Eigenschaften/WirkungenATC-Code
L04AA42
Wirkungsmechanismus
Siponimod ist ein Sphingosin-1-Phosphat (S1P) Rezeptormodulator. Siponimod bindet selektiv an zwei von fünf G-Protein-gekoppelten Rezeptoren (GPCRs) für S1P: S1P1 und S1P5. Siponimod wirkt als funktioneller Antagonist auf den S1P1-Rezeptor der Lymphozyten und verhindert so den Austritt der Lymphozyten aus den Lymphknoten. Dies vermindert die Rezirkulation von T-Zellen in das Zentralnervensystem (ZNS), und begrenzt so die Entzündung im ZNS. Siponimod passiert die Blut-Hirn-Schranke. Siponimod hat keinen nachhaltigen Einfluss auf die effektorischen T-Gedächtniszellen im peripheren Gewebe sowie im Blut und beeinträchtigt die Lymphozytenaktivierung nicht.
Pharmakodynamik
In tierexperimentellen Studien wurden direkte Effekte für Siponimod auf Nervenzellen, über S1P1 auf Astrozyten und S1P5 auf Oligodendrozyten nachgewiesen. In einem Mausmodell der experimentellen autoimmunen Enzephalomyelitis wurde eine direkte neuroprotektive Wirkung, unabhängig von Wirkungen auf Lymphozyten, auch für zentral angewendetes Siponimod (über intrazerebroventrikuläre Infusionen) nachgewiesen.
Immunsystem
Mayzent induziert eine dosisabhängige Reduktion der Lymphozytenzahl im peripheren Blut innerhalb von 6 Stunden nach der ersten Dosis, dies ist auf das reversible Zurückhalten (Sequestrierung) von Lymphozyten im Lymphgewebe zurückzuführen.
Bei andauernder täglicher Gabe nimmt die Lymphozytenzahl kontinuierlich ab und erreicht einen medianen Minimalwert (90 % KI) von ungefähr 0.560 (0.271 bis 1.08) Zellen/nl bei einem typischen, nicht aus Japan stammenden CYP2C9*1/*1 oder CYP2C9*1/*2-Patienten mit SPMS, was einer Verringerung von 20 bis 30 % gegenüber dem Ausgangswert entspricht. Bei täglicher Einnahme werden niedrige Lymphozytenzahlen aufrechterhalten.
Die Lymphozytenzahl kehrt bei der überwiegenden Mehrheit (90 %) der SPMS-Patienten innerhalb von 10 Tagen nach Beendigung der Behandlung in den Normalbereich zurück. Nach Beendigung der Mayzent-Behandlung kann die Reduzierung der peripheren Lymphozytenzahl bis zu 3 bis 4 Wochen nach der letzten Dosis anhalten.
Kardialelektrophysiologie
Herzfrequenz und Rhythmus
Mayzent führt bei Behandlungsbeginn zu einer vorübergehenden Abnahme der Herzfrequenz und der atrioventrikulären Überleitung (s. «Unerwünschte Wirkungen»). Dies ist ursächlich verbunden mit einer Aktivierung von G-Protein-gekoppelten, nach innen rektifizierenden Kalium-(GIRK-)Kanälen über die S1P1-Rezeptorstimulation, was zu einer zellulären Hyperpolarisation und verringerter Erregbarkeit führt. Aufgrund ihres funktionellen Antagonismus an S1P1-Rezeptoren desensibilisiert die Anfangstitration von Siponimod nacheinander die GIRK-Kanäle, bis die Erhaltungsdosis erreicht ist.
Potenzial zur Verlängerung des QT-Intervalls
Die Auswirkungen von therapeutischen (2 mg) und supratherapeutischen (10 mg) Siponimod-Dosierungen auf die kardiale Repolarisation wurden in einer ausführlichen QT-Studie untersucht. Die Ergebnisse deuten nicht auf ein arrhythmogenes Potenzial in Zusammenhang mit der QT-Verlängerung mit Siponimod hin, da Siponimod das Placebo-korrigierte Baseline-bereinigte mittlere QTcF-Intervall (ΔΔQTcF) um mehr als 5 ms erhöhte, mit einer maximalen mittleren Wirkung von 7.8 ms (2 mg) bzw. 7.2 ms (10 mg) 3 Stunden nach der Dosisgabe. Die Obergrenze des einseitigen 95 % KI für das ΔΔQTcF-Intervall blieb zu jedem Zeitpunkt unter 10 ms. Die kategorische Analyse ergab keine behandlungsbedingten QTc-Werte über 480 ms, Zunahmen des QTc-Intervalls von mehr als 60 ms gegenüber dem Ausgangswert und kein korrigierter oder unkorrigierter QT/QTc-Wert überstieg 500 ms.
