Eigenschaften/WirkungenATC-Code
L01XL05
Wirkungsmechanismus
CARVYKTI ist eine mit gentechnisch veränderten autologen T-Zellen durchgeführte und gegen BCMA gerichtete Immuntherapie, bei der eine Umprogrammierung der T-Zellen eines Patienten mit Hilfe eines Transgens erfolgt, das einen chimären Antigenrezeptor (Chimeric Antigen Receptor, CAR) codiert, um BCMA-exprimierende Zellen zu erkennen und zu eliminieren. BCMA wird hauptsächlich auf der Oberfläche von Zellen der B-Linie des Multiplem Myeloms sowie von B-Zellen im Spätstadium und Plasmazellen exprimiert. Das CAR-Protein in CARVYKTI besteht aus zwei gegen BCMA gerichteten Einzeldomänenantikörpern mit hoher Avidität für humanes BCMA, einer kostimulatorischen Domäne 4-1BB und einer zytoplasmatischen Signaldomäne CD3-zeta (CD3ζ). Bei Bindung an BCMA exprimierende Zellen übermittelt der CAR ein Signal, welches die Aktivierung und Expansion der T-Zellen und die Eliminierung von Zielzellen fördert.
In-vitro-Experimente mit Kokulturen zeigten, dass die durch Ciltacabtagen-Autoleucel vermittelte Zytotoxizität und die Zytokinfreisetzung (Interferon-gamma, [IFN-γ], Tumornekrosefaktor [TNF-α], Interleukin [IL]-2) BCMA-abhängig waren.
Pharmakodynamik
Nach einer Einzelinfusion von CARVYKTI geht die Expansion CARpositiver T-Zellen mit Verringerungen von löslichem BCMA im Serum, des M-Gradienten und/oder freier Leichtketten im Serum einher. Bei allen Patienten wurde nach der Infusion ein Anstieg der Konzentrationen von IL-6, IL-10, IFN-γ und IL-2-Rezeptor alpha festgestellt, der an Tag 7 bis 14 einen Höchstwert erreichte. Generell gingen die Serumkonzentrationen aller Zytokine innerhalb von 2 bis 3 Monaten nach der Infusion wieder bis auf die Anfangswerte zurück.
Immunogenität
Mithilfe eines validierten Assays zum Nachweis von Bindungsantikörpern gegen CARVYKTI wurde die Immunogenität von CARVYKTI vor der Infusion sowie zu mehreren Zeitpunkten nach der Infusion untersucht. In der Studie MMY2001 waren 19 von 97 Patienten (19,6%) positiv auf Anti-CAR-Antikörper. In der Studie MMY3002 waren 37 von 176 Patienten (21%) positiv auf Anti-CAR-Antikörper.
Es gab keine eindeutigen Hinweise darauf, dass die Anti-CAR-Antikörper die Kinetik von CARVYKTI bei der anfänglichen Expansion und Persistenz, die Wirksamkeit oder die Sicherheit beeinträchtigen.
Klinische Wirksamkeit
Studie MMY2001
MMY2001 war eine offene Studie zur Beurteilung von CARVYKTI zur Behandlung von Patienten mit rezidiviertem oder refraktärem Multiplem Myelom, die vorgängig einen Proteasom-Inhibitor, ein immunmodulatorisches Agens und einen Anti-CD38-Antikörper erhalten hatten und bei denen die Krankheit während oder nach der letzten Therapie fortgeschritten war.
Insgesamt wurde bei 113 Patienten eine Leukapherese durchgeführt. Für jeden Patienten wurde CARVYKTI hergestellt. Sechzehn Patienten wurden nicht mit CARVYKTI behandelt (n = 12 nach Leukapherese und n = 4 nach der lymphodepletierenden Chemotherapie), da entweder der Patient seine Einwilligung zurückzog (n = 5), eine Krankheitsprogression aufgetreten war (n = 2) oder der Patient verstarb (n = 9).
Von den 97 behandelten Patienten betrug der mediane Zeitraum vom Tag nach Eingang des Leukapheresematerials in der Herstellungseinrichtung bis zur Freigabe des Produkts für die Infusion 29 Tage (Bereich: 23 bis 64 Tage), und der mediane Zeitraum von der anfänglichen Leukapherese bis zur Infusion von CARVYKTI betrug 47 Tage (Bereich: 41 bis 167 Tage).
