Eigenschaften/WirkungenATC-Code: D11AH05
Wirkmechanismus/Pharmakodynamik
Dupilumab ist ein rekombinanter, humaner, monoklonaler IgG4-Antikörper, der die Signalwege von Interleukin 4 (IL-4) und Interleukin 13 (IL-13) hemmt. Dupilumab hemmt den IL-4-Signalweg über den Typ-I-Rezeptor (IL-4Rα/γc) und die Signalwege von IL-4 und IL-13 über den Typ-II-Rezeptor (IL-4Rα/IL-13Rα).
In klinischen Studien war die Behandlung mit Dupilumab mit einer Senkung der Konzentration von Biomarker der Immunität Typ 2 wie thymusaktivitätsregulierten Chemokinen (TARC/CCL17), dem Gesamt-IgE im Serum sowie dem allergenspezifischen IgE im Serum gegenüber den Ausgangswerten assoziiert. Unter der Behandlung mit Dupixent wurde eine Abnahme der Laktatdehydrogenase (LDH) beobachtet.
Während der Behandlung kommt es durch die Präsenz von Dupilumab zu einem Anstieg des IgG4 Spiegels. Die Auswirkungen einer Langzeittherapie mit einem monoklonalen IgG4 Antikörper wurden noch nicht ausreichend untersucht.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von Dupixent als Monotherapie und mit einer begleitenden Behandlung mit topischen Kortikosteroiden wurden in drei randomisierten, placebokontrollierten, doppelblinden Zulassungsstudien (SOLO 1, SOLO 2 und CHRONOS) untersucht. Eingeschlossen waren 2.119 Patienten ab 18 Jahren mit einer mittelschweren bis schweren atopischen Dermatitis (AD), definiert durch einen Investigator's Global Assessment-Score ≥3 (IGA), einen EASI-Score (Eczema Area and Severity Index) ≥16 und eine betroffene Körperoberfläche von 10% oder mehr. Die für diese drei Studien geeigneten und darin eingeschlossenen Patienten hatten vorher nur unzureichend auf eine topische Behandlung angesprochen.
In den drei Studien erhielten die Patienten 1) eine Anfangsdosis von 600 mg Dupixent (zwei Injektionen zu je 300 mg) an Tag 1, gefolgt von 300 mg einmal alle zwei Wochen (Q2W); 2) eine Anfangsdosis von 600 mg Dupixent an Tag 1, gefolgt von 300 mg einmal wöchentlich (QW; dieses Dosierungsschema ist in der Schweiz nicht zugelassen) oder 3) ein entsprechendes Placebo. Dupixent wurde in allen Studien als subkutane (s. c.) Injektion verabreicht. Um als unerträglich empfundene Symptome der atopischen Dermatitis zu lindern, konnten die Patienten nach Ermessen des Prüfarztes eine Rescue-Therapie erhalten (u.a. topische Steroide mit höherer Wirksamkeit oder systemische Immunsuppressiva). Patienten, die eine Rescue-Therapie erhielten, wurden als Non-Responder eingestuft.
In die Studie SOLO 1 wurden 671 Patienten eingeschlossen (224 in die Placebo-Gruppe, 224 in die Gruppe mit Dupixent 300 mg Q2W und 223 in die Gruppe mit Dupixent 300 mg QW). Der Behandlungszeitraum betrug 16 Wochen.
In die Studie SOLO 2 wurden 708 Patienten eingeschlossen (236 in die Placebo-Gruppe, 233 in die Gruppe mit Dupixent 300 mg Q2W und 239 in die Gruppe mit Dupixent 300 mg QW; (dieses Dosierungsschema ist in der Schweiz nicht zugelassen). Der Behandlungszeitraum betrug 16 Wochen.
In die Studie CHRONOS wurden 740 Patienten eingeschlossen (315 in die Gruppe Placebo + TCS, 106 in die Gruppe mit Dupixent 300 mg Q2W + TCS und 319 in die Gruppe mit Dupixent 300 mg QW + TCS) (QW; dieses Dosierungsschema ist in der Schweiz nicht zugelassen). Der Behandlungszeitraum betrug 52 Wochen. Die Patienten erhielten ab Baseline Dupixent oder ein Placebo sowie eine begleitende Therapie mit TCS gemäss standardisiertem Behandlungsschema. Die Patienten konnten auch topische Calcineurin-Inhibitoren (TCI) erhalten.
