ZusammensetzungWirkstoff: Letermovir.
Hilfsstoffe Filmtablette: Mikrokristalline Cellulose, Croscarmellose-Natrium, Povidon 25, kolloidales Siliciumdioxid, Magnesiumstearat und folgende Hilfsstoffe im Filmüberzug: Laktosemonohydrat, Hypromellose 2910, Titandioxid, Triacetin, Eisenoxid gelb, (nur 480-mg-Tabletten) Eisenoxid rot, Carnaubawachs als Poliermittel.
1 ml Infusionslösung enthält folgende Hilfsstoffe: Hydroxypropylbetadex (150 mg), Natriumchlorid (3,1 mg), Natriumhydroxid (1,2 mg), Aqua ad iniectabilia. Die Natriumhydroxidmenge kann angepasst werden, um einen pH-Wert von ca. 7,5 zu erreichen.
Galenische Form und Wirkstoffmenge pro EinheitPrevymis Filmtablette enthält 240 mg oder 480 mg Letermovir.
Prevymis Konzentrat zur Herstellung einer Infusionslösung ist eine klare, sterile Lösung ohne Konservierungsstoffe in Einzeldosis-Durchstechflaschen mit 240 mg oder 480 mg Letermovir je Durchstechflasche. 1 ml Lösung enthält 20 mg Letermovir. Prevymis Konzentrat zur Herstellung einer Infusionslösung muss vor der Verabreichung verdünnt werden. Nur als intravenöse (i.v.) Infusion verabreichen.
Indikationen/AnwendungsmöglichkeitenPrevymis ist zur Prophylaxe von Cytomegalovirus (CMV) Infektionen oder Erkrankungen bei erwachsenen, CMV-seropositiven Empfängern [R+] einer allogenen hämatopoetischen Stammzelltransplantation (HSZT) indiziert.
Dosierung/AnwendungDie empfohlene Dosis Prevymis beträgt 480 mg einmal täglich. Die Gabe von Prevymis ist nach der HSZT zu beginnen. Die Gabe von Prevymis kann am Tag der Transplantation und bis spätestens 28 Tage nach der Transplantation gestartet werden. Die Gabe von Prevymis kann vor oder nach dem Einwachsen der Stammzellen (Engraftment) begonnen werden. Die Behandlung mit Prevymis wird bis 100 Tage nach der Transplantation weitergeführt. Prevymis Infusionslösung, die Hydroxypropylbetadex enthält, sollte nur bei Patienten angewendet werden, die keine orale Therapie einnehmen können. Patienten sollten auf orales Prevymis umgestellt werden, sobald sie in der Lage sind, orale Medikamente einzunehmen. Siehe Abschnitt «Pharmakokinetik» von Letermovir nach i.v. und oraler Verabreichung.
Prevymis Filmtabletten und Konzentrat zur Herstellung einer Infusionslösung können abwechselnd nach Ermessen des Arztes angewendet werden; eine Dosisanpassung ist nicht erforderlich.
Die Tablette ist ganz zu schlucken und kann mit oder ohne Nahrung eingenommen werden. Die Tablette darf nicht geteilt, zerdrückt oder zerkaut werden.
Prevymis Konzentrat zur Herstellung einer Infusionslösung muss vor der Verabreichung verdünnt werden (siehe Abschnitt «Sonstige Hinweise»). Nur als intravenöse (i.v.) Infusion verabreichen. Nicht als intravenöse Schnellinfusion oder Bolus verabreichen. Verabreichen Sie Prevymis nach der Verdünnung durch intravenöse Infusion über einen peripheren oder zentralen Venenkatheter über einen Gesamtzeitraum von ca. 60 Minuten. Verabreichen Sie den gesamten Inhalt des Infusionsbeutels.
Wenn Prevymis zusammen mit Cyclosporin verabreicht wird, ist die Dosis von Prevymis auf 240 mg einmal täglich zu reduzieren (siehe Abschnitte «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»).
·Wird nach Beginn der Gabe von Prevymis eine Cyclosporin-Therapie eingeleitet, ist die nächste Dosis Prevymis auf 240 mg einmal täglich zu reduzieren.
·Wird Cyclosporin nach Beginn der Gabe von Prevymis abgesetzt, ist die nächste Dosis Prevymis auf 480 mg einmal täglich zu erhöhen.
·Wird die Cyclosporingabe aufgrund hoher Cyclosporinspiegel vorübergehend unterbrochen, ist keine Dosisanpassung von Prevymis erforderlich.
Vergessene Dosis
Weisen Sie die Patienten an, die Einnahme der vergessenen Dosis Prevymis nachzuholen, sobald sie sich daran erinnern. Falls der nächste Einnahmezeitpunkt unmittelbar bevorsteht, sollen die Patienten die Einnahme nicht nachholen, sondern mit dem gewohnten Einnahmeschema fortfahren. Weisen Sie die Patienten an, ihre nächste Dosis nicht zu verdoppeln und nicht mehr als die verordnete Dosis einzunehmen.
Spezielle Dosierungsempfehlungen
Prevymis wird bei Patienten mit mässiger Einschränkung der Leberfunktion in Kombination mit mässiger oder schwerer Einschränkung der Nierenfunktion nicht empfohlen (siehe Abschnitt «Interaktionen»).
Patienten mit eingeschränkter Nierenfunktion
Bei Patienten mit einer Kreatininclearance >10 ml/min ist keine Dosisanpassung von Prevymis erforderlich (siehe Abschnitt «Pharmakokinetik»). Es liegen keine Daten bei Patienten mit Nierenerkrankung im Endstadium (KrCl unter 10 ml/min) oder Dialysepatienten vor, so dass keine Dosierungsempfehlungen möglich sind. Prevymis Konzentrat zur Herstellung einer Infusionslösung enthält Hydroxypropylbetadex. Die erwartete klinische Exposition gegenüber Hydroxypropylbetadex bei intravenös verabreichtem Letermovir beträgt ca. 3600 mg/Tag bei einer Letermovirdosis von 480 mg. Bei Patienten mit einer eingeschränkten Nierenfunktion (Kreatininclearance <50 ml/min), die Prevymis Infsuionslösung erhalten, könnte eine Akkumulation von Hydroxypropylbetadex auftreten. Bei diesen Patienten ist eine engmaschige Überprüfung der Serumkreatininspiegel erforderlich.
Patienten mit eingeschränkter Leberfunktion
Bei leichter (Child-Pugh-Klasse A) bis mässiger (Child-Pugh-Klasse B) Einschränkung der Leberfunktion ist keine Dosisanpassung von Prevymis erforderlich. Prevymis wird bei Patienten mit schwerer Einschränkung der Leberfunktion (Child-Pugh-Klasse C) nicht empfohlen.
Prevymis wird bei Patienten mit mässiger Einschränkung der Leberfunktion in Kombination mit mässiger oder schwerer Einschränkung der Nierenfunktion nicht empfohlen (siehe Abschnitt «Pharmakokinetik»).
Ältere Patienten
Von den 373 in einer randomisierten, doppelblinden, placebokontrollierten Phase-3-Studie mit Prevymis behandelten Patienten (P001) waren 56 (15,0%) 65 Jahre und älter. Die Sicherheit und Wirksamkeit waren bei älteren und jüngeren Patienten ähnlich. Eine altersentsprechende Dosisanpassung von Prevymis ist nicht erforderlich.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Prevymis bei Patienten unter 18 Jahren sind nicht erwiesen. Es liegen keine Daten vor (siehe Abschnitt «Pharmakokinetik»).
KontraindikationenPrevymis ist bei Patienten mit Überempfindlichkeit gegenüber Letermovir oder einem der Hilfsstoffe kontraindiziert.
Pimozid
Die gleichzeitige Gabe von Prevymis mit Pimozid kann wegen der Hemmung von Cytochrom P450 (CYP3A) durch Letermovir zu erhöhten Pimozidkonzentrationen führen, die QT-Verlängerung und Torsades de pointes zur Folge haben (siehe Abschnitte «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»).
Ergotalkaloide
Die gleichzeitige Gabe von Prevymis mit Ergotalkaloiden kann wegen der Hemmung von CYP3A durch Letermovir zu erhöhten Konzentrationen von Ergotalkaloiden (Ergotamin und Dihydroergotamin) führen, was Ergotismus zur Folge haben kann (siehe Abschnitte «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»).
Cyclosporin mit Pitavastatin oder Simvastatin
Die gleichzeitige Verabreichung von Prevymis in Kombination mit Cyclosporin kann zu einer signifikant erhöhten Pitavastatin- oder Simvastatin-Konzentration führen, was zu Myopathie oder Rhabdomyolyse führen kann (siehe Abschnitt «Besondere Warnhinweise und Vorsichtsmassnahmen», Risiko von Nebenwirkungen oder verminderter therapeutischer Wirkung aufgrund von Arzneimittelwechselwirkungen und Abschnitt «Interaktionen» von Prevymis auf andere Medikamente).
