ZusammensetzungWirkstoffe
Dabrafenib als Dabrafenibmesylat.
Hilfsstoffe
Hartkapsel:
Mikrokristalline Cellulose, Magnesiumstearat, kolloidales Siliciumdioxid, rotes Eisenoxid (E 172), Titandioxid (E 171), Hypromellose (E 464), Schellac, schwarzes Eisenoxid (E172), Propylenglykol, Ammoniumhydroxid.
Tabletten zur Herstellung einer Suspension zum Einnehmen:
Mannitol (E421), mikrokristalline Cellulose (E460), Crospovidon (E1202), Hypromellose (E464), Acesulfam-Kalium (E950), Magnesiumstearat (E470b), künstliches Beerenaroma (Maltodextrin, Propylenglykol 0.036 mg [E1520], künstliche Aromen, Triethylcitrat [E1505], Benzylalkohol <0.00078 mg [E1519]), kolloidales Siliciumdioxid (E551).
Indikationen/AnwendungsmöglichkeitenBefristet zugelassene Indikation
Niedriggradiges Gliom (Low-grade-Gliom, LGG)
Tafinlar in Kombination mit Trametinib ist angezeigt zur Behandlung von pädiatrischen Patienten ab 1 Jahr mit niedriggradigem Gliom (Low grade Gliom, LGG) mit einer BRAF-V600E-Mutation, die eine systemische Therapie benötigen.
Aufgrund einer zum Zeitpunkt der Begutachtung des Gesuches unvollständigen klinischen Datenlage, wird/werden diese Indikation/en befristet zugelassen (Art. 9a Heilmittelgesetz). Die befristete Zulassung ist zwingend an die zeitgerechte Erfüllung von Auflagen gebunden. Nach deren Erfüllung kann die befristete Zulassung in eine ordentliche Zulassung überführt werden.
Ordentlich zugelassene Indiaktionen
Nicht resezierbares oder metastasiertes Melanom
·Tafinlar in Kombination mit Trametinib ist angezeigt zur Behandlung von erwachsenen Patienten mit nicht resezierbarem oder metastasiertem Melanom mit einer BRAF-V600-Mutation (V600E/K).
·Tafinlar ist angezeigt zur Behandlung von erwachsenen chemotherapienaiven Patienten mit nicht resezierbarem oder metastasiertem Melanom mit einer BRAF-V600E-Mutation (siehe «Eigenschaften/Wirkungen»). Zur Diagnose des Vorliegens einer V600E-Mutation ist die Anwendung eines validierten BRAF-Mutationstests erforderlich.
Adjuvante Behandlung des Melanoms
Tafinlar in Kombination mit Trametinib ist angezeigt zur adjuvanten Behandlung von Patienten mit Melanom im Stadium III mit einer BRAF-V600-Mutation nach vollständiger Resektion.
Fortgeschrittenes oder metastasiertes, nicht-kleinzelliges Lungenkarzinom
Tafinlar in Kombination mit Trametinib kann angewendet werden zur Behandlung von erwachsenen Patienten mit metastasiertem, nicht-kleinzelligem Lungenkarzinom (NSCLC; non-small cell lung cancer) mit einer BRAF-V600E Mutation.
Nicht-resezierbare oder metastasierte solide Tumore
Tafinlar in Kombination mit Trametinib ist zur Behandlung von erwachsenen Patienten indiziert, bei denen ein nicht resezierbarer oder metastasierter solider Tumor mit einer BRAF V600E-Mutation vorliegt, der nach einer früheren Behandlung fortgeschritten ist und für den es keine zufriedenstellenden alternativen Behandlungsmöglichkeiten gibt (siehe «Klinische Wirksamkeit»).
Tafinlar ist nicht indiziert bei Patienten mit BRAF-Wildtyp-Tumoren oder kolorektalem Karzinom, aufgrund dessen bekannter intrinsischer Resistenz gegenüber BRAF Inhibition (siehe «Wirkungsmechanismus»).
Dosierung/AnwendungDie Behandlung mit Tafinlar sollte von einem in der Anwendung onkologischer Arzneimittel erfahrenen Arzt eingeleitet und überwacht werden.
Tafinlar ist in zwei Darreichungsformen erhältlich: als Kapseln und als Tabletten zur Herstellung einer Suspension zum Einnehmen.
Vor der Einnahme von Tafinlar soll das Vorliegen einer BRAF-V600-Mutation gemäss der zugelassenen Indikation anhand eines validierten Tests bestätigt sein.
Übliche Dosierung
Bei erwachsenen Patienten beträgt die empfohlene Dosis Tafinlar, sowohl bei der Tafinlar Monotherapie als auch in Kombination mit Trametinib, 150 mg Tafinlar (zwei Kapseln à 75 mg) zweimal täglich (entspricht einer Tagesgesamtdosis von 300 mg), unabhängig vom Körpergewicht.
Bei pädiatrischen Patienten richtet sich die empfohlene Dosis für Tafinlar nach dem Körpergewicht (Tabelle 1 für Kapseln und Tabelle 2 für Tabletten).
Tabelle 1: Empfohlene gewichtsabhängige Dosierung für Tafinlar Kapseln für pädiatrische Patienten (Körpergewicht ≥26kg)
|
Körpergewicht
|
Empfohlene Dosis
| |
26 bis 37 Kilo
|
75 mg (eine Kapsel à 75 mg) zweimal täglich
| |
38 bis 50 Kilo
|
100 mg (zwei Kapseln à 50 mg) zweimal täglich
| |
≥51 Kilo
|
150 mg (zwei Kapseln à 75 mg) zweimal täglich
| |
Tafinlar Kapseln sind für Patienten mit einem Körpergewicht von weniger als 26 Kilo nicht geeignet.
|
Tabelle 2: Empfohlene gewichtsabhängige Dosierung für Tafinlar Tabletten zur Herstellung einer Suspension zum Einnehmen
|
|
Empfohlene Dosis
| |
Körpergewicht
|
Gesamttagesdosis
|
Anzahl der Tabletten für die orale Suspension
| |
8 bis 9 Kilo
|
20 mg zweimal täglich
|
zwei 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
| |
10 bis 13 Kilo
|
30 mg zweimal täglich
|
drei 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
| |
14 bis 17 Kilo
|
40 mg zweimal täglich
|
vier 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
| |
18 bis 21 Kilo
|
50 mg zweimal täglich
|
fünf 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
| |
22 bis 25 Kilo
|
60 mg zweimal täglich
|
sechs 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
| |
26 bis 29 Kilo
|
70 mg zweimal täglich
|
sieben 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
| |
30 bis 33 Kilo
|
80 mg zweimal täglich
|
acht 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
| |
34 bis 37 Kilo
|
90 mg zweimal täglich
|
neun 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
| |
38 bis 41 Kilo
|
100 mg zweimal täglich
|
zehn 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
| |
42 bis 45 Kilo
|
110 mg zweimal täglich
|
elf 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
| |
46 bis 50 Kilo
|
130 mg zweimal täglich
|
dreizehn 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
| |
≥51 Kilo
|
150 mg zweimal täglich
|
fünfzehn 10-mg-Tabletten zur Herstellung einer Suspension zum Einnehmen zweimal täglich
|
Verabreichungsschema
Tafinlar wird mindestens eine Stunde vor oder mindestens zwei Stunden nach einer Mahlzeit eingenommen, wobei zwischen den einzelnen Dosen ein Abstand von etwa 12 Stunden liegt.
Tafinlar soll jeden Tag ungefähr zur gleichen Zeit eingenommen werden.
Wenn Tafinlar und Trametinib in Kombination eingenommen werden, soll die einmal tägliche Dosis Trametinib täglich zur gleichen Zeit eingenommen werden wie die Morgen- oder Abenddosis von Tafinlar. Die Pharmakokinetik der abendlichen Einnahme von Trametinib wurde nicht untersucht, daher ist die morgendliche Einnahme vorzuziehen.
Zur Sicherstellung der Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln wird empfohlen, bei jeder Behandlung den Handelsnamen und die Chargennummer zu dokumentieren.
Hartkapseln
Tafinlar Hartkapseln sollen unzerkaut geschluckt werden. Die Kapseln dürfen nicht zerkaut oder zerkleinert werden.
Tabletten zur Herstellung einer Suspension zum Einnehmen
Tafinlar Tabletten zur Herstellung einer Suspension zum Einnehmen dürfen nur als Suspension eingenommen werden und sollen nicht im Ganzen geschluckt, gekaut oder zerkleinert werden.
Die orale Suspension wird in einem beigefügten Dosierbecher zubereitet. Tafinlar Suspensionen in Form von Tabletten zur Herstellung einer Suspension zum Einnehmen können auf drei verschiedene Arten eingenommen werden: durch Trinken der Suspension aus dem Dosierbecher, durch Schlucken der Suspension aus einer oralen Spritze, die mit der aus dem Dosierbecher entnommenen Suspension gefüllt ist, oder durch Aufnahme der Suspension über eine Magensonde.
Es ist darauf zu achten, dass die gesamte Dosis eingenommen wird. Es kann 3 Minuten (oder länger) dauern, bis die Tabletten vollständig aufgelöst sind. Sobald sie aufgelöst sind, sollte die Suspension trüb-weiss aussehen.
Die Suspension sollte nicht später als 30 Minuten nach dem Auflösen der Tabletten eingenommen werden. Sind mehr als 30 Minuten vergangen, ist die Suspension entsprechend den lokalen Bestimmungen zu entsorgen und die Anwendung erneut zu beginnen.
Eine vollständige und bebilderte Gebrauchsanweisung für die Tablette zur Herstellung einer Suspension zum Einnehmen finden Sie in der Rubrik «Gebrauchsanweisung».
Therapiedauer
Die Behandlung wird bis zur Feststellung einer Progression bzw. bis zum Auftreten nicht zumutbarer Toxizitätserscheinungen fortgesetzt (siehe Tabellen 3-6). Bei Auftreten einer Tumorprogression ist die Behandlung abzubrechen.
Bei einer adjuvanten Melanom-Behandlung sollten die Patienten maximal 12 Monate lang behandelt werden, es sei denn, es kommt zu einem Wiederauftreten der Krankheit oder zu einer inakzeptablen Toxizität.
Die empfohlene Behandlungsdauer für pädiatrische Patienten mit LGG dauert bis zur Krankheitsprogression oder bis zu inakzeptabler Toxizität. Dabei ist zu beachten, dass in den klinischen Studien bei pädiatrischen Patienten mit LGG deutliche Unterschiede in der Beurteilung der Wirksamkeit zwischen der unabhängigen Überprüfung und den Prüfzentren beobachtet wurden, die auch die Feststellung einer Krankheitsprogression umfassten (siehe auch «Klinische Wirksamkeit»). Darüber hinaus gibt es nur begrenzte Daten zu Langzeit-Anwendung und optimaler Therapiedauer der Behandlung mit Tafinlar in Kombination mit Trametinib in der pädiatrischen Population (siehe «Warnhinweise und Vorsichtsmassnahmen»). Die mediane Therapiedauer in den relevanten klinischen Studien betrug bisher ca. 24 Monate (siehe «Klinische Wirksamkeit»). Weiterhin liegen nur begrenzte Daten zu Patienten mit LGG im Alter von über 18 Jahren vor, die eine erste systemische Therapie benötigen. Daher ist die Fortsetzung der Behandlung bis ins Erwachsenenalter entsprechend der Beurteilung durch den Arzt von den Vorteilen und Risiken für den einzelnen Patienten abhängig zu machen.
Dosisanpassung aufgrund unerwünschter Wirkungen/Interaktionen
Monotherapie und in Kombination mit Trametinib
Bei unerwünschten Reaktionen kann eine Unterbrechung der Behandlung, eine Dosisreduktion oder der Behandlungsabbruch erforderlich werden (siehe Tabellen 3-6).
Bei unerwünschten Reaktionen in Form eines kutanen Plattenepithelkarzinoms (cuSCC) oder eines neuen Primärmelanoms werden keine Dosisanpassungen oder Behandlungsunterbrechungen empfohlen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Die Behandlung sollte unterbrochen werden, wenn die Körpertemperatur des Patienten ≥38,5 ºC beträgt. Die Patienten sollten auf Anzeichen und Symptome einer Infektion überwacht werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Die Empfehlungen zu den stufenweisen Dosisreduktionen und den Dosisanpassungen sind den Tabellen 3-6 zu entnehmen. Tafinlar als Kapsel ist dauerhaft abzusetzen, wenn 50 mg zweimal täglich nicht vertragen wird (Tabelle 3 und 4). Bei Patienten, die über mehrere Wochen ihre Dosis als Kapsel nicht auf > 50 mg BID erhöhen können, ist die Behandlung abzubrechen. Tafinlar als Tabletten zur Herstellung einer Suspension ist dauerhaft abzusetzen, wenn 10 mg zweimal täglich oder nach maximal drei Dosisreduktionen nicht vertragen wird (Tabelle 5).
Tabelle 3: Empfohlene Dosisreduktionsstufen von Tafinlar-Kapseln bei erwachsenen Patienten
|
Dosisreduktionen
|
Dosis/Verabreichungsschema
| |
Volle therapeutische Anfangsdosis
|
150 mg oral zweimal täglich
| |
Erste Dosisreduktion
|
100 mg oral zweimal täglich
| |
Zweite Dosisreduktion
|
75 mg oral zweimal täglich
| |
Dritte Dosisreduktion
|
50 mg oral zweimal täglich
| |
Dauerhaft absetzen, wenn Tafinlar 50 mg als Kapsel oral zweimal täglich nicht vertragen wird
|
Für die Trametinib Dosierungsanweisungen siehe Fachinformation Trametinib, «Dosierung / Anwendung».
Tabelle 4: Empfohlene Dosisreduktionen für Tafinlar-Kapseln bei pädiatrischen Patienten
|
Dosisreduktion
|
Empfohlene Anfangsdosis
| |
|
75 mg
oral zweimal täglich
|
100 mg
oral zweimal täglich
|
150 mg
oral zweimal täglich
| |
Erste Dosisreduktion
|
50 mg
oral zweimal täglich
|
75 mg
oral zweimal täglich
|
100 mg
oral zweimal täglich
| |
Zweite Dosisreduktion
|
-
|
50 mg
oral zweimal täglich
|
75 mg
oral zweimal täglich
| |
Dritte Dosisreduktion
|
-
|
-
|
50 mg
oral zweimal täglich
| |
Werden maximal drei Dosisreduktionen oder eine Tafinlar 50 mg Kapsel oral zweimal täglich nicht vertragen, muss es dauerhaft abgesetzt werden.
|
Tabelle 5: Empfohlene gewichtsabhängige Dosisreduktionsstufen von Tafinlar Tabletten zur Herstellung einer Suspension zum Einnehmen
|
Körpergewicht (kg)
(Empfohlene Anfangsdosierung)
|
Erste Dosisreduktion
|
Zweite Dosisreduktion
|
Dritte Dosisreduktion
| |
Tabletten zur oralen Suspension zweimal täglich
| |
8 bis 9 kg
(20 mg zweimal täglich)
|
20 mg zweimal täglich
|
N/A
|
N/A
| |
10 bis 13 kg
(30 mg zweimal täglich)
|
20 mg zweimal täglich
|
10 mg zweimal täglich
|
N/A
| |
14 bis 17 kg
(40 mg zweimal täglich)
|
30 mg zweimal täglich
|
20 mg zweimal täglich
|
10 mg zweimal täglich
| |
18 bis 21 kg
(50 mg zweimal täglich)
|
30 mg zweimal täglich
|
20 mg zweimal täglich
|
10 mg zweimal täglich
| |
22 bis 25 kg
(60 mg zweimal täglich)
|
40 mg zweimal täglich
|
30 mg zweimal täglich
|
20 mg zweimal täglich
| |
26 bis 29 kg
(70 mg zweimal täglich)
|
50 mg zweimal täglich
|
40 mg zweimal täglich
|
20 mg zweimal täglich
| |
30 bis 33 kg
(80 mg zweimal täglich)
|
50 mg zweimal täglich
|
40 mg zweimal täglich
|
30 mg zweimal täglich
| |
34 bis 37 kg
(90 mg zweimal täglich)
|
60 mg zweimal täglich
|
50 mg zweimal täglich
|
30 mg zweimal täglich
| |
38 bis 41 kg
(100 mg zweimal täglich)
|
70 mg zweimal täglich
|
50 mg zweimal täglich
|
30 mg zweimal täglich
| |
42 bis 45 kg
(110 mg zweimal täglich)
|
70 mg zweimal täglich
|
60 mg zweimal täglich
|
40 mg zweimal täglich
| |
46 bis 50 kg
(130 mg zweimal täglich)
|
90 mg zweimal täglich
|
70 mg zweimal täglich
|
40 mg zweimal täglich
| |
≥51 kg
(150 mg zweimal täglich)
|
100 mg zweimal täglich
|
80 mg zweimal täglich
|
50 mg zweimal täglich
| |
Dauerhaft absetzen, wenn Tafinlar 10 mg zweimal täglich als Tabletten zur Herstellung einer Suspension oder nach maximal drei Dosisreduktionen nicht vertragen wird.
|
Tabelle 6: Empfohlene Dosierungsänderungen für Tafinlar bei Nebenwirkungen
|
Schweregrad der Nebenwirkung [siehe Warnhinweise und Vorsichtsmassnahmen] a
|
Dosierungsänderung für Tafinlarb
| |
Neue primäre Malignome
| |
Nicht-kutane RAS-Mutations-positive Malignome
|
Tafinlar dauerhaft absetzen.
| |
Kardiomyopathie
| |
·Symptomatische kongestive Herzinsuffizienz
·Absolute Abnahme der linksventrikulären Ejektionsfraktion (LVEF) von mehr als 20 % gegenüber dem Ausgangswert, die unter der unteren Normgrenze (LLN) liegt.
|
Tafinlar aussetzen, bis sich die LVEF auf mindestens die institutionelle LLN verbessert hat und die absolute LVEF-Reduktion auf 10% oder weniger im Vergleich zum Ausgangswert zurückgegangen ist, dann die Behandlung mit derselben Dosis fortsetzen.
| |
Uveitis
| |
·Uveitis, einschliesslich Iritis und Iridozyklitis
|
Bei leichter oder mittelschwerer Uveitis, die nicht auf eine Therapie anspricht, oder bei schwerer Uveitis, Tafinlar bis zu 6 Wochen aussetzen.
·Bei Besserung auf Schweregrad 0-1 Tafinlar mit gleicher oder niedrigerer Dosis wieder aufnehmen.
·Wenn keine Besserung eintritt, setzen Sie Tafinlar dauerhaft ab.
| |
Fieberreaktionen
| |
·Fieber von 38°C bis 40°C (oder erste Symptome bei Rezidiv)
|
Unterbrechen Sie die Behandlung mit Tafinlar, bis das Fieber abgeklungen ist, und setzen Sie die Behandlung dann mit derselben oder einer niedrigeren Dosis fort.
| |
·Fieber über 40°C
·Fieber, kompliziert durch Schüttelfrost, Hypotonie, Dehydration oder Nierenversagen
|
·Unterbrechen Sie die Behandlung mit Tafinlar bis die fieberhaften Reaktionen für mindestens 24 Stunden abgeklungen sind, und setzen Sie die Behandlung dann mit einer niedrigeren Dosis fort.
Oder
·Setzen Sie Tafinlar dauerhaft ab.
| |
Hauttoxizitäten
| |
·Schweregrad 2, wenn nicht tolerierbar
·Schweregrad 3 oder 4
|
Unterbrechen Sie Tafinlar für bis zu 3 Wochen.
·Bei Besserung Tafinlar mit niedrigerer Dosis wieder aufnehmen.
·Wenn keine Besserung eintritt, setzen Sie Tafinlar dauerhaft ab.
| |
·Schwere kutane Nebenwirkungen (SCARs)
|
Tafinlar dauerhaft absetzen.
| |
Andere Nebenwirkungen, einschliesslich Blutungen
| |
·Schweregrad 2, wenn nicht tolerierbar
·Schweregrad 3
|
Tafinlar aussetzen.
·Bei Besserung auf Grad 0-1 Tafinlar mit niedrigerer Dosis wieder aufnehmen.
·Falls keine Besserung eintritt, Tafinlar dauerhaft absetzen.
| |
·Erstes Auftreten von Schweregrad 4
|
·Unterbrechen Sie die Behandlung mit Tafinlar, bis sich die Behandlung auf Grad 0-1 verbessert hat, und setzen Sie die Behandlung dann mit einer niedrigeren Dosis fort
Oder
·Tafinlar dauerhaft absetzen.
| |
·Rezidiv Schweregrad 4
|
Tafinlar dauerhaft absetzen.
|
a National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) Version 4.0.
b Siehe Tabelle 3-5 für empfohlene Dosisreduktionen von Tafinlar.
c Für die folgenden Nebenwirkungen werden keine Dosisanpassungen von Tafinlar empfohlen, wenn es zusammen mit Trametinib verabreicht wird: retinaler Venenverschluss (RVO), retinale Pigmentepithelablösung (RPED), interstitielle Lungenerkrankung/Pneumonitis und unkomplizierte venöse Thrombembolie. Eine Dosisanpassung von Tafinlar ist bei neuen primären Malignomen der Haut nicht erforderlich.
Beachten Sie die Trametinib-Fachinformation für Dosisanpassungen bei Nebenwirkungen im Zusammenhang mit Trametinib.
Wenn die unerwünschten Reaktionen bei einem Patienten effektiv kontrolliert sind, kann eine erneute Aufdosierung in entsprechenden Stufen wie bei der Dosisreduktion in Betracht gezogen werden. Die empfohlene Anfangsdosierung darf nicht überschritten werden, siehe Tabellen 3 und 4 für empfohlene Dosisreduktionsstufen für Tafinlar-Hartkapseln, und Tabelle 5 für empfohlene Dosisreduktionsstufen für Tafinlar dispergierbare Tabletten.