Primäre Endpunkte:
Die co-primären Endpunkte aller drei Zulassungsstudien waren der Anteil der Patienten, bei denen sich der Wert auf einer IGA-Skala (von 0 bis 4) von der Baseline bis Woche 16 um ≥2 Punkte auf dann 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei») verbesserte sowie der Anteil der Patienten, deren EASI-Score sich von der Baseline bis Woche 16 um mindestens 75% (EASI-75) verbesserte. Die weiteren untersuchten Resultate umfassten den Anteil der Patienten mit einer EASI-Verbesserung von mindestens 50% (EASI-50) bzw. 90% (EASI-90), einer Verringerung des Juckreizes gemessen an der numerischen Bewertungsskala für Pruritus (Peak Pruritus Numerical Rating Scale [NRS]) und einer prozentualen Veränderung des SCORAD (SCORing Atopic Dermatitis) von der Baseline bis Woche 16. Zusätzliche sekundäre Endpunkte umfassten die mittlere Veränderung des POEM (Patient-Oriented Eczema Measure), des DLQI (Dermatology Life Quality Index) sowie des HADS-Werts (Hospital Anxiety and Depression Scale) von der Baseline bis Woche 16. In der CHRONOS-Studie wurde die Wirksamkeit auch in Woche 52 bewertet.
Patientencharakteristika bei der Baseline:
In allen Behandlungsgruppen der Monotherapie-Studien (SOLO 1 und SOLO 2) lag das mittlere Alter bei 38,3 Jahren und das mittlere Gewicht bei 76,9 kg. 42,1% der Studienteilnehmer waren Frauen, 68,1% Weisse, 21,8% Asiaten und 6,8% Schwarze. In diesen Studien hatten 51,6% der Patienten einen Baseline-IGA-Score von 3 (mittelschwere AD), 48,3% einen Baseline-IGA-Score von 4 (schwere AD) und 32,4% der Patienten wurden in der Vergangenheit mit systemischen Immunsuppressiva behandelt. Bei Behandlungsbeginn betrug der mittlere EASI-Score 33,0, der wöchentliche Durchschnittswert gemäss Pruritus NRS 7,4, der mittlere SCORAD 67,8, der mittlere POEM 20,5, der mittlere DLQI 15,0 und der mittlere HADS-Gesamtwert 13,3.
In allen Behandlungsgruppen der Studie mit begleitender TCS-Therapie (CHRONOS) lag das mittlere Alter bei 37,1 Jahren und das mittlere Gewicht bei 74,5 kg. 39,7% der Studienteilnehmer waren Frauen, 66,2% Weisse, 27,2% Asiaten und 4,6% Schwarze. In dieser Studie hatten 53,1% der Patienten einen Baseline-IGA-Score von 3, 46,9% einen Baseline-IGA-Score von 4 und 33,6% der Patienten wurden in der Vergangenheit mit systemischen Immunsuppressiva behandelt. Bei Behandlungsbeginn betrug der mittlere EASI-Score 32,5, der wöchentliche Durchschnittswert gemäss Pruritus NRS 7,3, der mittlere SCORAD 66,4, der mittlere POEM 20,1, der mittlere DLQI 14,5 und der mittlere HADS-Gesamtwert 12,7.
Klinisches Ansprechen: 16-wöchige Monotherapie-Studien (SOLO 1 und SOLO 2)
Im Vergleich zu Placebo erreichte in den Studien SOLO 1 und SOLO 2 von der Baseline bis Woche 16 ein signifikant höherer Anteil an Patienten, denen randomisiert Dupixent zugewiesen worden war, einen IGA-Score von 0 oder 1, den EASI-75 und/oder eine Verbesserung um >4 Punkte gemäss Pruritus NRS (siehe Tabelle 1).
Im Vergleich zu Placebo erreichte ein signifikant höherer Anteil an Patienten, die randomisiert Dupixent erhielten, eine schnelle Verbesserung gemäss Pruritus NRS (definiert als Verbesserung um ≥4 Punkte bereits in Woche 2; p <0,01); im Laufe des Behandlungszeitraums stieg der Patientenanteil mit einem Ansprechen gemäss Pruritus NRS weiterhin an. Die Verbesserung gemäss Pruritus NRS trat parallel zu einer Verbesserung der objektiven Anzeichen der atopischen Dermatitis auf.
