ZusammensetzungWirkstoffe
Finerenonum.
Hilfsstoffe
Cellulosum microcristallinum, Carmellosum natricum conexum, Lactosum monohydricum, Magnesii stearas, Hypromellosum, Natrii laurilsulfas.
Filmüberzug: Hypromellosum, Talcum, Titanii dioxidum (E 171), Ferrum oxydatum rubrum (E 172) (nur in der 10 mg Dosisstärke enthalten), Ferrum oxydatum flavum (E 172) (nur in der 20 mg Dosisstärke enthalten).
Eine 10 mg Filmtablette enthält 0.44 mg Natrium und 42.75 mg Lactose.
Eine 20 mg Filmtablette enthält 0.47 mg Natrium und 38.00 mg Lactose.
Indikationen/AnwendungsmöglichkeitenKerendia ist indiziert zur Verzögerung der Progression einer chronischen Nierenerkrankung bei erwachsenen Patienten mit Typ-2-Diabetes mellitus (siehe Rubriken «Dosierung/Anwendung» und «Klinische Wirksamkeit»).
Für Studienergebnisse zu Auswirkungen auf kardiovaskuläre Ereignisse siehe Rubrik «Klinische Wirksamkeit».
Dosierung/AnwendungÜbliche Dosierung
Die empfohlene Zieldosis von Kerendia beträgt 20 mg einmal täglich (entspricht der maximalen Tagesdosis).
Therapieeinleitung
Kerendia sollte in Ergänzung zur Standardtherapie angewandt werden (siehe Rubrik «Klinische Wirksamkeit»).
Eine Therapieeinleitung mit Kerendia wird empfohlen, wenn der Serumkaliumspiegel ≤4.8 mmol/l beträgt. Zur Überwachung des Serumkaliums siehe «Erhaltungstherapie».
Wenn der Serumkaliumspiegel > 4.8 bis 5.0 mmol/l beträgt, kann je nach Patientenmerkmalen und Serumkaliumspiegel eine Behandlung mit Kerendia in Betracht gezogen werden, wobei in den ersten 4 Wochen eine zusätzliche Überwachung des Serumkaliums erfolgen sollte (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Wenn der Serumkaliumspiegel > 5.0 mmol/l beträgt, wird eine Behandlung mit Kerendia nicht empfohlen (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Zur Bestimmung der Anfangsdosis wird die geschätzte glomeruläre Filtrationsrate (eGFR) gemessen. Die Anfangsdosis von Kerendia beträgt:
·20 mg einmal täglich, wenn eGFR ≥60 ml/min/1.73 m2
·10 mg einmal täglich, wenn eGFR ≥25 bis < 60 ml/min/1.73 m2
Eine Therapieeinleitung mit Kerendia wird bei Patienten mit eGFR < 25 ml/min/1.73 m2 aufgrund der begrenzten klinischen Erfahrungen nicht empfohlen.
Erhaltungstherapie
Vier Wochen nach Einleitung, Wiederbeginn oder Auftitration der Behandlung mit Kerendia müssen Serumkalium und eGFR erneut gemessen werden. Um die Fortsetzung der Behandlung mit Kerendia und die Dosisanpassung zu bestimmen, siehe Tabelle 1. Danach soll die Messung des Serumkaliums periodisch und bei Bedarf auf der Grundlage der Patientenmerkmale und des Serumkaliumspiegels wiederholt werden (siehe Rubriken «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»).
Tabelle 1: Fortsetzung (4 Wochen nach Einleitung, Wiederbeginn oder Auftitration) der Behandlung mit Kerendia und Dosisanpassung
|
Serumkalium (mmol/l)
|
Dosierung von Kerendia (ab Woche 5)
| |
≤4.8
|
Dosierung von 20 mg einmal täglich beibehalten.
Bei Patienten mit einer Dosierung von 10 mg einmal täglich die Dosierung auf 20 mg einmal täglich steigern, wenn eGFR nicht um > 30 % gegenüber der vorherigen Messung abgenommen hat.
| |
> 4.8–5.5
|
Dosierung beibehalten.
| |
> 5.5
|
Gabe von Kerendia aussetzen.
Gabe mit 10 mg einmal täglich wieder aufnehmen, wenn Serumkalium ≤5.0 mmol/l.
|
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit schwerer Leberinsuffizienz (Child-Pugh C) ist eine Behandlung mit Kerendia zu vermeiden (siehe Rubriken «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Bei Patienten mit leichter bis mittelschwerer Leberinsuffizienz ist keine initiale Dosisanpassung erforderlich (Child-Pugh A oder B) (siehe Rubrik «Pharmakokinetik»).
Bei Patienten mit mittelschwerer Leberinsuffizienz (Child-Pugh B) ist eine zusätzliche Überwachung des Serumkaliums in Betracht zu ziehen und die Überwachung je nach Patientenmerkmalen anzupassen (siehe Rubriken «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Therapieeinleitung
Bei Patienten mit eGFR ≥25 bis < 60 ml/min/1.73 m2 beträgt die Anfangsdosis von Kerendia 10 mg einmal täglich.
Bei Patienten mit eGFR < 25 ml/min/1.73 m2 wird eine Therapieeinleitung mit Kerendia aufgrund der begrenzten klinischen Erfahrungen nicht empfohlen (siehe Rubriken «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Erhaltungstherapie
Bei Patienten mit leichter, mittelschwerer oder schwerer Nierenfunktionsstörung erfolgt die Weiterbehandlung mit Kerendia und die Dosisanpassung in Abhängigkeit vom Serumkalium.
Um zu bestimmen, ob die Anfangsdosis auf die empfohlene Tagesdosis erhöht werden kann, ist die eGFR 4 Wochen nach Einleitung der Behandlung zu bestimmen. Siehe Tabelle 1 und «Erhaltungstherapie».
Bei Patienten mit terminaler Niereninsuffizienz (eGFR < 15 ml/min/1.73 m2) ist die Behandlung mit Kerendia mit Blick auf den Serumkaliumspiegel vorsichtig fortzusetzen, da die klinischen Erfahrungen begrenzt sind (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Kombinationstherapie
Bei allen Patienten, die bereits mit einem mässigen CYP3A4-Inhibitor behandelt werden, ist die Therapie mit Kerendia mit einer Anfangsdosis von 10 mg einmal täglich einzuleiten (siehe Rubriken «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»).
Körpergewicht
Eine Dosisanpassung auf Basis des Körpergewichts ist nicht erforderlich (siehe Rubrik «Pharmakokinetik»).
Ältere Patienten
Eine Dosisanpassung auf Basis des Alters ist nicht erforderlich (siehe Rubrik «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Kerendia bei Patienten unter 18 Jahren wurde nicht untersucht.
Verspätete Dosisgabe
Eine versäumte Dosis sollte so bald wie möglich eingenommen werden, jedoch nur am selben Tag. Um eine versäumte Dosis nachzuholen, sollte nicht die doppelte Dosis am selben Tag eingenommen werden.
Art der Anwendung
Zur oralen Anwendung.
Die Tabletten können mit einem Glas Wasser unabhängig von den Mahlzeiten eingenommen werden (siehe Rubrik «Pharmakokinetik»).
Die Einnahme von Kerendia zusammen mit Grapefruits oder Grapefruitsaft ist zu vermeiden (siehe Rubriken «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»).
Für Patienten, die nicht in der Lage sind, ganze Tabletten zu schlucken, können die Tabletten unmittelbar vor der Anwendung zerkleinert und mit Wasser oder dickflüssiger Kost (wie z.B. Apfelmus) eingenommen werden (siehe Rubrik «Pharmakokinetik»).
KontraindikationenKerendia ist kontraindiziert bei Patienten:
·die eine Begleitbehandlung mit starken CYP3A4-Inhibitoren haben (z.B. Itraconazol, Ketoconazol, Ritonavir, Nelfinavir, Cobicistat, Clarithromycin, Telithromycin und Nefazodon) (siehe Rubrik «Interaktionen»).
·mit Morbus Addison.
·mit einer Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe (siehe Rubriken «Zusammensetzung» und «Warnhinweise und Vorsichtsmassnahmen»).
Warnhinweise und VorsichtsmassnahmenHyperkaliämie
Hyperkaliämie wurde bei Patienten, die mit Kerendia behandelt wurden, beobachtet (siehe Rubrik «Unerwünschte Wirkungen»).