Warnhinweise und VorsichtsmassnahmenRisiko von unerwünschten Reaktionen oder einer Verminderung der therapeutischen Wirkung aufgrund von Arzneimittelinteraktionen
Die gleichzeitige Anwendung von Prevymis und bestimmten Arzneimitteln kann zu bekannten oder potenziell signifikanten Arzneimittelinteraktionen führen. Mögliche Folgen können sein:
·Klinisch signifikante unerwünschte Reaktionen durch erhöhte Exposition gegenüber Begleitmedikamenten oder Prevymis.
·Signifikante Verringerung der Plasmakonzentration von Begleitmedikamenten, die zu einer Verminderung der therapeutischen Wirkung des Begleitmedikaments führen kann.
Massnahmen zur Vermeidung bzw. Beherrschung dieser bekannten oder potenziell signifikanten Arzneimittelinteraktionen einschliesslich Dosierungsempfehlungen sind Tabelle 1 zu entnehmen (siehe Abschnitte «Kontraindikationen» und «Interaktionen»).
Prevymis sollte mit Vorsicht zusammen mit Arzneimittel, die CYP3A-Substrate mit enger therapeutischer Breite sind (z.B. Alfentanil, Fentanyl und Chinidin) verwendet werden, da die gleichzeitige Gabe zu Erhöhungen der Plasmakonzentrationen von CYP3A-Substraten führen kann. Eine engmaschige Überwachung und/oder Dosisanpassung gleichzeitig verabreichter CYP3A-Substrate wird empfohlen. Die Angaben in der Fachinformation der betroffenen CYP3A-Substrate sind zu beachten. (Siehe Tabelle 1 und Abschnitt «Interaktionen»).
Laktoseintoleranz
Die Tabletten enthalten Laktosemonohydrat. Patienten mit der seltenen hereditären Problemen von Galaktose-Intoleranz, Lapp-Laktase-Mangel oder Glukose-Galaktose-Malabsorption sollten dieses Arzneimittel nicht anwenden.
Natrium
Prevymis 240 mg Konzentrat zur Herstellung einer Infusionslösung enthält 22,91 mg (oder 1,00 mmol) Natrium je Dosis. Dies ist bei Patienten, die eine kochsalzarme Diät einhalten müssen, zu berücksichtigen.
Prevymis 480 mg Konzentrat zur Herstellung einer Infusionslösung enthält 45,82 mg (oder 1,99 mmol) Natrium je Dosis. Dies ist bei Patienten, die eine kochsalzarme Diät einhalten müssen, zu berücksichtigen.
InteraktionenBei gesunden Probanden wurden Studien zu Arzneimittelinteraktionen mit Prevymis und Arzneimitteln, die wahrscheinlich gleichzeitig verabreicht werden, oder Arzneimitteln, die häufig als Prüfmedikamente für pharmakokinetische Interaktionen verwendet werden, durchgeführt (siehe Tabelle 1 und Tabelle 2).
Effekte anderer Medikamente auf Prevymis
Die In-vitro-Resultate zeigen, dass Letermovir ein Substrat von OATP1B1/3, P-gp, UGT1A1 und UGT1A3 ist. Inhibitoren von OATP1B1/3-Transportern können zu erhöhten Letermovirkonzentrationen im Plasma führen.
OATP1B1 oder 3 Inhibitoren
Wenn Prevymis gleichzeitig mit Cyclosporin (einem potenten OATP1B1/3-Inhibitor) verabreicht wird, beträgt die empfohlene Dosis Prevymis 240 mg einmal täglich (siehe Abschnitt «Dosierung und Art der Anwendung»). Weitere Beispiele für OATP1B1 Inhibitoren sind Gemfibrozil, Erythromycin, Clarithromycin sowie diverse Protease Inhibitoren (z.B. Atazanavir, Lopinavir Ritonavir, Simeprevir).
P-gp Inhibitoren
Es wird nicht erwartet, dass die Veränderungen der Letermovirkonzentrationen im Plasma aufgrund der Inhibition von P-gp klinisch relevant sind.
UGT1A1 oder 3 Inhibitoren
Es wird nicht erwartet, dass die Inhibition von UGT einen klinisch relevanten Effekt auf die Letermovirkonzentrationen im Plasma hat.
CYP Inhibitoren oder Induktoren
Zwar wurden CYP3A, CYP2D6 und CYP2J2 als Enzyme identifiziert, die in der Lage sind, den Metabolismus von Letermovir in vitro zu vermitteln, doch wird der oxidative Metabolismus auf Grundlage humaner In-vivo-Daten nur als unbedeutender Ausscheidungsweg betrachtet.
Effekt von Prevymis auf andere Medikamente
CYP Substrate
Letermovir ist ein zeitabhängiger Inhibitor und Induktor von CYP3A in vitro. Die gleichzeitige Gabe von Prevymis mit Midazolam führte zu einer verstärkten Midazolam-Exposition, was darauf hindeutet, dass der Nettoeffekt von Letermovir auf CYP3A eine mässige Inhibition ist (siehe Tabelle 2). Nach diesen Ergebnissen kann die gleichzeitige Gabe von Prevymis mit CYP3A-Substraten die Plasmakonzentrationen von CYP3A-Substraten erhöhen (siehe Abschnitte «Kontraindikationen», «Warnhinweise und Vorsichtsmassnahmen», «Interaktionen» und Tabelle 1).
Letermovir ist in vitro ein reversibler Inhibitor von CYP2C8. Die physiologisch basierte pharmakokinetische Modellierung sagt eine Erhöhung der Plasmakonzentrationen von CYP2C8-Substraten bei gleichzeitiger Gabe mit Prevymis voraus (siehe Tabelle 1 und Tabelle 2).
Die gleichzeitige Gabe von Prevymis reduzierte die Voriconazol-Exposition, höchstwahrscheinlich aufgrund der Induktion der Voriconazol-Ausscheidungswege CYP2C9 und CYP2C19. Durch die gleichzeitige Verabreichung von Prevymis mit CYP2C9- und CYP2C19-Substraten können die Plasmakonzentrationen der CYP2C9- und CYP2C19-Substrate verringert werden (siehe Tabelle 1). Weitere Beispiele für CYP2C9 oder 2C19 Substrate sind Warfarin Phenytoin, Diazepam, Lansoprazol, Omeprazol, Esomeprazol, Pantoprazol, Tolbutamid.
Letermovir ist in vitro ein Induktor von CYP2B6; die klinische Relevanz ist unbekannt.
Substrate von Transportern
Letermovir bewirkte in vitro eine Hemmung der Effluxtransporter P-gp, Breast Cancer Resistance Protein (BCRP), Gallensalz-Export-Pumpe (BSEP), Multidrug-Resistance-associated Protein 2 (MRP2), OAT3 und des hepatischen Aufnahmetransporters OATP1B1/3.
Die gleichzeitige Gabe von Prevymis mit Substraten von OATP1B1/3-Transportern (z.B. Atorvastatin, einem bekannten Substrat von CYP3A, OATP1B1/3 und möglicherweise BCRP) kann zu einer klinisch relevanten Erhöhung der Plasmakonzentrationen von OATP1B1/3-Substraten führen (siehe Tabelle 1). Das Ausmass von OATP1B1/3 vermittelten Interaktionen kann grösser sein, wenn Prevymis zusammen mit Cyclosporin verabreicht wird.
Klinisch relevante Veränderungen der Plasmakonzentrationen von Digoxin, einem P-gp-Substrat, oder Aciclovir, einem OAT3-Substrat, nach der gleichzeitigen oralen Verabreichung mit Prevymis in klinischen Studien lagen nicht vor (siehe Tabelle 2).
Der Effekt von Letermovir auf BCRP-, BSEP- und MRP2-Substrate wurde nicht in klinischen Studien beurteilt; die klinische Relevanz ist unbekannt.
Falls Dosisanpassungen von Begleitmedikamenten aufgrund einer Therapie mit Prevymis vorgenommen werden, sollten diese Dosen nach Abschluss der Therapie mit Prevymis erneut angepasst werden.
Wenn Prevymis zusammen mit Cyclosporin verabreicht wird, kann die kombinierte Wirkung auf CYP3A-Substrate ähnlich einem starken CYP3A-Inhibitor sein. Beachten Sie die Fachinformation für die Dosierung des CYP3A-Substrats mit einem starken CYP3A-Inhibitor.
Wenn Prevymis zusammen mit Cyclosporin verabreicht wird, kann die kombinierte Wirkung auf Wirkstoffe, die sowohl CYP3A- als auch OATP1B1/3-Substrate sind, anders sein als bei der alleinigen Verabreichung mit Prevymis. Beachten Sie die Fachinformation sowohl für das gemeinsam verabreichte Medikament als auch für Cyclosporin.
