ZusammensetzungWirkstoff: Dupilumab (aus gentechnisch veränderten Zellen des Chinesischen Hamsters).
Hilfsstoffe: L-Histidinum, Histidini hydrochloridum monohydricum, Arginini hydochloridum, Natrii Acetas Trihydricus, Acidum Aceticum Glaciale, Polysorbatum 80, Saccharum, Aqua ad iniectabilia q.s. ad solutionem 2 ml.
Galenische Form und Wirkstoffmenge pro EinheitDupixent 300 mg Injektionslösung zur subkutanen Injektion:
·Jede Fertigspritze enthält 300 mg Dupilumab in 2 ml Lösung (150 mg/ml).
·Jede Fertigspritze mit Sicherheitssystem enthält 300 mg Dupilumab in 2 ml Lösung (150 mg/ml).
Indikationen/AnwendungsmöglichkeitenDupixent wird angewendet zur Behandlung von mittelschwerer bis schwerer atopischer Dermatitis bei erwachsenen Patienten, wenn eine Therapie mit verschreibungspflichtigen topischen Medikamenten keine angemessene Krankheitskontrolle ermöglicht, oder nicht empfohlen wird.
Dupixent kann mit oder ohne topische Kortikosteroide verwendet werden.
Dosierung/AnwendungDie Behandlung ist durch in der Diagnose und Behandlung der atopischen Dermatitis erfahrenes medizinisches Fachpersonal einzuleiten.
Um die Rückverfolgbarkeit biotechnologisch hergestellter Arzneimittel sicherzustellen, sollten der Handelsname und die Chargennummer bei jeder Behandlung dokumentiert werden.
Dosierung
Die empfohlene Dosierung von Dupixent bei Erwachsenen ist eine Anfangsdosis von 600 mg als subkutane Injektion (zwei Injektionen zu je 300 mg), gefolgt von einer Dosis von 300 mg als subkutane Injektion alle zwei Wochen.
Dupixent kann mit oder ohne topische Kortikosteroide verwendet werden. Die Anwendung topischer Calcineurin-Inhibitoren ist möglich, muss aber auf Problemzonen wie Gesicht, Hals, intertriginöse Bereiche und den Genitalbereich beschränkt bleiben.
Falls eine Dosis versäumt wird, bitten Sie den Patienten, die Injektion innerhalb von 7 Tagen nach der eigentlichen Injektion nachzuholen, und die Injektionen dann gemäss dem regulären Dosierungsplan fortzusetzen. Wenn die vergessene Dosis nicht innerhalb der 7 Tage nachgeholt wird, bitten Sie den Patienten, die nächste im Dosierungsplan vorgesehene Dosis abzuwarten.
Bei Patienten, die nach 16 Behandlungswochen nicht auf die Behandlung ansprechen, ist ein Abbruch der Behandlung in Betracht zu ziehen.
Dosisanpassung:
Ältere Patienten (≥65 Jahre)
Es liegen nur sehr begrenzte Daten für Patienten ab 65 Jahren vor. Obwohl zwischen älteren und jüngeren Patienten keine Unterschiede hinsichtlich der Sicherheit oder Wirksamkeit beobachtet wurden, ist die Anzahl der Patienten ≥65 Jahre nicht ausreichend, um zu bestimmen, ob sich das Ansprechen dieser Gruppe vom Ansprechen jüngerer Patienten unterscheidet.
Patienten mit eingeschränkter Nierenfunktion
Bei Patienten mit leichter oder mittelschwerer Einschränkung der Nierenfunktion ist keine Dosisanpassung erforderlich. Zu Patienten mit einer schweren Einschränkung der Nierenfunktion liegen nur sehr begrenzte Daten vor (siehe Abschnitt «Pharmakokinetik»).
Eingeschränkte Leberfunktion
Es liegen keine Daten für Patienten mit einer Einschränkung der Leberfunktion vor (siehe Abschnitt «Pharmakokinetik»).
Pädiatrische Patienten
Die Anwendungssicherheit und Wirksamkeit von Dupixent bei Kindern unter 18 Jahren ist bisher nicht nachgewiesen. Es liegen keine Daten vor.
Art der Anwendung
Subkutane Anwendung.
Dupixent wird subkutan in den Oberschenkel oder das Abdomen ausserhalb eines Umkreises von 5 cm um den Bauchnabel herum injiziert. Falls die Injektion durch eine andere Person erfolgt, kommt als Injektionsstelle auch der Oberarm infrage.
Die Anfangsdosis von 600 mg ist in Form von zwei konsekutiven Injektionen zu je 300 mg Dupixent an zwei unterschiedlichen Injektionsstellen zu verabreichen.
Die Einstichstelle muss bei jeder Injektion gewechselt werden. Dupixent darf nicht an Stellen injiziert werden, an denen die Haut empfindlich oder verletzt ist, oder Quetschungen oder Narben vorliegen.
Dupixent kann durch den Patienten selbst oder durch eine Pflegeperson injiziert werden, wenn das medizinische Fachpersonal dies als angemessen erachtet. Vor der Anwendung sind der Patient und/oder die Pflegepersonen gemäss den Hinweisen zur Anwendung in der Packungsbeilage in der Vorbereitung und Verabreichung von Dupixent zu unterweisen.
Wie bei allen selbst verabreichten Therapeutika muss der Arzt/die Ärztin die Einhaltung der Behandlung durch den Patienten sorgfältig überwachen. In klinischen Phase-III-Studien vergassen Patienten, die sich die Injektionen selbst verabreichten, etwa ein Sechstel der Dosen. Dabei wurden 17,5% der Dosen Dupilumab und 26,2% der Placebo-Dosen versäumt.
Weitere Informationen zur Verabreichung dieses Arzneimittels sind dem Abschnitt «Hinweise für die Handhabung» zu entnehmen.
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der im Abschnitt «Zusammensetzung» genannten Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenÜberempfindlichkeitsreaktionen:
Bei einer (unmittelbaren oder verzögerten) allgemeinen systemischen Überempfindlichkeitsreaktion ist die Anwendung von Dupixent sofort zu beenden und eine geeignete Behandlung einzuleiten. In klinischen Studien wurde nach der Anwendung von Dupixent in seltenen Fällen über Serumkrankheit oder serumkrankheitsähnliche Symptome berichtet (siehe Abschnitt «Unerwünschte Wirkungen»).
Helminthose:
Patienten mit einer bekannten Helminthose wurden von den klinischen Studien ausgeschlossen. Dupixent kann durch Hemmung der IL-4/IL-13-Signalwege die Immunreaktion auf eine Helminthose beeinflussen. Patienten mit einer vorbestehenden Helminthose sind zu behandeln, bevor die Behandlung mit Dupixent eingeleitet wird. Wenn der Patient sich während der Behandlung mit Dupixent infiziert und nicht auf eine Behandlung gegen Helminthose anspricht, muss die Behandlung mit Dupixent ausgesetzt werden, bis die Infektion abgeklungen ist.
Ereignisse im Zusammenhang mit Konjunktivitis und Keratitis:
Patienten, die unter der Behandlung mit Dupixent eine Konjunktivitis entwickeln, die nach der Standardbehandlung nicht abklingt, sollten sich einer ophthalmologischen Untersuchung unterziehen.