Einige Patienten tragen ein erhöhtes Risiko für die Entwicklung einer Hyperkaliämie. Risikofaktoren beinhalten erniedrigte eGFR, erhöhtes Serumkalium und frühere Episoden von Hyperkaliämie. Bei diesen Patienten sind häufigere Kontrollen in Betracht zu ziehen.
Eine Therapieeinleitung mit Kerendia wird nicht empfohlen, wenn der Serumkaliumspiegel > 5.0 mmol/l beträgt. Wenn der Serumkaliumspiegel > 4.8 bis 5.0 mmol/l beträgt, kann je nach Patientenmerkmalen und Serumkaliumspiegel eine Behandlung mit Kerendia in Betracht gezogen werden, wobei in den ersten 4 Wochen eine zusätzliche Überwachung des Serumkaliums erfolgen sollte (siehe Rubrik «Dosierung/Anwendung»).
Die Gabe von Kerendia bei behandelten Patienten ist auszusetzen, wenn der Serumkaliumspiegel > 5.5 mmol/l beträgt. Lokale Leitlinien für die Behandlung von Hyperkaliämie sind zu beachten. Die Behandlung mit Kerendia wird mit einer Dosierung von 10 mg einmal täglich wiederaufgenommen, wenn das Serumkalium ≤5.0 mmol/l beträgt (siehe Rubrik «Dosierung/Anwendung»).
Bei allen Patienten müssen Serumkalium und eGFR vier Wochen nach Einleitung, Wiederbeginn oder Auftitration der Behandlung mit Kerendia erneut gemessen werden. Danach soll die Messung des Serumkaliums periodisch und bei Bedarf auf der Grundlage der Patientenmerkmale und des Serumkaliumspiegels wiederholt werden (siehe Rubrik «Dosierung/Anwendung»).
Begleitbehandlung
Das Risiko für eine Hyperkaliämie kann auch bei Einnahme von Begleitmedikamenten, die den Serumkaliumspiegel erhöhen können, ansteigen (siehe Rubrik «Interaktionen»). Siehe auch «Gleichzeitige Anwendung von Substanzen, welche die Finerenon-Exposition beeinflussen».
Die gleichzeitige Anwendung von Kerendia mit folgenden Arzneimitteln ist zu vermeiden:
·kaliumsparende Diuretika (z.B. Amilorid, Triamteren)
·andere Mineralokortikoid-Rezeptor-Antagonisten (MRA) (z.B. Eplerenon, Esaxerenon, Spironolacton, Canrenon)
Kerendia ist mit Vorsicht anzuwenden und der Serumkaliumspiegel ist zu überwachen, wenn Kerendia gleichzeitig mit folgenden Arzneimitteln eingenommen wird:
·Kaliumsupplemente
·Trimethoprim oder Trimethoprim-Sulfamethoxazol. Eine vorübergehende Unterbrechung der Behandlung mit Kerendia kann erforderlich sein.
Nierenfunktionsstörung
Das Risiko für eine Hyperkaliämie steigt mit abnehmender Nierenfunktion. Eine kontinuierliche Überwachung der Nierenfunktion sollte nach Bedarf gemäss der Standardpraxis durchgeführt werden (siehe Rubrik «Dosierung/Anwendung»).
Eine Therapieeinleitung mit Kerendia wird bei Patienten mit eGFR < 25 ml/min/1.73 m2 aufgrund der begrenzten klinischen Erfahrungen nicht empfohlen (siehe Rubriken «Dosierung/Anwendung» und «Pharmakokinetik»).
Bei Patienten mit terminaler Niereninsuffizienz (eGFR < 15 ml/min/1.73 m2) ist die Behandlung mit Kerendia vorsichtig mit Blick auf den Serumkaliumspiegel fortzusetzen, da die klinischen Erfahrungen begrenzt sind (siehe Rubrik «Dosierung/Anwendung»).
Leberfunktionsstörung
Patienten mit schwerer Leberinsuffizienz (Child-Pugh C) wurden nicht untersucht (siehe Rubrik «Pharmakokinetik»). Aufgrund eines erwarteten erheblichen Anstiegs der Finerenon-Exposition ist die Anwendung von Kerendia bei Patienten mit schwerer Leberinsuffizienz zu vermeiden (siehe Rubrik «Dosierung/Anwendung»).
Aufgrund eines Anstiegs der Finerenon-Exposition, ist bei Patienten mit mittelschwerer Leberinsuffizienz (Child-Pugh B) eine zusätzliche Überwachung des Serumkaliums in Betracht zu ziehen und die Überwachung je nach Patientenmerkmalen anzupassen (siehe Rubriken «Dosierung/Anwendung» und «Pharmakokinetik»).
Gleichzeitige Anwendung von Substanzen, welche die Finerenon-Exposition beeinflussen
Mässige und schwache CYP3A4-Inhibitoren
Bei gleichzeitiger Anwendung von Kerendia mit mässigen CYP3A4-Inhibitoren (z.B. Erythromycin und Verapamil) sowie schwachen CYP3A4-Inhibitoren (z.B. Amiodaron und Fluvoxamin) wird eine erhöhte Finerenon-Exposition erwartet (siehe Rubrik «Interaktionen»). Daher ist der Serumkaliumspiegel vor allem während der Einleitung der Behandlung mit oder bei Änderungen der Dosierung von Kerendia oder des CYP3A4-Inhibitors zu überwachen (siehe Rubrik «Dosierung/Anwendung»).
Starke und mässige CYP3A4-Induktoren
Die gleichzeitige Anwendung von Kerendia mit starken CYP3A4-Induktoren (z.B. Rifampicin, Carbamazepin, Phenytoin, Phenobarbital, Johanniskraut) oder mässigen CYP3A4-Induktoren (z.B. Efavirenz), die die Plasmakonzentrationen von Finerenon stark verringern und zu einer reduzierten therapeutischen Wirkung führen, ist zu vermeiden (siehe Rubrik «Interaktionen»). Die Anwendung eines alternativen Begleitmedikaments ohne oder mit schwachem Potenzial zur Induktion von CYP3A4 ist in Betracht zu ziehen.
Grapefruit
Die gleichzeitige Einnahme von Grapefruit oder Grapefruitsaft ist zu vermeiden, da dies die Plasmakonzentration von Finerenon erhöht (siehe Rubriken «Dosierung/Anwendung» und «Interaktionen»).
Embryofetale Toxizität
Kerendia darf während der Schwangerschaft nicht angewendet werden, es sei denn, der Nutzen für die Mutter und das Risiko für das ungeborene Kind wurden sorgfältig abgewogen. Wenn eine Frau während der Einnahme von Kerendia schwanger wird, ist sie über die potenziellen Risiken für das ungeborene Kind zu informieren. Frauen im gebärfähigen Alter sind darauf hinzuweisen, dass sie während der Behandlung mit Kerendia eine wirksame Verhütungsmethode anwenden müssen. Frauen sind darauf hinzuweisen, dass sie während der Behandlung mit Kerendia nicht stillen dürfen. Für weitere Informationen siehe Rubriken «Schwangerschaft, Stillzeit» und «Präklinische Daten».
Informationen zu den Hilfsstoffen
Kerendia beinhaltet Lactose. Patienten mit der seltenen hereditären Galactose-Intoleranz, völligem Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht anwenden.
Kerendia beinhaltet Natrium. Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Filmtablette, d.h. es ist nahezu «natriumfrei».
InteraktionenInteraktionsstudien wurden nur bei Erwachsenen durchgeführt. Finerenon wird fast ausschliesslich durch Cytochrom-P450- (CYP-)vermittelte oxidative Verstoffwechslung (hauptsächlich durch CYP3A4 [90 %] und zu einem geringen Anteil durch CYP2C8 [10 %]) eliminiert.
Wirkung anderer Arzneimittel auf Kerendia
Enzyminhibitoren
Starke CYP3A4-Inhibitoren
Die gleichzeitige Anwendung von Kerendia mit Itraconazol, Clarithromycin und anderen starken CYP3A4-Inhibitoren (z.B. Ketoconazol, Ritonavir, Nelfinavir (in der Schweiz nicht zugelassen), Cobicistat, Telithromycin (in der Schweiz nicht zugelassen) oder Nefazodon (in der Schweiz nicht zugelassen) ist kontraindiziert, da ein deutlicher Anstieg der Finerenon-Exposition zu erwarten ist (siehe Rubrik «Kontraindikationen»). Simulationen zeigen, dass die gleichzeitige Anwendung von Kerendia mit Itraconazol (200 mg zweimal täglich) die Finerenon-Exposition erhöht (geometrisches mittleres Verhältnis (GMR) und 90% Populationsintervall für AUC und Cmax: 6.31 [3.36-11.41] und 2.37 [1.76-3.31]). Clarithromycin (500 mg zweimal täglich) führt ebenfalls zum erwartungsgemässen Anstieg der Finerenon-Exposition (GMR und 90% Populationsintervall für AUC und Cmax: 5.28 [2.88-10.48] und 2.25 [1.76-2.98]).