Wenn unter gleichzeitiger Behandlung mit Tafinlar und Trametinib behandlungsbedingte Toxizitätserscheinungen auftreten, sollten die Dosen beider Arzneimittel gleichzeitig reduziert oder beide Behandlungen unterbrochen bzw. ganz abgebrochen werden, wobei die untenstehenden Ausnahmen gelten.
Ausnahmen, bei denen nur bei Tafinlar eine Dosisänderung erforderlich ist:
·Uveitis
Behandlung von Pyrexie
Die Therapie sollte unterbrochen werden (Tafinlar, wenn es als Monotherapie angewendet wird, bzw. sowohl Tafinlar als auch Trametinib, wenn sie in Kombination angewendet werden), wenn die Körpertemperatur eines Patienten ≥38 °C (≥100,4 °F) beträgt. Im Falle eines Rezidivs kann die Therapie auch beim ersten Symptom einer Pyrexie unterbrochen werden. Es wird eine antipyretische Behandlung, z.B. mit Ibuprofen oder mit Paracetamol, eingeleitet. Die Patienten müssen auf Anzeichen und Symptome einer Infektion überwacht werden (siehe «Warnhinweise und Vorsichtsmassnahmen»). Die Behandlung mit Tafinlar bzw. sowohl mit Tafinlar als auch mit Trametinib, wenn diese in Kombination angewendet werden, sollte wieder aufgenommen werden, wenn der Patient seit mindestens 24 Stunden symptomfrei ist, und zwar:
·mit derselben Dosisstufe
·oder Dosis um eine Stufe reduzieren, falls die Pyrexie wieder auftritt und/oder von schweren Symptomen wie Dehydration, Hypotonie oder Nierenversagen begleitet wird.
Die Anwendung von Kortikosterioden sollte in Fällen erwogen werden, in denen Antipyretika nicht ausreichen.
Behandlung von Uveitis:
Es sollte versucht werden, die Uveitis mit einer lokalen Therapie am Auge zu beherrschen, ohne dabei die Dosierung von Tafinlar zu verändern. Wenn die Uveitis nicht auf eine lokale okulare Therapie anspricht, ist die Behandlung mit Tafinlar bis zum Rückgang der Augenentzündung zu unterbrechen und anschliessend mit einer um eine Dosisstufe reduzierten Tafinlar-Dosis wiederaufzunehmen. Falls Tafinlar in Kombination mit Trametinib eingenommen wird, so ist keine Trametinib Dosisanpassung nötig.
Für die empfohlenen Dosisanpassungen für Trametinib, siehe Fachinformation Mekinist.
Kombinationstherapie
Wenn Tafinlar in Kombination mit Trametinib verabreicht wird, ist die Mekinist Fachinformation bezüglich der Dosierungsanweisungen zu Mekinist zu konsultieren (s. Fachinformation Mekinist «Dosierung / Anwendung»).
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit leichter Leberfunktionsbeeinträchtigung ist keine Dosisanpassung erforderlich. Auf Grundlage der populationspharmakokinetischen Analyse hat eine leichte Einschränkung der Leberfunktion keinen relevanten Einfluss auf die orale Clearance von Dabrafenib bzw. auf die Konzentration seiner Metaboliten (siehe «Pharmakokinetik»). Klinische Daten von Personen mit mässiger bis hochgradiger Leberfunktionsbeeinträchtigung liegen nicht vor, daher lassen sich bezüglich der Notwendigkeit einer Dosisanpassung keine Angaben machen. Dabrafenib und seine Metaboliten unterliegen hauptsächlich einem hepatischen Metabolismus und der biliären Ausscheidung. Patienten mit mässiger bis hochgradiger Leberfunktionsbeeinträchtigung sind daher möglicherweise stärker exponiert und könnten ein höheres Risiko für unerwünschte Wirkungen aufgrund einer höheren Exposition zu Dabrafenib haben. Bei der Verabreichung von Tafinlar an Patienten mit mässiger bis hochgradiger Leberfunktionsbeeinträchtigung ist Vorsicht geboten.
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter bis mässiger Beeinträchtigung der Nierenfunktion ist keine Dosisanpassung erforderlich. Auf Grundlage der populationspharmakokinetischen Analyse hat eine leichte bis mässige Einschränkung der Nierenfunktion keinen relevanten Einfluss auf die orale Clearance von Dabrafenib bzw. auf die Konzentration seiner Metaboliten (siehe «Pharmakokinetik»). Klinische Daten von Personen mit hochgradiger Nierenfunktionsbeeinträchtigung liegen nicht vor, daher lassen sich bezüglich der Notwendigkeit einer Dosisanpassung keine Angaben machen. Bei der Verabreichung von Tafinlar an Patienten mit hochgradiger Nierenfunktionsbeeinträchtigung ist Vorsicht geboten.
Ältere Patienten
Keine Dosierungsanpassungen sind erforderlich bei Patienten über 65 Jahren (siehe «Pharmakokinetik»).
Pädiatrische Population
Die Anwendung von Tafinlar bei pädiatrischen Patienten im Alter von weniger als 1 Jahr ist nicht zugelassen. Aufgrund der renalen Toxizität von Tafinlar in Untersuchungen zur juvenilen Toxizität bei Ratten wurden diese Patienten von klinischen Studien mit Tafinlar ausgeschlossen (siehe «Allgemeine Toxizität»). Für Kinder zwischen 1 und 2 Jahren liegen nur begrenzte Daten zur Sicherheit und Wirksamkeit vor (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Genotyp/Genetische Polymorphismen
Wirksamkeit und Sicherheit von Tafinlar sind bei Patienten mit BRAF-Wildtyp-Melanom oder BRAF-Wild-Typ NSCLC nicht erwiesen; Tafinlar darf daher bei Melanom- und NSCLC-Patienten mit BRAF-Wildtyp nicht angewendet werden (siehe «Eigenschaften/ Wirkungen»).
Verspätete Dosisgabe
Eine versäumte Tafinlar Dosis darf nicht nachgeholt werden, wenn bis zur nächsten planmässigen Dosis weniger als 6 Stunden verbleiben.
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile.
Warnhinweise und VorsichtsmassnahmenSchwerwiegende nichtinfektiöse febrile Ereignisse
Aus klinischen Studien mit Tafinlar Monotherapie als auch mit Tafinlar in Kombination mit Trametinib wurde das Auftreten von Fieber gemeldet (siehe «Unerwünschte Wirkungen»).
Häufigkeit und Schwere der Pyrexie waren unter der Kombinationstherapie erhöht. Die Mehrheit der Pyrexie-Ereignisse trat innerhalb des ersten Therapiemonats auf. Bei etwa einem Drittel der Patienten unter der Kombinationstherapie, bei denen Pyrexie auftrat, traten drei oder mehr Ereignisse ein.
Pyrexie kann von schwerem Schüttelfrost, Dehydratation und Hypotonie begleitet werden, was in manchen Fällen zu akuter Niereninsuffizienz führen kann. Während sowie nach einem schwerwiegenden Fieber-Ereignis sollten die Serum-Kreatininwerte kontrolliert werden und der Patient auf weitere Hinweise einer Niereninsuffizienz überprüft werden.
Es wurden schwerwiegende, nichtinfektiöse febrile Ereignisse beobachtet. Diese Ereignisse haben in klinischen Studien gut auf eine Unterbrechung der Behandlung und/oder eine Dosisreduktion und auf unterstützende Massnahmen angesprochen (inkl. der Verabreichung von nicht-steroidalen und steroidalen Antipyretika).
Ein studienübergreifender Vergleich bei 1810 Patienten, die mit der Kombinationstherapie behandelt wurden, zeigte eine Verringerung der Inzidenz von hochgradiger Pyrexie und anderen pyrexiebezogenen unerwünschten Ereignissen, wenn die Behandlung sowohl mit Tafinlar als auch mit Trametinib unterbrochen wurde, im Vergleich zu einer Unterbrechung nur von Tafinlar. Daher wird eine Unterbrechung sowohl von Tafinlar als auch von Trametinib empfohlen, wenn die Körpertemperatur des Patienten ≥38 °C (≥100,4 °F) beträgt. Im Falle eines Rezidivs kann die Therapie auch beim ersten Symptom einer Pyrexie unterbrochen werden (siehe «Dosierung/Anwendung» und «Klinische Wirksamkeit»).
Zur Behandlung von Pyrexie, siehe Leitlinien zur Dosisänderung (siehe «Dosierung/Anwendung»).
Glucose-6-phosphat-Dehydrogenase-Mangel
Tafinlar, das einen Sulfonamid-Anteil enthält, birgt ein potenzielles Risiko für eine hämolytische Anämie bei Patienten mit Glucose-6-phosphat-Dehydrogenase-Mangel (G6PD). Überwachen Sie Patienten mit G6PD-Mangel auf Anzeichen einer hämolytischen Anämie während der Einnahme von Tafinlar.
Hyperglykämie
In klinischen Studien wurden in einigen Patienten, welche mit Tafinlar behandelt worden sind, erhöhte Blutzuckerwerte gemessen. Bei Patienten mit Diabetes oder Hyperglykämie sollten die Blutzuckerwerte während der Therapie mit Tafinlar engmaschig überwacht werden.
Kutanes Plattenepithelkarzinom (cuSCC)
Sowohl bei Patienten unter Behandlung mit Tafinlar Monotherapie als auch mit Tafinlar in Kombination mit Trametinib wurden Fälle von cuSCC (einschliesslich als Keratoakanthom oder als Subtyp gemischtes Keratoakanthom klassifizierte Fälle) gemeldet (siehe «unerwünschte Wirkungen»).
In der Phase-III-Studie MEK115306 trat cuSCC bei 10 % (22/211) der Patienten auf, die Tafinlar als Monotherapie erhielten, wobei die mediane Zeit bis zur Diagnose des ersten Auftretens annähernd 8 Wochen betrug. cuSCC trat bei 3 % (6/209) der Patienten, die Tafinlar in Kombination mit Trametinib erhielten, auf und hier traten die Ereignisse später ein, wobei die mediane Zeitspanne bis zur Diagnose des ersten Auftretens 20 bis 32 Wochen betrug. Mehr als 90 % der Patienten, die Tafinlar erhielten und bei denen cuSCC auftrat, wurden ohne Dosierungsänderungen weiter mit Tafinlar behandelt. In der Phase-III-Studie zur adjuvanten Behandlung von Melanom trat bei 1% (6/435) der Patienten unter Tafinlar in Kombination mit Trametinib kutanes Plattenepithelkarzinom (cuSCC) auf, im Vergleich zu 1% (5/432) der Patienten, die ein Placebo erhielten. Die mediane Zeit bis zum ersten Auftreten von cuSCC war im Kombinationsarm ungefähr 18 Wochen.
Eine Hautuntersuchung wird vor Einleitung einer Tafinlar Therapie empfohlen sowie jeweils alle 2 Monate während der Behandlung. Regelmässige Hautkontrollen sollten alle 2-3 Monate bis zu sechs Monate nach Beendigung der Therapie mit Tafinlar durchgeführt werden bzw. bis zum Beginn einer neuen antineoplastischen Therapie.
Bei Vorliegen eines cuSCC sollte eine dermatologische Exzision erfolgen und die Behandlung mit Tafinlar bei unveränderter Dosierung fortgesetzt werden. Die Patienten sind anzuweisen, bei der Entwicklung neuer Läsionen unverzüglich ihren Arzt zu benachrichtigen.
Neues Primärmelanom
Aus klinischen Studien wurde das Auftreten neuer Primärmelanome unter Behandlung mit Tafinlar gemeldet. Diese wurden innerhalb der ersten 5 Behandlungsmonate festgestellt, per Exzision behandelt und erforderten keine Behandlungsänderung. In der klinischen Phase-III-Studie zur adjuvanten Behandlung von Melanom traten neue Primärmelanome bei <1% (1/435) der Patienten unter Kombinationstherapie von Tafinlar und Trametinib auf im Vergleich zu 1% (6/432) der Patienten, die ein Placebo erhielten.
Die Patienten sollten, wie bereits für das kutane Plattenepithelkarzinom beschrieben, auf das Auftreten von Hautläsionen hin überwacht werden.
Zusätzliche Informationen bezüglich Warnhinweisen und Vorsichtsmassnahmen in Zusammenhang mit einer Behandlung mit Trametinib finden Sie in der Mekinist Fachinformation.
Nichtkutane sekundäre/ rezidivierende Malignome
Aus In-vitro-Studien ist bei Exposition von NRAS mutierten BRAF-Wildtyp-Zellen gegenüber BRAF-Inhibitoren eine paradoxe Aktivierung des MAP-Kinase-Signalweges bekannt. Dies kann bei Patienten unter Tafinlar zu einem erhöhten Risiko nichtkutaner Malignome führen. Fälle von RAS-assoziierten Malignomen wurden unter BRAF Inhibitoren beobachtet.
Beim Vergleich der Kombination von Tafinlar und Trametinib zu Placebo wurden in der Phase-III-Studie zur adjuvanten Behandlung von Melanom nichtkutane sekundäre Malignome oder rezidivierte Malignome bei 1% (5/435) der Patienten unter aktiver Therapie beobachtet, im Vergleich zu 1% (3/432) der Patienten, die ein Placebo erhielten.
Die Patienten sind entsprechend den klinischen Gegebenheiten zu überwachen. Bei Patienten mit einer nicht kutanen malignen Erkrankung, die durch eine RAS-Mutation gekennzeichnet ist, sind vor Fortsetzung der Therapie mit Tafinlar Nutzen und Risiken abzuwägen. Unter der Kombinationstherapie von Tafinlar mit Trametinib sind keine Dosisanpassungen für Trametinib erforderlich.
Vor Beginn der Behandlung sollte bei den Patienten eine Untersuchung des Kopf- und Halsbereichs durchgeführt werden mit visueller Begutachtung der Mundschleimhaut und Palpation der Lymphknoten als minimale Massnahme, sowie eine CT-Aufnahme des Thorax/des Abdomens. Während der Behandlung sollten die Patienten wie klinisch geboten überwacht werden, einschliesslich Untersuchungen des Kopf- und Halsbereichs alle 3 Monate und CT-Aufnahmen des Thorax/des Abdomens alle 6 Monate. Rektale Untersuchungen und (bei Frauen) des Beckens werden vor Beginn und am Ende der Behandlung empfohlen, oder wie klinisch indiziert. Das grosse Blutbild sollte wie klinisch indiziert bestimmt werden.
Der Patient sollte bis zu 6 Monate nach Beendigung der Tafinlar -Therapie bzw. bis zum Beginn einer neuen antineoplastischen Therapie regelmässig auf nichtkutane sekundäre / wiederkehrende Malignome kontrolliert werden.
Uveitis
Die Behandlung mit Tafinlar wurde mit der Entwicklung von Uveitis (einschliesslich Iritis) assoziiert. Die Patienten sind unter der Behandlung routinemässig auf die Sehfunktion betreffende Anzeichen und Symptome (z.B. Sehstörungen, Photophobie und Augenschmerzen) zu überwachen (vgl. «Dosierung / Anwendung»). Bei Patienten, die mit Tafinlar in Kombination mit Mekinist behandelt wurden, wurden Fälle von biokularer Panuveitis oder biokularer Iridozyklitis berichtet, die auf ein Vogt-Koyanagi-Harada-ähnliches Syndrom hindeuten. In solchen Fällen kann eine systemische Kortikosteroidbehandlung in Betracht gezogen werden.
Pankreatitis
Eine Pankreatitis wurde bei < 1 % der mit Tafinlar behandelten Studienteilnehmer mit metastasiertem Melanom beobachtet. Eine akute Pankreatitis wurde bei 1% der mit Tafinlar behandelten Studienteilnehmer mit NSCLC beobachtet. Eines dieser Ereignisse trat am ersten Tag der Behandlung auf und erneut nach Wiederaufnahme der Therapie mit einer reduzierten Dosis. In der Studie zur adjuvanten Behandlung von Melanom wurde Pankreatitis bei 1% der Patienten unter Tafinlar in Kombination mit Trametinib berichtet sowie bei <1% der Patienten, die ein Placebo erhielten
Unerklärlichen Bauchschmerzen sollte unverzüglich nachgegangen werden, unter anderem durch die Bestimmung von Serumamylase und -lipase. Bei Wiederaufnahme der Behandlung mit Tafinlar nach einer pankreatitischen Episode sind die Patienten engmaschig zu überwachen.
Blutungen
Blutungsereignisse, darunter schwere Blutungsereignisse in kritische Körperbereiche und Organe, sind bei Patienten aufgetreten, die Tafinlar in Kombination mit Trametinib angewendet haben (siehe «Unerwünschte Wirkungen»). In klinischen Studien mit dieser Kombination wurden in 17% der Patienten Blutungen berichtet, 3% der Patienten hatten gastrointestinale Blutungen. Sechs von 559 Patienten (1%) mit nicht resezierbarem oder metastasiertem Melanom, die Tafinlar in Kombination mit Trametinib erhielten, hatten tödliche intrakranielle Blutungsereignisse. Drei Fälle wurden in Studie MEK115306 (COMBI-d) und drei in Studie MEK116513 (COMBI-v) beobachtet. Bei der adjuvanten Behandlung des Melanoms traten in der Phase-III-Studie keine tödlich verlaufenden Blutungsereignisse auf.
Zwei von 93 Teilnehmern (2 %), die Tafinlar in Kombination mit Trametinib in einer Phase-II-Studie zu nicht-kleinzelligem Lungenkarzinom (NSCLC; non-small cell lung cancer) erhielten, hatten tödliche Blutungsereignisse (einen Fall von Subarachnoidalblutung und einen Fall von retroperitonealer Blutung).
Wenn Patienten Symptome einer intrakraniellen Blutung entwickeln, ist eine umgehende medizinische Versorgung erforderlich.
Das Risiko von Blutungsereignissen kann durch die begleitende Anwendung einer Antiplatelet- oder Antikoagulans-Therapie erhöht werden. Bei Blutungen sollten Patienten gemäss klinischer Indikation behandelt werden.
Bei Blutungen vom Grad 4 ist Tafinlar dauerhaft abzusetzen, ebenso bei Grad 3 Blutungen, die sich nicht bessern.
Venöse Thromboembolien (VTE)
Bei Anwendung von Tafinlar in Kombination mit Trametinib können venöse Thromboembolien (VTE), einschliesslich der tiefen Venenthrombose (TVT) und der Lungenembolie (LE), auftreten. Die Patienten sollten angewiesen werden, sich beim Auftreten von Symptomen einer VTE umgehend in ärztliche Behandlung zu begeben. Im Fall einer lebensbedrohlichen Lungenembolie ist die Behandlung mit Trametinib und Dabrafenib dauerhaft abzusetzen.
Schwere unerwünschte Hautreaktionen (SCARs)
Berichte über schwere unerwünschte Hautreaktionen, inklusive Stevens-Johnson-Syndrom und Arzneimittelreaktion mit Eosinophilie und systemischen Symptomen (DRESS), welche lebensbedrohlich oder tödlich verlaufen können, sind während der Therapie mit Dabrafenib in Kombination mit Trametinib gemeldet worden. Vor Beginn der Behandlung sollen Patienten über Zeichen und Symptome aufgeklärt werden.
Während der Therapie sollten Patienten engmaschig auf Hautreaktionen untersucht werden. Falls der Verdacht auf eine schwere unerwünschte Hautreaktion aufkommt, müssen Dabrafenib und Trametinib abgesetzt werden.
InteraktionenDabrafenib ist ein Substrat von CYP2C8 und CYP3A4. Die gleichzeitige Verabreichung starker Induktoren dieser Enzyme sollte daher nach Möglichkeit vermieden werden, da dies die Wirksamkeit von Tafinlar beeinträchtigen könnte.
Dabrafenib ist ein Induktor metabolisierender Enzyme und kann Transporter induzieren.
Die gleichzeitige Anwendung von Tafinlar mit Arzneimitteln, die empfindliche Substrate für die Enzyme CYP3A4, CYP2C8, CYP2C9, CYP2C19 oder CYP2B6 (z.B. orale Kontrazeptiva) bzw. die Substrate für Transporter sind, kann zu einem Wirksamkeitsverlust dieser Arzneimittel führen und ist daher, sofern keine entsprechenden Dosisanpassungen möglich sind, generell zu vermeiden (siehe Abschnitt «Interaktionen»).
Die gleichzeitige Verabreichung von Dabrafenib mit Warfarin führt zu verminderter Warfarin Exposition. Die Einleitung oder Beendigung einer Behandlung mit Dabrafenib bei gleichzeitiger Gabe von Warfarin, Acenocoumarol und Phenprocoumon erfordert Vorsicht sowie eventuell eine strengere Überwachung des INR-Werts (International Normalized Ratio).
Hilfsstoffe von besonderem Interesse (Tabletten zur Herstellung einer Suspension zum Einnehmen.)
Jede Tablette zur Herstellung einer Suspension zum Einnehmen enthält <0,00078 mg Benzylalkohol. Benzylalkohol kann allergische Reaktionen hervorrufen.
Wenden Sie dieses Arzneimittel bei Kleinkindern (unter 3 Jahren) nicht länger als eine Woche an, ausser auf Anraten Ihres Arztes oder Apothekers bzw. Ihrer Ärztin oder Apothekerin. Bei Kleinkindern besteht aufgrund von Akkumulation ein erhöhtes Risiko.
