Eigenschaften/WirkungenATC-Code
L01FF05
Wirkungsmechanismus
Durch die Bindung von PD-L1 an PD-1- und B7.1-Rezeptoren auf T-Zellen wird die zytotoxische T-Zell-Aktivität über eine Hemmung der Proliferation und Zytokinproduktion der T-Zellen supprimiert. PD-L1 kann auf Tumorzellen (TC) und Tumor-infiltrierenden Immunzellen (IC) exprimiert werden und zur Hemmung der antitumoralen Immunantwort in der Mikroumgebung beitragen.
Atezolizumab ist ein humanisierter monoklonaler Immunglobulin G1 (IgG1)-Antikörper mit modifiziertem Fc-Teil, der direkt an PD-L1 bindet und die Interaktionen mit den PD-1- und B7.1-Rezeptoren blockiert. Dies führt zu einer Aufhebung der über den PD-L1/PD-1-Signalweg vermittelten Hemmung der Immunantwort einschliesslich Reaktivierung der antitumoralen Immunantwort. Atezolizumab lässt die PD-L2/PD-1-Interaktion intakt. In syngenen murinen Tumormodellen führte die Blockade der PD-L1-Aktivität zu vermindertem Tumorwachstum.
Die duale Hemmung der PD-1/PD-L1- und MAPK- sowie der BRAF- und MEK-Signalwege unterdrückt in Mauskrebsmodellen das Tumorwachstum und verbessert die Tumorimmunogenität durch eine im Vergleich zur alleinigen zielgerichteten Therapie erhöhte Antigenpräsentation und T-Zell-Infiltration und -Aktivierung.
Klinische Wirksamkeit
Nicht-kleinzelliges Lungenkarzinom
1L bei metastasiertem nicht-plattenepithelialem NSCLC
IMpower150
In einer unverblindeten randomisierten Phase-III-Studie, GO29436 (IMpower150), wurden die Wirksamkeit und Sicherheit von Tecentriq in Kombination mit Paclitaxel und Carboplatin, mit oder ohne Bevacizumab, bei Patienten mit metastasiertem nichtplattenepithelialem NSCLC ohne vorgängige Chemotherapiebehandlung bewertet. Es wurden insgesamt 1202 Patienten in die Studie aufgenommen und im Verhältnis 1:1:1 randomisiert, um eines der unten beschriebenen Behandlungsregimes zu erhalten. Die Randomisierung war nach Geschlecht, Vorhandensein von Lebermetastasen und PD-L1-Expression auf Tumorzellen (TC) und Tumor-infiltrierenden Immunzellen (IC) stratifiziert.
Die Patienten wurden in einen der folgenden drei Behandlungsarme randomisiert:
·Arm A: Tecentriq 1200 mg, Paclitaxel 175 mg/m² oder 200 mg/m² und Carboplatin AUC 6 mg/ml/min an Tag 1 jedes 21-Tage-Zyklus über maximal 4 oder 6 Zyklen.
·Arm B: Tecentriq 1200 mg, Bevacizumab 15 mg/kg, Paclitaxel 175 mg/m² oder 200 mg/m² und Carboplatin AUC 6 mg/ml/min an Tag 1 jedes 21-Tage-Zyklus für maximal 4 oder 6 Zyklen.
·Arm C: Bevacizumab 15 mg/kg, Paclitaxel 175 mg/m² oder 200 mg/m² und Carboplatin AUC 6 mg/ml/min an Tag 1 jedes 21-Tage-Zyklus für maximal 4 oder 6 Zyklen.
Carboplatin und Paclitaxel wurden bis zum Abschluss von 4 oder 6 Zyklen oder bis zum Eintreten einer Krankheitsprogression oder dem Auftreten inakzeptabler Toxizität gegeben, je nachdem, was zuerst der Fall ist. Die Paclitaxel-Anfangsdosis für Patienten asiatischer Ethnie betrug aufgrund des insgesamt höheren Grades an hämatologischen Toxizitäten bei Patienten aus asiatischen Ländern gegenüber solchen aus nicht-asiatischen Ländern 175 mg/m2.
Patienten, bei denen es nach Abschluss oder Absetzen einer platinbasierten Chemotherapie nicht zu einer Krankheitsprogression kam, erhielten:
·Arm A: Tecentriq 1200 mg intravenös an Tag 1 jedes 21-Tage-Zyklus bis zum Verlust des klinischen Nutzens nach Einschätzung des Prüfarztes.
·Arm B: Tecentriq 1200 mg und Bevacizumab 15 mg/kg intravenös an Tag 1 jedes 21-Tage-Zyklus. Die Gabe von Tecentriq wurde bis zum Verlust des klinischen Nutzens nach Einschätzung des Prüfarztes fortgesetzt, und die Gabe von Bevacizumab wurde bis zum Eintreten einer Krankheitsprogression oder dem Auftreten inakzeptabler Toxizität fortgesetzt.
·Arm C: Bevacizumab 15 mg/kg intravenös an Tag 1 jedes 21-Tage-Zyklus bis zum Eintreten einer Krankheitsprogression oder dem Auftreten inakzeptabler Toxizität.
Patienten waren von der Teilnahme ausgeschlossen bei Autoimmunerkrankung, Erhalt eines lebend-attenuierten Impfstoffes in den 28 Tagen vor der Randomisierung, Erhalt von systemischen Immunstimulanzien in den 4 Wochen vor der Randomisierung oder von systemischen Immunsuppressiva in den 2 Wochen vor der Randomisierung, aktiven oder unbehandelten ZNS-Metastasen, eindeutiger Tumorinfiltration in die grossen Thoraxgefässe oder eindeutiger Kavitation pulmonaler Läsionen in der Bildgebung. Tumorbeurteilungen wurden alle 6 Wochen in den ersten 48 Wochen nach Zyklus 1, Tag 1 und danach alle 9 Wochen durchgeführt.
Tumorproben wurden auf die PD-L1 Expression auf Tumorzellen (TC) und tumorinfiltrierenden Immunzellen (IC) untersucht und die Ergebnisse verwendet, um die PD-L1-Expressions-Subgruppen für die unten beschriebenen Analysen zu definieren.
Die demografischen Daten und die Krankheitsmerkmale bei Behandlungsbeginn im Studienkollektiv (ITT) waren zwischen beiden Behandlungsarmen ausgeglichen. In dieser Studie betrug das mediane Alter 63 Jahre (Spanne: 31 bis 90), und 60% der Patienten waren männlich. Die meisten Patienten waren weiss (82%). Ungefähr 10% der Patienten wiesen bekannte EGFR-Mutationen auf, 4% wiesen bekannte ALK-Translokationen auf, 14% hatten zu Studienbeginn Lebermetastasen und die meisten Patienten waren Raucher oder ehemalige Raucher (80%). Insgesamt wiesen 95% der Patienten ein nichtplattenepitheliales NSCLC mit Adenokarzinom-Histologie auf. Der ECOG-Performance-Status zum Studienbeginn betrug 0 (43%) oder 1 (57%).
Die demografischen Daten und die Krankheitsmerkmale bei Behandlungsbeginn bei Patienten mit einer PD-L1-Expression von ≥1% auf TC oder ≥1% auf IC (im Folgenden bezeichnet als PD-L1-Expression ≥1%), die keine EGFR-Mutationen oder ALK-Translokationen aufweisen, waren zwischen den Behandlungsarmen ausgeglichen. 51% der Tumoren der Patienten hatten eine PD-L1 Expression von ≥1% und 49% der Tumoren der Patienten hatten eine PD-L1 Expression von < 1% TC und < 1% IC.
Das ITT-WT-Kollektiv ist definiert als alle randomisierten Patienten mit Ausnahme jener mit genomischen EGFR- oder ALK-Tumoraberrationen.
Zum Zeitpunkt der finalen OS-Analyse für Tecentriq + Carboplatin + Paclitaxel (CP) gegenüber Bevacizumab + CP betrug die mediane Nachbeobachtungsdauer 39,3 Monate. Für das ITT-WT-Kollektiv mit Patienten, deren Tumoren eine PD-L1-Expression von ≥1% aufweisen, wurde in der Gruppe Tecentriq + CP im Vergleich zur Gruppe Bevacizumab + CP eine Verbesserung des OS nachgewiesen (nicht stratifizierte HR: 0,71; [95-%-KI: 0,55, 0,91] aufgrund des statistischen Plans nicht formell getestet), wobei das mediane OS 24,4 Monate betrug (8,4 Monate länger als das in der Gruppe Bevacizumab + CP beobachtete mediane OS von 16,0 Monaten). Dies entspricht einer relativen Reduktion des Sterberisikos um 29% in Verbindung mit Tecentriq + CP im Vergleich zu Bevacizumab + CP in dieser Population. Die Überlebensraten laut Landmark-Analyse betrugen im ITT-WT Kollektiv (PD-L1-Expression von ≥1%) nach einem Jahr in der Gruppe Tecentriq + CP 70,6% gegenüber 55,9% in der Gruppe Bevacizumab + CP und nach zwei Jahren 51,5% und 36,9%.
Zum Zeitpunkt der finalen PFS-Analyse betrug die nicht stratifizierte PFS Hazard Ratio 0,74 [95-%-KI: 0,58, 0,94] bei Patienten unter Tecentriq + CP im Vergleich zu Bevacizumab + CP (ITT-WT-Kollektivs mit einer PD-L1-Expression von ≥1%).
IMpower130
In einer offenen, randomisierten Phase-III-Studie, GO29537 (IMpower130), wurden die Wirksamkeit und Sicherheit von Tecentriq in Kombination mit Nab-Paclitaxel und Carboplatin bei chemotherapeutisch nicht vorbehandelten Patienten mit metastasiertem, nicht-plattenepithelialem NSCLC untersucht. Patienten, einschliesslich solcher mit genomischen EGFR- oder ALK-Tumoraberrationen, wurden nach der Aufnahme in die Studie im Verhältnis von 2:1 randomisiert, um eine der in Tabelle 5 beschriebenen Behandlungen zu erhalten. Die Randomisierung war nach Geschlecht, Vorhandensein von Lebermetastasen und PD-L1-Expression auf TC und IC stratifiziert. Patienten, die das Behandlungsregime B erhielten, hatten die Möglichkeit, nach Fortschreiten der Erkrankung auf eine Tecentriq-Monotherapie umgestellt zu werden.
Tabelle 5: Intravenöse Behandlungsregimes in der IMpower130
|
Behandlungsregime
|
Induktion
(vier oder sechs 21-Tage-Zyklen)
|
Erhaltung
(21-Tage-Zyklen)
| |
A
|
Tecentriq (1200 mg) a + Nab-Paclitaxel (100 mg/m2) b, c + Carboplatin (AUC 6) c
|
Tecentriq (1200 mg) a
| |
B
|
Nab-Paclitaxel (100 mg/m2) b + Carboplatin (AUC 6) c
|
Beste unterstützende Versorgung oder Pemetrexed
|
a Die Gabe von Tecentriq wird bis zum Verlust des klinischen Nutzens per Beurteilung durch den Prüfarzt fortgesetzt.
b Nab-Paclitaxel wird an den Tagen 1, 8 und 15 jedes Zyklus gegeben.
c Nab-Paclitaxel und Carboplatin werden gegeben, bis 4-6 Zyklen abgeschlossen sind, ein Fortschreiten der Krankheit oder inakzeptable Toxizität eintritt, je nachdem, was als Erstes der Fall ist.
