PharmakokinetikNach Anwendung von Paliperidon zeigt die Pharmakokinetik von Paliperidon innerhalb der empfohlenen klinischen Dosisspanne (zwischen 3 und 12 mg) ein dosisproportionales Verhalten.
Absorption
Nach Applikation einer Einzeldosis Paliperidon steigen die Paliperidon-Konzentrationen im Plasma stetig an, bis etwa 24 Stunden nach Gabe Plasmahöchstkonzentrationen (Cmax) erreicht werden. Bei einmal täglicher Anwendung von Paliperidon stellen sich bei den meisten Patienten innerhalb von 4–5 Behandlungstagen Steady-State-Konzentrationen ein.
Im Gegensatz zu Risperidon mit sofortiger Freisetzung ist Paliperidon aufgrund seiner spezifischen Freisetzungseigenschaften mit nur minimalen Schwankungen zwischen Höchst- und Mindestkonzentrationen verbunden. In einer Studie zum Vergleich der Steady-State-Pharmakokinetik nach einmal täglicher Anwendung von 12 mg Paliperidon (als Tablette mit retardierter Freisetzung) bzw. von 4 mg Risperidon (sofortige Freisetzung) bei schizophrenen Probanden lag der Schwankungsindex im Fall von Paliperidon mit modifizierter Freisetzung bei 38%, verglichen mit 125% im Fall von Risperidon mit sofortiger Freisetzung (siehe Abbildung 1).
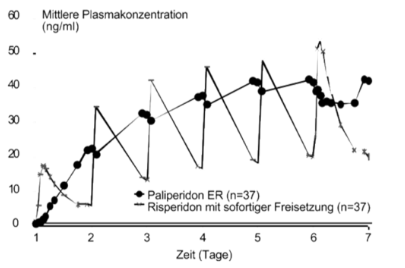
Abbildung 1: Steady-State-Konzentrationsprofil nach Verabreichung von 12 mg Paliperidon in Form von 6 Tabletten mit retardierter Freisetzung zu 2 mg einmal täglich über 6Tage (dargestellt sind die Konzentrationen von Paliperidon) im Vergleich zur Gabe von Risperidon mit sofortiger Freisetzung, verabreicht mit einer Dosis von 2 mg einmal täglich an Tag 1 und 4 mg einmal täglich an den Tagen 2 bis 6 (dargestellt sind die Konzentrationen von Paliperidon + Risperidon).
Nach Anwendung von Paliperidon kommt es zu einer Interkonversion der (+)- und (–)-Enantiomere von Paliperidon, wobei im Steady-State ein AUC(+)/(–)-Verhältnis von ungefähr 1,6 erreicht wird. Nach Verabreichung von Paliperidon liegt die absolute orale Bioverfügbarkeit von Paliperidon bei 28%.
Bei Einnahme von Paliperidon-Retardtabletten mit einer standardisierten Mahlzeit mit hohem Fett- und Kaloriengehalt liegen die Cmax- und AUC-Werte für Paliperidon im Vergleich zur Einnahme auf nüchternen Magen um bis zu 50–60% höher.
In den klinischen Studien zur Untersuchung der Sicherheit und Wirksamkeit von Paliperidon wurde der Einfluss durch die Nahrungsaufnahme nicht berücksichtigt (siehe «Dosierung/Anwendung»).
Distribution
Paliperidon wird rasch verteilt. Das scheinbare Distributionsvolumen liegt bei 487 l. Die Plasmaproteinbindung von Paliperidon beträgt 74%. Der Wirkstoff bindet vorwiegend an α1-saures Glykoprotein und Albumin.
Metabolismus
Eine Woche nach Verabreichung einer oralen Einzeldosis von 1 mg 14C-markiertem Paliperidon mit sofortiger Freisetzung wurden 59% der Dosis unverändert über den Urin ausgeschieden. Dies deutet darauf hin, dass Paliperidon in der Leber nicht extensiv metabolisiert wird. Ungefähr 80% der verabreichten Radioaktivität wurden im Urin wiedergefunden, 11% in den Fäzes. In vivo wurden vier Stoffwechselwege identifiziert, von denen keiner für mehr als 6,5% der Dosis verantwortlich war: Dealkylierung, Hydroxylierung, Dehydrogenierung und Benzisoxazol-Spaltung. Obwohl In-vitro-Versuche für eine Beteiligung von CYP2D6 und CYP3A4 an der Metabolisierung von Paliperidon sprechen, deutet in vivo nichts darauf hin, dass diese Isoenzyme bei der Verstoffwechslung von Paliperidon eine bedeutende Rolle spielen. Obwohl hinsichtlich der Fähigkeit zur Metabolisierung von CYP2D6-Substraten in der Allgemeinbevölkerung starke Unterschiede bestehen, haben Analysen der Populationspharmakokinetik mit Blick auf die scheinbare Clearance von Paliperidon nach Anwendung von Paliperidon keine deutlichen Unterschiede zwischen schnellen Metabolisierern (extensive metabolisers) und langsamen Metabolisierern (poor metabolisers) von CYP2D6-Substraten ergeben. In-vitro-Untersuchungen mit Mikrosomenpräparationen heterologer Systeme haben gezeigt, dass CYP1A2, CYP2A6, CYP2C9, CYP2C19 und CYP3A5 nicht an der Metabolisierung von Paliperidon beteiligt sind. Die terminale Eliminationshalbwertszeit von Paliperidon liegt bei rund 23 Stunden.
