ZusammensetzungWirkstoffe
Icatibantum (ut icatibantum acetas).
Hilfsstoffe
Natrii chloridum, acidum acetas glaciale (E260), natrii hydroxidum (E524), aqua ad injectabilia q.s. ad solutionem pro 3 ml.
1 Fertigspritze zu 3 ml enthält 9,9 mg Natrium.
Indikationen/AnwendungsmöglichkeitenIcatibant Spirig HC ist für die symptomatische Behandlung akuter Attacken eines hereditären Angioödems (HAE) bei Erwachsenen, Jugendlichen und Kindern ab 2 Jahren mit C1-Esterase-Inhibitor-Mangel indiziert.
Dosierung/AnwendungIcatibant Spirig HC ist für die Anwendung unter der Anleitung durch medizinisches Fachpersonal bestimmt.
Dosierung
Erwachsene
Die empfohlene Dosierung für Erwachsene ist eine subkutane Injektion von Icatibant Spirig HC 30 mg.
In den meisten Fällen ist eine einzelne Injektion von Icatibant Spirig HC ausreichend, um eine Attacke zu behandeln. Bei unzureichender Linderung oder Wiederauftreten der Symptome kann 6 Stunden später eine zweite Injektion von Icatibant Spirig HC erfolgen. Wenn auch die zweite Injektion keine ausreichende Symptomlinderung bewirkt bzw. ein Wiederauftreten der Symptome festgestellt wird, kann nach weiteren 6 Stunden eine dritte Injektion von Icatibant Spirig HC verabreicht werden. Innerhalb von 24 Stunden dürfen maximal 3 Injektionen von Icatibant Spirig HC verabreicht werden.
In den klinischen Studien sind maximal 8 Injektionen von Icatibant pro Monat angewendet worden.
Kinder und Jugendliche
Die anhand des Körpergewichts bestimmte empfohlene Dosis von Icatibant Spirig HC für Kinder und Jugendliche (im Alter von 2 bis 17 Jahren) ist in der Tabelle 1 unten angegeben.
Tabelle 1: Dosierungsschema für Kinder und Jugendliche
|
Körpergewicht
|
Dosis (Injektionsvolumen)
| |
12 kg bis 25 kg
|
10 mg (1,0 ml)
| |
26 kg bis 40 kg
|
15 mg (1,5 ml)
| |
41 kg bis 50 kg
|
20 mg (2,0 ml)
| |
51 kg bis 65 kg
|
25 mg (2,5 ml)
| |
> 65 kg
|
30 mg (3,0 ml)
|
In der klinischen Studie wurde nicht mehr als 1 Icatibant Injektion pro HAE Attacke verabreicht.
Empfehlungen zum Dosierungsschema für Kinder, die jünger als 2 Jahre sind oder deren Körpergewicht weniger als 12 kg beträgt, können nicht gegeben werden, da die Sicherheit und Wirksamkeit in dieser Altersgruppe nicht erwiesen ist.
Ältere Patienten
Für Patienten im Alter von über 65 Jahren liegen begrenzte Daten vor.
Es wurde festgestellt, dass die systemische Exposition gegenüber Icatibant bei älteren Personen höher ist. Die Bedeutung dieses Sachverhalts für die Sicherheit von Icatibant Spirig HC ist nicht bekannt (s. Pharmakokinetik).
Patienten mit Leberfunktionsstörungen
Bei Patienten mit Leberfunktionsstörung ist keine Dosisanpassung erforderlich.
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit Nierenfunktionsstörung ist keine Dosisanpassung erforderlich.
Art der Anwendung
Icatibant Spirig HC ist für die subkutane Anwendung, vorzugsweise im Abdominalbereich, bestimmt.
Wegen des zu verabreichenden Volumens sollte die Icatibant Spirig HC-Injektionslösung langsam injiziert werden.
Jede Icatibant Spirig HC-Spritze ist nur zum einmaligen Gebrauch bestimmt.
Anleitung zur Anwendung siehe Anhang dieser Fachinformation oder Patienteninformation als Packungsbeilage.
Anwendung durch Pflegepersonen/Selbstanwendung
Die Entscheidung zur Anwendung von Icatibant Spirig HC durch eine Pflegeperson oder durch den Patienten selbst sollte nur von einem Arzt getroffen werden, der über Erfahrungen in der Diagnose und Therapie des hereditären Angioödems verfügt (siehe Warnhinweise und Vorsichtsmassnahmen).
Erwachsene
Icatibant Spirig HC kann von den Patienten selbst oder von Pflegepersonen angewendet werden, sofern sie zuvor durch medizinisches Fachpersonal in der subkutanen Injektionstechnik geschult worden sind.
Kinder und Jugendliche im Alter von 2-17 Jahren
Patienten, die zuvor noch nicht mit Icatibant Spirig HC behandelt wurden, sollten die erste Behandlung in einer medizinischen Einrichtung oder unter der Anleitung eines Arztes erhalten. Icatibant Spirig HC darf von Pflegepersonen nur verabreicht werden, wenn sie zuvor durch medizinisches Fachpersonal ausreichend in der subkutanen Injektionstechnik inklusive der notwendigen Schritte zum Aufziehen der Lösung in eine leere Spritze geschult worden sind. Im Falle einer unzureichenden Linderung oder eines Wiederauftretens der Symptome nach der Verabreichung durch eine Pflegeperson soll ärztlicher Rat eingeholt werden.