In den 16-wöchigen Monotherapie-Studien lag der Anteil der Patienten, bei denen eine Keratitis berichtet wurde, in der Dupixent-Gruppe bei <1% (1 Fall pro 100 Patientenjahre) und in der Placebogruppe bei 0% (0 Fälle pro 100 Patientenjahre).
In der 52-wöchigen CHRONOS-Studie (Dupixent + topische Kortikosteroide) lag der Anteil der Patienten, bei denen eine Keratitis berichtet wurde, in der Gruppe Dupixent + TCS bei 4% (12 pro 100 Patientenjahre) und in der Gruppe Placebo + TCS bei 0% (0 pro 100 Patientenjahre). Die Mehrzahl der Patienten mit Keratitis wurde während der Behandlungsphase geheilt oder erholte sich.
Weisen Sie die Patienten darauf hin, dass sie das Auftreten oder eine Verschlimmerung von Augensymptomen ihrem Arzt/ihrer Ärztin mitteilen sollten.
Komorbides Asthma:
Die Sicherheit und Wirksamkeit von Dupixent bei der Behandlung von Asthma wurden bisher nicht nachgewiesen. Patienten mit einem komorbiden Asthma dürfen ihre Asthma-Behandlung ohne vorherige Absprache mit ihrem Arzt/ihrer Ärztin nicht anpassen oder absetzen. Asthmapatienten sind nach dem Absetzen der Behandlung mit Dupixent sorgfältig zu überwachen.
Hilfsstoffe:
Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro 300mgDosis.
InteraktionenInteraktionen mit CYP-Substraten:
Die Auswirkungen von Dupilumab auf die Pharmakokinetik von CYP-Substraten wurden im Rahmen einer klinischen Studie an Patienten mit atopischer Dermatitis untersucht. Die aus dieser Studie gewonnenen Daten zeigen keine klinisch relevanten Auswirkungen von Dupilumab auf die Aktivität von CYP1A2, CYP3A4, CYP2C19, CYP2D6 oder CYP2C9.
Impfungen:
Die Sicherheit und Wirksamkeit der gleichzeitigen Anwendung von Dupixent und Lebendimpfstoffen wurden nicht untersucht. Die Anwendung von Lebendimpfstoffen bei Patienten, die mit Dupixent behandelt werden, ist zu vermeiden.
Inaktivierte Impfstoffe: Die Immunreaktionen auf eine Impfung wurden in einer Studie untersucht, in der Patienten mit atopischer Dermatitis 16 Wochen lang einmal wöchentlich mit 300 mg Dupixent behandelt wurden (dies entspricht dem Doppelten der empfohlenen Dosierungsfrequenz). Nach einer zwölfwöchigen Anwendung von Dupixent wurden die Patienten mit einem Tdap-Impfstoff (Adacel®) und einem Meningokokken-Polysaccharid-Impfstoff (Menomune®) geimpft. Vier Wochen später wurden die Antikörperreaktionen auf das Tetanustoxoid und den Polysaccharid-Impfstoff gegen Meningokokken der Serogruppe C beurteilt. Die humorale Immunantwort auf den Tetanus-Impfstoff und auf den Meningokokken-Polysaccharid-Impfstoff war bei Patienten, die Dupixent erhielten, ähnlich wie bei Placebo-Patienten. Die Immunreaktionen auf die anderen Wirkstoffe der Impfstoffe Adacel und Menomune wurden nicht untersucht.
Schwangerschaft/StillzeitSchwangerschaft
Es liegen nur wenige Daten zur Anwendung von Dupilumab bei Schwangeren vor.
Tierexperimentelle Studien ergaben hinsichtlich der Reproduktionstoxizität keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen (siehe «Präklinische Daten»). Dupixent darf während der Schwangerschaft nicht angewendet werden, es sei denn, der potenzielle Nutzen übersteigt das potenzielle Risiko für den Fötus.
Stillzeit
Es ist nicht bekannt, ob Dupilumab in die Muttermilch übergeht oder ob es nach der Einnahme systemisch resorbiert wird. Es muss eine Entscheidung getroffen werden, ob das Stillen oder die Behandlung mit Dupixent unterbrochen werden soll. Dabei ist der Nutzen des Stillens für das Kind gegen den Nutzen der Therapie für die Mutter abzuwägen.
Fertilität
In tierexperimentellen Studien wurde keine Beeinträchtigung der Fertilität nachgewiesen (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDie Auswirkungen von Dupixent auf die Fahrtüchtigkeit oder das Bedienen von Maschinen wurden nicht direkt untersucht.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die häufigsten unerwünschten Nebenwirkungen waren Reaktionen an der Injektionsstelle, Konjunktivitis, Blepharitis und oraler Herpes.
In den Studien zur Anwendung von Dupixent in Monotherapie lag der Anteil der Patienten, die die Behandlung aufgrund unerwünschter Wirkungen abbrachen, in der Placebo-Gruppe bei 1,9%, in der Gruppe, die alle zwei Wochen (Q2W) 300 mg Dupixent erhielt, bei 1,9%, und in der Gruppe, die einmal wöchentlich (QW; dieses Dosierungsschema ist in der Schweiz nicht zugelassen) 300 mg Dupixent erhielt, bei 1,5%.
In der Studie mit gleichzeitiger Anwendung topischer Kortikosteroide (TCS) lag der Anteil der Patienten, die die Behandlung aufgrund unerwünschter Wirkungen abbrachen, in der Gruppe Placebo + TCS bei 7,6%, in der Gruppe die 300 mg Dupixent Q2W + TCS erhielt bei 1,8%, und in der Gruppe die 300 mg Dupixent QW + TCS (diese Dosierung ist in der Schweiz nicht zugelassen) erhielt bei 2,9%.
Übersicht über die Nebenwirkungen
Die Anwendungssicherheit von Dupixent wurde in vier randomisierten, placebokontrollierten, doppelblinden Studien und in einer Dosisfindungsstudie bei Patienten mit einer mittelschweren bis schweren atopischen Dermatitis untersucht. In diesen fünf Studien erhielten 1.689 Studienteilnehmer Dupixent als subkutane Injektion mit oder ohne begleitende Behandlung mit topischen Kortikosteroiden (TCS). Insgesamt wurden 305 Patienten mindestens ein Jahr lang mit Dupixent behandelt.
Die unerwünschten Wirkungen sind im Folgenden geordnet nach Systemorganklassen und nach Häufigkeit aufgeführt; hierbei gelten folgende Häufigkeitsangaben: Sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1'000, <1/100), selten (≥1/10'000, <1/1'000) oder sehr selten (<1/10'000). Innerhalb jeder Häufigkeitskategorie sind die unerwünschten Wirkungen nach abnehmendem Schweregrad aufgeführt.
Infektionen und parasitäre Erkrankungen:
Häufig: Konjunktivitis, oraler Herpes.
Gelegentlich: Infektion durch andere Herpes-Simplex-Viren (ausschliesslich Herpes-Ekzem).