Lungenfunktion
Eine Einzel- oder Mehrfachgabe von Mayzent über 28 Tage ist nicht mit einer klinisch relevanten Erhöhung des respiratorischen Widerstands, gemessen mittels forciertem exspiratorischem Fluss in 1 Sekunde (FEV1) bei 25 und 75 % des Lungenvolumens (FEF25-75 %) verbunden. Bei nicht-therapeutischen Einzeldosen (> 10 mg) wurde ein leichter Trend zu einem reduzierten FEV1 festgestellt. Die gleichzeitige Behandlung mit Mayzent und Propranolol führte zu einem minimalen Rückgang des FEV1 im Vergleich zu Propranolol allein, wobei die Veränderungen mit den einzelnen Medikamenten oder mit der Kombination innerhalb der physiologischen FEV1-Variabilität lagen und klinisch nicht signifikant waren.
Klinische Wirksamkeit
Die Wirksamkeit von Mayzent wurde in einer Phase-3-Studie untersucht, in der eine einmal tägliche Dosis von 2 mg Mayzent bei Patienten mit SPMS bewertet wurde. Eine Phase-2-Dosisfindungsstudie bei Patienten mit RRMS wies eine dosisabhängige Reduktion der entzündlichen Läsionen auf dem MRT nach und zeigte, dass Mayzent 2 mg eine nahezu maximale Wirkung erzielt.
Studie A2304 (EXPAND) bei SPMS
Die Studie A2304 war eine randomisierte, doppelblinde, placebokontrollierte, Ereignis- und Nachbeobachtungsdauer-getriebene Studie der Phase 3 bei Patienten mit SPMS, die in den letzten 2 Jahren in Abwesenheit oder unabhängig von Schüben eine nachgewiesene Progression, keine Hinweise auf einen Schub in den 3 Monaten vor Beginn der Studie und einen medianen EDSS-Score (Expanded Disability Status Scale) von 3.0 bis 6.5 bei Studienbeginn hatten.
Der mediane EDSS-Ausgangswert war 6.0. Patienten älter als 61 Jahre wurden nicht eingeschlossen. Im Hinblick auf die Krankheitsaktivität können die für die Entzündungsaktivität bei SPMS charakteristischen Merkmale Schübe- oder bildgebungsbezogen sein (d.h. Kontrastmittel-anreichernde T1-Läsionen oder aktive [neue oder sich neu vergrössernde] T2-Läsionen).
Die Patienten wurden im Verhältnis 2:1 für den Erhalt von Mayzent 2 mg einmal täglich oder Placebo randomisiert. Die klinischen Bewertungen wurden beim Screening, sowie alle 3 Monate und zum Zeitpunkt eines Schubes durchgeführt. MRT-Bewertungen wurden beim Screening und alle 12 Monate durchgeführt.
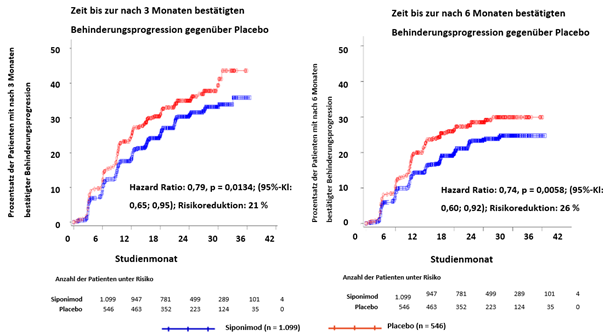
Der primäre Endpunkt der Studie war die Zeit bis zur 3-monatigen bestätigten Behinderungsprogression (confirmed disability progression, CDP), die als Zunahme um mindestens 1 Punkt gegenüber dem Ausgangswert des EDSS (0.5-Punkte-Zunahme für Patienten mit einem EDSS von 5.5 oder mehr) über 3 Monate hinweg ermittelt wurde. Wichtige sekundäre Endpunkte waren die Zeit bis zu einer bestätigten Verschlechterung nach 3 Monaten von mindestens 20 % gegenüber Baseline im zeitkontrollierten 25-Fuss-Gehtest (timed 25 foot walk test, T25FW) und die Veränderung gegenüber Baseline im T2-Läsionsvolumen. Weitere sekundäre Endpunkte waren die Zeit bis zur 6-monatigen CDP, prozentuale Veränderung des Gehirnvolumens und Messungen der entzündlichen Krankheitsaktivität (jährliche Schubrate, MRT-Läsionen). Die Veränderung der kognitiven Verarbeitungsgeschwindigkeit beim Symbol Digit Modalities Test war ein explorativer Endpunkt.
