ZusammensetzungWirkstoffe
Olipudase alfa (hergestellt mittels Ovarialzellen des Chinesischen Hamsters (CHO)).
Hilfsstoffe
Methionin, Natriummonohydrogenphosphat-Heptahydrat, Natriumdihydrogenphosphat-Monohydrat, Saccharose.
Eine Durchstechflasche zu 4 mg enthält 0,60 mg Natrium.
Eine Durchstechflasche zu 20 mg enthält 3,02 mg Natrium.
Indikationen/AnwendungsmöglichkeitenXenpozyme ist als Enzymersatztherapie zur Behandlung von Manifestationen eines Mangels an saurer Sphingomyelinase (ASMD) vom Typ A/B oder Typ B ausserhalb des Zentralnervensystems bei pädiatrischen und erwachsenen Patienten indiziert.
Dosierung/AnwendungDie Behandlung mit Xenpozyme sollte unter Aufsicht von medizinischem Fachpersonal erfolgen, das über Erfahrung mit der Behandlung von ASMD oder vergleichbaren angeborenen Stoffwechselerkrankungen verfügt und Zugriff auf geeignete medizinische Notfallmassnahmen hat, mit denen mögliche schwere Reaktionen wie schwerwiegende systemische Überempfindlichkeitsreaktionen behandelt werden können. Die Behandlung mit Xenpozyme muss immer mit einer Dosissteigerung gemäss untenstehender Schemata (siehe Tabellen 1 und 2) gefolgt von einer Erhaltungsdosis eingeleitet werden, um das Risiko infusionsbedingter Reaktionen, einschliesslich Akute-Phase-Reaktionen, und Erhöhungen der Transaminasewerte zu minimieren. Zum Vorgehen nach versäumter Dosisgabe siehe unten.
Während der Erhaltungsphase der Behandlung kann eine Heiminfusion unter Aufsicht von medizinischem Fachpersonal in Erwägung gezogen werden (s.u.).
Dosierung
Der rasche Abbau von angereichertem Sphingomyelin (SM) durch Xenpozyme führt zur Bildung von proinflammatorischen Abbauprodukten, die infusionsbedingte Reaktionen und/oder vorübergehend erhöhte Leberwerte verursachen können. Ein Therapieschema mit Dosissteigerung kann die meisten dieser unerwünschten Ereignisse minimieren (siehe «Präklinische Daten»).
Die Xenpozyme-Dosis hängt bei Patienten mit einem Body-Mass-Index (BMI) ≤30 vom tatsächlichen Körpergewicht und bei Patienten mit einem BMI > 30 vom angepassten Körpergewicht ab (siehe Abschnitt zu Patienten mit einem BMI > 30).
Um Dosierungsfehler einschliesslich Überdosierung (siehe «Überdosierung») zu vermeiden, sind alle Anweisungen zur Dosierung und Verabreichung (siehe unten) sowie zur Zubereitung und Handhabung (siehe «Sonstige Hinweise» und «Zubereitung der Infusionslösung gemäss Dosierung») zu befolgten. Es gilt zu beachten, dass sich die Dosissteigerung bei pädiatrischen Patienten von der bei Erwachsenen unterscheidet. Zusätzlich zum Schema zur Dosissteigerung muss die Infusionsrate bei jeder Dosis schrittweise erhöht werden (siehe Tabellen 4 und 5).
Erwachsene
Dosissteigerungsphase
Die empfohlene Anfangsdosis von Xenpozyme bei Erwachsenen beträgt 0,1 mg/kg* (für zusätzliche Hinweise siehe auch den Unterabschnitt zur verspäteten Dosisabgabe), daran anschliessend sollte die Dosis gemäss dem in Tabelle 1 aufgeführten Schema zur Dosissteigerung erhöht werden:
Tabelle 1: Schema zur Dosissteigerung bei Erwachsenen
|
Erwachsene Patienten (≥18 Jahre)
| |
Erste Dosis (Tag 1/Woche 0)
|
0,1 mg/kg*
| |
Zweite Dosis (Woche 2)
|
0,3 mg/kg*
| |
Dritte Dosis (Woche 4)
|
0,3 mg/kg*
| |
Vierte Dosis (Woche 6)
|
0,6 mg/kg*
| |
Fünfte Dosis (Woche 8)
|
0,6 mg/kg*
| |
Sechste Dosis (Woche 10)
|
1 mg/kg*
| |
Siebte Dosis (Woche 12)
|
2 mg/kg*
| |
Achte Dosis (Woche 14)
|
3 mg/kg* (empfohlene Erhaltungsdosis)
|
* Bei Patienten mit einem BMI ≤ 30 wird das tatsächliche Körpergewicht zugrunde gelegt. Bei Patienten mit einem BMI > 30 wird, wie weiter unten beschrieben, ein angepasstes Körpergewicht zugrunde gelegt.
Erhaltungsphase
Die empfohlene Erhaltungsdosis von Xenpozyme beträgt 3 mg/kg* alle 2 Wochen.
* Bei Patienten mit einem BMI ≤30 wird das tatsächliche Körpergewicht zugrunde gelegt. Bei Patienten mit einem BMI > 30 wird, wie weiter unten beschrieben, ein angepasstes Körpergewicht zugrunde gelegt.
Leberfunktionsstörungen
Bei Patienten mit Leberfunktionsstörungen wird keine Dosisanpassung empfohlen (siehe «Pharmakokinetik»).
Nierenfunktionsstörungen
Bei Patienten mit Nierenfunktionsstörungen wird keine Dosisanpassung empfohlen (siehe «Pharmakokinetik»).
Ältere Patienten
Bei Patienten über 65 Jahren wird keine Dosisanpassung empfohlen (siehe «Pharmakokinetik»).
Kinder und Jugendliche
Dosissteigerungsphase
Die empfohlene Anfangsdosis von Xenpozyme bei pädiatrischen Patienten beträgt 0,03 mg/kg*, daran anschliessend sollte die Dosis gemäss dem in Tabelle 2 aufgeführten Schema zur Dosissteigerung erhöht werden:
Tabelle 2: Schema zur Dosissteigerung bei pädiatrischen Patienten
|
Pädiatrische Patienten (0 bis < 18 Jahre)
| |
Erste Dosis (Tag 1/Woche 0)
|
0,03 mg/kg*
| |
Zweite Dosis (Woche 2)
|
0,1 mg/kg*
| |
Dritte Dosis (Woche 4)
|
0,3 mg/kg*
| |
Vierte Dosis (Woche 6)
|
0,3 mg/kg*
| |
Fünfte Dosis (Woche 8)
|
0,6 mg/kg*
| |
Sechste Dosis (Woche 10)
|
0,6 mg/kg*
| |
Siebte Dosis (Woche 12)
|
1 mg/kg*
| |
Achte Dosis (Woche 14)
|
2 mg/kg*
| |
Neunte Dosis (Woche 16)
|
3 mg/kg* (empfohlene Erhaltungsdosis)
|
* Bei Patienten mit einem BMI ≤ 30 wird das tatsächliche Körpergewicht zugrunde gelegt. Bei Patienten mit einem BMI > 30 wird, wie weiter unten beschrieben, ein angepasstes Körpergewicht zugrunde gelegt.
Erhaltungsphase
Die empfohlene Erhaltungsdosis von Xenpozyme beträgt 3 mg/kg* alle 2 Wochen.
* Bei Patienten mit einem BMI ≤ 30 wird das tatsächliche Körpergewicht zugrunde gelegt. Bei Patienten mit einem BMI > 30 wird, wie weiter unten beschrieben, ein angepasstes Körpergewicht zugrunde gelegt.
Besondere Patientengruppen
Patienten mit BMI > 30
Bei erwachsenen sowie pädiatrischen Patienten mit einem Body-Mass-Index (BMI) > 30 wird ein angepasstes Körpergewicht zur Berechnung der Xenpozyme-Dosis mithilfe der folgenden Methode (für Dosissteigerungs- und Erhaltungsphase) zugrunde gelegt.
Körpergewicht (kg) für die Dosisberechnung = 30 × (tatsächliche Körpergrösse in m)2
Beispiel:
Bei einem Patienten mit
einem BMI von 38,
einem Körpergewicht von 110 kg und
einer Grösse von 1,70 m
wird zur Berechnung der zu verabreichenden Dosis ein Körpergewicht von 30 × 1,702 = 86,7 kg zugrunde gelegt.
Versäumte Dosisgabe
Eine Dosis wird als versäumt betrachtet, wenn sie nicht innerhalb von 3 Tagen nach dem vorgesehenen Datum verabreicht wird. Wenn eine Dosis von Xenpozyme versäumt wird, sollte die nächste Dosis wie in Tabelle 3 beschrieben so schnell wie möglich verabreicht werden. Danach sollten die Gaben alle 2 Wochen ab dem Datum der letzten Verabreichung erfolgen.
Tabelle 3: Dosierungsempfehlung bei versäumter Dosisgabe*
|
Anzahl versäumter Infusionen
|
Dosissteigerungsphase
|
Erhaltungsphase
| |
Wenn eine Infusion versäumt wurde:
|
Die letzte vertragene Dosis sollte verabreicht werden, bevor die Dosissteigerung gemäss Schema für Erwachsene (Tabelle 1) oder für pädiatrische Patienten (Tabelle 2) wieder aufgenommen wird.
|
Es sollte die Erhaltungsdosis verabreicht und der Behandlungsplan entsprechend angepasst werden.
| |
Wenn 2 Infusionen in Folge versäumt wurden:
|
Es sollte eine Dosis mit einer Dosierungsstufe unterhalb der letzten vertragenen Dosis verabreicht werden (Mindestdosis 0,3 mg/kg), bevor die Dosissteigerung gemäss Tabelle 1 oder Tabelle 2 wieder aufgenommen wird.
|
Es sollte eine niedrigere Dosis als die Erhaltungsdosis (d.h. 2 mg/kg) verabreicht werden. Danach sollte bei den anschliessenden Infusionen die Erhaltungsdosis (3 mg/kg) alle 2 Wochen verabreicht werden.
| |
Wenn 3 oder mehr Infusionen in Folge versäumt wurden:
|
Bei Patienten, die die Dosissteigerungsphase nicht abgeschlossen haben, sollte die Dosissteigerung
·bei Erwachsenen wieder mit 0,1 mg/kg begonnen und gemäss Tabelle 1 fortgesetzt werden.