Mässige CYP3A4-Inhibitoren
Die gleichzeitige Anwendung von Erythromycin (500 mg dreimal täglich), eines mässigen CYP3A4-Inhibitors, erhöhte die mittlere Finerenon-Exposition (GMR und 90% Konfidenzintervall für AUC und Cmax: 3.48 [3.02-4.02] und 1.88 [1.63-2.17]). Ein anderer mässiger CYP3A4-Inhibitor, Verapamil (240 mg-Tablette mit verzögerter Wirkstoffabgabe einmal täglich), erhöhte die mittlere Finerenon-Exposition (GMR und 90% Konfidenzintervall für AUC und Cmax: 2.70 [2.43-3.01] und 2.22 [1.88-2.62]). Da das Serumkalium damit ansteigen kann, muss der Serumkaliumspiegel überwacht werden (siehe Rubriken «Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»).
Schwache CYP3A4-Inhibitoren
Simulationen deuten darauf hin, dass Fluvoxamin (100 mg zweimal täglich), ein schwacher CYP3A4-Inhibitor, die Finerenon-Exposition erhöht (GMR und 90% Populationsintervall für AUC und Cmax: 1.57 [1.25-2.08] und 1.38 [1.18-1.64]). Da das Serumkalium damit ansteigen kann, muss der Serumkaliumspiegel überwacht werden (siehe Rubriken «Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»).
Grapefruit
Die gleichzeitige Aufnahme von Grapefruit oder Grapefruitsaft erhöht wahrscheinlich die Plasmakonzentration von Finerenon und sollte vermieden werden (siehe Rubriken «Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»).
Enzyminduktoren
Die gleichzeitige Anwendung von Kerendia mit Rifampicin und anderen starken CYP3A4-Induktoren (z.B. Carbamazepin, Phenytoin, Phenobarbital, Johanniskraut) oder mit Efavirenz und anderen mässigen CYP3A4-Induktoren wird nicht empfohlen. Diese CYP3A4-Induktoren verringern die Plasmakonzentrationen von Finerenon deutlich und führen zu reduzierter therapeutischer Wirkung (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Simulationen deuten darauf hin, dass Rifampicin (600 mg einmal täglich), ein starker CYP3A4-Induktor, die Finerenon-Exposition verringert (GMR und 90% Populationsintervall für AUC und Cmax: 0.07 [0.05-0.11] und 0.14 [0.11-0.21]). Efavirenz (600 mg einmal täglich), ein mässiger CYP3A4-Induktor, verringert die Finerenon-Exposition (GMR und 90% Populationsintervall für AUC und Cmax: 0.19 [0.14-0.27] und 0.32 [0.23-0.43]).
Pharmakodynamische Interaktionen
Arzneimittel, die das Serumkalium erhöhen
Es ist zu erwarten, dass Arzneimittel, die das Serumkalium erhöhen, bei gleichzeitiger Anwendung mit Kerendia das Risiko für eine Hyperkaliämie erhöhen.
Die gleichzeitige Anwendung von Kerendia mit folgenden Arzneimitteln sollte vermieden werden:
·kaliumsparende Diuretika (z.B. Amilorid, Triamteren (in der Schweiz nicht zugelassen))
·andere Mineralokortikoid-Rezeptor-Antagonisten (MRA) (z.B. Eplerenon, Spironolacton, Esaxerenon (in der Schweiz nicht zugelassen), Canrenon (in der Schweiz nicht zugelassen))
Kerendia sollte bei gleichzeitiger Anwendung mit folgenden Arzneimitteln mit Vorsicht angewendet und der Serumkaliumspiegel muss während der Behandlung überwacht werden:
·Kaliumsupplemente
·Trimethoprim oder Trimethoprim-Sulfamethoxazol. Eine vorübergehende Unterbrechung der Behandlung mit Kerendia kann erforderlich sein (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Wirkung von Kerendia auf andere Arzneimittel
In vivo hatte ein Mehrdosisschema von 20 mg Finerenon einmal täglich keine Auswirkung auf die AUC des CYP3A4-Probesubstrats Midazolam. Finerenon bewirkt weder eine Hemmung noch eine Induktion von CYP3A4.
Eine Einzeldosis von 20 mg Finerenon hatte ebenfalls keine Auswirkung auf die AUC und Cmax des CYP2C8-Probesubstrats Repaglinid. Finerenon hemmt CYP2C8 nicht.
Es wurde keine gegenseitige pharmakokinetische Wechselwirkung zwischen Finerenon und dem CYP2C9-Substrat Warfarin sowie zwischen Finerenon und dem P-gp-Substrat Digoxin nachgewiesen.
Mehrfachdosen von 40 mg Finerenon einmal täglich hatten keine klinisch relevante Auswirkung auf die AUC oder Cmax des BCRP- und OATP-Substrats Rosuvastatin.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Frauen, die schwanger werden können, sollten während der Behandlung mit Kerendia eine wirksame Kontrazeption anwenden.
Schwangerschaft
Es liegen keine Daten zur Anwendung von Kerendia bei Schwangeren vor. Tierexperimentelle Studien haben eine embryofetale Entwicklungstoxizität nach Expositionen gezeigt, die über der maximalen Exposition beim Menschen liegen (siehe Rubrik «Präklinische Daten»).
Kerendia sollte nicht während der Schwangerschaft angewendet werden, es sei denn, dass eine Behandlung mit Finerenon aufgrund des klinischen Zustandes der Frau erforderlich ist.
Stillzeit
Es ist nicht bekannt, ob Finerenon oder seine Metaboliten in die menschliche Muttermilch übergehen. Verfügbare pharmakokinetische und toxikologische Daten bei Tieren haben eine Ausscheidung von Finerenon und seiner Metaboliten in die Milch gezeigt (siehe Rubrik «Präklinische Daten»). Ein Risiko für gestillte Neugeborene und Kleinkinder kann nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit Kerendia verzichtet werden soll. Dabei soll sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau berücksichtigt werden.
Fertilität
Es liegen keine Daten zur Wirkung von Kerendia auf die humane Fertilität vor. Tierexperimentelle Studien mit Finerenon ergaben keine klinisch relevanten Hinweise auf ein Risiko für eine beeinträchtigte Fertilität (siehe Rubrik «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenBasierend auf dem Sicherheitsprofil von Kerendia wird kein Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen, erwartet.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die Sicherheit von Kerendia bei Patienten mit chronischer Nierenerkrankung und Typ-2-Diabetes wurde in zwei randomisierten, doppelblinden, placebokontrollierten, multizentrischen Phase-III-Studien untersucht: in der FIDELIO-DKD bzw. FIGARO-DKD Studie erhielten 2818 bzw. 3671 Patienten Kerendia (10 mg oder 20 mg einmal täglich), wobei die mittlere Behandlungsdauer 2.2 bzw. 2.9 Jahre betrug.
Die am häufigsten (≥10 %) berichtete unerwünschte Arzneimittelwirkung von Kerendia war Hyperkaliämie (siehe «Beschreibung ausgewählter unerwünschter Arzneimittelwirkungen» unten und Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Liste der unerwünschten Arzneimittelwirkungen
Die mit Kerendia beobachteten unerwünschten Arzneimittelwirkungen sind in der folgenden Tabelle 2 nach MedDRA-Systemorganklassen und Häufigkeitskategorien zusammengefasst. Die unerwünschten Arzneimittelwirkungen werden nach Systemorganklasse und in absteigender Häufigkeitsreihenfolge unter Verwendung der folgenden Konvention aufgeführt: Sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1000, <1/100), selten (≥1/10'000, <1/1000) und sehr selten (<1/10'000).