Nach der Leukapherese und vor der Infusion von CARVYKTI erhielten 73 der 97 behandelten Patienten (75%) eine Überbrückungstherapie. Die am häufigsten als Überbrückungstherapie angewendeten Wirkstoffe (≥20% der Patienten) waren Dexamethason: 62 Patienten (64%), Bortezomib: 26 Patienten (27%), Cyclophosphamid: 22 Patienten (23%) und Pomalidomid: 21 Patienten (22%).
CARVYKTI wurde als einzelne intravenöse Infusion 5 bis 7 Tage nach Beginn einer lymphodepletierenden Chemotherapie (täglich Cyclophosphamid 300 mg/m2 intravenös und täglich Fludarabin 30 mg/m2 intravenös über 3 Tage) gegeben. Siebenundneunzig Patienten erhielten CARVYKTI in einer medianen Dosis von 0,71 × 106 CAR-positiven lebensfähigen T-Zellen/kg (Bereich: 0,51 bis 0,95 × 106 Zellen/kg). Alle Patienten wurden für die Infusion von CARVYKTI und für einen Zeitraum von mindestens 10 Tagen danach stationär aufgenommen.
Von den 113 Patienten, bei denen eine Leukapherese durchgeführt wurde, waren 58% männlich, 74% waren kaukasisch und 15% Afroamerikaner. Das mediane Alter der Patienten betrug 62 Jahre (Bereich: 29 bis 78 Jahre). Die Patienten hatten im Median 5 (Bereich: 3 bis 18) vorgängige Therapielinien erhalten, und 88% der Patienten hatten vorgängig eine Transplantation autologer Stammzellen (ASCT) erhalten. Neunundneunzig Prozent der Patienten waren gegen ihre letzte vorgängige Therapielinie refraktär, und 89% waren gegen einen Proteasom-Inhibitor (PI), ein immunmodulatorisches Agens und einen Anti-CD38-Antikörper refraktär.
Von den 97 behandelten Patienten waren 59% männlich, 71% waren kaukasisch und 18% waren Schwarz oder Afroamerikaner. Das mediane Alter der Patienten betrug 61 Jahre (Bereich: 43 bis 78 Jahre). Die Patienten hatten im Median 6 (Bereich: 3 bis 18) vorgängige Therapielinien erhalten, und 90% der Patienten hatten vorgängig eine Transplantation autologer Stammzellen (ASCT) erhalten. Neunundneunzig Prozent der Patienten waren gegen ihre letzte vorgängige Therapielinie refraktär, und 88% waren gegen einen Proteasom-Inhibitor (PI), ein immunmodulatorisches Agens und einen Anti-CD38-Antikörper refraktär.
Patienten mit bekannter aktiver oder anamnestisch erfasster signifikanter Erkrankung des Zentralnervensystems (ZNS), einschliesslich eines Multiplen Myeloms mit Manifestation im ZNS, allogener Stammzelltransplantation innerhalb von 6 Monaten vor der Apherese oder laufender Behandlung mit Immunsuppressiva, einer Kreatininclearance < 40 ml/min, einer absoluten Lymphozytenkonzentration < 300/µl, einem Anstieg der Lebertransaminasen auf über das 3-Fache der oberen Normgrenze, einer Ejektionsfraktion < 45% oder mit aktiver schwerwiegender Infektion wurden von der Teilnahme an der Studie ausgeschlossen.
Die Wirksamkeitsergebnisse beruhten auf der Gesamtansprechrate, die von einem unabhängigen Bewertungsgremium nach IMWG-Kriterien beurteilt wurde (siehe Tabelle 5).