·bei pädiatrischen Patienten wieder mit 0,03 mg/kg begonnen und gemäss Tabelle 2 fortgesetzt werden.
|
Die Dosissteigerung sollte wieder mit 0,3 mg/kg beginnen und gemäss Tabelle 1 oder Tabelle 2 fortgesetzt werden.
Bei Patienten, die Erhaltungsinfusionen über einen längeren Zeitraum versäumt haben, während dessen sich Sphingomyelin erneut angesammelt haben könnte, sollte der behandelnde Arzt
·bei erwachsenen Patienten die Wiederaufnahme der Dosis bei 0,1 mg/kg und eine Dosissteigerung gemäss Tabelle 1 in Erwägung ziehen.
·bei pädiatrischen Patienten die Wiederaufnahme der Dosis bei 0,03 mg/kg und eine Dosissteigerung gemäss Tabelle 2 in Erwägung ziehen.
|
* Wenn die verabreichte Dosis 0,3 oder 0,6 mg/kg beträgt, sollte diese Dosis bei der nächsten vorgesehenen Infusion nach einer ausgelassenen Dosis gemäss Tabelle 1 und Tabelle 2 zweimal verabreicht werden.
Überwachung der Transaminasewerte
Transaminasewerte (Alaninaminotransferase [ALT] und Aspartataminotransferase [AST]) sollten vor Einleitung der Therapie bestimmt und während jeder Dosissteigerungsphase überwacht werden (siehe «Warnhinweise und Vorsichtsmassnahmen»). Wenn die Transaminasewerte vor Infusionsbeginn über den Ausgangswert hinaus erhöht sind und mehr als das 2-fache der ULN (Upper Limit of Normal, oberer Grenzwert einer Standardnormalverteilung) betragen, kann die Xenpozyme-Dosis in Abhängigkeit vom Ausmass der Transaminaseerhöhung angepasst (erneute Gabe der vorherigen Dosis oder reduzierte Dosis) oder die Behandlung vorübergehend ausgesetzt werden. Wenn ein Patient eine Dosisanpassung benötigt oder eine Unterbrechung der Behandlung erforderlich ist, sollte die Wiederaufnahme der Behandlung nach dem in Tabelle 1 und Tabelle 2 für erwachsene bzw. pädiatrische Patienten beschriebenen Schema zur Dosissteigerung und den Empfehlungen für den Fall einer ausgelassenen Dosis erfolgen (siehe «Versäumte Dosisgabe»).
Art der Anwendung
Xenpozyme ist nur zur intravenösen Anwendung bestimmt. Die Infusionen sind schrittweise und vorzugsweise mit einer Infusionspumpe durchzuführen.
Hinweise zur Rekonstitution und Verdünnung des Arzneimittels vor der Anwendung siehe «Hinweise für die Handhabung».
Nach Rekonstitution und Verdünnung wird die Lösung als intravenöse Infusion verabreicht. Nur wenn infusionsbedingte Reaktionen ausbleiben, darf die Infusionsgeschwindigkeit während der Infusion schrittweise erhöht werden (bei infusionsbedingten Reaktionen siehe «Warnhinweise und Vorsichtsmassnahmen»). Die Infusionsgeschwindigkeit und die Infusionsdauer (+/- 5 min) eines jeden Infusionsschrittes sind in Tabelle 4 und Tabelle 5 aufgeführt. Bei der Bestimmung der Infusionsgeschwindigkeit in den Tabellen 4 und 5 ist die Dosis aus dem Schema zur Dosissteigerung zu verwenden, die entweder in Tabelle 1 (Erwachsene) oder in Tabelle 2 (pädiatrische Patienten) aufgeführt ist.
Tabelle 4: Infusionsgeschwindigkeit und Infusionsdauer bei erwachsenen Patienten
|
Dosis* (mg/kg)
|
Infusionsgeschwindigkeit
Infusionsdauer
|
Ungefähre Infusionsdauer
| |
|
Schritt 1
|
Schritt 2
|
Schritt 3
|
Schritt 4
|
| |
0,1
|
20 ml/h
über 20 min
|
60 ml/h
über 15 min
|
n. z.
|
n. z.
|
35 min
| |
0,3 bis 3
|
3,33 ml/h
über 20 min
|
10 ml/h
über 20 min
|
20 ml/h
über 20 min
|
33,33 ml/h
über 160 min
|
220 min
|
h: Stunde; min: Minute; n. z.: nicht zutreffend
* Dosis aus dem Schema zur Dosissteigerung in Tabelle 1
Tabelle 5: Infusionsgeschwindigkeit und Infusionsdauer bei pädiatrischen Patienten
|
Dosis* (mg/kg)
|
Infusionsgeschwindigkeit
Infusionsdauer
|
Ungefähre Infusionsdauer
| |
Schritt 1
|
Schritt 2
|
Schritt 3
|
Schritt 4
| |
0,03
|
0,1 mg/kg/h über die gesamte Dauer der Infusion
|
n. z.
|
n. z.
|
n. z.
|
18 min
| |
0,1
|
0,1 mg/kg/h über 20 min
|
ab 0,3 mg/kg/h
|
n. z.
|
n. z.
|
35 min
| |
0,3
|
0,1 mg/kg/h über 20 min
|
0,3 mg/kg/h
über 20 min
|
ab 0,6 mg/kg/h
|
n. z.
|
60 min
| |
0,6
|
0,1 mg/kg/h über 20 min
|
0,3 mg/kg/h über 20 min
|
0,6 mg/kg/h über 20 min
|
ab 1 mg/kg/h
|
80 min
| |
1
|
100 min
| |
2
|
160 min
| |
3
|
220 min
|
h: Stunde; min: Minute; n. z.: nicht zutreffend
* Dosis aus dem Schema zur Dosissteigerung in Tabelle 2
Während der Infusion sollte auf Anzeichen und Symptome von infusionsbedingten Reaktionen (IAR; infusion-associated reactions) wie Kopfschmerzen, Urtikaria, Fieber, Übelkeit und Erbrechen sowie andere Anzeichen oder Symptome einer Überempfindlichkeit geachtet werden. Je nach Schweregrad der Symptome kann die Infusion bei Bedarf verlangsamt, unterbrochen oder abgebrochen und geeignete medizinische Massnahmen können eingeleitet werden.
Bei schwerer Überempfindlichkeitsreaktion und/oder anaphylaktischer Reaktion sollte die Behandlung mit Xenpozyme unverzüglich abgebrochen werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Am Ende der Infusion (wenn die Spritze oder der Infusionsbeutel leer ist) sollte die Infusionsleitung mit Natriumchlorid-Injektionslösung 0,9 % (9 mg/ml) mit der gleichen Infusionsgeschwindigkeit wie im letzten Teil der Infusion gespült werden.
Heiminfusion während der Erhaltungsphase
Für Patienten, die mit der Erhaltungsdosis behandelt werden und ihre Infusionen gut vertragen, kann eine Heiminfusion unter Aufsicht von medizinischem Fachpersonal in Erwägung gezogen werden. Die Entscheidung für einen Wechsel der Patienten auf eine Heiminfusion sollte nach Beurteilung und auf Empfehlung des verschreibenden Arztes bzw. der verschreibenden Ärztin erfolgen.
Bei der Verabreichung von Xenpozyme muss eine geeignete medizinische Betreuung, einschliesslich in Notfallmassnahmen geschultes Personal, vorhanden sein. Bei anaphylaktischen oder anderen Akutreaktionen sind die Infusion von Xenpozyme unverzüglich abzubrechen, geeignete medizinische Massnahmen einzuleiten und ein Arzt bzw. eine Ärztin zu konsultieren. Bei schweren Überempfindlichkeitsreaktionen sollten nachfolgende Infusionen nur in einer Umgebung erfolgen, in der eine Ausrüstung für Wiederbelebungsmassnahmen verfügbar ist. Bei der Heiminfusion sollten die Dosis und Infusionsgeschwindigkeiten die gleichen bleiben wie in der überwachten klinischen Umgebung und nicht ohne Anordnung durch den verschreibenden Arzt bzw. die verschreibende Ärztin geändert werden. Bei ausgelassenen Dosen oder verspäteter Infusion sollte der verschreibende Arzt bzw. die verschreibende Ärztin kontaktiert werden, weil die nachfolgenden Infusionen vielleicht in einer überwachten klinischen Umgebung erfolgen.
KontraindikationenLebensbedrohliche Überempfindlichkeit (anaphylaktische Reaktion) gegen Olipudase alfa oder einen der sonstigen Bestandteile (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Warnhinweise und VorsichtsmassnahmenRückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Keine Überwindung der Blut-Hirn-Schranke
Es ist nicht zu erwarten, dass Xenpozyme die Blut-Hirn-Schranke überwindet oder Manifestationen der Erkrankung im Zentralnervensystem (ZNS) beeinflusst.
Infusionsbedingte Reaktionen (IAR; infusion-associated reactions)
In klinischen Studien traten bei ca. 60 % der mit Xenpozyme behandelten Patienten IAR auf. Zu diesen IAR zählten Überempfindlichkeitsreaktionen und Akute-Phase-Reaktionen (siehe «Unerwünschte Wirkungen»). Die häufigsten IAR waren Kopfschmerzen, Urtikaria, Fieber, Übelkeit und Erbrechen (siehe «Unerwünschte Wirkungen»). IAR traten normalerweise während der Dauer der Infusion bis zu 24 Stunden nach Abschluss der Infusion auf.