Tabelle 1 enthält eine Liste der erwiesenermassen oder potenziell klinisch signifikanten Arzneimittelinteraktionen. Die beschriebenen Arzneimittelinteraktionen beruhen auf Studien, die mit Prevymis durchgeführt wurden, oder es handelt sich um vorhergesagte Arzneimittelinteraktionen, die mit Prevymis auftreten können (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Tabelle 1: Interaktionen mit anderen Arzneimitteln und Dosierungsempfehlungen: Eine Dosisanpassung kann aufgrund der Ergebnisse von Arzneimittel-Interaktionsstudien oder vorhergesagten Interaktionen empfohlen werden*
|
Klasse und/oder Ausscheidungsweg der begleitend verabreichten Arzneimittel: Name des Wirkstoffs
|
Effekt auf die Konzentration†
Mittelwerte (90% Konfidenzintervall) für AUC, Cmax (wahrscheinlicher Wirkungsmechanismus)
|
Empfehlungen zur gleichzeitigen Anwendung mit Prevymis
| |
Antiarrhythmika
| |
Amiodaron§
|
↑ Amiodaron
|
Die gleichzeitige Verabreichung von Prevymis mit Amiodaron erhöht die Plasmakonzentration von Amiodaron. Eine enge klinische Überwachung auf unerwünschte Ereignisse im Zusammenhang mit Amiodaron wird während der gleichzeitigen Verabreichung empfohlen. Überwachen Sie häufig die Amiodaron-Konzentrationen.
| |
Antidiabetika
| |
Glyburid §
|
↑ Glyburid
|
Prevymis kann die Plasmakonzentration von Glyburid erhöhen. Eine häufige Überwachung der Glukosekonzentration wird empfohlen#.
| |
Antimykotika
| |
Fluconazol
(400 mg Einzeldosis PO/ Letermovir 480 mg Einzeldosis PO)
|
↔ Letermovir
AUC 1.11 (1.01, 1.23)
Cmax 1.06 (0.93, 1.21)
↔ Fluconazol
AUC 1.03 (0.99, 1.08)
Cmax 0.95 (0.92, 0.99)
|
Keine Dosisanpassung erforderlich.
| |
Posaconazol‡
(300 mg Einzeldosis)/ Letermovir (480 mg täglich)
|
↔ Posaconazol
AUC 0.98 (0.82, 1.17)
Cmax 1.11 (0.95, 1.29)
|
Keine Dosisanpassung erforderlich.
| |
Voriconazol‡
(200 mg zweimal täglich/ Letermovir (480 mg täglich)
|
↓ Voriconazol
AUC 0.56 (0.51, 0.62)
Cmax 0.61 (0.53, 0.71)
(CYP2C9/19-Induktion)
|
Wenn die gleichzeitige Verabreichung erforderlich ist, wird eine engmaschige Überwachung auf verminderte Wirksamkeit und ein therapeutisches Arzneimittel-Monitoring (TDM) von Voriconazol empfohlen.#
| |
Virostatika
| |
Aciclovir‡
(400 mg Einzeldosis)/ Letermovir (480 mg täglich)
|
↔ Aciclovir
AUC 1.02 (0.87, 1.2)
Cmax 0.82 (0.71, 0.93)
|
Keine Dosisanpassung erforderlich.
| |
HMG-CoA-Reduktase-Inhibitoren
| |
Pitavastatin§, Simvastatin§
|
↑Pitavastatin
↑Simvastatin
(CYP3A und/oder OATP1B1/3, und potentielle intestinale BCRP-Inhibition)
|
Die Behandlung mit HMG-CoA-Reduktase-Inhibitoren sollte während der Behandlung mit Prevymis# ausgesetzt werden.
Wenn Prevymis zusammen mit Cyclosporin verabreicht wird, ist die Verwendung von Pitavastatin oder Simvastatin kontraindiziert (siehe Abschnitt «Kontraindikationen»).
| |
Atorvastatin‡
(20 mg Einzeldose)/ Letermovir (480 mg täglich)
|
↑ Atorvastatin
AUC 3.29 (2.84, 3.82)
Cmax 2.17 (1.76, 2.67)
(CYP3A, OATP1B1/3 Inhibition)
|
Die Behandlung mit HMG-CoA Reduktase Inhibitoren sollte während der Behandlung mit Prevymis ausgesetzt werden.
| |
Sonstige HMG-CoA-Reduktase-Inhibitoren§
Beispiele: Fluvastatin,
Lovastatin,
Pravastatin,
Rosuvastatin
|
↑ Konzentrationen von HMG-CoA-Reduktase-Inhibitoren
(CYP3A- und/oder OATP1B1/3- und potenziell intestinale BCRP-Inhibition)
| |
Immunsuppressiva
| |
Cyclosporin
(50 mg Einzeldosis)/ Letermovir (240 mg Täglich)
|
↑ Cyclosporin
AUC 1.66 (1.51, 1.82)
Cmax 1.08 (0.97, 1.19)
(CYP3A-Inhibition)
|
Wenn Prevymis zusammen mit Cyclosporin verabreicht wird, sollte die Dosierung von Prevymis auf 240 mg einmal täglich reduziert werden (siehe die Abschnitte «Dosierung und Art der Anwendung» und «Pharmakokinetik»).
Während und bei Abbruch der Anwendung von Prevymis sollte eine häufige Überwachung der Cyclosporinkonzentrationen im Vollblut erfolgen und die Cyclosporindosis entsprechend angepasst werden.#
| |
Cyclosporine
(200 mg Einzeldosis)/ Letermovir (240 mg Täglich)
|
↑ Letermovir
AUC 2.11 (1.97, 2.26)
Cmax 1.48 (1.33, 1.65)
(OATP1B1/3 Inhibition)
| |
Mycophenolat-Mofetil
(1 g Einzeldosis)/ Letermovir (480 mg Täglich)
|
↔ Mycophenolsäure
AUC 1.08 (0.97, 1.20)
Cmax 0.96 (0.82, 1.12)
↔ Letermovir
AUC 1.18 (1.04, 1.32)
Cmax 1.11 (0.92, 1.34)
|
Keine Dosisanpassung für Mycophenolat Mofetil erforderlich.
| |
Sirolimus‡
(2 mg Einzeldosis)/ Letermovir (480 mg täglich)
|
↑ Sirolimus
AUC 3.40 (3.01, 3.85)
Cmax 2.76 (2.48, 3.06)
(CYP3A-Inhibition)
|
Während und bei Abbruch der Anwendung von Prevymis sollte eine häufige Überwachung der Sirolimuskonzentrationen im Vollblut erfolgen und die Sirolimusdosis entsprechend angepasst werden.#
Wenn Prevymis zusammen mit Cyclosporin verabreicht wird, beziehen Sie sich bitte auf die Fachinformation von Sirolimus, um spezifische Dosierungsempfehlungen für die Verwendung von Sirolimus mit Cyclosporin zu erhalten.#
| |
Tacrolimus
(5 mg Einzeldosis)/ Letermovir (480 mg täglich)
Tacrolimus
(5 mg Einzeldosis)/ Letermovir (80 mg zwei mal täglich)
|
↑ Tacrolimus
AUC 2.42 (2.04, 2.88)
Cmax 1.57 (1.32, 1.86)
(CYP3A-Inhibition)
↔ Letermovir
AUC 1.02 (0.97, 1.07)
Cmax 0.92 (0.84, 1.00)
|
Während und bei Abbruch der Anwendung von Prevymis sollte eine häufige Überwachung der Tacrolimuskonzentrationen im Vollblut erfolgen und die Tacrolimusdosis entsprechend angepasst werden
Wenn Prevymis zusammen mit Cyclosporin verabreicht wird, beziehen Sie sich auf die Tacrolimus-Fachinformation für spezifische Dosierungsempfehlungen für die Verwendung von Tacrolimus mit Cyclosporin.#
| |
Orale Kontrazeptiva
| |
Ethinylestradiol (EE) (0,03 mg)//Levonorgestrel (LNG)‡ (0,15 mg) Einzeldosis/ Letermovir (480 mg täglich)
|
↔ EE
AUC 1.42 (1.32, 1.52)
Cmax 0.89 (0.83, 0.96)
↔ LNG
AUC 1.36 (1.30, 1.43)
Cmax 0.95 (0.86, 1.04)
|
Prevymis kann mit hormonellen Verhütungsmitteln verwendet werden. Die klinische Relevanz der von Prevymis erwarteten Erhöhung der Ethinylesteradiol- und Levonorgestrelspiegel, bei mehrfacher Verabreichung dieser Wirkstoffe, ist nicht bekannt.
| |
Protonenpumpeninhibitoren
| |
Omeprazol§, Pantoprazol§
|
↓Omeprazol
↓Pantoprazol
|
Die gleichzeitige Verabreichung von Prevymis mit diesen Protonenpumpeninhibitoren (PPI) kann die Plasmakonzentration der PPIs verringern. Klinische Überwachung und Dosisanpassung können erforderlich sein, wenn sie gemeinsam mit Prevymis verabreicht werden#.
| |
CYP2C8-Substrate**
| |
Beispiele: Repaglinid§, Rosiglitazon§
|
↑ Konzentrationen von CYP2C8-Substraten
|
Prevymis kann die Plasmakonzentrationen von CYP2C8-Substraten erhöhen.