Abbildung 1 und Abbildung 2 stellen die mittlere prozentuale Veränderung der EASI- bzw. NRS-Scores von der Baseline bis Woche 16 dar.
Tabelle 1: Wirksamkeitsergebnisse der Dupixent-Monotherapie in Woche 16
|
|
SOLO 1 (FSA)a
|
SOLO 2 (FSA)a
| |
Placebo
|
Dupixent
300 mg Q2W
|
Placebo
|
Dupixent
300 mg Q2W
| |
Randomisierte Patienten
|
224
|
224
|
236
|
233
| |
IGA 0 oder 1b,
% der Responderc
|
10,3%
|
37,9%e
|
8,5%
|
36,1%e
| |
EASI-50,
% der Responderc
|
24,6%
|
68,8%e
|
22,0%
|
65,2%e
| |
EASI-75,
% der Responderc
|
14,7%
|
51,3%e
|
11,9%
|
44,2%e
| |
EASI-90,
% der Responderc
|
7,6%
|
35,7%e
|
7,2%
|
30,0%e
| |
Anzahl Patienten mit Pruritus NRS-Wert bei Baseline >4
|
212
|
213
|
221
|
225
| |
Pruritus gemäss NRS (Verbesserung ≥4 Punkte)
% der Responderc,d
|
12,3%
|
40,8%e
|
9,5%
|
36,0%e
|
LS = Least Squares [Methode der kleinsten Quadrate]; SE = Standard Error [Standardfehler]
a Die Full-Sample-Analyse (FSA) umfasst alle randomisierten Patienten.
b Als Responder angesehen wurden Patienten mit einem IGA-Score von 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei») mit einer Verbesserung um ≥2 Punkte auf einer von 0 bis 4 reichenden IGA-Skala.
c Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, wurden als Non-Responder eingestuft.
d In Woche 2 war der Anteil der Patienten, bei denen eine Verbesserung gemäss Pruritus NRS um ≥4 Punkte festzustellen war, in den Dupixent-Gruppen signifikant höher als in der Placebo-Gruppe (p <0,01).
e p <0,0001
Abbildung 1: Mittlere prozentuale Veränderung des EASI-Score gegenüber der Baseline in den Studien SOLO 1a und SOLO 2a (FSA)b
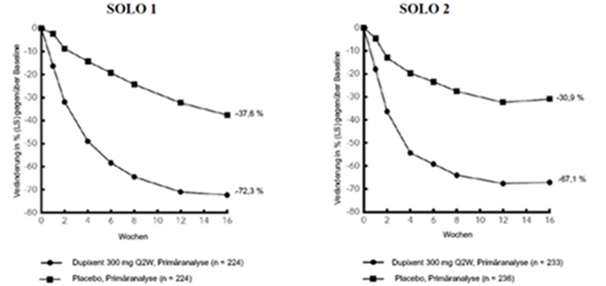
Abbildung 2: Mittlere prozentuale Veränderung gemäss Pruritus NRS gegenüber der Baseline in den Studien SOLO 1a und SOLO 2a (FSA)b
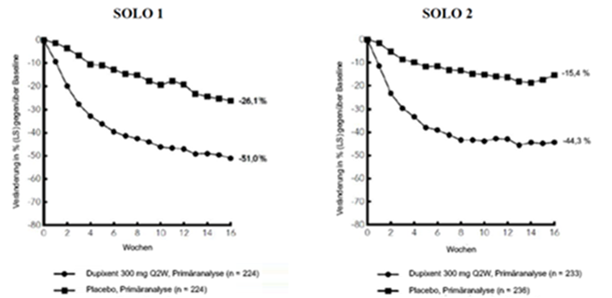
LS = Least Squares [Methode der kleinsten Quadrate]
a In den primären Analysen der Wirksamkeitsendpunkte wurden Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, als Non-Responder eingestuft.
b Die Full-Sample-Analyse (FSA) umfasst alle randomisierten Patienten.
In den Studien SOLO 1 und SOLO 2 stimmten die Wirksamkeit der Behandlung in den Untergruppen (Gewicht, Alter, Geschlecht, ethnische Herkunft sowie Hintergrundbehandlung einschliesslich Immunsuppressiva) mit den Ergebnissen der gesamten Studienpopulation überein.