Schwerwiegende unerwünschte Wirkungen, einschliesslich Todesfälle, sind nach Überdosierung während der Dosissteigerungsphase aufgetreten (siehe «Überdosierung»). Um das Risiko solcher Reaktionen zu vermindern, sind die Dosierungsempfehlungen gemäss «Dosierung/Anwendung» sowie die Anleitungen zur Zubereitung und Handhabung (siehe «Sonstige Hinweise» und «Zubereitung der Infusionslösung gemäss Dosierung») zu befolgen.
Überempfindlichkeit einschliesslich Anaphylaxie
Von Patienten, die mit Xenpozyme behandelt wurden, sind Überempfindlichkeitsreaktionen einschliesslich Anaphylaxie berichtet worden (siehe «Unerwünschte Wirkungen»). In klinischen Studien traten Überempfindlichkeitsreaktionen bei 9 (22,5 %) erwachsenen und 9 (45 %) pädiatrischen Patienten auf, darunter ein pädiatrischer Patient mit Anaphylaxie.
Leichte bis mittelschwere Überempfindlichkeitsreaktionen wie Urtikaria, Erythem, Pruritus, Hautausschlag und Angioödem wurden bei mehr als einem erwachsenen Patienten berichtet. Bei pädiatrischen Patienten war mehr als ein Patient von leichten bis mittelschweren Überempfindlichkeitsreaktionen wie Urtikaria, Erythem, Hautausschlag und Pruritus betroffen.
Die Patienten sollten während der Infusion und für einen gemäss klinischer Beurteilung angemessenen Zeitraum danach engmaschig überwacht werden. Die Patienten sind über die möglichen Symptome einer Überempfindlichkeit/Anaphylaxie aufzuklären und anzuweisen, bei Auftreten von Symptomen unverzüglich medizinische Hilfe zu suchen. Die Behandlung der IAR richtet sich nach dem Schweregrad der Anzeichen und Symptome und kann eine vorübergehende Unterbrechung der Infusion von Xenpozyme, Senkung der Infusionsgeschwindigkeit und/oder eine geeignete medizinische Behandlung umfassen.
Bei schwerer Überempfindlichkeitsreaktion oder Anaphylaxie ist Xenpozyme unverzüglich abzusetzen und eine geeignete medizinische Behandlung ist einzuleiten. Der Patient, bei dem es in der klinischen Studie zu Anaphylaxie kam, unterzog sich einer individuell angepassten Desensibilisierungstherapie, die es ihm ermöglichte, die langfristige Behandlung mit Xenpozyme in der empfohlenen Erhaltungsdosis wieder aufzunehmen. Der verschreibende Arzt bzw. die verschreibende Ärztin muss die Risiken und den Nutzen der erneuten Gabe von Xenpozyme nach einer Anaphylaxie oder schweren Überempfindlichkeitsreaktion abwägen. Wird eine erneute Verabreichung von Xenpozyme nach einer Anaphylaxie in Erwägung gezogen, sollte sich der verschreibende Arzt bzw. die verschreibende Ärztin hinsichtlich der erneuten Verabreichung mit dem lokalen Vertreter von Sanofi beraten. Bei solchen Patienten ist bei der erneuten Verabreichung von Xenpozyme äusserste Vorsicht geboten, und es muss eine geeignete Ausrüstung für Wiederbelebungsmassnahmen verfügbar sein.
Bei leichten oder mittelschweren IAR können die Infusionsgeschwindigkeit verlangsamt oder die Infusion vorübergehend unterbrochen, die Dauer der einzelnen Schritte bei einer Einzelinfusion verlängert und/oder die Dosis von Xenpozyme verringert werden. Wenn bei einem Patienten eine Dosisreduktion erforderlich ist, sollte die erneute Dosissteigerung gemäss dem in Tabelle 1 für Erwachsene und in Tabelle 2 für Kinder und Jugendliche beschriebenen Schema erfolgen (siehe «Dosierung/Anwendung»).
Patienten können zur Vermeidung oder Verringerung allergischer Reaktionen mit Antihistaminika, Antipyretika und/oder Glukokortikoiden vorbehandelt werden.
Immunogenität
Bei erwachsenen sowie pädiatrischen Patienten wurde davon berichtet, dass während der klinischen Studien behandlungsbedingte Antikörper gegen das Arzneimittel (ADA, anti-drug antibodies) auftraten (siehe «Unerwünschte Wirkungen»). IAR und Überempfindlichkeitsreaktionen können unabhängig von sich entwickelnden ADA auftreten. Die Mehrheit der IAR und Überempfindlichkeitsreaktionen war von leichter oder mittelschwerer Ausprägung und mit klinischen Standardverfahren zu behandeln.
Bei Patienten, bei denen eine schwere Überempfindlichkeitsreaktion auf Olipudase alfa auftrat, kann ein IgE-Test auf ADA in Erwägung gezogen werden.
Auch wenn in den klinischen Studien kein Verlust der Wirksamkeit berichtet wurde, kann ein IgG-Test auf ADA in Erwägung gezogen werden für den Fall, dass das Ansprechen auf die Therapie nachlässt.
Vorübergehende Erhöhungen der Transaminasewerte
Während der Dosissteigerungsphase von Xenpozyme wurden bei 4 erwachsenen und 7 pädiatrischen Patienten vorübergehende Erhöhungen der Transaminasewerte (ALT oder AST) innerhalb von 24 bis 48 Stunden nach der Infusion berichtet (siehe «Unerwünschte Wirkungen»). Zum Zeitpunkt der nächsten vorgesehenen Infusion waren diese erhöhten Transaminasewerte im Allgemeinen auf die Werte vor der Infusion von Xenpozyme zurückgegangen.
Die Transaminasewerte (ALT und AST) sollten einen Monat vor Einleitung der Xenpozyme-Therapie bestimmt werden (siehe «Dosierung/Anwendung»). Während der Dosissteigerung oder bei Wiederaufnahme der Behandlung nach ausgelassenen Dosen, sollten die Transaminasewerte innerhalb von 72 Stunden vor der nächsten Xenpozyme-Infusion bestimmt werden. Wenn die Transaminasewerte zu Beginn bzw. vor einer Infusion während der Dosissteigerung mehr als das 2-fache der ULN (Upper Limit of Normal, oberer Grenzwert einer Standardnormalverteilung) betragen, sollten die Transaminasewerte zusätzlich innerhalb von 72 Stunden nach dem Ende der Infusion bestimmt werden. Wenn die Transaminasewerte über den Ausgangswert hinaus erhöht sind und mehr als das 2-fache der ULN betragen, kann die Xenpozyme-Dosis gemäss klinischer Beurteilung angepasst (erneute Gabe der vorherigen Dosis oder reduzierte Dosis) oder die Behandlung vorübergehend ausgesetzt werden.
Nach Erreichen der empfohlenen Erhaltungsdosis kann die Untersuchung der Transaminasewerte im Rahmen der routinemässigen klinischen Behandlung der ASMD erfolgen.
Natrium
Dieses Arzneimittel enthält 0,60 mg Natrium pro 4-mg-Durchstechflasche oder 3,02 mg Natrium pro 20-mg-Durchstechflasche, entsprechend 0,03 % beziehungsweise 0,15 % der von der WHO für einen Erwachsenen oder Jugendlichen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g und ≤0,08 % beziehungsweise ≤0,38 % der für ein Kind unter 16 Jahren maximal vertretbaren täglichen Natriumaufnahme mit der Nahrung.
InteraktionenEs wurden keine Studien zur Erfassung von Arzneimittelinteraktionen durchgeführt. Da es sich bei Olipudase alfa um ein rekombinantes humanes Protein handelt, sind keine Cytochrom-P450-vermittelten Arzneimittelinteraktionen zu erwarten.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Bei Frauen im gebärfähigen Alter kann vor Beginn der Behandlung zum Ausschluss einer Schwangerschaft ein Schwangerschaftstest erwogen werden.
Frauen im gebärfähigen Alter sind anzuweisen, während der Behandlung und für 14 Tage nach der letzten Dosis, falls Xenpozyme abgesetzt wird, eine wirksame Verhütungsmethode anzuwenden.
Schwangerschaft
Bisher liegen nur begrenzte Erfahrungen mit der Anwendung von Olipudase alfa bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe «Präklinische Daten»). Die Anwendung von Xenpozyme während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen, es sei denn, der mögliche Nutzen für die Mutter überwiegt die möglichen Risiken, einschliesslich derer für das ungeborene Kind.
Stillzeit
Es ist nicht bekannt, ob Olipudase alfa in die Muttermilch übergeht. Olipudase alfa wurde in der Milch von laktierenden Tieren nachgewiesen (siehe «Präklinische Daten»). Ein Risiko für Neugeborene/Kinder kann nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen oder die Behandlung mit Xenpozyme zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zur berücksichtigen.
Fertilität
Es liegen keine Daten zu den Wirkungen von Olipudase alfa auf die männliche und weibliche Fertilität beim Menschen vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte schädliche Auswirkungen in Bezug auf die Fertilität (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDa in klinischen Studien von Hypotonie berichtet wurde, kann Xenpozyme einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen haben (siehe «Unerwünschte Wirkungen»).
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Schwerwiegende unerwünschte Wirkungen wurden bei mit Xenpozyme behandelten Patienten berichtet und kamen in Form von Extrasystolen im Kontext einer anamnestisch bekannten Kardiomyopathie bei einem erwachsenen Patienten (2,5 %) und in Form von anaphylaktischer Reaktion, Urtikaria, Ausschlag, Überempfindlichkeit und erhöhtem Alaninaminotransferasewert bei jeweils einem pädiatrischen Patienten (5 %) vor. Die Inzidenz schwerwiegender IAR aufgrund von Überempfindlichkeit war bei pädiatrischen Patienten höher als bei Erwachsenen. Bei einem erwachsenen Patienten kam es zu einem dauerhaften Abbruch der Behandlung in Folge wiederkehrenden unerwünschten Wirkungen in Form von Hautausschlag.