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile.
Warnhinweise und VorsichtsmassnahmenLaryngeale Attacken
Patienten mit laryngealen Attacken sollten nach der Injektion in einer geeigneten medizinischen Einrichtung behandelt werden, bis der Arzt eine Entlassung als sicher erachtet.
Ischämische Herzkrankheit
Unter ischämischen Bedingungen kann sich durch Blockierung des Bradykinin-Rezeptors Typ 2 eine Verschlechterung der Herzfunktion und eine Verminderung der Durchblutung der Herzkranzgefässe ergeben. Bei der Anwendung von Icatibant Spirig HC bei Patienten mit akuter ischämischer Herzkrankheit oder instabiler Angina pectoris ist daher Vorsicht angezeigt (s. Präklinische Daten).
Schlaganfall
Obgleich es Hinweise auf einen günstigen Effekt einer B2-Rezeptorblockade unmittelbar nach einem Schlaganfall gibt, besteht theoretisch die Möglichkeit, dass Icatibant die positive neuroprotektive Spätphasenwirkung von Bradykinin abschwächt. Entsprechend ist bei der Anwendung von Icatibant bei Patienten in den Wochen nach einem Schlaganfall Vorsicht angezeigt.
Anwendung durch Pflegepersonen/Selbstanwendung
Patienten, die zuvor noch nicht mit Icatibant Spirig HC behandelt wurden, sollten die erste Behandlung in einer medizinischen Einrichtung oder unter der Anleitung eines Arztes erhalten.
Im Falle einer unzureichenden Linderung oder eines Wiederauftretens der Symptome nach der Selbstbehandlung oder Verabreichung durch eine Pflegeperson wird empfohlen, dass der Patient oder die Pflegeperson ärztlichen Rat einholt. Bei Erwachsenen sollte die Anwendung mehrerer nacheinander gegebener Dosen, die zur Behandlung einer Attacke erforderlich sein können, in einer medizinischen Einrichtung erfolgen (siehe Dosierung/Anwendung). Zur Anwendung mehrerer nacheinander gegebener Dosen zur Behandlung einer Attacke bei Jugendlichen oder Kindern liegen keine Daten vor.
Patienten mit laryngealen Attacken sollten grundsätzlich medizinischen Rat einholen und in einer medizinischen Einrichtung beobachtet werden, auch wenn sie die Injektion zu Hause erhalten haben.
Kinder und Jugendliche
Über die Anwendung von Icatibant Spirig HC zur Behandlung von mehr als einer HAE-Attacke bei Kindern und Jugendlichen liegen nur begrenzte Erfahrungen vor.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Fertigspritze, d.h. es ist nahezu natriumfrei.
InteraktionenEs werden keine pharmakokinetischen Arzneimittelwechselwirkungen in Verbindung mit CYP450 erwartet (s. Pharmakokinetik).
Die gleichzeitige Anwendung von Icatibant Spirig HC und Angiotension Converting Enzyme (ACE)-Hemmern wurde nicht untersucht. ACE-Hemmer sind bei HAE-Patienten infolge einer möglichen Erhöhung des Bradykininspiegels kontraindiziert.
Schwangerschaft, StillzeitSchwangerschaft
Für Icatibant liegen keine klinischen Daten über exponierte Schwangere vor. Tierstudien zeigten Auswirkungen auf die Einnistung in den Uterus und während der Geburt (s. Präklinische Daten), aber das mögliche Risiko beim Menschen ist nicht bekannt.
Icatibant Spirig HC sollte während einer Schwangerschaft nur angewendet werden, wenn der mögliche Nutzen gegenüber dem möglichen Risiko für den Fetus überwiegt (z.B. zur Behandlung potenziell lebensgefährlicher laryngealer Attacken).
Stillzeit
Es ist nicht bekannt, ob Icatibant in die menschliche Muttermilch übertritt, aber es wird empfohlen, dass stillende Mütter, die Icatibant Spirig HC anwenden möchten, in den 12 Stunden nach der Behandlung nicht stillen.
Icatibant wird in die Milch laktierender Ratten in ähnlichen Konzentrationen wie im mütterlichen Blut sezerniert.
Fertilität
In einer Studie mit 39 gesunden Männern und Frauen, die insgesamt 9 Dosierungen zu je 30 mg alle 6 Stunden, verteilt auf jeweils 3 Dosierungen alle 3 Tage erhalten hatten, gab es gegenüber dem Ausgangswert weder bei Männern noch bei Frauen klinisch signifikante Veränderungen der basalen und GnRH-stimulierten Konzentration der Reproduktionshormone. Icatibant hatte bei Frauen keine signifikanten Auswirkungen auf die Progesteron-Konzentration in der Lutealphase und die Lutealfunktion sowie auf die Länge des Menstruationszyklus. Bei Männern hatte Icatibant keine signifikanten Auswirkungen auf die Anzahl, Motilität und Morphologie der Spermien. Es ist unwahrscheinlich, dass das für diese Studie verwendete Dosierungsschema im klinischen Einsatz beibehalten wird. Tierexperimentelle Studien zeigten Hinweise auf eine Reproduktionstoxizität. Icatibant hatte jedoch keine Auswirkungen auf die Fertilität (s. Präklinische Daten).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenIcatibant Spirig HC hat einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Nach Anwendung von Icatibant Spirig HC sind Abgeschlagenheit, Lethargie, Müdigkeit, Schläfrigkeit und Schwindel berichtet worden. Diese Symptome können als Ergebnis einer HAE-Attacke auftreten. Patienten sollte empfohlen werden, sich nicht ans Steuer eines Fahrzeugs zu setzen oder Maschinen zu bedienen, wenn sie sich müde fühlen oder ein Schwindelgefühl haben.