Tabelle 2: Unerwünschte Arzneimittelwirkungen, die mit Kerendia in Phase-III-Studien (gepoolte Daten der Studien FIDELIO-DKD und FIGARO-DKD) berichtet wurden
|
MedDRA-
Systemorganklasse
|
Sehr häufig
|
Häufig
| |
Stoffwechsel- und Ernährungsstörungen
|
Hyperkaliämie1
|
Hyponatriämie2
Hyperurikämie3, 4
| |
Gefässerkrankungen
|
|
Hypotonie5, 6
| |
Untersuchungen
|
|
Glomeruläre Filtrationsrate verringert7
| |
1
Umfasst erhöhtes Blutkalium und Hyperkaliämie
2 Umfasst verringertes Blutnatrium und Hyponatriämie
3 Umfasst erhöhte Harnsäure im Blut und Hyperurikämie
4 Asymptomatische Hyperurikämie wurde beobachtet. In der Studie FIGARO-DKD wurde in der Kerendia-Gruppe im Vergleich zu Placebo eine Erhöhung des Harnsäurespiegels im Serum um bis zu 0.3 mg/dl gegenüber dem Ausgangswert festgestellt, die sich im Zeitverlauf abschwächte. Es wurden keine Behandlungsabbrüche im Zusammenhang mit Hyperurikämie gemeldet.
5 Umfasst verminderter Blutdruck, diastolisch verminderter Blutdruck, diastolische Hypotonie und Hypotonie
6 Bei den mit Kerendia behandelten Patienten nahm nach 1 Monat der mittlere systolische Blutdruck (SBP) um 3 mmHg und der mittlere diastolische Blutdruck (DBP) um 1–2 mmHg ab und blieb danach stabil. Die meisten hypotensiven Ereignisse waren leicht oder mittelschwer ausgeprägt und klangen spontan ab. Mit Hypotonie assoziierte Ereignisse, wie z.B. Schwindel, Synkope oder Sturz, traten bei Patienten unter Kerendia nicht häufiger auf als unter Placebo.
7 Eine initiale Abnahme der eGFR (im Mittel um 2 ml/min/1.73 m2) wurde im Laufe der Zeit schwächer im Vergleich zum Placebo. Diese Abnahme hat sich nach Absetzen der Behandlung als reversibel erwiesen (siehe Rubrik «Eigenschaften/Wirkungen»).
|
Beschreibung ausgewählter unerwünschter Arzneimittelwirkungen
Hyperkaliämie
In der Studie FIDELIO-DKD an Patienten mit chronischer Nierenerkrankung (mittlere eGFR 44.4 ml/min/1.73 m2) und Typ-2-Diabetes wurden bei 18.2 % der mit Kerendia behandelten Patienten Hyperkaliämie-Ereignisse berichtet, verglichen mit 9.0 % der mit Placebo behandelten Patienten. Ein Anstieg des mittleren Serumkaliumspiegels von ca. 0.2 mmol/l gegenüber dem Ausgangswert wurde im ersten Behandlungsmonat im Kerendia-Arm verglichen mit Placebo beobachtet, der danach stabil blieb (siehe Rubrik «Eigenschaften/Wirkungen»). Der Anteil an Hospitalisierungen wegen Hyperkaliämie lag für die Kerendia-Gruppe bei 1.4 % versus 0.3 % in der Placebogruppe. Die Häufigkeit von Hyperkaliämie, die zum dauerhaften Absetzen des Prüfpräparates führte, betrug bei Patienten unter Kerendia 2.3 % versus 0.9 % in der Placebogruppe.
In der FIGARO-DKD Studie an Patienten mit chronischer Nierenerkrankung (mittlere eGFR 67.8 ml/min/1.73 m2) und Typ-2-Diabetes wurden bei 10.7 % der mit Kerendia behandelten Patienten Hyperkaliämie-Ereignisse berichtet, verglichen mit 5.3 % der mit Placebo behandelten Patienten. Ein Anstieg des mittleren Serumkaliumspiegels von ca. 0.15 mmol/l gegenüber dem Ausgangswert wurde im ersten Behandlungsmonat im Kerendia-Arm verglichen mit Placebo beobachtet, der danach stabil blieb (siehe Rubrik «Eigenschaften/Wirkungen»). Der Anteil an Hospitalisierungen wegen Hyperkaliämie lag für die Kerendia-Gruppe bei 0.6 % versus <0.1 % in der Placebogruppe. Die Häufigkeit von Hyperkaliämie, die zum dauerhaften Absetzen des Prüfpräparates führte, betrug bei Patienten unter Kerendia 1.3 % versus 0.4 % in der Placebogruppe.
In beiden Studien waren die meisten Hyperkaliämie-Ereignisse bei den mit Kerendia behandelten Patienten leicht bis mittelschwer ausgeprägt.
Zu besonderen Empfehlungen siehe Rubriken «Dosierung/Anwendung» sowie «Warnhinweise und Vorsichtsmassnahmen».
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurden keine Fälle von unerwünschten Ereignissen im Zusammenhang mit Überdosierung von Kerendia beim Menschen berichtet. Als wahrscheinlichstes Anzeichen einer Überdosierung ist Hyperkaliämie zu erwarten. Bei Entwicklung einer Hyperkaliämie sollte die Standardbehandlung gemäss lokalen Leitlinien eingeleitet werden. Die Behandlung mit Kerendia sollte gemäss Tabelle 1 weitergeführt werden (siehe Rubrik «Dosierung/Anwendung»).
Es ist unwahrscheinlich, dass Finerenon durch Hämodialyse effizient eliminiert wird, da die an Plasmaproteine gebundenen Fraktion etwa 90 % beträgt.
Eigenschaften/WirkungenATC-Code
C03DA05
Wirkungsmechanismus
Finerenon ist ein nichtsteroidaler, selektiver Antagonist des Mineralokortikoid-Rezeptors (MR) und schwächt die durch eine MR-Überaktivierung vermittelte Entzündung und Fibrose wirksam ab. Der MR wird in Nieren, Herz und Blutgefässen exprimiert, wo Finerenon auch der Natriumretention und hypertrophen Prozessen entgegenwirkt. Finerenon hat aufgrund seiner nichtsteroidalen Struktur und seines sperrigen Bindungsmodus eine hohe Potenz und Selektivität für den MR. Finerenon hat keine relevante Affinität für Androgen-, Progesteron-, Östrogen- sowie Glukokortikoidrezeptoren und verursacht daher keine geschlechtshormonbedingten Nebenwirkungen (wie z.B. Gynäkomastie). Seine Bindung an den MR führt zu einem spezifischen Rezeptor-Ligand-Komplex, der die Rekrutierung von transkriptionellen Koaktivatoren blockiert, die an der Expression von proinflammatorischen und profibrotischen Mediatoren beteiligt sind.
Pharmakodynamik
Wirkungen bei gesunden Probanden
Mehrdosisschemata (Tagesdosen von 20 mg oder 40 mg über 10 Tage) führten zur Aktivierung des Renin-Angiotensin-Aldosteron-Systems (RAAS), d.h. zu reversiblen Anstiegen der Plasma-Renin-Aktivität und der Serumaldosteronkonzentrationen, wobei innerhalb von 48 Stunden nach der letzten Dosis wieder die Ausgangswerte erreicht wurden.
Nach Aktivierung des MR mit seinem Agonisten Fludrocortison zeigten Einzeldosen Finerenon von bis zu 20 mg dosisabhängige natriuretische Wirkungen, sowie eine verringerte Kaliumausscheidung im Urin verglichen mit Placebo.
Einzel- oder Mehrfachdosen von Finerenon hatten bei gesunden Probanden keinen Einfluss auf die Vitalzeichen.
Wirkungen bei Patienten mit chronischer Nierenerkrankung und Typ-2-Diabetes
In den zwei Phase-III-Studien FIDELIO-DKD und FIGARO-DKD betrug die placebokorrigierte relative Reduktion des Albumin-Kreatinin-Quotienten im Urin (UACR) bei Patienten, die randomisiert einer Behandlung mit Finerenon zugeteilt waren, nach 4 Monaten 31 % bzw. 32 %. Die Senkung des UACR persistierte in beiden Studien.
In der Studie ARTS DN, einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen Phase-IIb-Dosisfindungsstudie an Erwachsenen mit chronischer Nierenkrankheit und Typ-2-Diabetes betrug die placebokorrigierte relative Reduktion des UACR nach 90 Tagen 25 % bzw. 38 % bei den mit Finerenon 10 mg bzw. 20 mg einmal täglich behandelten Patienten.