Patienten mit einer Autoimmunerkrankung in der Vorgeschichte und Patienten mit Immunisierung mit einem lebend-attenuierten Impfstoff innerhalb von 28 Tagen vor der Randomisierung, Erhalt von Immunstimulanzien innerhalb von 4 Wochen oder systemischen Immunsuppressiva innerhalb von 2 Wochen vor der Randomisierung sowie solche mit aktiven oder unbehandelten ZNS-Metastasen waren von der Teilnahme ausgeschlossen. Die Tumorbeurteilungen wurden in den ersten 48 Wochen nach Zyklus 1 alle 6 Wochen und danach alle 9 Wochen durchgeführt.
Die demografischen Merkmale und Grunderkrankungen des Studienkollektivs (n=723) waren zwischen den Behandlungsarmen gut ausgewogen. Das Durchschnittsalter betrug 64 Jahre (Spanne: 18 bis 86 Jahre). Die Mehrheit der Patienten war männlich (57%) und weiss (90%). Zu Beginn der Studie hatten 14,8% der Patienten Lebermetastasen, und die meisten Patienten waren aktuell oder vorgängig Raucher (88%). Die meisten Patienten hatten einen ECOG-Leistungsstatus von 1 (58,7%).
Die primäre Analyse wurde mit allen Patienten durchgeführt, ausser bei solchen mit genomischen EGFR- oder ALK-Tumoraberrationen (n=685), definiert als ITT-WT-Kollektiv. Die beiden primären Wirksamkeitsendpunkte waren das OS und das vom Prüfarzt beurteilte PFS. Nachstehend sind die wichtigsten Ergebnisse dieser Studie bei einer medianen Nachbeobachtung des Überlebens über 18,6 Monate zusammengefasst.
Die Behandlung mit Atezo + Carboplatin + Nab-Paclitaxel (Atezo + CnP) war mit einer statistisch signifikanten Verbesserung des OS gegenüber Carboplatin und Nab-Paclitaxel (CnP) verbunden. Bei den Patienten im ITT-WT-Kollektiv war das mediane OS 4,7 Monate länger als im Behandlungsarm mit Atezo und CnP: 18,6 Monate (95%-KI: 15,8; 21,2) im Behandlungsarm mit Atezo + CnP versus 13,9 Monate (95%-KI: 12,0; 18,7) im CnP-Arm (stratifizierter p-Wert = 0,030). Die stratifizierte HR betrug 0,79 (95%-KI: 0,64; 0,98), was einer relativen Reduzierung des mit Atezo + CnP verbundenen Sterberisikos um 21% gegenüber CnP entspricht.
Die Behandlung mit Atezo + CnP war mit einer statistisch signifikanten Verbesserung des vom Prüfarzt beurteilten PFS (nach RECIST v1.1) gegenüber CnP verbunden. Bei den Patienten im ITT-WT Kollektiv war das mediane PFS zahlenmässig länger als im Behandlungsarm mit Atezo + CnP: 7,0 Monate (95%-KI: 6,3; 7,3) im Behandlungsarm mit Atezo + CnP versus 5,5 Monate (95%-KI: 4,4; 5,9) im CnP-Arm (stratifizierter p-Wert < 0,0001). Die stratifizierte HR betrug 0,64 (95%-KI: 0,54; 0,76).
Der Anteil der ITT-WT Patienten mit einem vom Prüfarzt beurteilten, bestätigten objektiven Ansprechen nach RECIST v1.1 war im Behandlungsarm mit Atezo + CnP um 17,6% höher als im CnP-Arm (49,3% bzw. 31,7%).
In allen PD-L1-Subgruppen, unabhängig von der Expression, wurde mit Ausnahme der Patienten mit Lebermetastasen ein Nutzen in Bezug auf OS und PFS erzielt. Wie eine Subgruppenanalyse einer begrenzten Anzahl von Patienten (n=101) mit Lebermetastasen ergab, scheint mit Atezolizumab, nab-Paclitaxel und Carboplatin in diesem Teilkollektiv keine Verbesserung der Behandlungswirkung im Vergleich zu nab-Paclitaxel und Carboplatin feststellbar zu sein (HR: 0,96; 95%-KI: 0,61; 1,51 für PFS und HR: 1,21; 95%-KI: 0,73; 2,00 für OS).
NSCLC im Frühstadium
IMpower010
Eine offene, multizentrische, randomisierte Phase-III-Studie, GO29527 (IMpower010), wurde zur Beurteilung der Wirksamkeit und Sicherheit von Tecentriq zur adjuvanten Behandlung von Patienten mit NSCLC im Stadium IB (Tumoren ≥4 cm) bis IIIA (gemäss UICC/AJCC-Klassifikationssystem, 7. Ausgabe) durchgeführt. Insgesamt 1280 der in die Studie aufgenommenen Patienten, bei denen eine vollständige Tumorresektion vorgenommen worden war, kamen dafür infrage, bis zu 4 Zyklen einer cisplatinbasierten Chemotherapie zu erhalten. In Tabelle 6 sind die Schemata der cisplatinbasierten Chemotherapien beschrieben.
Tabelle 6: Behandlungsschemata der intravenösen Chemotherapie in der Studie IMpower010
|
Adjuvante cisplatin-basierte Chemotherapie
Cisplatin 75 mg/m2 i.v. an Tag 1 jedes 21-Tage-Zyklus mit einem dieser Behandlungsschemata
|
Vinorelbin 30 mg/m2 i.v., Tag 1 und 8
| |
Docetaxel 75 mg/m2 i.v., Tag 1
| |
Gemcitabin 1250 mg/m2 i.v., Tag 1 und 8
| |
Pemetrexed 500 mg/m2 i.v., Tag 1
|
Nach Abschluss der cisplatinbasierten Chemotherapie (bis zu 4 Zyklen) wurden insgesamt 1005 Patienten im Verhältnis 1:1 randomisiert und erhielten Tecentriq (Arm A) oder die bestmögliche unterstützende Behandlung (Arm B). Tecentriq wurde in einer Fixdosis von 1200 mg als i.v.-Infusion alle 3 Wochen über 16 Zyklen verabreicht, ausser bei Auftreten eines Krankheitsrezidivs oder inakzeptabler Toxizität. Die Randomisierung erfolgte stratifiziert nach Geschlecht, Krankheitsstadium, Histologie und PD-L1-Expression (beurteilt mit dem VENTANA PD-L1 [SP142] Assay).
Ausgeschlossen aus der Studie wurden Patienten mit einer Autoimmunerkrankung in der Anamnese, Verabreichung eines attenuierten Lebendimpfstoffs innert 28 Tagen vor der Randomisierung oder Verabreichung systemischer immunstimulierender Wirkstoffe innert 4 Wochen bzw. systemischer immunsuppressiver Arzneimittel innert 2 Wochen vor der Randomisierung. Die Tumorbeurteilungen erfolgten zu Beginn der Randomisierungsphase und im ersten Jahr nach Zyklus 1, Tag 1 alle 4 Monate, danach alle 6 Monate bis Jahr 5 und danach jährlich.
Die demografischen Daten und die Krankheitsmerkmale waren zwischen den Behandlungsarmen gut ausgeglichen. Das mediane Alter betrug 62 Jahre (Bereich: 26 bis 84 Jahre), 67% der Patienten waren männlich. Die Mehrzahl der Patienten war weisser Hautfarbe (73%), 24% waren Asiaten. Die meisten Patienten waren Raucher oder ehemalige Raucher (78%), der ECOG-Performance-Status zu Studienbeginn betrug 0 (55%) oder 1 (44%). Die Krankheit wurde bei insgesamt 12% der Patienten als Stadium IB, bei 47% als Stadium II und bei 41% als Stadium IIIA eingestuft. Beurteilt mit dem VENTANA PD-L1 (SP263) Assay, betrug der Anteil der Patienten mit Tumoren mit einer PD-L1-Expression von ≥1% auf TC 55% und der Anteil der Patienten mit Tumoren mit einer PD-L1-Expression von ≥50% auf TC 26%.
Der primäre Wirksamkeitsendpunkt war das krankheitsfreie Überleben (DFS) gemäss Beurteilung durch den Prüfarzt. Das DFS war definiert als die Zeit von der Randomisierung bis zum Auftreten eines der folgenden Ereignisse: erstes dokumentiertes Krankheitsrezidiv, neues primäres NSCLC oder Tod jeglicher Ursache, je nachdem, was zuerst auftrat. Das primäre Wirksamkeitsziel lautete, das DFS in der Patientenpopulation mit einer PD-L1-Expression von ≥1% auf TC (laut SP263) und einer Krankheit im Stadium II bis IIIA zu bewerten. Wichtige sekundäre Wirksamkeitsendpunkte waren die Bewertung des DFS in der Patientenpopulation mit einer PD-L1-Expression ≥50% auf TC (laut SP263) und einer Krankheit im Stadium II bis IIIA sowie das Gesamtüberleben (OS) in der ITT-Population.
Bei Patienten mit einer PD-L1-Expression ≥50% auf TC und einer Erkrankung im Stadium II bis IIIA (n=229) wurde eine klinisch relevante Verbesserung des DFS mit einer unstratifizierten HR von 0,48 (95%-KI: 0,32; 0,72) gezeigt. Das mediane DFS wurde bei Patienten im Tecentriq-Arm nicht erreicht (95%-KI: NE; NE) und betrug bei Patienten im Arm mit der bestmöglichen unterstützenden Behandlung 41,1 Monate (95%-KI: 29,7; NE). Die OS-Daten waren zum Zeitpunkt der Abschlussanalyse des DFS- und der zweiten Zwischenanalyse des OS noch unvollständig, wobei in der Population mit einer PD-L1-Expression ≥50% auf TC und einer Erkrankung im Stadium II bis IIIA im Tecentriq Arm eine Mortalität von etwa 20,9% und in dem Arm mit der bestmöglichen unterstützenden Behandlung eine Mortalität von 37,7% gemeldet wurde. Eine explorative Analyse des OS wies auf eine Tendenz zugunsten von Tecentriq gegenüber der bestmöglichen unterstützenden Behandlung in dieser Patientenpopulation hin (unstratifizierte HR=0,47; [95%-KI: 0,28; 0,77]).
Chemotherapie-vorbehandeltes NSCLC
OAK
Eine offene, multizentrische, internationale, randomisierte Phase III-Studie, GO28915 (OAK), wurde zur Beurteilung der Wirksamkeit und Sicherheit von Tecentriq im Vergleich zu Docetaxel bei Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC durchgeführt, deren Erkrankung während oder nach einer platinhaltigen Chemotherapie fortgeschritten ist. Insgesamt wurden 1'225 Patienten in die Studie aufgenommen, von denen die ersten 850 randomisierten Patienten in die Primäranalyse zur Wirksamkeit eingeschlossen wurden; die sekundäre Wirksamkeitsanalyse beruhte auf 1'225 Patienten. Geeignete Patienten wurden hinsichtlich des PD-L1-Expressionsstatus der tumorinfiltrierenden ICs, der Anzahl der vorausgegangenen Chemotherapie-Regimes und des histologischen Typs stratifiziert. Die Patienten erhielten randomisiert (1:1) entweder Tecentriq oder Docetaxel. Patienten mit anamnestisch bekannter Autoimmunerkrankung, aktiven oder kortikosteroidabhängigen Hirnmetastasen, Anwendung einer attenuierten Lebendvakzine in den 28 Tagen vor der Aufnahme in die Studie sowie Anwendung von systemischen immunstimulierenden Wirkstoffen in den 4 Wochen oder von systemischen immunsuppressiven Arzneimitteln in den 2 Wochen vor der Aufnahme in die Studie wurden von der Teilnahme an der Studie ausgeschlossen. Tumorbeurteilungen erfolgten in den ersten 36 Wochen alle 6 Wochen, danach alle 9 Wochen. In Tumorgewebeproben wurde prospektiv die PD-L1-Expression auf TC und IC beurteilt; die Ergebnisse wurden zur Definition der Untergruppen mit PD-L1-Expression für die unten beschriebenen Analysen herangezogen.