Unerwünschte WirkungenIn den für die Zulassung verwendeten klinischen Studien wurden insgesamt 999 HAE-Attacken mit 30 mg Icatibant behandelt, die subkutan durch medizinisches Fachpersonal verabreicht wurden. Icatibant 30 mg s.c. wurden durch medizinisches Fachpersonal an 129 gesunden Probanden und an 236 Patienten mit HAE verabreicht.
Fast alle Studienteilnehmer, die in klinischen Studien mit Icatibant subkutan behandelt worden sind, entwickelten Reaktionen an der Injektionsstelle (gekennzeichnet durch Hautirritation, Schwellung, Schmerzen, Erythem oder Brennen). Diese Reaktionen waren im Allgemeinen ihrem Schweregrad nach leicht bis mässig ausgeprägt, vorübergehend und klangen ohne weitere Massnahmen ab.
Die Häufigkeit der unten aufgeführten unerwünschten Wirkungen ist wie folgt definiert: Sehr häufig (≥1/10); häufig (> 1/100, < 1/10); gelegentlich (> 1/1'000, < 1/100); selten (> 1/10'000, < 1/1'000); sehr selten (< 1/10'000); nicht bekannt (die Häufigkeit kann anhand der verfügbaren Daten nicht geschätzt werden).
Erkrankungen des Nervensystems
Häufig: Schwindelgefühl, Kopfschmerzen
Erkrankungen des Gastrointestinaltrakts
Häufig: Übelkeit
Erkrankungen der Haut und des Unterhautgewebes
Häufig: Ausschlag, Erythem, Pruritus
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Reaktionen an der Injektionsstelle*
Häufig: Fieber
Untersuchungen
Häufig: Transaminasen erhöht
* blauer Fleck an der Injektionsstelle, Injektionsstelle Hämatom, Brennen an der Injektionsstelle, Erythem an der Injektionsstelle, Injektionsstelle Hypästhesie, Injektionsstelle gereizt, Injektionsstelle Taubheitsgefühl, Injektionsstelle Ödem, Schmerzen an der Injektionsstelle, Injektionsstelle Druckgefühl, Injektionsstelle juckend, Schwellung an der Injektionsstelle, Urtikaria an der Injektionsstelle und Wärme an der Injektionsstelle.
Kinder und Jugendliche
In den klinischen Studien erhielten insgesamt 32 pädiatrische Patienten mit HAE (8 Kinder im Alter von 2 bis 11 Jahren und 24 Jugendliche im Alter von 12 bis 17 Jahren) eine Behandlung mit Icatibant. Einunddreissig Patienten erhielten eine Einzeldosis Icatibant und 1 Patient (ein Jugendlicher) erhielt Icatibant zur Behandlung von zwei HAE-Attacken (zwei Dosen insgesamt). Icatibant wurde als subkutane Injektion in einer Dosierung von 0,4 mg/kg Körpergewicht bis zu einer Maximaldosis von 30 mg verabreicht.
Bei der Mehrzahl der pädiatrischen Patienten traten nach subkutaner Injektion von Icatibant Reaktionen an der Injektionsstelle auf (z.B. Erythem, Schwellung, Brennen, Schmerzen der Haut und Juckreiz/Pruritus). Diese waren in der Regel leicht bis mässig stark ausgeprägt und entsprachen den bei Erwachsenen berichteten Reaktionen (wie Erythem, Schwellung, Brennen, Juckreiz, Wärmegefühl und Schmerzen der Haut). Bei 2 pädiatrischen Patienten traten als schwer bewertete Reaktionen an der Injektionsstelle auf, die innerhalb von 6 Stunden vollständig abklangen. Dabei handelte es sich um Erythem, Schwellung, Brennen und Wärmegefühl.
Während der klinischen Studien wurden keine klinisch signifikanten Veränderungen der Reproduktionshormone beobachtet.
Unerwünschte Wirkungen nach Markteinführung
Erkrankungen der Haut und des Unterhautgewebes
Nicht bekannt: Urtikaria
Immunogenität
In den kontrollierten Studien der Phase III wurden bei wiederholter Behandlung bei Erwachsenen in seltenen Fällen vorübergehend positive Testergebnisse auf Anti-Icatibant-Antikörper beobachtet. Die Wirksamkeit blieb bei allen Patienten erhalten. Ein mit Icatibant behandelter Patient wurde vor und nach der Icatibant–Therapie positiv auf Anti-Icatibant-Antikörper getestet. Der Patient wurde 5 Monate lang nachuntersucht; dabei waren die späteren Tests auf Anti-Icatibant-Antikörper negativ. Unter Icatibant Spirig HC wurden keine Hypersensibilitätsreaktionen oder anaphylaktische Reaktionen berichtet.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs liegen keine klinischen Daten zu Überdosierungen vor.