Die am häufigsten berichteten unerwünschten Arzneimittelwirkungen (UAW bei ≥10 % der Patienten, die Xenpozyme erhielten) waren Kopfschmerzen (31,7 %), Urtikaria (26,7 %), Fieber (25 %), Übelkeit (20 %), Abdominalschmerzen (16,7 %), Erbrechen (16,7 %), Pruritus (13,3 %), Myalgie (13,3 %), Ausschlag (11,7 %), Erhöhung des C-reaktiven Proteins (11,7 %), Schmerzen im Oberbauch (10 %), und Erythem (10 %).
Die aus 4 klinischen Studien (eine Verträglichkeitsstudie mit erwachsenen Patienten, ASCEND, ASCEND-Peds und eine Verlängerungsstudie mit erwachsenen sowie pädiatrischen Patienten) gepoolte Sicherheitsanalyse umfasste insgesamt 60 Patienten (40 erwachsene sowie 20 pädiatrische Patienten), die Xenpozyme in einer Dosis von bis zu 3 mg/kg alle 2 Wochen erhielten.
Liste der unerwünschten Wirkungen
Unerwünschte Wirkungen, die in der gepoolten Sicherheitsanalyse der abgeschlossenen klinischen Studien berichtet wurden, sind nach Systemorganklasse angegeben, wobei folgende Häufigkeitskategorien verwendet wurden: sehr häufig (≥1/10), häufig (≥1/100 bis < 1/10).
Tabelle 6: Unerwünschte Wirkungen, die in der gepoolten Sicherheitsanalyse der abgeschlossenen klinischen Studien berichtet wurden
|
Systemorganklasse
|
Häufigkeit
| |
Sehr häufig
|
Häufig
| |
Erkrankungen des Immunsystems
|
|
Anaphylaxie und Überempfindlichkeit
| |
Erkrankungen des Nervensystems
|
Kopfschmerzen (31,7 %)
|
| |
Augenerkrankungen
|
|
Okuläre Hyperämie, Augenbeschwerden, Augenjucken
| |
Herzerkrankungen
|
|
Palpitationen, Tachykardie
| |
Gefässerkrankungen
|
|
Hypotonie, Hitzewallung, Flush
| |
Erkrankungen der Atemwege, des Brustraums und des Mediastinums
|
|
Pharynxödem, Pharyngeale Schwellung, Engegefühl des Halses, Giemen, Kehlkopfirritation, Dyspnoe, Rachenreizung
| |
Erkrankungen des Gastrointestinaltrakts
|
Übelkeit (20 %), Abdominalschmerz (15 %), Erbrechen (16,7 %), Schmerzen im Oberbauch (10 %)
|
Diarrhoe, abdominale Beschwerden, gastrointestinale Schmerzen
| |
Leber- und Gallenerkrankungen
|
|
Leberschmerzen
| |
Erkrankungen der Haut und des Unterhautgewebes
|
Urtikaria (21,7 %), Pruritus (10 %), Ausschlag (11,7 %), Erythem (10 %)
|
Angioödem, Fixes Exanthem, Papulöser Ausschlag, Makulöser Ausschlag, Makulopapulöser Ausschlag, Erythematöser Hautausschlag, Ausschlag mit Juckreiz, Morbilliformer Ausschlag, Papel, Makula
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
|
Myalgie (11,7 %)
|
Knochenschmerzen, Arthralgie, Rückenschmerzen
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Fieber (25 %)
|
Schmerzen, Schüttelfrost, Schmerzen an der Katheterstelle, Katheterstellenbedingte Reaktion, Pruritus an der Katheterstelle, Schwellung an der Katheterstelle, Ermüdung, Asthenie
| |
Untersuchungen
|
C-reaktives Protein erhöht (11,7 %)
|
Alaninaminotransferase erhöht, Aspartataminotransferase erhöht, Ferritin im Serum erhöht, C-reaktives Protein anomal, Körpertemperatur erhöht
|
Beschreibung ausgewählter unerwünschter Wirkungen
Infusionsbedingte Reaktionen (IAR) einschliesslich Überempfindlichkeit/anaphylaktische Reaktionen
IAR wurden bei 57,5 % der erwachsenen und bei 65 % der pädiatrischen Patienten berichtet. Symptome einer IAR, die bei erwachsenen Patienten am häufigsten berichtet wurden, waren Kopfschmerzen (25 %), Übelkeit (17,5 %), Urtikaria (17,5 %), Myalgie (12,5 %), Arthralgie (10 %), Fieber (10 %), Pruritus (10 %), Erbrechen (7,5 %), Abdominalschmerzen (7,5 %), Erythem (7,5 %) und Ermüdung (7,5 %). Symptome einer IAR, die bei pädiatrischen Patienten am häufigsten berichtet wurden, waren Fieber (40 %), Urtikaria (40 %), Erbrechen (30 %), C-reaktives Protein erhöht (20 %), Kopfschmerzen (20 %), Übelkeit (20 %), Erythem (15 %), Ausschlag (15 %), Ferritin im Serum erhöht (15 %), Abdominalschmerz (10 %) und Pruritus (10 %). IAR traten normalerweise während der Dauer der Infusion bis 24 Stunden nach Abschluss der Infusion auf. Die meisten IAR wurden als leicht oder moderat eingestuft.
IAR in Zusammenhang mit einer Überempfindlichkeit, einschliesslich Anaphylaxie, traten in klinischen Studien bei 30 % der Patienten auf, darunter 22,5 % erwachsene und 45 % pädiatrische Patienten. Die am häufigsten berichteten Symptome einer IAR in Zusammenhang mit einer Überempfindlichkeit waren Urtikaria (25 %), Pruritus (10 %), Erythem (10 %) und Ausschlag (8,3 %).
Ein pädiatrischer Patient in den klinischen Studien erlitt eine schwere anaphylaktische Reaktion. Darüber hinaus kam es ausserhalb des klinischen Studienprogramms bei einem 16 Monate alten und mit Xenpozyme behandelten Patienten mit ASMD Typ A zu 2 anaphylaktischen Reaktionen. Bei beiden Patienten wurden IgE-Antikörper gegen Olipudase alfa nachgewiesen.
Bei 2 erwachsenen und 3 pädiatrischen Patienten waren die Symptome der IAR mit Veränderungen der Laborwerte (z.B. C-reaktives Protein, Ferritin) verbunden, was auf eine Akute-Phase-Reaktion hinweist. Alle Ereignisse können wie andere IAR behandelt werden.
Erhöhte Transaminasewerte
In den klinischen Studien wurden bei manchen der mit Xenpozyme behandelten Patienten während der Dosissteigerungsphase vorübergehende Erhöhungen der Transaminasewerte (ALT oder AST) innerhalb von 24 bis 48 Stunden nach einer Infusion berichtet. Diese erhöhten Werte gingen im Allgemeinen bis zur nächsten vorgesehenen Infusion auf die Transaminasewerte vor der vorherigen Infusion zurück.
Insgesamt gingen die mittleren ALT-Werte nach 52-wöchiger Behandlung mit Xenpozyme um 46,9 % und die AST-Werte um 40,2 % im Vergleich zu den Ausgangswerten zurück. Bei erwachsenen Patienten wiesen alle 16 Patienten, die zu Studienbeginn erhöhte ALT-Werte hatten, und 10 von 12 Patienten, die zu Studienbeginn erhöhte AST-Werte hatten, Werte im Normbereich auf.
Immunogenität
Insgesamt bildeten 19 von 40 (47,5 %) erwachsenen Patienten und 15 von 20 (75 %) pädiatrischen Patienten, die mit Xenpozyme behandelt wurden, während der Behandlung Antikörper gegen das Arzneimittel (ADA). Die mediane Zeit zur Serokonversion nach der ersten Infusion von Xenpozyme betrug bei erwachsenen Patienten ungefähr 52 Wochen und bei pädiatrischen Patienten 12 Wochen. Die Mehrzahl der ADA-positiven Patienten (16 von 19 erwachsenen und 10 von 15 pädiatrischen Patienten) zeigten ein geringfügiges ADA-Ansprechen (Spitzentiter ≤400) oder wurden wieder ADA-negativ. Drei erwachsene und 4 pädiatrische ADA-positive Patienten zeigten ein intermediäres ADA-Ansprechen (Spitzentiter im Bereich von 800-6400). Acht der 19 erwachsenen ADA-positiven Patienten und 9 der 15 ADA-positiven pädiatrischen Patienten hatten neutralisierende Antikörper (NAb), welche die Aktivität von Olipudase alfa hemmten. Zwei erwachsene und 3 pädiatrische Patienten bildeten zu mehr als einem Zeitpunkt neutralisierende Antikörper. Keiner der Patienten entwickelte NAb, welches die zelluläre Aufnahme von Olipudase alfa hemmte. Bei einem pädiatrischen Patienten kam es zu einer anaphylaktischen Reaktion und der Bildung von IgE-ADA sowie IgG-ADA mit einem Spitzentiter von 1600.
Es wurden keine Auswirkungen von ADA auf die Pharmakokinetik und Wirksamkeit von Xenpozyme bei erwachsenen und pädiatrischen Patienten beobachtet. Bei Patienten, die behandlungsbedingte ADA bildeten, traten bei einem grösseren Anteil behandlungsbedingte IAR (einschliesslich Überempfindlichkeitsreaktionen) auf als bei Patienten ohne behandlungsbedingte ADA (70,6 % vs. 46,2 %). Die IAR waren kontrollierbar und führten nicht zu einem Abbruch der Behandlung.
Pädiatrische Population
Abgesehen von einer höheren Inzidenz von IAR in Zusammenhang mit einer Überempfindlichkeit bei pädiatrischen im Vergleich zu erwachsenen Patienten war das Sicherheitsprofil von Xenpozyme bei pädiatrischen und erwachsenen Patienten ähnlich.