Die demografischen Daten und die Krankheitsmerkmale zu Behandlungsbeginn waren bei den ersten 850 randomisierten Patienten, die in die primäre Wirksamkeitsanalyse eingeschlossen wurden, in den beiden Behandlungsarmen gut ausgewogen. Das mediane Alter betrug 64 Jahre (Bereich: 33 bis 85); 61% der Patienten waren männlich. Die Mehrzahl der Patienten waren Weisse (70%). Fast drei Viertel der Patienten (74%) hatten eine nicht-plattenepitheliale Histologie, 10% hatten eine bekannte EGFR-Mutation, 0,2% hatten bekannte ALK-Translokation, bei 10% lagen ZNS-Metastasen bei Behandlungsbeginn vor, und die meisten Patienten waren Raucher oder ehemalige Raucher (82%). Der ECOG-Performance-Status bei Behandlungsbeginn betrug 0 (37%) oder 1 (63%). 75% der Patienten hatten zuvor nur ein platinbasiertes Therapieregime erhalten. Die bei der sekundären Wirksamkeitsanalyse beobachteten demografischen Daten und Krankheitsmerkmale stehen im Einklang mit den Beobachtungen bei der primären Wirksamkeitsanalyse.
Tecentriq wurde in einer Fixdosis von 1200 mg als i.v. Infusion alle 3 Wochen verabreicht. Dosisreduktionen waren nicht zulässig. Die Patienten wurden bis zum Verlust des klinischen Nutzens gemäss Beurteilung durch den Prüfarzt behandelt. Docetaxel wurde in der Dosierung 75 mg/m2 als i.v. Infusion an Tag 1 jedes 21-tägigen Zyklus bis zum Fortschreiten der Erkrankung verabreicht. Bezogen auf alle behandelten Patienten betrug die mediane Behandlungsdauer bei der primären Wirksamkeitsanalyse 2,1 Monate im Docetaxel-Arm bzw. 3,4 Monate im Tecentriq-Arm.
Primärer Wirksamkeitsendpunkt war das Gesamtüberleben (overall survival, OS). Die wichtigsten Ergebnisse dieser Studie, die bei der primären Wirksamkeitsanalyse für die ersten 850 randomisierten Patienten mit einer medianen Nachbeobachtung hinsichtlich des Überlebens von 21 Monaten gemeldet wurden, werden im Folgenden zusammengefasst (Cutoff-Datum: 07.07.2016). Die Behandlung mit Tecentriq war im Vergleich zu Docetaxel mit einer klinisch bedeutsamen und statistisch signifikanten Verlängerung des OS verbunden. Das mediane OS der ersten 850 randomisierten Patienten der ITT (intention-to-treat)-Population war im Tecentriq-Arm 4,2 Monate länger: Es betrug 9,6 Monate (95%-KI: 8,6; 11,2) im Docetaxel-Arm gegenüber 13,8 Monaten (95%-KI: 11,8; 15,7) im Tecentriq-Arm (stratifizierter p-Wert = 0,0003). Die Hazard Ratio (HR) betrug 0,73 (95%-KI: 0,62; 0,87; dies entspricht einer relativen Reduktion des Mortalitätsrisikos unter Tecentriq von 27% im Vergleich zu Docetaxel (stratifizierte Analyse). Die Kaplan–Meier-Kurven zeigten ab etwa 3 Monaten eine klare Trennung zugunsten des Tecentriq-Armes, welche in der Folge bestehen blieb In gleicher Weise war auch in der Patientenuntergruppe mit einer PD-L1-Expression ≥1% auf TC oder IC die Behandlung mit Tecentriq im Vergleich zu Docetaxel mit einer klinisch bedeutsamen und statistisch signifikanten Verlängerung des OS verbunden. Das mediane OS der Patienten war im Tecentriq-Arm 5,4 Monate länger: Es betrug 10,3 Monate (95%-KI: 8,8; 12,0) im Docetaxel-Arm gegenüber 15,7 Monaten (95%-KI: 12,6; 18,0) im Tecentriq-Arm (stratifizierter p-Wert = 0,0102). Die HR betrug 0,74 (95%-KI: 0,58; 0,93); dies entspricht einer relativen Reduktion des Mortalitätsrisikos unter Tecentriq von 26% im Vergleich zu Docetaxel (stratifizierte Analyse).
Das OS in den Patientenuntergruppen mit einer PD-L1-Expression ≥50% auf TC oder ≥10% auf IC, einer PD-L1-Expression ≥5% auf TC oder IC bzw. einer PD-L1-Expression < 1% auf TC und IC zeigte, dass die mediane Dauer des OS in allen untersuchten PD-L1-Untergruppen länger war. Die Werte waren wie folgt: PD-L1-Expression ≥50% auf TC oder ≥10% auf IC: medianes OS von 20,5 Monaten (95%-KI: 17,5; NA) im Tecentriq-Arm gegenüber 8,9 Monaten (95%-KI: 5,6; 11,6) im Docetaxel-Arm (HR=0,41, 95%-KI: 0,27; 0,64); PD-L1-Expression ≥5% auf TC oder IC: medianes OS von 16,3 Monaten (95%-KI: 13,3; 20,1) im Tecentriq-Arm gegenüber 10,8 Monaten (95%-KI: 8,8; 12,7) im Docetaxel-Arm (HR=0,67, 95%-KI: 0,49; 0,90); PD-L1-Expression < 1% auf TC und IC: medianes OS von 12,6 Monaten (95%-KI: 9,6; 15,2) im Tecentriq-Arm gegenüber 8,9 Monaten (95%-KI: 7,7; 11,5) im Docetaxel-Arm (HR=0,75, 95%-KI: 0,59; 0,96).
Insgesamt zeigten die Ergebnisse für das Gesamtüberleben in der primären Wirksamkeitsanalyse (850 Patienten) in der ITT-Gruppe und den PD-L1-Untergruppen unter Tecentriq einen OS-Nutzen in allen Untergruppen, einschliesslich derjenigen mit einer PD-L1-Expression < 1% auf TC und IC.
Der Anteil der ITT-Patienten mit einem durch den Prüfarzt beurteilten bestätigten Ansprechen gemäss RECIST V1.1 war zwischen beiden Armen vergleichbar: 13,4% (95%-KI: 10,32; 17,02) im Docetaxel-Arm und 13,6% (95%-KI: 10,53; 17,28) im Tecentriq-Arm. Die Behandlung mit Tecentriq war mit dem Erreichen eines dauerhaften Ansprechens verbunden. Unter den Respondern war die mediane DOR im Tecentriq-Arm signifikant länger (16,3 Monate) als im Docetaxel-Arm (6,2 Monate). In der ITT-Gruppe betrug das mediane PFS 2,8 Monate (95%-KI: 2,6; 3,0) im Tecentriq-Arm bzw. 4,0 Monate (95%-KI: 3,3; 4,2) im Docetaxel-Arm bei einer HR von 0,95 (95%-KI 0,82; 1,10).
Eine Verbesserung des OS unter Tecentriq im Vergleich zu Docetaxel wurde sowohl bei den Patienten mit nicht-plattenepithelialem NSCLC (Hazard-Ratio [HR] 0,73, 95%-KI: 0,60; 0,89; medianes OS von 15,6 Monaten unter Tecentriq im Vergleich zu 11,2 Monaten unter Docetaxel) als auch bei den Patienten mit plattenepithelialem (Plattenepithel) NSCLC (HR: 0,73, 95%-KI: 0,54; 0,98; medianes OS von 8,9 Monaten unter Tecentriq im Vergleich zu 7,7 Monaten unter Docetaxel) beobachtet.
Bei der sekundären Wirksamkeitsanalyse (Cutoff-Datum: 23. Januar 2017) mit einer medianen Nachbeobachtungszeit von 26 Monaten war die Behandlung mit Tecentriq weiterhin mit einer statistisch signifikanten Verlängerung des OS im Vergleich zu Docetaxel assoziiert. Das mediane OS aller randomisierten 1'225 Patienten war im Tecentriq-Arm 3,5 Monate länger: 13,3 Monate (95%-KI: 11,3; 14,9) im Tecentriq-Arm bzw. 9,8 Monate (95%-KI: 8,8; 11,3) im Docetaxel-Arm. Die stratifizierte HR betrug 0,80 (95%-KI: 0,70; 0,92).
POPLAR
Eine multizentrische, internationale, randomisierte, offene, kontrollierte Phase II-Studie, GO28753 (POPLAR), wurde bei Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC durchgeführt bei welchen es während oder nach einer platinhaltigen Chemotherapie zu einer Progression gekommen war. Primäre Wirksamkeitsvariable war das Gesamtüberleben. Insgesamt 287 Patienten erhielten randomisiert im Verhältnis 1:1 entweder Tecentriq oder Docetaxel. Die Randomisierung wurde hinsichtlich der PD-L1-Expression, der Anzahl der vorausgegangenen Chemotherapie-Regimes und des histologischen Typs stratifiziert. Eine aktualisierte Analyse mit insgesamt 200 verzeichneten Todesfällen und einer medianen Nachbeobachtung hinsichtlich des Überlebens von 22 Monaten ergab ein medianes OS von 12,6 Monaten bei mit Tecentriq behandelten Patienten im Vergleich zu 9,7 Monaten bei mit Docetaxel behandelten Patienten (HR: 0,69, 95%-KI: 0,52; 0,92). Die ORR betrug 15,3% unter Tecentriq im Vergleich zu 14,7% unter Docetaxel; die mediane DOR betrug 18,6 Monate unter Tecentriq im Vergleich zu 7,2 Monaten unter Docetaxel.
Erstlinien-Therapie beim kleinzelligen Lungenkarzinom im ausgedehnten Stadium (1L ES – SCLC)
IMpower133
Die randomisierte, multizentrische, doppelblinde, Placebo-kontrollierte Phase I/III-Studie GO30081 (IMpower133) wurde durchgeführt, um die Wirksamkeit und Sicherheit von Tecentriq in Kombination mit Carboplatin und Etoposid bei Patienten mit kleinzelligem Lungenkarzinom im ausgedehnten Stadium (ES-SCLC) ohne bisherige Chemotherapie und mit einem ECOG-Performance-Status 0 oder 1 zu beurteilen. Es wurden insgesamt 403 Patienten randomisiert (1:1), um eines der Behandlungsregimes zu erhalten: Arm A: Tecentriq 1200 mg + Carboplatin AUC 5 + Etoposid 100 mg/m2 durch intravenöse Infusion alle 3 Wochen über vier Induktionszyklen, gefolgt von einer Erhaltungstherapie mit Tecentriq 1200 mg alle 3 Wochen; oder Arm B: Placebo + Carboplatin AUC 5 + Etoposid 100 mg/m2 durch intravenöse Infusion alle 3 Wochen über vier Induktionszyklen, gefolgt von einer Erhaltungstherapie mit einem Placebo alle 3 Wochen. Tecentriq und Carboplatin wurden an Tag 1, und Etoposid wurde an den Tagen 1, 2 und 3 jedes Induktionszyklus verabreicht. Carboplatin und Etoposid wurden bis zum Abschluss von vier Zyklen oder bis zum Fortschreiten der Erkrankung oder bis zum Auftreten inakzeptabler Toxizität verabreicht, je nachdem, was zuerst auftrat. Tecentriq wurde verabreicht, bis nach Einschätzung des Prüfarztes kein klinischer Nutzen mehr vorlag. Die Randomisierung erfolgte stratifiziert nach Geschlecht, ECOG-Performance-Status (0 oder 1) und Vorhandensein von Hirnmetastasen.