Kardiale Elektrophysiologie
Eine eingehende QT-Studie mit 57 gesunden Probanden ergab keine Hinweise auf eine QT/QTc-verlängernde Wirkung von Finerenon nach Einzeldosen von 20 mg (therapeutisch) oder 80 mg (supratherapeutisch), was darauf hindeutet, dass Finerenon keine Wirkung auf die kardiale Repolarisation hat.
Klinische Wirksamkeit
Finerenon wurde in den zwei randomisierten, doppelblinden, placebokontrollierten, multizentrischen Phase-III-Studien FIDELIO-DKD und FIGARO-DKD untersucht.
Die FIDELIO-DKD Studie untersuchte die Wirkung von Finerenon verglichen mit Placebo auf renale und kardiovaskuläre Ergebnisse bei erwachsenen Patienten mit Typ-2-Diabetes und chronischer Nierenerkrankung (Einschlusskriterien: a) moderate Albuminurie [UACR ≥30 - < 300 mg/g] und eGFR 25 - 60 ml/min/1.73 m2 bei gleichzeitigem Vorliegen einer diabetischen Retinopathie ODER b) schwere Albuminurie [UACR ≥300 mg/g] und eGFR 25 – 75 ml/min/1.73 m2). In die Studie eingeschlossene Patienten mussten einen Serumkaliumspiegel von ≤4.8 mmol/l aufweisen und eine Vorbehandlung mit einer Standardtherapie, inklusive einer maximal verträglichen Dosis eines Angiotensin-Converting-Enzyme-Hemmers (ACEI [34 %]) oder eines Angiotensinrezeptorblockers (ARB [66%]) erhalten Die Indikation für eine Behandlung mit einem MRA gemäss Leitlinien (wie symptomatische chronische Herzinsuffizienz mit reduzierter Auswurffraktion) war ein Ausschlusskriterium.
Der primäre Endpunkt der FIDELIO-DKD-Studie war zusammengesetzt aus der Zeit bis zum ersten Auftreten eines Nierenversagens (definiert als chronische Dialyse, Nierentransplantation oder Abfall der eGFR auf < 15 ml/min/1.73 m2 über mindestens vier Wochen), einer Abnahme der eGFR um ≥40 % gegenüber dem Ausgangswert über mindestens vier Wochen oder eines nierenbedingten Todes. Sekundärer Schlüsselendpunkt war ein zusammengesetzter kardiovaskulärer Endpunkt bestehend aus der Zeit bis zum ersten Auftreten von kardiovaskulärem (KV) Tod, nichttödlichem Myokardinfarkt (MI), nichttödlichem Schlaganfall oder Hospitalisierung wegen Herzinsuffizienz.
Die Studie untersuchte 5662 Patienten, welche im Verhältnis 1:1 für die Behandlung mit Finerenon einmal täglich (n = 2824) oder Placebo (n = 2838) randomisiert wurden. Die Anfangsdosis betrug entweder 10 mg [bei einer eGFR von 25 - < 60 ml/min/1.73 m2] oder 20 mg [bei einer eGFR von ≥60 ml/min/1.73 m2]. Die Dosisstärke wurde im Verlauf der Studie auf 10 mg oder 20 mg QD, hauptsächlich auf Grund des Serumkaliumspiegels, angepasst. Die mediane Beobachtungsdauer betrug 2.6 Jahre. Die Studienpopulation war zu 63 % weisser, zu 25 % asiatischer und zu 5 % schwarzer Abstammung. Das mittlere Alter bei der Rekrutierung betrug 66 Jahre, und 70 % der Patienten waren Männer. Der mittlere eGFR-Ausgangswert betrug 44.4 ml/min/1.73 m2 und 55 % der Patienten hatten eine eGFR < 45 ml/min/1.73 m2. Der mediane UACR betrug 853 mg/g, das mittlere glykierte Hämoglobin A1c (HbA1c) betrug 7.7 %. Zirka 46 % der Studienteilnehmer hatten eine atherosklerotische kardiovaskuläre Vorerkrankung, 30 % eine koronare Herzkrankheit und 8 % eine Herzinsuffizienz in der Anamnese. Der mittlere Blutdruck betrug 138/76 mmHg. Die mittlere Dauer seit Diagnose des Typ-2-Diabetes bei Aufnahme in die Studie betrug 16.6 Jahre, und bei Studienbeginn erhielten nahezu alle Studienteilnehmer (97%) ein oder mehrere Antidiabetika (Insulin [64 %], Biguanide [44 %], Glucagon-like-Peptid-1-[GLP-1-]Rezeptoragonisten [7 %], Natrium-Glukose-Cotransporter-2-[SGLT2-]Inhibitoren [5 %]). Ausserdem wiesen 47 % bzw. 26 % der Patienten zu Studienbeginn eine vorbestehende diabetische Retinopathie und diabetische Neuropathie auf. Der Grossteil der Patienten erhielt zusätzlich ein Statin (74 %) und/oder einen Calciumantagonisten (63 %).
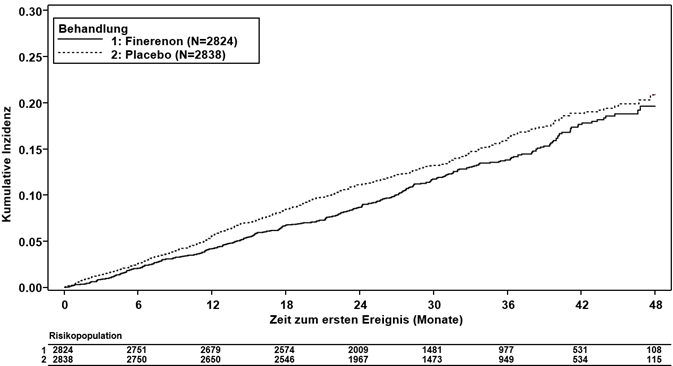
Die FIDELIO-DKD Studie zeigte eine Überlegenheit der Behandlung mit Finerenon gegenüber Placebo für den kombinierten (renalen) primären Endpunkt (HR 0.82, 95%-KI 0.73–0.92, p = 0.0009; siehe Tabelle 3 und Abbildung 1).
Ferner reduzierte Finerenon signifikant das Risiko für den kombinierten (kardiovaskulären) sekundären Schlüsselendpunkt (HR 0.86, 95%-KI 0.75–0.99, p = 0.0344; siehe Tabelle 3 und Abbildung 2). Im Finerenon-Arm ergaben sich im Vergleich zum Placebo-Arm niedrigere Inzidenzraten in Bezug auf Herzinsuffizienz, nichttödlichem MI und kardiovaskulär bedingtem Tod. Nichttödliche Schlaganfälle traten in beiden Behandlungsarmen mit ähnlicher Inzidenz auf (siehe Tabelle 3).
Tabelle 3: Analyse der primären und wichtigsten sekundären Ereigniszeit-Endpunkte (und ihrer Einzelkomponenten) in der Phase-III-Studie FIDELIO-DKD
|
|
Patienten mit chronischer Nierenerkrankung und Typ-2-Diabetes
| |
|
Finerenon*
10 oder 20 mg 1x täglich
n = 2824
|
Placebo*
n = 2838
|
Behandlungseffekt
Finerenon/Placebo
| |
Primäre und sekundäre Ereigniszeit-Endpunkte:
|
n (%)
|
Ereignisrate (100 Pt.j.)
|
n (%)
|
Ereignisrate (100 Pt.j.)
|
Hazard Ratio
(95%-KI)
|
p-Wert
| |
Primärer kombinierter Endpunkt «Nierenversagen, anhaltende eGFR-Abnahme ≥40 % oder nierenbedingter Tod»
|
498 (17.6 %)
|
7.53
|
600 (21.1 %)
|
9.09
|
0.82
[0.73; 0.92]
|
0.0009
| |
Nierenversagen
|
205 (7.3 %)
|
2.96
|
235 (8.3 %)
|
3.39
|
0.86
[0.72; 1.05]
|
-
| |
Anhaltende eGFR-Abnahme ≥40 %
|
473 (16.7 %)
|
7.15
|
577 (20.3 %)
|
8.74
|
0.81
[0.72; 0.91]
|
-
| |
Nierenbedingter Tod
|
2 (< 0.1 %)
|
-
|
2 (< 0.1 %)
|
-
|
-
|
-
| |
Sekundärer kombinierter Endpunkt «KV Tod, nichttödlicher MI, nichttödlicher Schlaganfall oder Hospitalisierung wegen Herzinsuffizienz»
|
366 (13.0 %)
|
5.11
|
420 (14.8 %)
|
5.93
|
0.86
[0.75; 0.99]
|
0.0344
| |
KV Tod
|
128 (4.5 %)
|
1.70
|
150 (5.3 %)
|
1.99
|
0.86
[0.68; 1.09]
|
-
| |
Nichttödlicher MI
|
70 (2.5 %)
|
0.94
|
87 (3.1 %)
|
1.18
|
0.80
[0.58; 1.09]
|
-
| |
Nichttödlicher Schlaganfall
|
90 (3.2 %)
|
1.22
|
87 (3.1 %)
|
1.18
|
1.03
[0.77; 1.38]
|
-
| |
Hospitalisierung wegen Herzinsuffizienz
|
138 (4.9 %)
|
1.88
|
162 (5.7 %)
|
2.22
|
0.85
[0.68; 1.07]
|
-
| |
* Behandlung zusätzlich zu maximal verträglichen zugelassenen Dosen von ACEI oder ARB.