Erkrankungen des Blutes und des Lymphsystems:
Häufig: Eosinophilie.
Erkrankungen des Immunsystems:
Selten: Serumkrankheit/serumkrankheitsähnliche Reaktionen.
Erkrankungen des Nervensystems:
Häufig: Kopfschmerzen.
Augenerkrankungen:
Häufig: Konjunktivitis; Augenjucken; Blepharitis.
Gelegentlich: Keratitis.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort:
Sehr häufig: Reaktionen an der Injektionsstelle (9,6%).
Beschreibung einzelner Nebenwirkungen:
Überempfindlichkeitsreaktionen:
Nach der Anwendung von Dupixent wurde in seltenen Fällen über Serumkrankheit oder serumkrankheitsähnliche Reaktionen berichtet (siehe Abschnitt «Unerwünschte Wirkungen»).
Eczema herpeticum:
In den 16-wöchigen Studien zur Anwendung von Dupixent als Monotherapie lag der Anteil der Patienten, bei denen ein Eczema herpeticum berichtet wurde, in den Dupixent-Gruppen bei <1% und in der Placebo-Gruppe bei <1%.
In der 52-wöchigen Studie mit Dupixent + TCS wurde ein Eczema herpeticum in der mit Dupixent + TCS behandelten Gruppe bei 0,2% der Patienten gemeldet und in der Gruppe unter Placebo + TCS bei 1,9%.
Eosinophilie:
Eine vorübergehende Eosinophilie wurde bei <2% der mit Dupixent behandelten Patienten berichtet.
Infektionen:
In den 16-wöchigen klinischen Monotherapie-Studien lag der Anteil der Patienten, bei denen schwerwiegende Infektionen berichtet wurden, in der Placebo-Gruppe bei 1,0% und in der Dupixent-Gruppe bei 0,5%. In der 52-wöchigen CHRONOS-Studie lag der Anteil der Patienten, bei denen schwerwiegende Infektionen berichtet wurden, in der Placebo-Gruppe bei 0,6% und in der Dupixent-Gruppe bei 0,2%.
Herpes zoster:
Herpes zoster wurde in den 16-wöchigen Monotherapie-Studien in den Dupixent-Gruppen bei <0,1% und in der Placebo-Gruppe bei <1% der Patienten berichtet. In der 52-wöchigen Studie mit Dupixent + TCS wurde Herpes zoster in der mit Dupixent + TCS behandelten Gruppe bei 1% der Patienten gemeldet und in der Gruppe unter Placebo + TCS bei 2%.
Immunogenität:
Wie alle therapeutischen Proteine ist Dupixent mit einem Immunogenitätsrisiko verbunden.
In der 52-wöchigen Studie wurden bei rund 3% der Patienten in der Placebo-Gruppe und bei 2% der Patienten in der Dupixent-Gruppe ADA-Reaktionen festgestellt, die länger als zwölf Wochen anhielten.
Rund 1% aller Studienteilnehmer, die Dupixent + TCS erhielten, wiesen als neutralisierend eingestufte Antikörper auf.
Bei den Studienteilnehmern, die Dupixent erhielten, war die Entwicklung von Antikörpern gegen Dupilumab mit niedrigeren Serumkonzentrationen von Dupilumab verbunden. Manche Studienteilnehmer, die hohe Antikörpertiter aufwiesen, hatten zusätzlich keine nachweisbare Serumkonzentration von Dupilumab. Zwei Studienteilnehmer entwickelten während der Behandlung mit Dupixent eine Serumkrankheit oder serumkrankheitsähnliche Reaktionen und hohe Antikörpertiter gegen Dupilumab.
ÜberdosierungEs gibt keine besondere Behandlung für einer Überdosierung mit Dupixent. Im Falle einer Überdosierung ist der Patient auf Anzeichen oder Symptome von Nebenwirkungen zu überwachen und im Bedarfsfall umgehend eine entsprechende symptomatische Behandlung einzuleiten.
Eigenschaften/WirkungenATC-Code: D11AH05
Wirkmechanismus/Pharmakodynamik
Dupilumab ist ein rekombinanter, humaner, monoklonaler IgG4-Antikörper, der die Signalwege von Interleukin 4 (IL-4) und Interleukin 13 (IL-13) hemmt. Dupilumab hemmt den IL-4-Signalweg über den Typ-I-Rezeptor (IL-4Rα/γc) und die Signalwege von IL-4 und IL-13 über den Typ-II-Rezeptor (IL-4Rα/IL-13Rα).
In klinischen Studien war die Behandlung mit Dupilumab mit einer Senkung der Konzentration von Biomarker der Immunität Typ 2 wie thymusaktivitätsregulierten Chemokinen (TARC/CCL17), dem Gesamt-IgE im Serum sowie dem allergenspezifischen IgE im Serum gegenüber den Ausgangswerten assoziiert. Unter der Behandlung mit Dupixent wurde eine Abnahme der Laktatdehydrogenase (LDH) beobachtet.
Während der Behandlung kommt es durch die Präsenz von Dupilumab zu einem Anstieg des IgG4 Spiegels. Die Auswirkungen einer Langzeittherapie mit einem monoklonalen IgG4 Antikörper wurden noch nicht ausreichend untersucht.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von Dupixent als Monotherapie und mit einer begleitenden Behandlung mit topischen Kortikosteroiden wurden in drei randomisierten, placebokontrollierten, doppelblinden Zulassungsstudien (SOLO 1, SOLO 2 und CHRONOS) untersucht. Eingeschlossen waren 2.119 Patienten ab 18 Jahren mit einer mittelschweren bis schweren atopischen Dermatitis (AD), definiert durch einen Investigator's Global Assessment-Score ≥3 (IGA), einen EASI-Score (Eczema Area and Severity Index) ≥16 und eine betroffene Körperoberfläche von 10% oder mehr. Die für diese drei Studien geeigneten und darin eingeschlossenen Patienten hatten vorher nur unzureichend auf eine topische Behandlung angesprochen.
In den drei Studien erhielten die Patienten 1) eine Anfangsdosis von 600 mg Dupixent (zwei Injektionen zu je 300 mg) an Tag 1, gefolgt von 300 mg einmal alle zwei Wochen (Q2W); 2) eine Anfangsdosis von 600 mg Dupixent an Tag 1, gefolgt von 300 mg einmal wöchentlich (QW; dieses Dosierungsschema ist in der Schweiz nicht zugelassen) oder 3) ein entsprechendes Placebo. Dupixent wurde in allen Studien als subkutane (s. c.) Injektion verabreicht. Um als unerträglich empfundene Symptome der atopischen Dermatitis zu lindern, konnten die Patienten nach Ermessen des Prüfarztes eine Rescue-Therapie erhalten (u.a. topische Steroide mit höherer Wirksamkeit oder systemische Immunsuppressiva). Patienten, die eine Rescue-Therapie erhielten, wurden als Non-Responder eingestuft.
In die Studie SOLO 1 wurden 671 Patienten eingeschlossen (224 in die Placebo-Gruppe, 224 in die Gruppe mit Dupixent 300 mg Q2W und 223 in die Gruppe mit Dupixent 300 mg QW). Der Behandlungszeitraum betrug 16 Wochen.