Langzeitanwendung
Die mediane Expositionsdauer war 4,95 Jahre (Bereich: 0,4 bis 9,6 Jahre) bei erwachsenen und 6,15 Jahre (Bereich: 4,3 bis 8,2 Jahre) bei pädiatrischen Patienten. Insgesamt war das Muster der unerwünschten Ereignisse, die bei erwachsenen und pädiatrischen Patienten bei der Langzeitanwendung beobachtet wurden, ähnlich dem im ersten Jahr der Behandlung.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungAnzeichen und Symptome
Während der Dosissteigerung wurde eine begrenzte Anzahl von Fällen einer Überdosierung von Xenpozyme bei pädiatrischen Patienten berichtet. Bei einigen dieser Patienten traten innerhalb von 24 Stunden nach Behandlungsbeginn schwerwiegende unerwünschte Ereignisse auf, darunter auch Todesfälle. Die wichtigsten klinischen Befunde umfassten Atemstillstand, Hypotonie, deutlich erhöhte Werte bei Leberfunktionstests und gastrointestinale Blutungen.
Behandlung
Es ist kein spezifisches Antidot bei einer Überdosierung von Xenpozyme bekannt. Im Falle einer Überdosierung muss die Infusion sofort abgebrochen und der Patient im Krankenhaus engmaschig auf die Entwicklung von IAR, einschliesslich Akute-Phase-Reaktionen, überwacht werden. Zur Behandlung von unerwünschten Wirkungen siehe «Warnhinweise und Vorsichtsmassnahmen».
Eigenschaften/WirkungenATC-Code
A16AB25
Wirkungsmechanismus
Olipudase alfa (rekombinante humane saure Sphingomyelinase) ist ein rekombinantes Enzym, welches die Anreicherung von Sphingomyelin (SM) in den Organen von Patienten mit einem Mangel an saurer Sphingomyelinase (ASMD) verringert. Es ist nicht zu erwarten, dass Xenpozyme die Blut-Hirn-Schranke passiert oder zentralnervöse Manifestationen der Erkrankung beeinflusst.
Pharmakodynamik
Für die Bewertung der pharmakodynamischen Aktivität von Xenpozyme bei ASMD-Patienten wurden Ceramid und Lyso-Sphingomyelin (eine deacetylierte SM-Form) herangezogen.
Nach wiederholter Verabreichung von Xenpozyme an erwachsene und pädiatrische Patienten stiegen die Ceramid-Plasmaspiegel nach jeder Dosis (nach der Infusion) vorübergehend an, wobei die Plasmaspiegel im Verlauf des Behandlungszeitraums graduell abnahmen. In der DFI12712/ASCEND-Studie betrug die prozentuale Veränderung der LS-Mittelwerte (Least-Squares-Methode) von Studienbeginn bis Woche 52 (Standardfehler, SE) beim Ceramid-Plasmaspiegel vor der Infusion in der Behandlungsgruppe unter Xenpozyme -36,4 % (5,3) und in der Placebogruppe -0,2 % (5,6). Bei pädiatrischen Patienten gingen die LS-Mittelwerte (Least Squares Means) beim Ceramid-Plasmaspiegel vor der Infusion zwischen Studienbeginn und Woche 52 (SE: 5,1) um 57 % zurück.
Lyso-Sphingomyelin ist im Plasma von erwachsenen und pädiatrischen ASMD-Patienten erheblich erhöht. Nach wiederholter Gabe von Xenpozyme gingen die Plasmawerte von Lyso-Sphingomyelin signifikant zurück, was eine Abnahme des Sphingomyelingehalts in den Geweben widerspiegelt. In der DFI12712/ASCEND-Studie betrug die prozentuale Veränderung der LS-Mittelwerte von Studienbeginn bis Woche 52 (SE) beim Lyso-Sphingomyelin-Plasmaspiegel vor der Infusion in der Behandlungsgruppe unter Xenpozyme -77,7 % (3,9) und in der Placebogruppe -5,0 % (4,2). Bei pädiatrischen Patienten gingen die LS-Mittelwerte beim Lyso-Sphingomyelin-Plasmaspiegel vor der Infusion zwischen Studienbeginn und Woche 52 (SE: 1,3) um 87,2 % zurück.
Der histopathologisch beurteilte Sphingomyelingehalt in der Leber nahm bei erwachsenen Patienten zwischen Studienbeginn und Woche 52 in der Behandlungsgruppe unter Xenpozyme um 92,0 % (SE: 8,1) ab (verglichen mit +10,3 % [SE: 7,8] in der Placebogruppe).
Klinische Wirksamkeit
Die Wirksamkeit von Xenpozyme wurde in 3 klinischen Studien (ASCEND bei erwachsenen Patienten, ASCEND-Peds bei pädiatrischen Patienten und eine Verlängerungsstudie bei erwachsenen und pädiatrischen Patienten) an insgesamt 61 Patienten mit ASMD untersucht.
Klinische Studie bei erwachsenen Patienten
Die Studie ASCEND ist eine multizentrische, randomisierte, doppelblinde, placebokontrollierte Studie der Phase II/III mit wiederholter Dosisgabe bei erwachsenen Patienten mit ASMD Typ A/B und B. Insgesamt 36 Patienten wurden randomisiert im Verhältnis 1:1 einer Behandlung mit Xenpozyme oder Placebo zugewiesen. Die Behandlung wurde in beiden Gruppen als intravenöse Infusion einmal alle 2 Wochen verabreicht. Bei Patienten, die Xenpozyme erhielten, wurde die Dosis von 0,1 mg/kg auf eine Zieldosis von 3 mg/kg erhöht. Die Studie wurde in 2 aufeinanderfolgende Phasen eingeteilt: eine randomisierte, placebokontrollierte, doppelblinde Phase zur Primäranalyse (PAP, primary analysis period) bis Woche 52, an die eine verlängerte Behandlungsphase (ETP, extension treatment period) von bis zu 4 Jahren anschloss.
Patienten, die in der PAP in den Placeboarm randomisiert wurden, wechselten in der ETP auf die aktive Behandlung und wurden auf die Zieldosis von 3 mg/kg auftitriert, während die Patienten im ursprünglichen Xenpozyme-Arm die Behandlung fortsetzten.
Patienten, die in die Studie aufgenommen wurden, hatten eine Diffusionskapazität der Lunge für Kohlenmonoxid (DLco) ≤70 % des Sollwerts, ein Milzvolumen von ≥6-fach der Norm gemäss Magnetresonanztomographie (MRT) und Werte von ≥5 auf der Splenomegalie-bezogenen Skala (SRS). Insgesamt waren die demografischen und krankheitsspezifischen Merkmale zu Studienbeginn bei den beiden Behandlungsgruppen ähnlich. Das mediane Patientenalter betrug 30 Jahre (Spanne: 18-66 Jahre). Das mittlere (Standardabweichung, SD) Alter zum Zeitpunkt der ASMD-Diagnose betrug 18 (18,4) Jahre. Zu Studienbeginn waren bei 9 von 36 erwachsenen Patienten (25 %) neurologische Manifestationen zu erkennen, die mit der klinischen Diagnose ASMD Typ A/B übereinstimmten. Die anderen 27 Patienten wiesen eine klinische Diagnose auf, die mit ASMD Typ B übereinstimmte.
Diese Studie beinhaltete zwei voneinander getrennte primäre Wirksamkeitsendpunkte: die prozentuale Veränderung der DLCO (in % des Sollwerts) und des Milzvolumens (MN; multiples of normal) gemäss MRT von Studienbeginn bis Woche 52.
Sekundäre Wirksamkeitsendpunkte waren die prozentuale Veränderung des Lebervolumens (MN) und der Thrombozytenzahl von Studienbeginn bis Woche 52.
In der Xenpozyme-Gruppe wurden in der 52-wöchigen Phase zur Primäranalyse im Vergleich zur Placebogruppe Verbesserungen bei der mittleren prozentualen Veränderung der DLCO (% des Sollwerts) (p = 0,0004) und dem Milzvolumen (p < 0,0001) sowie dem mittleren Lebervolumen (p < 0,0001) und der Thrombozytenzahl (p = 0,0185) beobachtet. In Woche 26 der Behandlung, dem ersten Zeitpunkt der Endpunktbeurteilung nach Dosisgabe, wurde eine signifikante Verbesserung der mittleren prozentualen Veränderung der DLCO (% des Sollwerts), des Milzvolumens, des Lebervolumens und der Thrombozytenzahl festgestellt.
Die Ergebnisse der Phase zur Primäranalyse bis Woche 52 sind in Tabelle 7 aufgeführt.
Tabelle 7: Mittelwerte (SD) für Wirksamkeitsendpunkte zu Studienbeginn und prozentuale Veränderung der LS-Mittelwerte (SE) von Studienbeginn bis Woche 52
|
|
Placebo (N = 18)
|
Xenpozyme (N = 18)
|
Differenz
[95 %-KI]
|
p-Wert*
| |
Primäre Endpunkte
|
| |
Mittlere DLCO bei Studienbeginn (% des Sollwerts)
Veränderung der DLCO von Studienbeginn bis Woche 52 (in % des Sollwerts)
|
48,5 (10,8)
3 (3,4)
|
49,4 (11,0)
22 (3,3)
|
n. z.
19 (4,8)
[9,3, 28,7]
|
n. z.
0,0004
| |
Mittleres Milzvolumen zu Studienbeginn (MN)
Prozentuale Veränderung des Milzvolumens vom Studienbeginn bis Woche 52
|
11,2 (3,8)
0,5 (2,5)
|
11,7 (4,9)
-39,4 (2,4)
|
n. z.
-39,9 (3,5)
[-47,1, -32,8]
|
n. z.
< 0,0001
| |
Sekundäre Endpunkte
|
| |
Mittleres Lebervolumen zu Studienbeginn (MN)
Prozentuale Veränderung des Lebervolumens von Studienbeginn bis Woche 52
|
1,6 (0,5)
-1,5 (2,5)
|
1,4 (0,3)
-28,1 (2,5)
|
n. z.