Während der gleichzeitigen Anwendung mit Repaglinid oder Rosiglitazon wird eine häufige Überwachung der Glukosekonzentrationen empfohlen. Wenn Prevymis gemeinsam mit Cyclosporin verabreicht wird, wird erwartet, dass der Anstieg der Plasmakonzentrationen von Repaglinid aufgrund der OATP1B-Inhibition durch Cyclosporin grösser sein wird als bei Prevymis allein. Spezifische Dosierungsempfehlungen finden Sie in der Repaglinid-Fachinformation.#
| |
CYP2C9/19-Substrate
| |
Beispiele: Phenytoin§, Warfarin§
|
↓ Konzentrationen von CYP2C9/19-Substraten
|
Prevymis kann die Plasmakonzentrationen von CYP2C9/19-Substraten vermindern.
Bei gleichzeitiger Anwendung von Phenytoin mit Prevymis sollte eine häufige Überwachung der Phenytoinkonzentrationen erfolgen.#
Bei gleichzeitiger Anwendung von Warfarin mit Prevymis sollte eine häufige Überwachung der INR erfolgen.#
| |
CYP3A-Substrate††
| |
Midazolam
(1 mg Einzeldosis IV)/ Letermovir (240 mg einmal täglich PO)
Midazolam (2 mg Einzeldosis PO) / Letermovir (240 mg einmal täglich PO)
|
↑ Midazolam
IV:
AUC 1.47 (1.37, 1.58)
Cmax 1.05 (0.94, 1.17)
PO:
AUC 2.25 (2.04, 2.49)
Cmax 1.72 (1.55, 1.92)
(CYP3A Inhibition)
|
Prevymis kann die Plasmakonzentrationen von CYP3A-Substraten erhöhen.
Wenn Letermovir allein mit einem CYP3A-Substrat verabreicht wird, ist die Fachinformation für die Dosierung des CYP3A-Substrats mit einem moderaten CYP3A-Inhibitor zu konsultieren.
Bei gleichzeitiger Anwendung wird eine häufige Überwachung auf unerwünschte Reaktionen im Zusammenhang mit diesen Arzneimitteln empfohlen. Eventuell ist eine Dosisanpassung von CYP3A-Substraten erforderlich# (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»)
| |
Andere Beispiele: Alfentanil§, Fentanyl§, Chinidin§
|
↑ Konzentrationen von CYP3A-Substraten
| |
P-gp-Substrate
| |
Digoxin‡
(0.5 mg Einzeldosis)/ Letermovir (240 mg Zweimal täglich)
|
↔ Digoxin
AUC 0.88 (0.80, 0.96)
Cmax 0.75 (0.63, 0.89)
(P-gp Induktion)
|
Keine Dosisanpassung erforderlich.
| |
* Diese Tabelle ist nicht allumfassend.
† ↓ =verringert, ↑=erhöht, ↔ =keine klinisch relevante Veränderung
‡ Einweg-Interaktionsstudie zur Beurteilung des Effekts von Letermovir auf die Begleitmedikation.
§ Diese Interaktionen wurden nicht untersucht.
# Siehe die jeweilige Fachinformation.
** Aufgrund der physiologisch basierten pharmakokinetischen Modellierung.
†† Auf der Basis von In-vivo-Studien mit Midazolam.
|
Schwangerschaft/StillzeitSchwangerschaft
Es liegen keine klinischen Daten zur Anwendung von Prevymis bei Schwangeren vor. Embryofetale Toxizität wurde bei Ratten und Kaninchen bei maternal-toxischen systemischen AUC Expositionen des ca. 11- bzw. 2-Fachen der AUC unter der empfohlenen Humandosis (RHD) beobachtet. In tierexperimentellen Studien wurde Reproduktionstoxizität bei maternal-toxischen Dosen beobachtet (siehe Abschnitt «Präklinische Daten»).
Das potenzielle Risiko für den Menschen ist unbekannt. Prevymis sollte während der Schwangerschaft nicht verabreicht werden, es sei denn dies ist eindeutig erforderlich.
Stillzeit
Es ist nicht bekannt, ob Letermovir beim Menschen in die Muttermilch übergeht, die Milchbildung beeinflusst oder Auswirkungen auf das gestillte Kind hat.
Bei Verabreichung an laktierende Ratten war Letermovir in der Milch nachweisbar, hatte aber keine Auswirkungen auf das Wachstum und die Entwicklung der gesäugten Jungtiere. Der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau sind bei einer Entscheidung zu berücksichtigen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenPrevymis hat wahrscheinlich keinen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen.
Unerwünschte WirkungenErfahrungen aus klinischen Prüfungen
Die Sicherheit von Prevymis wurde in einer randomisierten, doppelblinden, placebokontrollierten Phase-3-Studie (P001) beurteilt, in der 565 Patienten randomisiert und bis Woche 14 nach der Transplantation mit Prevymis (N=373) oder Placebo (N=192) behandelt und bis Woche 24 nach der Transplantation einer Nachbeobachtung der Sicherheit unterzogen wurden (siehe Abschnitt «Pharmakokinetik»).
Häufig auftretende unerwünschte Ereignisse
Die Häufigkeit von unerwünschten Ereignissen (unabhängig von der vom Prüfer bewerteten Kausalität), die bei mindestens 10% der Probanden der Prevymis-Gruppe und bei einer Häufigkeit, die mindestens 2% höher ist als die von Placebo, aufgetreten ist, ist in Tabelle 2 nach Systemorganklasse und Häufigkeit aufgeführt. Die Häufigkeit ist wie folgt definiert: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1'000, <1/100); selten (≥1/10'000, <1/1'000); sehr selten (<1/10'000).
Tabelle 2: Studie P001 Unerwünschte Ereignisse in ≥10% der Prevymis-behandelten HSCT-Empfänger mit einer Häufigkeit, mindestens 2% grösser als Placebo.
|
Häufigkeit
|
Unerwünschte Ereignisse
| |
Erkrankungen des Nervensystems
| |
sehr häufig
|
Kopfschmerzen (14% vs. 9% Placebo)
| |
Erkrankungen der Atemwege
| |
sehr häufig
|
Husten (14% vs. 10% Placebo)
| |
Erkrankungen des Gastrointestinaltrakts
| |
sehr häufig
|
Übelkeit (27% vs. 23% Placebo), Diarrhö (26% vs. 24% Placebo), Erbrechen (19% vs. 14% placebo), Abdominalschmerzen (12% vs. 9% Placebo)
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
| |
Häufig
|
Erschöpfung (13% vs. 11% Placebo), periphes Ödem (14% vs. 9% placebo)
|
Insgesamt stellten ähnliche Anteile der Probanden in beiden Gruppe die Studienmedikation aufgrund eines unerwünschten Ereignisses ein (13% der Prevymis Patienten vs. 12% der Placebo Patienten). Das am häufigsten gemeldete unerwünschte Ereignis, das zum Abbruch der Studie führte, war Übelkeit, die bei 2% der Prevymis Patienten und 1% der Placebo Patienten auftrat.
Kardiale Ereignisse
Die Frequenz der kardialen Ereignisse (unabhängig von der vom Prüfer bewerteten Kausalität) war bei den Probanden, die Prevymis erhielten, höher (13%) als bei den Probanden, die Placebo erhielten (6%). Die häufigsten kardialen Ereignisse waren Tachykardie (berichtet in 4% der Prevymis-Patienten und in 2% der Placebo-Patienten) und Vorhofflimmern (berichtet in 3% der Prevymis-Patienten und in 1% der Placebo-Patienten). Unter den 6 Probanden, die eine oder mehrere kardiale Ereignisse hatten, wiesen 85% der Prevymis- und 92% der Placebo-Patienten Ereignisse auf, die als leicht oder mittelmässig eingestuft wurden.
Beschreibung ausgewählter unerwünschter Reaktionen
Unter Prevymis wurde bei einem Patienten über Überempfindlichkeit als nicht schwerwiegende unerwünschte Reaktion berichtet.
Abnorme Laborwerte
Insgesamt war der Anteil der Patienten mit potenziell klinisch signifikanten Veränderungen der Laborwerte (z.B. Hämatologie, Chemie, Nieren- und Leberfunktion) in der Prevymis- und der Placebo-Gruppe ähnlich. Es gab keine Unterschiede in der Inzidenz oder der Zeit bis zum Einwachsen der Stammzellen (Engraftment) zwischen der Prevymis- und der Placebo-Gruppe.
In P001 wurden bei männlichen Patienten Biomarker der testikulären Toxizität bewertet (siehe Abschnitt «Präklinische Daten»). Veränderungen der männlichen Geschlechtshormone (Serum-Inhibin B, luteinisierendes Hormon (LH), follikelstimulierendes Hormon (FSH) und Testosteron) gegenüber dem Ausgangswert waren in der Prevymis- und der Placebo-Gruppe ähnlich.
ÜberdosierungEs liegen keine Erfahrungen zur Überdosierung von Prevymis beim Menschen vor. In klinischen Studien der Phase 1 erhielten 86 gesunde Probanden bis zu 14 Tage Prevymis-Dosen von 720 mg/Tag bis 1'440 mg/Tag. Das Profil der unerwünschten Reaktionen entsprach etwa dem Profil unter der klinischen Dosis von 480 mg/Tag. Es steht kein spezifisches Antidot für den Fall einer Überdosierung von Prevymis zur Verfügung. Bei einer Überdosierung wird empfohlen, den betroffenen Patienten engmaschig auf unerwünschte Reaktionen zu überwachen. Gegebenenfalls muss eine geeignete symptomatische Therapie eingeleitet werden.