Klinisches Ansprechen: 52-wöchige Studie mit begleitender Therapie mit TCS (CHRONOS)
Im Vergleich zu Placebo + TCS erreichte in der Studie CHRONOS von der Baseline bis Woche 16 und 52 ein signifikant höherer Anteil an Patienten, denen randomisiert Dupixent 300 mg Q2W + TCS zugewiesen worden war, einen IGA-Score von 0 oder 1, den EASI-75 und/oder eine Verbesserung um >4 Punkte gemäss Pruritus NRS (siehe Tabelle 2).
Im Vergleich zu Placebo erreichte ein signifikant höherer Anteil an Patienten, die randomisiert Dupixent + TCS erhielten, eine schnelle Verbesserung gemäss Pruritus NRS (definiert als Verbesserung um >4 Punkte bereits in Woche 2; p <0,05); im Laufe des Behandlungszeitraums stieg der Patientenanteil mit einem Ansprechen gemäss Pruritus NRS weiterhin an. Die Verbesserung gemäss Pruritus NRS trat im Zusammenhang mit einer Verbesserung der objektiven Anzeichen der atopischen Dermatitis auf.
Abbildung 3 und Abbildung 4 stellen die mittlere prozentuale Veränderung der EASI- bzw. NRS-Scores in der CHRONOS-Studie von der Baseline bis Woche 52 dar.
Tabelle 2: Wirksamkeitsergebnisse von Dupixent mit begleitenden TCSa in Woche 16 und Woche 52 in der CHRONOS-Studie
|
|
Woche 16 (FSA)b
|
Woche 52 (FSA Woche 52)b
| |
Placebo +
TCS
|
Dupixent
300 mg
Q2W + TCS
|
Placebo +
TCS
|
Dupixent
300 mg
Q2W + TCS
| |
Randomisierte Patienten
|
315
|
106
|
264
|
89
| |
IGA 0 oder 1c,
% der Responderd
|
12,4%
|
38,7%f
|
12,5%
|
36,0%f
| |
EASI-50,
% der Responderd
|
37,5%
|
80,2%f
|
29,9%
|
78,7%f
| |
EASI-75,
% der Responderd
|
23,2%
|
68,9%f
|
21,6%
|
65,2%f
| |
EASI-90,
% der Responderd
|
11,1%
|
39,6%f
|
15,5%
|
50,6%f
| |
Anzahl Patienten mit Pruritus NRS-Wert bei Baseline > 4
|
299
|
102
|
249
|
86
| |
Pruritus NRS-Wert
(Verbesserung ≥4 Punkte)
% der Responderd,e
|
19,7%
|
58,8%f
|
12,9%
|
51,2%f
|
LS = Least Squares [Methode der kleinsten Quadrate]; SE = Standard Error [Standardfehler]
a Alle Patienten erhielten eine Hintergrundbehandlung mit TCS. Die Patienten konnten topische Calcineurin-Inhibitoren anwenden.
b Die Full-Sample-Analyse (FSA) umfasst alle randomisierten Patienten. Die FSA in Woche 52 umfasst alle Patienten, die mindestens ein Jahr vor dem Enddatum der Primäranalyse randomisiert wurden.
c Als Responder angesehen wurden Patienten mit einem IGA-Score von 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei») mit einer Verbesserung um ≥2 Punkte auf einer von 0 bis 4 reichenden IGA-Skala.
d Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, wurden als Non-Responder eingestuft.
e In Woche 2 war der Anteil der Patienten, bei denen eine Verbesserung gemäss Pruritus NRS um ≥4 Punkte festzustellen war, unter Dupixent signifikant höher
als unter Placebo (p <0,05).