Ausgeschlossen aus dieser Studie wurden Patienten mit aktiven oder unbehandelten Metastasen im ZNS, Autoimmunerkrankung in der Anamnese, Verabreichung von attenuiertem Lebendimpfstoff innerhalb von 4 Wochen vor der Randomisierung oder Verabreichung von systemischen immunsuppressiven Medikamenten innerhalb von 1 Woche vor der Randomisierung. Die Tumor-Beurteilungen erfolgten alle 6 Wochen in den ersten 48 Wochen nach Zyklus 1 Tag 1 und danach alle 9 Wochen. Bei Patienten, die nach Krankheitsprogression weiter behandelt wurden, erfolgte die Tumorbeurteilung alle 6 Wochen bis zum Abbruch der Behandlung.
Die demografischen Daten und die Krankheitsmerkmale der Populationen für die Primäranalyse waren zwischen den Behandlungsarmen gut ausgeglichen. Es wurden insgesamt 403 Patienten in die Studie randomisiert: 201 Patienten in den Arm A mit Tecentriq, Carboplatin und Etoposid und 202 Patienten in Arm B mit Placebo, Carboplatin und Etoposid. Das mediane Alter betrug 64 Jahre (Bereich: 26 bis 90 Jahre). Die Mehrzahl der Patienten (65%) war männlich und weisser Hautfarbe (80%), 9% hatten Hirnmetastasen und die meisten Patienten (97%) waren derzeit oder früher Raucher. Der Ausgangswert (Basislinie) des ECOG Performance-Status war 0 (35%) oder 1 (65%).
Die beiden primären Endpunkte waren das Gesamtüberleben (Overall Survival, OS) und das vom Prüfarzt beurteilte progressionsfreie Überleben (Progression-Free Survival, PFS). Nachstehend sind die wichtigsten Ergebnisse dieser Studie bei einer medianen Nachbeobachtung der Restlebensdauer über 13,9 Monate zusammengefasst.
Die Behandlung mit Tecentriq + Carboplatin und Etoposid (CE) war mit einer statistisch signifikanten Verbesserung des OS im Vergleich zur Anwendung von Placebo + CE verbunden. Von den Patienten im ITT-Kollektiv war das mediane OS im Tecentriq + CE-Behandlungsarm 2 Monate länger, d.h. 12,3 Monate (95-%-KI: 10,8; 15,9), im Vergleich zu 10,3 Monaten (95-%-KI: 9,3; 11,3) im Placebo + CE-Behandlungsarm (stratifizierter p-Wert = 0,0069). Die stratifizierte HR betrug 0,70 (95-%-KI: 0,54; 0,91), was einer relativen Reduzierung des Sterberisikos um 30% bei Anwendung von Tecentriq + CE im Vergleich zu Placebo + CE entspricht. Nach 12 Monaten waren im Tecentriq + CE-Behandlungsarm noch 51,7% und im Placebo + CE-Behandlungsarm noch 38,2% der Patienten am Leben. Bei Patienten < 65 Jahren betrug das mediane OS im Tecentriq + CE-Arm 12,1 Monate gegenüber 11,5 Monaten im Placebo + CE-Arm (nicht-stratifizierte HR: 0,92; 95-%-KI: 0,64; 1,32). Bei Patienten ≥65 Jahren betrug das mediane OS im Tecentriq + CE-Arm 12,5 Monate gegenüber 9,6 Monaten im Placebo + CE-Arm (nicht-stratifizierte HR: 0,53; 95-%-KI: 0,36; 0,77).
Daten aus den explorativen Analysen der PD-L1-Untergruppen auf der Grundlage einer begrenzten Anzahl Proben von Patienten in der Studie (34% des «intention-to-treat» (ITT)-Kollektivs) ergaben für PD-L1-negative Patienten (<1% TC und IC) eine OS HR (Gesamtüberleben Hazard Ratio) von 0,51 (95%-KI: 0,30; 0,89) mit einem mittleren Gesamtüberleben im Tecentriq + CE-Studienarm (10,2 Monate) im Vergleich zum PBO + CE-Studienfarm (8,3 Monate). Für PD-L1-positive Patienten (≥1% TC oder IC) betrug die OS HR 0,87 (95%-KI: 0,51; 1,49) mit einem mittleren Gesamtüberleben im Tecentriq + CE-Studienarm (9,7 Monate) im Vergleich zum PBO + CE-Studienarm (10,6 Monate). Diese Daten sind aber zu begrenzt, um eine Schlussfolgerung zu ermöglichen. Die Inzidenz von während der Behandlung auftretenden Anti-Drug-Antikörpern (ADAs) gegen Atezolizumab im Tecentriq+CE-Arm betrug 18,6% (35/188 Patienten). In einer unadjustierten Analyse betrug das mediane OS in der ADA-negativen 12,6 Monate und in der ADA-positiven Gruppe 10,9 Monate. In der Placebo+CE-Gruppe betrug das mediane OS 10,3 Monate. Aufgrund der geringen Fallzahl in der ADA-positiven Gruppe sind keine definitiven Schlussfolgerungen zum möglichen Einfluss von ADAs auf die Wirksamkeit möglich.
Die Behandlung mit Tecentriq + CE war mit einer statistisch signifikanten Verbesserung des vom Prüfarzt beurteilten PFS (per RECIST v1.1) im Vergleich zur Anwendung von Placebo + CE verbunden. Bei den Patienten im ITT-Kollektiv war die mediane Dauer des PFS im Tecentriq + CE-Behandlungsarm 5,2 Monate (95-%-KI: 4,4; 5,6) gegenüber 4,3 Monaten (95-%-KI: 4,2; 4,5) im Placebo + CE-Behandlungsarm (stratifizierter p-Wert = 0,0170). Die stratifizierte HR betrug 0,77 (95-%-KI: 0,62; 0,96). Nach 6 Monaten waren im Tecentriq + CE-Behandlungsarm 30,9% und im Placebo + CE-Behandlungsarm 22,4% der Patienten frei von PFS-relevanten Ereignissen (Progress oder Tod). Nach 12 Monaten waren im Tecentriq + CE-Behandlungsarm 12,6% und im Placebo + CE-Behandlungsarm 5,4% der Patienten PFS-Ereignis-frei.
Die Anteile der Patienten mit bestätigtem objektivem Ansprechen per Prüfarztbeurteilung nach RECIST v1.1 im ITT-Kollektiv betrugen: 60,2% im Tecentriq + CE-Behandlungsarm gegenüber 64,4% im Placebo + CE-Behandlungsarm.
IMforte (GO43104)
Die Wirksamkeit von Tecentriq + Lurbinectedin als Erhaltungstherapie wurde in der Studie IMforte (GO43104), einer randomisierten multizentrischen offenen Studie, bei 483 Patienten mit 1L ES-SCLC untersucht. Die Patienten kamen für die Randomisierung infrage, wenn die Krankheit nach Abschluss von 4 Zyklen Induktionsbehandlung mit Tecentriq, Carboplatin und Etoposid nicht fortgeschritten war und sie einen ECOG-Performancestatus von 0 oder 1 aufwiesen. Die Patienten wurden im Verhältnis 1:1 randomisiert, um eine Erhaltungsbehandlung mit Tecentriq + Lurbinectedin oder mit Tecentriq zu erhalten. Rund 84% der Patienten im Behandlungsarm mit Tecentriq + Lurbinectedin erhielten prophylaktisch G-CSF.
Von der Studie ausgeschlossen waren Patienten mit ECOG Performance-Status >1, ZNS-Metastasen (in der Vorgeschichte oder aktuell), Patienten mit einer Autoimmunerkrankung in der Anamnese und Patienten, die innerhalb von 1 Woche vor der Aufnahme in die Studie systemische Immunsuppressiva erhalten haben, Patienten mit einer leptomeningealen Erkrankung, Patienten mit unzureichender hämatologischer und Endorganfunktion, Patienten mit einer geplanten konsolidierenden Brustbestrahlung, Patienten mit unkontrolliertem Pleura- oder Perikarderguss / Aszites, Patienten mit Läsionen, die eine palliative Strahlentherapie erfordern und Patienten mit anhaltenden klinisch signifikanten Toxizitäten aus der Induktionstherapie mit Grad >1. Die Randomisierung erfolgte stratifiziert nach ECOG-Performancestatus (0 vs. 1), LDH-Wert (≤ ULN vs. > ULN), Vorliegen von Lebermetastasen und vorhergehender prophylaktischer Schädelbestrahlung (ja vs. nein).
Tabelle 7: Schema der intravenösen Behandlung in der Studie IMforte
|
Behandlungsschema
|
Erhaltung
(21-Tage-Zyklen)
| |
A
|
Tecentriq als intravenöse Infusion (1200 mg) + Lurbinectedin 3,2 mg/m2 als intravenöse Infusion an Tag 1 jedes 21-Tage-Zyklus
| |
B
|
Tecentriq als intravenöse Infusion (1200 mg) an Tag 1 jedes 21-Tage-Zyklus
|
Die primären Endpunkte der Studie waren das OS und das PFS nach Bewertung durch eine unabhängige Prüfeinrichtung (Independent Review Facility, IRF) per RECIST v1.1 im randomisierten Kollektiv (siehe Tabelle 8).
Es wurden insgesamt 483 Patienten randomisiert, davon 242 in den Behandlungsarm mit Tecentriq und Lurbinectedin und 241 in den Behandlungsarm mit Tecentriq. Das mediane Alter betrug 66 Jahre (Bereich: 35 bis 85 Jahre). Die Mehrheit der Patienten waren weisser Hautfarbe (81,6 %); 12,8 % waren asiatisch, 6,6 % hispanischer oder lateinamerikanischer Abstammung und < 1 % Schwarze oder Afroamerikaner. Die meisten Patienten waren männlich (62,5 %) und 97,5 % waren Raucher oder ehemalige Raucher. Der ECOG-Performancestatus zu Studienbeginn war 0 (42,9 %) oder 1 (57,1 %).
Zum Zeitpunkt der primären Analyse betrug die mediane Nachbeobachtung 15 Monate. Unter Behandlung mit Tecentriq in Kombination mit Lurbinectedin ergab sich eine statistisch signifikante Verbesserung des OS (stratifizierte HR: 0,73 [95%-KI: 0,57; 0,95]; p = 0,0174, medianes OS: 13,2 Monate vs. 10,6 Monate) und des PFS per IRF-Bewertung (stratifizierte HR: 0,54 [95%-KI: 0,43; 0,67]; p ≤0,0001, medianes PFS: 5,4 Monate vs. 2,1 Monate) verglichen mit Tecentriq als Monotherapie. Die wichtigsten Studienergebnisse zum Zeitpunkt der Primäranalyse sind in Tabelle 8 zusammengefasst.
