ZusammensetzungWirkstoffe
Mepolizumab.
Hilfsstoffe
Saccharose, Dinatriumhydrogenphosphat-Heptahydrat, Citronensäure-Monohydrat, Polysorbat 80, EDTA-Dinatrium-Dihydrat, Wasser für Injektionszwecke.
Indikationen/AnwendungsmöglichkeitenSchweres eosinophiles Asthma
Nucala ist angezeigt als Zusatztherapeutikum bei Erwachsenen und Jugendlichen ab 12 Jahren mit schwerem eosinophilem Asthma, gekennzeichnet durch folgende Kriterien:
·mindestens zwei Exazerbationen in den vorausgegangenen 12 Monaten unter aktueller Standardtherapie (hochdosierte inhalative Kortikosteroide plus zusätzliche Erhaltungstherapie) und/oder Notwendigkeit zur Behandlung mit systemischen Kortikosteroiden.
·Eosinophilenzahl im Blut von ≥0.15 G/L* (entspricht ≥150 Zellen/μL) bei Behandlungsbeginn oder von ≥0.3 G/L (entspricht ≥300 Zellen/μL) in den vorausgegangenen 12 Monaten.
* Für genauere Angaben zur Patientenpopulation siehe Rubrik «Eigenschaften/Wirkungen», Klinische Wirksamkeit und Sicherheit
Chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
Nucala ist angezeigt als Zusatztherapeutikum zu intranasalen Kortikosteroiden bei Erwachsenen ab 18 Jahren mit schwerer chronischer Rhinosinusitis mit Nasenpolypen (CRSwNP), die mit intermittierenden systemischen Kortikosteroiden und/oder chirurgischem Eingriff nicht ausreichend kontrolliert werden kann.
Eosinophile Granulomatose mit Polyangiitis (EGPA)
Nucala ist angezeigt als Zusatztherapeutikum bei Erwachsenen ab 18 Jahren mit eosinophiler Granulomatose mit Polyangiitis (EGPA), gekennzeichnet durch folgende Kriterien:
·Rezidivierende oder therapieresistente EGPA
·Vorgängige Stabilisierung der Krankheit mit systemischen Kortikosteroiden
·eine Erhaltungs-Behandlung mit systemischen Kortikosteroiden und allenfalls Steroid-sparenden Immunsuppressiva ist notwendig
Hypereosinophilie-Syndrom (HES)
Nucala ist angezeigt als Zusatztherapeutikum zur Behandlung von Erwachsenen und Jugendlichen ab 12 Jahren mit Hypereosinophilie-Syndrom ohne FIP1L1-PDGFRα Fusion (F/P negatives HES) (siehe Rubrik «Eigenschaften/Wirkungen»).
Nucala sollte nur von Ärzten angewendet werden, welche mit der Behandlung von schwerem Asthma, CRSwNP, EGPA oder HES vertraut sind.
Dosierung/AnwendungDosierung
Schweres eosinophiles Asthma
Erwachsene und Jugendliche ab 12 Jahren
Die empfohlene Dosis beträgt 100 mg Nucala und wird einmal alle 4 Wochen subkutan verabreicht.
Kinder unter 12 Jahren
Die Sicherheit und Wirksamkeit von Nucala bei Kindern unter 12 Jahren wurde in kontrollierten Studien nicht untersucht.
Dauer der Behandlung:
Nach spätestens 8 Gaben Nucala sollte der Therapieerfolg beurteilt und über die Fortführung der Behandlung entschieden werden. Zur Beurteilung des Ansprechens auf die Zusatz-Therapie sind eine sorgfältige Erfassung der Asthmakontrolle, des Bedarfs an systemischen Kortikosteroiden und der Exazerbationshäufigkeit vor und unter der Behandlung notwendig. Im Falle eines Ansprechens ist Nucala für die Langzeitbehandlung vorgesehen. Der Nutzen und die Notwendigkeit einer Fortführung der Therapie sollte mindestens jährlich überprüft werden, basierend auf der ärztlichen Beurteilung des Krankheitsschweregrads und der Exazerbationskontrolle.
Chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
Erwachsene ab 18 Jahren
Die empfohlene Dosis beträgt 100 mg Nucala und wird einmal alle 4 Wochen subkutan verabreicht.
Kinder und Jugendliche unter 18 Jahren
Die Sicherheit und Wirksamkeit von Nucala bei Kindern und Jugendlichen mit CRSwNP unter 18 Jahren wurde nicht untersucht.
Eosinophile Granulomatose mit Polyangiitis (EGPA)
Es wurden keine EGPA-spezifischen klinischen Dosisfindungsstudien durchgeführt. (siehe Rubrik Eigenschaften/Wirkungen, Studie MEA115921).
Die einzelnen Injektionsstellen sollten einen Abstand von wenigstens 5 cm voneinander haben (s. «Sonstige Hinweise», Hinweise für die Handhabung).
Erwachsene ab 18 Jahren
Die empfohlene Dosis beträgt 300 mg Nucala und wird einmal alle 4 Wochen subkutan verabreicht.
Kinder und Jugendliche unter 18 Jahren
Die Sicherheit und Wirksamkeit von Nucala bei Kindern und Jugendlichen mit EGPA unter 18 Jahren wurde nicht untersucht.
Dauer der Behandlung
Im Falle eines Ansprechens ist Nucala für die Langzeitbehandlung vorgesehen. Die Notwendigkeit einer Fortführung der Therapie sollte mindestens einmal jährlich, auf Basis der ärztlichen Beurteilung des Krankheitsschweregrads und der Verbesserung der Symptomkontrolle des Patienten, überprüft werden.
Bei Patienten, die lebensbedrohliche Manifestationen der EGPA entwickeln, sollte ebenfalls überprüft werden, ob eine Fortführung der Therapie erforderlich ist, da Nucala in dieser Patientengruppe nicht untersucht wurde.
Hypereosinophilie-Syndrom (HES)
Die einzelnen Injektionsstellen sollten einen Abstand von wenigstens 5 cm voneinander haben (s. «Sonstige Hinweise», Hinweise für die Handhabung).
Erwachsene und Jugendliche ab 12 Jahren
Die empfohlene Dosis beträgt 300 mg Nucala und wird einmal alle 4 Wochen subkutan verabreicht.
Kinder unter 12 Jahren
Die Sicherheit und Wirksamkeit von Nucala bei Kindern unter 12 Jahren wurde nicht untersucht.
Dauer der Behandlung
Im Falle eines Ansprechens ist Nucala für die Langzeitbehandlung vorgesehen. Die Notwendigkeit einer Fortführung der Therapie sollte mindestens einmal jährlich, auf Basis der ärztlichen Beurteilung des Krankheitsschweregrads und der Verbesserung der Symptomkontrolle des Patienten, überprüft werden.
Bei Patienten, die lebensbedrohliche Manifestationen des HES entwickeln, sollte ebenfalls überprüft werden, ob eine Fortführung der Therapie erforderlich ist, da Nucala in dieser Patientengruppe nicht untersucht wurde.
Besondere Patientengruppen
Ältere Patienten (> 65 Jahre)
Bei älteren Patienten ist keine Anpassung der Dosierung erforderlich (vgl. «Pharmakokinetik»).
Eingeschränkte Nierenfunktion
Bei Patienten mit eingeschränkter Nierenfunktion ist keine Dosisanpassung erforderlich (vgl. «Pharmakokinetik»).
Eingeschränkte Leberfunktion
Bei Patienten mit eingeschränkter Leberfunktion ist keine Dosisanpassung erforderlich (vgl. «Pharmakokinetik»).
Art der Anwendung (Hinweise für die Handhabung s. «Sonstige Hinweise»)
Nucala Fertigpen und Fertigspritze darf nur zur subkutanen Injektion angewendet werden (siehe separate Gebrauchsanweisung).
Nucala kann durch den Patienten selbst oder durch eine Betreuungsperson verabreicht werden, wenn der Arzt dies für angemessen hält, und der Patient bzw. die Betreuungsperson in der entsprechenden Injektionstechnik geschult wurde.
Die Injektionsstellen bei Selbstinjektion sind Bauch oder Oberschenkel. Eine Betreuungsperson kann die Injektion auch in den Oberarm verabreichen.
Die Injektionen dürfen nicht in empfindliche, verletzte, gerötete oder verhärtete Hautbezirke verabreicht werden.
Eine genaue Anleitung zur Anwendung des Fertigpens bzw. der Fertigspritze befindet sich in der der Packung beigelegten Gebrauchsanweisung.
Bei der Anwendung von Nucala können lokale oder systemische Hypersensitivitäts-Reaktionen auftreten. Die Patienten müssen darüber informiert werden, dass solche Reaktionen möglich sind und sofortige medizinische Behandlung erforderlich machen können. Nach jeder Verabreichung sollten Patienten während mindestens 30 Minuten auf Anzeichen und Symptome von Überempfindlichkeitsreaktionen achten.