Weisen Sie Patientinnen, die schwanger sind oder werden könnten, auf das potenzielle Risiko für den Fötus durch den Hilfsstoff Benzylalkohol hin, der sich mit der Zeit anreichern und eine metabolische Azidose verursachen kann. Bei Patienten mit eingeschränkter Leber- oder Nierenfunktion mit Vorsicht anwenden, da sich Benzylalkohol mit der Zeit anreichern und eine metabolische Azidose verursachen kann.
Bei Umstellung der Therapie auf eine andere Darreichungsform und/oder ein anderes Arzneimittel mit gleichem Wirkstoff ist Vorsicht geboten. Der Patient sollte adäquat kontrolliert werden.
Hemophagocytic lymphohistiocytosis (HLH)
In der Erfahrung nach der Markteinführung von Tafinlar in Kombination mit Mekinist (trametinib) wurde HLH beobachtet. HLH ist ein potentiell lebensbedrohliches Syndrom mit pathologischer Aktivierung der Immunabwehr. Falls die HLH nicht frühzeitig erkannt und behandelt wird, verläuft sie häufig letal. Die Erkrankung ist gekennzeichnet durch klinische Anzeichen und Symptome einer schweren systemischen Entzündung wie Fieber, Hautausschlag, Hepatosplenomegalie, Zytopenien, Lymphadenopathie, neurologische Symptome, hohes Serum-Ferritin, Hypertriglyceridämie sowie Störungen der Leberfunktion und der Koagulation. Die Symptome treten in der Regel innerhalb von 2 Monaten nach Beginn der Behandlung auf, ein späteres Auftreten ist aber möglich. Bei Verdacht auf HLH sollte die Behandlung unterbrochen werden. Nach Bestätigung von HLH sollte die Behandlung abgebrochen und eine geeignete HLH-Behandlung eingeleitet werden.
Tumorlysesyndrom (TLS)
Bei Patienten, die mit Mekinist in Kombination mit Tafinlar behandelt wurden, sind Fälle von TLS, einschliesslich tödlich verlaufender Fälle, berichtet worden. Zu den Risikofaktoren für TLS gehören schnell wachsende Tumore, eine hohe Tumorlast, Nierenfunktionsstörungen und Dehydrierung. Patienten mit Risikofaktoren für TLS sollten engmaschig überwacht werden. Eine Prophylaxe sollte in Betracht gezogen werden (z.B. intravenöse Flüssigkeitszufuhr und Behandlung hoher Harnsäurespiegel vor Beginn der Behandlung) und Patienten sollten je nach klinischer Indikation behandelt werden.
Pädiatrische Population
Die sich in Entwicklung befindlichen pädiatrische Patienten unterliegen einer potentiellen Langzeittherapie mit Tafinlar in Kombination mit Trametinib. Gleichzeitig wirken beide Arzneimittel auf eine Signaltransduktionskette, die eine wichtige Rolle bei der Regulierung von Zell- und Gewebeentwicklung spielt (siehe «Wirkungsmechanismus»). Vor diesem Hintergrund weisen die in der pädiatrischen Population vorliegenden Daten Limitationen auf (siehe auch «Dosierung/Anwendung»).
So liegen keine ausreichenden Daten zu Sicherheit und Wirksamkeit der Langzeit-Anwendung und optimaler Therapiedauer der Behandlung mit Tafinlar in Kombination mit Trametinib in der pädiatrischen Population vor. Die mediane Therapie- und Nachbeobachtungsdauer betrug in den relevanten klinischen Studien bisher ca. 24 respektive 26 Monate (siehe «Klinische Wirksamkeit»). Die Langzeitfolgen der unter der Kombination sehr häufig beobachteten unerwünschten Gewichtszunahmen sind derzeit unklar. Obwohl bisher keine Zunahme der Häufigkeiten sekundärer maligner Neoplasien in der pädiatrischen Population berichtet wurde, ist eine abschliessende Bewertung zum jetzigen Zeitpunkt nicht möglich (siehe «Unerwünschte Wirkungen»).
Für Kinder zwischen 1 und 2 Jahren liegen nur begrenzte Daten zur Sicherheit und Wirksamkeit vor.
Der Beitrag von Tafinlar und Trametinib zur Wirksamkeit der Kombination bei pädiatrischen Patienten mit LGG ist unklar, da ein direkter Vergleich zwischen Kombination und Monotherapien in einer ausreichend grossen Patientenpopulation fehlt.
Von klinischen Studien ausgeschlossene Patienten
Die gemeinsamen Ausschlusskriterien für alle Studien zu Tafinlar waren: Wildtyp-BRAF oder eine Nicht-V600-Mutation; Vorgeschichte oder Hinweise auf ein kardiovaskuläres Risiko; niedriger Leistungsstatus; bekannter Glucose-6-Phosphat-Dehydrogenase (G6PD)-Mangel. Studienspezifische Ausschlusskriterien finden Sie im Abschnitt «Klinische Wirksamkeit».
Interaktionen
Tafinlar Monotherapie
Wirkung von Tafinlar auf andere Arzneimittel
Dabrafenib führte bei humanen Hepatozyten zu einem dosisabhängigen Konzentrationsanstieg der CYP2B6- und CYP3A4-mRNA bis zum 32-fachen der Kontrollen und erwies sich in vivo als CYP3A4- und CYP2C9-Induktor. In einer klinischen Studie mit 12 Teilnehmern waren bei Verabreichung mit einer Einzeldosis des CYP3A4-Substrats Midazolam unter regelmässiger Einnahme von Dabrafenib die AUC und die Cmax von Midazolam um 74 % und um 61% herabgesetzt gegenüber einer Einzeldosis Midazolam ohne Dabrafenib. In einer weiteren Studie mit 14 Teilnehmern war bei wiederholter Verabreichung von Dabrafenib die AUC nach Einzeldosen von S-Warfarin (CYP2C9 Substrat) und R-Warfarin (CYP3A4/CYP1A2 Substrat) um 37 % bzw. 33 % vermindert, wobei es zu einem minimalen Anstieg der Cmax um 18 % bzw. 19 % kam. Dabrafenib kann auch andere Enzyme, unter anderem CYP2B6, CYP2C8 und CYP2C19, UDP Glucuronosyltransferase (UGT) und Transporter (z.B. P-Glycoprotein, P-gp) induzieren.
Die gleichzeitige Verabreichung von Tafinlar mit Arzneimitteln, die durch die Induktion dieser Enzyme unter Tafinlar beeinflusst werden, z.B. hormonale Kontrazeptiva (siehe «Schwangerschaft/ Stillzeit»), Antikoagulantien (z.B. Acenocoumarol, Warfarin, Rivaroxaban), Kortikosteroide (z.B. Dexamethason), antiretrovirale Substanzen (z.B. Amprenavir, Atazanavir, Darunavir), durch CYP3A4 metabolisierte Statine (z.B. Atorvastatin, Simvastatin) oder Immunsuppressiva (z.B. Cyclosporin, Tacrolimus, Sirolimus) kann zu verminderten Konzentrationen und einem Wirksamkeitsverlust führen. Nach Möglichkeit ist eine Substitution dieser Arzneimittel in Betracht zu ziehen. Der Effekt von Dabrafenib auf die Exposition von Phenprocoumon und Acenocoumarol ist bisher nicht untersucht worden. Wenn die gleichzeitige Verabreichung unumgänglich ist, sind die betroffenen Patienten auf einen etwaigen Wirksamkeitsverlust hin zu überwachen.
Dabrafenib ist ein in vitro Hemmer des humanen organischen Anionen-Transporterpolypeptids (OATP) 1B1 (OATP1B1) und des OATP1B3; eine klinische Relevanz dieser Eigenschaft kann nicht ausgeschlossen werden. Aus diesem Grund ist Vorsicht geboten bei der gleichzeitigen Verabreichung von Dabrafenib mit OATP1B1- bzw. OATP1B3-Substraten, z.B. Statinen.
Obwohl Dabrafenib und seine Metaboliten Hydroxy-Dabrafenib, Carboxy-Dabrafenib und Desmethyl-Dabrafenib sich in vitro als Inhibitoren des humanen organischen Anionen-Transporterpolypeptids (OATP) 1B1 (OATP1B1), des OATP1B3, des organischen Anionentransporters (OAT) 1 und des OAT3 erwiesen haben, ist das Risiko einer Arzneimittelwechselwirkung bei klinischer Exposition minimal. Dabrafenib und Desmethyl-Dabrafenib haben sich ausserdem als moderate Hemmer des humanen Brustkrebs-Resistenzproteins (BCRP) erwiesen; bei klinischer Exposition ist das Risiko einer Arzneimittelwechselwirkung jedoch minimal.
Weder für Dabrafenib noch für seine 3 Metaboliten konnte in vitro eine Hemmwirkung auf das P-Glycoprotein (Pgp) nachgewiesen werden. Wechselwirkungen sind mit vielen Arzneimitteln zu erwarten, die über Verstoffwechselung oder mittels aktiven Transports eliminiert werden. Falls deren therapeutische Wirkung von grosser Bedeutung für den Patienten ist und Dosisanpassungen nicht einfach auf Basis der Überwachung der Wirksamkeit oder von Plasmakonzentrationen durchgeführt werden können, sind diese Arzneimittel nur mit Vorsicht anzuwenden. Es wird vermutet, dass das Risiko für eine Leberschädigung nach Gabe von Paracetamol möglicherweise bei Patienten höher ist, die gleichzeitig mit Enzyminduktoren behandelt werden.
Die Zahl der von möglichen Wechselwirkungen betroffenen Arzneimittel wird als sehr hoch eingeschätzt, obwohl die Grössenordnung der Wechselwirkungen variieren kann. Gruppen von möglicherweise betroffenen Arzneimitteln beinhalten, sind aber nicht beschränkt auf:
·Analgetika (z.B. Fentanyl, Methadon)
·Antibiotika (z.B. Clarithromycin, Doxycyclin)
·Antineoplastische Arzneimittel (z. B.Cabazitaxel)
·Antikoagulanzien (z.B. Acenocoumarol, Phenprocoumon, Warfarin)
·Antiepileptika (z.B. Carbamazepin, Phenytoin, Primidon, Valproinsäure)
·Antipsychotika (z.B. Haloperidol)
·Kalziumkanalblocker (z.B. Diltiazem, Felodipin, Nicardipin, Nifedipin, Verapamil)
·Herzglykoside (z. B. Digoxin)
·Kortikosteroide (z. B. Dexamethason, Methylprednisolon)
·Antivirale Mittel gegen HIV (z.B. Amprenavir, Atazanavir, Darunavir, Delavirdin, Efavirenz, Fosamprenavir, Indinavir, Lopinavir, Nelfinavir, Saquinavir, Tipranavir)
·Hormonelle Kontrazeptiva
·Hypnotika (z.B. Diazepam, Midazolam, Zolpidem)
·Immunsuppressiva (z.B. Cyclosporin, Tacrolimus, Sirolimus)
·Statine, die über CYP3A4 verstoffwechselt werden (z.B. Atorvastatin, Simvastatin)
Der Eintritt der induzierenden Wirkung tritt wahrscheinlich nach 3 Tagen wiederholter Gabe von Dabrafenib auf. Nach Absetzen von Dabrafenib erfolgt die Kompensation der Induktion schrittweise, die Konzentrationen der sensitiven CYP3A4-, CYP2B6-, CYP2C8-, CYP2C9- und CYP2C19-Enzyme, der UDP-Glucuronosyltransferase (UGT) und der Transportsubstrate können ansteigen, auch sollten die Patienten auf Toxizitäten hin überwacht und, falls erforderlich, die Dosis dieser Arzneimittel angepasst werden. Die Kombination mit Arzneimitteln, welche den pH-Wert im Magen erhöhen wie Protonenpumpeninhibitoren, H2-Rezeptorantagonisten und Antazida ist nicht untersucht worden. Die Kombination mit solchen Arzneimitteln könnte aufgrund der pH-abhängigen Löslichkeit von Dabrafenib die Absorption von Dabrafenib beeinträchtigen und ist deshalb zu vermeiden.
Wirkung anderer Arzneimittel auf Tafinlar
In den präklinischen in-vitro-Untersuchungen wurde nachgewiesen, dass Dabrafenib hauptsächlich über CYP2C8 und CYP3A4 metabolisiert wird, während es sich bei Hydroxy-Dabrafenib und Desmethyl-Dabrafenib um CYP3A4-Substrate handelt. Die pharmakokinetischen Daten haben bei gleichzeitiger Verabreichung von Dabrafenib (wiederholte Dosen) mit Ketoconazol (CYP3A4 Inhibitor) einen Anstieg der Dabrafenib Cmax um 33 % und der AUC um 71 %, sowie einen Anstieg der AUC von Hydroxy- und Desmethyl-Dabrafenib um 82 bzw. 68 % ergeben. Bei Carboxy-Dabrafenib wurde eine Abnahme der AUC um 16 % festgestellt. Bei der gleichzeitigen Verabreichung von Gemfibrozil (CYP2C8 Inhibitor) ergab sich in dieser Studie ein Anstieg der Dabrafenib AUC um 47 %, aber ohne Anstieg der Cmax. Gemfibrozil hatte keine klinisch relevante Wirkung auf die systemische Exposition der Dabrafenib-Metaboliten (≤13 %).
Starke Hemmer oder Induktoren des CYP2C8 oder des CYP3A4 führen daher wahrscheinlich zu einer Erhöhung bzw. Abnahme der Dabrafenib-Konzentrationen. Nach Möglichkeit sollten bei gleichzeitiger Verabreichung mit Dabrafenib alternative Wirkstoffe zum Einsatz kommen. Bei der gleichzeitigen Verabreichung von Dabrafenib mit starken Hemmern (z.B. Ketoconazol, Nefazodon, Clarithromycin, Ritonavir, Saquinavir, Telithromycin, Itraconazol, Voriconazol, Posaconazol, Atazanavir) oder Induktoren (z.B. Rifampicin, Phenytoin, Carbamazepin, Phenobarbital, Johanniskraut (Hypericum perforatum)) von CYP2C8 oder CYP3A4 ist Vorsicht geboten.
Dabrafenib ist in vitro ein Substrat des humanen Pgp und des murinen BCRP 1. Diese Transportmoleküle haben jedoch minimale Auswirkungen auf die orale Bioverfügbarkeit und die Elimination von Dabrafenib; aus diesem Grund ist das Risiko einer klinisch relevanten Arzneimittelwechselwirkung mit Pgp- oder BCRP-Hemmern gering.
Wirkung von Arzneimitteln, welche den pH-Wert im Magen verändern
Arzneimittel, welche den pH-Wert im Magen beeinflussen (z.B. Protonenpumpenhemmer, H2-Rezeptorantagonisten, Antazida) können die Löslichkeit von Dabrafenib verändern und damit die Bioverfügbarkeit reduzieren. Es wurden jedoch keine entsprechenden spezifischen klinischen Studien durchgeführt. Die Auswirkung auf die Wirksamkeit von Tafinlar ist nicht bekannt.
Tafinlar und Trametinib Kombinationstherapie
Die gleichzeitige Verabreichung wiederholter Dosen Dabrafenib 150 mg zweimal täglich und Trametinib 2 mg einmal täglich führte nicht zu klinisch relevanten Cmax- und AUC-Änderungen bei Dabrafenib oder Trametinib, mit Anstiegen der Cmax und der AUC von Dabrafenib um 16 % bzw. 23 %. Anhand einer populationspharmakokinetischen Analyse wurde für die gleichzeitige Verabreichung von Dabrafenib mit Trametinib ein geringfügiger Rückgang der Bioverfügbarkeit von Trametinib, entsprechend einer 12 %igen Abnahme der AUC, geschätzt.
Schwangerschaft, StillzeitSchwangerschaft
Weibliche und männliche Patienten müssen wirksame Empfängnisverhütungsmethoden anwenden:
Frauen: Potenziell gebärfähige Frauen müssen während der Behandlung und in den ersten zwei Wochen nach Behandlungsende mit Tafinlar und mindestens 16 Wochen nach der letzten Einnahme von Trametinib wirksame Methoden der Empfängnisverhütung anwenden (Methoden, die zu Schwangerschaften von weniger als 1% führen).
Wird Tafinlar als Monotherapie oder in Kombination mit Trametinib in der Schwangerschaft verabreicht oder wird eine Patientin während der Behandlung schwanger, so muss sie über das potenzielle Risiko für das Ungeborene unterrichtet werden.
Frauen in gebärfähigen Alter, die Tafinlar in Kombination mit Trametinib erhalten, sollten darüber aufgeklärt werden, dass Tafinlar die Wirksamkeit von Hormon-Verhütungsmitteln senken kann und dass alternative Empfängnisverhütungsmethoden angewendet werden sollten (siehe «Interaktionen»).
Männer: Männliche Patienten (auch solche, bei denen eine Vasektomie durchgeführt wurde) mit Sexualpartnerinnen, die schwanger sind, möglicherweise schwanger sind oder schwanger werden könnten, müssen während der Monotherapie mit Tafinlar sowie für mindestens 2 Wochen nach Beendigung der Behandlung mit Tafinlar während des Geschlechtsverkehrs Kondome verwenden. Wenn die männlichen Patienten eine Kombinationstherapie mit Tafinlar und Trametinib erhalten, müssen sie während der Behandlungszeit sowie für mindestens 16 Wochen nach Beendigung der Behandlung mit Tafinlar während des Geschlechtsverkehrs Kondome verwenden.
Stillzeit
Es liegen keine Daten über die Wirkung von Tafinlar auf das gestillte Kind oder die Wirkung von Tafinlar auf die Milchproduktion vor. Zur Ausscheidung von Dabrafenib in der Muttermilch ist nichts bekannt. Da viele Arzneimittel in der Muttermilch ausgeschieden werden, kann ein Risiko für das gestillte Kind nicht ausgeschlossen werden. Bei stillenden Müttern sollte Tafinlar als Monotherapie oder in Kombination mit Trametinib nicht angewendet werden. Unter Abwägung der Wichtigkeit des Stillens für das Kind gegen den Nutzen der Behandlung für die Mutter sollte entweder abgestillt oder die Behandlung mit Tafinlar als Monotherapie oder in Kombination mit Trametinib beendet werden.
Fertilität
Daten zur Fertilität beim Menschen mit Dabrafenib als Monotherapie oder in Kombination mit Trametinib liegen nicht vor. Am Tier wurden unerwünschte Wirkungen auf die männlichen und weiblichen Fortpflanzungsorgane beobachtet (siehe «Präklinische Daten»). Männliche Patienten sollten auf das potenzielle Risiko einer möglicherweise irreversiblen Beeinträchtigung der Spermatogenese hingewiesen werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zu den Auswirkungen von Tafinlar auf die Verkehrstüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen durchgeführt. Angesichts der Pharmakologie von Tafinlar dürfte es zu keiner ungünstigen Beeinflussung derartiger Tätigkeiten kommen. Ob ein Patient in der Lage ist, Aufgaben zu bewältigen, die Urteilsvermögen und uneingeschränkte motorische bzw. kognitive Leistungsfähigkeit erfordern, sollte unter Berücksichtigung des klinischen Status des Patienten und des Nebenwirkungsprofils von Tafinlar beurteilt werden.
Unerwünschte WirkungenNicht resezierbares oder metastasiertes Melanom
Tafinlar Monotherapie
Die im Folgenden beschriebenen unerwünschten Arzneimittelwirkungen (UAW) berücksichtigen verschiedene Quellen von Sicherheitsinformationen, einschliesslich klinischer Studien, Berichte nach der Markteinführung und Literaturberichte.
Die Häufigkeit der unten beschriebenen UAW basiert auf Daten aus fünf klinischen Monotherapie-Studien an insgesamt 578 Patienten mit Melanom. Ungefähr 30 % der Patienten erhielten eine Tafinlar Therapie von mehr als 6 Monaten. Die am häufigsten unter Tafinlar beobachteten und gemeldeten unerwünschten Arzneimittelwirkungen (UAW) (≥ 15 %) waren Hyperkeratose, Kopfschmerzen, Fieber, Muskel- / Gelenkschmerzen, Abgeschlagenheit, Übelkeit, Durchfall, Papillom, Haarausfall, Hautausschlag und Erbrechen.
Nachstehend sind die bei Melanompatienten gemeldeten UAWs gemäss MedDRA-Terminologie nach Systemorganklasse und Häufigkeit aufgeführt. Häufigkeiten: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1'000, <1/100); selten (≥1/10'000, <1/1'000); sehr selten (<1/10'000), einschliesslich Einzelberichte.
Infektionen und parasitäre Erkrankungen
Häufig: Nasopharyngitis.
Gutartige, bösartige und unspezifische Neubildungen (einschl. Zysten und Polypen)
Sehr häufig: Papilloma (24 %), kutanes Plattenepithelkarzinomb cuSCC (11 %).
Häufig: Seborrhoische Keratose, Akrochordon (Fibrome).
Gelegentlich: Neues Primärmelanom.
Erkrankung des Immunsystems
Gelegentlich: Überempfindlichkeit.
Nicht bekannt: Hämophagozytische Lymphohistiozytose
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: verminderter Appetit (14 %).
Häufig: Hypophosphatämie, Hyperglykämie.
Erkrankung des Nervensystems
Sehr häufig: Kopfschmerzen (30 %).
Häufig: Periphere Neuropathie.
Augenerkrankungen
Gelegentlich: Uveitis.
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Sehr häufig: Husten (13 %).
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Übelkeit (25 %), Erbrechen (18 %), Durchfall (16 %), Verstopfung (10 %).
Gelegentlich: Pankreatitis.
Erkrankungen der Haut und des Unterhautgewebes
Sehr häufig: Hyperkeratose (32 %), Haarausfall (23 %), Hautausschlag (20 %), Palmar-plantares Erythrodysästhesie-Syndrom (14 %).