Es ist nicht bekannt, ob Prevymis durch Dialyse in bedeutendem Umfang aus dem systemischen Kreislauf entfernt wird.
Eigenschaften/WirkungenATC-Code: J05AX18
Prevymis ist ein antivirales Arzneimittel gegen CMV.
Wirkungsmechanismus
Letermovir ist ein antivirales Arzneimittel gegen CMV.
Letermovir hemmt den Terminase-Komplex der CMV-DNA, der für die virale Replikation erforderlich ist. Biochemische Charakterisierung und Elektronenmikroskopie haben gezeigt, dass Letermovir die Bildung von Genomen mit geeigneter Einheitslänge beeinflusst und die Virion-Reifung beeinträchtigt.
Antivirale Aktivität
Der mittlere EC50-Wert von Letermovir gegen eine Reihe von klinischen CMV-Isolaten in einem Zellkultur-Infektionsmodell betrug 2,1 nM (Spanne 0,7 nM bis 6,1 nM n=74).
Virale Resistenz
In Zellkulturen
Die CMV-Gene UL56 und UL89 kodieren Untereinheiten der CMV-DNA-Terminase. CMV-Mutanten mit reduzierter Empfindlichkeit gegenüber Letermovir sind in Zellkultur selektiert worden. Die Mutationen korrespondieren mit UL56 und treten in Aminosäureresten zwischen 231 und 369 auf (V231A, V231L, V236L, V236M, E237D, L241P, T244K, T244R, L257I, F261C, F261L, F261S, Y321C, C325F, C325R, C325Y, M329T, R369G, R369M, R369S). Die EC50-Werte für diese Mutationen sind um das 13- bis 5'870-Fache höher als beim Wildtyp-Referenzvirus. Es korrespondieren keine bekannten Letermovir-Resistenzmutationen mit UL89.
In klinischen Studien
In einer Phase-2b-Studie zur Beurteilung von Letermovir-Dosen von 60, 120 oder 240 mg/Tag oder Placebo über einen Zeitraum von bis zu 84 Tagen bei 131 HSZT-Empfängern wurde eine DNA-Sequenzanalyse einer ausgewählten UL56-Region (Aminosäuren 231 bis 369) an Proben von 12 mit Letermovir behandelten Studienteilnehmern durchgeführt, bei denen es zu Prophylaxeversagen kam und von denen Proben zur Analyse verfügbar waren. Bei einem Studienteilnehmer (der 60 mg/Tag erhielt) lag eine Letermovir-resistente Genotyp-Variante (GV) (V236M) vor.
In einer Phase-3-Studie (P001) wurde eine DNA-Sequenzanalyse der gesamten kodierenden Regionen von UL56 und UL89 an Proben von 22 mit Letermovir behandelten Patienten der FAS-Population durchgeführt, bei denen es zu Prophylaxeversagen kam und von denen Proben zur Analyse verfügbar waren. Bei einem Patienten lag eine Letermovir-resistente GV (V236M) vor.
Kreuzresistenz
Eine Kreuzresistenz mit Arzneimitteln ausserhalb dieser Klasse ist nicht wahrscheinlich. Letermovir ist voll wirksam gegen Viruspopulationen mit Substitutionen, die Resistenz gegen CMV-DNA-Polymerase-Hemmer (Ganciclovir, Cidofovir und Foscarnet) verleihen. Diese DNA-Polymerase-Hemmer sind voll wirksam gegen Viruspopulationen mit Substitutionen, die Resistenz gegen Letermovir verleihen.
Pharmakogenomik
Die Auswirkung genetischer Varianten im OATP1B1-Gen SLCO1B1 (rs4149056, rs2306283, rs4149032) und UGT1A1 (rs4148323 und Promoter-TA-Repeat-Varianten) auf die Pharmakokinetik von Letermovir wurde bei 299 Studienteilnehmern beurteilt. Klinisch relevante Auswirkungen dieser Varianten auf die Letermovir-Expositionen lagen nicht vor.
Kardiale Elektrophysiologie
Die Auswirkung von Letermovir-Dosen bis zu 960 mg i.v. auf das QTc-Intervall wurde in einer randomisierten, sowohl Placebo- als auch aktiv (Moxifloxacin 400 mg oral) kontrollierten, vierphasigen Einzeldosis-Crossover-Studie zur QT-Zeit an 38 gesunden Probanden untersucht. Letermovir verlängert das QTc-Intervall nicht in klinisch relevantem Umfang nach der i.v.-Dosis von 960 mg mit Plasmakonzentrationen, die etwa doppelt so hoch sind wie bei der i.v.-Dosis von 480 mg.
Klinische Wirksamkeit und Sicherheit
Erwachsene CMV-seropositive Empfänger [R+] eines allogenen hämatopoetischen Stammzelltransplantats
Zur Beurteilung der Letermovir-Prophylaxe als Präventionsstrategie gegen eine CMV-Infektion oder -Erkrankung wurde die Wirksamkeit von Letermovir in einer multizentrischen, doppelblinden, placebokontrollierten Phase-3-Studie (P001) mit erwachsenen CMV-seropositiven Empfängern [R+] eines allogenen HSZT untersucht. Die Patienten erhielten per Randomisierung (2:1) oder Placebo. Die Randomisierung war nach Studienzentrum und Risiko einer CMV-Reaktivierung (hoch gegenüber niedrig) bei Eintritt in die Studie stratifiziert. Die Letermovir-Therapie wurde nach HSZT (Tag 0-28 nach der Transplantation) eingeleitet und bis Woche 14 nach der Transplantation fortgesetzt. Letermovir wurde entweder oral oder i.v. verabreicht. Bis Woche 24 nach der Transplantation wurden die Patienten im Hinblick auf den primären Wirksamkeitsendpunkt überwacht; bis Woche 48 nach der Transplantation wurde eine kontinuierliche Nachbeobachtung durchgeführt.
Von den 565 behandelten Patienten erhielten 373 Letermovir (einschliesslich 99 Patienten, die mindestens eine i.v.-Dosis erhielten) und 192 Placebo (einschliesslich 48 Patienten, die mindestens eine i.v.-Dosis erhielten). Die mediane Zeit bis zum Beginn der Letermovir-Behandlung betrug 9 Tage nach der Transplantation. Bei 37% der Patienten war das Einwachsen der Stammzellen (Engraftment) zu Studienbeginn erfolgt. Das mediane Alter betrug 54 Jahre (Spanne 18 bis 78 Jahre). Zu Studienbeginn erhielten 50% der Patienten eine myeloablative Therapie, 52% bekamen Cyclosporin und 42% Tacrolimus. Die häufigsten primären Gründe für die Transplantation waren akute myeloische Leukämie (38%), myeloblastisches Syndrom (15%) und Lymphom (13%). Zwölf Prozent (12%) der Patienten waren zu Studienbeginn positiv auf CMV-DNA getestet.
Zu Studienbeginn bestand bei 31% der Patienten ein hohes Risiko einer Reaktivierung, definiert durch mindestens eines der folgenden Kriterien: Humanes-Leukozytenantigen-(HLA-)verwandter Spender (Bruder oder Schwester) mit mindestens einem Mismatch an einem der folgenden drei HLA-Genloci: HLA-A, -B oder -DR, haploidentischer Spender; nicht verwandter Spender mit mindestens einem Mismatch an einem der folgenden vier HLA-Genloci: HLA-A, -B, -C und -DRB1; Verwendung von Nabelschnurblut als Stammzellquelle; Verwendung von Ex-vivo-T-Zellerschöpften Transplantaten; Graft-versus-Host-Krankheit (GVHD) Grad 2 oder höher, die systemische Kortikosteroide erforderlich machte.
Wirksamkeit
Klinisch signifikante CMV-Infektion
Der primäre Wirksamkeitsendpunkt von P001 war das Auftreten einer klinisch signifikanten CMV-Infektion bis Woche 24 nach der Transplantation. Als klinisch signifikante CMV-Infektion wurde entweder das Auftreten einer CMV-Endorgan-Erkrankung oder die Einleitung einer präemptiven Therapie (PET) gegen CMV aufgrund einer dokumentierten CMV-Virämie (unter Verwendung des Roche COBAS AmpliPrep/COBAS TaqMan Assay,.
Letermovir zeigte in der Analyse des primären Endpunkts eine überlegene Wirksamkeit gegenüber Placebo, wie in Tabelle 3 dargestellt. Der geschätzte Behandlungsunterschied von -23,5% war statistisch signifikant (einseitiger p-Wert <0,0001).