KontraindikationenÜberempfindlichkeit gegenüber Mepolizumab oder einem der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenNucala darf nicht zur Behandlung akuter Asthma-Exazerbationen eingesetzt werden.
Unter der Behandlung mit Nucala können asthma-bedingte unerwünschte Ereignisse oder Exazerbationen eintreten. Die Patienten sollten angewiesen werden, ärztliche Hilfe in Anspruch zu nehmen, wenn ihr Asthma nach Einleitung der Behandlung mit Nucala weiterhin unkontrolliert ist oder sich verschlechtert.
Das abrupte Absetzen von Kortikosteroiden nach Einleitung der Behandlung mit Nucala wird nicht empfohlen. Erforderlichenfalls sollten unter Aufsicht eines Arztes die Kortikosteroiddosen stufenweise verringert werden.
Überempfindlichkeits- und verabreichungsbedingte Reaktionen
Nach Verabreichung von Nucala traten systemische Reaktionen vom Sofort- und vom Spättyp auf, darunter auch Überempfindlichkeitsreaktionen (z.B. Anaphylaxie, Nesselausschlag, Angioödem, Rash, Bronchospasmus, Hypotonie). Solche Reaktionen treten in der Regel in den ersten Stunden nach der Verabreichung, in manchen Fällen jedoch auch verzögert (d.h. nach mehreren Tagen) auf.
Parasitäre Erkrankungen
Eosinophile können an der Immunantwort auf einen Befall mit bestimmten Helminthen beteiligt sein. Patienten, bei denen ein Helminthenbefall vorliegt, sollten vor der Verabreichung von Mepolizumab entsprechend behandelt werden. Wenn Patienten, bei denen unter der Behandlung mit Mepolizumab ein Helminthenbefall eintritt, nicht auf eine antihelminthische Behandlung ansprechen, ist ein vorübergehendes Aussetzen von Mepolizumab in Erwägung zu ziehen.
Organgefährdende oder lebensbedrohliche EGPA
Nucala wurde bei Patienten mit organgefährdenden oder lebensbedrohlichen Manifestationen von EGPA nicht untersucht (siehe «Dosierung/Anwendung»).
Lebensbedrohliche HES
Nucala wurde bei Patienten mit lebensbedrohlichen Manifestationen von HES nicht untersucht (siehe «Dosierung/Anwendung»).
InteraktionenEs wurden keine Wechselwirkungsstudien mit Mepolizumab und anderen Arzneimitteln durchgeführt.
Klinische Erfahrungen zur Impfantwort bei Impfungen während einer Therapie mit Nucala liegen nicht vor.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine bzw. nur eingeschränkte Daten zur Anwendung von Mepolizumab in der Schwangerschaft vor.
Aus tierexperimentellen Studien gehen keine Hinweise auf eine Reproduktionstoxizität hervor (vgl. «Präklinische Daten»).
Nucala soll während der Schwangerschaft nicht verabreicht werden, es sei denn, dies ist klar notwendig.
Stillzeit
Für den Menschen liegen keine Daten zur Ausscheidung von Mepolizumab in der Muttermilch vor. In der Milch von Cynomolgus-Affen wurde Mepolizumab jedoch in Konzentrationen von weniger als 0,5 % der entsprechenden Plasmakonzentrationen ausgeschieden.
Unter Abwägung der Vorteile des Stillens für das Kind und des Nutzens der Behandlung für die Mutter muss entweder abgestillt oder die Behandlung mit Nucala eingestellt werden.
Fertilität
Daten zur Fertilität beim Menschen liegen nicht vor. In tierexperimentellen Studien wurden keine unerwünschten Wirkungen einer anti-IL-5-Behandlung auf die Fertilität festgestellt (vgl. «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine spezifischen Studien zum Einfluss von Nucala auf die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt.
Unerwünschte WirkungenSchweres eosinophiles Asthma
Zusammenfassung des Sicherheitsprofils
In klinischen Studien an Patienten mit hochgradigem eosinophilem Asthma waren die am häufigsten gemeldeten unerwünschten Wirkungen während der Behandlung Kopfschmerzen, Reaktionen an der Injektionsstelle und Rückenschmerzen. Das Sicherheitsprofil war über alle Behandlungsgruppen hinweg ähnlich, mit Ausnahme der Reaktionen an der Injektionsstelle, die im Vergleich zu Placebo (3 %) in der Gruppe mit Mepolizumab 100 mg s.c. häufiger (8 %) auftraten. Reaktionen an der Injektionsstelle traten in der Regel zu Beginn der Behandlung, bis zur dritten Injektion, auf; für spätere Injektionen liegen weniger Meldungen vor.
Auflistung der unerwünschten Arzneimittelwirkungen
Die Sicherheit von Nucala wurde an insgesamt 1'327 Erwachsenen und Jugendlichen ab 12 Jahren mit schwerem eosinophilem Asthma untersucht, die im Rahmen klinischer Studien von 24- bis 52-wöchiger Dauer entweder eine subkutane (s.c.) oder eine intravenöse (i.v.) Dosis erhielten. In der nachstehenden Tabelle sind die unerwünschten Wirkungen aufgeführt, die in den zwei placebokontrollierten Studien bei Patienten unter subkutanem Mepolizumab 100 mg (n = 263) auftraten.
Die Häufigkeiten der Nebenwirkungen sind konventionsgemäss folgendermassen definiert: sehr häufig (≥1/10); häufig (≥1/100 bis < 1/10); gelegentlich (≥1/1'000 bis < 1/100); selten (≥1/10'000 bis < 1/1'000); sehr selten (< 1/10'000) und nicht bekannt (Häufigkeit kann auf Basis der vorhandenen Daten nicht angegeben werden).
Erkrankungen des Gastrointestinaltrakts
Häufig: Schmerzen im Oberbauch.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Fieber, Reaktionen an der Injektionsstelle*.
Infektionen und parasitäre Erkrankungen
Häufig: Pharyngitis, Infektionen der unteren Atemwege, Harnwegsinfektionen.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Rückenschmerzen.
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (20 %).
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Häufig: Verstopfte Nase.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Ekzem.
* Zu den häufigsten Beschwerden im Zusammenhang mit subkutanen Injektionen gehören Schmerzen, Erythem, Schwellung, Juckreiz und Brennen.
Das Sicherheitsprofil von Nucala bei Patienten mit schwerem Asthma (n=998), die in offenen Verlängerungsstudien über einen Median von 2,8 Jahren (Spanne von 4 Wochen bis 4,5 Jahren) behandelt wurden, zeigte im Vergleich zu den placebo-kontrollierten Studien keine zusätzlichen unerwünschten Wirkungen. Die häufigsten unerwünschten Wirkungen waren Kopfschmerzen (22%), Rückenschmerzen (14%) und Reaktionen an der Injektionsstelle (8%).
CRSwNP
In einer randomisierten, doppelblinden, placebokontrollierten 52-wöchigen Studie an Probanden mit CRSwNP (100 mg Mepolizumab n= 206, Placebo n= 201) wurden im Vergleich zu den Studien bei schwerem Asthma keine weiteren unerwünschten Wirkungen festgestellt.
EGPA
In einer doppelblinden, placebokontrollierten Studie an Patienten mit EGPA (300 mg Mepolizumab n = 68, Placebo n = 68) wurden im Vergleich zu den Studien bei schwerem Asthma keine weiteren unerwünschten Wirkungen festgestellt.
HES
In einer randomisierten, doppelblinden, Placebo-kontrollierten 32-wöchigen Studie an HES-Patienten (300 mg Mepolizumab n = 54, Placebo n = 54) zeigten sich unter Mepolizumab gegenüber Placebo mehr Infekte (37/54 vs 28/54). Die Mehrzahl davon waren Infektionen der oberen Atemwege. Das Sicherheitsprofil von Mepolizumab in HES-Patienten (n=102) in der offenen 20-wöchigen Verlängerungsstudie war ähnlich wie das Sicherheitsprofil in der pivotalen Placebo-kontrollierten Studie. Im Vergleich zu den Studien bei schwerem Asthma wurden keine weiteren unerwünschten Wirkungen festgestellt.
Post-Marketing Daten:
Erkrankungen des Immunsystems
Selten: Hypersensitivitätsreaktionen einschliesslich Anaphylaxie
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Arthralgie
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungZur Überdosierung von Mepolizumab liegen keine klinischen Erfahrungen vor.
Im Rahmen einer klinischen Studie wurden Patienten mit eosinophiler Erkrankung Einzeldosen bis 1'500 mg intravenös verabreicht, ohne dass Hinweise auf dosisabhängige Toxizitäten erkennbar waren.
Behandlung
Für den Fall einer Überdosierung von Mepolizumab ist keine spezifische Behandlung verfügbar. Im Falle einer Überdosierung muss der Patient die jeweils angemessene unterstützende Behandlung erhalten und entsprechend überwacht werden.
Das Vorgehen richtet sich nach den klinischen Erfordernissen bzw., sofern verfügbar, nach den Empfehlungen des jeweiligen toxikologischen Informationszentrums.