Tabelle 8: Zusammenfassung der Wirksamkeitsdaten aus IMforte (Primäranalyse)
|
Wichtige Wirksamkeitsendpunkte
|
Arm A
(Tecentriq mit Lurbinectedin)
|
Arm B
(Tecentriq)
| |
Primäre Endpunkte
|
N = 242
|
N = 241
| |
OS-Analyse1, 2
|
|
| |
Anzahl der Todesfälle (%)
|
113 (46,7)
|
136 (56,4)
| |
Mediane Dauer des OS (Monate)
|
13,2
|
10,6
| |
95%-KI
|
(11,9; 16,4)
|
(9,5; 12,2)
| |
Stratifizierte Hazard-Ratio‡ (95%-KI)
|
0,73 (0,57; 0,95)
| |
p-Wert3, 4
|
0,0174
| |
OS-Rate nach 12 Monaten (%)
|
56,3
|
44,1
| |
PFS per IRF-Bewertung (RECIST v1.1)2
|
|
| |
Anzahl Ereignisse (%)
|
174 (71,9)
|
202 (83,8)
| |
Mediane Dauer des PFS (Monate)
|
5,4
|
2,1
| |
95%-KI
|
(4.2, 5,8)
|
(1,6, 2,7)
| |
Stratifizierte Hazard-Ratio‡ (95%-KI)
|
0,54 (0,43; 0,67)
| |
p-Wert3, 5
|
< 0,0001
| |
PFS-Rate nach 6 Monaten (%)
|
41,2
|
18,7
| |
PFS-Rate nach 12 Monaten (%)
|
20,5
|
12
|
1 Auf der Grundlage der OS-Zwischenanalyse.
2 Gemessen ab dem Zeitpunkt der Randomisierung.
3 Auf der Grundlage des zweiseitigen stratifizierten Log-rank-Tests.
4 Im Vergleich zum zugewiesenen alpha α von 0,0313 (zweiseitig) für diese OS-Zwischenanalyse.
5 Im Vergleich zum zugewiesenen alpha α von 0,001 (zweiseitig) für diese PFS-Abschlussanalyse.
‡ Stratifiziert nach ECOG-Performancestatus, LDH-Wert, Vorliegen von Lebermetastasen und vorhergehender prophylaktischer Schädelbestrahlung.
PFS = progressionsfreies Überleben (progression-free survival); RECIST = Response Evaluation Criteria in Solid Tumors v1.1. (Beurteilungskriterien des Ansprechens bei soliden Tumoren, Version 1.1); KI = Konfidenzintervall, OS = Gesamtüberleben (overall survival).
In der deskriptiven OS-Endauswertung, ca. 6,5 Monate nach der Primäranalyse, betrug die Hazard Ratio (95-%-KI) 0,81 (0,65, 1,01) [Todesfälle 159 (65,7%) im Behandlungsarm mit Tecentriq + Lurbinectedin Arm und 169 (70,1%) im Tecentriq Arm]. Die Ergebnisse der explorativen OS-Subgruppenanalyse waren generell konsistent mit denen der Studienpopulation (N=483).
Die OS Hazard Ratio (95-%-KI) in der Subgruppe von Patienten mit ECOG PS 0 zum Zeitpunkt des Studieneinschlusses in die Phase der Induktionsbehandlung und zum Zeitpunkt der Randomisierung in die Phase der Erhaltungsbehandlung betrug jeweils 1,21 (0,84, 1,74) und 0,99 (0,70, 1,40), und betrug 1,03 (0,73, 1,46) bei Patienten <65 Jahre [Subgruppengrössen 40 - 43% der Studienpopulation].
Mehr Patienten im Tecentriq mit Lurbinectedin Arm der IMforte Studie hatten zum Zeitpunkt der Primäranalyse eine Erstprogression im Gehirn (16,1%) im Vergleich zum Tecentriq Arm (7,9%). Bei Patienten mit Erstprogression im Gehirn gab es keinen Hinweis auf eine schnellere Krankheitsprogression im Tecentriq mit Lurbinectedin Arm. Eine Folgeradiotherapie des Gehirns wurde bei 24,0% im Tecentriq mit Lurbinectedin Arm und bei 14,5%% im Tecentriq Arm erfasst und ein operativer Folgeeingriff am Gehirn bei 2,1% vs, 0% der Patienten. Aufgrund der geringen Patientenzahl und des explorativen Charakters dieser Analyse können jedoch keine endgültigen Schlussfolgerungen aus diesen Daten gezogen werden.
Erstlinien-Therapie bei triple-negativem Mammakarzinom (1L TNBC)
IMpassion130
Die Phase-III, doppelblinde, zweiarmige, randomisierte, Placebo-kontrollierte Studie WO29522 (IMpassion130) wurde durchgeführt, um die Wirksamkeit und Sicherheit von Tecentriq in Kombination mit Nab-Paclitaxel bei Patienten mit nicht resezierbarem, lokal fortgeschrittenem oder metastasierendem triple-negativem Mammakarzinom (TNBC) ohne vorhergehende Chemotherapie oder zielgerichtete systemische Therapie wegen inoperablem, lokal fortgeschrittenem oder metastasiertem TNBC zu evaluieren. Insgesamt 902 Patienten wurden in die Studie aufgenommen und stratifiziert nach dem Vorhandensein von Lebermetastasen, vorhergehender Behandlung mit einem Taxan und dem PD-L1-Expressions-Status in Tumor-infiltrierenden Immunzellen (IC) (PD-L1-gefärbte Tumor-infiltrierende Immunzellen [IC] < 1% der Tumorfläche vs. ≥1% der Tumorfläche), beurteilt mit dem VENTANA PD-L1 (SP142) Assay.
Die Patienten wurden randomisiert und mit Tecentriq (840 mg) oder i.v. Placebo-Infusionen an den Tagen 1 und 15 plus Nab-Paclitaxel (100 mg/m2) als i.v. Infusion an den Tagen 1, 8 und 15 eines jeden 28-tägigen Zyklus behandelt. Die Patienten erhielten die Behandlung bis zur radiologischen Krankheitsprogression, gemäss RECIST Version 1.1, oder bis zu einer nicht akzeptablen Toxizität. Die Behandlung mit Tecentriq wurde fortgesetzt, wenn Nab-Paclitaxel wegen inakzeptabler Toxizität abgesetzt wurde.
Von der Studie ausgeschlossen waren unter anderem Patienten mit einer Autoimmunkrankheit in der Anamnese; Verabreichung eines attenuierten Lebendimpfstoffs innerhalb von 4 Wochen vor der Randomisierung; Applikation systemischer immunstimulierender Wirkstoffe innerhalb von 4 Wochen oder systemischer immunsuppressiver Arzneimittel innerhalb von 2 Wochen vor der Randomisierung; mit unbehandelten oder Kortikosteroid-abhängigen Hirnmetastasen; vorgängige Transplantation allogener Stammzellen oder eines festen Organs; anamnestisch erhobene oder aktive idiopathische Lungenfibrose und Pneumonitis (ausgenommen Strahlenpneumonitis im Bestrahlungsfeld) und organisierende Pneumonie; aktive Infektionen, z.B. HIV, Hepatitis B und C und Tuberkulose. Die Beurteilung des Tumors erfolgte alle 8 Wochen in den ersten 12 Monaten und alle 12 Wochen danach.
Die Studienpopulation war bezüglich demographischer Daten und Krankheitsmerkmalen beim Studienbeginn, zwischen den Studienarmen, vergleichbar. Die meisten Patienten (99,6%) waren weiblichen Geschlechts. 67,5% der Patienten waren weisser Hautfarbe, 17,8% asiatisch, 6,5% Schwarze oder Afroamerikaner und 4,4% Indianer oder Ureinwohner Alaskas. Das mediane Alter betrug 55 (Bereich 20-86) Jahre. Der Ausgangswert des ECOG-Leistungsstatus war 0 (58,4%) oder 1 (41,3%). Insgesamt hatten bei Aufnahme in die Studie 41% der eingeschlossenen Patienten eine PD-L1-Expression ≥1%, 27% hatten Lebermetastasen und 7% Hirnmetastasen. Annähernd die Hälfte der Patienten hatte ein Taxan (51%) oder Anthracyclin (54%) als (neo) adjuvante Therapie erhalten. Die demographischen Daten der Patienten und die Tumorkrankheit beim Einschluss in die Studie waren in der Population mit PD-L1-Expression ≥1% im Allgemeinen repräsentativ für die gesamte Studienpopulation.
Die in Tabelle 9 und im nachfolgenden Text zusammengefassten PFS (progressionsfreies Überleben), ORR (objektive Ansprechrate) und DOR (Ansprechdauer)-Ergebnisse dieser Studie gelten für Patienten mit PD-L1-Expression ≥1% mit einer medianen Nachbeobachtung überlebender Patienten von 13 Monaten.
Es wurde eine finale OS-Analyse mit einer PD-L1 Expression von ≥1% mit einer medianen Nachbeobachtung über 19,12 Monate durchgeführt. Die OS-Ergebnisse werden in Tabelle 9 gezeigt.
Bei Patienten, bei denen zwischen der letzten Operation und der Diagnose einer metastasierten oder lokal fortgeschrittenen, inoperablen Erkrankung ≥24 Monate lagen, wurde für Atezolizumab + nab-Paclitaxel im Vergleich zu Placebo + nab-Paclitaxel kein Nutzen im Hinblick auf das OS beobachtet.
Tabelle 9: Zusammenfassung der Wirksamkeit bei Patienten mit PD-L1-Expression ≥ 1% (IMpassion130)
|
Wichtige Wirksamkeitsendpunkte
|
Tecentriq + Nab-Paclitaxel
|
Placebo + Nab-Paclitaxel
| |
Co-primäre Endpunkte
|
|
| |
Prüfarzt-beurteiltes progressionsfreies Überleben (PFS) (RECIST Ver. 1.1 – Primäranalyse 3)
|
n=185
|
n=184
| |
Anzahl Ereignisse (%)
|
138 (74,6%)
|
157 (85,3%)
| |
Mediane Dauer des progressionsfreien Überlebens (Monate)
|
7,5
|
5,0
| |
95%-KI
|
(6,7; 9,2)
|
(3,8; 5,6)
| |
Stratifizierte Hazard-Ratio ‡ (95%-KI)
|
0,62 (0,49; 0,78)
| |
p-Wert 1
|
<0,0001
| |
12 Monate progressionsfreies Überleben (%)
|
29,1
|
16,4
| |
Prüfarzt-beurteiltes PFS (RECIST Vers. 1.1) – Aktualisierte explorative Analyse 4
|
|
| |
Anzahl Ereignisse (%)
|
149 (80,5%)
|
163 (88,6%)
| |
Mediane Dauer des progressionsfreien Überlebens (Monate)
|
7,5
|
5,3
| |
95% KI
|
(6,7; 9,2)
|
(3,8; 5,6)
| |
Stratifizierte Hazard-Ratio ‡ (95%-KI)
|
0,63 (0,50-0,80)
| |
12 Monate progressionsfreies Überleben (%)
|
30,3
|
17,3
| |
Analyse des Gesamtüberlebens 1, 2, 5
|
n=185
|
n=184
| |
Anzahl Todesfälle (%)
|
120 (64,9%)
|
139 (75,5%)
| |
Mediane Zeit bis Ereignis (Monate)
|
25,4
|
17,9
| |
95%-KI
|
(19,6; 30,7)
|
(13,6; 20,3)
| |
Stratifizierte Hazard-Ratio ‡ (95%-KI)
|
0,67 (0,53; 0,86)
|
1 Auf der Grundlage des stratifizierten Log-rank-Tests2 Entsprechend der im Voraus spezifizierten Analysen-Hierarchie wurden Gesamtüberleben-Vergleiche zwischen den Behandlungsarmen bei Patienten mit PD-L1-Expression ≥1% nicht formal getestet3 Laut PFS-, ORR- und DOR-Endanalyse und erster OS-Zwischenanalyse zum Zeitpunkt des klinischen Cut-offs am 17. April 20184 Laut explorativer PFS-Analyse zum Zeitpunkt des klinischen Cut-offs am 2. Januar 20195 Laut finaler OS-Analyse zum Zeitpunkt des klinischen Cut-offs am 14. April 2020
‡ Stratifiziert nach dem Vorliegen von Lebermetastasen und vorhergehender Behandlung mit Taxan
PFS = progressionsfreies Überleben (progression-free survival); RECIST = Response Evaluation Criteria in Solid Tumors v1.1. (Beurteilungskriterien des Ansprechens bei soliden Tumoren, Version 1.1); CI = Konfidenzintervall (confidence interval); OS = Gesamtüberleben (overall survival), NE = nicht schätzbar (not estimable)
Bei PD-L1-positiven Patienten wurde im Tecentriq- und Nab-Paclitaxel-Arm (58,9%) eine zahlenmässig höhere Gesamtansprechrate (Overall Response Rate, ORR) festgestellt als im Placebo- und Nab-Paclitaxel-Arm (42,6%). Unter den Respondern war die Anzahl derer mit anhaltendem Ansprechen im Tecentriq- und Nab-Paclitaxel-Arm höher (35,8%) als im Placebo- und Nab-Paclitaxel-Arm (24,4%). Die mediane geschätzte Ansprechdauer (Duration of Response, DOR), war im Tecentriq- und Nab-Paclitaxel-Arm 3 Monate länger (8,5 Monate vs. 5,5 Monaten im Placebo- und Nab-Paclitaxel-Arm, nicht-stratifizierte HR: 0,60; 95%-KI; 0,43; 0,86).