Tabelle 5: Wirksamkeitsergebnisse für Studie MMY2001
|
|
Alle behandelten Patienten (n = 97)
|
Alle Patienten mit Leukapherese (n = 113)
| |
Gesamtansprechrate (Overall Response Rate (sCRa + VGPR + PR), n (%)
|
95 (97,9)
|
95 (84,1)
| |
95%-KI (%)
|
(92,7; 99,7)
|
(76,0; 90,3)
| |
Stringentes komplettes Ansprechen (sCRa) n (%)
|
80 (82,5)
|
80 (70,8)
| |
Sehr gutes partielles Ansprechen (VGPR) n (%)
|
12 (12,4)
|
12 (10,6)
| |
Partielles Ansprechen PR) n (%)
|
3 (3,1)
|
3 (2,7)
| |
Dauer des Ansprechens (DOR)b
| |
Anzahl der Responder
DOR (Monate): Median (95%-KI):
|
95
NE (23,3; NE)
|
-
| |
Anzahl der Responder mit sCRa
DOR, wenn das beste Ansprechen sCRa ist (Monate): Median (95%-KI)
|
80
NE (28,3; NE)
|
-
| |
Anzahl der Responder mit VGPR oder besser DOR, wenn das beste Ansprechen VGPR oder besser ist (Monate): Median (95%-KI):
|
92
NE (24,4; NE)
|
-
| |
Dauer des Zeitraums bis zum Ansprechen (Monate)
| |
Anzahl der Responder
Median
Bereich
|
95
0,95
(0,9; 10,7)
|
-
| |
Dauer des Zeitraums bis zur sCRa (Monate)
| |
Anzahl der Responder mit sCRa
Median
Bereich
|
80
2,89
(0,9; 17,8)
|
-
|
Hinweise: Basierend auf einer medianen Nachbeobachtungsdauer von 27,7 Monaten.
a Bei jeder vollständigen Remission handelte es sich um ein stringentes CR.
NE = not estimable (nicht abschätzbar)
b Die geschätzte DOR-Rate betrug 60,3% (95%-KI: 49,6%; 69,5%) nach 24 Monaten und 51,2% (95%-KI: 39,0%; 62,1%) nach 30 Monaten.
Tabelle 6: Zusammenfassung der Häufigkeit von MRD-Negativität
|
|
Alle behandelten Patienten (n = 97)
|
Alle Patienten mit Leukapherese (n = 113)
| |
Anteil der Patienten mit MRD-Negativität, n (%)
|
56 (57,7)
|
56 (49,6)
| |
95%-KI (%)
|
(47,3; 67,7)
|
(40.0, 59,1)
| |
MRD-negative Patienten mit sCR n (%)a
|
42 (43,3)
|
42 (37,2)
| |
95%-KI (%)
|
33,3; 53,7
|
(28,3; 46,8)
| |
|
Auswertbare Patienten (n = 61)
| |
Anteil der Patienten mit MRD-Negativität, n (%)
|
56 (91,8)
|
-
| |
95%-KI (%)
|
(81,9; 97,3)
|
-
|
MRD = Minimale Resterkrankung
Hinweise: Basierend auf einer medianen Nachbeobachtungsdauer von 27.7 Monaten.
a Es wurden nur MRD-Beurteilungen (10 -5 Testschwelle) innerhalb von 3 Monaten nach Erreichen einer CR/sCR bis Tod/Progression/Anschlusstherapie (ausschliesslich) berücksichtigt. Bei jeder vollständigen Remission handelte es sich um ein stringentes CR.
Bei einer medianen Nachbeobachtungsdauer von 27,7 Monaten wurde das mediane progressionsfreie Überleben (Progression Free Survival, PFS) nicht erreicht (95%-KI: 24,5, nicht abschätzbar). Die PFS-Rate nach 12 Monaten (95%-KI) betrug 76,3% (66,5%, 83,6%). Die 24-Monats-PFS-Rate (95%-KI) betrug 62,7% (52,2%; 71,5%).
Bei Patienten mit sCR (bei jeder vollständigen Remission handelte es sich um ein stringentes CR) wurde das mediane PFS nicht erreicht (95%-KI: 30,1%, nicht abschätzbar), wobei die geschätzte PFS-Rate nach 12 Monaten 88,8% (95%-KI: 79,5%, 94,0%) betrug. Die 24-Monats-PFS-Rate betrug 73,5% (95%-KI: 62,3%; 81,9%).