Eigenschaften/WirkungenATC-Code
R03DX09
Wirkungsmechanismus
Mepolizumab ist ein humanisierter monoklonaler Antikörper (IgG1, kappa), der sich hochaffin und spezifisch gegen das humane Interleukin-5 (IL-5) richtet. IL-5 ist das für Wachstum und Differenzierung, Rekrutierung, Aktivierung und Überleben der Eosinophilen wichtigste Zytokin. Mepolizumab hemmt im nanomolaren Konzentrationsbereich die biologischen Wirkungen von IL-5, indem es verhindert, dass IL-5 an die Alpha-Kette des auf der Zelloberfläche von Eosinophilen exprimierten IL-5-Rezeptorkomplexes bindet; auf diese Weise wird der IL-5-Signalweg blockiert und damit die Bildung von Eosinophilen und deren Überleben reduziert.
Pharmakodynamik
In klinischen Studien wurde nach einer Behandlung mit Mepolizumab eine Verminderung der Eosinophilenzahl im Blut beobachtet. Grössenordnung und Dauer dieser Verminderung waren nach subkutaner Gabe von 12.5 mg-125 mg dosisabhängig.
Bei Patienten mit schwerem Asthma ging nach subkutaner Verabreichung von 100 mg alle 4 Wochen über einen Zeitraum von 32 Wochen die Eosinophilenzahl im Blut auf einen geometrischen Mittelwert von 40 Zellen/µL zurück. Dies entspricht einer geometrisch gemittelten Reduktion um 84% vs Placebo. Dieser Rückgang wurde grössenordnungsmässig bereits innerhalb der ersten 4 Behandlungswochen beobachtet. Dieses Ausmass der Verringerung der Eosinophilen im Blut blieb bei Patienten mit schwerem Asthma (n=998), die in offenen Verlängerungsstudien über einen Median von 2,8 Jahren (Spanne von 4 Wochen bis 4,5 Jahren) behandelt wurden, erhalten.
Bei Patienten mit CRSwNP ging nach subkutaner Verabreichung von100 mg alle 4 Wochen über einen Zeitraum von 52 Wochen die Eosinophilenzahl im Blut auf einen geometrischen Mittelwert von 60 Zellen/µL zurück. Dies entspricht einer geometrisch gemittelten Reduktion um 83% vs Placebo. Dieser Rückgang wurde innerhalb der ersten 4 Behandlungswochen beobachtet und blieb während der Therapiedauer erhalten.
Bei Patienten mit EGPA ging nach subkutaner Verabreichung von 300 mg alle 4 Wochen über einen Zeitraum von 52 Wochen die Eosinophilenzahl im Blut auf einen geometrischen Mittelwert von 38 Zellen/µL zurück. Dies entspricht einer geometrisch gemittelten Reduktion um 83% vs Placebo.
Bei Patienten mit HES ging nach subkutaner Verabreichung von 300 mg alle 4 Wochen über einen Zeitraum von 32 Wochen die Zahl der Eosinophilen im Blut auf einen geometrischen Mittelwert von 70 Zellen/µl zurück. Das entspricht einer geometrisch gemittelten Reduktion um 92% vs Placebo. Diese Reduktionsgrössenordnung wurde für weitere 20 Wochen beibehalten in Patienten, die mit der Mepolizumab-Behandlung in der offenen Verlängerung weiterfuhren.
Immunogenität
Aufgrund des immunogenen Potenzials von protein- oder peptidbasierten Therapeutika können die Patienten nach der Behandlung Antikörper gegen Mepolizumab entwickeln.
Insgesamt entwickelten 15/260 (6 %) der mit mindestens einer subkutanen Dosis in Höhe von 100 mg behandelten Patienten mit schwerem Asthma Antikörper gegen Mepolizumab. Das Immunogenitätsprofil von Nucala bei Patienten mit schwerem Asthma (n=998), die in offenen Verlängerungsstudien über einen Median von 2,8 Jahren (Spanne von 4 Wochen bis 4,5 Jahren) behandelt wurden, war ähnlich wie in den placebo-kontrollierten Studien.
Insgesamt entwickelten 6/196 (3 %) der mit mindestens einer subkutanen Mepolizumab-Dosis von 100 mg behandelten CRSwNP-Patienten Antikörper gegen Mepolizumab.
Insgesamt entwickelten 1/68 (1 %) der mit mindestens einer subkutanen Mepolizumab-Dosis von 300 mg behandelten EGPA-Patienten Antikörper gegen Mepolizumab.
Insgesamt entwickelten 1/53 (2 %) der mit mindestens einer subkutanen Mepolizumab-Dosis von 300 mg behandelten HES-Patienten Antikörper gegen Mepolizumab.
Über alle Indikationen wurden bei einer erwachsenen Person (mit schwerem Asthma) neutralisierende Antikörper festgestellt.
Das Vorliegen von Antikörpern gegen Mepolizumab hatte bei der Mehrzahl der Patienten keine merklichen Auswirkungen auf die PK oder PD von Mepolizumab; Hinweise auf einen Zusammenhang zwischen den Antikörpertitern und einer Änderung der Eosinophilenzahl lagen nicht vor.
Klinische Wirksamkeit
Schweres eosinophiles Asthma
Die Wirksamkeit von Mepolizumab bei hochgradigem eosinophilem Asthma wurde im Rahmen von 3 randomisierten, doppelblinden klinischen Parallelgruppenstudien von 24- bis 52-wöchiger Dauer an Patienten ab 12 Jahren untersucht. Diese Studien waren auf die Beurteilung der Wirksamkeit von Mepolizumab bei Verabreichung in vierwöchigen Abständen als subkutane oder intravenöse Injektion bei Patienten angelegt, die unter ihrer laufenden Standardbehandlung (z.B. inhalative Kortikosteroide [ICS], Kombination aus ICS und langwirkenden Beta2-Sympathomimetika [LABA], Leukotrienmodifikatoren und kurzwirkenden Beta2-Sympathomimetika [SABA]) nicht kontrolliert waren. In MEA112997 und MEA115588 sind klinisch relevante Asthma-Exazerbationen wie folgt definiert: Verschlechterung der Asthmasymptome, die die Anwendung von oralen/systemischen Kortikosteroiden und/oder eine Hospitalisierung bzw. eine Notfallbehandlung erfordert.
Placebokontrollierte Studien
Dosisfindungsstudie MEA112997 (DREAM-Studie)
Die Ergebnisse der randomisierten, doppelblinden, placebokontrollierten, multizentrischen, 52-wöchigen Parallelgruppenstudie MEA112997 an 616 Patienten zeigten, dass Mepolizumab (75 mg, 250 mg oder 750 mg) bei intravenöser Verabreichung zu einer signifikanten Reduktion der Asthma-Exazerbationen gegenüber Placebo führte. Hinsichtlich der Wirksamkeit konnte zwischen den 3 untersuchten Dosierungen kein signifikanter Unterschied festgestellt werden (siehe Tabelle 1).
Tabelle 1: Häufigkeit klinisch relevanter Exazerbationen nach 52 Wochen bei der Intent-to-treat-Population
|
|
Mepolizumab IV
|
Placebo
| |
75 mg
n = 153
|
250 mg
n = 152
|
750 mg
n = 156
|
n = 155
| |
Exazerbationen/Jahr
|
1,24
|
1,46
|
1,15
|
2,40
| |
Prozent Senkung
|
48 %
|
39 %
|
52 %
|
| |
Verhältnisrate (95%-KI)
|
0,52 (0,39; 0,69)
|
0,61 (0,46; 0,81)
|
0,48 (0,36; 0,64)
|
| |
p-Wert
|
< 0,001
|
< 0,001
|
< 0,001
|
-
|
Die Ergebnisse dieser Studie weisen darauf hin, dass eine Eosinophilenzahl im Blut von ≥150 Zellen/µL in der Voruntersuchungsphase bzw. von ≥300 Zellen/µL in den vorausgegangenen 12 Monaten als Biomarker herangezogen werden kann, um vorherzusagen, welche Patienten von der Behandlung mit Mepolizumab profitieren können. Allerdings wurden bei einem Drittel der so ausgewählten Patienten auch unter Placebo während der einjährigen Behandlungsphase keine Exazerbationen mehr beobachtet.
Bei Erhöhung des «Cut off» Wertes der Eosinophilenzahl im Blut wurde bei entsprechend selektionierten Patienten eine verstärkte Abnahme der Exazerbationshäufigkeit beobachtet. Es ist aber nicht untersucht, in wie weit bei höheren «Cut off» Werten Patienten ausgeschlossen wurden, welche von der Mepolizumab-Zusatz-Behandlung profitierten.