-26,6 (3,6)
[-33,9, -19,3]
|
n. z.
< 0,0001
| |
Mittlere Thrombozytenzahl zu Studienbeginn (109/l)
Prozentuale Veränderung der Thrombozytenzahl von Studienbeginn bis Woche 52
|
115,6 (36,3)
2,5 (4,2)
|
107,2 (26,9)
16,8 (4,0)
|
n. z.
+14,3 (5,8)
[2,6, 26,1]
|
n. z.
0,0185
|
* Statistisch signifikant nach Korrektur für multiples Testen
Siebzehn von 18 Patienten, die zuvor Placebo erhielten, und 18 von 18 Patienten, die zuvor in der PAP 52 Wochen lang mit Xenpozyme behandelt wurden, begannen eine bis zu 4-jährige Behandlung mit Xenpozyme bzw. setzten diese bis zu 4 Jahre lang fort. Die anhaltenden Auswirkungen von Xenpozyme auf die Wirksamkeitsendpunkte bis Woche 104 sind in den Abbildungen 1 und 2 sowie Tabelle 8 aufgeführt.
Abbildung 1: Darstellung der LS-Mittelwerte (95 %-KI) der prozentualen Veränderung der DLCO (% des Sollwerts) von Studienbeginn bis Woche 104 – mITT (modified intent-to-treat) Population
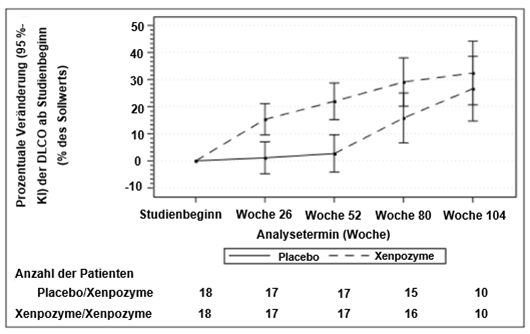
Die senkrechten Balken stellen die 95 %-KI der LS-Mittelwerte dar.
Die LS-Mittelwerte und 95 %-KI basieren auf einem gemischten Modell mit wiederholten Messungen (MMRM, mixed model repeated measures) für die Daten bis zu Woche 104.
Die Patienten der Gruppe Placebo/Xenpozyme erhielten bis Woche 52 Placebo und wechselten danach zu Xenpozyme.
Abbildung 2: Darstellung der LS-Mittelwerte (95 %-KI) der prozentualen Veränderung des Milzvolumens (MN) von Studienbeginn bis Woche 104 – mITT (modified intent-to-treat) Population
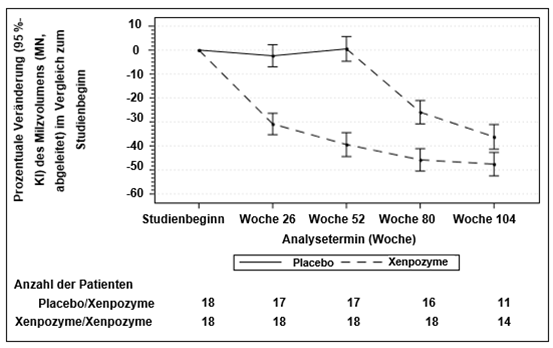
Die senkrechten Balken stellen die 95 %-KI der LS-Mittelwerte dar.
Die LS-Mittelwerte und 95 %-KI basieren auf einem gemischten Modell mit wiederholten Messungen (MMRM) für die Daten bis zu Woche 104.
Die Patienten der Gruppe Placebo/Xenpozyme erhielten bis Woche 52 Placebo und wechselten danach zu Xenpozyme.
Tabelle 8: Prozentuale Veränderung (SE) der LS-Mittelwerte von Studienbeginn bis Woche 104 des Lebervolumens (MN) und der Thrombozytenzahl (109/l) bei mit Xenpozyme über 104 Wochen behandelten Patienten
|
|
Vorherige Olipudase-alfa-Gruppe
| |
|
Woche 52 (Beginn ETP)
|
Woche 104
| |
N
Prozentuale Veränderung des Lebervolumens (SD)
|
17
-27,8 (2,5)
|
14
-33,4 (2,2)
| |
N
Prozentuale Veränderung der Thrombozytenzahl (SD)
|
18
16,6 (4,0)
|
13
24,9 (6,9)
|
N: Anzahl der Patienten
Verlängerungsstudie bei erwachsenen Patienten
Fünf erwachsene Patienten, die an einer unverblindeten Dosissteigerungsstudie mit ASMD-Patienten teilnahmen, setzten die Behandlung in einer unverblindeten Verlängerungsstudie fort und erhielten Xenpozyme für eine Dauer von bis zu > 9 Jahren.
Im Studienverlauf wurden bei Erwachsenen anhaltende Verbesserungen der DLCO (% des Sollwerts), des Milz- und Lebervolumens sowie der Thrombozytenzahl im Vergleich zum Ausgangswert festgestellt (siehe Tabelle 9).
Tabelle 9: Mittlere prozentuale Veränderung (SD) der Wirksamkeitsparameter von Studienbeginn bis Monat 78
|
|
Monat 78
(N = 5)
| |
Prozentuale Veränderung der DLCO (% des Sollwerts) (SD)
|
55,3 % (48,1)
| |
Prozentuale Veränderung des Milzvolumens (SD)
|
-59,5 % (4,7)
| |
Prozentuale Veränderung des Lebervolumens (SD)
|
-43,7 % (16,7)
| |
Prozentuale Veränderung der Thrombozytenzahl (SD)
|
38,5 % (14,7)
|
N: Anzahl der Patienten
Pädiatrische Population
Die Studie ASCEND-Peds (klinische Studie der Phase I/II) ist eine multizentrische, unverblindete Studie mit wiederholter Verabreichung zur Untersuchung der Sicherheit und Verträglichkeit der Gabe von Xenpozyme über 64 Wochen bei pädiatrischen Patienten < 18 Jahren mit ASMD (Typ A/B und B). Darüber hinaus wurden in Woche 52 exploratorische Wirksamkeitsendpunkte in Bezug auf Organomegalie, Lungen- und Leberfunktion sowie Längenwachstum beurteilt.
Bei insgesamt 20 Patienten (4 Jugendliche im Alter von 12 bis < 18 Jahren, 9 Kinder im Alter von 6 bis < 12 Jahren und 7 Säuglinge/Kleinkinder im Alter von < 6 Jahren) wurde die Xenpozyme-Dosis im Rahmen eines Schemas zur Dosissteigerung von 0,03 mg/kg auf eine Zieldosis von 3 mg/kg erhöht. Die Behandlung wurde bis zu 64 Wochen lang als intravenöse Infusion einmal alle 2 Wochen verabreicht. Die in die Studie aufgenommenen Patienten hatten ein Milzvolumen von ≥5-fach der Norm gemäss MRT. Die Patienten waren über alle Altersgruppen von 1,5 bis 17,5 Jahren verteilt und beide Geschlechter waren gleichermassen vertreten. Das mittlere (SD) Alter zum Zeitpunkt der ASMD-Diagnose betrug 2,5 (2,5) Jahre. Zu Studienbeginn waren bei 8 von 20 pädiatrischen Patienten (40 %) neurologische Manifestationen zu erkennen, die mit der klinischen Diagnose ASMD Typ A/B übereinstimmten. Die anderen 12 Patienten wiesen eine klinische Diagnose auf, die mit ASMD Typ B übereinstimmte.
Die Behandlung mit Xenpozyme führte in Woche 52 zu Verbesserungen bei der mittleren prozentualen Veränderung des Sollwerts (%) der DLCO, des Milz- und Lebervolumens, der Thrombozytenzahl und der Entwicklung des Längenwachstums (laut Z-Scores für Körpergrösse) im Vergleich zum Studienbeginn (siehe Tabelle 10).
Tabelle 10: Prozentuale Veränderung (SE) der LS-Mittelwerte oder Veränderung (SD) der Wirksamkeitsparameter von Studienbeginn bis Woche 52 (alle Alterskohorten)
|
|
Ausgangswert
(N = 20)
|
Woche 52
(N = 20)
| |
Mittlere DLCO in % des Sollwerts (SD)
Prozentuale Veränderung des DLCO*-Sollwerts
95 %-KI
|
54,8 (14,2)
|
71,7 (14,8)
32,9 (8,3)
13,4; 52,5
| |
Mittleres Milzvolumen (MN) (SD)
Veränderung des Milzvolumens (MN)
95 %-KI
|
19,0 (8,8)
|
9,3 (3,9)
-49,2 (2,0)
-53,4, -45,0
| |
Mittleres Lebervolumen (MN) (SD)
Prozentuale Veränderung des Lebervolumens (MN)
95 %-KI
|
2,7 (0,7)
|
1,5 (0,3)
-40,6 (1,7)
-44,1, -37,1
| |
Mittlere Thrombozytenzahl (109/l) (SD)
Prozentuale Veränderung der Thrombozytenzahl
95 %-KI
|
137,7 (62,3)
|
173,6 (60,5)
34,0 (7,6)
17,9, 50,1
| |
Mittlere Körpergrösse als Z-Scores (SD)
Veränderung der Z-Scores der Körpergrösse*
95 %-KI
|
-2,1 (0,8)
|
-1,6 (0,8)
0,6 (0,4)
(0,38, 0,73)
|
* Die DLCO wurde bei 9 pädiatrischen Patienten im Alter von ≥5 Jahren untersucht, bei denen der Test durchgeführt werden konnte. Die Veränderung der Körpergrösse (Z-Score) wurde bei 19 pädiatrischen Patienten untersucht.
Die Wirkungen von Xenpozyme auf Milz- und Lebervolumina sowie Thrombozytenzahl und die Körpergrösse (Z-Scores) wurden bei allen in die Studie eingeschlossenen pädiatrischen Alterskohorten beobachtet.