Eine Dosis von 3,2 mg/kg intravenös (etwa das 8-fache der therapeutischen Dosis) verursachte bei gesunden Personen ein transientes Erythem, Jucken, Hitzegefühl oder Hypotonie. Ein therapeutisches Eingreifen war nicht erforderlich.
Eigenschaften/WirkungenATC-Code
B06AC02
Wirkungsmechanismus
HAE (eine autosomal dominante Krankheit) wird durch Fehlen bzw. eine Funktionsstörung des C1- Esteraseinhibitors verursacht. HAE-Attacken gehen mit einer erhöhten Ausschüttung von Bradykinin einher, bei dem es sich um den wichtigsten Faktor bei der Entwicklung klinischer Symptome handelt.
HAE äussert sich in Form intermittierender Attacken eines subkutanen und/oder submukosalen Ödems der oberen Atemwege, der Haut und des Magendarmtraktes. Eine Attacke dauert üblicherweise 2 bis 5 Tage.
Icatibant ist ein selektiver kompetitiver Antagonist des Bradykininrezeptors Typ 2 (B2). Es handelt sich um ein synthetisches Dekapeptid mit einer ähnlichen Struktur wie Bradykinin, aber mit 5 nicht proteinogenen Aminosäuren. Bei HAE sind erhöhte Bradykininkonzentrationen die wichtigsten Einflussfaktoren bei der Entwicklung klinischer Symptome.
Pharmakodynamik
Bei gesunden jungen Personen wurden durch Anwendung von Icatibant in einer Dosierung von 0,8 mg/kg über 4 Stunden, von 1,5 mg/kg/Tag oder 0,15 mg/kg/Tag für 3 Tage die Entwicklung einer bradykinininduzierten Hypotonie, Vasodilatation und Reflextachykardie verhindert. Icatibant erwies sich als kompetitiver Antagonist, wenn die Bradykinin-Testdosis auf das 4-fache erhöht wurde.
Klinische Wirksamkeit
Daten zur Wirksamkeit stammten aus einer ersten offenen Phase-II-Studie und drei kontrollierten Phase-III-Studien.
Die klinischen Studien der Phase III (FAST-1 und FAST-2) waren randomisierte, kontrollierte Doppelblindstudien mit – bis auf das verwendete Vergleichspräparat – gleichem Design (eine mit oraler Tranexamsäure als Vergleichspräparat und eine placebokontrolliert).
Es wurden insgesamt 130 Patienten randomisiert und erhielten entweder eine Icatibant-Dosis von 30 mg (63 Patienten) oder ein Vergleichspräparat (entweder Tranexamsäure, 38 Patienten, oder ein Placebo, 29 Patienten). Spätere HAE-Attacken wurden in einer offenen Anschlussstudie behandelt. Patienten mit Symptomen eines laryngealen Ödems erhielten eine offene Behandlung mit Icatibant. In den Phase-III-Studien war der primäre Wirksamkeits-endpunkt die Zeit bis zum Einsetzen der Symptomlinderung, was mithilfe einer visuellen Analogskala (VAS) festgestellt wurde. Die Tabelle 2 zeigt die Wirksamkeitsergebnisse für diese Studien.
FAST-3 war eine randomisierte, placebokontrollierte Parallelgruppenstudie mit 98 erwachsenen Patienten im Alter von durchschnittlich 36 Jahren. Die Patienten wurden randomisiert auf zwei Gruppen verteilt, die entweder Icatibant 30 mg oder Placebo als subkutane Injektion erhielten. Eine Untergruppe der Patienten in dieser Studie entwickelte akute HAE-Attacken unter der Behandlung mit Androgenen, Antifibrinolytika bzw. C1-Inhibitoren. Der primäre Wirksamkeitsendpunkt war die Zeit bis zum Einsetzen der Symptomlinderung. Die Beurteilung erfolgte dabei mithilfe einer dreiteiligen zusammengesetzten visuellen Analogskala (VAS-3), auf der die Schwellung der Haut, Hautschmerzen und Bauchschmerzen beurteilt wurden. Die Tabelle 3 zeigt die Wirksamkeitsergebnisse für FAST-3.
In diesen Studien war der mittlere Zeitraum bis zum Einsetzen der Symptomlinderung bei Patienten unter Icatibantbehandlung kürzer (2,0 h, 2,5 h und 2.0 h) als bei Gabe von Tranexamsäure (12,0 h) und Placebo (4,6 h und 19,8 h). Der Behandlungseffekt von Icatibant wurde durch die sekundären Wirksamkeitsendpunkte bestätigt.
Unabhängig von der Altersgruppe, vom Geschlecht, der Rasse und dem Gewicht sowie von der Anwendung oder Nichtanwendung von Androgenen oder Antifibrinolytika waren die Zeit bis zum Einsetzen der Symptomlinderung und die Zeit bis zum Einsetzen der Linderung des Primärsymptoms in der zusammenfassenden Analyse dieser kontrollierten Phase-III-Studien gleich.
In den kontrollierten Phase-III-Studien war die Reaktion auch über wiederholte Attacken hinweg gleichbleibend. Insgesamt 237 Patienten erhielten 1'386 Dosierungen mit 30 mg Icatibant zur Behandlung von 1'278 akuten HAE-Attacken. Bei den ersten 15 mit Icatibant behandelten Attacken (1'114 Dosen für 1'030 Attacken) waren die medianen Zeiten bis zum Einsetzen der Symptomlinderung bei den Attacken ähnlich (2,0 bis 2,5 h). Dabei wurden 92,4 % dieser HAE-Attacken mit einer einzigen Dosis Icatibant behandelt.