Das mediane Gesamtüberleben (Overall Survival, OS) wurde nicht erreicht (95%-KI: nicht abschätzbar, nicht abschätzbar). Die OS-Rate nach 12 Monaten betrug 87,6% (95%-KI: 79,2%, 92,8%). Die 24-Monats-OS-Rate betrug 76,2% (95%-KI: 66,5%; 83,5%).
Die gesundheitsbezogene Lebensqualität (Health-related quality of life, HRQoL) wurde zu Beginn der Studie (n = 63) und nach der Infusion mithilfe des Fragebogens EORTC QLQ-C30 erfasst. Die adjustierte mittlere (95%-KI) Veränderung gegenüber dem Anfangsergebnis auf der Schmerz-Subskala des EORTC QLQ-C30 betrug -1,9 (-8,5; -4,6) an Tag 7, -9,9 (-16,5; -3,3) an Tag 28, -6,3 (-12,9; -0,4) an Tag 56, -9,4 (-16,3; -2,5) an Tag 78 und -10,5 (-17,3; -3,8) an Tag 100, was insgesamt auf eine Verringerung der Schmerzen nach der Infusion von CARVYKTI hindeutete. Bei 72,2% der Patienten waren hinsichtlich des Ergebnisses auf der Schmerz-Subskala an Tag 100 klinisch bedeutsame Verbesserungen festzustellen, bei 53,8% war dies hinsichtlich des Ergebnisses auf der Fatigue-Subskala der Fall, bei 57,7% hinsichtlich des Ergebnisses auf der Subskala für die körperliche Funktion und bei 53,7% hinsichtlich des Ergebnisses auf der Subskala für den Gesundheitszustand insgesamt.
Studie MMY3002
MMY3002 ist eine randomisierte, offene, multizentrische Phase-3-Studie zur Bewertung der Wirksamkeit von CARVYKTI für die Behandlung von Patienten mit rezidiviertem und Lenalidomid-refraktärem multiplem Myelom, die zuvor mindestens eine vorherige Therapielinie mit einem Proteasom-Inhibitor und einem immunmodulatorischen Wirkstoff erhalten haben. Insgesamt wurden 419 Patienten randomisiert und erhielten entweder eine Abfolge von Leukapherese, Überbrückungstherapie, Lymphozytendepletion und CARVYKTI (n = 208) oder die Standardbehandlung, die nach Ermessen des Arztes entweder Daratumumab, Pomalidomid und Dexamethason oder Bortezomib, Pomalidomid und Dexamethason (n = 211) beinhaltete.
Patienten mit bekannter aktiver oder anamnestisch erfasster Beteiligung des zentralen Nervensystems, Patienten mit klinischen Anzeichen einer Meningealbeteiligung des multiplen Myeloms und Patienten mit Morbus Parkinson oder einer anderen neurodegenerativen Erkrankung in der Vorgeschichte wurden von der Studie ausgeschlossen.
Von den 419 Patienten, die randomisiert wurden (208 für CARVYKTI und 211 für die Standardbehandlung), waren 57% männlich, 75% waren kaukasisch, 3% waren Schwarz oder afroamerikanisch und 7% waren hispanisch oder lateinamerikanisch. Das mediane Alter der Patienten betrug 61 Jahre (Bereich: 27 bis 80 Jahre). Die Patienten hatten im Median 2 vorgängige Therapielinien (Bereich: 1 bis 3) erhalten, und 85% der Patienten hatten vorgängig eine Transplantation autologer Stammzellen (ASCT) erhalten. Neunundneunzig Prozent der Patienten waren gegen ihre letzte vorgängige Therapielinie refraktär. Achtundvierzig Prozent waren gegen einen Proteasom-Inhibitor (PI) und 100% gegen ein immunmodulatorisches Agens refraktär.
Bei allen 208 Patienten, die dem CARVYKTI-Arm zugeteilt wurden, wurde eine Leukapherese durchgeführt. Nach der Leukapherese und vor der Verabreichung von CARVYKTI erhielten alle 208 randomisierten Patienten die gemäss Studienprotokoll vorgesehene Überbrückungstherapie (Standardbehandlung). Von diesen 208 Patienten wurden 12 aufgrund von Krankheitsprogression (n = 10) oder Tod (n = 2) nicht mit CARVYKTI behandelt; bei 20 Patienten trat vor der Infusion mit CARVYKTI eine Krankheitsprogression auf, sie konnten CARVYKTI jedoch als weiterführende Therapie erhalten.