In die Studie SOLO 2 wurden 708 Patienten eingeschlossen (236 in die Placebo-Gruppe, 233 in die Gruppe mit Dupixent 300 mg Q2W und 239 in die Gruppe mit Dupixent 300 mg QW; (dieses Dosierungsschema ist in der Schweiz nicht zugelassen). Der Behandlungszeitraum betrug 16 Wochen.
In die Studie CHRONOS wurden 740 Patienten eingeschlossen (315 in die Gruppe Placebo + TCS, 106 in die Gruppe mit Dupixent 300 mg Q2W + TCS und 319 in die Gruppe mit Dupixent 300 mg QW + TCS) (QW; dieses Dosierungsschema ist in der Schweiz nicht zugelassen). Der Behandlungszeitraum betrug 52 Wochen. Die Patienten erhielten ab Baseline Dupixent oder ein Placebo sowie eine begleitende Therapie mit TCS gemäss standardisiertem Behandlungsschema. Die Patienten konnten auch topische Calcineurin-Inhibitoren (TCI) erhalten.
Primäre Endpunkte:
Die co-primären Endpunkte aller drei Zulassungsstudien waren der Anteil der Patienten, bei denen sich der Wert auf einer IGA-Skala (von 0 bis 4) von der Baseline bis Woche 16 um ≥2 Punkte auf dann 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei») verbesserte sowie der Anteil der Patienten, deren EASI-Score sich von der Baseline bis Woche 16 um mindestens 75% (EASI-75) verbesserte. Die weiteren untersuchten Resultate umfassten den Anteil der Patienten mit einer EASI-Verbesserung von mindestens 50% (EASI-50) bzw. 90% (EASI-90), einer Verringerung des Juckreizes gemessen an der numerischen Bewertungsskala für Pruritus (Peak Pruritus Numerical Rating Scale [NRS]) und einer prozentualen Veränderung des SCORAD (SCORing Atopic Dermatitis) von der Baseline bis Woche 16. Zusätzliche sekundäre Endpunkte umfassten die mittlere Veränderung des POEM (Patient-Oriented Eczema Measure), des DLQI (Dermatology Life Quality Index) sowie des HADS-Werts (Hospital Anxiety and Depression Scale) von der Baseline bis Woche 16. In der CHRONOS-Studie wurde die Wirksamkeit auch in Woche 52 bewertet.
Patientencharakteristika bei der Baseline:
In allen Behandlungsgruppen der Monotherapie-Studien (SOLO 1 und SOLO 2) lag das mittlere Alter bei 38,3 Jahren und das mittlere Gewicht bei 76,9 kg. 42,1% der Studienteilnehmer waren Frauen, 68,1% Weisse, 21,8% Asiaten und 6,8% Schwarze. In diesen Studien hatten 51,6% der Patienten einen Baseline-IGA-Score von 3 (mittelschwere AD), 48,3% einen Baseline-IGA-Score von 4 (schwere AD) und 32,4% der Patienten wurden in der Vergangenheit mit systemischen Immunsuppressiva behandelt. Bei Behandlungsbeginn betrug der mittlere EASI-Score 33,0, der wöchentliche Durchschnittswert gemäss Pruritus NRS 7,4, der mittlere SCORAD 67,8, der mittlere POEM 20,5, der mittlere DLQI 15,0 und der mittlere HADS-Gesamtwert 13,3.
In allen Behandlungsgruppen der Studie mit begleitender TCS-Therapie (CHRONOS) lag das mittlere Alter bei 37,1 Jahren und das mittlere Gewicht bei 74,5 kg. 39,7% der Studienteilnehmer waren Frauen, 66,2% Weisse, 27,2% Asiaten und 4,6% Schwarze. In dieser Studie hatten 53,1% der Patienten einen Baseline-IGA-Score von 3, 46,9% einen Baseline-IGA-Score von 4 und 33,6% der Patienten wurden in der Vergangenheit mit systemischen Immunsuppressiva behandelt. Bei Behandlungsbeginn betrug der mittlere EASI-Score 32,5, der wöchentliche Durchschnittswert gemäss Pruritus NRS 7,3, der mittlere SCORAD 66,4, der mittlere POEM 20,1, der mittlere DLQI 14,5 und der mittlere HADS-Gesamtwert 12,7.
Klinisches Ansprechen: 16-wöchige Monotherapie-Studien (SOLO 1 und SOLO 2)
Im Vergleich zu Placebo erreichte in den Studien SOLO 1 und SOLO 2 von der Baseline bis Woche 16 ein signifikant höherer Anteil an Patienten, denen randomisiert Dupixent zugewiesen worden war, einen IGA-Score von 0 oder 1, den EASI-75 und/oder eine Verbesserung um >4 Punkte gemäss Pruritus NRS (siehe Tabelle 1).
Im Vergleich zu Placebo erreichte ein signifikant höherer Anteil an Patienten, die randomisiert Dupixent erhielten, eine schnelle Verbesserung gemäss Pruritus NRS (definiert als Verbesserung um ≥4 Punkte bereits in Woche 2; p <0,01); im Laufe des Behandlungszeitraums stieg der Patientenanteil mit einem Ansprechen gemäss Pruritus NRS weiterhin an. Die Verbesserung gemäss Pruritus NRS trat parallel zu einer Verbesserung der objektiven Anzeichen der atopischen Dermatitis auf.
Abbildung 1 und Abbildung 2 stellen die mittlere prozentuale Veränderung der EASI- bzw. NRS-Scores von der Baseline bis Woche 16 dar.
Tabelle 1: Wirksamkeitsergebnisse der Dupixent-Monotherapie in Woche 16
|
|
SOLO 1 (FSA)a
|
SOLO 2 (FSA)a
| |
Placebo
|
Dupixent
300 mg Q2W
|
Placebo
|
Dupixent
300 mg Q2W
| |
Randomisierte Patienten
|
224
|
224
|
236
|
233
| |
IGA 0 oder 1b,
% der Responderc
|
10,3%
|
37,9%e
|
8,5%
|
36,1%e
| |
EASI-50,
% der Responderc
|
24,6%
|
68,8%e
|
22,0%
|
65,2%e
| |
EASI-75,
% der Responderc
|
14,7%
|
51,3%e
|
11,9%
|
44,2%e
| |
EASI-90,
% der Responderc
|
7,6%
|
35,7%e
|
7,2%
|
30,0%e
| |
Anzahl Patienten mit Pruritus NRS-Wert bei Baseline >4
|
212
|
213
|
221
|
225
| |
Pruritus gemäss NRS (Verbesserung ≥4 Punkte)
% der Responderc,d
|
12,3%
|
40,8%e
|
9,5%
|
36,0%e
|
LS = Least Squares [Methode der kleinsten Quadrate]; SE = Standard Error [Standardfehler]
a Die Full-Sample-Analyse (FSA) umfasst alle randomisierten Patienten.
b Als Responder angesehen wurden Patienten mit einem IGA-Score von 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei») mit einer Verbesserung um ≥2 Punkte auf einer von 0 bis 4 reichenden IGA-Skala.
c Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, wurden als Non-Responder eingestuft.
d In Woche 2 war der Anteil der Patienten, bei denen eine Verbesserung gemäss Pruritus NRS um ≥4 Punkte festzustellen war, in den Dupixent-Gruppen signifikant höher als in der Placebo-Gruppe (p <0,01).
e p <0,0001
Abbildung 1: Mittlere prozentuale Veränderung des EASI-Score gegenüber der Baseline in den Studien SOLO 1a und SOLO 2a (FSA)b
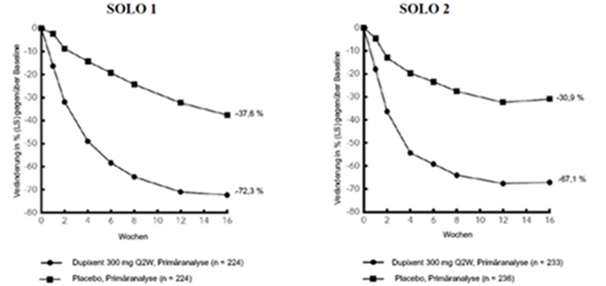
Abbildung 2: Mittlere prozentuale Veränderung gemäss Pruritus NRS gegenüber der Baseline in den Studien SOLO 1a und SOLO 2a (FSA)b
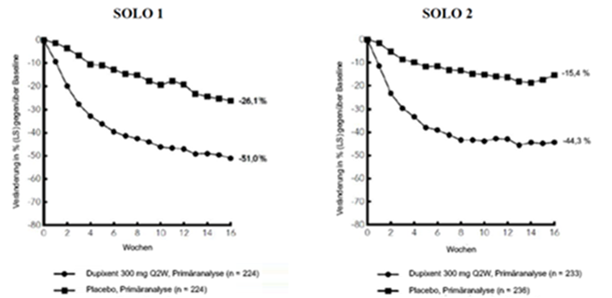
LS = Least Squares [Methode der kleinsten Quadrate]
a In den primären Analysen der Wirksamkeitsendpunkte wurden Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, als Non-Responder eingestuft.
b Die Full-Sample-Analyse (FSA) umfasst alle randomisierten Patienten.
In den Studien SOLO 1 und SOLO 2 stimmten die Wirksamkeit der Behandlung in den Untergruppen (Gewicht, Alter, Geschlecht, ethnische Herkunft sowie Hintergrundbehandlung einschliesslich Immunsuppressiva) mit den Ergebnissen der gesamten Studienpopulation überein.
Klinisches Ansprechen: 52-wöchige Studie mit begleitender Therapie mit TCS (CHRONOS)
Im Vergleich zu Placebo + TCS erreichte in der Studie CHRONOS von der Baseline bis Woche 16 und 52 ein signifikant höherer Anteil an Patienten, denen randomisiert Dupixent 300 mg Q2W + TCS zugewiesen worden war, einen IGA-Score von 0 oder 1, den EASI-75 und/oder eine Verbesserung um >4 Punkte gemäss Pruritus NRS (siehe Tabelle 2).
Im Vergleich zu Placebo erreichte ein signifikant höherer Anteil an Patienten, die randomisiert Dupixent + TCS erhielten, eine schnelle Verbesserung gemäss Pruritus NRS (definiert als Verbesserung um >4 Punkte bereits in Woche 2; p <0,05); im Laufe des Behandlungszeitraums stieg der Patientenanteil mit einem Ansprechen gemäss Pruritus NRS weiterhin an. Die Verbesserung gemäss Pruritus NRS trat im Zusammenhang mit einer Verbesserung der objektiven Anzeichen der atopischen Dermatitis auf.
Abbildung 3 und Abbildung 4 stellen die mittlere prozentuale Veränderung der EASI- bzw. NRS-Scores in der CHRONOS-Studie von der Baseline bis Woche 52 dar.
Tabelle 2: Wirksamkeitsergebnisse von Dupixent mit begleitenden TCSa in Woche 16 und Woche 52 in der CHRONOS-Studie
|
|
Woche 16 (FSA)b
|
Woche 52 (FSA Woche 52)b
| |
Placebo +
TCS
|
Dupixent
300 mg
Q2W + TCS
|
Placebo +
TCS
|
Dupixent
300 mg
Q2W + TCS
| |
Randomisierte Patienten
|
315
|
106
|
264
|
89
| |
IGA 0 oder 1c,
% der Responderd
|
12,4%
|
38,7%f
|
12,5%
|
36,0%f
| |
EASI-50,
% der Responderd
|
37,5%
|
80,2%f
|
29,9%
|
78,7%f
| |
EASI-75,
% der Responderd
|
23,2%
|
68,9%f
|
21,6%
|
65,2%f
| |
EASI-90,
% der Responderd
|
11,1%
|
39,6%f
|
15,5%
|
50,6%f
| |
Anzahl Patienten mit Pruritus NRS-Wert bei Baseline > 4
|
299
|
102
|
249
|
86
| |
Pruritus NRS-Wert
(Verbesserung ≥4 Punkte)
% der Responderd,e
|
19,7%
|
58,8%f
|
12,9%
|
51,2%f
|
LS = Least Squares [Methode der kleinsten Quadrate]; SE = Standard Error [Standardfehler]
a Alle Patienten erhielten eine Hintergrundbehandlung mit TCS. Die Patienten konnten topische Calcineurin-Inhibitoren anwenden.
b Die Full-Sample-Analyse (FSA) umfasst alle randomisierten Patienten. Die FSA in Woche 52 umfasst alle Patienten, die mindestens ein Jahr vor dem Enddatum der Primäranalyse randomisiert wurden.
c Als Responder angesehen wurden Patienten mit einem IGA-Score von 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei») mit einer Verbesserung um ≥2 Punkte auf einer von 0 bis 4 reichenden IGA-Skala.
d Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, wurden als Non-Responder eingestuft.
e In Woche 2 war der Anteil der Patienten, bei denen eine Verbesserung gemäss Pruritus NRS um ≥4 Punkte festzustellen war, unter Dupixent signifikant höher
als unter Placebo (p <0,05).
f p <0,0001
g p = 0,0015
h p = 0,0003
i p = 0,0005
Abbildung 3: Mittlere Veränderung (in %) des EASI-Score gegenüber der Baseline in der CHRONOS-Studiea (FSA in Woche 52)b
CHRONOS
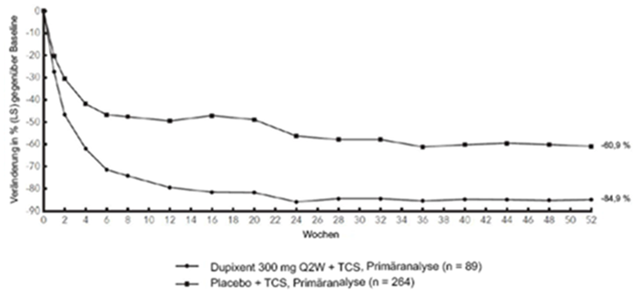
Abbildung 4: Mittlere Veränderung gemäss Pruritus NRS gegenüber der Baseline in der CHRONOS-Studiea (FSA in Woche 52)b
CHRONOS
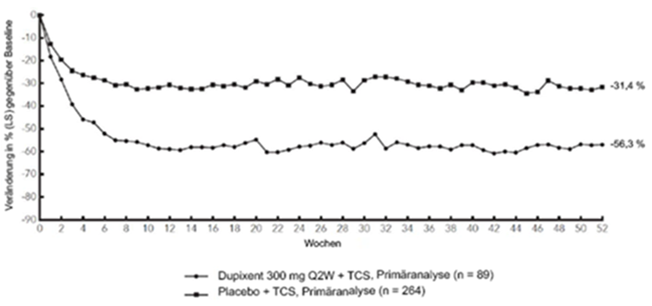
LS = Least Squares
a In den primären Analysen der Wirksamkeitsendpunkte wurden Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, als Non-Responder eingestuft.
b Die FSA in Woche 52 umfasst alle Patienten, die mindestens ein Jahr vor dem Enddatum der Primäranalyse randomisiert wurden.
