die Durchstechflasche nicht zu schütteln.
Hinweis: Falls das verdünnte Arzneimittel nicht sofort verwendet wird, sollte die Spritze nicht vom Adapter für die Durchstechflasche entfernt werden, damit die mikrobiologische Reinheit erhalten bleibt.
Entfernen Sie anschliessend die leere Spritze vom Adapter der Durchstechflasche. Fahren Sie mit Schritt 9 fort.
Zulassungsnummer61541 (Swissmedic)
PackungenNplate 500 Mikrogramm (mcg): Jede Packung enthält
1 Durchstechflasche mit Pulver,
1 Fertigspritze mit 1,2 ml Wasser für Injektionszwecke für die Rekonstitution,
1 Kolbenstange für die Fertigspritze,
1 sterilen Adapter für die Durchstechflasche,
1 sterile 1 ml Luer-Lock-Spritze,
1 sterile Injektionsnadel mit Sicherheitssystem,
4 Alkoholtupfer (A).
Nplate 250 Mikrogramm (mcg): Jede Packung enthält
1 Durchstechflasche mit Pulver,
1 Fertigspritze mit 0,72 ml Wasser für Injektionszwecke für die Rekonstitution,
1 Kolbenstange für die Fertigspritze,
1 sterilen Adapter für die Durchstechflasche,
1 sterile 1 ml Luer-Lock-Spritze,
1 sterile Injektionsnadel mit Sicherheitssystem,
4 Alkoholtupfer (A).
ZulassungsinhaberinAmgen Switzerland AG, Risch
Domizil: 6343 Rotkreuz
Stand der InformationFebruar 2025
Version#160824
|