Tabelle 2: Berechnete Reduktion der Rate klinisch signifikanter Exazerbationen bei Baseline-Grenzwerten der Eosinophilenzahl im Blut
|
Studie
|
Baseline Eosinophilenzahl im Blut
|
Verhältnisrate (95%-KI)
| |
MEA112997
|
150 Zellen/µL
|
0,70 (0,53; 0,93)
| |
|
300 Zellen/µL
|
0,52 (0,41; 0,65)
| |
|
500 Zellen/µL
|
0,42 (0,32; 0,54)
| |
MEA115588
|
150 Zellen/µL
|
0,61 (0,45; 0,82)
| |
|
300 Zellen/µL
|
0,49 (0,38; 0,63)
| |
|
500 Zellen/µL
|
0,42 (0,31; 0,55)
|
Auf Grundlage der Ergebnisse dieser Studie wurden die in späteren Studien zur subkutanen Verabreichung von Mepolizumab zu untersuchenden Dosen ausgewählt. Nucala ist ein Präparat, das nicht intravenös angewendet werden darf, sondern ausschliesslich subkutan verabreicht werden muss.
Reduktion von Exazerbationen (MEA115588), MENSA-Studie
MEA115588 (MEpolizumab as adjunctive therapy iN patients with Severe Asthma) war eine randomisierte, doppelblinde, placebokontrollierte, multizentrische Parallelgruppenstudie zur Beurteilung der Wirksamkeit und Sicherheit von Mepolizumab als Zusatztherapeutikum an 576 Patienten mit hochgradigem eosinophilem Asthma.
Die Patienten waren überwiegend 18 Jahre oder älter, hatten in den vorausgegangenen 12 Monaten mindestens zwei Asthma-Exazerbationen durchgemacht und waren unter ihrer laufenden Pharmakotherapie gegen Asthma (hochdosierte inhalative Kortikosteroide [ICS] in Kombination mit mindestens einem anderen Kontrollmedikament, z.B. langwirkenden Beta2-Sympathomimetika [LABA] oder Leukotrienmodifikatoren) nicht kontrolliert. Die Patienten durften orale Kortikosteroide verwenden und erhielten während der Studie weiterhin ihre gewohnten Medikamente gegen Asthma.
Schweres eosinophiles Asthma lag definitionsgemäss vor bei einer Eosinophilenzahl im peripheren Blut von ≥150 Zellen/μL innerhalb der 6 Wochen vor der Randomisierung (erste Dosis) oder einer Eosinophilenzahl im Blut von ≥300 Zellen/μL innerhalb des der Randomisierung vorangegangenen Jahres. Die Patienten erhielten über 32 Wochen hinweg jeweils einmal alle vier Wochen entweder Mepolizumab 100 mg subkutan (s.c.), Mepolizumab 75 mg intravenös (i.v.) oder ein Placebo. Die Ergebnisse für den primären Studienendpunkt, die Verminderung der Häufigkeit klinisch relevanter Asthma-Exazerbationen, fielen statistisch signifikant aus (p < 0,001).
Tabelle 3 fasst die Ergebnisse für den primären und die sekundären Endpunkte aus MEA115588 zusammen.
Tabelle 3: Ergebnisse für den primären und die sekundären Endpunkte nach 32 Wochen in der Intent-to-treat-Population (MEA115588)
|
|
Mepolizumab
(100 mg s.c.)
n = 194
|
Placebo
n = 191
| |
Primärer Endpunkt
| |
Häufigkeit klinisch relevanter Exazerbationen
| |
Exazerbationen/Jahr
|
0,83
|
1,74
| |
Prozent Senkung
Verhältnisrate (95%-KI)
|
53 %
0,47 (0,35; 0,64)
|
-
| |
p-Wert
|
< 0,001
|
| |
Sekundäre Endpunkte
| |
Häufigkeit von Exazerbationen mit erforderlicher Hospitalisierung/Notfallbehandlung
| |
Exazerbationen/Jahr
|
0,08
|
0,20
| |
Prozent Senkung
Verhältnisrate (95%-KI)
|
61 %
0,39 (0,18; 0,83)
|
_
| |
p-Wert
|
0,015
|
| |
Häufigkeit hospitalisierungsbedürftiger Exazerbationen
| |
Exazerbationen/Jahr
|
0,03
|
0,10
| |
Prozent Senkung
Verhältnisrate (95%-KI)
|
69 %
0,31 (0,11; 0,91)
|
_
| |
p-Wert
|
0,034
|
| |
FEV1 (mL) vor Bronchodilatator nach 32 Wochen
| |
Mittlere Änderung gegenüber Ausgangswert (Standardfehler)
|
183 (31,1)
|
86 (31,4)
| |
Unterschied (Mepolizumab vs. Placebo)
|
98
|
| |
95%-KI
|
(11, 184)
|
| |
p-Wert
|
0,028
|
| |
St. George's Respiratory Questionnaire (SGRQ) nach 32 Wochen
| |
Mittlere Änderung gegenüber Ausgangswert (Standardfehler)
|
-16,0 (1,13)
|
-9,0 (1,16)
| |
Unterschied (Mepolizumab vs. Placebo)
|
-7,0
|
| |
95%-KI
|
(-10,2; -3,8)
|
| |
p-Wert
|
< 0,001
|
|
Studie zur Reduktion oraler Kortikosteroide (SIRIUS)
MEA115575 beurteilte die Wirkung von Mepolizumab 100 mg s.c. in Bezug auf die Verminderung des Bedarfs an oralen Kortikosteroiden (OCS) zur Erhaltungstherapie, unter Aufrechterhaltung der Asthmakontrolle bei auf systemische Kortikosteroide angewiesenen Patienten mit schwerem eosinophilem Asthma. Die Patienten wiesen in den 12 Monaten vor dem Screening eine Eosinophilenzahl im peripheren Blut von ≥300/μL bzw. vor Behandlungsbeginn eine Eosinophilenzahl im peripheren Blut von ≥150/μL auf und erhielten im Behandlungszeitraum in vierwöchigen Abständen Mepolizumab oder Placebo. Die OCS-Dosis wurde während der OCS-Reduktionsphase (Wochen 4–20) alle 4 Wochen vermindert, solange die Asthmakontrolle dabei aufrechterhalten wurde. Während der Studie setzten die Patienten ihre Ausgangstherapie gegen Asthma fort (hochdosierte inhalative Kortikosteroide [ICS] in Kombination mit mindestens einem weiteren Kontrollmedikament, z.B. langwirkenden Beta2-Sympathomimetika [LABA] oder Leukotrienmodifikatoren).
Die Studienpopulation aus insgesamt 135 Patienten war wie folgt charakterisiert: Durchschnittsalter 50 Jahre, 55 % der Teilnehmer waren weiblich, 48 % nahmen seit mindestens 5 Jahren orale Kortikosteroide und erhielten bei Studienbeginn ein mittleres Prednisolonäquivalent von ca. 13 mg pro Tag.
Primärer Studienendpunkt war die Verminderung der OCS-Tagesdosis (Wochen 20–24) unter Aufrechterhaltung der Asthmakontrolle im Vergleich zu Patienten unter Placebo (siehe Tabelle 4).
Tabelle 4: Ergebnisse für den primären und die sekundären Endpunkte in der Intent-to-treat-Population von MEA115575
|
|
Mepolizumab
(100 mg s.c.)
n = 69
|
Placebo
n = 66
| |
Primärer Endpunkt
| |
OCS-Reduktion gegenüber der Ausgangsdosis nach 20–24 Wochen (%)
| |
90 %–100 %
|
16 (23 %)
|
7 (11 %)
| |
75 %–< 90 %
|
12 (17 %)
|
5 (8 %)
| |
50 %–< 75 %
|
9 (13 %)
|
10 (15 %)
| |
> 0 %–< 50 %
|
7 (10 %)
|
7 (11 %)
| |
Keine OCS-Reduktion/unzureichende Asthmakontrolle/ Behandlungsabbruch
|
25 (36 %)
|
37 (56 %)
| |
Odds Ratio (95%-KI)
|
2,39 (1,25; 4,56)
|
| |
p-Wert
|
0,008
|
| |
Sekundäre Endpunkte
| |
Reduktion der OCS-Tagesdosis (%)
| |
Mindestens 50%ige Reduktion
|
37 (54 %)
|
22 (33 %)
| |
Odds Ratio (95%-KI)
|
2,26 (1,10; 4,65)
|
| |
p-Wert
|
0,027
|
| |
Reduktion der OCS-Tagesdosis (%)
| |
Auf ≤5 mg/Tag
|
37 (54 %)
|
21 (32 %)
| |
Odds Ratio (95%-KI)
|
2,45 (1,12; 5,37)
|
| |
p-Wert
|
0,025
|
| |
Reduktion der OCS-Tagesdosis (%)
| |
Auf null
|
10 (14 %)
|
5 (8 %)
| |
Odds Ratio (95%-KI)
|
1,67 (0,49; 5,75)
|
| |
p-Wert
|
0,414
|
| |
Mediane prozentuale Reduktion der OCS-Tagesdosis
| |
Mediane Reduktion (%) gegenüber Ausgangsdosis (95%-KI)
|
50,0 (20,0; 75,0)
|
0,0 (-20,0; 33,3)
| |
Medianer Unterschied (95%-KI)
|
-30,0 (-66,7; 0,0)
|
| |
p-Wert
|
0,007
|
|
Ausserdem wurde anhand des SGRQ die gesundheitsbezogene Lebensqualität ermittelt. Nach 24 Wochen war in Bezug auf den mittleren SGRQ-Score unter Nucala eine signifikante Verbesserung gegenüber Placebo feststellbar: -5,8 (95%-KI: -10,6; -1,0; p = 0,019). In Woche 24 war der Anteil der Patienten mit klinisch bedeutsamer Abnahme des SGRQ-Score (definiert als Rückgang um mindestens 4 Einheiten gegenüber dem Ausgangswert) unter Nucala höher (58 %, 40/69) als unter Placebo (41 %, 27/66).