Tabelle 2: Wirksamkeitsergebnisse von FAST-1 und FAST-2
|
Kontrollierte klinische Studie von ICATIBANT im Vergleich zu Tranexamsäure bzw. Placebo: Wirksamkeitsergebnisse
| |
FAST-2
|
FAST-1
| |
|
Icatibant
|
Tranexam-säure
|
|
Icatibant
|
Placebo
| |
Anzahl der Patienten in der ITT-Population
|
36
|
38
|
Anzahl der Patienten in der ITT-Population
|
27
|
29
| |
Basis-VAS-Wert (mm)
|
63,7
|
61,5
|
Basis-VAS-Wert (mm)
|
69,3
|
67,7
| |
Änderung zwischen Basis-Wert und 4 Stunden
|
-41,6
|
-14,6
|
Änderung zwischen Basis-Wert und 4 Stunden
|
-44,8
|
-23,5
| |
Unterschied zwischen den Behandlungen (95 % CI, p-Wert)
|
-27,8 (-39,4, -16,2) p < 0,001
|
Unterschied zwischen den Behandlungen (95 % CI, p-Wert)
|
-23,3 (-37,1, -9,4) p = 0,002
| |
Änderung zwischen Basis-Wert und 12 Stunden
|
-54,0
|
-30,3
|
Änderung zwischen Basis-Wert und 12 Stunden
|
-54,2
|
-42,4
| |
Unterschied zwischen den Behandlungen (95 % CI, p-Wert)
|
-24,1 (-33,6, -14,6) p < 0,001
|
Unterschied zwischen den Behandlungen (95 % CI, p-Wert)
|
-15,2 (-28,6, -1,7) p = 0,028
| |
Mittlere Zeit bis zum Einsetzen der Symptomlinderung (Stunden)
|
|
|
Mittlere Zeit bis zum Einsetzen der Symptomlinde-rung (Stunden)
|
|
| |
Alle Episoden
(N = 74)
|
2,0
|
12,0
|
Alle Episoden
(N = 56)
|
2,5
|
4,6
| |
Ansprechrate (%, VI) 4 Stunden nach Beginn der Behandlung
|
|
|
Ansprechrate (%, VI) 4 Stunden nach Beginn der Behandlung
|
|
| |
Alle Episoden
(N = 74)
|
80,0
(63,1, 91,6)
|
30,6
(16,3, 48,1)
|
Alle Episoden
(N = 56)
|
66,7
(46,0, 83,5)
|
46,4
(27,5, 66,1)
| |
Mittlere Zeit bis zum Einsetzen der Symptomlinde-rung: alle Symptome (Std.):
|
|
|
Mittlere Zeit bis zum Einsetzen der Symptomlinde-rung: alle Symptome (Std.):
|
|
| |
Bauchschmerzen
|
1,6
|
3,5
|
Bauchschmerzen
|
2,0
|
3,3
| |
Hautschwellung
|
2,6
|
18,1
|
Hautschwellung
|
3,1
|
10,2
| |
Hautschmerzen
|
1,5
|
12,0
|
Hautschmerzen
|
1,6
|
9,0
| |
Mittlere Zeit bis zum fast vollständigen Abklingen der Symptome (Std.)
|
|
|
Mittlere Zeit bis zum fast vollständigen Abklingen der Symptome (Std.)
|
|
| |
Alle Episoden
(N = 74)
|
10,0
|
51,0
|
Alle Episoden
(N = 56)
|
8,5
|
19,4
| |
Mittlere Zeit bis zur Regression der Symptome, nach Patient (Std.)
|
|
|
Mittlere Zeit bis zur Regression der Symptome, nach Patient (Std.)
|
|
| |
Alle Episoden
(N = 74)
|
0,8
|
7,9
|
Alle Episoden
(N = 56)
|
0,8
|
16,9
| |
Mittlere Zeit bis zur Verbesserung der Gesamtverfas-sung des Patienten, nach Arzt (Std.)
|
|
|
Mittlere Zeit bis zur Verbesserung der Gesamtverfas-sung des Patienten, nach Arzt (Std.)