f p <0,0001
g p = 0,0015
h p = 0,0003
i p = 0,0005
Abbildung 3: Mittlere Veränderung (in %) des EASI-Score gegenüber der Baseline in der CHRONOS-Studiea (FSA in Woche 52)b
CHRONOS
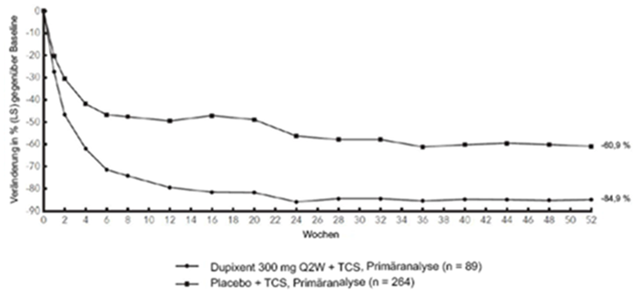
Abbildung 4: Mittlere Veränderung gemäss Pruritus NRS gegenüber der Baseline in der CHRONOS-Studiea (FSA in Woche 52)b
CHRONOS
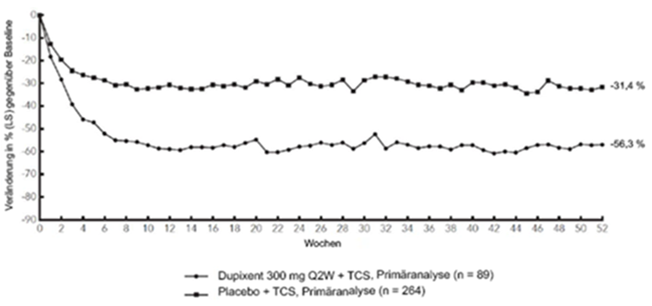
LS = Least Squares
a In den primären Analysen der Wirksamkeitsendpunkte wurden Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, als Non-Responder eingestuft.
b Die FSA in Woche 52 umfasst alle Patienten, die mindestens ein Jahr vor dem Enddatum der Primäranalyse randomisiert wurden.
In der CHRONOS-Studie stimmte die Wirksamkeit der Behandlung in den Untergruppen (Gewicht, Alter, Geschlecht, ethnische Herkunft sowie Hintergrundbehandlung einschliesslich Immunsuppressiva) mit den Ergebnissen der gesamten Studienpopulation überein.
Klinisches Ansprechen: Patienten, die unter einer Ciclosporin-Behandlung unzureichend eingestellt waren, eine Unverträglichkeit gegenüber Ciclosporin aufwiesen oder für die diese Behandlung medizinisch nicht indiziert war (CAFE-Studie)
Im Rahmen der CAFE-Studie wurde die Wirksamkeit von Dupixent mit begleitender TCS-Therapie gegenüber Placebo innerhalb eines 16-wöchigen Behandlungszeitraums bewertet. Bei den Studienteilnehmern handelte es sich um erwachsene Patienten mit schwerer atopischer Dermatitis, die unter einem oralen Ciclosporin unzureichend eingestellt waren, dieses nicht vertrugen oder für die diese Behandlung derzeit kontraindiziert oder medizinisch nicht angezeigt ist.
Insgesamt wurden 325 Patienten eingeschlossen, von denen 210 bereits in der Vergangenheit mit Ciclosporin behandelt wurden, während 115 Patienten noch nie Ciclosporin erhalten hatten, da für sie eine Ciclosporin-Behandlung medizinisch nicht angezeigt war. Das mittlere Alter lag bei 38,4 Jahren, 38,8% der Patienten waren Frauen. Bei Behandlungsbeginn betrug der mittlere EASI-Score 33,1, die durchschnittliche betroffene Körperoberfläche 55,7, der wöchentliche Durchschnittswert gemäss Pruritus NRS 6,4, der mittlere SCORAD 67,2 und der mittlere DLQI 13,8.
Primärer Endpunkt war der Anteil der Patienten mit einem EASI-75 in Woche 16.
In Tabelle 3 sind sowohl die primären als auch die sekundären Endpunkte der 16-wöchigen CAFE-Studie zusammengefasst.