Das langfristige Wirksamkeitsprofil von Nucala bei Patienten mit schwerem Asthma (n=998), die in den offenen Verlängerungsstudien MEA115666, MEA115661 und 201312 über einen Median von 2,8 Jahren (Spanne von 4 Wochen bis 4,5 Jahren) behandelt wurden, stimmte im Allgemeinen mit den drei placebo-kontrollierten Studien überein.
Chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
Bei der Studie 205687 handelte es sich um eine 52-wöchige, randomisierte, doppelblinde, placebokontrollierte Studie, in der 407 Patienten im Alter über 18 Jahren mit CRSwNP untersucht wurden.
Die in die Studie aufgenommenen Patienten mussten für die nasale Obstruktion einen VAS-Symptom-Score (visuelle Analogskala) von >5 (von max. 10 Punkten), einen Gesamt-VAS-Symptom-Score >7 (von max. 10 Punkten) und einen endoskopischen bilateralen NP-Score von 5 (von max. 8 möglichen Punkten; mit einem Mindest-Scorewert von 2 pro Nasenhöhle) aufweisen. Ferner mussten die Patienten in den vorangegangenen 10 Jahren mindestens eine Operation infolge von Nasenpolypen erhalten haben.
Die Patienten erhielten eine Dosis 100 mg Mepolizumab oder Placebo, die alle 4 Wochen zusätzlich zur Basistherapie mit intranasalen Kortikosteroiden subkutan verabreicht wurde.
Die demografischen Daten und Baseline-Eigenschaften der an Studie 205687 teilnehmenden Patienten sind unten in Tabelle 5 aufgeführt:
Tabelle 5: Demographische Daten und Baseline-Eigenschaften der CRSwNP
|
|
N = 407
| |
Alter (Jahre) der Patienten, Mittelwert (SD)
|
49 (13)
| |
Weiblich, n (%)
|
143 (35)
| |
Europäischstämmig, n (%)
|
379 (93)
| |
Dauer (Jahre) der CRSwNP, Mittelwert (SD)
|
11,4 (8,39)
| |
Patienten mit ≥1 vorangegangener Operation, n (%)
|
407 (100)
| |
Patienten mit ≥3 vorangegangenen Operationen, n (%)
|
124 (30)
| |
OCS-Anwendung wegen NP (≥1 Verlauf) in den vergangenen 12 Monaten, n (%)
|
197 (48)
| |
Gesamter endoskopischer NP-Scorea b c, Mittelwert (SD), max. Scorewert = 8
|
5,5 (1,29)
| |
VAS-Scorea d der nasalen Obstruktion, Mittelwert (SD), max. Scorewert = 10
|
9,0 (0,83)
| |
Gesamt-VAS-Symptom-Scorea d, Mittelwert (SD), max. Scorewert = 10
|
9,1 (0,74)
| |
SNOT-22-Gesamtscoree, Mittelwert (SD), Bereich 0-110
|
64,1 (18,32)
| |
Zusammengesetzter VAS-Symptome-Scorea, Mittelwert (SD), max. Scorewert = 10
|
9,0 (0,82)
| |
VAS-Scorea,d des Geruchsverlusts, Mittelwert (SD), max. Scorewert = 10
|
9,7 (0,72)
| |
Asthma, n (%)
|
289 (71)
| |
AERD, n (%)
|
108 (27)
| |
Geometrisches Mittel der Eosinophilen-Zahl bei Baseline, Zellen/mcl (95%-KI)
|
390 (360, 420)
|
CRSwNP = chronische Rhinosinusitis mit Nasenpolypen, SD = Standardabweichung, OCS = orales Kortikosteroid, NP = Nasenpolypen, VAS = visuelle Analogskala, SNOT-22 = Sino-Nasal-Outcome-Test, AERD = Aspirin-exazerbierte Atemwegserkrankung
a Höhere Scorewerte weisen auf eine höheren Schweregrad der Erkrankung hin.
b Wie von unabhängigen verblindeten Bewertern bewertet
c Der NP-Score ist die Summe der Scorewerte beider Nasenlöcher (auf einer Skala 0-8), bei denen jedes Nasenloch bewertet wurde (0=keine Polypen; 1=kleine Polypen im mittleren Gehörgang, die nicht unter den unteren Rand der mittleren Nasenmuschel ragen; 2=Polypen, die bis unter den unteren Rand der mittleren Nasenmuschel ragen; 3=grosse Polypen, die den unteren Rand der unteren Nasenmuschel oder Polypen medial zur mittleren Ohrmuschel überragen; 4=grosse Polypen, die eine fast vollständige Verstopfung/Obstruktion des unteren Gehörgangs bewirken).
d Täglich von den Patienten anhand einer Skala von 0 bis 10 bewertet (0=kein; 10=extrem schlimm).
e SNOT-22 ist ein Test zur Bewertung der gesundheitsbezogenen Lebensqualität und umfasst 22 Punkte in 6 Bereichen von Symptomen und Auswirkungen im Zusammenhang mit CRSwNP (nasal, nicht-nasal, Ohr/Gesicht, Schlaf, Müdigkeit, emotionale Folgen). Höhere Scorewerte deuten auf eine schlechtere gesundheitsbezogene Lebensqualität hin.
Die koprimären Endpunkte waren die Veränderung des gesamten endoskopischen NP-Scores in Woche 52 gegenüber Baseline und die Veränderung des mittleren VAS-Wertes der nasalen Obstruktion in den Wochen 49-52 gegenüber Baseline.
Patienten, die Mepolizumab erhalten hatten, wiesen im Vergleich zu Placebo signifikant grössere Verbesserungen (Reduktionen) beim endoskopischen NP-Gesamtscore in Woche 52 und beim VAS-Score bei nasaler Obstruktion in den Wochen 49-52 auf (siehe Tabelle 6).
Tabelle 6: Analysen der koprimären Endpunkte (Intent-To-Treat-Population)
|
|
Placebo
(N=201)
|
Mepolizumab
100 mg s.c.
(N=206)
| |
Endoskopischer Gesamt-Score in Woche 52 a
| |
Medianer Score an Baseline (min, max)
|
6,0 (0, 8)
|
5,0 (2, 8)
| |
Mediane Änderung gegenüber Baseline
|
0,0
|
-1,0
| |
p-Wert b
|
|
<0,001
| |
Korrigierter Behandlungsunterschied bei den Medianen (95%-KI) c
|
|
-0,73 (-1,11, -0,34)
| |
≥1-Punkt-Verbesserung, n (%)
|
57 (28)
|
104 (50)
| |
≥2-Punkt-Verbesserung, n (%)
|
26 (13)
|
74 (36)
| |
VAS-Score für die nasale Obstruktion (Wochen 49 bis 52) a
| |
Medianer Score an Baseline (min, max)
|
9,14 (5,31, 10,00)
|
9,01 (6,54, 10,00)
| |
Mediane Änderung gegenüber Baseline
|
-0,82
|
-4,41
| |
p-Wert b
|
|
<0,001
| |
Korrigierter Behandlungsunterschied bei den Medianen (95%-KI) c
|
|
-3,14 (-4,09, -2,18)
| |
>1-Punkt-Verbesserung, n (%)
|
100 (50)
|
146 (71)
| |
≥3-Punkt-Verbesserung, n (%)
|
73 (36)
|
124 (60)
|
a Studienteilnehmer mit einer Nasenoperation/Sinuplastik vor dem Studientermin wiesen den schlechtesten beobachteten Scorewert der Situation vor der Nasenoperation/Sinuplastik zu. Teilnehmer, die ohne Nasenoperation/Sinuplastik aus der Studie ausschieden, wiesen ihren schlechtesten beobachteten Scorewerte der Situation vor dem Studienabbruch zu.
b Auf Basis des Wilcoxon-Rangsummentests.
c Quantile Regression mit Kovariaten der Behandlungsgruppe, der geografischen Region, des Baseline-Scorewerts und der log(e)-Eosinophilenzahl im Blut an Baseline.