Die Studiendauer war für einzelne Patienten variabel (mediane Studiendauer 21 Monate, Bereich 1 Tag bis 37 Monate).
In dieser Studie wurden 1'651 Patienten auf entweder den Erhalt von Mayzent 2 mg (N = 1'105) oder Placebo (N = 546) randomisiert; 82 % der mit Mayzent behandelten Patienten und 78 % der mit Placebo behandelten Patienten schlossen die Studie ab. Das mediane Alter betrug 49.0 Jahre, die mediane Krankheitsdauer 16.0 Jahre und der mediane EDSS-Score 6.0 zu Studienbeginn; 64 % der Patienten hatten in den 2 Jahren vor Studienbeginn keine Schübe und 76 % hatten keine Gadolinium (Gd)-anreichernden Läsionen auf ihrem Ausgangs-MRT-Scan; 78 % der Patienten waren zuvor mit einer Therapie für ihre MS behandelt worden.
Die Zeit bis zum Auftreten der nach 3 und 6 Monaten bestätigten Behinderungsprogression war unter Siponimod signifikant verzögert, mit einer Risikoreduktion für eine nach 3 Monaten bestätigte Behinderungsprogression um 21 % im Vergleich zu Placebo (Hazard Ratio [HR] 0.79, p = 0.0134) und einer Risikoreduktion für eine nach 6 Monaten bestätigte Behinderungsprogression um 26 % gegenüber Placebo (HR 0.74, p = 0.0058).
Die Ergebnisse dieser Studie sind in Tabelle 3 und Abbildung 1 und 2 zusammengefasst.
Tabelle 3: Klinische und MRT-Ergebnisse der Studie A2304
|
Endpunkte
|
A2304 (EXPAND)
| |
Siponimod
2 mg
(n = 1.099)
|
Placebo
(n = 546)
| |
Klinische Endpunkte
| |
Primärer Wirksamkeitsendpunkt: Anteil der Patienten mit nach 3 Monaten bestätigter Behinderungsprogression (primärer Endpunkt)
|
26.3 %
|
31.7 %
| |
Risikoreduktion1
|
21 % (p = 0.0134)
| |
Anteil der Patienten mit nach 3 Monaten bestätigter 20%iger Zunahme beim
25-Fuss-Gehtest
|
39.7 %
|
41.4 %
| |
Risikoreduktion1
|
6 % (p = 0.4398)
| |
Anteil der Patienten mit nach 6 Monaten bestätigter Behinderungsprogression
|
19.9 %
|
25.5 %
| |
Risikoreduktion1
|
26 % [(p = 0.0058)] 6
| |
Jährliche Schubrate (ARR)
|
0.071
|
0.152
| |
Verringerung der Rate2
|
55 % [(p < 0.0001)] 6
| |
MRT-Endpunkte
| |
Veränderung des Volumens der
T2-Läsionen (mm3) gegenüber dem Ausgangswert3
|
+184 mm3
|
+879 mm3
| |
Unterschied bei der Änderung des Volumens der T2-Läsionen
|
–695 mm3 (p < 0.0001)7
| |
Prozentuale Veränderung des Gehirnvolumens gegenüber dem Ausgangswert (95%-KI)3
|
–0.497 %
|
–0.649 %
| |
Unterschied bei der prozentualen Veränderung des Gehirnvolumens
|
0.152 % [(p = 0.0002)] 6
| |
Durchschnittliche kumulative Anzahl der Gd-anreichernden, T1-gewichteten Läsionen (95%-KI)4
|
0.081
|
0.596
| |
Verringerung der Rate
|
86 % [(p < 0.0001)] 6
| |
Anteil der Patienten mit einer Verschlechterung um 4 Punkte beim Symbol Digit Modalities Test5
|
16.0 %
|
20.9 %
| |
Risikoreduktion1
|
25 % [(p = 0.0163)] 6
| |
1
Aus Cox-Modellen für die Zeit bis zur Progression
2 Aus einem Modell für wiederkehrende Ereignisse
3 Durchschnitt aus Monat 12 und Monat 24
4 Bis Monat 24
5 Bestätigt nach 6 Monaten
6 [Der nominale p-Wert für Endpunkte, die nicht in den hierarchischen Tests enthalten und nicht an die Multiplizität eingestellt sind]
7 Nicht bestätigender p-Wert; das hierarchische Testverfahren wurde vor Erreichen des Endpunkts beendet
|
Abbildung 1 Patienten mit nach 3 und 6-Monaten bestätigte Behinderungsprogession anhand von EDSS-Kaplan-Meier-Kurven (vollständiger Analysensatz, Studie A2304)
Die Ergebnisse der Studie zeigten eine durchgängige Risikoreduktion bezüglich der Zeit bis zur 3-Monats und 6-Monats-Behinderungsprogression mit Mayzent im Vergleich zu Placebo in Untergruppen, die nach Geschlecht, Alter, vorheriger Therapie der multiplen Sklerose, Schubaktivität vor der Studie, MRT-Krankheitsaktivität bei Baseline, Krankheitsdauer und Behinderungsgrad bei Baseline definiert wurden.