Tabelle 3: P001: Wirksamkeitsergebnisse bei HSZT-Empfängern (NC=F-Ansatz, FAS-Population)
|
|
Letermovir
|
Placebo
| |
|
(N=325)
|
(N=170)
| |
Parameter
|
n (%)
|
n (%)
| |
Primärer Endpunkt
(Anteil der Patienten mit Prophylaxeversagen)
|
122 (37,5)
|
103 (60,6)
| |
Gründe für Versagen†
|
|
| |
Klinisch signifikante CMV-Infektion bis Woche 24‡
|
57 (17,5)
|
71 (41,8)
| |
Einleitung der präemptiven Therapie auf Basis der dokumentierten CMV-Virämie
|
52 (16,0)
|
68 (40,0)
| |
CMV-Endorganerkrankung
|
5 (1,5)
|
3 (1,8)
| |
Vor Woche 24 aus Studie ausgeschieden
|
56 (17,2)
|
27 (15,9)
| |
Fehlendes Outcome in Besuchsfenster Woche 24
|
9 (2,8)
|
5 (2,9)
| |
Stratum-adjustierter Behandlungsunterschied (Letermovir-Placebo)§
|
|
| |
Unterschied (95%-KI)
|
-23,5 (-32,5; -14,6)
|
| |
p-Wert
|
<0,0001
|
| |
† Die Kategorien für das Versagen schliessen sich gegenseitig aus und beruhen auf der Hierarchie der Kategorien in der aufgeführten Reihenfolge.
‡ Eine klinisch signifikante CMV-Infektion wurde definiert als CMV-Endorganerkrankung oder Einleitung einer präemptiven Therapie auf Basis einer dokumentierten CMV-Virämie und des klinischen Zustands des Patienten.
§ 95%-KIs und p-Wert für die Behandlungsunterschiede im prozentualen Ansprechen wurden unter Verwendung der Stratum-adjustierten Mantel-Haenszel-Methode berechnet, wobei der Unterschied nach dem harmonischen Mittelwert der Stichprobengrösse je Arm für jede Schicht (hohes oder niedriges Risiko) gewichtet wurde. Zur Erklärung der statistischen Signifikanz wurde ein einseitiger p-Wert von ≤0,0249 verwendet.
Hinweis: FAS= komplettes Analyse-Set; FAS umfasst die randomisierten Patienten, die mindestens eine Dosis des Studienmedikaments erhalten haben; ausgeschlossen sind Patienten mit nachweisbarer CMV-DNA zu Studienbeginn. Ansatz für die Behandlung fehlender Werte: Ansatz Abbruch=Versagen (Non-Completer=Failure - NC=F). Bei dem NC=F-Ansatz wurde Versagen definiert als alle Patienten, die eine klinisch signifikante CMV-Infektion entwickelten oder vorzeitig aus der Studie ausschieden oder bei denen bis zum Besuchsfenster in Woche 24 nach der Transplantation kein Outcome vorlag.
N = Anzahl der Patienten in jeder Behandlungsgruppe.
n (%) = Anzahl (Prozent) der Patienten in jeder Unterkategorie.
|
In Woche 24 nach der Transplantation betrug die Kaplan-Meier-(KM)-Ereignisrate für klinisch signifikante CMV-Infektion 18,9% in der Letermovir-Gruppe im Vergleich zu 44,3% in der Placebo-Gruppe (nomineller zweiseitiger stratifizierter Log-Rank-p-Wert <0,0001) (siehe Abb. 1). Die folgenden Faktoren standen bei den mit Letermovir behandelten Patienten mit einer klinisch signifikanten CMV-Infektion zwischen Woche 14 und Woche 24 nach der Transplantation in Zusammenhang:
·hohes Risiko einer CMV-Reaktivierung zu Studienbeginn,
·bestehende GVHD, und
·Anwendung eines Steroids zu irgendeinem Zeitpunkt nach der Randomisierung.
Abb. 1: P001: Kaplan-Meier-Plot der Zeit bis zum Beginn einer klinisch signifikanten CMV-Infektion bis Woche 24 nach der Transplantation bei HSZT-Empfängern (FAS-Population)
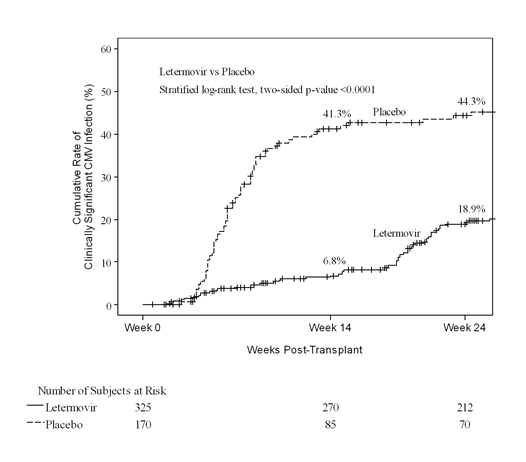
Im Hinblick auf die Wirksamkeit war Letermovir in allen Untergruppen, einschliesslich der folgenden, deutlich vorteilhafter:
·geringes oder hohes Risiko einer CMV-Reaktivierung,
·Konditionierungstherapien, und
·Begleittherapien mit Immunsuppressiva.
Mortalität
Die KM-Ereignisrate für die Gesamtmortalität in der Letermovir- gegenüber der Placebo-Gruppe betrug 12,1% gegenüber 17,2% in Woche 24 nach der Transplantation (nomineller zweiseitiger stratifizierter Log-Rank-p-Wert = 0,0401) bzw. 23,8% gegenüber 27,6% in Woche 48 nach der Transplantation (nomineller zweiseitiger stratifizierter Log-Rank-p-Wert = 0,2117). Die K-M-Ereignisrate für die Gesamtmortalität nach Geschlecht (Männer vs. Frauen) in der Woche 24 nach der Transplantation betrug 16,4% und 6,6% in der Letermovir-Gruppe und lag bei 14,2% und 25,4% in der Placebo-Gruppe; diese nach Geschlecht aufgeschlüsselten Ereignisraten sind mit Vorsicht zu interpretieren, da die Randomisierung nicht nach Geschlecht stratifiziert wurde, was bei Baseline zu Ungleichgewichten beim geschlechtsspezifischen Mortalitätsrisiko zwischen den Behandlungsgruppen führte.
PharmakokinetikAllgemeine Einleitung
Die Pharmakokinetik von Letermovir wurde nach oraler und intravenöser Verabreichung an gesunde Probanden und HSZT-Empfänger charakterisiert.
Bei gesunden Probanden stieg die Letermovir-Exposition bei beiden Verabreichungswegen (oral oder i.v.) nach Einzel- und Mehrfachdosen von 240 mg und 480 mg überproportional zur Dosis an. Letermovir wurde mit einer medianen Zeit bis zur maximalen Plasmakonzentration (Tmax) von 1.5 bis 3 Stunden schnell absorbiert, und die Plasmakonzentrationen fielen biphasisch ab. Die geometrischen Mittelwerte von AUC und Cmax im Steady State betrugen bei gesunden Probanden bei 480 mg Prevymis einmal täglich oral 71'500 ng•h/ml bzw.13'000 ng/ml. Das Plasmakonzentrations-Zeit-Profil nach Absorption von Letermovir nach oraler Gabe ähnelte dem nach intravenöser Gabe. Letermovir erreichte in 9 bis 10 Tagen einen Gleichgewichtszustand (Steady State) mit einem Akkumulationsverhältnis von 1,23 für AUC und 1,09 für Cmax.
Bei HSZT-Empfängern wurde die Letermovir-AUC anhand von populationspharmakokinetischen Analysen unter Verwendung von Phase-3-Daten geschätzt (siehe Tabelle 4). Die unterschiedliche Exposition in den einzelnen Therapieschemen ist klinisch nicht relevant; die Wirksamkeit war über den gesamten in P001 beobachteten Expositionsbereich konsistent.
Tabelle 4: AUC-Werte von Letermovir (ng•h/ml) bei HSZT-Empfängern
|
Therapieschema
|
Median (90% Prognoseintervall)*
| |
480 mg oral, kein Cyclosporin
|
34'400 (16'900, 73'700)
| |
480 mg i.v., kein Cyclosporin
|
100'000 (65'300, 148'000)
| |
240 mg oral, mit Cyclosporin
|
60'800 (28'700, 122'000)
| |
240 mg i.v., mit Cyclosporin
|
70'300 (46'200, 106'000)
| |
* Mediane und 90% Prognoseintervall basieren auf Simulationen unter Verwendung des Phase-3-Bevölkerungs-PK-Modells mit interindividueller Variabilität.
|
Absorption
Auf der Basis von populationspharmakokinetischen Analysen wurde die absolute Bioverfügbarkeit von Letermovir bei gesunden Probanden über den Dosisbereich von 240 mg bis 480 mg auf ca. 94% geschätzt. Bei HSZT-Empfängern wurde die Bioverfügbarkeit von Letermovir auf ca. 35% geschätzt, wenn 480 mg Prevymis einmal täglich oral ohne Cyclosporin verabreicht wurden. Die interindividuelle Variabilität für die Bioverfügbarkeit wurde auf ca. 37% geschätzt.
Effekt von Cyclosporin
Bei HSZT-Empfängern erhöhte die gleichzeitige Gabe von Cyclosporin die Plasmakonzentrationen von Letermovir. Die Bioverfügbarkeit von Letermovir wurde auf ca. 85% geschätzt, wenn 240 mg Prevymis einmal täglich oral zusammen mit Cyclosporin verabreicht wurden. Wenn Prevymis gleichzeitig mit Cyclosporin verabreicht wird, beträgt die empfohlene Dosis Prevymis 240 mg einmal täglich (siehe Abschnitt «Dosierung und Art der Anwendung»).