Abbildung 1: Abbildung des gesamten endoskopischen Nasenpolypen-Scores (zentral abgelesen) Responders per Kontrolle
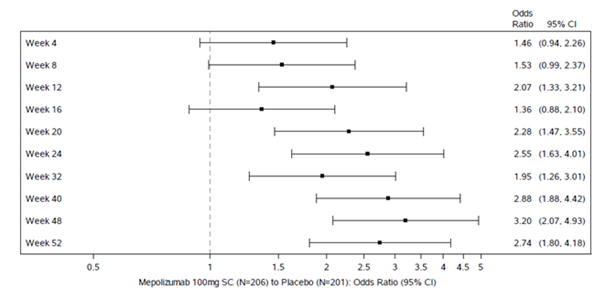
Alle sekundären Endpunkte waren statistisch signifikant und unterstützten die koprimären Endpunkte. Der wichtigste sekundäre Endpunkt war die Zeit bis zur ersten NP-Operation bis Woche 52 (siehe Abbildung 1). Daten zu den anderen sekundären Endpunkten sind in Tabelle 7 aufgeführt.
Zeit bis zur ersten NP-Operation
Über den gesamten Behandlungszeitraum von 52 Wochen war bei den Patienten der Mepolizumab-Gruppe die Wahrscheinlichkeit geringer, sich einer NP-Operation unterziehen zu müssen, als bei den Patienten in der Placebo-Gruppe (als Operation wurde jeder Eingriff definiert, bei dem Instrumente eingesetzt wurden, die zu einer Inzision und Entfernung von Gewebe [Polypektomie] in der Nasenhöhle führten).
Bis Woche 52 hatten sich 18 Patienten (9 %) in der Mepolizumab-Gruppe einer NP-Operation unterzogen, wohingegen dieser Wert in der Placebo-Gruppe bei 46 Patienten (23 %) lag.
Bei Patienten, die Mepolizumab erhielten, war die Zeit bis zur ersten NP-Operation im Vergleich zu Placebo länger. Das Operationsrisiko während des Behandlungszeitraums war bei Patienten, die mit Mepolizumab behandelt wurden, im Vergleich zu Patienten, die mit Placebo behandelt wurden, um 57 % niedriger und somit signifikant geringer (Hazard Ratio: 0,43; 95%-KI 0,25, 0,76; unbereinigt/bereinigt p=0,003), eine Post-hoc-Analyse ergab eine 61%-ige Verringerung der Wahrscheinlichkeit einer Operation (Odds Ratio: 0,39, 95%-KI: 0,21, 0,72; p= 0,003.
Abbildung 2: Kaplan-Meier-Kurve für die Zeit bis zur ersten Nasenpolypen-OP

Tabelle 7: Ergebnisse der anderen sekundären Endpunkte in der Intent-To-Treat-Population
|
|
Placebo
(N=201)
|
Mepolizumab
(N=206)
| |
Gesamter VAS-Score (Wochen 49-52) a
| |
Medianer Score an Baseline (min, max)
|
9,20 (7,21, 10,00)
|
9,12 (7,17, 10,00)
| |
Mediane Änderung gegenüber Baseline
|
-0,90
|
-4,48
| |
Unbereinigter/bereinigter p-Wert b,c
|
|
<0,001/0,003
| |
Korrigierter Behandlungsunterschied bei den Medianen (95%-KI) d
|
|
-3,18 (-4,10, -2,26)
| |
≥2,5-Punkte-Verbesserung (%)
|
40
|
64
| |
SNOT-22-Gesamtscore in Woche 52 a, g
| |
n
|
198
|
205
| |
Medianer Score an Baseline (min, max)
|
64,0 (19, 110)
|
64,0 (17, 105)
| |
Mediane Änderung gegenüber Baseline
|
-14,0
|
-30,0
| |
Unbereinigter/bereinigter p-Wert b,c
|
|
<0,001/0,003
| |
Korrigierter Behandlungsunterschied bei den Medianen (95%-KI) d
|
|
-16,49 (-23,57, -9,42)
| |
≥28-Punkte-Verbesserung (%) g
|
32
|
54
| |
Patienten, die wegen Nasenpolypen bis Woche 52 systemische Steroide benötigen
| |
Anzahl der Patienten mit ≥1 Verlauf
|
74 (37)
|
52 (25)
| |
Odds Ratio zu Placebo (95%-KI)
|
|
0,58 (0,36, 0,92)
| |
Unbereinigter/bereinigter p-Wert c, e
|
|
0,020/0,020
| |
Zusammengesetzter VAS-Score – nasale Symptome (Wochen 49-52) a,f
| |
Medianer Score an Baseline (min, max)
|
9,18 (6,03, 10,00)
|
9,11 (4,91, 10,00)
| |
Mediane Änderung gegenüber Baseline
|
-0,89
|
-3,96
| |
Unbereinigter/bereinigter p-Wert b,c
|
|
<0,001/0,020
| |
Korrigierter Behandlungsunterschied bei den Medianen (95%-KI) d
|
|
-2,68 (-3,44, -1,91)
| |
≥2-Punkte-Verbesserung (%) h
|
40
|
66
| |
VAS-Score für den Geruchsverlust (Wochen 49-52) a
| |
Medianer Score an Baseline (min, max)
|
9,97 (6,69, 10,00)
|
9,97 (0,94, 10,00)
| |
Mediane Änderung gegenüber Baseline
|
0,00
|
-0,53
| |
Unbereinigter/bereinigter p-Wert b,c
|
|
<0,001/0,020
| |
Korrigierter Behandlungsunterschied bei den Medianen (95%-KI) d
|
|
-0,37 (-0,65, -0,08)
| |
≥3-Punkte-Verbesserung (%) h
|
19
|
36
|
a Patienten mit einer Nasenoperation/Sinuplastik vor dem Studientermin wiesen den schlechtesten beobachteten Scorewert der Situation vor der Nasenoperation/Sinuplastik zu. Teilnehmer, die ohne Nasenoperation/Sinuplastik aus der Studie ausschieden, wiesen ihren schlechtesten beobachteten Scorewerte der Situation vor dem Studienabbruch zu.
b Auf Basis des Wilcoxon-Rangsummentests.
c Multiplizität kontrolliert mithilfe von Tests der sekundären Endpunkte gemäss einer vordefinierten Hierarchie.
d Quantile Regression mit Kovariaten der Behandlungsgruppe, der geografischen Region, des Baseline-Scorewerts und der log(e)-Eosinophilenzahl im Blut an Baseline.
e Analyse unter Verwendung eines logistischen Regressionsmodells mit Kovariaten der Behandlungsgruppe, der geografischen Region, der Anzahl der OCS-Verläufe für NP in den letzten 12 Monaten (0, 1, >1 als Ordinalzahl), des ENP-Gesamtscores an Baseline (zentral gelesen), des VAS-Scores für nasale Obstruktion und der log(e)-Eosinophilenzahl im Blut an Baseline.
f Zusammengesetzter VAS-Score für nasale Obstruktion, Nasenausfluss, Rachenschleim und Geruchsverlust.
g In allen 6 Bereichen der Symptome und Auswirkungen im Zusammenhang mit CRSwNP wurde eine Verbesserung festgestellt.
h Der Schwellenwert für die Verbesserung jedes einzelnen Endpunktes wurde als sinnvolle intraindividuelle Veränderung bestimmt.
Endpunkte der Subgruppe von Patienten mit komorbidem Asthma
Bei 289 (71 %) Patienten mit komorbidem Asthma ergaben vordefinierte Analysen im Vergleich zu Placebo bei den Patienten, die 100 mg Mepolizumab erhielten, Verbesserungen bei den koprimären Endpunkten, die mit denen der Gesamtpopulation übereinstimmten.
Eosinophile Granulomatose mit Polyangiitis (EGPA)
MEA115921 war eine randomisierte, doppelblinde, placebokontrollierte, 52-wöchige Studie, in der 136 Patienten ab 18 Jahren mit rezidivierender oder therapierefraktärer EGPA unter stabil dosierten oralen Kortikosteroiden (OCS; ≥7,5 bis ≤50 mg/Tag Prednisolon/Prednison) untersucht wurden. 53% (n = 72) der Teilnehmer erhielten gleichzeitig Immunsuppressiva in stabiler Dosierung.
Die Patienten erhielten eine Dosis von 300 mg Mepolizumab oder Placebo subkutan einmal alle vier Wochen, ergänzend zu ihrer Prednisolon/Prednison-Basistherapie mit oder ohne Immunsuppressiva. Die OCS-Dosis wurde nach dem Ermessen des Prüfarztes stufenweise reduziert.
Koprimäre Endpunkte waren die kumulative Gesamtremissionsdauer, wobei Remission definiert war als Birmingham Vasculitis Activity Score (BVAS) von 0 (keine aktive Vaskulitis) plus Prednisolon/Prednison-Dosis ≤4 mg/Tag, sowie der Anteil der Teilnehmer in Remission nach 36 sowie nach 48 Behandlungswochen.
Remission
Im Vergleich zu Placebo erreichten die Teilnehmer unter Mepolizumab 300 mg eine signifikant längere Gesamtremissionsdauer. Darüber hinaus war der Anteil der Teilnehmer in Remission sowohl in Woche 36 als auch in Woche 48 unter Mepolizumab 300 mg signifikant höher als unter Placebo (Tabelle 8).