Melanom
IMspire150
CO39262 (IMspire150) ist eine doppelblinde, zweiarmige, randomisierte, placebokontrollierte Phase-III-Studie. In dieser wurde die Wirksamkeit und Sicherheit von Tecentriq in Kombination mit Cobimetinib und Vemurafenib bei Patienten mit BRAF-V600- Mutations-positivem, metastasiertem oder nicht resezierbarem, lokal fortgeschrittenem Melanom untersucht. Die BRAF V600-Mutation des Melanoms wurde mit einem vor Ort verfügbaren Test nachgewiesen und zentral mit dem FoundationOneTM-Assay bestätigt. Die Patienten hatten zuvor keine systemische Melanombehandlung in diesem Setting erhalten. Es wurden insgesamt 514 Patienten randomisiert, um eines der unten beschriebenen Behandlungsregime zu erhalten. Die Randomisierung wurde nach geografischem Standort und Lactatdehydrogenase(LDH)-Anfangswert stratifiziert.
Die Behandlung wurde in 28-tägigen Zyklen durchgeführt. In Zyklus 1 erhielten alle Patienten 21 Tage lang zweimal täglich 960 mg Vemurafenib oral und einmal täglich 60 mg Cobimetinib oral, sowie anschliessend für 7 Tage entweder zweimal täglich 720 mg Vemurafenib + Placebo in der Tecentriq-Gruppe oder 960 mg in der Kontrollgruppe. Ab Zyklus 2 erhielten die Patienten in der Tecentriq-Gruppe an den Tagen 1 und 15 840 mg Tecentriq intravenös sowie zweimal täglich 720 mg Vemurafenib + Placebo (über 28 Tage) und einmal täglich 60 mg Cobimetinib (mit Medikationsgabe über 21 Tage, gefolgt von einer 7-tägigen Einnahmepause). Die Patienten in der Kontrollgruppe erhielten an den Tagen 1 und 15 intravenös ein Placebo sowie zweimal täglich 960 mg Vemurafenib und einmal täglich Cobimetinib über 21 Tage, gefolgt von einer 7-tägigen Einnahmepause. Bei Vemurafenib und Cobimetinib waren Dosisreduktionen und Behandlungsunterbrechungen, bei Tecentriq ausschliesslich Behandlungsunterbrechungen zum Management unerwünschter Ereignisse zulässig. Die Behandlung wurde bis zum Eintreten einer Krankheitsprogression, Tod oder inakzeptabler Toxizität nach Feststellung durch den Prüfarzt fortgesetzt. Zum Zeitpunkt des Fortschreitens war ein Crossover nicht zulässig. Patienten wurden von der Teilnahme ausgeschlossen, wenn sie eine Autoimmunerkrankung in der Vorgeschichte hatten, bei Erhalt eines attenuierten Lebensimpfstoffs innerhalb von 28 Tagen vor der Randomisierung, bei Erhalt eines systemischen Immunstimulans innerhalb von 4 Wochen oder eines systemischen Immunsuppressivums innerhalb von 2 Wochen vor der Randomisierung. Die Tumorbeurteilungen wurden in den ersten 24 Monaten alle 8 Wochen (± 1 Woche) und danach alle 12 Wochen (± 1 Woche) durchgeführt.
Die demografischen Daten und die Krankheitsmerkmale des Studienkollektivs zur Baseline waren zwischen den Behandlungsarmen ausgewogen. Das mediane Alter betrug 54 Jahre (Bereich: 22–88), 58% der Patienten waren männlich. Die meisten Patienten waren weiss (95%). 2,5% der Patienten hatten zuvor eine Behandlung wegen Hirnmetastasen erhalten, 30% der Patienten hatten beim Eintritt in die Studie Lebermetastasen und 14% der Patienten waren mit einer adjuvanten systemischen Therapie behandelt worden. Das Melanom wurde bei 5,8% der Patienten als Stadium IIIC und bei 93,8% der Patienten als Stadium IV (M1A, 14,8%; M1B, 19,1%; M1C, 59,9%) gemäss der Klassifikation des American Joint Committee on Cancer (AJCC), siebte Fassung, eingestuft.
Der ECOG-Leistungsstatus zum Studienbeginn betrug 0 (76,5%) oder 1 (22,8%). Der Anteil der Patienten mit erhöhter LDH betrug 32,8% im Behandlungsarm mit Tecentriq und 32,9% im Behandlungsarm ohne Tecentriq.
Ausgehend von zentral durchgeführten Tests lag bei 74% die Mutation V600E vor, bei 11% die Mutation V600K und 1% die Mutation V600D oder V600R.
Der primäre Wirksamkeitsendpunkt war das progressionsfreie Überleben (PFS) gemäss Bewertung durch den Prüfarzt. Am Ende des Zeitraums der Datenerfassung (Primäranalyse) betrug der mediane Beobachtungszeitraum 18,9 Monate.
In der Tecentriq-V600E-Subgruppe betrug das mediane PFS gemäss Bewertung durch den Prüfarzt 16,2 Monate, in der Kontroll-V600E-Subgruppe dagegen 10,0 Monate (unstratifizierte Hazard Ratio: 0,68, 95% CI (0,53, 0,88)). Zu den sekundären Endpunkten zählte das mediane PFS gemäss Bewertung durch einen unabhängigen Prüfausschuss (IRC), das in der Tecentriq-V600E-Subgruppe 17,6 Monate und in der Kontroll-V600E-Subgruppe 11,4 Monate betrug (unstratifizierte Hazard Ratio 0,71, 95% KI (0,55, 0,93)). Eine Zwischenanalyse des Gesamtüberlebens (OS) ergab eine unstratifizierte Hazard Ratio von 0,69 (95% KI 0,50, 0,95). Die objektiven Ansprechraten (bewertet durch den Prüfarzt) waren in den beiden Gruppen ähnlich und betrugen 68,9% in der Tecentriq-V600E-Subgruppe und 63,2% in der Kontroll-V600E-Subgruppe. Die mediane Dauer des Ansprechens (bewertet durch den Prüfarzt) betrug 20,4 Monate in der Tecentriq-V600E-Subgruppe im Vergleich zu 12,6 Monate in der Kontroll-V600E-Subgruppe.
Tecentriq in Kombination mit Vemurafenib und Cobimetinib wurde nicht bei Patienten mit Melanom mit Wildtyp-BRAF untersucht.
HCC
IMbrave150
Es wurde eine globale, randomisierte, multizentrische, unverblindete Phase-III-Studie, YO40245 (IMbrave150), durchgeführt, um die Wirksamkeit und Sicherheit von Tecentriq in Kombination mit Bevacizumab bei Patienten mit lokal fortgeschrittenem oder metastasiertem und/oder inoperablem HCC ohne vorgängige systemische Behandlung zu bewerten. Es wurden insgesamt 501 Patienten randomisiert (2:1), um entweder alle 3 Wochen 1200 mg Tecentriq und 15 mg/kg Bevacizumab per intravenöse Infusion oder zweimal täglich 400 mg Sorafenib oral zu erhalten. Die Randomisierung wurde nach geografischer Region (Asien ohne Japan vs. Rest der Welt), makrovaskulärer Invasion und/oder extrahepatischer Ausbreitung (vorhanden vs. nicht vorhanden), Baseline-AFP (< 400 vs. ≥400 ng/ml) und ECOG-Leistungsstatus (0 vs. 1) stratifiziert. In beiden Armen wurden die Patienten bis zum Verlust des klinischen Nutzens oder bis zum Auftreten inakzeptabler Toxizität behandelt. Die Patienten konnten Tecentriq oder Bevacizumab (z.B. aufgrund von Nebenwirkungen) absetzen und mit einer Monotherapie fortfahren, bis kein klinischer Nutzen mehr damit erzielt wurde oder inakzeptable Toxizität im Zusammenhang mit dem Monotherapeutikum auftrat.
Die Studienteilnehmer waren Erwachsene (Child-Pugh A, ECOG 0/1) ohne systemische Vorbehandlung. Blutung (einschliesslich tödlicher Vorkommnisse) ist eine bekannte unerwünschte Reaktion auf Bevacizumab, und Blutung im oberen Magendarmtrakt ist eine häufige und lebensbedrohliche Komplikation bei Patienten mit HCC. Daher mussten die Patienten innerhalb von 6 Monaten vor der Behandlung auf Vorhandensein von Varizen untersucht werden. Eine Varizenblutung in den 6 Monaten vor der Behandlung, unbehandelte oder unvollständig behandelte Varizen mit Blutung oder hohem Blutungsrisiko waren Ausschlusskriterien für die Teilnahme. Ausschlusskriterien waren Varizenblutung innerhalb 6 Monate vor Behandlung, unbehandelte Varizen, mässiger bis starker Aszites, hepatische Enzephalopathie in der Vorgeschichte, anamnestisch bekannte Autoimmunerkrankung, Erhalt eines lebend-attenuierten Impfstoffes innerhalb von 4 Wochen, von systemischen Immunstimulatoren innerhalb von 4 Wochen oder systemischen Immunsuppressiva innerhalb von 2 Wochen vor der Randomisierung sowie unbehandelte oder Kortikosteroid-abhängige Gehirnmetastasen. Die Tumorbeurteilungen erfolgten in den ersten 54 Wochen alle 6 Wochen, danach alle 9 Wochen.