Häufig: Hauttrockenheit, Aktinische Keratose, Hautläsion, Erythem, Pruritus, Lichtempfindlichkeitsreaktion.
Gelegentlich: Panniculitis.
Nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden): Recall-Phänomen
Skelettmuskulatur-Bindegewebs- und Knochenerkrankungen
Sehr häufig: Arthralgie (29 %), Myalgie (15 %), Gliederschmerzen (16 %).
Erkrankungen der Nieren und Harnwege
Häufig: Niereninsuffizienz, akute Niereninsuffizienz.
Gelegentlich: Tubulointerstitielle Nephritis.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Pyrexie (30 %), Abgeschlagenheit (26 %), Schüttelfrost (13 %), Asthenie (11 %).
Häufig: Grippeähnliche Beschwerden.
Untersuchungen
Häufig: Abnahme der LVEF, QT- Intervallverlängerung.
Zusammenfassende Bezeichnungen
a Papillom, Hautpapillom
b Kutanes Plattenepithelkarzinom: SCC, SCC der Haut, In-situ-SCC (Morbus Bowen) und Keratoakanthom
c Fälle von Lichtempfindlichkeitsreaktion wurden auch nach Markteinführung beobachtet. Alle Fälle, über welche in den klinischen Studien berichtet wurde, waren vom Schweregrad 1 und erforderten keine Dosisanpassung.
Tafinlar und Trametinib Kombinationstherapie
Die im Folgenden beschriebenen unerwünschten Arzneimittelwirkungen (UAW) berücksichtigen verschiedene Quellen von Sicherheitsinformationen, einschliesslich klinischer Studien, Berichte nach der Markteinführung und Literaturberichte.
Die Häufigkeit der unten beschriebenen UAW basiert auf der integrierten Sicherheitspopulation von 1228 erwachsenen Patienten mit BRAF V600-mutiertem inoperablem oder metastasiertem Melanom Stadium III, BRAF V600-mutiertem Melanom nach vollständiger Resektion mit adjuvanter Behandlung, fortgeschrittenem NSCLC und fortgeschrittenen soliden Tumoren.
Alle Patienten wurden mit 2 mg Trametinib einmal täglich und 150 mg Tafinlar zweimal täglich behandelt. Von diesen Patienten wurden 559 in zwei randomisierten Phase-III-Studien, MEK115306 (COMBI-d) und MEK116513 (COMBI-v)a, für BRAF V600-mutiertes-Melanom behandelt. 435 in der adjuvanten Behandlung von BRAF V600-mutiertem Melanom im Stadium III nach vollständiger Resektion in einer randomisierten Phase-III-Studie BRF115532 (COMBI-AD), 93 für BRAF-V600-mutiertes NSCLC in einer nicht-randomisierten Phase-II-Multi-Kohortenstudie BRF113928 und 141 in einer nicht-randomisierten Phase-II-Studie in fortgeschrittenen BRAF-V600E-mutierten soliden Tumoren (Studie BRF117019) behandelt.
Die häufigsten Nebenwirkungen (Inzidenz >20 %) für Tafinlar in Kombination mit Trametinib waren: Pyrexie, Müdigkeit, Übelkeit, Schüttelfrost, Kopfschmerzen, Durchfall, Erbrechen, Arthralgie und Hautausschlag.
Häufigkeiten: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1'000, <1/100); selten (≥1/10'000, <1/1'000); sehr selten (<1/10'000), einschliesslich Einzelberichte.
Infektionen und parasitäre Erkrankungen
Sehr häufig: Nasopharyngitis (11%)
Häufig: Harnwegsinfektion, Cellulitis, Follikulitis, Paronychie, pustulärer Hautausschlag
Gutartige, bösartige und unspezifische Neubildungen (einschl. Zysten und Polypen)
Häufig: Kutanes Plattenepithelkarzinom (squamous cell carcinoma, SCC)b, Papillomec, seborrhoische Keratose
Gelegentlich: Neues Primärmelanomd, Acrochordon (Fibrome)
Erkrankungen des Blutes- und des Lymphsystems
Häufig: Neutropenie, Anämie, Thrombozytopenie, Leukopenie
Erkrankungen des Immunsystems
Gelegentlich: Überempfindlichkeite, Sarkoidose
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: Verminderter Appetit (14 %)
Häufig: Dehydration, Hyponatriämie, Hypophosphatämie, Hyperglykämie
Nicht bekannt: Tumorlysesyndrom
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (33%), Schwindelgefühl (11%)
Häufig: Periphere Neuropathie
Gelegentlich: Guillain-Barré syndrom
Augenerkrankungen
Häufig: Verschwommene Sicht, Sehverschlechterung, Uveitis
Gelegentlich: Chorioretinopathie, Ablösung des Netzhautpigmentepithels/ Netzhautablösung (RPED), periorbitales Ödem
Herzerkrankungen
Häufig: Verminderte Ejektionsfraktion, Atrioventrikulärer Blockl
Gelegentlich: Bradykardie, Bündelzweigblockm
Selten: Myokarditis*
Gefässerkrankungen
Sehr häufig: Hypertonie (17 %), Hämorrhagie (19 %)f
Häufig: Hypotonie, Lymphödem
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Sehr häufig: Husten (20 %)
Häufig: Dyspnoe
Gelegentlich: Pneumonitis
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Übelkeit (38 %), Diarrhö (31 %), Erbrechen (29 %), Bauchschmerzen (17 %)g, Verstopfung (14 %),
Häufig: Mundtrockenheit, Stomatitis
Gelegentlich: Pankreatitis, Kolitis
Selten: Gastrointestinale Perforation
Leber und Gallenerkrankungen
Sehr häufig: Erhöhte Alaninaminotransferase (14 %), Erhöhte Aspartataminotransferase (13 %)
Häufig: Erhöhte alkalische Phosphatase im Blut, Erhöhte Konzentrationen der Gamma-Glutamyltransferase
Erkrankungen der Haut und des Unterhautgewebes
Sehr häufig: Ausschlag (25 %), Trockene Haut (13 %), Pruritus (11 %), Erythemh (10%)
Häufig: akneiformeDermatitis, aktinische Keratose, Nachtschweiss, Hyperkeratose, Alopezie, Erythrodysästhesie-Syndrom der Handflächen und Fusssohlen, Hautläsion, Hyperhidrose, Hautfissuren, Pannikulitis, Lichtempfindlichkeit
Sehr selten: Stevens- Johnson-Syndrom, Arzneimittelreaktion mit Eosinophilie und systemischen Symptomen (DRESS), Generalisierte exfoliative Dermatitis.
Nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden): Recall-Phänomen, Akute febrile neutrophile Dermatose (Sweet-Syndrom)
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Arthralgie (26 %), Myalgie (15 %), Schmerzen in den Extremitäten (11 %), Muskelkrämpfei (10%)
Häufig: Erhöhte Kreatinphosphokinase im Blut
Erkrankungen der Nieren und Harnwege
Häufig: Nierenversagen
Gelegentlich: Nephritis
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Pyrexie (57 %), Müdigkeit (38 %), Schüttelfrost (31 %), peripheres Ödem (16 %), Asthenie (15 %), grippaler Infekt (10%)
Häufig: Schleimhautentzündung, Gesichtsödem
a Das Sicherheitsprofil von MEK116513 ähnelt im Allgemeinen dem von MEK115306, mit folgenden Ausnahmen: 1) Die folgenden Nebenwirkungen haben eine höhere Häufigkeitskategorie im Vergleich zu MEK115306: Muskelkrämpfe (sehr häufig); Nierenversagen und Lymphödem (häufig); akutes Nierenversagen (gelegentlich); 2) Die folgenden Nebenwirkungen traten in MEK116513 auf, aber nicht in MEK115306: Herzinsuffizienz, linksventrikuläre Dysfunktion, interstitielle Lungenerkrankung (gelegentlich). 3) Die folgende Nebenwirkung ist in MEK116513 und BRF115532 aufgetreten, aber nicht in MEK115306 und BRF113928: Rhabdomyolyse (gelegentlich)
b schliesst die SCC der Haut, SCC in-situ (Morbus Bowen) und Keratoakanthom ein
c schliesst Papillome, Hautpapillome ein
d schliesst bösartiges Melanom; metastasiertes, bösartiges Melanom und oberflächlich spreitendes Melanom im Stadium III ein.
e schliesst Überempfindlichkeit gegenüber Arzneimittel ein
f Blutungen von verschiedenen Stellen, einschliesslich intrakranielle Blutungen und tödliche Blutungen
g schliesst Abdominalschmerz, Oberbauchschmerzen und Unterbauchschmerzen ein.
h schliesst Erythem, generalisiertes Erythem ein.
i schliesst Muskelkrämpfe und muskuloskelettale Steifigkeit ein
lDer atrioventrikuläre Block umfasst den atrioventrikulären Block, den atrioventrikulären Block ersten Grades, den atrioventrikulären Block zweiten Grades und den vollständigen atrioventrikulären Block
m Der Bündelzweigblock umfasst den rechten Bündelzweigblock und den linken Bündelzweigblock
*Häufigkeit basiert auf dem Bericht nach der Markteinführung
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Pyrexie
Über Fieber wurde in klinischen Studien berichtet. Bei 1 % der Patienten in klinischen Studien wurden ernsthafte nicht-infektiöse fiebrige Ereignisse als Fieber identifiziert, begleitet von schwerem Rigor, Dehydration, niedrigem Blutdruck und/oder akuter Niereninsuffizienz prärenalen Ursprungs bei Patienten mit normalen Nierenfunktionsausgangswerten. Das Auftreten dieser ernsthaften, nicht-infektiösen fiebrigen Ereignisse erfolgte typischerweise innerhalb des ersten Therapiemonats. Patienten mit ernsthaften nichtinfektiösen fiebrigen Ereignissen sprachen gut auf eine Behandlungsunterbrechung und/oder Dosisreduktion mit unterstützender Behandlung an.
Kutanes Plattenepithelkarzinom
Kutane Plattenepithelkarzinome (einschliesslich jener, die als Keratoakanthom- oder gemischte Keratoakanthom-Subtypen klassifiziert wurden) traten bei 9 % der Patienten unter der Behandlung mit Tafinlar auf. Ungefähr 70 % dieser Ereignisse traten innerhalb der ersten 12 Behandlungswochen auf mit einer medianen Zeit bis zum Auftreten von 8 Wochen. Sechsundneunzig Prozent der Patienten, die ein cuSCC entwickelten, konnten die Behandlung ohne Dosismodifikation fortsetzen.
Neue primäre Melanoma
In klinischen Studien mit Tafinlar wurde über neue primäre Melanome berichtet. Die Fälle wurden durch Exzision behandelt, eine Dosisanpassung von Dabrafenib war nicht erforderlich.
Nicht-kutane maligne Erkrankungen
Die Aktivierung des MAP-Kinase-Signalübertragungsweges in Zellen vom BRAF-Wildtyp, die BRAF Inhibitoren exponiert waren, kann zu einem erhöhten Risiko von nicht-kutanen malignen Erkrankungen, einschliesslich solchen mit RAS-Mutationen führen. In klinischen Studien wurden Fälle von RAS-getriebenen Malignitäten unter Tafinlar berichtet, wenn in Kombination mit Trametinib (MEK Inhibitor) verabreicht. Die Patienten sollten wie klinisch geboten überwacht werden.
Verringerung der LVEF
Über eine Verringerung der LVEF, die in den meisten Fällen asymptomatisch und reversibel war, wurde bei 1 % der Patienten berichtet. Patienten mit einer LVEF unterhalb des unteren Grenzwerts für den Normbereich der jeweiligen Einrichtung wurden nicht in die klinischen Studien mit Dabrafenib eingeschlossen.
Arthralgien
In klinischen Studien mit Tafinlar wurde sehr häufig (25 %) über Arthralgien berichtet, wobei diese hauptsächlich vom Schweregrad 1 oder 2 waren; nur gelegentlich (< 1 %) wurde vom Grad 3 berichtet, der Grad 4 wurde nicht beobachtet.
Hypophosphatämie
In klinischen Studien mit Tafinlar wurde häufig über Hypophosphatämie (7 %) berichtet. Ungefähr die Hälfte dieser Fälle (4 %) waren vom Schweregrad 3.
Pankreatitis
Über Pankreatitis wurde bei Patienten unter der Behandlung mit Tafinlar berichtet. Ungeklärte Bauchschmerzen sollten umgehend untersucht werden, einschliesslich einer Bestimmung der Serum-Amylase und Lipase. Bei einer Wiederaufnahme der Tafinlar-Behandlung sollten die Patienten engmaschig überwacht werden.
Nierenversagen
Nierenversagen infolge mit Pyrexie verbundener prärenaler Azotämie oder granulomatöser Nephritis waren selten, jedoch wurde Tafinlar nicht bei Patienten mit Niereninsuffizienz (definiert als Kreatinin > 1.5 x des oberen Grenzwertes des Normbereichs [ULN]) untersucht. Bei dieser Patientenpopulation ist Vorsicht geboten.
Besondere Patientengruppen
Pädiatrische Patienten
Tafinlar in Kombination mit Trametinib
Die Sicherheit von Tafinlar in Kombination mit Trametinib wurde bei 171 pädiatrischen Patienten in zwei Studien untersucht (Studie DRB436G2201; n=123 und Studie TMT212X2101; n=48), davon 159 mit BRAF V600E-Mutation-positivem Gliom.
Das allgemeine Sicherheitsprofil in der pädiatrischen Population war ähnlich wie das bei Erwachsenen beobachtete Sicherheitsprofil. Die am häufigsten gemeldeten unerwünschten Arzneimittelwirkungen (≥20 %) waren Fieber (Pyrexie) (65%), Hautausschlag (47%), Kopfschmerzen (39%), Erbrechen (38%), trockene Haut (34%), Müdigkeit (Fatigue) (33%), Diarrhoe (30%), Blutungen (29%), Neutropenie (25%), Übelkeit (25%), akneiforme Dermatitis (25%), Bauchschmerzen (23%), Husten (21%), und Transaminasen erhöht (21.6%).
Im pädiatrischen Sicherheitspool wurde eine unerwünschte Arzneimittelwirkung in Form einer Gewichtszunahme mit einer Häufigkeit von 16 % (sehr häufig) festgestellt. Bei 61 von 171 Patienten (36 %) war der BMI im Vergleich zu Baseline um ≥2 BMI-Perzentilkategorien gestiegen.
In Studie DRB436G2201 wurde bei 20,5% (15 von 73) der pädiatrischen Patienten, die mit Tafinlar in Kombination mit Trametinib behandelt wurden, eine COVID-19 Infektion berichtet, in einem Fall vom Schweregrad ≥3.
Im pädiatrischen Sicherheitspool wurde bei 8,7% (14 von 161) der pädiatrischen Patienten unter kombinierter Behandlung mit Tafinlar und Trametinib eine Abnahme der linksventrikulären Ejektionsfraktion (LVEF) von 10 % oder mehr gegenüber dem Ausgangswert und unterhalb der institutionellen Untergrenze des Normalwerts (LLN) beobachtet.
Weitere unerwünschte Arzneimittelwirkungen, die bei pädiatrischen Patienten im Vergleich zu erwachsenen Patienten häufiger auftraten, waren Neutropenie, akneiforme Dermatitis, Paronychie, Anämie, Leukopenie (sehr häufig); Papillom der Haut (sehr häufig), generalisierte exfoliative Dermatitis, Überempfindlichkeit und Pankreatitis (häufig). Darüber hinaus war in der LGG-Kohorte der Studie G2201 die relative Inzidenz von Lymphozytenerhöhungen, Magnesiumerhöhungen und niedrigem systolischem Blutdruck bei zielgerichteter Therapie höher als bei Chemotherapie.
Tabelle 7: Häufigste unerwünschte Arzneimittelwirkungen vom Grad 3/4 (≥2 %) bei Tafinlar in Kombination mit Trametinib bei pädiatrischen Patienten
|
Unerwünschte Arzneimittelwirkungen
|
Tafinlar in Kombination mit Trametinib
N = 171
| |
Grad 3/4
n (%)
| |
Neutropenie1
|
25 (15)
| |
Fieber (Pyrexie)
|
19 (11)
| |
Transaminasen erhöht2
|
11 (6)
| |
Gewichtszunahme (Gewicht erhöht)
|
9 (5)
| |
Kopfschmerzen
|
5 (3)
| |
Erbrechen
|
5 (3)
| |
Hypotension
|
4 (2)
| |
Ausschlag3
|
4 (2)
| |
Alkalische Phosphatase im Blut erhöht
|
4 (2)
| |
1
Neutropenie beinhaltet Neutrophilenzahl erniedrigt, Neutropenie und febrile Neutropenie.
2 Erhöhte Transaminasen umfassen erhöhte Aspartat-Aminotransferase, erhöhte Alanin-Aminotransferase, Hypertransaminasämie und erhöhte Transaminasen
3 Ausschlag umfasst Ausschlag, Ausschlag makulo-papulös, Ausschlag papulös, erythematöser Hautausschlag, und Ausschlag makulös.
|
Bei pädiatrischen Patienten unter 6 Jahren wurden unter kombinierter Behandlung mit Tafinlar und Trametinib mehr schwerwiegende Ereignisse berichtet als bei Patienten im Alter zwischen 6 und 12 Jahren. Fieber trat bei Kindern unter 6 Jahre häufiger auf als bei älteren pädiatrischen Patienten.
Der pädiatrische Sicherheitspool enthält nur vier Kinder im Alter zwischen 1 und 2 Jahren, so dass das Sicherheitsprofil in dieser Alterskategorie unvollständig charakterisiert ist.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungDie Erfahrungen bezüglich einer Überdosierung von Tafinlar sind derzeit sehr eingeschränkt. Die maximal verabreichte Dosis von Dabrafenib während klinischen Studien war 600 mg (300 mg zweimal täglich).
Behandlung
Für den Fall einer Überdosierung von Tafinlar ist keine spezifische Behandlung verfügbar. Im Falle einer Überdosierung sollte der Patient die jeweils angemessene unterstützende Behandlung erhalten und entsprechend überwacht werden. Weiteres Patientenmanagement soll gemäss der klinischen Indikation oder gegebenenfalls unter Einbeziehung des nationalen toxikologischen Informationszentrums erfolgen.
Eigenschaften/WirkungenATC-Code
L01EC02
Wirkungsmechanismus
Tafinlar Monotherapie
Dabrafenib ist ein potenter, selektiver, ATP-kompetitiver Hemmer der RAF-Kinasen mit IC50-Werten von 0.65, 0.5 bzw. 1.84 nM für die Enzyme BRAF V600E, BRAF V600K und BRAF V600D. Onkogene BRAF-Mutationen führen zu einer konstitutiven Aktivierung der RAS/RAF/MEK/ERK-Signaltransduktionskette und zur Stimulierung des Tumorzellwachstums. BRAF-Mutationen werden bei bestimmten Krebsarten sehr häufig festgestellt, unter anderem bei ungefähr 50 % aller Melanome. Die am häufigsten beobachtete BRAF-Mutation V600E macht ca. 90 % aller bei Melanompatienten beobachteten BRAF-Mutationen aus. Ausserdem kommen eine Reihe seltener Substitutionen vor, z.B. die Mutation V600K, V600D, V600G, V600M und V600R.
Dabrafenib hemmt ausserdem Wildtyp-BRAF- und -CRAF-Enzyme mit IC50-Werten von 3.2 bzw. 5.0 nM. Dabrafenib hemmt das Wachstum BRAF-V600-Mutation-positiver Melanomzellen in vitro und in vivo.
Für Dabrafenib ist bei BRAF-V600-mutierten Melanom-Zelllinien die Suppression eines pharmakodynamischen Downstream-Biomarkers (phosphoryliertes ERK) in vitro und in Tiermodellen belegt.
Bei Teilnehmern mit BRAF-V600-Mutation-positivem Melanom führte die Verabreichung von Dabrafenib gegenüber dem Wert vor der Behandlung zu einer Hemmung der phosphorylierten Tumor-ERK.
Tafinlar und Trametinib Kombinationstherapie - metastasiertes Melanom und NSCLC
Trametinib ist ein reversibler, hoch selektiver, allosterischer Hemmer der Aktivierung und Kinaseaktivität von MEK 1 und MEK 2 (MEK= Mitogen-aktivierte, durch extrazelluäre Signale regulierte Kinasen). MEK-Proteine sind kritische Komponenten des durch extrazelluläre Signale regulierten Kinase (ERK)-Wegs.
Trametinib und Dabrafenib hemmen innerhalb dieser Signaltransduktionskette die beiden Kinasen MEK und BRAF; die Kombination beider Wirkstoffe führt zu einer dualen, wirksamen Hemmung der Signaltransduktionskette. Die Kombination aus Trametinib und Dabrafenib hat sich bei BRAF-V600-mutierten Melanom- und NSCLC-Zelllinien in vitro als synergistisch erwiesen und führt zu einer Verzögerung der Resistenzentwicklung in vivo bei BRAF-V600-mutierten Melanom-Heterotransplantaten.
Studie MEK111054
QT Verlängerung
Eine QTc Intervallverlängerung von schlimmstenfalls > 60 ms wurde in 3 % der mit Tafinlar behandelten Patienten beobachtet (darunter ein Fall mit > 500 ms in der Gesamt-Sicherheitspopulation).
Das QT-verlängernde Potential von Dabrafenib wurde in einer speziell darauf abgestimmten QT-Studie mit Mehrfachdosierung untersucht. Den 32 Teilnehmern mit BRAF-V600-Mutations-positiven Tumoren wurde zweimal täglich eine supratherapeutische Dosis von 300 mg Dabrafenib verabreicht. Es wurden keine klinisch relevanten Effekte von Dabrafenib oder Metaboliten von Dabrafenib auf das QTc-Intervall beobachtet.