Einfluss von Nahrungsmitteln
Im Vergleich zum Nüchternzustand hatte die orale Verabreichung einer Einzeldosis von 480 mg Prevymis zusammen mit einer fett- und kalorienreichen Standardmahlzeit keinerlei Auswirkungen auf die Gesamtexposition (AUC) und führte zu einer Erhöhung der Spitzenkonzentrationen (Cmax) von Letermovir um ca. 30%. Prevymis kann oral mit oder ohne Nahrung eingenommen werden (siehe Abschnitt «Dosierung und Art der Anwendung»).
Distribution
Auf der Grundlage von populationspharmakokinetischen Analysen wird das mittlere Verteilungsvolumen im Gleichgewichtszustand (Steady State) nach intravenöser Verabreichung bei HSZT-Patienten auf 45,5 l geschätzt.
Letermovir wird in vitro stark an humane Plasmaproteine gebunden (98,7%). Bei In-vitro-Beurteilung beträgt die Blut/Plasma-Verteilung von Letermovir 0,56 und ist unabhängig vom Konzentrationsbereich (0,1 bis 10 mg/l).
In präklinischen Studien zur Distribution wurde Letermovir in Organen und Geweben verteilt, wobei die höchsten Konzentrationen im Gastrointestinaltrakt, im Gallengang und in der Leber und niedrige Konzentrationen im Gehirn beobachtet wurden.
Elimination
Die mittlere scheinbare terminale Halbwertszeit von Letermovir beträgt nach einer i.v.-Dosis von 480 mg Prevymis bei gesunden Probanden ca. 12 Stunden.
Auf Grundlage von populationspharmakokinetischen Analysen beträgt die Steady-State-CL von Letermovir nach intravenöser Verabreichung bei HSZT-Patienten schätzungsweise 4,84 l/h. Die interindividuelle Variabilität für die CL wird auf 24,6% geschätzt.
Metabolismus
Der Grossteil der arzneimittelbezogenen Komponenten im Plasma ist unveränderte Muttersubstanz (96,6%). Im Plasma wurden keine wesentlichen Metaboliten nachgewiesen. .
Die wichtigsten Ausscheidungswege von Letermovir sind die biliäre Ausscheidung und die direkte Glucuronidierung. Der Prozess beinhaltet die hepatischen Aufnahme-Transporter OATP1B1 und 3, gefolgt von UGT1A1/3 katalysierter Glucuronidierung.
Ausscheidung
Nach oraler Verabreichung von radioaktiv markiertem Letermovir wurden 93,3% der Radioaktivität im Stuhl nachgewiesen. Der Grossteil des Arzneimittels wurde als unveränderte Muttersubstanz mit einer geringen Menge (6% der Dosis) als Acrylglucuronid-Metabolit im Stuhl ausgeschieden. Die Ausscheidung von Letermovir im Urin war vernachlässigbar (<2% der Dosis).
Besondere Patientengruppen
Kinder und Jugendliche
Die Pharmakokinetik von Letermovir bei Kindern und Jugendlichen unter 18 Jahren wurde nicht untersucht.
Ältere Patienten
Populationspharmakokinetische Analysen ergaben keinen Effekt des Alters auf die Pharmakokinetik von Letermovir. Eine altersentsprechende Dosisanpassung ist nicht erforderlich.
Geschlecht
Populationspharmakokinetische Analysen zeigen keinen Unterschied der Pharmakokinetik von Letermovir bei Frauen im Vergleich zu Männern.
Gewicht
Auf Grundlage von populationspharmakokinetischen Analysen ist die AUC von Letermovir bei Probanden, die 80-100 kg wiegen, im Vergleich zu Probanden mit einem Gewicht von 67 kg schätzungsweise 18.7% niedriger. Diese Veränderung ist klinisch nicht relevant.
Ethnische Zugehörigkeit
Auf Grundlage von populationspharmakokinetischen Analysen ist die AUC von Letermovir bei Asiaten schätzungsweise 33,2% höher als bei Weissen. Diese Veränderung ist klinisch nicht relevant.
Eingeschränkte Nierenfunktion
Die AUC von Letermovir war bei Patienten mit mässiger (eGFR 30 bis 59 ml/min/1,73 m2) und schwerer (eGFR kleiner als 30 ml/min/1,73 m2) Einschränkung der Nierenfunktion ca. 1,9- bzw. 1,4-fach höher als bei gesunden Probanden. Die Veränderungen der Letermovir-Exposition aufgrund einer Einschränkung der Nierenfunktion sind klinisch nicht relevant. Für Patienten mit eGFR unter 10 ml/min/1,73 m2 oder für Dialysepatienten liegen keine Daten vor.
Eingeschränkte Leberfunktion
Die AUC von Letermovir war bei Patienten mit mässiger (Child-Pugh-Klasse B [CP-B], Score von 7-9) und schwerer (Child-Pugh-Klasse C [CP-C], Score von 10-15) Einschränkung der Leberfunktion ca. 1,6- bzw. 3,8-fach höher als bei gesunden Probanden. Die Veränderungen der Letermovir-Exposition bei Patienten mit mässiger Einschränkung der Leberfunktion sind klinisch nicht relevant.
Klinisch relevante Erhöhungen der Letermovir-Exposition werden bei Patienten mit schwerer Einschränkung der Leberfunktion oder bei Patienten mit mässiger Einschränkung der Leberfunktion in Kombination mit mässiger bis schwerer Einschränkung der Nierenfunktion erwartet.
Präklinische DatenAllgemeine Toxizität
Testikuläre Toxizität wurde bei Ratten bei systemischen Expositionen (AUC) festgestellt, die der ≥3-fachen Exposition beim Menschen unter der RHD entsprachen. Diese Toxizität war durch Degeneration der Tubuli seminiferi, Oligospermie und Zellfragmente in den Epididymiden mit verminderten Gewichten von Testikeln und Epididymiden gekennzeichnet. Der NOAEL (No Observed Adverse Effect Level) für testikuläre Toxizität bei Ratten wurde bei ähnlichen Expositionen (AUC) wie bei Menschen unter der RHD beobachtet. Diese testikuläre Toxizität scheint artspezifisch zu sein; testikuläre Toxizität wurde bei Mäusen und Affen in den höchsten getesteten Dosen bei Expositionen bis zum 4-Fachen bzw. 2-Fachen der AUC unter der RHD nicht beobachtet. Die Relevanz für Menschen ist nicht bekannt.
Mit Ausnahme der Vakuolenbildung, die in den Nieren von Ratten festgestellt wurde, denen mit 1'500 mg/kg/Tag des Cyclodextrin-Hilfsstoffs Hydroxypropylbetadex formuliertes Letermovir i. v. verabreicht wurde, war das Toxizitätsprofil von Letermovir in Studien an Ratten und Affen mit oraler und intravenöser Applikation generell ähnlich. Es ist bekannt, dass Hydroxypropylbetadex bei Ratten Vakuolenbildung in den Nieren verursachen kann, wenn es in Dosen von mehr als 50 mg/kg/Tag verabreicht wird.
Mutagenität
In einer Reihe von In-vitro- und In-vivo-Tests, einschliesslich Tests zur mikrobiellen Mutagenese, zu Chromosomenabberationen in CHO Zellen sowie eines In-vivo-Mikrokerntests an Mäusen, zeigte Letermovir keine genotoxischen Eigenschaften.
Karzinogenität
Es wurden keine Untersuchungen zur Kanzerogenität von Letermovir durchgeführt.
Reproduktion
Fertilität
In den Studien zur Fertilität und frühen Embryonalentwicklung bei Ratten wurden in der höchsten untersuchten Dosis von 240 mg/kg/Tag (ca. das 5-Fache der AUC beim Menschen unter der RHD) keine Effekte von Letermovir auf die weibliche Fertilität festgestellt. Bei männlichen Ratten wurden reduzierte Spermienkonzentration, reduzierte Spermienmotilität und verminderte Fertilität bei systemischen Expositionen von dem ≥3-Fachen der AUC beim Menschen unter der RHD beobachtet (siehe Allgemeine Toxikologie).
Entwicklung
Letermovir wurde trächtigen Ratten in Dosen von 0, 10, 50 oder 250 mg/kg/Tag an den Trächtigkeitstagen 6 bis 17 oral verabreicht. Maternale Toxizität (einschliesslich verringerter Zunahme des Körpergewichts) wurde unter 250 mg/kg/Tag beobachtet (ca. 11-fache AUC unter der RHD); bei den Nachkommen wurden reduziertes Fetalgewicht mit verzögerter Ossifikation, leicht ödematöse Feten und eine erhöhte Inzidenz von Verkürzung der Nabelschnur und Variationen sowie Fehlbildungen von Wirbeln, Rippen und Becken beobachtet. Bei Dosen von bis zu 50 mg/kg/Tag (ca. 2,5-fache AUC unter der RHD) wurden keine maternalen oder Entwicklungseffekte festgestellt.