Verlängerungsstudie bei pädiatrischen Patienten
Zwanzig pädiatrische Patienten, die an der ASCEND-Peds-Studie teilnahmen, setzten die Behandlung in einer unverblindeten Verlängerungsstudie fort und erhielten Xenpozyme bis zu > 8 Jahre.
Im Studienverlauf wurden bei pädiatrischen Patienten anhaltende Verbesserungen der Wirksamkeitsparameter (DLCO (% des Sollwerts), Milz- und Lebervolumen, Thrombozytenzahlen, Körpergrösse (Z-Scores) und Knochenalter) bis Monat 48 beobachtet (siehe Tabelle 11).
Tabelle 11: Prozentuale Veränderung der LS-Mittelwerte oder Veränderung (SD) der Wirksamkeitsparameter von Studienbeginn bis Monat 48
|
|
Monat 48
| |
N
Prozentuale Veränderung der DLCO (% des Sollwerts) (SD)
|
5
60,3 (58,5)
| |
N
Prozentuale Veränderung des Milzvolumens (SD)
|
7
-69,1 (4,1)
| |
N
Prozentuale Veränderung des Lebervolumens (SD)
|
7
-55,4 (11,0)
| |
N
Prozentuale Veränderung der Thrombozytenzahl (SD)
|
5
35,8 (42,4)
| |
N
Veränderung der Körpergrösse (Z-Scores) (SD)
|
5
2,3 (0,8)
| |
N
Veränderung des Knochenalters (Monate) (SD)
|
7
18,5 (19,0)
|
N: Anzahl der Patienten
PharmakokinetikDie Pharmakokinetik von Olipudase alfa wurde bei 49 erwachsenen Patienten mit ASMD aus allen klinischen Studien untersucht, die eine oder mehrere Dosen erhalten hatten. Bei der Dosis von 3 mg/kg einmal alle 2 Wochen betrugen die mittlere (prozentualer Variationskoeffizient, %VK) Spitzenkonzentration (Cmax) und die Fläche unter der Konzentrations-Zeit-Kurve über ein Dosierungsintervall (AUC0-τ) im Steady State 30,2 µg/ml (17 %) bzw. 607 µg·h/ml (20 %).
Absorption
Es erfolgt keine Absorption, da Xenpozyme intravenös angewendet wird.
Distribution
Das geschätzte mittlere (%VK) Verteilungsvolumen von Olipudase alfa beträgt 13,1 l (18 %).
Metabolismus
Olipudase alfa ist ein rekombinantes humanes Enzym und wird vermutlich proteolytisch zu kleinen Peptiden und Aminosäuren abgebaut.
Elimination
Die mittlere (%VK) Clearance von Olipudase alfa beträgt 0,331 l/h (22 %). Die mittlere terminale Halbwertszeit (t½) reichte von 31,9 bis 37,6 Stunden.
Linearität/Nicht Linearität
Olipudase alfa zeigte über den Dosisbereich von 0,03 bis 3 mg/kg eine lineare Pharmakokinetik. Nach einem Schema der Dosissteigerung von 0,1 mg/kg bis zur Erhaltungsdosis von 3 mg/kg einmal alle 2 Wochen war die Akkumulation von Olipudase alfa im Plasma minimal.
Kinetik spezieller Patientengruppen
Es gab keine klinisch bedeutsamen geschlechtsspezifischen Unterschiede bei der Pharmakokinetik von Olipudase alfa.
Leberfunktionsstörungen
Olipudase alfa ist ein rekombinantes Protein und wird vermutlich proteolytisch abgebaut. Daher ist nicht zu erwarten, dass eine Leberfunktionsstörung die Pharmakokinetik von Olipudase alfa beeinflusst.
Nierenfunktionsstörungen
Vier Patienten (11,1 %) mit leichter Nierenfunktionsstörung (60 ml/min ≤ Kreatinin-Clearance < 90 ml/min) wurden in die ASCEND-Studie aufgenommen. Die Pharmakokinetik von Olipudase alfa zeigte bei Patienten mit leichter Nierenfunktionsstörung keine klinisch bedeutsamen Unterschiede. Die Auswirkungen einer mittelschweren bis schweren Nierenfunktionsstörung auf die Pharmakokinetik von Olipudase alfa sind nicht bekannt. Es ist nicht zu erwarten, dass Olipudase alfa über die Nieren ausgeschieden wird. Daher ist nicht zu erwarten, dass eine Nierenfunktionsstörung die Pharmakokinetik von Olipudase alfa beeinflusst.
Ältere Patienten
Populationspharmakokinetische Analysen wiesen auf keinen Unterschied bei der Exposition von älteren Patienten hin (in den klinischen Studien zu Xenpozyme waren nur 2 Patienten im Alter zwischen 65 und 75 Jahren eingeschlossen).
Kinder und Jugendliche
Die Pharmakokinetik von Olipudase alfa wurde bei 20 pädiatrischen Patienten, darunter 4 Jugendliche, 9 Kinder und 7 Kleinkinder/Säuglinge, beurteilt (Tabelle 12). Die Exposition gegenüber Olipudase alfa war bei pädiatrischen Patienten geringer als bei erwachsenen Patienten. Diese Unterschiede wurden jedoch nicht als klinisch bedeutsam erachtet.
Tabelle 12: Mittelwerte (%VK) pharmakokinetischer Parameter von Olipudase alfa nach Gabe von 3 mg/kg alle 2 Wochen bei Jugendlichen, Kindern und Kleinkindern/Säuglingen mit ASMD
|
Altersgruppe
|
Alter (Jahre)
|
Cmax (µg/ml)
|
AUC0-τ (µg.h/ml)
| |
Jugendliche (N = 4)
|
12, < 18
|
27,5 (8)
|
529 (7)
| |
Kinder (n = 9)
|
6, < 12
|
24,0 (10)
|
450 (15)
| |
Kleinkinder/Säuglinge (N = 7)
|
< 6
|
22,8 (8)
|
403 (11)
| |
Beschreibende Statistiken stellen die Post-hoc-Schätzungen zur Exposition im Steady State anhand populationspharmakokinetischer Analyse dar.
AUC0-τ: Fläche unter der Plasmakonzentrations-Zeit-Kurve über ein Dosierungsintervall; Cmax: maximale Plasmakonzentration; N: Gesamtzahl der Patienten.
|
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei einmaliger Gabe und Toxizität bei wiederholter Gabe, die an Wildtyp-Tieren (Mäusen, Ratten, Kaninchen, Hunden und Affen) durchgeführt wurden, lassen die präklinischen Daten bei Dosen, welche die für den Menschen empfohlene maximale Dosis (MRHD) um das 10-fache überschreiten, die dem bis zu 4-fachen der Exposition beim Menschen (basierend auf der AUC) unter der empfohlenen therapeutischen Erhaltungsdosis und Häufigkeit entsprechen, keine besonderen Gefahren für den Menschen erkennen.
Toxizität bei wiederholter Gabe
Bei Knockout-Mäusen für die saure Sphingomyelinase (ASMKO-Mäuse, ein Tiermodell für die ASMD) trat nach Verabreichung von Einzeldosen von Olipudase alfa in Form von intravenöser Bolusinjektion, die ≥3,3-mal höher waren als die MRHD, Mortalität auf. Allerdings zeigen Studien zur Toxizität bei wiederholter Gabe, dass die Verabreichung von Olipudase alfa über ein Dosissteigerungsschema zu keiner substanzbezogenen Mortalität führte und den Schweregrad anderer Toxizitätsbefunde reduzierte, und zwar bis zur höchsten getesteten Dosis (30 mg/kg), die 10-mal höher war als die MRHD.
Genotoxizität
Studien zur Beurteilung der Mutagenität von Olipudase alfa wurden nicht durchgeführt.
Kanzerogenität
Studien zur Beurteilung des kanzerogenen Potenzials von Olipudase alfa wurden nicht durchgeführt.
Reproduktionstoxizität
Bei trächtigen Mäusen, die täglich mit Olipudase alfa in einer Dosierung behandelt wurden, die der Exposition beim Menschen unter der empfohlenen therapeutischen Erhaltungsdosis und Häufigkeit entsprach, wurde eine erhöhte Inzidenz der Exenzephalie beobachtet. Diese Inzidenz war geringfügig höher als die aus früheren Kontrolldaten. Die Relevanz dieser Beobachtung für den Menschen ist nicht bekannt. Die tägliche intravenöse Anwendung von Olipudase alfa bei trächtigen Kaninchen führte bei Dosierungen, welche dem 10,5-fachen der Exposition (basierend auf der AUC) beim Menschen in der empfohlenen therapeutischen Erhaltungsdosis und Häufigkeit entsprachen, nicht zu fetalen Fehlbildungen oder Abweichungen.
Nach Verabreichung von 3 mg/kg Olipudase alfa an Mäuse am 7. postpartalen Tag wurde Olipudase alfa 2 Tage später in der Milch nachgewiesen.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Rekonstituiertes Arzneimittel
Die chemische und physikalische Haltbarkeit von Xenpozyme nach Rekonstitution mit sterilem Wasser für Injektionszwecke wurde für bis zu 48 Stunden bei Lagerung bei 2-8 °C oder bei Raumtemperatur (bis zu 25 °C) nachgewiesen. Aus mikrobiologischer Sicht sollte das rekonstituierte Arzneimittel sofort verwendet werden. Falls die Verdünnung nicht sofort erfolgt, ist der Anwender für die Lagerungsdauer und -bedingungen nach Anbruch verantwortlich, welche normalerweise 24 Stunden bei 2-8 °C oder 6 Stunden bei Raumtemperatur (bis zu 25 °C) nicht überschreiten sollten.
Verdünntes Arzneimittel
Die chemische und physikalische Haltbarkeit nach Verdünnung mit 9 mg/ml (0,9 %) Natriumchlorid-Injektionslösung auf 0,1 mg/ml bis 3,5 mg/ml wurde für 48 Stunden bei Lagerung bei 2-8 °C oder bei Raumtemperatur (bis zu 25 °C) nachgewiesen.