Tabelle 8: Analysen zu den koprimären Endpunkten (ITT-Population)
|
|
Anzahl (%) Teilnehmer
| |
Placebo n = 68
|
Mepolizumab 300 mg n = 68
| |
Gesamtremissionsdauer während 52 Wochen
| |
0 Wochen
|
55 (81)
|
32 (47)
| |
> 0 bis < 12 Wochen
|
8 (12)
|
8 (12)
| |
12 bis < 24 Wochen
|
3 (4)
|
9 (13)
| |
24 bis < 36 Wochen
|
0
|
10 (15)
| |
≥36 Wochen
|
2 (3)
|
9 (13)
| |
Odds Ratio (Mepolizumab/Placebo)
|
|
5,91
| |
95%-KI
|
----
|
2,68; 13,03
| |
p-Wert
|
----
|
< 0,001
| |
Teilnehmer in Remission in Woche 36 und 48
|
2 (3)
|
22 (32)
| |
Odds Ratio (Mepolizumab/Placebo)
|
|
16,74
| |
95%-KI
|
----
|
3,61; 77,56
| |
p-Wert
|
----
|
< 0,001
|
Odds Ratio > 1 spricht zugunsten von Mepolizumab
Teilnehmer unter Mepolizumab 300 mg erreichten eine signifikant längere Gesamtremissionsdauer (p < 0,001), und der Anteil der Teilnehmer in Remission war bei Anwendung der Remissionsdefinition für den sekundären Endpunkt (BVAS = 0 plus Prednisolon/Prednison ≤7,5 mg/Tag) sowohl in Woche 36 als auch in Woche 48 unter Mepolizumab 300 mg höher als unter Placebo (p < 0,001).
Rezidive
Im Vergleich zu Placebo war die Zeit bis zum ersten Rezidiv (definiert als durch Vaskulitis, Asthma oder sinonasale Symptome bedingte Verschlechterung, die eine Dosiserhöhung der Kortikosteroide bzw. der Immunsuppressiva oder eine Hospitalisierung erfordert), bei Teilnehmern unter Mepolizumab 300 mg signifikant länger (p < 0,001). Darüber hinaus war die annualisierte Rezidivrate bei Teilnehmern unter Mepolizumab um 50 % geringer als bei Teilnehmern unter Placebo: 1,14 vs. 2,27.
Dosisreduktion der oralen Kortikosteroide
Im Vergleich zu Teilnehmern unter Placebo erhielten Teilnehmer unter Mepolizumab 300 mg in den Wochen 48 bis 52 eine geringere mittlere Tagesdosis an oralen Kortikosteroiden (p < 0,001). In der Gruppe mit Mepolizumab 300 mg konnten 12 Teilnehmer (18 %) die Behandlung mit oralen Kortikosteroiden gänzlich ausschleichen, gegenüber nur 2 Teilnehmern (3 %) in der Placebogruppe.
Hypereosinophilie-Syndrom (HES)
Studie 200622 war eine randomisierte, doppelblinde, placebokontrollierte, 32-wöchige Studie, in der 108 HES-Patienten im Alter von ≥12 Jahren untersucht wurden. Patienten mit nicht-hämatologischem sekundärem HES (z.B. Medikamentenüberempfindlichkeit, parasitäre Infektion, HIV-Infektion, nicht-hämatologisches Malignom) oder F/P positivem HES wurden von der Studie ausgeschlossen. Die Patienten erhielten subkutan einmal alle 4 Wochen 300 mg Mepolizumab bzw. Placebo, unter Beibehaltung ihrer stabilen HES-Therapie. Einer der 4 eingeschlossenen Jugendlichen erhielt 300 mg Mepolizumab, die anderen 3 erhielten Placebo, alle jeweils 32 Wochen lang. Die Standard-HES-Therapie konnte OCS und Immunsuppressiva oder Zytotoxika umfassen. Die Studienteilnehmer hatten in den letzten12 Monaten mindestens zwei HES-Schübe erlitten und wiesen beim Screening eine Blut-Eosinophilenzahl von ≥1000 Zellen/µl auf.
Primärer Endpunkt der Studie 200622 war der Anteil der Studienteilnehmer, die während des 32-wöchigen Behandlungszeitraums einen HES-Schub erlitten. Ein HES-Schub war definiert als Verschlechterung der klinischen Anzeichen und Symptome des HES oder als Anstieg der Eosinophilen (zu mindestens 2 Zeitpunkten), die eine OCS-Erhöhung oder die Dosiserhöhung bzw. die zusätzliche Verabreichung einer zytotoxischen oder immunsuppressiven HES-Therapie erforderlich machten.
Die Primäranalyse verglich Patienten der Mepolizumab- und der Placebogruppe, die einen HES-Schub erlitten oder aus der Studie ausschieden. Im Vergleich zur Placebogruppe erlitten in der Gruppe mit 300 mg Mepolizumab während des 32-wöchigen Behandlungszeitraums 50 % weniger Patienten einen HES-Schub oder brachen die Studie ab; 28 % versus 56 % (OR 0,28, 95%-KI 0,12–0,64) (siehe Tabelle 9).
Sekundäre Endpunkte waren die Zeit bis zum ersten HES-Schub, der Anteil der Studienteilnehmer, die in Woche 20 bis Woche 32 einen HES-Schub erlitten, die Rate der HES-Schübe und die Änderung des Fatigue-Schweregrads gegenüber dem Ausgangswert. Alle sekundären Endpunkte waren statistisch signifikant und untermauerten den primären Endpunkt (siehe Abbildung 3 und Tabelle 10).
Tabelle 9: Ergebnisse für den primären Endpunkt / Ergebnisse der Primäranalyse in der Intent-to-Treat-Population (Studie 200622)
|
|
Mepolizumab
N = 54
|
Placebo
N = 54
| |
Anteil der Studienteilnehmer, die einen HES-Schub erlitten
| |
Studienteilnehmer mit ≥1 HES-Schub oder Studienabbruch (%)
|
15 (28)
|
30 (56)
| |
Studienteilnehmer mit ≥1 HES-Schub (%)
|
14 (26)
|
28 (52)
| |
Studienteilnehmer ohne HES-Schub, die die Studie abbrachen (%)
|
1 (2)
|
2 (4)
| |
Odds Ratio (95%-KI)
|
0,28 (0,12–0,64)
|
| |
p-Wert im CMH
|
0,002
|
|
CMH = Cochran-Mantel-Haenszel
Zeit bis zum ersten Schub
Bei Studienteilnehmern unter 300 mg Mepolizumab war die Zeit bis zum ersten HES-Schub im Vergleich zu Patienten unter Placebo signifikant länger. Das Risiko eines ersten HES-Schubs während des Behandlungszeitraums war bei Patienten unter Mepolizumab um 66 % niedriger als bei Patienten unter Placebo (Hazard Ratio: 0,34, 95%-KI 0,18–0,67, p = 0,002).
Abbildung 3: Kaplan-Meier-Kurve für die Zeit bis zum ersten HES-Schub
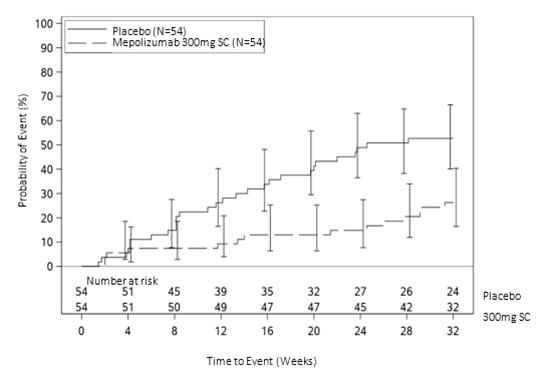
Tabelle 10 Ergebnisse für andere sekundäre Endpunkte in der Intent-to-Treat-Population (Studie 200622)
|
|
Mepolizumab
N = 54
|
Placebo
N = 54
| |
HES-Schübe in Woche 20 und bis einschliesslich Woche 32
| |
Studienteilnehmer mit ≥1 HES-Schub oder Studienabbruch (%)
|
9 (17)
|
19 (35)
| |
Odds Ratio (95%-KI)
|
0,33 (0,13–0,85)
| |
CMH-p-Wert (nicht bereinigt/bereinigt)a
|
0,02/0,02
| |
Rate der HES-Schübe
| |
Geschätzte jährliche Durchschnittsrate
|
0,50
|
1,46
| |
Ratenverhältnis (95%-KI)
|
0,34 (0,19–0,63)
| |
Wilcoxon-p-Wert (nicht bereinigt/bereinigt)a
|
0,002/0,02
| |
Änderung des Fatigue-Schweregrads gegenüber dem Ausgangswert, basierend auf Item 3 des Brief Fatigue Inventory (BFI) (schwerste Fatigue-Ausprägung während der letzten 24 Stunden) in Woche 32b
| |
Mediane Änderung beim BFI-Item 3
|
-0,66
|
0,32
| |
Vergleich (Mepolizumab vs. Placebo) der p-Werte (nicht bereinigt/bereinigt)a
|
0,036/0,036
|
|
a Bereinigte p-Werte, basierend auf einer vorab festgelegten Hierarchie von Endpunkten.
b Patienten mit fehlenden Daten wurden eingeschlossen unter Verwendung des schlechtesten beobachteten Werts.