Bei den 176 Patienten, die CARVYKTI als Studienbehandlung erhielten, betrug der mediane Zeitraum vom Tag nach Eingang des Leukapheresematerials in der Herstellungseinrichtung bis zur Freigabe des Produkts für die Infusion 44 Tage (Bereich: 25 bis 127 Tage) und der mediane Zeitraum von der anfänglichen Leukapherese bis zur Infusion von CARVYKTI betrug 79 Tage (Bereich: 45 bis 246 Tage).
CARVYKTI wurde als einmalige intravenöse Infusion 5 bis 7 Tage nach Beginn einer lymphodepletierenden Chemotherapie (täglich Cyclophosphamid 300 mg/m2 intravenös und täglich Fludarabin 30 mg/m2 intravenös über 3 Tage) mit einer medianen Dosis von 0,71 × 106 CAR-positiven lebensfähigen T-Zellen/kg (Bereich: 0,39 bis 1,07 × 106 Zellen/kg) verabreicht.
Der primäre Messwert für die Wirksamkeit war das progressionsfreie Überleben (PFS), das anhand des Intent-To-Treat-Analysesets analysiert wurde (siehe Tabelle 7 und Abbildung 1). Nach einer medianen Nachbeobachtungszeit von 15,9 Monaten betrug das mediane PFS 11,8 Monate (95%-KI: 9,7; 13,8) im Vergleichsarm und war nicht abschätzbar (95%-KI: 22,8, nicht abschätzbar) im CARVYKTI-Arm (Hazard Ratio: 0,26 [95%-KI: 0,18; 0,38]). Die geschätzte PFS-Rate nach 12 Monaten betrug 75,9% (95%-KI: 69,4%; 81,1%) im CARVYKTI-Arm und 48,6% (95%-KI: 41,5%; 55,3%) im Vergleichsarm. Im CARVYKTI-Arm wurde die geschätzte mediane Ansprechdauer (DOR) nicht erreicht. Im Vergleichsarm betrug die geschätzte mediane Ansprechdauer 16,6 Monate (95%-KI: 12,9; nicht abschätzbar).
Tabelle 7: Wirksamkeitsergebnisse für Studie MMY3002 (Intent-To-Treat-Analyseset)
|
|
CARVYKTI (n = 208)
|
Vergleichsbehandlung
(n = 211)
| |
Progressionsfreies Überlebena
|
|
| |
Anzahl der Ereignisse, n (%)
|
65 (31,3)
|
122 (57,8)
| |
Median, Monate [95%-KI]b
|
NA [22,8; NE]
|
11,8 [9,7; 13,8]
| |
Hazard Ratio [95%-KI]c
|
0,26 [0,18; 0,38]
| |
p-Wertd
|
< 0,0001
| |
Rate eines vollständigen Ansprechens oder bessera, % [95%-KI]
|
73,1 [66,5; 79,0]
|
21,8 [16,4; 28,0]
| |
p-Werte
|
< 0,0001
| |
Gesamtansprechrate (ORR)a, % [95%-KI]
|
84,6 [79,0; 89,2]
|
67,3 [60,5; 73,6]
| |
p-Werte
|
< 0,0001
| |
MRD-Gesamtnegativitätsrate, % [95%-KI]
|
60,6 [53,6; 67,3]
|
15,6 [11,0; 21,3]
| |
p-Wertf
|
< 0,0001
|
NE = not estimable (nicht abschätzbar); KI = Konfidenzintervall; MRD = Minimale Resterkrankung
Hinweis: Basierend auf einer medianen Nachbeobachtungszeit von 15,9 Monaten.