Aus mikrobiologischer Sicht sollte das verdünnte Arzneimittel sofort verwendet werden. Falls es nach der Verdünnung nicht sofort verwendet wird, ist der Anwender für die Lagerungsdauer und -bedingungen nach Anbruch verantwortlich, welche normalerweise 24 Stunden bei 2-8 °C, gefolgt von 12 Stunden (einschliesslich Infusionsdauer) bei Raumtemperatur (bis zu 25 °C) nicht überschreiten sollten.
Besondere Lagerungshinweise
Im Kühlschrank (2-8 °C) lagern.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Xenpozyme ist ein Pulver für ein Konzentrat zur Herstellung einer Infusionslösung in einer Durchstechflasche. Die Durchstechflaschen sind nur für den einmaligen Gebrauch bestimmt.
Die Infusionen sind schrittweise und vorzugsweise mit einer Infusionspumpe durchzuführen.
Gebrauchsanweisungen finden Sie am Ende der Fachinformation.
Das Pulver für ein Konzentrat zur Herstellung einer Infusionslösung muss mit sterilem Wasser für Injektionszwecke rekonstituiert, mit 9 mg/ml (0,9 %) Natriumchlorid-Injektionslösung verdünnt und dann mittels intravenöser Infusion verabreicht werden.
Rekonstitution und Verdünnung müssen unter aseptischen Bedingungen erfolgen. Bei der Zubereitung der Infusionslösung dürfen zu keiner Zeit Filtervorrichtungen verwendet werden. Bei der Rekonstitution und Verdünnung ist Schaumbildung zu vermeiden.
Die verdünnte Lösung muss bei der Verabreichung durch einen 0,2- μm-Inlinefilter mit geringer Proteinbindung gefiltert werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den lokalen Anforderungen zu beseitigen.
Zulassungsnummer67287 (Swissmedic)
PackungenXenpozyme 4 mg, Pulver für ein Konzentrat zur Herstellung einer Infusionslösung
·Packung mit 1 Einweg-Durchstechflasche zu 4 mg [A]
·Packung mit 10 Einweg-Durchstechflaschen zu 4 mg [A]
Xenpozyme 20 mg, Pulver für ein Konzentrat zur Herstellung einer Infusionslösung
·Packung mit 1 Einweg-Durchstechflasche zu 20 mg [A]
·Packung mit 10 Einweg-Durchstechflaschen zu 20 mg [A]
Zulassungsinhaberinsanofi-aventis (schweiz) ag, 1214 Vernier/GE
Stand der InformationJuni 2025
Die folgenden Informationen sind nur für medizinisches Fachpersonal bestimmt:
Zubereitung der Infusionslösung gemäss Dosierung
Das Pulver für ein Konzentrat zur Herstellung einer Infusionslösung muss mit sterilem Wasser für Injektionszwecke rekonstituiert, mit 9 mg/ml (0,9 %) Natriumchlorid-Injektionslösung verdünnt und dann mittels intravenöser Infusion verabreicht werden.
Rekonstitution und Verdünnung müssen unter aseptischen Bedingungen erfolgen. Bei der Zubereitung der Infusionslösung dürfen zu keiner Zeit Filtervorrichtungen verwendet werden. Bei der Rekonstitution und Verdünnung ist Schaumbildung zu vermeiden.
Die verdünnte Lösung muss bei der Verabreichung durch einen 0,2- μm-Inlinefilter mit geringer Proteinbindung gefiltert werden.
1) Anzahl der zu rekonstituierenden Durchstechflaschen auf Grundlage des individuellen Patientengewichts und der verschriebenen Dosis bestimmen.
Patientengewicht (kg) × Dosis (mg/kg) = Patientendosis (in mg). Beispiel bei Verwendung von Durchstechflaschen mit 20 mg: Patientendosis (in mg) geteilt durch 20 mg/Durchstechflasche = Anzahl der zu rekonstituierenden Durchstechflaschen. Wenn die Anzahl an Durchstechflaschen einen Bruchteil enthält, zur nächsten ganzen Zahl aufrunden.
2) Erforderliche Anzahl an Durchstechflaschen aus dem Kühlschrank nehmen und ungefähr 20 bis 30 Minuten beiseitestellen, damit sie Raumtemperatur annehmen können.
3) Jede Durchstechflasche durch die Injektion der folgenden Mengen rekonstituieren:
- 1,1 ml steriles Wasser für Injektionszwecke in die Durchstechflasche mit 4 mg
- 5,1 ml steriles Wasser für Injektionszwecke in die Durchstechflasche mit 20 mg
Mit langsamer, tropfenweiser Zuführung an der Innenseite in die Durchstechflasche geben.
4) Jede Durchstechflasche schräg halten und vorsichtig hin- und herrollen. Schaumbildung vermeiden. Jede Durchstechflasche ergibt eine klare, farblose Lösung mit einer Konzentration von 4 mg/ml.
5) Rekonstituierte Lösung in den Durchstechflaschen visuell auf Partikel und Verfärbung prüfen. Die Xenpozyme-Lösung muss klar und farblos sein. Durchstechflaschen mit lichtundurchlässigen Partikeln oder Verfärbungen dürfen nicht verwendet werden.
6) Das der verschriebenen Dosis entsprechende Volumen der rekonstituierten Lösung aus der verordneten Anzahl an Durchstechflaschen entnehmen und mit 9 mg/ml (0,9 %) Natriumchlorid- Injektionslösung je nach Infusionsvolumen in einer Spritze oder einem Infusionsbeutel verdünnen (siehe Tabelle 1 für empfohlenes Gesamtvolumen der Infusion abhängig vom Alter und/oder Gewicht des Patienten).
Tabelle 1: Empfohlene Volumina für die Infusion
|
|
Körpergewicht ≥3 kg bis < 10 kg
|
Körpergewicht ≥10 kg bis < 20 kg
|
Körpergewicht ≥20 kg (pädiatrische Patienten < 18 Jahre)
|
Erwachsene Patienten (≥18 Jahre)
| |
Dosis (mg/kg)
|
Gesamtvolumen der Infusion (ml)
|
Gesamtvolumen der Infusion (ml)
|
Gesamtvolumen der Infusion (ml)
|
Gesamtvolumen der Infusion (ml)
| |
0,03
|
Variables Volumen abhängig vom Körpergewicht
|
Variables Volumen abhängig vom Körpergewicht
|
5
|
n. z.
| |
0,1
|
Variables Volumen abhängig vom Körpergewicht
|
5
|
10
|
20
| |
0,3
|
5
|
10
|
20
|
100
| |
0,6
|
10
|
20
|
50
|
100
| |
1,0
|
20
|
50
|
100
|
100
| |
2,0
|
50
|
75
|
200
|
100
| |
3,0
|
50
|
100
|
250
|
100
|
·Variable Infusionsendvolumina nach Körpergewicht bei pädiatrischen Patienten (siehe Tabelle 1):
- Infusionslösung mit 0,1 mg/ml zubereiten, indem 0,25 ml (1 mg) der in Schritt 3) rekonstituierten Lösung und 9,75 ml der 9 mg/ml (0,9 %) Natriumchlorid-Injektionslösung in einer leeren 10-ml-Spritze gemischt werden.
- Erforderliches Volumen (ml) für die Patientendosis (mg) berechnen.
Beispiel: 0,3 mg ÷ 0,1 mg/ml = 3 ml
·Übertragen Sie das erforderliche Volumen der 0,1 mg/ml-Infusionslösung in eine leere sterile Spritze mit der nächsten dem Infusionsvolumen angemessenen Grösse.
·Anweisungen zur Verdünnung für 5 ml ≤ Gesamtvolumen ≤20 ml mit einer Spritze:
·Das erforderliche Volumen der rekonstituierten Lösung langsam an der Innenseite der leeren Spritze abgeben.
·Langsam eine ausreichende Menge an 9 mg/ml (0,9 %) Natriumchlorid-Injektionslösung zugeben, um das erforderliche Gesamtinfusionsvolumen zu erhalten (Schaumbildung in der Spritze vermeiden).
·Anweisungen zur Verdünnung für ein Gesamtvolumen ≥50 ml mit einem Infusionsbeutel:
·Leerer Infusionsbeutel:
·Das erforderliche Volumen der rekonstituierten Lösung aus Schritt 3) langsam in den sterilen Infusionsbeutel geeigneter Grösse geben.
·Langsam eine ausreichende Menge an 9 mg/ml (0,9 %) Natriumchlorid-Injektionslösung zugeben, um das erforderliche Gesamtinfusionsvolumen zu erhalten (Schaumbildung im Beutel vermeiden).
·Vorgefüllter Infusionsbeutel:
·Aus dem mit 9 mg/ml (0,9 %) Natriumchlorid-Injektionslösung vorgefüllten Infusionsbeutel ein entsprechendes Volumen der Natriumchlorid-Injektionslösung entnehmen, um ein Endvolumen wie in Tabelle 1 angegeben zu erhalten.
·Langsam das erforderliche Volumen der rekonstituierten Lösung aus Schritt 3) in den Infusionsbeutel geben (Schaumbildung im Beutel vermeiden).
7) Spritze oder Infusionsbeutel zum Mischen vorsichtig umdrehen. Nicht schütteln. Da es sich um eine Proteinlösung handelt, tritt nach dem Verdünnen gelegentlich eine leichte Ausflockung (beschrieben als dünne transparente Fäden) auf.
8) Die verdünnte Lösung muss bei der Verabreichung durch einen 0,2- μm-Inlinefilter mit geringer Proteinbindung gefiltert werden.
9) Nach Abschluss der Infusion sollte die Infusionsleitung mit 9 mg/ml (0,9 %) Natriumchlorid-Injektionslösung mit der gleichen Infusionsgeschwindigkeit wie im letzten Teil der Infusion gespült werden.
|