In der Phase-III-Studie MEK115306 trat bei keinem der mit Trametinib in Kombination mit Dabrafenib behandelten Patienten der ungünstigste Fall einer QTcB-Verlängerung auf > 500 ms auf; bei 1 % (3/209) der Patienten war die QTcB-Strecke um mehr als 60 ms im Vergleich zum Ausgangswert verlängert. In der Phase-III-Studie MEK116513 hatten vier mit Trametinib in Kombination mit Dabrafenib behandelte Patienten (1 %) einen QTcB-Anstieg vom Grad 3 auf > 500 ms. Zwei dieser Patienten hatten einen QTcB-Anstieg vom Grad 3 auf > 500 ms, der gleichzeitig einen Anstieg um > 60 ms im Vergleich zum Ausgangswert darstellte.
Pharmakodynamik
Klinische Wirksamkeit
Tafinlar Monotherapie
Die Wirksamkeit von Tafinlar bei der Behandlung erwachsener Patienten mit BRAF-V600-Mutation-positivem, nicht resezierbarem oder metastasiertem Melanom wurde in 3 Studien (BRF113683 [BREAK-3], BRF113929 [BREAK-MB] und BRF113710 [BREAK-2]) an Patienten mit BRAF-V600E- und/oder BRAF-V600K-Mutation untersucht. Die pivotale Studie BREAK-3 wurde ausschliesslich bei Patienten mit V600E-Mutation durchgeführt. In diese Studien waren insgesamt 402 Patienten mit BRAF-V600E-Mutation und 49 Patienten mit BRAF-V600K-Mutation eingeschlossen. Bei Vorliegen einer V600K-Mutation ist nach den Ergebnissen der Phase 2 Studien die Wirksamkeit geringer als bei V600E-Mutation-positiven Tumoren.
Die Wirksamkeit von Tafinlar bei mit einem Proteinkinasehemmer vorbehandelten Patienten ist nicht untersucht.
Nicht vorbehandelte Patienten (Ergebnisse der Phase-III-Studie BREAK-3)
Wirksamkeit und Sicherheit von Tafinlar wurden in einer randomisierten, offenen Phase-III-Studie (BREAK 3) zum Vergleich von Tafinlar mit Dacarbazin (DTIC) an behandlungsnaiven Patienten mit BRAF-V600E-Mutation-positivem, fortgeschrittenem (nicht resezierbar, Stadium III) oder metastasiertem (Stadium IV) Melanom untersucht.
Primäres Studienziel war die Beurteilung der Wirksamkeit von Tafinlar im Vergleich zu DTIC in Bezug auf das progressionsfreie Überleben (progression-free survival, PFS). Die Patienten im DTIC-Arm konnten nach der ersten unabhängigen radiographischen Bestätigung einer Progression Tafinlar erhalten. Die Behandlungsgruppen zeigten in Bezug auf die Ausgangsmerkmale eine ausgeglichene Zusammensetzung. Sechzig Prozent der Patienten waren männlich, 99.6 % vom kaukasischen Typ; das Alter lag im Median bei 52 Jahren, wobei 21 % der Patienten ≥65 Jahre alt waren; 98.4 % hatten einen ECOG-Status von 0 oder 1, und bei 97 % der Patienten lag eine metastasierte Erkrankung vor.
Bei der im Voraus festgelegten Analyse der Daten, welche bis zum 19. Dezember 2011 erhoben wurden, ergab sich eine signifikante Verbesserung des primären Endpunktes PFS (HR = 0.30; 95 % Cl 0.18, 0.51; p < 0.0001). Die Wirksamkeitsergebnisse einer post-hoc Analyse mit zusätzlichen 6 Monaten Nachfolgeuntersuchungen sind in Tabelle 8 zusammengefasst. Das Gesamtüberleben (OS) aus einer weiteren post-hoc Analyse basierend auf einer Datenerhebung vom 18. Dezember 2012 ist in Tabelle 9 und Abbildung 1 dargestellt.
Tabelle 8: Wirksamkeit in nicht vorbehandelten Patienten gemäss Prüfärzten (BREAK-3-Studie, 25. Juni 2012)
|
|
Intention-to-treat-Population
| |
|
Tafinlar
N = 187
|
DTIC
N = 63
| |
Progressionsfreies Überleben (gemäss Prüfärzten)
| |
Median (Monate) (95 %-KI)
|
6.9 (5.2; 9.0)
|
2.7 (1.5; 3.2)
| |
HR (95 %-KI)
|
0.37 (0.24; 0.58)
P < 0.0001
| |
Gesamtansprechrate a
| |
% (95 %-KI)b
|
59 (51.4;66.0)
|
24 (21.4; 36.2)
| |
P < 0.0001
| |
Dauer des Ansprechens
| |
|
N = 110
|
N = 15
| |
Median (Monate) (95 %-KI)
|
8.0 (6.6; 11.5)
|
7.6 (5.0; 9.7)
| |
Abkürzungen: KI: Konfidenzintervall; DTIC: Dacarbazin; HR: Hazard Ratio
a Definiert als vollständiges + partielles Ansprechen.
b Gesichertes Ansprechen.
|
Bei der Datenerhebung vom 25. Juni 2012 wechselten 35 von 63 Teilnehmer (55.6 %), die per Randomisierung DTIC erhalten hatten, zur Gruppe mit Tafinlar über. Das mediane PFS nach dem Behandlungsgruppenwechsel betrug 4.4 Monate.
Tabelle 9: Überlebensdaten aus der post-hoc Analyse (18. Dezember 2012).
|
Behandlung
|
Anzahl Todesfälle (%)
|
12-monatige Gesamtüberlebens-Rate
|
Hazard Ratio
(95 % CI)
| |
DTIC
|
28 (44 %)
|
63 %
|
0.76 (0.48, 1.21) (a)
| |
Tafinlar
|
78 (42 %)
|
70 %
|
(a) Patienten waren nicht zensiert zur Zeit des Behandlungsgruppenwechsels
Abbildung 1: Kaplan-Meier Kurve des Gesamtüberlebens (BREAK-3) (18. Dezember 2012)

Patienten mit Hirnmetastasen (Ergebnisse der Phase II Studie BREAK-MB)
BREAK-MB war eine multizentrische, offene Zwei-Kohorten-Studie der Phase II und dazu konzipiert, das intrakranielle Ansprechen auf Tafinlar bei Studienteilnehmern mit histologisch bestätigtem (Stadium IV) Melanom mit Hirnmetastasen und der BRAF (V600E oder V600K)-Mutation zu beurteilen. Die Studienteilnehmer wurden entweder in die Kohorte A (Studienteilnehmer ohne vorangegangene Lokaltherapie der Hirnmetastasen) oder in die Kohorte B (Studienteilnehmer mit vorangegangener Lokaltherapie der Hirnmetastasen) eingeschlossen.
Der primäre Endpunkt dieser Studie war das intrakranielle Gesamt-Ansprechen (Overall Intracranial Response Rate, OIRR) nach Einschätzung der Prüfärzte in der Population mit der V600E-Mutation. Das bestätigte OIRR sowie andere Ergebnisse zur Wirksamkeit sind in Tabelle 10 dargestellt.
Tabelle 10: Daten zur Wirksamkeit bei Patienten mit Hirnmetastase (BREAK-MB Studie)
|
Gesamtpopulation aller behandelten Studienteilnehmer
| |
|
BRAF V600E (Primär)
|
BRAF V600K
| |
|
Kohorte A
N = 74
|
Kohorte B
N = 65
|
Kohorte A
N = 15
|
Kohorte B
N = 18
| |
Intrakranielle Gesamtansprechrate, % (95 % KI)a
| |
|
39 % (28.0, 51.2)
P < 0.001b
|
31 % (19.9, 43.4)
P < 0.001b
|
7 % (0.2, 31.9)
|
22 % (6.4, 47.6)
| |
Dauer des intrakraniellen Ansprechens, Median, Monate (95 % KI)
| |
|
N = 29
4.6 (2.8, NR)
|
N = 20
6.5 (4.6, 6.5)
|
N = 1
2.9 (NR, NR)
|
N = 4
3.8 (NR, NR)
| |
Gesamtansprechrate, % (95 % KI)a
| |
|
38 % (26.8, 49.9)
|
31 % (19.9, 43.4)
|
0 (0, 21.8)
|
28 % (9.7, 53.5)
| |
Dauer des Ansprechens, Median, Monate (95 % KI)
| |
|
N = 28
5.1 (3.7, NR)
|
N = 20
4.6 (4.6, 6.5)
|
NA
|
N = 5
3.1 (2.8, NR)
| |
Progressionsfreies Überleben, Median, Monate (95% KI)
| |
|
3.7 (3.6, 5.0)
|
3.8 (3.6, 5.5)
|
1.9 (0.7, 3.7)
|
3.6 (1.8, 5.2)
| |
Gesamtüberleben, Median, Monate (95% KI)
| |
Median, Monate
|
7.6 (5.9, NR)
|
7.2 (5.9, NR)
|
3.7 (1.6, 5.2)
|
5.0 (3.5, NR)
|
Abkürzungen: KI: Konfidenzintervall; NR: nicht erreicht; NA: nicht anwendbar
a – Bestätigtes Ansprechen.
b – Diese Studie wurde konzipiert, um bei Studienteilnehmern mit der BRAF-V600E-Mutation die Null-Hypothese einer OIRR ≤10 % (basierend auf historischen Ergebnissen) entweder zu unterstützen oder zugunsten der Alternativ-Hypothese einer OIRR ≥30 % abzulehnen
Patienten ohne vorangegangene Therapie oder nach Versagen mindestens einer vorangegangenen systemischen Therapie (Ergebnisse der Phase II [BREAK-2])
BRF113710 (BREAK-2) war eine multizentrische einarmige Studie, in der 92 Studienteilnehmer mit metastasiertem Melanom (Stadium IV) und bestätigter BRAF-V600E- oder V600K-Mutation eingeschlossen wurden.
Die bestätigte Ansprechrate nach Einschätzung der Prüfärzte bei Patienten mit metastasiertem Melanom und der BRAF-V600E-Mutation (n = 76) betrug 59 % (95 % KI: 48.2; 70.3) und die mediane Dauer des Ansprechens 5.2 Monate (95 % KI: 3.9; nicht abschätzbar), basierend auf einer medianen Nachbeobachtungszeit von 6.5 Monaten seit Beginn der Behandlung. Bei Patienten mit metastasiertem Melanom und der BRAF-V600K Mutation (n = 16) betrug die Ansprechrate 13 % (2 Patienten von 16) (95 % KI: 0.0; 28.7) mit einer medianen Dauer des Ansprechens von 5.3 Monaten (95 % KI: 3.7; 6.8).
Tafinlar und Trametinib Kombinationstherapie
Wirksamkeit und Sicherheit der empfohlenen Tafinlar-Dosis (zweimal täglich 150 mg) in Kombination mit Trametinib (einmal täglich 2 mg) zur Behandlung von erwachsenen Patienten mit nicht resezierbarem oder metastasiertem Melanom mit einer BRAF-V600-Mutation wurden in zwei pivotalen Studien der Phase III untersucht.
Die Kombination von Tafinlar 150 mg BID und Trametinib 2 mg QD zeigte in einer frühen Studie begrenzte klinische Aktivität bei einer geringen Anzahl von 26 Patienten, bei denen es nach einer Therapie mit einem BRAF-Inhibitor zur Krankheitsprogression kam. Die vom Prüfarzt bewertete, bestätigte Ansprechrate lag bei 15 % (95-%-KI: 4.4, 34.9) und mittlere PFS lag bei 3.6 Monaten (95-%-KI: 1.9, 5.2). Ähnlich waren die Resultate bei den 45 Patienten, die von der Tafinlar -Monotherapie in die Kombination von Tafinlar 150 mg BID und Trametinib 2 mg QD in Teil C der Studie wechselten. Bei diesen Patienten wurde eine bestätigte Ansprechrate von 13 % (95-%-KI: 5.0, 27.0) mit einem mittleren PFS von 3.6 Monaten beobachtet (95-%-KI: 2, 4). Diese Ansprechrate ist deutlich tiefer als bei Patienten, die keine Krankheitsprogression unter Vorbehandlung mit einem BRAF-Inhibitor gezeigt hatten, so dass eine Anwendung von Tafinlar in Kombination mit Trametinib bei diesen Patienten erst nach Abwägen weiterer Therapieoptionen in Betracht gezogen werden sollte.
MEK115306 (COMBI-d)
MEK115306 (COMBI-d) war eine randomisierte, doppelblinde Phase-III-Studie zum Vergleich der Kombination von Tafinlar und Trametinib gegenüber Tafinlar und Placebo in der Erstlinientherapie bei Patienten mit nicht resezierbarem (Stadium IIIC) oder metastasiertem (Stadium IV) BRAF-V600E/K-Mutations-positivem kutanem Melanom. Der primäre Endpunkt der Studie war das vom Prüfarzt bewertete progressionsfreie Überleben (PFS). Sekundäre Endpunkte waren das mediane Gesamtüberleben (overall survival, OS), die Gesamtansprechrate (overall response rate, ORR) und die Dauer des Ansprechens (duration of response, DOR). Die Studienteilnehmer wurden nach Laktatdehydrogenase (LDH)-Aktivität (> obere Normgrenze (ONG) versus ≤ ONG) und BRAF-Mutation (V600E versus V600K) stratifiziert.
Es wurden insgesamt 423 Studienteilnehmer im Verhältnis von 1:1 randomisiert und dem Kombinationstherapie-Arm (zweimal täglich Tafinlar 150 mg und einmal täglich Trametinib 2 mg) (N = 211) bzw. dem Tafinlar -Monotherapie-Arm (zweimal täglich 150 mg) (N = 212) zugeordnet. Die Merkmale bei Baseline waren in den beiden Behandlungsgruppen ausgeglichen. Bei den meisten Patienten lag eine BRAF-V600E-Mutation vor (85 %); bei den übrigen 15 % der Patienten lag eine BRAF-V600K-Mutation vor.
Zum Zeitpunkt der Primäranalyse des PFS wurden ein medianes PFS von 9.3 Monaten unter der Kombination von Tafinlar und Trametinib beobachtet und von 8.8 Monaten unter der Tafinlar -Monotherapie (HR = 0.75, 95 % CI: 0.57, 0.99, p = 0.035). ORR war 67 % vs. 51 % (p = 0.0014) und DOR betrug 9.2 vs. 10.2 Monate bei der Kombination gegenüber der Tafinlar -Monotherapie. In einer späteren Analyse, welche zusammenfiel mit der Hauptanalyse des OS (siehe weiter unten) wurde ein noch deutlicherer Unterschied zugunsten der Kombination von Tafinlar und Trametinib als in der vorausgegangenen Hauptanalyse dieses Parameters beobachtet, mit 11.0 Monaten im PFS unter der Kombination (95 % CI: 8.0, 13.9) und weiter von 8.8 Monaten (95 % CI: 5.9, 9.3) unter der Tafinlar-Monotherapie (HR = 0.67, 95 % CI: 0.53, 0.84, p<0.001). In dieser Analyse betrug die ORR 69 % gegenüber 53 % (p = 0.0014) und die DOR betrug 12.9 gegenüber 10.6 Monaten bei der Kombinationstherapie respektive bei Behandlung mit der Tafinlar -Monotherapie.
Zum Zeitpunkt der Hauptanalyse des OS-Analyse wurden 222 Todesfälle (52.5 %) aus der randomisierten (oder ITT-) Population berichtet [Kombination 99 Todesfälle (47 %) und Tafinlar 123 Todesfälle (58 %)]. Die mediane Nachbeobachtungszeit für die Studienbehandlung betrug 20 Monate im Kombinationstherapie-Arm und 16 Monate im Tafinlar -Monotherapie-Arm. Die Studie MEK115306 zeigte eine statistisch signifikante Senkung des Todesrisikos von 29 % im Kombinationstherapie-Arm im Vergleich zum Tafinlar -Monotherapie-Arm (HR = 0.71, 95%-KI: 0.55, 0.92; p = 0.011). Das mediane OS betrug 25.1 Monate im Kombinationstherapie-Arm und 18.7 Monate im Tafinlar-Monotherapie-Arm. Die ermittelten OS-Werte für 12 Monate (74 %) und 24 Monate (51.4 %) waren zudem im Kombinationsarm höher als die im Tafinlar-Monotherapie-Arm (67.6 bzw. 42.1 %).
Eine Analyse des Gesamtüberlebens (OS) nach 5 Jahren ergab, dass das mediane Gesamtüberleben (OS) unter der Kombinationstherapie etwa 7 Monate länger war als das mediane Gesamtüberleben (OS) unter der Tafinlar -Monotherapie (25,8 Monate (95%-KI: 19.2, 38.2) gegenüber 18.7 Monaten (95%-KI: 15.2, 23.1)) mit einer Hazard Ratio von 0,80 (95% KI: 0.63, 1.01). Die 5-Jahres-Gesamtüberlebensrate betrug 32% (95%-KI: 25.1, 38.3) unter der Kombinationstherapie gegenüber 27% (95%-KI: 20.7, 33.0) unter der Tafinlar -Monotherapie. Nach 5 Jahren war das mediane progressionsfreie Überleben (PFS) für die Kombinationstherapie 10.2 Monate (95% Kl: 8.1, 12.8) gegenüber 8.8 Monate (95% Kl: 5.9, 9.3) für Tafinlar-Monotherapie mit einer Hazard Ratio von 0,73 (95% KI: 0.59, 0.91).
MEK116513 (COMBI-v)
Die Studie MEK116513 war eine 2-armige, randomisierte, offene Phase-III-Studie zum Vergleich von Tafinlar und Trametinib als Kombinationstherapie mit Vemurafenib als Monotherapie bei BRAF-V600-Mutations-positivem metastasiertem Melanom. Der primäre Endpunkt der Studie war das Gesamtüberleben (OS). Die Studienteilnehmer wurden nach Laktatdehydrogenase (LDH)-Aktivität (> obere Normgrenze (ONG) versus ≤ ONG) und BRAF-Mutation (V600E versus V600K) stratifiziert.
Insgesamt wurden 704 Studienteilnehmer im Verhältnis 1:1 randomisiert und entweder dem Kombinationstherapie-Arm (zweimal täglich Tafinlar 150 mg und einmal täglich Trametinib 2 mg) oder dem Vemurafenib-Monotherapie-Arm (zweimal täglich 960 mg) zugewiesen. Bei der Mehrzahl der Studienteilnehmer lag eine BRAF-V600E-Mutation vor (89 %). 10 % der Patienten hatten eine BRAF-V600K Mutation und 1 Patient (<1 %) zeigte beide Mutationen (BRAF V600E/K).
Die OS-Analyse wurde durchgeführt, als insgesamt 222 Todesfälle aufgetreten waren (77 % der erforderlichen Ereignisse für die finale Analyse). Die unabhängige Monitorengruppe (Independent Data Monitoring Committee, IDMC) empfahl den Abbruch der Studie, da die OS-Ergebnisse die zuvor festgelegte statistische Wirksamkeitsgrenze überschritten hatten. In der Folge galt die OS-Zwischenanalyse als finale OS-Vergleichsanalyse.
Die OS-Analyse für die Studie MEK116513 basierte auf 222 Todesfällen (32 %) [Kombination; 100 Todesfälle (28 %) und Vemurafenib 122 Todesfälle (35 %)]. Die mediane Nachbeobachtungszeit für die Studienbehandlung betrug 11 Monate im Kombinationstherapie-Arm und 9 Monate im Vemurafenib-Arm. Die Studie MEK116513 zeigte eine statistisch signifikante Senkung des Todesrisikos von 31 % im Kombinationstherapie-Arm im Vergleich zu Vemurafenib (HR = 0.69, 95%-KI: 0.53, 0.89; p = 0.005). Das mediane OS wurde für den Kombinationstherapie-Arm noch nicht erreicht und betrug für den Vemurafenib-Monotherapie-Arm 17.2 Monate.
Das beobachtete mediane PFS betrug 11.4 Monate bei der Kombinationstherapie von Trametinib und Tafinlar und 7.3 Monate unter Vemurafenib-Monotherapie (HR = 0.56, 95 % CI: 0.46, 0.69, p < 0.001). Die ORR betrug 64 % versus 51 % (p = 0.0005) und die DOR betrug 13.8 versus 7.5 Monate bei der Kombinationstherapie gegenüber der Vemurafenib-Monotherapie.
Eine Analyse des Gesamtüberlebens (OS) nach 5 Jahren ergab, dass das mediane Gesamtüberleben (OS) unter der Kombinationstherapie etwa 8 Monate länger war als das mediane Gesamtüberleben (OS) unter der Vemurafenib-Monotherapie (26.0 Monate (95%-KI: 22.1, 33.8) gegenüber 17.8 Monaten (95%-KI: 15.6, 20.7), mit einer Hazard ratio von 0.70 (95%-KI: 0.58, 0.84). Die 5-Jahres-Gesamtüberlebensrate betrug 36% (95%-KI: 30.5, 40.9) unter der Kombinationstherapie gegenüber 23% (95%-KI: 18.1, 27.4) unter der Vemurafenib-Monotherapie.
BRF117277 / DRB436B2204 (COMBI-MB) - Patienten mit Hirnmetastasen eines metastasierten Melanoms
Die Wirksamkeit und Sicherheit von Tafinlar in Kombination mit Trametinib bei Patienten mit BRAF-Mutation-positivem Melanom mit Hirnmetastasen wurde in einer nicht-randomisierten offenen, multizentrischen Phase-II-Studie (COMBI-MB-Studie) untersucht. Es wurden insgesamt 125 Patienten in vier Kohorten aufgenommen:
·Kohorte A: Melanom-Patienten mit BRAFV600E-Mutation und asymptomatischen Hirnmetastasen ohne vorherige lokale zielgerichtete Therapie der Hirnmetastasen bei einem ECOG-Leistungsstatus von 0 oder 1.