|
|
| |
Alle Episoden
(N = 74)
|
1,5
|
6,9
|
Alle Episoden
(N = 56)
|
1,0
|
5,7
|
Tabelle 3: Wirksamkeitsergebnisse für FAST-3
|
Wirksamkeitsergebnisse: FAST-3, kontrollierte Phase – ITT-Population
| |
Endpunkt
|
Statistische Masszahl
|
Icatibant
|
Placebo
|
p-Werte
| |
|
|
(n = 43)
|
(n = 45)
|
| |
Primärer Endpunkt
| |
Zeit bis zum Einsetzen der Symptomlinderung – mehrteilige VAS (Stunden)
|
Medianwert
|
2,0
|
19,8
|
<0,001
| |
Andere Endpunkte
| |
Zeit bis zum Einsetzen der Linderung des Primärsymptoms (Stunden)
|
Medianwert
|
1,5
|
18,5
|
<0,001
| |
Veränderung des zusammengesetzten VAS-Wertes 2 Std. nach der Behandlung
|
Mittelwert
|
-19,74
|
-7,49
|
<0,001
| |
Veränderung des zusammengesetzten Probanden-Eigenbewertungswerts nach 2 Stunden
|
Mittelwert
|
-0,53
|
-0,22
|
<0,001
| |
Veränderung des zusammengesetzten Prüfarzt-Bewertungs-werts nach 2 Stunden
|
Mittelwert
|
-0,44
|
-0,19
|
<0,001
| |
Zeit bis zur nahezu vollständigen Symptomlinderung (Stunden)
|
Medianwert
|
8,0
|
36,0
|
0,012
| |
Zeit bis zur ersten Symptomverbesserung gemäss Probanden-Eigenbewertung (Stunden)
|
Medianwert
|
0,8
|
3,5
|
<0,001
| |
Zeit bis zur ersten visuellen Symptom-verbesserung gemäss Prüfarztbewertung (Stunden)
|
Medianwert
|
0,8
|
3,4
|
<0,001
|
Insgesamt 66 Patienten wurden in diesen kontrollierten klinischen Studien der Phase III wegen HAE-Attacken behandelt, die den Kehlkopf betrafen. Die Ergebnisse waren in Bezug auf die Zeit bis zum Einsetzen der Symptomlinderung ähnlich wie bei Patienten mit nicht den Kehlkopf betreffenden HAE-Attacken.
Sicherheit und Wirksamkeit bei pädiatrischen Patienten
Mit insgesamt 32 Patienten wurde eine offene, nicht randomisierte einarmige Studie (HGT-FIR-086) durchgeführt. Mit einer Ausnahme erhielten alle Patienten einmalig subkutan eine Dosis Icatibant (0,4 mg/kg Körpergewicht, bis zu einer Maximaldosis von 30 mg). Die Mehrzahl der Patienten wurde mindestens 6 Monate lang nachbeobachtet. Elf Patienten waren im präpubertären Alter und 21 Patienten entweder im pubertären oder postpubertären Alter.
Die für die Beurteilung der Wirksamkeit relevante Population bestand aus 22 Patienten (11 prä-pubertäre und 11 pubertäre/postpubertäre), die wegen einer akuten HAE-Attacke mit Icatibant behandelt worden waren.
Primärer Wirksamkeitsendpunkt war die Zeit bis zum Einsetzen einer Symptomlinderung (time to onset of symptom relief, TOSR). Die Beurteilung des Schweregrads der Symptome erfolgte durch den Prüfarzt gemäss einer vordefinierten zusammengesetzten Symptom-Bewertungsskala (0 bis 5). Die Zeit bis zum Einsetzen einer Symptomlinderung war definiert als die Zeitdauer (in Stunden) bis zum Eintreten einer Verbesserung der Symptome in einer Grössenordnung von 20 %.
Insgesamt betrug die mediane Zeit bis zum Einsetzen einer Symptomlinderung 1,0 Stunden (95 % Konfidenzintervall, 1,0 1,1 Stunden). An den Zeitpunkten 1 bzw. 2 Stunden nach der Behandlung hatte bei etwa 50 % bzw. 90 % der Patienten eine Symptomlinderung eingesetzt.
Insgesamt betrug die mediane Zeit bis zu einer grösstmöglichen Symptomlinderung (frühester posttherapeutischer Zeitpunkt, zu dem alle Symptome entweder leicht ausgeprägt oder abgeklungen waren) 1,1 Stunden (95 % Konfidenzintervall, 1,0 2,0 Stunden).
PharmakokinetikDie Pharmakokinetik von Icatibant wurde in Studien mit intravenöser und subkutaner Anwendung bei gesunden Freiwilligen und Patienten charakterisiert. Das pharmakokinetische Profil von Icatibant bei Patienten mit HAE ist dem bei gesunden Freiwilligen ähnlich.
Absorption
Nach subkutaner Anwendung beträgt die absolute Bioverfügbarkeit von Icatibant 97 %. Es dauert etwa 30 Minuten, bis eine maximale Konzentration erreicht ist.
Distribution
Das Verteilungsvolumen (Vss) von Icatibant liegt bei etwa 20 bis 25 Liter. Die Bindung an Plasmaproteine liegt bei 44 %.
Metabolismus
Der Grossteil von Icatibant wird von proteolytischen Enzymen in inaktive Metaboliten umgewandelt, die überwiegend im Harn ausgeschieden werden.
In vitro-Studien haben bestätigt, dass Icatibant nicht durch oxidative Stoffwechselwege abgebaut wird, kein Inhibitor wichtiger Cytochrom P450 (CYP)-Isoenzyme (CYP 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 und 3A4) ist und CYP 1A2 und 3A4 nicht induziert.
Elimination
Icatibant wird hauptsächlich durch Verstoffwechselung eliminiert, während weniger als 10 % einer Icatibantdosis unverändert mit dem Harn ausgeschieden werden. Die Clearance beträgt ungefähr 15-20 l/h und ist von der Dosis unabhängig. Die terminale Halbwertszeit im Plasma beträgt etwa 1 bis 2 Stunden.
Kinetik spezieller Patientengruppen
Ältere Patienten
Die Daten deuten auf einen altersbezogenen Rückgang bei der Clearance hin, woraus sich eine um 50 - 60 % höhere Exposition bei älteren Patienten (75 - 80 Jahre) im Vergleich zu Patienten im Alter von 40 Jahren ergibt.