a Gemäss dem Konsens der International Myeloma Working Group (IMWG), bewertet mittels eines computergestützten Algorithmus
b Kaplan-Meier-Schätzung
c Basierend auf einem stratifizierten Cox-Proportional-Hazard-Modell, das ausschliesslich PFS-Ereignisse berücksichtigt, die mehr als 8 Wochen nach der Randomisierung auftraten. Eine Hazard Ratio < 1 gibt einen Vorteil für den CARVYKTI-Arm an. Bei allen stratifizierten Analysen erfolgte die Stratifizierung auf Grundlage der Entscheidung des Prüfarztes (PVd oder DPd), des ISS-Stagings (I, II, III) und der Anzahl der vorherigen Therapielinien (1 vs. 2 oder 3), die randomisiert wurden.
d Stratifizierter gewichteter Log-Rank-Test (Gewichtung von 0 in der Log-Rank-Statistik bei den ersten 8 Wochen nach der Randomisierung und 1 danach)
e Stratifizierter Cochran-Mantel-Haenszel-Chi-Quadrat-Test
f Exakter Test nach Fisher
Abbildung 1: Kaplan-Meier-Kurve des PFS in Studie MMY3002 (Intent-To-Treat-Analyseset)
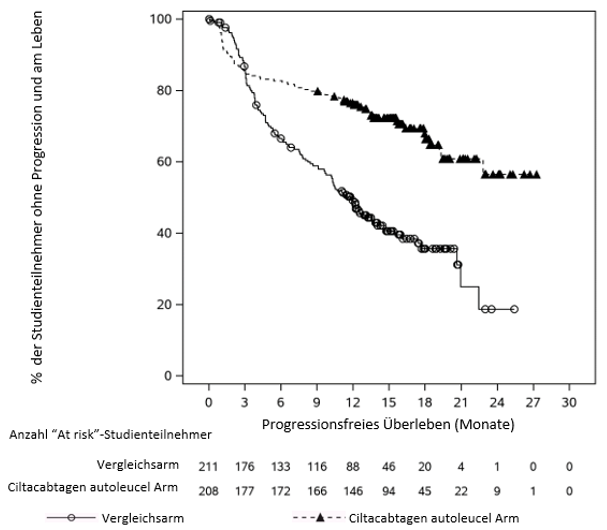
Hinweis: Das Intent-to-treat-Analyseset besteht aus Patienten, die in die Studie randomisiert wurden.
Bei den 176 Patienten, die CARVYKTI als Studienbehandlung erhielten, war das mediane PFS nicht abschätzbar (95%-KI: nicht abschätzbar, nicht abschätzbar), wobei die 12-Monats-PFS-Rate 89,7% betrug. Die ORR bei diesen Patienten betrug 99,4% (95%-KI: 96,9%; 100,0%). Die CR/sCR-Rate betrug 86,4% (95%-KI: 80,4%; 91,1%).Bei den 20 Patienten, bei denen es zu einer frühzeitigen und schnellen Krankheitsprogression kam und die CARVYKTI als weiterführende Therapie erhielten, betrug das mediane PFS nach der Infusion von CARVYKTI 7,39 Monate (95%-KI: 1,61; nicht abschätzbar) mit einer 12-Monats-PFS-Rate von 39,4% (95%-KI: 18,6; 59,7), einer ORR von 65% (95%-KI: 40,8%; 84,6%) und einer CR/sCR von 40% (95%-KI: 19,1%; 63,9%). In der zweiten Zwischenanalyse (IA2) der randomisierten Studie MMY3002 (Stichtag 1. Mai 2024) wurde bei einer medianen Nachbeobachtungszeit von 33,6 Monaten das mediane Gesamtüberleben (Overall Survival, OS) in keinem der Studienarme erreicht. Die HR (CARVYKTI versus Vergleichstherapie) betrug 0,55 (95%-KI: 0,39; 0,79), und der p-Wert von 0,0009 überschritt die vordefinierte Abbruchgrenze von 0,0108. Eine einmalige Infusion von CARVYKTI zeigt eine statistisch signifikante und klinisch relevante Verbesserung des OS mit einer 45%igen Verringerung des Mortalitätsrisikos bei den mit CARVYKTI behandelten Teilnehmern verglichen mit der Vergleichstherapie. Die 30-Monats-OS-Rate betrug 76,4% (95%-KI: 70,0; 81,6) im CARVYKTI-Arm und 63,8% (95%-KI: 56,9; 69,9) für die Vergleichstherapie. Die aktualisierten OS-Resultate sind in Tabelle 8 und in Abbildung 2 dargestellt.