|
Abbildung 1: Zeit bis zum ersten Auftreten von Nierenversagen, anhaltender eGFR-Abnahme ≥40 % vs. Ausgangswert oder nierenbedingtem Tod in der FIDELIO-DKD-Studie
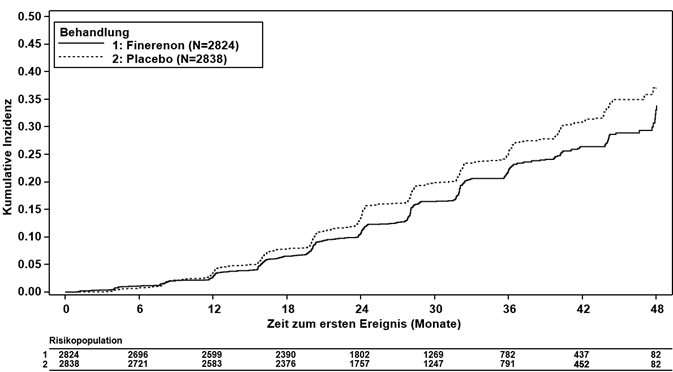
Abbildung 2: Zeit bis zum ersten Auftreten von KV Tod, nichttödlichem MI, nichttödlichem Schlaganfall oder Hospitalisierung wegen Herzinsuffizienz in der FIDELIO-DKD-Studie
Die FIGARO-DKD Studie untersuchte die Wirkung von Finerenon gegenüber Placebo auf das Eintreten kardiovaskulärer und renaler Ereignisse bei erwachsenen Patienten mit Typ-2-Diabetes und chronischer Nierenerkrankung (Einschlusskriterien: a) moderate Albuminurie [UACR ≥30 - < 300 mg/g] und eGFR 25 - 90 ml/min/1.73 m2 ODER b) schwere Albuminurie [UACR ≥300 mg/g] und eGFR ≥60 ml/min/1.73 m2). Um in die Studie eingeschlossen zu werden, mussten die Patienten ausserdem einen Serumkaliumspiegel von ≤4.8 mmol/l aufweisen sowie bereits eine Vorbehandlung mit einer Standardtherapie, inklusive einer maximal verträglichen Dosis eines Angiotensin-Converting-Enzyme-Hemmers (ACEI [43 %]) oder eines Angiotensinrezeptorblockers (ARB [57%]) erhalten. Die Indikation für eine Behandlung mit einem MRA gemäss Leitlinien (wie symptomatische chronische Herzinsuffizienz mit reduzierter Auswurffraktion) war ein Ausschlusskriterium.
Der primäre Endpunkt in der FIGARO-DKD-Studie war ein zusammengesetzter kardiovaskulärer (KV) Endpunkt bestehend aus der Zeit bis zum ersten Auftreten von KV Tod, nichttödlichem MI, nichttödlichem Schlaganfall oder Hospitalisierung wegen Herzinsuffizienz. Sekundärer Schlüsselendpunkt war ein zusammengesetzter renaler Endpunkt aus der Zeit bis zum Nierenversagen, einer Abnahme der eGFR um ≥40 % gegenüber dem Ausgangswert über mindestens vier Wochen oder eines nierenbedingten Todes.
Die Studie untersuchte 7328 Patienten, welche im Verhältnis 1:1 für die Behandlung mit Finerenon (n = 3674) oder Placebo (n = 3654) randomisiert wurden. Die Anfangsdosis betrug entweder 10 mg [bei einer eGFR von 25 - < 60 ml/min/1.73 m2] oder 20 mg [bei einer eGFR von ≥60 ml/min/1.73 m2]. Die Dosisstärke wurde im Verlauf der Studie auf 10 mg oder 20 mg QD, hauptsächlich auf Grund des Serumkaliumspiegels, angepasst. Die mediane Beobachtungsdauer betrug 3.4 Jahre. Die Studienpopulation war zu 72 % weisser, zu 20 % asiatischer und zu 4 % schwarzer Abstammung. Das mittlere Alter bei der Rekrutierung betrug 64 Jahre, und 69 % der Patienten waren Männer. Der mittlere eGFR-Ausgangswert betrug 67.8 ml/min/1.73 m2 und 62 % der Patienten hatten eine eGFR ≥60 ml/min/1.73 m2. Der mediane UACR betrug 309 mg/g, das mittlere glykierte Hämoglobin A1c (HbA1c) betrug 7.7 %. Zirka 45 % der Studienteilnehmer hatten eine atherosklerotische kardiovaskuläre Erkrankung in der Anamnese, 8 % hatten eine Herzinsuffizienz in der Anamnese. Der mittlere Blutdruck betrug 136/77 mmHg. Die mittlere Dauer des Typ-2-Diabetes bei Aufnahme in die Studie betrug 14.5 Jahre und bei Studienbeginn erhielten nahezu alle Studienteilnehmer (98%) ein oder mehrere Antidiabetika (Insulin [54 %], Biguanide [69 %], GLP-1-Rezeptoragonisten [8 %], SGLT2-Inhibitoren [8 %]). Ausserdem wiesen 31 % bzw. 28% der Patienten zu Studienbeginn eine vorbestehende diabetische Retinopathie und diabetische Neuropathie auf. Der Grossteil der Patienten erhielt zusätzlich ein Statin (71 %).
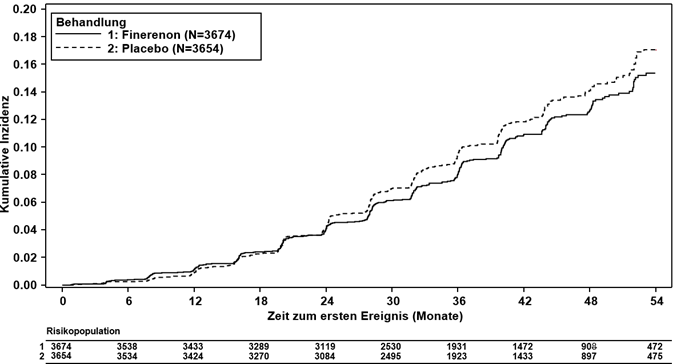
Finerenon reduzierte signifikant das Risiko für den primären (kardiovaskulären) kombinierten Endpunkt verglichen mit Placebo (HR 0.87, 95%-KI 0.76–0.98, p = 0.0254) (siehe Abbildung 3 und Tabelle 4). Der Behandlungseffekt für den primären Endpunkt war in allen Untergruppen, darunter Region, eGFR, UACR, systolischer Blutdruck und HbA1c zur Baseline, einheitlich. In dem Finerenon-Arm wurde im Vergleich zum Placebo-Arm eine niedrigere Inzidenzrate des kombinierten sekundären (renalen) Endpunkts aus Nierenversagen, anhaltender eGFR-Abnahme ≥40 % oder nierenbedingtem Tod beobachtet; dieser Unterschied erreichte jedoch keine statistische Signifikanz (HR 0.87, 95%-KI 0.75–1.01, p = 0.0635) (siehe Abbildung 4 und Tabelle 4).