In der CHRONOS-Studie stimmte die Wirksamkeit der Behandlung in den Untergruppen (Gewicht, Alter, Geschlecht, ethnische Herkunft sowie Hintergrundbehandlung einschliesslich Immunsuppressiva) mit den Ergebnissen der gesamten Studienpopulation überein.
Klinisches Ansprechen: Patienten, die unter einer Ciclosporin-Behandlung unzureichend eingestellt waren, eine Unverträglichkeit gegenüber Ciclosporin aufwiesen oder für die diese Behandlung medizinisch nicht indiziert war (CAFE-Studie)
Im Rahmen der CAFE-Studie wurde die Wirksamkeit von Dupixent mit begleitender TCS-Therapie gegenüber Placebo innerhalb eines 16-wöchigen Behandlungszeitraums bewertet. Bei den Studienteilnehmern handelte es sich um erwachsene Patienten mit schwerer atopischer Dermatitis, die unter einem oralen Ciclosporin unzureichend eingestellt waren, dieses nicht vertrugen oder für die diese Behandlung derzeit kontraindiziert oder medizinisch nicht angezeigt ist.
Insgesamt wurden 325 Patienten eingeschlossen, von denen 210 bereits in der Vergangenheit mit Ciclosporin behandelt wurden, während 115 Patienten noch nie Ciclosporin erhalten hatten, da für sie eine Ciclosporin-Behandlung medizinisch nicht angezeigt war. Das mittlere Alter lag bei 38,4 Jahren, 38,8% der Patienten waren Frauen. Bei Behandlungsbeginn betrug der mittlere EASI-Score 33,1, die durchschnittliche betroffene Körperoberfläche 55,7, der wöchentliche Durchschnittswert gemäss Pruritus NRS 6,4, der mittlere SCORAD 67,2 und der mittlere DLQI 13,8.
Primärer Endpunkt war der Anteil der Patienten mit einem EASI-75 in Woche 16.
In Tabelle 3 sind sowohl die primären als auch die sekundären Endpunkte der 16-wöchigen CAFE-Studie zusammengefasst.
Tabelle 3: Ergebnisse der primären und sekundären Endpunkte der CAFE-Studie
|
|
Placebo + TCS
|
Dupixent
300 mg Q2W + TCS
| |
Randomisierte Patienten
|
108
|
107
| |
EASI-75, Responder (in %)
|
29,6%
|
62,6%
| |
EASI, mittlere prozentuale Veränderung (LS) gegenüber den Ausgangswerten (+/– SE)
|
-46,6
(2,76)
|
-79,8
(2,59)
| |
Wert gemäss Pruritus NRS, mittlere prozentuale Veränderung (LS) gegenüber den Ausgangswerten (+/– SE)
|
-25,4% (3,39)
|
-53,9% (3,14)
| |
SCORAD, mittlere prozentuale Veränderung (LS) gegenüber den Ausgangswerten (+/– SE)
|
-29,5% (2,55)
|
-62,4% (2,48)
| |
DLQI, mittlere Veränderung (LS) gegenüber den Ausgangswerten (SE)
|
-4,5
(0,49)
|
-9,5
(0,46)
|
In der Patientenuntergruppe der 52-wöchigen CHRONOS-Studie, die der Studienpopulation der CAFE-Studie ähnelte, erreichten bis Woche 16 69,6% der mit Dupixent 300 mg Q2W behandelten Patienten ein EASI-75-Ansprechen, während es bei den mit Placebo behandelten Patienten 18,0% waren. Bis Woche 52 erreichten 52,4% der mit Dupixent 300 mg Q2W behandelten Patienten ein EASI-75-Ansprechen gegenüber 18,6% in der Placebo-Gruppe. In dieser Untergruppe lag die mittlere prozentuale Veränderung des Pruritus NRS-Werts von der Baseline bis Woche 16 bei –51,4% unter Dupixent 300 mg Q2W und bei –30,2% unter Placebo, beziehungsweise bis Woche 52 bei –54,8% in der Gruppe mit Dupixent 300 mg Q2W und bei –30,9% in der Placebo-Gruppe.
Aufrechterhaltung und Dauer des Ansprechens (Studie SOLO CONTINUE)
Um Aufrechterhaltung und Dauer des Ansprechens zu untersuchen, wurden die Studienteilnehmer, die in den Studien SOLO 1 und SOLO 2 16 Wochen lang mit Dupixent behandelt wurden und einen IGA-Wert von 0 oder 1 oder ein EASI-75-Ansprechen erreichten, im Rahmen der Studie SOLO CONTINUE erneut randomisiert. Diese Studie umfasste eine zusätzliche 36-wöchige Behandlung mit Dupixent oder Placebo, sodass sich die Gesamtdauer der Studienbehandlung auf 52 Wochen belief. Die Beurteilung der Endpunkte erfolgte in Woche 51 oder 52.
Die co-primären Endpunkte waren der Unterschied zwischen der Baseline (Woche 0) und Woche 36 gemessen an der prozentualen Veränderung des EASI-Score gegenüber der Baseline der Studien SOLO 1 und SOLO 2 sowie der prozentuale Anteil an Patienten mit einem EASI-75-Ansprechen in Woche 36 bei Patienten, die bereits bei Behandlungsbeginn ein EASI-75-Ansprechen hatten.
Bei Patienten, die dasselbe Behandlungsschema beibehielten, mit dem sie in den Studien SOLO 1 und SOLO 2 behandelt worden waren (300 mg Q2W oder 300 mg QW; letzteres Dosierungsschema ist in der Schweiz nicht zugelassen), konnte ein optimaler Erhaltungseffekt des klinischen Ansprechens nachgewiesen werden, wohingegen sich die Wirksamkeit bei anderen Dosierungsschemata dosisabhängig verringerte.
Sowohl die primären als auch die sekundären Endpunkte der 52-wöchigen Studie SOLO CONTINUE sind in Tabelle 4 zusammengefasst.