CMH = Cochran-Mantel-Haenszel
Offene Verlängerungsphase bei HES
Geeignete Patienten, darunter 4 Jugendliche, fuhren nach Abschluss der Studie 200622 mit der 20-wöchigen offenen Verlängerungsstudie 205203 fort, in der das langfristige Sicherheitsprofil untersucht wurde und zusätzliche Daten zum klinischen Nutzen von Mepolizumab bei HES-Patienten über 32 Wochen hinaus gewonnen wurden.
Die in Studie 200622 beobachtete Wirkung der Mepolizumab-Behandlung auf die Reduktion der HES-Schübe wurde aufrechterhalten bei den Patienten, welche die Mepolizumab-Behandlung in Studie 205203 fortsetzten, in der bei 94 % (47/50) der Patienten keine Schübe auftraten.
In den Wochen 16 bis 20 hatten 28 % aller Patienten, die in den Wochen 0 bis 4 eine durchschnittliche OCS-Dosis von > 0 mg/Tag (Prednison oder Äquivalent) erhalten hatten, eine durchschnittliche tägliche OCS-Dosisreduktion von ≥50 % erreicht. Wirksamkeitsdaten aus dieser Studie deuten darauf hin, dass der klinische Nutzen von Mepolizumab über 52 Wochen anhält und eine Verringerung der OCS-Behandlung bei HES-Patienten ermöglicht.
PharmakokinetikNach subkutaner Verabreichung an Patienten mit mittel- bis hochgradigem Asthma zeigte Mepolizumab im Bereich zwischen 12,5 mg und 250 mg eine dosisproportionale Pharmakokinetik. Die Pharmakokinetik von Mepolizumab stimmte bei Patienten mit Asthma, CRSwNP, EGPA oder HES überein.
Die systemische Exposition bei subkutaner Verabreichung von 300 mg Mepolizumab betrug ungefähr das Dreifache der Exposition bei 100 mg.
In einer pharmakokinetischen Vergleichsstudie an gesunden Probanden erwies sich die Pharmakokinetik von Mepolizumab für die unterschiedlichen Formulierungen (kürzlich hergestellt) nach Verabreichung einer subkutanen Einzeldosis von 100 mg als vergleichbar.
Absorption
Nach subkutaner Verabreichung an gesunde Personen oder Asthma-Patienten wurde Mepolizumab langsam resorbiert, mit einer Zeit bis zum Erreichen der maximalen Plasmakonzentration (Tmax) von im Median 4 bis 8 Tagen.
Nach einmaliger subkutaner Verabreichung an Bauch, Oberschenkel oder Oberarm betrug die absolute Bioverfügbarkeit von Mepolizumab bei gesunden Personen 64 %, 71 % bzw. 75 %. Bei Patienten mit Asthma lag die absolute Bioverfügbarkeit von Mepolizumab nach subkutaner Verabreichung am Oberarm bei 74%. Nach subkutaner Verabreichung in vierwöchigen Abständen wird der Steady State innerhalb von 16 Wochen erreicht. Nach wiederholter subkutaner Verabreichung in vierwöchigen Abständen kommt es am Steady State zu einer Akkumulation um den Faktor 2. Die Pharmakokinetik von Mepolizumab ist unabhängig von der Zeit.
Distribution
Nach Verabreichung einer intravenösen Einzeldosis an Patienten mit leichtem Asthma verteilt sich Mepolizumab mit einem mittleren Verteilungsvolumen von 55 bis 85 mL/kg.
Metabolismus
Mepolizumab ist ein humanisierter monoklonaler IgG1-Antikörper und wird von proteolytischen Enzymen abgebaut, die nicht nur in hepatischem Gewebe, sondern verbreitet im ganzen Körper vorkommen.
Elimination
Nach Verabreichung einer intravenösen Einzeldosis an Asthma-Patienten reichte die mittlere systemische Clearance (CL) von 1,9 bis 3,3 mL/Tag/kg, mit einer mittleren terminalen Halbwertszeit von ca. 20 Tagen. Nach subkutaner Verabreichung von Mepolizumab lag die mittlere terminale Halbwertszeit (t1/2) zwischen 16 und 22 Tagen.
Kinetik spezieller Patientengruppen
Ältere Patienten (> 65 Jahre)
Es wurden keine formalen Studien an älteren Patienten durchgeführt. Die populationspharmakokinetische Analyse ergab jedoch keine Hinweise auf einen Einfluss des Alters (12–82 Jahre) auf die Pharmakokinetik von Mepolizumab.
Eingeschränkte Nierenfunktion
Zum Einfluss einer Nierenfunktionsbeeinträchtigung auf die Pharmakokinetik von Mepolizumab wurden keine formalen Studien durchgeführt. Auf Grundlage der populationspharmakokinetischen Analysen ist bei Patienten mit einer Creatinin-Clearance zwischen 50 und 80 mL/min keine Dosisanpassung erforderlich. Zu Patienten mit einer Creatinin-Clearance unter 50 mL/min liegen nur eingeschränkte Daten vor.
Eingeschränkte Leberfunktion
Zum Einfluss einer Leberfunktionsbeeinträchtigung auf die Pharmakokinetik von Mepolizumab wurden keine formalen Studien durchgeführt. Da Mepolizumab durch verbreitet vorkommende proteolytische Enzyme abgebaut wird, die nicht auf das hepatische Gewebe beschränkt sind, dürfte die Elimination von Mepolizumab durch eine Leberfunktionsstörung kaum beeinflusst werden.
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie und Toxizität bei wiederholter Gabe an Affen lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. Die intravenöse und subkutane Verabreichung an Affen ging mit einer Abnahme der Eosinophilenzahl im peripheren Blut und in der Lunge, aber ohne toxikologisch relevante Befunde einher.
Eosinophile spielen offenbar eine Rolle bei den Reaktionen des Immunsystems auf einen Befall mit bestimmten Parasiten. In Studien an mit Antikörpern gegen IL-5 behandelten Mäusen oder Mäusen mit genetisch induziertem IL-5- oder Eosinophilenmangel war keine Beeinträchtigung der Abwehrfähigkeit gegenüber Parasiten feststellbar.
Da es sich bei Mepolizumab um einen monoklonalen Antikörper handelt, wurden keine Mutagenitäts- oder Karzinogenitätsstudien durchgeführt.
Reproduktionstoxikologie
Trächtigkeit
Bei Affen hatte Mepolizumab keinen Einfluss auf die Trächtigkeit oder die embryofetale bzw. postnatale Entwicklung (einschliesslich Immunfunktion). Untersuchungen auf innere oder skelettale Fehlbildungen wurden nicht durchgeführt. Daten von Cynomolgus-Affen belegen die Plazentagängigkeit von Mepolizumab. Die Mepolizumab-Konzentrationen waren noch mehrere Monate nach der Geburt bei den Nachkommen ca. 1,2- bis 2,4-mal höher als bei den Müttern und beeinträchtigten ihr Immunsystem nicht.
Fertilität
Mit einem analogen, IL-5 bei Mäusen hemmenden Antikörper wurde in einer Studie an Mäusen zur Fertilität und zur allgemeinen Reproduktionstoxizität keine Beeinträchtigung der Fertilität festgestellt. Diese Studie schloss keine Nachkommen und keine funktionelle Beurteilung der Filialgeneration ein.
Sonstige HinweiseInkompatibilitäten
Keine bekannt.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2°C bis 8°C) und bis zum Gebrauch in der Originalpackung vor Licht geschützt aufbewahren. Nicht einfrieren!
Der Zeitraum ausserhalb des Kühlschranks darf maximal 7 Tage betragen, wenn der Fertigpen resp. die Fertigspritze vor Licht geschützt und nicht über 30°C aufbewahrt wurden. Sie sind zu entsorgen, wenn sie länger als 7 Tage ausserhalb des Kühlschranks gelagert wurden.
Der Fertigpen resp. die Fertigspritze dürfen nicht mehr verwendet werden, wenn sie länger als 8 Stunden ausserhalb der Verpackung geblieben sind.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Eine genaue Anleitung zur Anwendung des Fertigpens bzw. der Fertigspritze befindet sich in der der Packung beigelegten Gebrauchsanweisung.
Der Fertigpen und die Fertigspritze sind zum einmaligen Gebrauch bestimmt.
Entsorgung
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
ZulassungsnummerNucala Fertigpen 67350 (Swissmedic)
Nucala Fertigspritze 67351 (Swissmedic)
PackungenFertigpen:
Packung mit 1 Fertigpen zu 100 mg/mL (B)
Fertigspritze:
Packung mit 1 Fertigspritze zu 100 mg/mL (B)
ZulassungsinhaberinGlaxoSmithKline AG, 6340 Baar
Stand der InformationNovember 2024
|