Tabelle 4: Analyse der primären und sekundären Ereigniszeit-Endpunkte (und ihrer Einzelkomponenten) in der Phase-III-Studie FIGARO-DKD
|
|
Patienten mit
chronischer Nierenerkrankung und Typ-2-Diabetes
| |
|
Finerenon*
10 oder 20 mg 1x täglich
n = 3674
|
Placebo*
n = 3654
|
Behandlungseffekt
Finerenon/Placebo
| |
Primäre und sekundäre Ereigniszeit-Endpunkte:
|
n (%)
|
Ereignisrate (100 Pt.j.)
|
n (%)
|
Ereignisrate (100 Pt.j.)
|
Hazard Ratio
(95%-KI)
|
p-Wert
| |
Primärer kombinierter Endpunkt «KV Tod, nichttödlicher MI, nichttödlicher Schlaganfall oder Hospitalisierung wegen Herzinsuffizienz»
|
457 (12.4 %)
|
3.88
|
518 (14.2 %)
|
4.46
|
0.87
[0.76; 0.98]
|
0.0254
| |
KV Tod
|
193 (5.3 %)
|
1.56
|
214 (5.9 %)
|
1.75
|
0.89
[0.73; 1.08]
|
-
| |
Nichttödlicher MI
|
103 (2.8 %)
|
0.85
|
101 (2.8 %)
|
0.84
|
1.00
[0.76; 1.32]
|
-
| |
Nichttödlicher Schlaganfall
|
108 (2.9 %)
|
0.89
|
111 (3.0 %)
|
0.93
|
0.97
[0.74; 1.26]
|
-
| |
Hospitalisierung wegen Herzinsuffizienz
|
117 (3.2 %)
|
0.97
|
163 (4.5 %)
|
1.36
|
0.71
[0.56; 0.90]
|
-
| |
Kombinierter Endpunkt «Nierenversagen, anhaltende eGFR-Abnahme ≥40 % oder nierendbedingter Tod»
|
350 (9.5 %)
|
3.17
|
395 (10.8 %)
|
3.59
|
0.87
[0.75; 1.01]
|
0.0635**
| |
Nierenversagen
|
46 (1.3 %)
|
0.40
|
62 (1.7 %)
|
0.55
|
0.72
[0.49; 1.05]
|
-
| |
Anhaltende eGFR-Abnahme ≥40 %
|
338 (9.2 %)
|
3.06
|
385 (10.5 %)
|
3.50
|
0.86
[0.74; <1.00]
|
-
| |
Nierenbedingter Tod
|
0
|
-
|
2 (<0.1 %)
|
-
|
-
|
-
| |
* Behandlung zusätzlich zu maximal verträglichen zugelassenen Dosen von ACEI oder ARB.
** Nicht signifikant.
|
Abbildung 3: Zeit bis zum ersten Auftreten von KV Tod, nichttödlichem MI, nichttödlichem Schlaganfall oder Hospitalisierung wegen Herzinsuffizienz in der FIGARO-DKD-Studie
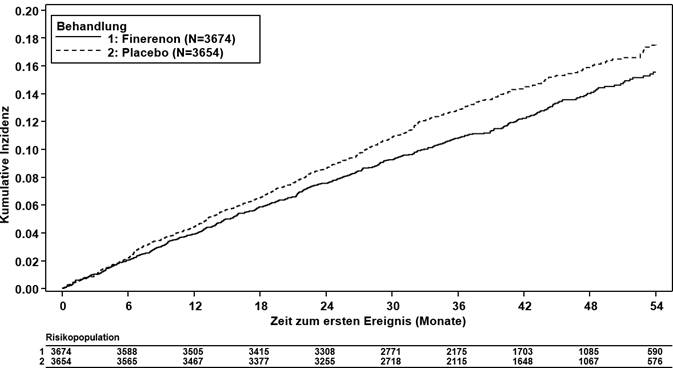
Abbildung 4: Zeit bis zum ersten Auftreten von Nierenversagen, anhaltender eGFR-Abnahme ≥40 % vs. Ausgangswert oder nierenbedingtem Tod in der FIGARO-DKD-Studie
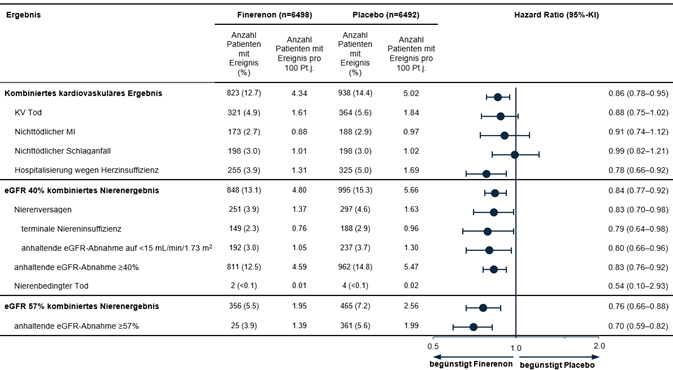
In einer vorgegebenen gepoolten Analyse der Studien FIDELIO-DKD und FIGARO-DKD reduzierte Finerenon das Risiko für den kombinierten kardiovaskulären Endpunkt «Zeit bis zum Eintritt von KV Tod, nichttödlichem MI, nichttödlichem Schlaganfall oder Hospitalisierung wegen Herzinsuffizienz» verglichen mit Placebo (HR 0.86 [95%-KI 0.78; 0.95]) (siehe Abbildung 5). Das Risiko für den kombinierten renalen Endpunkt «Zeit bis zum Eintritt von Nierenversagen, anhaltender eGFR-Abnahme ≥40 % gegenüber dem Ausgangswert oder nierenbedingtem Tod» war ebenfalls reduziert mit Finerenon verglichen mit Placebo (HR 0.84 [95%-KI 0.77; 0.92]), ebenso wie der kombinierte Endpunkt aus Zeit bis zum Eintritt von Nierenversagen, anhaltender eGFR-Abnahme ≥57 % (entspricht etwa einer Verdoppelung des Serumkreatinins) gegenüber dem Ausgangswert oder nierenbedingtem Tod (HR 0.76 [95%-KI 0.66; 0.88]) (siehe Abbildung 5).
Abbildung 5: Kombinierte kardiovaskuläre und renale Endpunkte in der gepoolten Analyse von FIDELIO-DKD und FIGARO-DKD
PharmakokinetikAbsorption
Finerenon wird nach oraler Verabreichung fast vollständig absorbiert. Die Absorption erfolgt rasch, und die maximalen Plasmakonzentrationen (Cmax) stellen sich 0.5 bis 1.25 Stunden nach Einnahme der Tablette im Nüchternzustand ein. Die absolute Bioverfügbarkeit von Finerenon beträgt 43.5 % aufgrund eines First-Pass-Metabolismus in Darmwand und Leber. Finerenon ist kein Substrat des Efflux-Transporters P-gp in vivo.
Die Aufnahme mit fett- und kalorienreicher Nahrung erhöhte die AUC von Finerenon um 21 %, reduzierte Cmax um 19 % und verlängerte die Zeit bis zum Erreichen von Cmax auf 2.5 Stunden. Dies ist klinisch nicht relevant. Daher kann Finerenon unabhängig von den Mahlzeiten eingenommen werden (siehe Rubrik «Dosierung/Anwendung»).
Distribution
Das Verteilungsvolumen von Finerenon im Steady State (Vss) beträgt 52.6 l. Die menschliche Plasmaproteinbindung von Finerenon in vitro beträgt 91.7 %; das Hauptbindungsprotein ist Serumalbumin.
Metabolismus
Etwa 90 % des Finerenon-Stoffwechsels werden durch CYP3A4 und 10 % durch CYP2C8 vermittelt. Vier Hauptmetaboliten (M-1a, M-1b, M-2a und M-3a) wurden im Plasma gefunden. Alle Metaboliten sind pharmakologisch inaktiv.
Elimination
Die Elimination von Finerenon aus dem Plasma erfolgt rasch mit einer Eliminationshalbwertszeit (t1/2) von etwa 2 bis 3 Stunden. Die Ausscheidung von unverändertem Finerenon stellt einen untergeordneten Ausscheidungsweg dar (< 1 % der Dosis im Urin durch glomeruläre Filtration, < 0.2 % in den Fäzes). Etwa 80 % der verabreichten Dosis wurden über den Urin und rund 20 % der Dosis wurden über die Fäzes ausgeschieden, und zwar fast ausschliesslich in Form von Metaboliten. Angesichts einer systemischen Clearance von etwa 25 l/h kann Finerenon als Wirkstoff mit niedriger Clearance eingestuft werden.