Die demografischen Merkmale und die Krankheitsmerkmale in dem Studienkollektiv waren in beiden Behandlungsarmen zur Baseline gut ausgewogen. Das mediane Alter war 65 Jahre (Bereich: 26 bis 88 Jahre), und 83% waren männlich. Die meisten Patienten waren asiatisch (57%) und weiss (35%). 40% stammten aus Asien (ohne Japan), 60% aus dem Rest der Welt. Bei ungefähr 75% der Patienten lag zum Zeitpunkt der Vorstellung eine makrovaskuläre Invasion und/oder eine extrahepatische Ausbreitung vor, und 37% hatten einen AFP-Wert ≥400 ng/ml zur Baseline. Der ECOG-Performance-Status zu Studienbeginn war 0 (62%) oder 1 (38%). Die primären Risikofaktoren für die Entwicklung eines HCC waren eine Hepatitis-B-Virusinfektion bei 48% der Patienten, eine Hepatitis-C-Virusinfektion bei 22% der Patienten und eine nicht-virale Krankheit bei 31% der Patienten. 82% der Patienten hatten ein HCC des BCLC (Barcelona Clinic Liver Cancer)-Stadiums C, 16% hatten ein HCC des BCLC-Stadiums B und 3% hatten ein HCC des BCLC-Stadiums A.
Die beiden co-primären Wirksamkeitsendpunkte waren das Gesamtüberleben (OS) und das unabhängig beurteilte progressionsfreie Überleben (PFS) gemäss RECIST v1.1. Zum Zeitpunkt der primären Analyse betrug die mediane Nachbeobachtungsdauer 8,6 Monate. Mit Tecentriq + Bevacizumab wurde im ITT-Kollektiv im Vergleich zu Sorafenib eine statistisch signifikante Verbesserung beider coprimärer Wirksamkeitsendpunkte erzielt. Das mediane OS wurde im Behandlungsarm mit Tecentriq + Bevacizumab nicht erreicht (NR) (95%-KI: NR; NR) und betrug im Sorafenib-Arm 13,2 Monate (95%-KI: 10,4; NR). Die stratifizierte HR betrug 0,58 (95%-KI: 0,42; 0,79; stratifizierter p-Wert = 0,0006), was einer Verringerung des mit Tecentriq + Bevacizumab verbundenen Sterberisikos um 42% im Vergleich zu Sorafenib entspricht.
Das mediane PFS betrug im Behandlungsarm mit Tecentriq + Bevacizumab 6,8 Monate (95%-KI: 5,8; 8,3) und im Sorafenib-Arm 4,3 Monate (95%-KI: 4,0; 5,6). Die stratifizierte HR betrug 0,59 (95%-KI: 0,47; 0,76; stratifizierter p-Wert < 0,0001), was einer Verringerung des mit Tecentriq + Bevacizumab verbundenen Krankheitsprogressions- oder Sterberisikos um 41% gegenüber Sorafenib entspricht.
Es zeigte sich eine statistisch signifikante Verbesserung der ORR per IRF-Bewertung gemäss RECIST v1.1 von 27,3% (95%-KI: 22,5%, 32,5%) im Behandlungsarm mit Tecentriq + Bevacizumab gegenüber 11,9% (95%-KI: 7,4%, 18,0%) im Sorafenib-Arm. Die Rate derer mit vollständigem Ansprechen betrug 5,5% unter Tecentriq + Bevacizumab gegenüber 0% im Sorafenib-Arm.
Eine deskriptive, aktualisierte Wirksamkeitsanalyse wurde mit einer medianen Nachbeobachtungszeit des Überlebens von 15,6 Monaten durchgeführt. Das mediane OS betrug im Behandlungsarm mit Tecentriq + Bevacizumab 19,2 Monate (95%-KI: 17,0; 23,7) und im Sorafenib-Arm 13,4 Monate (95%-KI: 11,4; 16,9). Die stratifizierte HR betrug 0,66 (95%-KI: 0,52; 0,85), was einer Verringerung des mit Tecentriq + Bevacizumab verbundenen Sterberisikos um 34% im Vergleich zu Sorafenib entspricht.
GO30140
Es wurde auch eine globale, unverblindete, multizentrische, mehrarmige Phase-Ib-Studie (GO30140) bei Patienten mit solidem Tumor durchgeführt. In Arm F der Studie wurde ein randomisiertes Design verwendet, um die Sicherheit und Wirksamkeit der Anwendung von Tecentriq in Kombination mit Bevacizumab gegenüber einer Monotherapie mit Tecentriq bei Patienten mit fortgeschrittenem oder metastasiertem und/oder inoperablem HCC ohne vorgängige systemische Behandlung zu bewerten. Der primäre Wirksamkeitsendpunkt war das unabhängig beurteilte PFS per RECIST v1.1. Es wurden insgesamt 119 Patienten 1:1 randomisiert, um entweder alle 3 Wochen Tecentriq (1200 mg) und Bevacizumab (15 mg/kg) durch intravenöse Infusion oder alle 3 Wochen nur Tecentriq (1200 mg) zu erhalten. Zum Zeitpunkt der primären Analyse betrug die mediane Dauer der Nachbeobachtung in Bezug auf das Überleben 6,6 Monate. Für die Kombination Tecentriq und Bevacizumab ergab sich im Vergleich zu einer Monotherapie mit Tecentriq ein statistisch signifikanter Nutzen in Bezug auf das PFS (HR: 0,55, 80%-KI: 0,40; 0,74, p-Wert = 0,0108), mit einem medianen PFS von 5,6 Monaten bei Patienten unter Behandlung mit Tecentriq und Bevacizumab vs. 3,4 Monate bei Patienten unter Behandlung mit einer Monotherapie mit Tecentriq.
Urothelkarzinom
Anwendung von Tecentriq bei Urothelkarzinompatienten mit ungünstigen Prognosefaktoren, die mit platin-basierter Chemotherapie vorbehandelt wurden
Ärzte sollten den verzögerten Effekt der Behandlung mit Tecentriq berücksichtigen bevor eine Behandlung mit Tecentriq bei Patienten mit ungünstigen Prognosefaktoren oder aggressiver Krankheit begonnen wird. In der Studie IMvigor211 wurde bei Urothelkarzinompatienten eine höhere Anzahl Todesfälle in den ersten 4 Monaten bei Behandlung mit Tecentriq gegenüber einer Behandlung mit Chemotherapie beobachtet. Faktoren, die mit frühen Todesfällen assoziiert waren, sind ≥2 Bellmunt Risikofaktoren (bestehend aus ECOG >0, Lebermetastasen und Hämoglobin < 10 g/dL), erhöhte ALP und/oder Lebermetastasen bei Eintritt in die Studie.
IMvigor211
Es wurde eine open-label, multizentrische, internationale, randomisierte Phase-III-Studie (GO29294, IMvigor211) durchgeführt, um die Wirksamkeit und Sicherheit von Tecentriq im Vergleich zu einer Chemotherapie (Vinflunin, Docetaxel oder Paclitaxel nach Auswahl des Prüfarztes) bei Patienten mit inoperablem, lokal fortgeschrittenem oder metastasiertem Urothelkarzinom zu bewerten, bei denen es während oder nach einer platinhaltigen Therapie zu einem Fortschreiten der Erkrankung gekommen war. Patienten mit folgender Vorgeschichte waren von der Studienteilnahme ausgeschlossen: Patienten mit Autoimmunkrankheit, aktiven oder kortikosteroidabhängigen Gehirnmetastasen, Erhalt eines attenuierten Lebendimpfstoffes in den 28 Tagen vor der Aufnahme und Erhalt systemischer Immunstimulanzien in den 4 Wochen oder eines systemischen immunsupprimierenden Arzneimittels in den 2 Wochen vor der Aufnahme. Tumorbeurteilungen wurden in den ersten 54 Wochen alle 9 Wochen und danach alle 12 Wochen durchgeführt. Tumorproben wurden prospektiv auf Expression von PD-L1 auf tumorinfiltrierenden ICs untersucht, und anhand der Ergebnisse wurden Untergruppen der PD-L1-Expression für die nachstehend beschriebenen Analysen definiert.
Insgesamt wurden 931 Patienten aufgenommen. Die Patienten wurden randomisiert (1:1), um entweder Tecentriq oder eine Chemotherapie zu erhalten. Die Randomisierung wurde nach Chemotherapie (Vinflunin vs. Taxan), PD-L1-Expressionsstatus auf ICs (< 5% vs. ≥5%), Anzahl an prognostischen Risikofaktoren (0 vs. 1-3) und Lebermetastasen (ja vs. nein) stratifiziert. Die prognostischen Risikofaktoren umfassten einen Zeitraum seit der vorhergehenden Chemotherapie < 3 Monaten, einen ECOG-Performance-Status > 0 und Hämoglobin < 10 g/dl.
Tecentriq wurde alle 3 Wochen als i.v. Infusion in einer Fixdosis von 1200 mg gegeben. Eine Reduzierung der Tecentriq-Dosis war nicht zulässig. Die Patienten wurden bis zum Verlust des klinischen Nutzens per Beurteilung durch den Prüfarzt oder bis zum Auftreten inakzeptabler Toxizität behandelt. Vinflunin wurde an Tag 1 jedes 3-Wochen-Zyklus in einer Dosierung von 320 mg/m2 bis zur Progression der Krankheit oder bis zum Auftreten inakzeptabler Toxizität als i.v. Infusion verabreicht. Paclitaxel wurde über 3 Stunden an Tag 1 jedes 3-Wochen-Zyklus in einer Dosierung von 175 mg/m2 bis zur Progression der Krankheit oder bis zum Auftreten inakzeptabler Toxizität als i.v. Infusion verabreicht. Docetaxel wurde an Tag 1 jedes 3-Wochen-Zyklus in einer Dosierung von 75 mg/m2 bis zur Progression der Krankheit oder bis zum Auftreten inakzeptabler Toxizität als i.v. Infusion verabreicht. Bei den behandelten Patienten betrug die mediane Dauer der Behandlung im Tecentriq-Arm 2,8 Monate, im Vinflunin- und im Paclitaxel-Arm 2,1 Monate und im Docetaxel-Arm 1,6 Monate.
Die demographischen und zum Studienbeginn vorhandenen Krankheitsmerkmale im primären Analysekollektiv waren im Vergleich der Behandlungsarme ausgewogen. Das mediane Alter betrug 67 Jahre (Bereich: 31 bis 88) und 77,1% der Patienten waren männlich. 53,9% der Patienten im Chemotherapie-Arm erhielten Vinflunin, bei 71,4% der Patienten lag mindestens ein ungünstiger prognostischer Risikofaktor vor und 28,8% hatten zum Studienbeginn Lebermetastasen. Der ECOG-Performance-Status zum Studienbeginn betrug 0 (45,6%) oder 1 (54,4%). Bei 71,1% der Patienten befand sich der Primärtumor in der Blase und 25,4% der Patienten hatten ein Urothelkarzinom in den oberen Harnwegen. 24,2% der Patienten hatten nur eine vorherige platinhaltige adjuvante oder neoadjuvante Therapie erhalten, und es war innerhalb von 12 Monaten zu einer Krankheitsprogression gekommen.