·Kohorte B: Melanom-Patienten mit BRAFV600E-Mutation und asymptomatischen Hirnmetastasen mit vorheriger lokaler zielgerichteter Therapie der Hirnmetastasen bei einem ECOG-Leistungsstatus von 0 oder 1.
·Kohorte C: Melanom-Patienten mit BRAFV600D/K/R-Mutation und asymptomatischen Hirnmetastasen mit oder ohne vorherige lokale zielgerichtete Therapie der Hirnmetastasen bei einem ECOG-Leistungsstatus von 0 oder 1.
·Kohorte D: Melanom-Patienten mit BRAFV600D/E/K/R-Mutation und symptomatischen Hirnmetastasen mit oder ohne vorherige lokale zielgerichtete Therapie der Hirnmetastasen bei einem ECOG-Leistungsstatus von 0 oder 1 oder 2.
Insgesamt hatten 104/125 Patienten eine V600E Mutation, 18 Patienten eine V600K Mutation und nur 3 Patienten eine V600R Mutation. Kein Patient hatte eine V600D Mutation.
Der primäre Endpunkt der Studie war das intrakranielle Ansprechen in Kohorte A, definiert als der Prozentsatz der Patienten mit einem bestätigten intrakraniellen Ansprechen, das vom Prüfarzt unter Verwendung der RECIST-Kriterien (Response Evaluation Criteria in Solid Tumors, Kriterien für die Bewertung des Ansprechens der Behandlung bei soliden Tumoren), Version 1.1, bewertet wurde. Die Wirksamkeitsergebnisse sind in Tabelle 11 zusammengefasst. Die sekundären Endpunkte waren die Dauer des intrakraniellen Ansprechens, die Gesamtansprechrate sowohl der intrakraniellen wie auch der extrakraniellen Tumormanifestationen, das PFS und das OS. Die Wirksamkeitsergebnisse sind in Tabelle 11 zusammengefasst.
Tabelle 11: COMBI-MB - Wirksamkeitsergebnisse nach Einschätzung des Prüfarztes
|
|
Alle behandelten Patienten
| |
Endpunkte/Beurteilung
|
Kohorte A
N=76
|
Kohorte B
N=16
|
Kohorte C
N=16
|
Kohorte D
N=17
| |
Intrakranielle Ansprechrate, % (95 % KI)
|
| |
|
59 %
(47.3, 70.4)
|
56 %
(29.9, 80.2)
|
44 %
(19.8, 70.1)
|
59 %
(32.9, 81.6)
| |
Dauer des intrakraniellen Ansprechens, Medianwert, Monate (95 % KI)
| |
|
6.5
(4.9, 8.6)
|
7.3
(3.6, 12.6)
|
8.3
(1.3, 15.0)
|
4.5
(2.8, 5.9)
| |
Gesamtansprechrate (intra- und extrakraniell), ORR, % (95% KI)
| |
|
59 %
(47.3, 70.4)
|
56 %
(29.9, 80.2)
|
44 %
(19.8, 70.1)
|
65 %
(38.3, 85.8)
| |
Medianwert PFS, Monate (95 % KI)
| |
|
5.7
(5.3, 7.3)
|
7.2
(4.7, 14.6)
|
3.7
(1.7, 6.5)
|
5.5
(3.7, 11.6)
| |
Medianwert OS, Monate (95 % KI)
| |
Medianwert, Monate
|
10.8
(8.7, 17.9)
|
24.3
(7.9, NR)
|
10.1
(4.6, 17.6)
|
11.5
(6.8, 22.4)
| |
KI = Konfidenzintervall
NR = Ohne Angabe
|
Adjuvante Behandlung des Melanoms
Studie BRF115532/CDRB436F2301 (COMBI-AD)
Die Wirksamkeit und Sicherheit von Tafinlar in Kombination mit Trametinib wurde in einer multizentrischen, randomisierten, doppelblinden, Placebo-kontrollierten Phase-III-Studie bei Patienten mit Melanom im Stadium III mit einer BRAF-V600-Mutation nach vollständiger Resektion untersucht.
Die Patienten wurden im Verhältnis 1:1 randomisiert und erhielten entweder eine Kombinationstherapie mit Tafinlar 150 mg zweimal täglich und Trametinib 2 mg einmal täglich oder zwei Placebos über einen Zeitraum von 12 Monaten. Nur Patienten mit einer vollständigen Melanomresektion und vollständiger Lymphadenektomie innerhalb von 12 Wochen vor der Randomisierung wurden in die Studie aufgenommen. Eine vorherige systemische Krebsbehandlung, einschliesslich Strahlentherapie, war nicht erlaubt. Teilnahmeberechtigt waren Patienten mit einer Vorgeschichte früherer Malignität, wenn sie mindestens 5 Jahre krankheitsfrei waren. Patienten mit bösartigen Tumoren mit bestätigten aktivierenden RAS-Mutationen waren nicht teilnahmeberechtigt. Die Patienten wurden nach dem BRAF-Mutationsstatus (V600E oder V600K) und dem Erkrankungsstadium vor der Operation stratifiziert (nach Stadium-III-Substadium, d.h. unterschiedliche Lymphknotenbeteiligungen, primäre Tumorgrösse und Ulzeration). Der primäre Endpunkt war das vom Prüfer beurteilte rückfallfreie Überleben (relapse-free survival, RFS), definiert als der Zeitraum ab Randomisierung bis zum Wiederauftreten des Tumors oder Tod jeglicher Ursache. Eine radiologische Tumorbeurteilung wurde in den ersten zwei Jahren alle 3 Monate und danach alle 6 Monate durchgeführt, bis der erste Rückfall beobachtet wurde. Sekundäre Endpunkte waren das Gesamtüberleben (OS; wichtiger sekundärer Endpunkt) und das fernmetastasenfreie Überleben (distant metastasis-free survival, DMFS).
Insgesamt 870 Patienten wurden zur Kombinationstherapie (n = 438) und zu Placebo (n = 432) randomisiert. Die Studie nahm Patienten mit Erkrankung aller Stadium-III-Substadien vor der Resektion auf; 18 % dieser Patienten wiesen nur mikroskopisch nachweisbare Lymphknotenbeteiligung und keine primäre Tumorulzeration auf. Die Mehrheit der Patienten hatte eine BRAF-V600E-Mutation (91 %). Zum Zeitpunkt der Primäranalyse, die mediane Dauer der Nachbeobachtung (Zeit ab Randomisierung bis zum letzten Kontakt oder Tod) betrug 2,83 Jahre in der Behandlungsgruppeund 2,75 Jahre in der Placebo-gruppe.
Die Ergebnisse der Primäranalyse sind in Tabelle 12 dargestellt. Die Studie zeigte einen statistisch signifikanten Unterschied für den primären Endpunkt hinsichtlich des RFS zwischen den Behandlungsarmen. Bei der Behandlungsgruppe, die Tafinlar und Trametinib als Kombinationstherapie erhielt, lag die Risikoreduktion bei 53 % im Vergleich zur Placebo-Gruppe.
Tabelle 12: COMBI-AD Primäranalyse – Ergebnisse des rückfallfreien Überlebens
|
|
Tafinlar + Trametinib
|
Placebo
| |
RFS-Parameter
|
N = 438
|
N = 432
| |
Anzahl der Ereignisse - n (%)
|
166 (38 %)
|
248 (57 %)
| |
Rezidiv
|
163 (37 %)
|
247 (57 %)
| |
Rezidiviert mit
Fernmetastasen
|
103 (24 %)
|
133 (31 %)
| |
Tod
|
3 (< 1 %)
|
1 (< 1 %)
| |
Medianwert (Monate)
|
NE
|
16,6
| |
(95 % KI)
|
(44,5, nicht schätzbar (NE))
|
(12,7; 22,1)
| |
Hazard Ratio[1]
|
0,47
| |
(95 % KI)
|
(0,39; 0,58)
| |
P-Wert[2]
|
1,53×10-14
| |
1-Jahres-Rate (95 % KI)
|
0,88 (0,85, 0,91)
|
0,56 (0,51, 0,61)
| |
2-Jahres-Rate (95 % KI)
|
0,67 (0,63, 0,72)
|
0,44 (0,40, 0,49)
| |
3-Jahres-Rate (95 % KI)
|
0,58 (0,54, 0,64)
|
0,39 (0,35, 0,44)
| |
[1]
Die Hazard Ratio ergibt sich aus dem stratifizierten Pike-Modell.
[2] Der P-Wert wird aus dem zweiseitigen stratifizierten Logrank-Test ermittelt (Stratifikationsfaktoren waren Krankheitsstadium, IIIA vs. IIIB vs. IIIC, und BRAF- V600-Mutationstyp - V600E vs. V600K).
NE = nicht schätzbar
|
Basierend auf 153 Ereignissen (60 (14 %) im Kombinationstherapie-Arm und 93 (22 %) im Placebo-Arm), was einem Informationsanteil von 26 % des Gesamtziels von 597 OS-Ereignissen entspricht, betrug die geschätzte Hazard Ratio für OS 0,57 (95 % KI: 0,42, 0,79; p = 0,0006). Diese Ergebnisse entsprachen nicht der präspezifizierten Grenze, um bei dieser ersten OS-Zwischenanalyse statistische Signifikanz zu erlangen (HR = 0,50; p = 0,000019). Schätzungen zum Überleben 1 und 2 Jahre nach Randomisierung betrugen 97 % und 91 % im Kombinationstherapie-Arm und 94 % und 83 % im Placebo-Arm.
Aufgrund der aktualisierten Daten aus weiterer 29-monatigen Nachbeobachtungszeit im Vergleich zur Primäranalyse (Mindestnachbeobachtung von 59 Monaten) wurde der Vorteil des rezidivfreies Überleben mit einer geschätzten Hazard Ratio von 0.51 (95% KI: 0,42, 0,61) aufrechterhalten. 5-jährige rezidivfreie Überlebensrate betrug 52 % (95 % Ki: 48, 58) im Kombinationstherapie-Arm im Vergleich zu 36 % (95 % KI: 32, 41) im Placebo-Arm.
Fortgeschrittenes NSCLC
Studie BRF113928
Die Wirksamkeit und Sicherheit von Tafinlar in Kombination mit Trametinib wurden in einer multizentrischen, nicht-randomisierten, offenen Phase-II-Studie bei 57 mit Chemotherapie vorbehandelten Patienten mit metastasiertem BRAF-V600E-Mutations-positivem NSCLC untersucht. Der primäre Endpunkt war die vom Prüfarzt beurteilte Gesamtansprechrate (overall response rate, ORR) unter Verwendung der RECIST-Kriterien (Response Evaluation Criteria in Solid Tumors, Kriterien für die Bewertung des Ansprechens der Behandlung bei soliden Tumoren). Die sekundären Endpunkte umfassten die Dauer des Ansprechens (duration of response, DoR), das progressionsfreie Überleben (PFS), das und Gesamtüberleben (overall survival, OS). ORR, DoR und PFS wurden von einem unabhängigen Prüfungsausschuss (Independent Review Committee, IRC) in Form einer Empfindlichkeitsanalyse beurteilt.
Zum Zeitpunkt der Primäranalysis, für den primären Endpunkt der vom Prüfarzt beurteilten Gesamtansprechrate (overall response rate, ORR) betrug die ORR in der zuvor bereits behandelten Population 66.7% (95% CI, 52.9%, 78.6%). Damit wurde die statistische Signifikanz in Ablehnung der Nullhypothese erfüllt, dass die ORR von Tafinlar in Kombination mit Trametinib für diese NSCLC-Population höchstens 30 % betrug. Die Ergebnisse für die von einem unabhängigen Prüfungsausschuss (Independent Review Committee, IRC) beurteilte Gesamtansprechrate (overall response rate, ORR) entsprachen der Prüfarztbewertung (Tabelle 13).
Tabelle 13: ORR nach Einschätzung des Prüfarztes und durch unabhängige Röntgenuntersuchung laut der Abschlussanalyse nach 5 Jahren
|
Gruppe, bei der Röntgenuntersuchung durchgeführt wurde
|
Prüfer:
|
Unabhängig
| |
Kategorie
|
N = 57*
|
N = 57*
| |
Gesamtansprechen – n (%)
| |
ORR (CR + PR)
|
39 (68.4)
|
36 (63,2)
| |
(95% KI)
|
(54.8, 80.1)
|
(49,3; 75,6)
| |
Dauer des Ansprechens
| |
Anzahl der Responder
|
39
|
36
| |
Anzahl der Patienten mit Krankheitsprogress oder Todesfälle - n (%)
|
27 (71)
|
20 (56)
| |
Medianwert DoR, Monate
(95% KI)
|
9.8
(6,9; 18,3)
|
12.6
(5,8; 26,2)
| |
Progressionsfreies Überleben
| |
Krankheitsprogress oder Tod - n (%)
|
41 (72)
|
38 (67)
| |
Medianwert PFS, Monate
(95% KI)
|
10,2
(6,9; 16.7)
|
8,6
(5,2; 16,8)
| |
Gesamtüberleben
| |
Anzahl der Todesfälle - n (%)
|
33 (58)
| |
Medianwert, Monate
(95% KI)
|
18,2
(14,3; 28,6)
| |
Konfidenzintervall (KI) errechnet mit der exakten Methode (das sogenannte Clopper-Pearson-Intervall).
CR=komplette Remission; PR=partielle Remission; SD=stabile Erkrankung; PD=Krankheitsprogress
|
In die Studie BRF113928 wurden auch 36 Patienten mit BRAF V600E-mutationspositivem NSCLC eingeschlossen, die Dabrafenib in Kombination mit Trametinib als Erstlinientherapie bei einer metastasierenden Erkrankung erhielten. Bei der Abschlussanalyse nach 5 Jahren für alle Studienteilnehmer, die mit der Erstlinientherapie behandelt wurden, lag der primäre Endpunkt der vom Prüfarzt beurteilten Gesamtansprechrate (overall response rate, ORR) bei 63.9 % (95%-KI, 46.2 %, 79.2 %). Damit wurde die statistische Signifikanz in Ablehnung der Nullhypothese erfüllt, dass die ORR von Dabrafenib in Kombination mit Trametinib für diese NSCLC-Population höchstens 30 % betrug. Die vom Prüfarzt beurteilte mediane Dauer der Remission (DoR) betrug 10.2 Monate (95%-KI: 8.3, 15.2), das vom Prüfarzt beurteilte mediane progressionsfreie Überleben betrug 10.8 Monate (95%-KI: 7.0, 14.5), das vom Prüfarzt beurteilte mediane Gesamtüberleben betrug 17.3 Monate (95%-KI: 12.3, 40.2), wobei 61 % der Todesfälle zum Zeitpunkt der Analyse vorlagen.
Niedriggradiges Gliom (Low-grade-Gliom, LGG)
Studie DRB436G2201
Die klinische Wirksamkeit und Sicherheit der Kombinationstherapie von Tafinlar plus Trametinib bei pädiatrischen Patienten im Alter von 1 bis <18 Jahren mit BRAF V600E-mutation-positivem Gliom wurden in der multizentrischen, offenen klinischen Phase II Studie CDRB436G2201 untersucht. Patienten mit niedriggradigem Gliom (Low-grade-Gliom) (WHO 2016-Grad 1 und 2)), die eine systemische Ersttherapie benötigten und zuvor keine Strahlentherapie erhalten hatten, wurden im Verhältnis 2:1 zu Dabrafenib plus Trametinib (D+T) oder Carboplatin plus Vincristin (C+V) randomisiert. Etwa 83% aller Patienten hatten sich einem vorgängigen chirurgischen Eingriff unterzogen; nur zwei Patienten (beide im D+T Arm) hatten nach dem Eingriff keine Resterkrankung. Patienten mit einem Karnofsky/Lansky-Performance-Score von < 50 %, Patienten ohne ausreichende Knochenmark-, Nieren-, Leber- und Herzfunktion sowie Patienten mit unkontrollierten oder signifikanten Erkrankungen, einschliesslich Herzerkrankungen, Diabetes mellitus, Bluthochdruck, Lebererkrankungen oder Infektionen von der Teilnahme ausgeschlossen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Der BRAF-Mutationsstatus wurde prospektiv durch einen lokalen Test oder, wenn ein lokaler Test nicht verfügbar war, durch einen Echtzeit-PCR-Test des Zentrallabors ermittelt. Darüber hinaus wurden retrospektive Tests von verfügbaren Tumorproben durch das Zentrallabor durchgeführt, um die BRAF-V600E-Mutation zu bestätigen.
Die Dosierung von Tafinlar und Trametinib war alters- und gewichtsabhängig, wobei Tafinlar oral in einer Dosierung von 2,625 mg/kg zweimal täglich für die Altersgruppe < 12 Jahre und 2,25 mg/kg zweimal täglich für die Altersgruppe 12 Jahre und älter verabreicht wurde; Trametinib wurde oral in einer Dosierung von 0,032 mg/kg einmal täglich für die Altersgruppe < 6 Jahre und von 0,025 mg/kg einmal täglich für die Altersgruppe 6 Jahre und älter verabreicht. Die Höchstdosis von Tafinlar wurde auf 150 mg zweimal täglich und die von Trametinib auf 2 mg einmal täglich begrenzt. Carboplatin und Vincristin wurden je nach Alter und Körperoberfläche in einer Dosierung von 175 mg/m2 bzw. 1,5 mg/m2 als 10-wöchige Induktionstherapie, gefolgt von acht 6-wöchigen Zyklen einer Erhaltungstherapie, verabreicht.
Der primäre Wirksamkeitsendpunkt in beiden Kohorten war die Gesamtansprechrate (ORR, Summe der Patienten mit bestätigter kompletter Remission/CR und partieller Remission/PR) durch unabhängige Überprüfung auf der Grundlage der RANO-Kriterien (2017). Die primäre Analyse wurde durchgeführt, als alle Patienten in beiden Kohorten mindestens 32 Wochen der Therapie abgeschlossen hatten.
In der Kohorte der niedriggradigen Gliome (LGG) der Studie G2201 wurden 110 Patienten nach dem Zufallsprinzip für D+T (n = 73) oder C+V (n = 37) ausgewählt. Das Durchschnittsalter betrug 9,5 Jahren, wobei 34 Patienten (30,9 %) zwischen 12 Monaten und < 6 Jahren, 36 Patienten (32,7 %) zwischen 6 und < 12 Jahren und 40 Patienten (36,4 %) zwischen 12 und < 18 Jahren alt waren; 60 % waren weiblich. Zum Zeitpunkt der Primäranalyse betrug die mediane Therapiedauer im D+T Arm 76 Wochen, die mediane Nachbeobachtungszeit in der LGG Kohorte 18,9 Monate. Die ORR im D+T-Arm (46,6 %) zeigte eine statistisch signifikante Verbesserung gegenüber dem C+V-Arm (10,8 %), mit einer Odds Ratio (95% KI) von 7,19 (2.3, 22.4) und einem einseitigen p-Wert < 0,001 (Tabelle 14). Die anschliessende hierarchische Prüfung ergab ebenfalls eine Verbesserung des progressionsfreien Überlebens (PFS) gegenüber der Chemotherapie, mit einer hazard Ratio (95% KI) von 0,31 (0.17,0.55) (einseitiger log-rank p-Wert des Log-Rang-Tests < 0,001). Es wurden Unterschiede in der Beurteilung der Wirksamkeit zwischen der unabhängigen Überprüfung und den Prüfzentren beobachtet, welche auch die Feststellung von kompletten und partiellen Remissionen sowie Krankheitsprogressionen umfassten. Die Rate der Übereinstimmung betrug im D+T Arm insgesamt 52%.
Als funktionelle Parameter wurden Veränderungen der Krampfanfallhäufigkeit («seizure activity») und des Visus («visual acuity») vor und nach Therapie untersucht. Für beide Parameter war keine Verbesserung unter Behandlung mit D+T im Vergleich zum Kontrollarm nachweisbar.
Tabelle 14: Ansprechen und progressionsfreies Überleben basierend auf unabhängiger Überprüfung in der Studie G2201 (LGG-Kohorte, Primäranalyse)
|
|
Dabrafenib + Trametinib
N = 73
|
Carboplatin plus Vincristin
N = 37
| |
Bestes Gesamtansprechen
|
|
| |
Komplette Remission (CR), n (%)
|
2 (2,7)
|
1 (2,7)
| |
Partielle Remission (PR), n (%)
|
32 (43,8)
|
3 (8,1)
| |
Stabile Erkrankung (SD), n (%)
|
30 (41,1)
|
15 (40,5)
| |
Krankheitsprogress (PD), n (%)
|
8 (11,0)
|
12 (32,4)
| |
Unbekannt, n (%)
|
1 (1,4)
|
6 (16,2) 1
| |
Gesamtansprechrate (ORR)
|
|
| |
ORR (CR+PR), (95-%-KI)2, p-Wert
|
46,6% (34,8 – 58,6 %), p < 0,001
|
10,8% (3,0 – 25,4 %)
| |
Odds Ratio3 (95% KI)
|
7,19 (2,3 – 22,4)
| |
Progressionsfreies Überleben
|
|
| |
Medianwert (Monate) (95% KI)4
|
20,1 (12,8; NA)
|
7,4 (3,6; 11,8)
| |
Hazard Ratio (95-%-KI)5, p-Wert
|
0,31 (0,17 – 0,55), p < 0,001
| |
KI= Konfidenzintervall; CR= komplettes Ansprechen; NE= nicht abschätzbar; ORR=overall response rate; PD= progrediente Erkrankung; PR= partielles Ansprechen; SD= stabile Erkrankung.