Geschlecht
Gewichtsbereinigte Daten deuten darauf hin, dass hinsichtlich der Clearance kein Unterschied zwischen Männern und Frauen besteht.
Leber- und Nierenfunktionsstörung
Limitierte Daten deuten an, dass eine Icatibant-Behandlung durch eine Leber- oder Nierenfunktionsstörung nicht beeinflusst wird.
Ethnische Zugehörigkeit
Die Datenlage zur Wirkung bei einzelnen Ethnien ist begrenzt. Die verfügbaren Daten weisen nicht auf Unterschiede hinsichtlich der Clearance zwischen den verschiedenen Ethnien hin.
Kinder und Jugendliche
In der Studie HGT-FIR-086 wurden die pharmakokinetischen Eigenschaften von Icatibant bei pädiatrischen HAE-Patienten beschrieben (siehe Abschnitt Pharmakodynamik, klinische Wirksamkeit). Nach Verabreichung einer einmaligen subkutanen Dosis (0,4 mg/kg bis maximal 30 mg) beträgt die Zeit bis zur höchsten Konzentration etwa 30 Minuten. Die terminale Halbwertszeit beträgt etwa 2 Stunden. Hinsichtlich der Exposition gegenüber Icatibant wurden keine Unterschiede zwischen HAE-Patienten mit bzw. ohne eine Attacke beobachtet. Wie die Modellierung der Populationspharmakokinetik zeigte, – für die sowohl Daten von Erwachsenen als auch von pädiatrischen Patienten verwendet wurden – besteht zwischen der Clearance von Icatibant und dem Körpergewicht ein Zusammenhang. Dabei wurden in der pädiatrischen HAE-Population niedrigere Clearance-Werte bei geringerem Körpergewicht beobachtet. Die Modellierung anhand einer Dosierung nach Körpergewicht zeigte, dass die erwartete Exposition gegenüber Icatibant in der pädiatrischen HAE-Population (siehe Abschnitt Dosierung/Anwendung) geringer als die Exposition ist, die in den mit erwachsenen HAE-Patienten durchgeführten Studien beobachtet wurde.
Präklinische DatenSicherheitspharmakologie
Untersuchungen in vitro (hERG Kanal) und an verschiedenen Modellen in vivo (ventrikuläre Stimulation, körperliche Belastung sowie Koronarligatur am Hund im Vergleich zur gesunden Kontrolle) ergaben keine Hinweise auf eine Beeinflussung hämodynamischer Parameter durch Icatibant. An mehreren Tiermodellen wurde jedoch beobachtet, dass Icatibant eine kardiale Ischämie verstärkt, obgleich bei akuter Ischämie ein nachteiliger Effekt nicht einheitlich aufgezeigt werden konnte.
Toxizität bei wiederholter Verabreichung
Bei Ratten und Hunden sind Studien mit Mehrfachdosierung über einen Zeitraum von bis zu 6 Monaten bzw. bis zu 9 Monaten durchgeführt worden. Bei Ratten wie bei Hunden wurde eine dosisabhängige Verminderung der Spiegel der zirkulierenden Sexualhormone beobachtet. Zudem kam es bei wiederholter Anwendung von Icatibant zu einer reversiblen Verzögerung der Geschlechtsreife.
Die anhand der Fläche unter der Kurve (AUC) definierte maximale tägliche Exposition bei der höchsten Dosis, bei der noch keine schädliche Wirkung erkennbar ist (NOAEL-Konzentration), betrug in der über 9 Monate laufenden Studie bei Hunden das 2,3-fache des AUC-Werts bei erwachsenen Menschen nach einer subkutanen Dosis von 30 mg. Eine NOAEL-Konzentration war in der Studie an Ratten nicht messbar, jedoch zeigten sämtliche aus dieser Studie gewonnenen Ergebnisse vollständige oder partiell reversible Auswirkungen bei den behandelten Ratten. Bei allen an Ratten getesteten Dosen wurde eine Nebennierenhypertrophie beobachtet, die nach Einstellung der Behandlung mit Icatibant zurückging. Die klinische Relevanz der Nebennierenbefunde ist nicht bekannt.
Reproduktionstoxizität
Icatibant hatte keine Auswirkungen auf die Fertilität von männlichen Mäusen (Höchstdosis 80,8 mg/kg/Tag) und Ratten (Höchstdosis 10 mg/kg/Tag).
Bei Ratten (Höchstdosis 25 mg/kg/Tag) und Kaninchen (Höchstdosis 10 mg/kg/Tag) war Icatibant nach subkutaner Injektion während der frühen embryonalen und fetalen Entwicklung nicht teratogen. Icatibant ist ein wirksamer Antagonist von Bradykinin, und eine Behandlung in hoher Dosierung kann daher in der Frühphase einer Trächtigkeit Auswirkungen auf den Einnistungsvorgang in den Uterus und auf die anschliessende Stabilität des Keims im Uterus haben. Diese uterinen Effekte äussern sich auch in einem späten Trächtigkeitsstadium, da Icatibant bei Ratten tokolytisch wirkt und zu einem verzögerten Geburtseintritt führt mit erhöhter Belastung für den Fetus (fetaler Distress) und Tod unter der Geburt bei hoher Dosis (10 mg/kg/Tag). Es wurden keine Auswirkungen auf die postnatale Entwicklung der neugeborenen Ratten festgestellt.