Elimination
Siehe «Metabolismus».
Kinetik spezieller Patientengruppen
Ethnische Zugehörigkeit
In der populationspharmakokinetischen Analyse ergaben sich keine Anzeichen für ethnische Unterschiede in der Pharmakokinetik von Paliperidon nach Anwendung von Paliperidon. Die apparente Clearance von Paliperidon nach der Gabe von Paliperidon lag bei dunkelhäutigen Personen um annähernd 38% niedriger als bei Personen anderer ethnischer Herkunft. Dieser Unterschied hat vermutlich keine klinische Relevanz. Die Dosisempfehlung für Paliperidon gilt unabhängig von der ethnischen Herkunft des Patienten, da Dosisanpassungen nach Behandlungsbeginn auf der Basis klinischer Untersuchung erfolgen sollten.
In einer Studie mit Japanern und Kaukasiern wurden keine Unterschiede in der Pharmakokinetik beobachtet.
Geschlecht
Nach Anwendung von Paliperidon ist die scheinbare Clearance von Paliperidon bei Frauen um etwa 19% geringer als bei Männern. Diese Abweichung lässt sich grösstenteils durch die geschlechtsspezifischen Unterschiede in der fettfreien Körpermasse und Kreatinin-Clearance erklären.
Raucherstatus
Aus In-vitro-Studien mit Enzymen der menschlichen Leber geht hervor, dass Paliperidon kein Substrat von CYP1A2 ist; Rauchen sollte daher keinen Einfluss auf die Pharmakokinetik von Paliperidon haben. Eine populationspharmakokinetische Auswertung zeigte eine leicht niedrigere Exposition mit Paliperidon bei Rauchern im Vergleich zu Nichtrauchern. Der Unterschied ist jedoch vermutlich nicht von klinischer Relevanz.
Leberfunktionsstörungen
Paliperidon wird nicht extensiv in der Leber metabolisiert. In einer Studie an Prüfungsteilnehmern mit mittelschwerer Einschränkung der Leberfunktion (Child-Pugh-Klasse B) waren die Plasmakonzentrationen von freiem Paliperidon ähnlich denen gesunder Prüfungsteilnehmer. Zu Patienten mit schwerer Einschränkung der Leberfunktion (Child- Pugh-Klasse C) liegen keine Erkenntnisse vor.
Nierenfunktionsstörungen
Die Elimination von Paliperidon nahm mit abnehmender Nierenfunktion ab. Die Gesamt-Clearance von Paliperidon war bei Prüfungsteilnehmern mit eingeschränkter Nierenfunktion um 32% bei leichter Einschränkung der Nierenfunktion (CrCl = 50 bis <80 ml/min), um 64% bei mässiger (CrCl = 30 bis <50 ml/min) und um 71% bei schwerer Einschränkung der Nierenfunktion (CrCl = 10 bis <30 ml/min) verringert. Die mittlere terminale Eliminationshalbwertzeit von Paliperidon betrug bei Prüfungsteilnehmern mit leichter, mässiger und schwerer Einschränkung der Nierenfunktion jeweils 24, 40 bzw. 51 Stunden, im Vergleich zu 23 Stunden bei Prüfungsteilnehmern mit normaler Nierenfunktion (CrCl ≥80 ml/min).
Ältere Patienten
Eine Dosisanpassung auf Grundlage des Alters wird nicht empfohlen. Daten aus einer Pharmakokinetik-Studie an älteren Patienten (≥65 Jahre, n= 26) haben ergeben, dass die scheinbare Steady-State-Clearance von Paliperidon nach Gabe von Paliperidon gegenüber einer Anwendung bei erwachsenen Probanden (18–45 Jahre, n= 28) um 20% vermindert war. Allerdings liess sich in der Untersuchung der Populationspharmakokinetik an Schizophrenie-Patienten nach Adjustierung für die altersbedingte Verminderung der CrCl kein erkennbarer Alterseffekt nachweisen.
|