1 4 Patienten, die für C+V randomisiert wurden, brachen die Behandlung vorzeitig ab.
2 Basierend auf dem exakten Clopper-Pearson-Konfidenzintervall.
3 Odds Ratio (D+T vs. C+V) und 95%-KI stammen aus einer logistischen Regression mit der Behandlung als einziger Kovariate, d. h., es handelt sich um die Wahrscheinlichkeit, ein Ansprechen im D+T-Arm zu beobachten, verglichen mit der Wahrscheinlichkeit, ein Ansprechen im C+V-Arm zu beobachten. Ein Odds Ratio >1 begünstigt D+T.
4 Basierend auf der Kaplan-Meier-Methode.
5 Basierend auf dem Proportional-Hazard-Modell.
|
Zum Zeitpunkt der endgültigen Analyse (mediane Dauer der Nachbeobachtung: 39,0 Monate) betrug die ORR 54,8 % im D+T-Arm und 16,2 % im C+V-Arm mit einem Odds Ratio von 6,26. Das mediane PFS betrug 24,9 Monate in der D+T-Gruppe und 7,2 Monate in der C+V-Gruppe.
Nicht resezierbare oder metastasierte solide Tumore
Die Sicherheit und Wirksamkeit von Tafinlar in Kombination mit Trametinib zur Behandlung von BRAF-V600E-Mutation-positiven nicht resezierbaren oder metastasierten soliden Tumoren wurde in den Studien BRF117019 und NCI-MATCH bei erwachsenen Patienten untersucht.
Patienten in beiden Studien erhielten Tafinlar 150 mg zweimal täglich und Trametinib 2 mg einmal täglich. Die wichtigsten Ergebnisse der Wirksamkeitsmessung waren die ORR (komplette (CR) oder partielle Remission (PR)) nach den RECIST v1.1-, RANO 2010-[HGG]- oder den modifizierten RANO 2017-[LGG]-Kriterien und die Dauer des Ansprechens (DoR). Minor Responses wurden von der ORR ausgeschlossen.
BRF117019-Studie und NCI-MATCH-Studie
Die BRF117019- Studie ist eine multizentrische, nicht-randomisierte, offene Studie mit mehreren Kohorten bei erwachsenen Patienten mit ausgewählten seltenen Tumoren mit der BRAF-V600E- Mutation. Die Studie umfasste 7 solide Tumorkohorten: hochgradiges Gliom (HGG) (n = 45), Gallengangskarzinom (BTC) (n = 43), anaplastisches Schilddrüsenkarzinom (ATC) (n = 36), niedriggradiges Gliom (LGG) (n = 13), Adenokarzinom des Dünndarms (ASI) (n = 3), und gastrointestinaler Stromatumor (GIST) (n = 1) sowie nicht-seminomatöse Keimzelltumoren (keine Patienten eingeschlossen). Es musste sich dabei um histologisch bestätigte, nach vorheriger Therapie fortgeschrittene Tumore (metastasiert oder lokal, nicht resezierbar) ohne alternative Behandlungsmöglichkeiten handeln. Die Patienten wurden auf der Grundlage lokaler Beurteilungen des BRAF-V600E-Mutationsstatus in die Studie aufgenommen; ein zentrales Labor bestätigte die BRAF-Mutation bei 126 der vorgenannten 141 Patienten. 34 der 141 Patienten (24%) wurden von einem einzigen Studienzentrum rekrutiert (beschränkt auf die Tumortypen HGG, BTC und ATC), was die Verallgemeinerbarkeit der Studienergebnisse beeinflusst haben könnte.
Die wichtigsten studienspezifischen Ausschlusskriterien umfassten das bekannte Vorliegen einer aktivierenden RAS-Mutation; vorhergehende Behandlung mit BRAF und/oder MEK-Inhibitoren; das Vorliegen von Gehirnmetastasen (ausser in den Gliom-Kohorten); interstitielle Lungenerkrankung oder Pneumonitis; Vorgeschichte eines Netzhautvenenverschlusses; und klinisch signifikante gastrointestinale Störungen.
Der Arm H (EAY131-H) der NCI-MATCH-Studie ist eine einarmige, offene Studie, in die erwachsene Patienten mit einer BRAF V600E-Mutation- positiven malignen Erkrankung aufgenommen wurden. Patienten mit Melanom, Schilddrüsenkarzinom oder Kolorektalkarzinom (KRK) wurden ausgeschlossen. Der BRAF-V600E-Mutationsstatus für die Aufnahme in die Studie wurde entweder durch einen zentralen oder lokalen Labortest bestimmt. Tabelle 15 schliesst die primäre Wirksamkeitspopulation der Studie ein, darunter gastrointestinale Tumoren (n = 14), Lungentumoren mit Ausnahme von NSCLC (n = 1), gynäkologische oder peritoneale Tumore (n = 6), ZNS-Tumore (n = 4) und Ameloblastom des Unterkiefers (n = 1).
Die Ausgangscharakteristika der 167 Patienten mit den in Tabelle 15 dargestellten Tumorarten, die in die BRF117019- und NCI-MATCH-Studien aufgenommen wurden, waren: mittleres Alter von 56 Jahren mit 32% im Alter von 65 Jahren oder älter; 56% weiblich; 77% weiss, 17% asiatisch, 2% schwarz, 4% andere; und 31% mit ECOG 0, 63% mit ECOG 1 und 6% mit ECOG 2. 85% der 167 Patienten hatten eine vorherige systemische Therapie erhalten. Die mittlere Expositionsdauer gegenüber der Kombinationstherapie betrug in beiden Studien 8,0 Monate.
Tabelle 15: Wirksamkeitsergebnisse basierend auf unabhängiger Überprüfung in der Studie BRF117019 und NCI-MATCH Arm H
|
Art des Tumors
|
N
|
Objektive Ansprechrate
(ORR)
|
Dauer des Ansprechens
(DoR)
| |
%
|
95% KI
|
Spanne (Monate)
| |
Gallenwegskarzinomb
|
48
|
46
|
(31, 61)
|
1.8d, 40d
| |
Hochgradiges Gliomc
|
48
|
33
|
(20, 48)
|
3.9, 44
| |
Glioblastom
|
32
|
25
|
(12, 43)
|
3.9, 27
| |
Anaplastisches pleomorphes Xanthoastrozytom
|
6
|
67
|
(22, 96)
|
6, 43
| |
Anaplastisches Astrozytom
|
5
|
20
|
(0.5, 72)
|
15
| |
Astroblastom
|
2
|
100
|
(16, 100)
|
15, 23d
| |
Undifferenziert
|
1
|
PR
|
(2.5, 100)
|
6
| |
Anaplastisches Gangliogliom
|
1
|
0
|
KA
|
KA
| |
Anaplastisches Oligodendrogliom
|
1
|
0
|
KA
|
KA
| |
ATC
|
36
|
53
|
(35.5, 69.6)
|
(0.9 d, 43.0 d)
| |
Niedriggradiges Gliom
|
14
|
50
|
(23, 77)
|
6, 29d
| |
Astrozytom
|
4
|
50
|
(7, 93)
|
7, 23
| |
Gangliogliom
|
4
|
50
|
(7, 93)
|
6, 13
| |
Pleomorphes Xanthoastrozytom
|
2
|
50
|
(1.3, 99)
|
6
| |
Pilozytisches Astrozytom
|
2
|
0
|
KA
|
KA
| |
Papillom des Plexus choroideus
|
1
|
PR
|
(2.5, 100)
|
29d
| |
Gangliozytom/Gangliogliom
|
1
|
PR
|
(2.5, 100)
|
18d
| |
Niedriggradiges seröses Ovarialkarzinom
|
5
|
80
|
(28, 100)
|
12, 42d
| |
Adenokarzinom im Dünndarm
|
4
|
50
|
(7, 93)
|
7, 8
| |
Adenokarzinom im Pankreas
|
3
|
0
|
KA
|
KA
| |
Gemischtes duktales / adenoneuroendokrines Karzinom
|
2
|
0
|
KA
|
KA
| |
Neuroendokrines Karzinom des Dickdarms
|
2
|
0
|
KA
|
KA
| |
Ameloblastom des Unterkiefers
|
1
|
PR
|
(2.5, 100)
|
30
| |
Kombiniertes kleinzellig-squamöses Karzinom der Lunge
|
1
|
PR
|
(2.5, 100)
|
5
| |
Papillär-muzinöses seröses Adenokarzinom des Peritoneums
|
1
|
PR
|
(2.5, 100)
|
8
| |
Adenokarzinom des Anus
|
1
|
0
|
KA
|
KA
| |
Gastrointestinaler Stromatumor
|
1
|
0
|
KA
|
KA
| |
Abkürzungen: PR, partielle Remission.
aAusgeschlossen sind NSCLC (n = 6).
b Mediane DoR 9,8 Monate (95% KI: 5,3, 20,4).
c Mediane DoR 13,6 Monate (95% KI: 5,5, 26,7).
d Bezeichnet eine rechtszensierte DoR.
|
Es wurden Unterschiede in der Beurteilung der Wirksamkeit zwischen der unabhängigen Überprüfung und den Prüfzentren beobachtet, welche auch die Feststellung von kompletten und partiellen Remissionen sowie Krankheitsprogressionen umfassten. In der BRF117019-Studie betrug die Rate der Übereinstimmung für ORR über die Tumorkohorten hinweg zwischen 46% und 67%; in NCI-MATCH Arm H bei 61%.
CTMT212X2101 (X2101) Studie
In der Studie X2101, einer multizentrische, offenen Studie bei pädiatrischen Patienten mit refraktären oder rezidivierenden soliden Tumoren wurde die Wirksamkeit von Tafinlar in Kombination mit Trametinib wurde bei 48 Patienten untersucht, darunter 34 Patienten mit LGG und 2 Patienten mit HGG. Das Durchschnittsalter der LGG-Patienten lag bei 10 Jahren (Spanne: 1 bis 17)).
Die Dosierung von Tafinlar und Trametinib war in der klinischen Studie alters- und gewichtsabhängig, wobei Tafinlar bei Altersgruppen < 12 Jahren zweimal täglich mit 2,625 mg/kg und bei Altersgruppen ab 12 Jahren mit 2,25 mg/kg zweimal täglich dosiert wurde; Trametinib wurde oral in einer Dosierung von 0,032 mg/kg einmal täglich für Kinder unter 6 Jahren und in einer Dosierung von 0,025 mg/kg einmal täglich für Kinder ab 6 Jahren verabreicht. Die Tafinlar-Dosen wurden auf 150 mg zweimal täglich und die Trametinib-Dosen auf 2 mg einmal täglich begrenzt.
Die ORR gemäss unabhängiger Überprüfung betrug 25% (95% KI: 12%, 42%), wobei es sich in allen Fällen um partielle Remissionen handelte. Eine Schätzung der DoR war bei 2 DoR-Ereignissen in 9 Respondern nicht möglich.
PharmakokinetikDie Pharmakokinetik von Tafinlar wurde an Patienten mit BRAF-V600-Mutation-positivem metastasiertem Melanom nach einer Einzeldosis sowie nach wiederholter Verabreichung von 150 mg zweimal täglich in ca. 12-stündigen Abständen bestimmt.
Absorption
Dabrafenib wird oral mit einer medianen Zeit bis zum Erreichen der Plasma-Spitzenkonzentration von 2 Stunden nach der Verabreichung resorbiert. Die mittlere absolute Bioverfügbarkeit von oralem Dabrafenib beträgt 95 % (90%-KI: 81; 110 %). Die Dabrafenib-Exposition (Cmax und AUC) nahm nach Verabreichung einer Einzeldosis zwischen 12 und 300 mg dosisabhängig zu; nach wiederholter zweimaliger täglicher Verabreichung war der Anstieg dagegen unterproportional zur Dosis. Bei wiederholter Verabreichung wurde eine Abnahme der Exposition beobachtet, vermutlich aufgrund der Eigeninduktion des Metabolismus. Das mittlere Akkumulationsverhältnis für die AUC betrug 0.73 für Tag 18/Tag 1. Nach Verabreichung von 150 mg zweimal täglich betrugen Cmax, AUC(0-τ) und die Konzentration vor Verabreichung (Cτ) im geometrischen Mittel 1478 ng/ml, 4341 ng*hr/ml bzw. 26 ng/ml.
Nach Verabreichung von Trametinib in Kombination mit Dabrafenib 150 mg zweimal täglich betrugen die geometrischen Mittelwerte von Trametinib Cmax, AUC(0-τ) und der Trametinib Konzentration vor Verabreichung 22.6 ng/ml, 356 ng*hr/ml bzw. 10.9 ng/ml.
Die Verabreichung einer Dabrafenib-Kapsel mit Speisen reduzierte die Bioverfügbarkeit (Cmax und AUC nahmen um 51 % bzw. 31 % ab) und verzögerte die Resorption der Dabrafenib Kapseln im Vergleich zur Nüchternverabreichung. Dabrafenib-Kapseln und -Tabletten zur Herstellung einer Suspension zum Einnehmen sind Darreichungsformen mit sofortiger Freisetzung, die ähnliche Auswirkungen auf die PK haben dürften.
Distribution
Dabrafenib bindet zu 99.7 % an humane Plasmaproteine. Das Verteilungsvolumen im Steady State beträgt nach intravenöser Verabreichung 46 l.
Metabolismus
Der Metabolismus von Dabrafenib wird in erster Linie durch CYP2C8 und CYP3A4 vermittelt; das so entstandene Hydroxy-Dabrafenib wird über das CYP3A4 weiter zu Carboxy-Dabrafenib oxidiert. Carboxy-Dabrafenib kann in einem nichtenzymatischen Prozess zu Desmethyl-Dabrafenib decarboxyliert werden. Carboxy-Dabrafenib wird über Galle und Urin ausgeschieden. Desmethyl-Dabrafenib kann auch im Darm gebildet und rückresorbiert werden. Desmethyl-Dabrafenib wird durch das CYP3A4 zu oxidierenden Metaboliten abgebaut. Die terminale Halbwertszeit von Hydroxy-Dabrafenib entspricht mit 10 h derjenigen der Muttersubstanz, während die Carboxy- und Desmethyl-Metaboliten längere Halbwertszeiten aufweisen (21 – 22 Stunden). Das mittlere AUC-Verhältnis zwischen Metaboliten und Muttersubstanz nach wiederholter Verabreichung betrug für Hydroxy-Dabrafenib 0.9, für Carboxy-Dabrafenib 11 und für Desmethyl-Dabrafenib 0.7. Auf Grundlage der Exposition, der relativen Wirkstärke und der pharmakokinetischen Eigenschaften tragen sowohl Hydroxy- als auch Desmethyl-Dabrafenib vermutlich zur klinischen Aktivität von Dabrafenib bei, wohingegen Carboxy-Dabrafenib hinsichtlich seiner Aktivität wahrscheinlich keine Relevanz besitzt.
Elimination
Die terminale Halbwertszeit nach einer intravenösen Mikrodosis beträgt 2.6 Stunden. Die terminale Halbwertszeit von Dabrafenib nach oraler Verabreichung beträgt aufgrund einer verzögerten terminalen Phase 8 Stunden. Die Plasma-Clearance nach intravenöser Verabreichung beträgt 12 l/h.
Die Ausscheidung mit den Fäzes ist der wichtigste Eliminationsweg nach oraler Verabreichung; 71 % einer radioaktiven Dosis wurden in den Fäzes wiedergefunden, lediglich 23 % der radioaktiven Dosis wurden mit dem Urin ausgeschieden.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Die Pharmakokinetik von Tafinlar wurde bei 65 in die klinischen Studien eingeschlossenen Patienten mit leichter Leberfunktionsbeeinträchtigung (auf Grundlage der Klassifizierung des National Cancer Institutes [NCI-Klassifizierung]) anhand einer Populationsanalyse bestimmt. Die orale Clearance von Dabrafenib unterschied sich bei diesen Patienten nicht signifikant von den lebergesunden Teilnehmern (4 % Unterschied). Eine leichte Leberfunktionsbeeinträchtigung hatte ausserdem keinen signifikanten Einfluss auf die Plasmakonzentrationen der Dabrafenib-Metaboliten. Es liegen keine Daten bei Patienten mit mässiger bis hochgradiger Leberfunktionsbeeinträchtigung vor (siehe «Dosierung/ Anwendung»).
Nierenfunktionsstörungen
Die Pharmakokinetik von Tafinlar wurde in den klinischen Studien an 233 Patienten mit leichter Nierenfunktionsbeeinträchtigung (GFR 60 – 89 ml/min/1.73 m2) sowie an 30 Patienten mit mässiger Nierenfunktionsbeeinträchtigung (GFR 30 – 59 ml/min/1.73 m2) anhand einer Populationsanalyse untersucht. Der Einfluss einer leichten bis mässigen Nierenfunktionsbeeinträchtigung auf die orale Clearance von Dabrafenib war gering (< 6 % in beiden Kategorien) und nicht von klinischer Relevanz. Eine leichte bis mässige Nierenfunktionsbeeinträchtigung hatte ausserdem keinen signifikanten Einfluss auf die Plasmakonzentrationen der Hydroxy-, Carboxy- und Desmethyl-Metaboliten von Dabrafenib. Zu Patienten mit hochgradiger Nierenfunktionsbeeinträchtigung liegen keine Daten vor (siehe «Dosierung/ Anwendung»).
Pädiatrische Population
Die Pharmakokinetik von Dabrafenib bei Gliomen und anderen soliden Tumoren wurde bei 243 pädiatrischen Patienten (1 bis < 18 Jahre alt) nach einmaliger oder wiederholter gewichtsangepasster Dosierung untersucht. Die pharmakokinetischen Eigenschaften (Absorptionsrate, Metabolitenverhältnisse, Arzneimittel-Clearance) von Dabrafenib bei pädiatrischen Patienten sind mit denen von Erwachsenen vergleichbar. Es zeigte sich, dass das Gewicht Auswirkungen auf die orale Clearance von Dabrafenib hat. Die pharmakokinetische Exposition gegenüber Dabrafenib in der empfohlenen gewichtsangepassten Dosierung bei pädiatrischen Patienten lag im Bereich der bei Erwachsenen beobachteten Exposition.
Ältere Patienten
Auf Grundlage der populationspharmakokinetischen Analyse hatte das Alter keinen signifikanten Einfluss auf die Pharmakokinetik von Dabrafenib. Ein Lebensalter von über 75 Jahren war ein signifikanter Prädiktor hinsichtlich der Plasmakonzentrationen von Carboxy- und Desmethyl-Dabrafenib, wobei Teilnehmer im Alter von 75 Jahren und darüber eine um 40 % höhere Exposition aufwiesen als Teilnehmer unter 75 Jahren.
Körpergewicht und Geschlecht:
Im Rahmen der populationspharmakokinetischen Analyse an Erwachsenen wurde ein Einfluss von Geschlecht und Körpergewicht auf die orale Clearance von Dabrafenib festgestellt; das Gewicht hatte ausserdem einen Einfluss auf das Verteilungsvolumen nach oraler Verabreichung und die Verteilungsclearance. Diese pharmakokinetischen Unterschiede wurden nicht als klinisch relevant eingestuft.
Ethnische Abstammung
Die Populationsanalyse der Pharmakokinetik von Dabrafenib zeigte keine signifikanten Unterschiede zwischen Asiatischen und Kaukasischen Patienten. Dosierungsanpassungen bei Patienten mit asiatischer Abstammung sind nicht erforderlich.
Es liegen keine hinreichenden Daten vor, um den möglichen Einfluss der Ethnizität auf die Pharmakokinetik von Tafinlar zu beurteilen.
Präklinische DatenDabrafenib in Kombination mit Trametinib
Bei Hunden, denen vier Wochen lang Dabrafenib und Trametinib in Kombination verabreicht wurden, wurden ähnliche Toxizitätserscheinungen festgestellt wie in präklinischen Studien, die mit den einzelnen Substanzen allein durchgeführt wurden. In Hunden, denen die Kombination aus Dabrafenib und Trametinib verabreicht wurde, wurden dieselben testikulären Wirkungen, bestehend aus Degeneration und sekundärer Nebenhoden Oligospermie, beobachtet, wie in Hunden, denen entweder Dabrafenib oder Trametinib allein verabreicht wurde. Siehe dazu auch die Fachinformation für Trametinib.
Sicherheitspharmakologie
Bei der Ratte und beim Hund wurde in Einzeldosenstudien ein dosisabhängiger Anstieg der Herzfrequenz beobachtet; Auffälligkeiten an der EKG-Kurve bzw. Arrhythmien oder Auswirkungen auf den arteriellen Blutdruck oder die Körpertemperatur konnten allerdings nicht festgestellt werden. Dabrafenib bewirkte eine leichte Hemmung der Repolarisation über hERG-Kanäle in vitro (IC25 von 11.7 μM; 6.1 μg/ml), wobei jedoch seine 3 wichtigsten Metaboliten nicht zu einer hERG-Kanal-Hemmung (IC50> 30 μM; 15.6 μg/ml) führten. In einem Perfusionsassay an linksventrikulären Myokardabschnitten von Kaninchen (Wedge-Assay) wurde eine Verkürzung des QT-Intervalls (30 % bei 30 μM) ohne Änderungen von QRS-Intervall oder torsadogenem Potential beobachtet. In Studien mit Einzel- und wiederholten Dosen an Hunden wurde kein Einfluss auf das QTc-Intervall beobachtet (Dauer bis 13 Wochen; ≥9-faches der humanen klinischen Exposition, bezogen auf die AUC).