Tabelle 4: Ergebnisse der primären und sekundären Endpunkte der Studie SOLO CONTINUE
|
|
Placebo
|
Dupilumab 300 mg
| |
N = 83
|
Q8W
N = 84
|
Q2W/QW
N = 169
| |
Co-primäre Endpunkte
| |
Mittlerer Unterschied (LS) (+/– SE) zwischen der Baseline und Woche 36 in prozentualer EASI-Veränderung gegenüber der Baseline der Vorläufer-Studien
|
21,7
(3,13)
|
6,8***
(2,43)
|
0,1***
(1,74)
| |
Prozentualer Anteil an Patienten mit EASI-75-Ansprechen in Woche 36 bei Patienten mit EASI-75-Ansprechen bei der Baseline, (n [%])
|
24/79
(30,4%)
|
45/82*
(54,9%)
|
116/162***
(71,6%)
| |
Wichtige sekundäre Endpunkte:
| |
Prozentualer Anteil an Patienten, deren IGA-Wert in Woche 36 um nicht mehr als 1 Punkt vom Baseline-Wert abwich, innerhalb der Patientengruppe mit IGA 0 oder 1 bei der Baseline, (n [%])
|
18/63
(28,6)
|
32/64†
(50,0)
|
89/126***
(70,6)
| |
Prozentualer Anteil an Patienten mit einem IGA-Wert von 0 oder 1 in Woche 36 unter den Patienten mit IGA von 0 oder 1 bei der Baseline, (n [%])
|
9/63
(14,3)
|
21/64†
(32,8)
|
68/126***
(54,0)
| |
Prozentualer Anteil an Patienten, deren maximaler Pruritus NRS-Wert von der Baseline bis Woche 35 um ≥3 Punkte anstieg, unter den Patienten mit einem maximalen Baseline-Pruritus
NRS-Wert ≤7, (n [%])
|
56/80
(70,0)
|
45/81
(55,6)
|
57/168***
(33,9)
|
†p <0,05; *p <0,01; **p <0,001; ***p ≤0,0001
In der Studie SOLO CONTINUE wurde bei verlängerten Dosierungsintervallen eine Tendenz zu einer behandlungsbedingten stärkeren Produktion von Antikörpern gegen den Wirkstoff (Anti-Drug Antibodies, ADA) beobachtet.
Behandlungsbedingte ADA-Reaktionen: QW: 1,2%; Q2W: 4,3%; Q4W: 6,0%; Q8W: 11,7%. ADA-Reaktionen, die länger als 12 Wochen andauerten: QW: 0,0%; Q2W: 1,4%; Q4W: 0,0%; Q8W: 2,6%.
Lebensqualität/von Patienten berichtete Ergebnisse (Patient Reported Outcomes, PRO)
In den Studien SOLO 1 und SOLO 2 wurde mit Dupixent im Vergleich zu Placebo gemäss der POEM-, der DLQI- und der HADS-Gesamtbewertung eine signifikante Verbesserung der Symptome der AD, der gesundheitsbezogenen Lebensqualität sowie der Angst- und Depressionssymptome erzielt.
In der CHRONOS-Studie wurde mit Dupixent im Vergleich zu Placebo gemäss der POEM- und der DLQI-Gesamtbewertung nach 16 Wochen eine signifikante Verbesserung der Symptome der AD und der gesundheitsbezogenen Lebensqualität erzielt.
In den Studien SOLO 1 und SOLO 2 waren die mittleren Veränderungen (± SE) der Least Squares der POEM-, der DLQI- und der HADS-Gesamtbewertung von der Baseline bis Woche 16 bei Patienten, die Dupilumab erhielten, signifikant höher als bei Placebo-Patienten (p <0,0001 für alle Vergleiche ausser HADS in der Studie SOLO 2, p <0,001), siehe Tabelle 5.
In der Studie CHRONOS waren die mittleren Veränderungen (± SE) der Least Squares der POEM- und der DLQI-Gesamtbewertung von der Baseline bis Woche 16 bei Patienten, die Dupilumab erhielten, signifikant höher als bei Placebo-Patienten (p <0,0001 für alle Vergleiche), siehe Tabelle 5.
Tabelle 5: Zusätzliche Ergenisse der sekundären Endpunkte der Dupixent-Behandlung mit oder ohne TCS in Woche 16
|
|
Monotherapie in Woche 16
|
Begleitende Therapie mit TCS in Woche 16
| |
SOLO 1
|
SOLO 2
|
CHRONOS
| |
Placebo
|
Dupixent
300 mg Q2W
|
Placebo
|
Dupixent
300 mg Q2W
|
Placebo
|
Dupixent
300 mg Q2W
| |
Randomisierte Patienten
|
224
|
224
|
236
|
233
|
315
|
106
| |
POEM, mittlere Veränderung (LS) gegenüber der Baseline (SE)
|
-5,1
(0,67)
|
-11,6a
(0,49)
|
-3,3
(0,55)
|
-10,2a
(0,49)
|
-5,3
(0,41)
|
-12,7a
(0,64)
| |
DLQI, mittlere Veränderung (LS) gegenüber der Baseline (SE)
|
-5,3
(0,50)
|
-9,3a
(0,40)
|
-3,6
(0,50)
|
-9,3a
(0,38)
|
-5,8
(0,34)
|
-10,0a
(0,50)
|
LS = Least Squares [Methode der kleinsten Quadrate]; SE = Standard Error [Standardfehler]
a p <0,0001
PharmakokinetikResorption
Nach einer subkutan (s. c.) verabreichten Anfangsdosis von 600 mg Dupilumab betrug die Dauer bis zur maximalen Plasmakonzentration ± Standardabweichung (Cmax) von 70,1 ± 24,1 μg/ml etwa eine Woche.
Basierend auf einer populationsbezogenen pharmakokinetischen Analyse lag die geschätzte absolute Bioverfügbarkeit von Dupilumab nach einer s. c. Dosis bei 64%.
Nach der Anwendung einer Anfangsdosis von 600 mg, gefolgt von einer Dosis von 300 mg alle zwei Wochen, wurden die Steady-State-Konzentrationen bis Woche 16 erreicht. In den klinischen Studien lagen die mittleren minimalen Serumkonzentrationen im Steady State (± SD) bei einer Dosis von 300 mg alle zwei Wochen zwischen 73,3 ± 40,0 μg/ml und 79,9 ± 41,4 μg/ml.
Verteilung
Basierend auf der populationsbezogenen pharmakokinetischen Analyse wurde das Verteilungsvolumen von Dupilumab auf etwa 4,6 l geschätzt.
Biotransformation
Spezifische Stoffwechselstudien wurden nicht durchgeführt, da Dupilumab ein Protein ist. Es wird erwartet, dass Dupilumab zu kleinen Peptiden und einzelnen Aminosäuren abgebaut wird.
Elimination
Dupilumab wird sowohl auf linearen als auch auf nichtlinearen Wegen parallel eliminiert. Bei höheren Konzentrationen wird Dupilumab vorwiegend über eine nicht sättigbare Proteolyse eliminiert, während bei geringeren Konzentrationen die nichtlineare sättigbare zielvermittelte IL-4R-α-Elimination überwiegt.
Gemäss den Schätzungen einer populationsbezogenen pharmakokinetischen Analyse betrug die mittlere Dauer der Konzentrationsabnahme von Dupilumab bis unter die untere Nachweisgrenze ab der letzten Steady-State-Dosis für das Behandlungsschema mit 300 mg Q2W 10 Wochen.