Tabelle 8: Zusammenfassung der aktualisierten Resultate des Gesamtüberlebens (Stichtag 1. Mai 2024); für Studie MMY3002 (IA2) (Intent-To-Treat-Analyseset)
|
|
CARVYKTI
(N = 208)
|
Vergleichsbehandlung
(N = 211)
| |
Gesamtüberleben (OS)
|
|
| |
Anzahl der Ereignisse (%)
|
50 (24,0 %)
|
83 (39,3 %)
| |
Anzahl der Zensierten (%)
|
158 (76,0 %)
|
128 (60,7 %)
| |
Kaplan-Meier-Schätzung (Monate)
|
|
| |
Median (95%-KI)
|
NE (NE; NE)
|
NE (37,75; NE)
| |
|
|
| |
p-Werta
|
0,0009
|
| |
Hazard Ratio (95%-KI)b
|
0,55 (0,39; 0,79)
|
| |
|
|
| |
12-Monats-Überlebensrate, % (95%-KI)
|
84,1 (78,4; 88,4)
|
83,6 (77,9; 88,0)
| |
24-Monats-Überlebensrate, % (95%-KI)
|
78,8 (72,6; 83,8)
|
66,2 (59,3; 72,2)
| |
30-Monats-Überlebensrate, % (95%-KI)
|
76,4 (70,0; 81,6)
|
63,8 (56,9; 69,9)
|
Erläuterung der Abkürzungen: NE = nicht abschätzbar; KI = Konfidenzintervall
a Der p-Wert basiert auf dem Log-Rank-Test, der nach Wahl des Prüfarztes (PVd oder DPd), ISS-Stadium (I, II, III) und Anzahl der vorherigen Therapielinien (1 versus 2 oder 3) gemäss der Randomisierung stratifiziert ist.
b Hazard Ratio und 95%-KI aus einem Cox-Proportional-Hazards-Modell mit Behandlung als einziger explanatorischer Variable und nach Wahl des Prüfers (PVd oder DPd), ISS-Stadium (I, II, III) und Anzahl der vorherigen Therapielinien (1 versus 2 oder 3) gemäss der Randomisierung stratifiziert. Eine Hazard Ratio <1 weist auf einen Vorteil für den CARVYKTI-Arm hin.
Hinweis: Das Intent-to-Treat-Analyseset besteht aus Patienten, die in der Studie randomisiert wurden.
Abbildung 2: Kaplan-Meier-Kurve der aktualisierten Ergebnisse des Gesamtüberlebens (Stichtag: 1. Mai 2024); Intent-to-Treat-Analyseset (Studie MMY3002 (IA2))

Erläuterung der Abkürzungen: Arm A = PVd oder DPd; Arm B = Abfolge von Apherese, Überbrückungstherapie (PVd oder DPd), Konditionierung (Cyclophosphamid und Fludarabin) und Cilta-Cel-Infusion
Erläuterung der Abkürzungen: PVd = Pomalidomid-Bortezomib-Dexamethason; DPd = Daratumumab-Pomalidomid-Dexamethason.
Hinweis: Der Intent-to-Treat-Analysensatz besteht aus Patienten, die in der Studie randomisiert wurden.
Die Resultate der Gesamtsymptomskala des Multiple Myeloma Symptom and Impact Questionnaire (MySIm-Q), einem Fragebogen zur Beurteilung der von den Patienten gemeldeten Ergebnisse (Patient Reported Outcomes) zum Schweregrad von Schmerzen, Neuropathie, Müdigkeit, Verdauungs- und kognitiven Symptomen, zeigten eine Verzögerung der Verschlechterung der Symptome unter CARVYKTI. Die mediane Zeit bis zu einer anhaltenden Verschlechterung der Symptome des Multiplen Myeloms betrug 34,3 Monate (95%-KI: 32,2; nicht abschätzbar) unter der Vergleichstherapie und war nicht abschätzbar unter CARVYKTI (95%-KI: nicht abschätzbar; nicht abschätzbar) (HR = 0,38 [95%-KI: 0,24; 0,61]; p <0,0001).
|