Mutagenität / Karzinogenität
Dabrafenib hat sich in vitro an Bakterien und Säugerzellkulturen sowie in vivo im Mikronukleustest bei Nagetieren als nicht mutagen oder klastogen erwiesen. Karzinogenitätsstudien mit Dabrafenib wurden nicht durchgeführt.
Reproduktionstoxizität
In kombinierten Studien zur weiblichen Fertilität sowie zur frühen embryonalen und embryofetalen Entwicklung an Ratten war die Zahl der Corpora lutea in den Ovarien trächtiger Weibchen bei 300 mg/kg/Tag (entspricht, bezogen auf die AUC, ungefähr der dreifachen klinischen Exposition beim Menschen) verringert, Auswirkungen auf den Zyklus, das Paarungsverhalten oder die Fertilität konnten jedoch nicht festgestellt werden. Entwicklungstoxische Erscheinungen, z.B. Embryonensterblichkeit und Ventrikelseptumdefekte, wurden bei Dosen von 300 mg/kg/Tag, verzögerte Skelettentwicklung und reduziertes Fetalgewicht ab Dosen von 20 mg/kg/Tag (entspricht, bezogen auf die AUC, dem ≥0,5-fachen der klinischen Exposition beim Menschen) beobachtet.
Studien zur männlichen Fertilität unter Dabrafenib wurden nicht durchgeführt. In Studien mit wiederholter Verabreichung wurde bei der Ratte und beim Hund allerdings eine Hodendegeneration/-atrophie beobachtet (bei Dosen, die, bezogen auf die AUC, dem ≥0,2-fachen der klinischen Exposition beim Menschen entsprechen). Die testikulären Veränderungen bei der Ratte und beim Hund waren auch nach einer vierwöchigen Erholungsphase noch vorhanden.
Allgemeine Toxizität
Beim Hund wurden bei Dosen, die, bezogen auf die AUC, dem ≥2-fachen der klinischen Exposition beim Menschen entsprachen, kardiovaskuläre Wirkungen beobachtet, unter anderem degenerative, nekröse und/oder hämorrhagische Veränderungen der Koronararterien, Atrioventrikularklappenhypertrophie/-hämorrhagie und atriale fibrovaskuläre Proliferation. Bei Mäusen wurden in verschiedenen Gewebearten Herde arterieller/perivaskulärer Entzündungsgeschehen beobachtet und bei Ratten wurden gehäuft eine Degeneration der Leberarterie sowie eine spontane Kardiomyozytendegeneration mit entzündlichen Veränderungen (spontane Kardiomyopathie) beobachtet (bei Dosen, die bei Ratten dem ≥0.5-fachen resp. bei Mäusen dem 0.6-Fachen der klinischen Exposition beim Menschen entsprachen).
Bei Mäusen wurden Auswirkungen auf die Leber, einschliesslich hepatozelluläre Nekrose und Entzündung, beobachtet (≥0.6-fache klinische Exposition).
Bei mehreren Hunden trat bei Dosen ≥20 mg/kg/Tag (entspricht, bezogen auf die AUC, dem ≥9-fachen der klinischen Exposition beim Menschen) eine bronchoalveoläre Entzündung der Lungen auf, die mit flacher Atmung und/oder Dyspnoe einherging.
Epitheliale Läsionen, die in erster Linie durch epitheliale Hyperplasie, Degeneration und/oder Hyperkeratose gekennzeichnet waren, wurden in der Haut von Ratten und Hunden sowie im nichtglandulären Vormagen von Ratten beobachtet (≥0.7-faches der humanen klinischen Exposition bei der Ratte; 1.5-faches der humanen klinischen Exposition beim Hund, jeweils bezogen auf die AUC).
Reversible hämatologische Wirkungen wurden bei Hunden und Ratten unter Dabrafenib beobachtet. In Studien von bis zu 13-wöchiger Dauer wurde bei Hunden und Ratten eine Abnahme der Retikulozytenzahl und/oder des Volumens der zirkulierenden Erythrozyten festgestellt (bei Dosen, die dem ≥10-fachen bzw. dem 1.4-fachen der klinischen Exposition beim Menschen entsprachen).
In Studien zur juvenilen Toxizität bei Ratten wurden Auswirkungen auf das Wachstum (verminderte Länge der Röhrenknochen), renale Toxizität (Ablagerungen in den Nierenkanälchen, erhöhte Inzidenz von kortikalen Nierenzysten und tubuläre Basophilie sowie ein reversibler Anstieg von Harnstoff und/oder der Kreatininkonzentrationen)undtestikuläre Toxizität (Degeneration und Dilatation der Tubuli) beobachtet (≥0.2-fache klinische Exposition der Erwachsenendosis basierend auf AUC).
Dabrafenib erwies sich in vitro in einem 3T3 NRU (Neutral Red Uptake)-Test mit einer Fibroblastenzelllinie der Maus als phototoxisch, sowie in vivo in einer Phototoxizitätsstudie mit haarlosen Mäusen bei oral verabreichten Dosierungen ≥100 mg/kg (> 44-fache der klinischen Exposition basierend auf Cmax). Obgleich sich Dabrafenib in nicht klinischen Studien als phototoxisch erwies, besteht anhand der klinischen Daten zur Sicherheit für Patienten, die Dabrafenib einnehmen, ein geringes Risiko für Phototoxizität.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Hartkapseln
Nicht über 30°C lagern.
Tabletten zur Herstellung einer Suspension zum Einnehmen
Nicht über 30°C lagern In der Originalflasche aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen. Das Trockenmittel nicht entfernen.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer62781 (Swissmedic)
69135 (Swissmedic)
PackungenHartkapseln 50 mg: 28 (A)
Hartkapseln 50 mg: 120 (A)
Hartkapseln 75 mg: 28 (A)
Hartkapseln 75 mg: 120 (A)
10 mg Tabletten zur Herstellung einer Suspension zum Einnehmen: 210 Tabletten und 2 Dosierbecher (A)
ZulassungsinhaberinNovartis Pharma Schweiz AG, Risch; Domizil: 6343 Rotkreuz.
Stand der InformationFebruar 2025
Sonstige Hinweise - Hinweise für die Anwendung und Handhabung
Hinweise für die Anwendung und Handhabung
|
GEBRAUCHSANWEISUNG
Tafinlar®
(Dabrafenib)
Tabletten zur Herstellung einer Suspension zum Einnehmen
| |
Diese «Gebrauchsanweisung» enthält Informationen zur Einnahme oder Verabreichung von Tafinlar Tabletten zur Herstellung einer Suspension zum Einnehmen als Suspension zum Einnehmen
| |
Wichtige Informationen, die Sie vor der Einnahme oder Verabreichung von Tafinlar-Tabletten zur Herstellung einer Suspension zum Einnehmen (Tafinlar-Tabletten) beachten müssen.
| |
·Lesen Sie diese Gebrauchsanweisung sorgfältig durch, bevor Sie Tafinlar zum ersten Mal anwenden und jedes Mal, wenn Sie eine Nachfüllpackung erhalten. Sie kann neue Informationen enthalten.
·Diese Anwendungshinweise ersetzen nicht das Gespräch mit Ihrem Arzt oder Apotheker über Ihre Erkrankung oder die Ihres Kindes und deren Behandlung.
·Ihr Arzt oder Apotheker sollte Ihnen zeigen, wie Sie eine Dosis von Tafinlar richtig einnehmen oder geben. Sie sollten Tafinlar immer genau so einnehmen oder verabreichen, wie Ihr Arzt es Ihnen sagt.
·Wenn Sie Fragen zur Zubereitung, Einnahme oder Gabe von Tafinlar haben, wenden Sie sich an Ihren Arzt oder Apotheker.
·Verwenden Sie stets den Dosierbecher, der Ihrer Tafinlar-Packung beiliegt. Wenn Ihre Packung keinen Dosierbecher enthält, wenden Sie sich an Ihren Arzt oder Apotheker.
·Wenn Tafinlar-Suspension zum Einnehmen zu irgendeinem Zeitpunkt auf Ihre Haut oder die Ihres Kindes gelangt, waschen Sie die betroffene Stelle gründlich mit Wasser und Seife.
·Wenn Tafinlar-Suspension zum Einnehmen zu irgendeinem Zeitpunkt in Ihre Augen oder die Ihres Kindes gelangt, spülen Sie die Augen gründlich mit kaltem Wasser aus.
·Wenn Sie Tafinlar-Suspension zum Einnehmen verschütten, befolgen Sie die Anweisungen am Ende dieser Gebrauchsanweisung im Abschnitt «Reinigung nach Verschütten».
Sie erhalten Tafinlar oder das ihres Kindes in einer versiegelten Flasche, die lösliche Tabletten enthält. Sie müssen die Tabletten in Wasser auflösen, bevor Sie Tafinlar einnehmen oder geben. Befolgen Sie die nachstehenden Anweisungen, um die Tabletten in Wasser aufzulösen.
·Ihre Tafinlar-Packung sollte enthalten:
|
1.1 Flasche mit Tafinlar Tabletten
2.2 wiederverwendbare Dosierbecher
Beipackzettel und Patienteninformation (dieses Dokument)
Ausserdem benötigen Sie Trinkwasser.
Für die Anwendung durch Schlucken gehen Sie zu Abschnitt A. Für die Anwendung über eine Ernährungssonde oder eine orale Spritze gehen Sie zu Abschnitt B.
|
|
ABSCHNITT A. Zubereitung und Gabe einer Dosis durch Schlucken direkt aus dem Dosierbecher
Falls die Tafinlar-Lösung verschüttet wurde oder mit der Haut oder den Augen in Berührung gekommen ist, befolgen Sie die Hinweise in Abschnitt E, Beseitigen von verschütteter Tafinlar-Suspension zum Einnehmen.
Für die Zubereitung und Gabe von Tafinlar benötigen Sie:
·die vorgeschriebene Anzahl von Tabletten
·1 Dosierbecher
·1 Teelöffel (aus Edelstahl)
·Stilles Trinkwasser
Zur oralen Anwendung (d.h. zum Schlucken der Lösung) können Sie
oder Ihr Kind diese direkt aus dem Dosierbecher trinken.
| |
Schritt 1. Waschen und trocknen Sie sich die Hände, bevor Sie Tafinlar zubereiten.
Fügen Sie die nachstehend angegebene Menge an kühlem Trinkwasser bis zu den Markierungen auf dem Dosierbecher hinzu (Hinweis: Die Wassermenge muss nicht genau abgemessen werden):
·Wenn die verschriebene Dosis von Ihnen oder Ihrem Kind 1 bis 4 Tabletten beträgt, benötigen Sie etwa 5 ml Wasser.
·Wenn die verschriebene Dosis von Ihnen oder Ihrem Kind 5 bis 15 Tabletten beträgt, benötigen Sie etwa 10 ml Wasser.
|
|
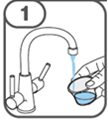
|
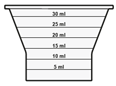
| |
Schritt 2. Entfernen Sie die Kappe, indem Sie sie nach unten drücken und gegen den Uhrzeigersinn drehen (siehe Abbildung).
Werfen Sie die Kappe nicht weg.
Wenn Sie die Flasche zum ersten Mal öffnen, entfernen Sie die Versiegelung von der Flasche.
|
|
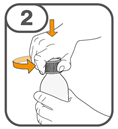
| |
Schritt 3. Zählen Sie die verordnete Anzahl Tabletten in Ihre Hand ab.
|
|
| |
Geben Sie die vorgeschriebene Anzahl Tabletten in das Wasser im Dosierbecher.
|
|
| |
Die Flasche enthält 2 Kunststoffbehälter, um die Tabletten trocken zu halten. Wenn einer der beiden Behälter herausfällt, wenn Sie Ihre Tabletten herausnehmen, setzen Sie ihn wieder in die Flasche ein.
|
|
| |
Schritt 4. Setzen Sie die Kappe wieder auf die Flasche und drehen Sie sie wie gezeigt im Uhrzeigersinn in Pfeilrichtung, um sie zu schliessen
|
|
| |
Schritt 5. Kippen Sie mit einer Hand den Dosierbecher.
Rühren Sie mit der anderen Hand das Wasser und die Tabletten vorsichtig mit dem Stiel eines Teelöffels um, bis die Tabletten vollständig aufgelöst sind.
·Es kann 3 Minuten (oder länger) dauern, bis die Tabletten vollständig aufgelöst sind. Sobald sie aufgelöst sind, sollte die Lösung trüb-weiss aussehen, kann aber kleine Stücke enthalten.
Geben Sie die Lösung nicht später als 30 Minuten nach dem Auflösen der Tabletten.
Sind mehr als 30 Minuten vergangen, entsorgen Sie die Lösung entsprechend den lokalen Bestimmungen und beginnen Sie wieder am Anfang von Abschnitt A. Wenn Sie sich nicht sicher sind, wie die Tafinlar-Suspension zum Einnehmen zu entsorgen ist, fragen Sie Ihren Arzt oder Apotheker.
|
|
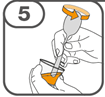
|
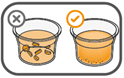
| |
Schritt 6. Trinken Sie die Lösung aus dem Dosierbecher.
WICHTIG: Nach dem Trinken bleiben Arzneimittelreste im Becher zurück. Diese Reste sind möglicherweise nur schwer zu erkennen. Führen Sie die Schritte 7 – 9 aus, um alle Reste einzunehmen und eine vollständige Dosis zu erhalten.
|
|
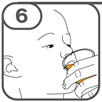
| |
Schritt 7. Geben Sie etwa 5 ml Wasser in den leeren Dosierbecher.
|
|
| |
Schritt 8. Rühren Sie mit dem Stiel eines Teelöffels um, um die verbleibenden Reste zu lösen.
|
|
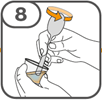
| |
Schritt 9. Trinken Sie die Lösung.
|
|
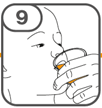
| |
·Bei 1 – 3 Tabletten: Schritte 7 – 9 einmal ausführen
·Bei 4 – 15 Tabletten: Schritte 7 – 9 zweimal ausführen
Es ist wichtig, alle Arzneimittelrückstände einzunehmen oder zu verabreichen, damit Sie oder Ihr Kind die volle Dosis TAFINLAR erhalten
| |
Schritt 10. Fahren Sie mit den Reinigungsschritten in Abschnitt C fort.
| |
ABSCHNITT B. Zubereitung und Anwendung von Tafinlar über eine Ernährungssonde oder eine orale Spritze
| |
WICHTIGE ANWENDUNGSHINWEISE
Stellen Sie sicher, dass alle Tabletten vollständig aufgelöst sind, bevor Sie die Lösung geben.
Die Mindestgrösse der Ernährungssonde, die Sie verwenden können, um die Tafinlar-Lösung zu verabreichen:
·Wenn Ihre verschriebene Dosis 1 bis 3 Tabletten beträgt, beträgt die Mindestgrösse der zu verwendenden Ernährungssonde 10 Charrière.
·Wenn Ihre verschriebene Dosis 4 bis 15 Tabletten beträgt, beträgt die Mindestgrösse der zu verwendenden Ernährungssonde 12 Charrière.
·Falls Tafinlar-Lösung mit Ihrer Haut oder Ihren Augen in Kontakt kommt, während Sie die nachstehenden Schritte befolgen, befolgen Sie die Anweisungen im Abschnitt «Wichtige Informationen, die Sie vor der Einnahme oder Verabreichung von Tafinlar beachten müssen».
·Falls Tafinlar-Lösung verschüttet wird, befolgen Sie die Anweisungen im Abschnitt «Reinigung nach Verschütten».
·Waschen und trocknen Sie Ihre Hände, bevor Sie eine Dosis Tafinlar verabreichen.
| |
Schritt 1. Führen Sie die Schritte 1 – 5 in Abschnitt A aus, um die Tabletten aufzulösen. Bei Verwendung einer Ernährungssonde spülen Sie die Sonde mit stillem Trinkwasser und gehen dann zu Schritt 2 in Abschnitt B über.
| |
Schritt 2. Ziehen Sie die gesamte TAFINLAR-Lösung aus dem Dosierbecher in eine Spritze auf, indem Sie den Kolben zurückziehen. Achten Sie darauf, eine Spritze zu verwenden, die mit der Ernährungssonde verwendet werden kann, oder um die TAFINLAR-Suspension zum Einnehmen oral zu verabreichen. Fragen Sie Ihren Arzt, wenn Sie sich nicht sicher sind, welche Spritze Sie verwenden sollen.
|
|
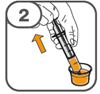
| |
Schritt 3. Bei Gabe über eine Ernährungssonde geben Sie die Lösung entsprechend den Angaben des Sondenherstellers in die Ernährungssonde.
Bei oraler Gabe platzieren Sie das Ende der Spritze in den Mund, so dass die Spitze die Innenseite einer Wange berührt. Achten Sie bei der Gabe an ein Kind darauf, dass es aufrecht sitzt.
Drücken Sie den Kolben langsam ganz hinein, um die volle Dosis abzugeben.
Achtung: Die direkte Gabe von Tafinlar in den Rachen oder ein zu schnelles Eindrücken des Kolbens kann zu Erstickung führen.
|
|

| |
Schritt 4. Geben Sie etwa 5 ml Wasser in den leeren Dosierbecher.
|
|
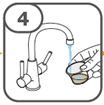
| |
Schritt 5. Umrühren, um die Reste zu lösen.
|
|
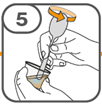
| |
Schritt 6. Die Lösung aufziehen.
|
|
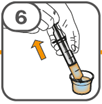
| |
Schritt 7. Die Lösung in die Ernährungssonde oder in die Innenseite der Wange abgeben.
|
|
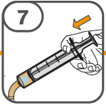
| |
3 Mal wiederholen
Es ist der gesamte Arzneimittelrest einzunehmen. Wiederholen Sie Schritt 4 bis Schritt 7 insgesamt dreimal, um eine vollständige Dosis zu geben.
| |
Schritt 8. Nachdem Sie Schritt 4 bis Schritt 7 insgesamt dreimal wiederholt haben, spülen Sie die Ernährungssonde mit stillem Trinkwasser aus. Fahren Sie dann mit den Reinigungsschritten in Abschnitt C fort.
| |
ABSCHNITT C. Reinigung des Dosierbechers und der Spritze (sofern verwendet)
| |
VERWENDEN SIE ZUM REINIGEN DES DOSIERBECHERS NUR SAUBERES WASSER. VERWENDEN SIE ZUM REINIGEN DES DOSIERBECHERS KEINE SEIFE ODER GESCHIRRSPÜLMITTEL.
| |
Schritt 1. Spülen Sie den Dosierbecher sofort nach der Dosierung unter sauberem, kühlem Wasser aus.
Überschüssiges Wasser abschütteln und mit sauberen Papiertüchern abtrocknen.
Hinweis: Halten Sie den Dosierbecher immer von anderen Küchenutensilien fern.
|
|
| |
Schritt 2. Spülen Sie den Teelöffel mit warmem Wasser mit Spülmittel von Hand oder in einer Geschirrspülmaschine.
|
|
| |
Schritt 3. Reinigen Sie die Applikationsspritze (sofern verwendet) entsprechend den Herstellerangaben.
|
|
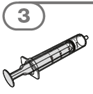
| |
ABSCHNITT D. Wie sind abgelaufene oder nicht mehr benötigte Tafinlar-Tabletten oder alte Dosierbecher zu entsorgen
| |
Entsorgen Sie alle Tafinlar-Tabletten, deren Haltbarkeitsdatum abgelaufen ist oder die Sie nicht mehr benötigen, auf sichere Weise.
Fragen Sie Ihren Arzt oder Apotheker, wie Sie Tafinlar-Tabletten sicher entsorgen können, wenn Sie sich nicht sicher sind.
| |
ABSCHNITT E: Was soll ich tun, wenn die Tafinlar-Lösung zum Einnehmen mit meiner Haut oder der meines Kindes in Berührung kommt oder in die Augen gelangt? REINIGUNG NACH VERSCHÜTTEN
| |
Wenn Tafinlar-Lösung auf Ihre Haut oder die Haut Ihres Kindes gelangt, waschen Sie die Stelle gründlich mit Wasser und Seife ab.
Wenn Tafinlar-Lösung in Ihre Augen oder die Augen Ihres Kindes gelangt, spülen Sie die Augen gründlich mit kaltem Wasser aus.
Führen Sie die folgenden Schritte aus, wenn Sie Tafinlar Lösung zum Einnehmen verschütten:
1.Ziehen Sie Handschuhe aus Kunststoff an.
2.Saugen Sie die Lösung mit einem saugfähigen Material vollständig auf, z.B. mit Papiertüchern, die entweder mit einer Mischung aus Wasser und Haushaltsdesinfektionsmittel oder mit 70%igem Ethanol (oder stärker) getränkt sind.
3.Wiederholen Sie die Reinigung mit frisch getränktem, saugfähigem Material mindestens dreimal, bis der Bereich sauber ist.
4.Trocknen Sie den Bereich mit Papiertüchern.
5.Geben Sie alle zur Reinigung der verschütteten Flüssigkeit verwendeten Einwegmaterialien in einen verschliessbaren Kunststoffbeutel.
6.Entsorgen Sie den Beutel entsprechend den lokalen Bestimmungen.
7.Waschen Sie Ihre Hände gründlich mit Wasser und Seife.
| |
Wie sollte ich Tafinlar aufbewahren
| |
·Bewahren Sie die Tafinlar-Flasche so auf, dass sich die beiden Kunststoffbehälter im Inneren befinden und die Kappe fest verschlossen ist. Die Behälter halten das Arzneimittel trocken und schützen es vor Feuchtigkeit.
·Bewahren Sie die Flasche und die Dosierbecher in der Originalverpackung auf.
·Lagern nicht über 30°C.
·Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf.
|
|