Linearität/Nicht-Linearität
Aufgrund der nichtlinearen Clearance erhöht sich die Dupilumab-Exposition, gemessen an der der Fläche unter der Kurve, nach s. c. Einzeldosen von 75 – 600 mg mit steigender Dosis überproportional.
Besondere Patientengruppen
Geschlecht, ethnische Herkunft und Gewicht:
Im Rahmen einer populationsbezogenen pharmakokinetischen Analyse wurden keine klinisch bedeutsamen Auswirkungen des Geschlechts und der ethnischen Herkunft auf die systemische Exposition von Dupilumab festgestellt.
Die Talspiegel von Dupilumab waren bei Patienten mit einem höheren Körpergewicht niedriger. Dabei wurden keine signifikanten Auswirkungen auf die Wirksamkeit von Dupilumab festgestellt.
Alter:
Ältere Patienten: Von den 1'472 Patienten mit atopischer Dermatitis, die Dupixent in einer Phase-II-Dosisfindungsstudie oder in placebokontrollierten Phase-III-Studien erhielten, waren insgesamt 67 Patienten mindestens 65 Jahre alt. Obwohl zwischen älteren und jüngeren Patienten keine Unterschiede hinsichtlich der Sicherheit oder Wirksamkeit festgestellt wurden, ist die Anzahl der Patienten ab 65 Jahren nicht ausreichend, um zu bestimmen, ob sich deren Ansprechen von dem jüngerer Patienten unterscheidet.
Im Rahmen einer populationsbezogenen pharmakokinetischen Analyse wurden keine klinisch bedeutsamen Auswirkungen des Alters auf die systemische Exposition von Dupilumab festgestellt. Allerdings umfasste diese Studie nur 61 Patienten ab 65 Jahren.
Kinder und Jugendliche:
Die Pharmakokinetik von Dupilumab bei Kindern und Jugendlichen wurde nicht untersucht.
Nierenfunktionsstörung:
Da Dupilumab ein monoklonaler Antikörper ist, dürfte die Elimination über die Nieren nicht signifikant sein. Es wurden keine klinischen Studien zu den Auswirkungen einer Nierenfunktionsstörung auf die Pharmakokinetik von Dupilumab durchgeführt. Im Rahmen der populationsbezogenen pharmakokinetischen Analyse wurden keine klinisch bedeutsamen Auswirkungen einer leichten bis mittelschweren Nierenfunktionsstörung auf die systemische Exposition von Dupilumab festgestellt. Es liegen nur sehr begrenzte Daten für Patienten mit einer schweren Nierenfunktionsstörung vor.
Leberfunktionsstörung:
Da Dupilumab ein monoklonaler Antikörper ist, dürfte die Elimination über die Leber nicht signifikant sein. Es wurden keine klinischen Studien zu den Auswirkungen einer Leberfunktionsstörung auf die Pharmakokinetik von Dupilumab durchgeführt.
Präklinische DatenBasierend auf den konventionellen Studien zur Toxizität bei wiederholter Gabe (einschliesslich der Kriterien zur Evaluation der Sicherheitspharmakologie) und zur Reproduktions- und Entwicklungstoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Das mutagene Potenzial von Dupilumab wurde noch nicht untersucht, es wird jedoch nicht erwartet, dass monoklonale Antikörper die DNA oder Chromosomen verändern.
Es wurden keine Karzinogenitätsstudien mit Dupilumab durchgeführt. Sowohl die Bewertung der vorliegenden Nachweise zur IL-4Rα-Inhibition als auch die tierexperimentellen Toxikologiedaten mit Surrogat-Antikörpern lieferten keine Hinweise auf ein erhöhtes Karzinogenitätspotenzial von Dupilumab.
Während der Reproduktionstoxizitätsstudie, die an Affen durchgeführt wurde, wurde ein affenspezifischer IL-4Rα-Surrogat-Antikörper verwendet. Bei Dosierungen, die IL-4Rα saturieren, wurden hierbei keine fetalen Fehlbildungen beobachtet.
Eine erweiterte prä- und postnatale Entwicklungsstudie ergab keine Nebenwirkungen bei Muttertieren oder deren Jungen bis sechs Monate nach der Geburt.
Fertilitätsstudien bei männlichen und weiblichen Mäusen, in denen ein Surrogat-Antikörper gegen IL-4Rα verwendet wurde, zeigten keine Beeinträchtigung der Fertilität (siehe Abschnitt «Schwangerschaft/Stillzeit»).
Sonstige HinweiseDie folgenden Hinweise gelten für alle erhältlichen Darreichungsformen.
Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Dupixent darf nur bis zu dem auf der Packung oder dem Etikett mit «EXP» bezeichneten Datum verwendet werden.
Hinweise zur Lagerung
Ausserhalb der Reichweite von Kindern aufbewahren.
Fertigspritze mit oder ohne Sicherheitssystem lichtgeschützt in der Originalverpackung aufbewahren.
Im Kühlschrank lagern (2–8 °C). Arzneimittel nicht einfrieren oder erhitzen.
Nach Entnahme aus dem Kühlschrank kann Dupixent bei Bedarf bis zu 14 Tage lang bei Raumtemperatur (unter 25 °C) gelagert werden. Nach diesem Zeitraum muss die Spritze unbedingt entsorgt werden.
Hinweise zur Handhabung:
Dupixent wird als Fertigspritze mit oder ohne Sicherheitssystem angeboten. Die Hinweise zur Anwendung befinden sich am Ende der Patienteninformation.
Klare bis leicht schimmernde, farblose bis blassgelbe Injektionslösung ohne sichtbare Partikel zur subkutanen Injektion.
Fertigspritze aus dem Kühlschrank entnehmen und auf Raumtemperatur erwärmen lassen (ca. 45 min.), bevor die Dupixent-Injektion vorgenommen wird.
Fertigspritze vor Wärme und direktem Sonnenlicht schützen und nicht schütteln.
Vor Gebrauch eine Sichtprüfung der Fertigspritze vornehmen. Wenn die Lösung trübe oder verfärbt ist oder Partikel erkennbar sind oder ein Teil der Vorrichtung beschädigt zu sein scheint, darf das Arzneimittel nicht verwendet werden.
Nicht aufgebrauchte Arzneimittel oder Abfälle müssen gemäss den vor Ort geltenden Bestimmungen entsorgt werden. Nach Gebrauch die Fertigspritze in einen durchstichsicheren Behälter geben und gemäss den vor Ort geltenden Bestimmungen entsorgen. Behälter nicht wiederverwenden.
Zulassungsnummer66649, 66874 (Swissmedic).
PackungenDupixent 300 mg, Injektionslösung in einer Fertigspritze mit Sicherheitssystem: Packung mit 2 Fertigspritzen (B)
Dupixent 300 mg, Injektionslösung in einer Fertigspritze: Packung mit 2 Fertigspritzen (B)
Zulassungsinhaberinsanofi-aventis (schweiz) ag, 1214 Vernier/GE.
Stand der InformationApril 2019.
|