Linearität/Nicht Linearität
Die Pharmakokinetik von Finerenon ist über den untersuchten Dosisbereich von 1.25 bis 80 mg linear.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Bei Zirrhosepatienten mit leichter Leberinsuffizienz (Child-Pugh A) zeigte sich keine Veränderung der Finerenon-Exposition.
Bei Zirrhosepatienten mit mittelschwerer Leberinsuffizienz (Child-Pugh B) war die mittlere AUC von Finerenon um 38 % erhöht und Cmax war unverändert im Vergleich zu gesunden Kontrollprobanden (siehe Rubrik «Dosierung/Anwendung»).
Es liegen keine Daten für Patienten mit schwerer Leberinsuffizienz (Child-Pugh C) vor (siehe Rubriken «Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»).
Nierenfunktionsstörungen
Eine leichte Nierenfunktionsstörung (Kreatinin-Clearance [CLCR] 60 bis < 90 ml/min) hatte keinen Einfluss auf die AUC und Cmax von Finerenon. Im Vergleich zu Probanden mit normaler Nierenfunktion (CLCR ≥90 ml/min) war die Auswirkung einer mittelschweren (CLCR 30 bis < 60 ml/min) oder schweren (CLCR < 30 ml/min) Niereninsuffizienz auf die AUC von Finerenon mit Anstiegen von 34–36 % ähnlich hoch. Eine mittelschwere oder schwere Nierenfunktionsstörung hatte keine Auswirkungen auf die Cmax (siehe Rubrik «Dosierung/Anwendung»).
Aufgrund der hohen Plasmaproteinbindung ist zu erwarten, dass Finerenon nicht dialysierbar ist.
Ältere Patienten
58 % der 2818 Patienten, die Finerenon in der FIDELIO-DKD-Studie erhielten, waren 65 Jahre alt oder älter, und 15 % waren 75 Jahre alt oder älter. Zwischen diesen Patienten und jüngeren Patienten wurden insgesamt keine Unterschiede in der Sicherheit oder Wirksamkeit beobachtet.
53 % der 3671 Patienten, die Finerenon in der FIGARO-DKD-Studie erhielten, waren 65 Jahre alt oder älter, und 14 % waren 75 Jahre alt oder älter. Zwischen diesen Patienten und jüngeren Patienten wurden insgesamt keine Unterschiede in der Sicherheit oder Wirksamkeit beobachtet.
Ältere Patienten (≥65 Jahre) wiesen höhere Plasmakonzentrationen von Finerenon auf als jüngere Patienten (≤45 Jahre), wobei die mittleren AUC- und Cmax-Werte bei den Älteren um 34 % bzw. 51 % höher waren (siehe Rubrik «Dosierung/Anwendung»).
Körpergewicht
In populationspharmakokinetischen Analysen erwies sich das Körpergewicht als Kovariate für Vc/F, was bei Probanden mit geringerem Körpergewicht zu höheren Finerenon Cmax-Werten führte. Die Cmax einer Person mit einem Körpergewicht von 50 kg war schätzungsweise um 38 % bis 51 % höher als bei einer Person mit 100 kg. Eine Dosisanpassung auf Basis des Körpergewichts ist nicht erforderlich (siehe Rubrik «Dosierung/Anwendung»).
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, akuten Toxizität und Phototoxizität, lassen die präklinischen Daten keine besonderen Gefahren bei klinisch relevanten Konzentrationen für den Menschen erkennen.
Wirkungen, die in Studien zur Toxizität bei wiederholter Verabreichung beobachtet wurden, waren hauptsächlich auf übersteigerte pharmakodynamische Aktivitäten von Finerenon und sekundäre adaptive Reaktionen zurückzuführen.
Reproduktionstoxizität
In Ratten wurde die männliche Fertilität durch Dosen bis zu 30 mg/kg/Tag Finerenon nicht beeinflusst (16-fach die AUCunbound beim Menschen). Finerenon verursachte eine reduzierte Fertilität in weiblichen Ratten (verringerte Anzahl an Corpora lutea und Implantationsstellen) sowie Zeichen einer frühen embryonalen Toxizität (erhöhte Postimplantationsverluste und verminderte Anzahl an lebensfähigen Feten) bei etwa dem 21-Fachen der AUCunbound beim Menschen. Ausserdem wurden bei etwa dem 17-Fachen der humanen AUCunbound reduzierte Ovarialgewichte festgestellt. Beim 10-Fachen der humanen AUCunbound wurden keine Wirkungen auf die weibliche Fertilität und die frühe embryonale Entwicklung beobachtet.
In den Studien zur embryofetalen Toxizität bei Ratten führte die orale Gabe von Finerenon zu reduziertem Plazentagewicht und Zeichen von embryofetaler Toxizität, darunter reduziertes Fetalgewicht und verzögerte Ossifikation ab der maternal toxischen Dosis von 10 mg/kg/Tag. Dies entsprach einer AUCunbound, die 19-fach über der klinischen Exposition liegt. Bei 30 mg/kg/Tag war die Inzidenz von viszeralen und skelettalen Veränderungen erhöht (leichtes Ödem, verkürzte Nabelschnur, leicht vergrösserte Fontanelle), und ein Fetus zeigte komplexe Fehlbildungen, einschliesslich einer seltenen Fehlbildung (doppelter Aortenbogen). Die AUCunbound liegt etwa 25-fach über der klinischen Exposition. Der NOAEL von 3 mg/kg/Tag (niedrige Dosis) bei Ratten ergab eine Sicherheitsspanne, die dem 10-Fachen der AUCunbound entsprach. Bei Kaninchen ergab der NOAEL von 2.5 mg/kg/Tag (hohe Dosis) eine Sicherheitsspanne, die dem 13-Fachen der AUCunbound entsprach.
In der Studie zur prä- und postnatalen Entwicklungstoxizität an Ratten wurden bei einer Dosis ab 3 mg/kg/Tag eine erhöhte Jungtiersterblichkeit und andere adverse Befunde (niedrigeres Jungtiergewicht, verzögerte Ohrmuschelentfaltung) beobachtet. Ausserdem zeigten die Jungtiere in diesen Dosisgruppen eine leicht gesteigerte lokomotorische Aktivität, jedoch keine weiteren neurologisch-verhaltensbezogenen Veränderungen ab einer AUCunbound, die etwa 4-fach über der klinischen Exposition lag. Der NOAEL von 1 mg/kg/Tag ergab eine Sicherheitsspanne, die etwa dem Zweifachen für die AUCunbound entsprach. Die erhöhte lokomotorische Aktivität bei Nachkommen könnte auf ein potenzielles Risiko für den Fetus hinweisen.
Genotoxizität
Im bakteriellen Mutagenesetest (Ames-Test) induzierte Finerenon keine Mutationen. Finerenon induzierte in vitro keine Chromosomenaberrationen in V79-Lungenzellen des Chinesischen Hamsters. Im Mikrokerntest bei männlichen Mäusen in vivo war Finerenon intraperitoneal in Konzentrationen von bis zu 1000 mg/kg/Tag nicht klastogen. Insgesamt zeigte Finerenon kein genotoxisches Potenzial.
Kanzerogenität
In 2-jährigen Kanzerogenitätsstudien zeigte eine orale Gabe von Finerenon kein kanzerogenes Potenzial bei männlichen und weiblichen Ratten sowie bei weiblichen Mäusen. Bei männlichen Mäusen führte Finerenon zu einem Anstieg von Leydigzelladenomen in einer Dosis von 30 mg/kg/Tag, welche einer AUCunbound entsprach, die 26-fach über der klinischen Exposition lag. Eine Dosis von 10 mg/kg/Tag, welche einer AUCunbound entsprach, die 17-fach über der klinischen Exposition lag, verursachte keine Tumore. Aufgrund der bekannten Empfindlichkeit von Nagern für die Entwicklung dieser Tumore sowie des pharmakologisch basierten Mechanismus bei supratherapeutischen Dosen und der ausreichenden Sicherheitsspannen wird die Zunahme von Leydigzelltumoren bei männlichen Mäusen nicht als klinisch relevant bewertet.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Nicht über 30°C lagern.
In der Originalverpackung aufbewahren.
Zulassungsnummer68130 (Swissmedic).
Packungen10 mg Filmtabletten: Packungen zu 28 oder 98 Filmtabletten (B)
20 mg Filmtabletten: Packungen zu 28 oder 98 Filmtabletten (B)
ZulassungsinhaberinBayer (Schweiz) AG, Zürich
Stand der InformationJanuar 2025
|