Tabelle 3: Ergebnisse der primären und sekundären Endpunkte der CAFE-Studie
|
|
Placebo + TCS
|
Dupixent
300 mg Q2W + TCS
| |
Randomisierte Patienten
|
108
|
107
| |
EASI-75, Responder (in %)
|
29,6%
|
62,6%
| |
EASI, mittlere prozentuale Veränderung (LS) gegenüber den Ausgangswerten (+/– SE)
|
-46,6
(2,76)
|
-79,8
(2,59)
| |
Wert gemäss Pruritus NRS, mittlere prozentuale Veränderung (LS) gegenüber den Ausgangswerten (+/– SE)
|
-25,4% (3,39)
|
-53,9% (3,14)
| |
SCORAD, mittlere prozentuale Veränderung (LS) gegenüber den Ausgangswerten (+/– SE)
|
-29,5% (2,55)
|
-62,4% (2,48)
| |
DLQI, mittlere Veränderung (LS) gegenüber den Ausgangswerten (SE)
|
-4,5
(0,49)
|
-9,5
(0,46)
|
In der Patientenuntergruppe der 52-wöchigen CHRONOS-Studie, die der Studienpopulation der CAFE-Studie ähnelte, erreichten bis Woche 16 69,6% der mit Dupixent 300 mg Q2W behandelten Patienten ein EASI-75-Ansprechen, während es bei den mit Placebo behandelten Patienten 18,0% waren. Bis Woche 52 erreichten 52,4% der mit Dupixent 300 mg Q2W behandelten Patienten ein EASI-75-Ansprechen gegenüber 18,6% in der Placebo-Gruppe. In dieser Untergruppe lag die mittlere prozentuale Veränderung des Pruritus NRS-Werts von der Baseline bis Woche 16 bei –51,4% unter Dupixent 300 mg Q2W und bei –30,2% unter Placebo, beziehungsweise bis Woche 52 bei –54,8% in der Gruppe mit Dupixent 300 mg Q2W und bei –30,9% in der Placebo-Gruppe.
Aufrechterhaltung und Dauer des Ansprechens (Studie SOLO CONTINUE)
Um Aufrechterhaltung und Dauer des Ansprechens zu untersuchen, wurden die Studienteilnehmer, die in den Studien SOLO 1 und SOLO 2 16 Wochen lang mit Dupixent behandelt wurden und einen IGA-Wert von 0 oder 1 oder ein EASI-75-Ansprechen erreichten, im Rahmen der Studie SOLO CONTINUE erneut randomisiert. Diese Studie umfasste eine zusätzliche 36-wöchige Behandlung mit Dupixent oder Placebo, sodass sich die Gesamtdauer der Studienbehandlung auf 52 Wochen belief. Die Beurteilung der Endpunkte erfolgte in Woche 51 oder 52.
Die co-primären Endpunkte waren der Unterschied zwischen der Baseline (Woche 0) und Woche 36 gemessen an der prozentualen Veränderung des EASI-Score gegenüber der Baseline der Studien SOLO 1 und SOLO 2 sowie der prozentuale Anteil an Patienten mit einem EASI-75-Ansprechen in Woche 36 bei Patienten, die bereits bei Behandlungsbeginn ein EASI-75-Ansprechen hatten.
Bei Patienten, die dasselbe Behandlungsschema beibehielten, mit dem sie in den Studien SOLO 1 und SOLO 2 behandelt worden waren (300 mg Q2W oder 300 mg QW; letzteres Dosierungsschema ist in der Schweiz nicht zugelassen), konnte ein optimaler Erhaltungseffekt des klinischen Ansprechens nachgewiesen werden, wohingegen sich die Wirksamkeit bei anderen Dosierungsschemata dosisabhängig verringerte.
Sowohl die primären als auch die sekundären Endpunkte der 52-wöchigen Studie SOLO CONTINUE sind in Tabelle 4 zusammengefasst.
Tabelle 4: Ergebnisse der primären und sekundären Endpunkte der Studie SOLO CONTINUE
|
|
Placebo
|
Dupilumab 300 mg
| |
N = 83
|
Q8W
N = 84
|
Q2W/QW
N = 169
| |
Co-primäre Endpunkte
| |
Mittlerer Unterschied (LS) (+/– SE) zwischen der Baseline und Woche 36 in prozentualer EASI-Veränderung gegenüber der Baseline der Vorläufer-Studien
|
21,7
(3,13)
|
6,8***
(2,43)
|
0,1***
(1,74)
| |
Prozentualer Anteil an Patienten mit EASI-75-Ansprechen in Woche 36 bei Patienten mit EASI-75-Ansprechen bei der Baseline, (n [%])
|
24/79
(30,4%)
|
45/82*
(54,9%)
|
116/162***
(71,6%)
| |
Wichtige sekundäre Endpunkte:
| |
Prozentualer Anteil an Patienten, deren IGA-Wert in Woche 36 um nicht mehr als 1 Punkt vom Baseline-Wert abwich, innerhalb der Patientengruppe mit IGA 0 oder 1 bei der Baseline, (n [%])
|
18/63
(28,6)
|
32/64†
(50,0)
|
89/126***
(70,6)
| |
Prozentualer Anteil an Patienten mit einem IGA-Wert von 0 oder 1 in Woche 36 unter den Patienten mit IGA von 0 oder 1 bei der Baseline, (n [%])
|
9/63
(14,3)
|
21/64†
(32,8)
|
68/126***
(54,0)
| |
Prozentualer Anteil an Patienten, deren maximaler Pruritus NRS-Wert von der Baseline bis Woche 35 um ≥3 Punkte anstieg, unter den Patienten mit einem maximalen Baseline-Pruritus
NRS-Wert ≤7, (n [%])
|
56/80
(70,0)
|
45/81
(55,6)
|
57/168***
(33,9)
|
†p <0,05; *p <0,01; **p <0,001; ***p ≤0,0001
In der Studie SOLO CONTINUE wurde bei verlängerten Dosierungsintervallen eine Tendenz zu einer behandlungsbedingten stärkeren Produktion von Antikörpern gegen den Wirkstoff (Anti-Drug Antibodies, ADA) beobachtet.