Mayzent hat eine positive Wirkung im Symbol Digit Modalities Test (SDMT) gezeigt. Die Veränderung gegenüber den Basiswerten war für Mayzent stabil oder besser und verschlechterte sich für Placebo mit einem signifikanten Unterschied zwischen den Gruppen von 1.1 Punkten im Monat 12 (p = 0.0132) bzw. 2.3 Punkten im Monat 24 (p = 0.0002). In einer Orientierungsuntersuchung senkte Mayzent das Risiko nach 6 Monaten einer bestätigten Verschlechterung um 4 Punkte im SDMT um 25 % (p = 0.0163) im Placebo-Vergleich. Eine Verschlechterung um 4 Punkte hat sich hierbei schon als klinisch relevant erwiesen.
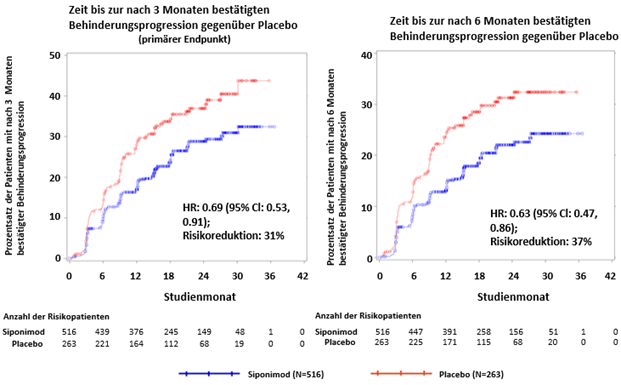
In der Subgruppe der Patienten (47.1 %, n = 779) mit Krankheitsaktivität (definiert als Patienten mit Schub in den 2 Jahren vor der Studie und/oder Vorhandensein von Gd-verstärkenden T1-Läsionen zu Studienbeginn) waren die Baseline-Eigenschaften ähnlich der Gesamtpopulation. Das mediane Alter betrug 47 Jahre, die mediane Krankheitsdauer 15 Jahre und der mediane EDSS-Wert zu Studienbeginn 6.0 (s. «Pharmakokinetik»).
Die Zeit bis zum Auftreten der nach 3 und 6 Monaten bestätigten Behinderungsprogression war bei mit Siponimod behandelten Patienten mit Krankheitsaktivität signifikant verzögert, um 31 % im Vergleich zu Placebo (Hazard Ratio [HR] 0.69; 95%-KI: 0.53; 0.91) und um 37 % im Vergleich zu Placebo (HR 0.63; 95%-KI: 0.47; 0.86). Die ARR (bestätigte Schübe) war im Vergleich zu Placebo um 46 % reduziert (ARR-Verhältnis 0.54; 95%-KI: 0.39; 0.77). Die relative Ratenreduktion der kumulativen Anzahl von Gd-verstärkenden T1-gewichteten Läsionen über 24 Monate betrug 85 % (Ratenverhältnis 0.155; 95%-KI: 0.104; 0.231) im Vergleich zu Placebo. Die Unterschiede in der Veränderung des T2-Läsionsvolumens und im Prozentsatz der Veränderung des Gehirnvolumens (Durchschnitt über die Monate 12 und 24) im Vergleich zu Placebo betrugen 1163 mm3 (95%-KI: 1484, 843 mm3) beziehungsweise 0.141 % (95%-KI: 0.020; 0.261 %).
In der Subgruppe der Patienten (n=827) ohne Anzeichen oder Symptome von Krankheitsaktivität (definiert als Patienten ohne Schub in den 2 Jahren vor der Studie und ohne Vorhandensein von Kontrastmittel anreichernden T1-Läsionen zu Studienbeginn), waren die Auswirkungen auf die nach 3 und 6 Monaten bestätigte Behinderungsprogression gering (die Risikoreduktionen betrugen 7 bzw. 13 %).
Abbildung 2 Patienten mit nach 3 und 6 Monaten bestätigter Behinderungsprogression anhand von EDSS-Kaplan-Meier-Kurven- Subgruppe mit entzündlicher Krankheitsaktivität (vollständiger Analysensatz, Studie A2304)
|