Letermovir wurde trächtigen Kaninchen in Dosen von 0, 25, 75 oder 225 mg/kg/Tag an den Trächtigkeitstagen 6 bis 20 oral verabreicht. Bei einer Dosis von 225 mg/kg/Tag (ca. 2-fache AUC unter der RHD) wurde maternale Toxizität (einschliesslich Mortalität und Aborte) festgestellt; in der Nachkommenschaft war eine erhöhte Inzidenz von Fehlbildungen und Variationen von Wirbeln und Rippen zu beobachten. Unter der Dosis von 75 mg/kg/Tag (weniger als die AUC unter der RHD) wurden keine maternalen oder Entwicklungseffekte festgestellt.
In der Studie zur prä- und postnatalen Entwicklung in trächtigen Ratten mit Dosen von 0, 10, 45 oder 180 mg/kg Letermovir (oral) pro Tag (bis etwa zum 2-fachen der AUC unter der RHD) wurde keine Entwicklungstoxizität beobachtet.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Nicht über 30 °C lagern. Tabletten in der Originalverpackung (Blister) aufbewahren, um sie vor Feuchtigkeit zu schützen. Durchstechflaschen mit Prevymis Konzentrat zur Herstellung einer Infusionslösung im Originalkarton aufbewahren, um sie vor Licht zu schützen.
Zubereitung der verdünnten Lösung
Prevymis Konzentrat zur Herstellung einer Infusionslösung wird in Einzeldosis-Durchstechflaschen mit 30 ml geliefert, die entweder 240 mg (12 ml je Durchstechflasche) oder 480 mg (24 ml je Durchstechflasche) enthalten. Die Anweisungen für die Zubereitung und Verabreichung sind für beide Dosen identisch.
Prevymis Durchstechflaschen sind nur zur einmaligen Anwendung bestimmt. Nicht verwendetes Arzneimittel ist zu entsorgen.
Zubereitung
·Prevymis ist vor der intravenösen (i.v.) Anwendung zu verdünnen.
Den Inhalt der Durchstechflasche vor der Verdünnung auf Verfärbung und vorhandene Partikel prüfen. Prevymis Konzentrat zur Herstellung einer Infusionslösung ist eine klare, farblose Lösung. Die Durchstechflasche nicht verwenden, wenn die Lösung Verfärbungen oder sichtbare Partikel aufweist.
·Die Prevymis Durchstechflasche nicht schütteln.
·Eine Einzeldosis-Durchstechflasche Prevymis Konzentrat zur Herstellung einer Infusionslösung in einen vorgefüllten 250-ml-Infusionsbeutel geben, der entweder 0,9% Natriumchlorid oder 5% Dextrose enthält, und den Beutel vorsichtig mischen. Nicht schütteln.
·Nach der Verdünnung ist die Prevymis Lösung klar und die Färbung reicht von farblos bis gelb. Farbliche Schwankungen innerhalb dieses Bereichs beeinträchtigen die Produktqualität nicht. Wenn die Lösung und der Behälter es zulassen, sollte die verdünnte Lösung vor der Verabreichung einer Sichtkontrolle auf Partikel und Verfärbung unterzogen werden. Die Lösung ist zu verwerfen, wenn sie Verfärbungen oder sichtbare Partikel aufweist.
Aufbewahrung der verdünnten Lösung
·Aus mikrobiologischen Gründen sollte die verdünnte Lösung, die keine Konservierungsstoffe enthält, sofort verwendet werden.
·Die chemische und physikalische Stabilität im Gebrauch wurde für 48 Stunden bei 25 °C und 48 Stunden bei 2 bis 8 °C nachgewiesen.
·Dieser Zeitraum umfasst die Aufbewahrung der verdünnten Lösung im Infusionsbeutel und die Infusionsdauer.
Verabreichung
·Nur als intravenöse Infusion verabreichen. Nicht als intravenöse Schnellinfusion oder Bolus geben.
·Prevymis nach der Verdünnung durch intravenöse Infusion über einen peripheren oder zentralen Venenkatheter über einen Gesamtzeitraum von ca. 60 Minuten verabreichen. Den gesamten Inhalt des Infusionsbeutels verabreichen.
Kompatible Verdünnungsmittel
Prevymis Konzentrat zur Herstellung einer Infusionslösung ist mit 0,9%igen Natriumchlorid- und 5%igen Dextroselösungen kompatibel.
Kompatible Arzneimittel
Es wurde eine Studie zur Bewertung der physikalischen Kompatibilität von Prevymis Konzentrat zur Herstellung einer Infusionslösung mit injizierbaren Arzneimitteln durchgeführt. Die Kompatibilität wurde durch visuelle Beobachtungen, Trübung und Partikelmessung bestimmt. Kompatible Arzneimittel sind unten aufgeführt.
Mit Ausnahme der unten genannten Präparate darf Prevymis nicht durch dieselbe intravenöse Leitung (oder Kanüle) gleichzeitig mit anderen Arzneimitteln oder Verdünnngsmittelkombinationen verabreicht werden.
Die folgenden kompatiblen Arzneimittel† können zusammen mit PREVYMIS zur Injektion verabreicht werden, nur wenn beide Arzneimittel in 0,9% Natriumchlorid zubereitet werden und über Y- Verbinder gemäss den genehmigten Anweisungen der jeweiligen Fachinformation verabreicht werden.
Antithymozytenglobulin, Caspofungin, Daptomycin, Fentanylcitrat, Fluconazol, Furosemid, Humaninsulin, Magnesiumsulfat, Methotrexat, Micafungin.
† Diese Injektionspräparate sind in der Schweiz erhältlich.
Die folgenden kompatiblen Arzneimittel† können zusammen mit Prevymis zur Injektion verabreicht werden, nur wenn beide Arzneimittel in 5%iger Dextrose zubereitet werden und über Y-Verbinder gemäss den genehmigten Anweisungen der jeweiligen Fachinformation verabreicht werden.
Amphotericin B (Lipidkomplex)*, Anidulafungin, Cefazolin-Natrium, Ceftarolin, Ceftriaxon-Natrium, Ganciclovir-Natrium, Morphinsulfat, Noradrenalin-Bitartrat, Pantoprazol-Natrium, Kaliumchlorid, Kaliumphosphat, Tacrolimus, Telavancin, Tigecyclin.
† Diese Injektionspräparate sind in der Schweiz erhältlich.
* Amphotericin B (Lipidkomplex) ist mit Prevymis kompatibel. Allerdings ist Amphotericin B (liposomal) inkompatibel (siehe «Sonstige Hinweise»).
Kompatible Materialien für Infusionsbeutel und Infusionssets
Prevymis ist mit den folgenden Materialien für Infusionsbeutel und Infusionssets kompatibel. Unten nicht genannte Materialien für Infusionsbeutel oder Infusionssets dürfen nicht verwendet werden.
Materialien für Infusionsbeutel
Polyvinylchlorid (PVC), Ethylenvinylacetat (EVA) und Polyolefin (Polypropylen und Polyethylen)
Materialien für Infusionssets
PVC, Polyethylen (PE), Polybutadien (PBD), Silikonkautschuk (SR), Styrol-Butadien-Copolymer (SBC), Styrol-Butadien-Styrol-Copolymer (SBS), Polystyrol (PS)
Weichmacher
Diethylhexylphthalat (DEHP), Tris(2-ethylhexyl)trimellitat (TOTM), Butylbenzylphthalat (BBP)
Katheter
Radioopakes Polyurethan.
Inkompatible Arzneimittel
Prevymis Konzentrat zur Herstellung einer Infusionslösung ist physikalisch inkompatibel mit Amiodaronhydrochlorid, Amphotericin B (liposomal), Aztreonam, Cefepimhydrochlorid, Ciprofloxacin, Cyclosporin, Diltiazemhydrochlorid, Filgrastim Amgen, Gentamicinsulfat, Levofloxacin, Linezolid, Lorazepam, Midazolam-HCl, Mycophenolat-Mofetilhydrochlorid, Ondansetron, Palonosetron.
Inkompatible Materialien für Infusionsbeutel und Infusionssets
Prevymis ist inkompatibel mit polyurethanhaltigen Schläuchen von Infusionssets.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Zulassungsnummer66652, 66653 (Swissmedic).
PackungenPrevymis 240 mg Filmtablette: 28 Filmtabletten in nicht-perforierten Aluminium-Blisterpackungen. (A)
Prevymis 480 mg Filmtablette: 28 Filmtabletten in nicht-perforierten Aluminium-Blisterpackungen. (A)
Prevymis 12 ml (240 mg) Konzentrat zur Herstellung einer Infusionslösung: Jeder Karton enthält 1 Durchstechflasche. (A)
Prevymis 24 ml (480 mg) Konzentrat zur Herstellung einer Infusionslösung: Jeder Karton enthält 1 Durchstechflasche. (A)
ZulassungsinhaberinMSD MERCK SHARP & DOHME AG, Luzern.
Stand der InformationJanuar 2018.
CCDS-MK8228-MF-032017/ MK8228-CHE-2018-017039
|