Behandlungsbedingte ADA-Reaktionen: QW: 1,2%; Q2W: 4,3%; Q4W: 6,0%; Q8W: 11,7%. ADA-Reaktionen, die länger als 12 Wochen andauerten: QW: 0,0%; Q2W: 1,4%; Q4W: 0,0%; Q8W: 2,6%.
Lebensqualität/von Patienten berichtete Ergebnisse (Patient Reported Outcomes, PRO)
In den Studien SOLO 1 und SOLO 2 wurde mit Dupixent im Vergleich zu Placebo gemäss der POEM-, der DLQI- und der HADS-Gesamtbewertung eine signifikante Verbesserung der Symptome der AD, der gesundheitsbezogenen Lebensqualität sowie der Angst- und Depressionssymptome erzielt.
In der CHRONOS-Studie wurde mit Dupixent im Vergleich zu Placebo gemäss der POEM- und der DLQI-Gesamtbewertung nach 16 Wochen eine signifikante Verbesserung der Symptome der AD und der gesundheitsbezogenen Lebensqualität erzielt.
In den Studien SOLO 1 und SOLO 2 waren die mittleren Veränderungen (± SE) der Least Squares der POEM-, der DLQI- und der HADS-Gesamtbewertung von der Baseline bis Woche 16 bei Patienten, die Dupilumab erhielten, signifikant höher als bei Placebo-Patienten (p <0,0001 für alle Vergleiche ausser HADS in der Studie SOLO 2, p <0,001), siehe Tabelle 5.
In der Studie CHRONOS waren die mittleren Veränderungen (± SE) der Least Squares der POEM- und der DLQI-Gesamtbewertung von der Baseline bis Woche 16 bei Patienten, die Dupilumab erhielten, signifikant höher als bei Placebo-Patienten (p <0,0001 für alle Vergleiche), siehe Tabelle 5.
Tabelle 5: Zusätzliche Ergenisse der sekundären Endpunkte der Dupixent-Behandlung mit oder ohne TCS in Woche 16
|
|
Monotherapie in Woche 16
|
Begleitende Therapie mit TCS in Woche 16
| |
SOLO 1
|
SOLO 2
|
CHRONOS
| |
Placebo
|
Dupixent
300 mg Q2W
|
Placebo
|
Dupixent
300 mg Q2W
|
Placebo
|
Dupixent
300 mg Q2W
| |
Randomisierte Patienten
|
224
|
224
|
236
|
233
|
315
|
106
| |
POEM, mittlere Veränderung (LS) gegenüber der Baseline (SE)
|
-5,1
(0,67)
|
-11,6a
(0,49)
|
-3,3
(0,55)
|
-10,2a
(0,49)
|
-5,3
(0,41)
|
-12,7a
(0,64)
| |
DLQI, mittlere Veränderung (LS) gegenüber der Baseline (SE)
|
-5,3
(0,50)
|
-9,3a
(0,40)
|
-3,6
(0,50)
|
-9,3a
(0,38)
|
-5,8
(0,34)
|
-10,0a
(0,50)
|
LS = Least Squares [Methode der kleinsten Quadrate]; SE = Standard Error [Standardfehler]
a p <0,0001
|