Der primäre Wirksamkeitsendpunkt in der IMvigor211 war das Gesamtüberleben (OS). Die sekundären Wirksamkeitsendpunkte sind die anhand der RECIST-Kriterien (Response Evaluation Criteria in Solid Tumors) v1.1 vom Prüfarzt bewertete objektive Ansprechrate (ORR), das progressionsfreie Überleben (PFS) sowie die Dauer des Ansprechens (DOR). Vergleiche des OS zwischen dem Behandlungsarm und dem Kontrollarm wurden mit einem hierarchischen Verfahren mit festgelegter Abfolge auf Basis eines stratifizierten Log-Rank-Tests mit einem zweiseitigen Niveau von 5% wie folgt getestet: Schritt 1) Untergruppe mit PD-L1-Expression ≥5%, Schritt 2) Untergruppe mit PD-L1-Expression ≥1%, Schritt 3) alle Patienten, welche die Aufnahmekriterien erfüllen («AllComers»). Die OS-Ergebnisse für die Schritte 2 und 3 konnten jeweils nur dann formal getestet werden, wenn das Ergebnis im vorhergehenden Schritt statistisch signifikant war.
Die mediane Nachbeobachtung in Bezug auf das Überleben betrug 17 Monate. Die Studie IMvigor211 erreichte ihren primären Endpunkt für OS nicht. In der Untergruppe der Patienten mit Tumoren mit einer PD-L1-Expression ≥5% bewirkte Tecentriq keinen statistisch signifikanten Überlebensvorteil im Vergleich zur Chemotherapie mit einem OS HR von 0,87 (95%-KI: 0,63; 1,21; medianes OS von 11,1 vs. 10,6 Monaten für Tecentriq bzw. Chemotherapie). Der p-Wert im stratifizierten Log-Rank-Test betrug 0,41. Daher wurden in der Untergruppe mit PD-L1-Expression ≥1% bzw. in der ITT Population keine formalen statistischen Tests hinsichtlich des OS durchgeführt, und die Ergebnisse jener Analysen werden als exploratorisch betrachtet.
Das mittlere progressionsfreie Überleben betrug 2,1 Monate (95%-KI 2,1-2,2) im Tecentriq-Studienarm und 4,0 Monate (95%-KI: 3,4; 4,2) im Chemotherapie-Studienarm mit einer Hazard Ratio von 1,10 (95%-KI: 0,95; 1,26). Der Anteil ITT-Patienten mit einem vom Prüfarzt bestätigten Ansprechen gemäss RECIST Version 1.1 war in beiden Studienarmen ähnlich: 13,4% (95%-KI: 10,45; 16,87) im Tecentriq-Studienarm und 13,4% (95%-KI: 10,47; 16,91) im Chemotherapie-Studienarm. Bei den Respondern war die mediane Dauer des Ansprechens auf die Therapie im Tecentriq-Studienarm signifikant länger (21,7 Monate) als im Chemotherapie-Studienarm (7,4 Monate).
In der exploratorischen, finalen Analyse (cut off Datum: 8. November 2018) zur Gesamtüberleben mit einer medianen Nachbeobachtungsdauer von 34 Monaten im ITT-Kollektiv betrug die mediane Überlebensdauer im Tecentriq-Arm 8,6 Monate (95%-KI: 7,8; 9,6) und im Chemotherapie-Arm 8,0 Monate (95%-KI: 7,2; 8,6) bei einer Hazard-Ratio von 0,82 (95%-KI: 0,71; 0,94). Bei Patienten im Tecentriq-Arm im Vergleich zum Chemotherapie-Arm im ITT-Kollektiv wurden nach 12, 24 und nach 30 Monaten zahlenmässig höhere OS-Raten festgestellt. Der prozentuale Anteil der Patienten, die nach 12 Monaten am Leben waren (KM-Schätzung), betrug im Chemotherapie-Arm 32,5% und im Tecentriq-Arm 39,2%. Im Chemotherapie-Arm waren nach 24 Monaten 12,7% und im Tecentriq-Arm 22,5% der Patienten am Leben (KM-Schätzung). Nach 30 Monaten (KM-Schätzung) waren es 9,8% im Chemotherapie-Arm und 18,1% im Tecentriq-Arm.
Supportive Studie bei Patienten mit einem lokal fortgeschrittenen oder metastasierten Urothelkarzinom, die zuvor mit platinbasierter Chemotherapie behandelt wurden: IMvigor210 Kohorte 2
Es wurde eine multizentrische, internationale, einarmige klinische Phase-II-Studie mit zwei Kohorten, GO29293 (IMvigor210), bei Patienten mit lokal fortgeschrittenem oder metastasiertem Urothelkarzinom durchgeführt. Es wurden insgesamt 438 Patienten in die Studie aufgenommen, welche aus zwei Patientenkohorten bestand. Kohorte 2 bestand aus Patienten, die mindestens eine platinbasierte Chemotherapie gegen ein lokal fortgeschrittenes oder metastasiertes Urothelkarzinom erhalten hatten oder bei denen innerhalb von 12 Monaten nach der Behandlung mit einer platinhaltigen neoadjuvanten oder adjuvanten Chemotherapie eine Krankheitsprogression aufgetreten war.
Die koprimären Wirksamkeitsendpunkte in Kohorte 2 waren das bestätigte ORR IRF-RECIST v1.1 sowie die vom Prüfarzt beurteilte ORR nach den modifizierten RECIST-Kriterien (mRECIST). Es wurden 310 Patienten alle 3 Wochen bis zum Verlust des klinischen Nutzens mit 1200 mg Tecentriq per i.v. Infusion behandelt. Die primäre Analyse von Kohorte 2 wurde durchgeführt, nachdem bei allen Patienten eine mindestens 24-wöchige Nachbeobachtung stattgefunden hatte. Die Studie erreichte in Kohorte 2 die beiden primären Endpunkte, d.h. statistisch signifikante ORRs bei IRF-Beurteilung per RECIST v1.1 und bei Beurteilung durch den Prüfarzt per mRECIST im Vergleich zu einer vorab bestimmten historischen Kontrollremissionsrate von 10%.
Darüber hinaus wurde auch in Kohorte 2 eine Analyse nach einer medianen überlebensbezogenen Nachbeobachtungsdauer von 21,1 Monaten durchgeführt. Die bestätigten ORRs per IRF-RECIST v1.1 betrugen 15,8% (95%-KI: 11,9; 20,4) in der ITT Population, die bestätigte ORR bei Beurteilung durch den Prüfarzt per mRECIST betrug 19,7% (95%-KI: 15,4; 24,6) und die OS Rate nach 12 Monaten betrug 37%. Die mediane DOR wurde noch nicht erreicht in der ITT Population.
Kinder und Jugendliche
Es wurde eine multizentrische Open-Label-Studie einer frühen Phase bei pädiatrischen Patienten (< 18 Jahre, n=69) und bei jungen erwachsenen Patienten (18–30 Jahre, n=18) mit rezidivierten oder progredienten soliden Tumoren oder mit Hodgkin- oder Non-Hodgkin-Lymphom zur Bewertung der Sicherheit und Pharmakokinetik von Tecentriq durchgeführt. Die Patienten erhielten alle 3 Wochen 15 mg Atezolizumab i.v. pro kg. Die primären Indikatoren für die Wirksamkeit waren die objektive Remissionsrate (objective response rate, ORR), das progressionsfreie Überleben (progression-free survival, PFS) und die Rate derer mit einer Remission mit klinischem Nutzen (clinical benefit response rate, CBRR; nur bei Patienten mit Osteosarkom).
Zum Zeitpunkt der primären Analyse betrug die ORR 4,6% (95%-KI: 1,59; 10,89) und die mittlere Dauer des PFS betrug 1,3 Monate (95%-KI: 1,2; 1,4). Da bei keinem der Patienten in der Osteosarkomkohorte eine komplette oder partielle Remission erreicht wurde oder eine mindestens 6 Monate anhaltende Stabilisierung der Krankheit eintrat, betrug die CBRR 0. Der klinische Nutzen von Atezolizumab bei pädiatrischen Patienten mit rezidivierten/refraktären soliden Tumoren wurde nicht aufgezeigt.
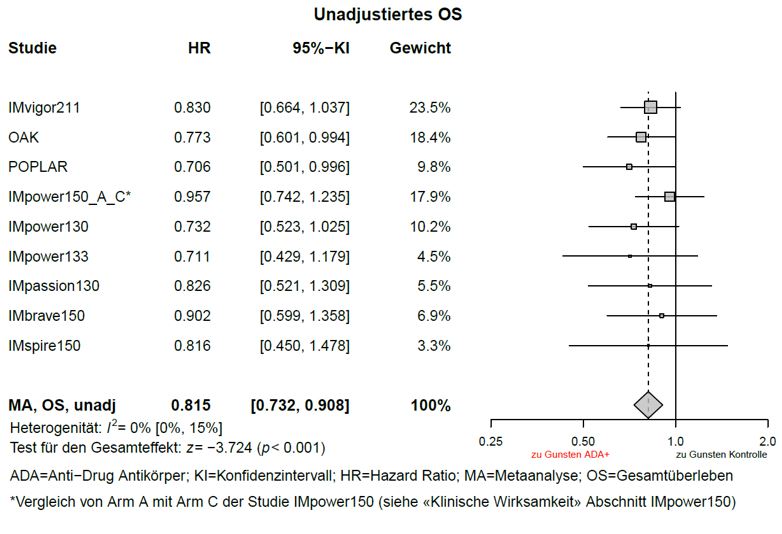
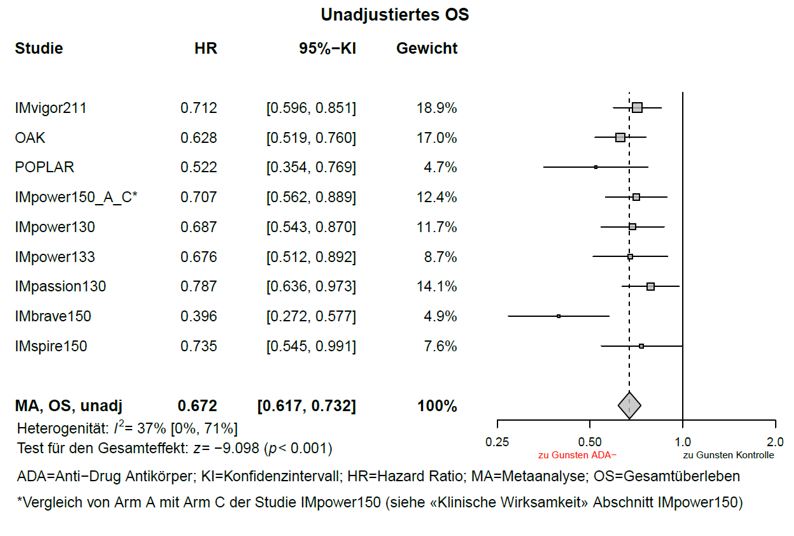
Wirksamkeit von Atezolizumab bei Vorliegen von Anti-Drug-Antiköpern
Explorative, nicht-adjustierte Metaanalysen zur Wirksamkeit (ohne Anpassungen von Ungleichgewichten der Baseline Charakteristika) wurden durchgeführt. Gemäss den Metaanalysen betrug der Schätzwert der HR für das Gesamtüberleben (OS) zwischen der ADA-positiven Subgruppe und dem Kontrollarm 0,815 (95-%-KI: 0,732; 0,908), sowie der Schätzwert der HR für das OS zwischen der ADA-negativen Subgruppe und dem Kontrollarm 0,672 (95-%-KI: 0,617; 0,732), siehe Graphiken 1 und 2.
Graphik 1: Meta-Analyse OS (unadjustiert) ADA – Positive Patienten im Vergleich zur Kontrolle
Graphik 2: Meta-Analyse OS (unadjustiert) ADA – Negative Patienten im Vergleich zur Kontrolle
|