In der Pivotalstudie zur Toxizität bei Jungtieren, in der nicht geschlechtsreife Ratten 7 Wochen lang täglich 3 mg/kg/Tag erhielten, wurde eine Atrophie der Hoden und Nebenhoden beobachtet. Die beobachteten Mikroskopiebefunde waren teilweise reversibel. Bei geschlechtsreifen Ratten und Hunden wurden ähnliche Wirkungen von Icatibant auf das reproduktive Gewebe beobachtet. Diese Befunde entsprachen den berichteten Wirkungen auf die Gonadotropine und erwiesen sich in den anschliessenden behandlungsfreien Zeiträumen als reversibel.
Karzinogenität
Zur Beurteilung des karzinogenen Potentials von Icatibant wurden 2-Jahres-Studien bei Mäusen und Ratten durchgeführt. Bei subkutanen Icatibant Dosen bis zu 15 mg/kg/Tag (zweimal wöchentlich) resp. 6 mg/kg/Tag (täglich) (ungefähr 3-fach und 2-fach höher als die für den Menschen höchste maximale empfohlene Tagesdosis [drei 30 mg Dosen an einem einzigen Tag] auf der Basis der AUC) wurde kein Hinweis auf Kanzerogenität beobachtet.
Mutagenität
In einer Reihe von Standardtests in vitro und in vivo war Icatibant nicht genotoxisch.
Sonstige HinweiseEine Fertigspritze zu 3 ml enthält Icatibant-Acetat entsprechend 30 mg Icatibant.
Die Lösung sollte klar und farblos und frei von Schwebeteilchen sein.
Wenn Sie verfärbt ist oder Klumpen, Flocken oder Schwebeteilchen enthält, darf sie nicht verwendet werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Nicht über 30°C lagern. Nicht einfrieren.
In der Originalverpackung aufbewahren.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Die Lösung sollte klar und farblos und frei von Schwebeteilchen sein.
Wenn sie verfärbt ist oder Klumpen, Flocken oder Partikel enthält, darf sie nicht verwendet werden.
Anwendung bei Kindern und Jugendlichen
Die angemessene anzuwendende Dosis wird anhand des Körpergewichts bestimmt (siehe Dosierung/Anwendung).
Wenn die erforderliche Dosis weniger als 30 mg (3 ml) beträgt, werden zur Entnahme und Verabreichung der geeigneten Dosis zusätzlich folgende Gegenstände benötigt:
·Adapter (proximaler und/oder distaler weiblicher Luer-Lock-Anschluss/Verbinder)
·3-ml-Spritze (empfohlen) mit Graduierung
Die Icatibant-Fertigspritze und alle übrigen Teile sind zum einmaligen Gebrauch bestimmt.
Nicht verwendetes Arzneimittel oder Abfallmaterial sind entsprechend zu entsorgen.
Alle Nadeln und Spritzen müssen nach Gebrauch in einem durchstichsicheren Behältnis entsorgt werden.
Zulassungsnummer68263 (Swissmedic)
PackungenFertigspritze (3-ml-Fertigspritze, Typ I Glas) mit Kolbenstopfen (mit Fluorcarbonpolymer beschichtetes Brombutyl).
Packung mit
1 Fertigspritze zu 3 ml
1 Injektionsnadel (25 G; 16 mm).
[B]
ZulassungsinhaberinSpirig HealthCare AG, 4622 Egerkingen.
Stand der InformationDezember 2019
Anhang 1
Anweisungsschritte für die Injektion
1) Allgemeine Informationen
·Reinigen Sie vorher den Arbeitsbereich (Oberfläche), den Sie benutzen wollen.
·Waschen Sie vorher Ihre Hände mit Wasser und Seife.
·Öffnen Sie die Schale, indem Sie den Verschluss abziehen.
·Nehmen Sie die Icatibant Spirig HC Fertigspritze aus der Schale heraus.
·Schrauben Sie die Kappe von dem einen Ende der Fertigspritze ab.
·Nach dem Abschrauben der Kappe legen Sie die Fertigspritze beiseite.
2a) Vorbereiten der Spritze für
Kinder und Jugendliche (2-17 Jahre)
mit einem Gewicht von 65 kg oder weniger:
Wichtige Hinweise für medizinisches Fachpersonal und Pflegepersonen:
Wenn die Dosis weniger als 3 ml (30 mg) beträgt, werden zur Entnahme und Verabreichung der geeigneten Dosis folgende Gegenstände benötigt:
a.Icatibant Spirig HC-Fertigspritze (mit Icatibant-Lösung)
b.Anschlussstück (Luer-Lock-Adapter)
c.3-ml-Spritze mit Graduierung
Das benötigte Injektionsvolumen in ml muss in eine leere 3-ml-Spritze mit Graduierung aufgezogen werden (siehe Tabelle 1).
Tabelle 1: Dosierungsschema für Kinder und Jugendliche
|
Körpergewicht
|
Injektionsvolumen (Dosis)
| |
12 kg bis 25 kg
|
1,0 ml
| |
26 kg bis 40 kg
|
1,5 ml
| |
41 kg bis 50 kg
|
2,0 ml
| |
51 kg bis 65 kg
|
2,5 ml
|
Patienten, die mehr als 65 kg wiegen, verwenden den gesamten Inhalt der Icatibant Spirig HC Fertigspritze (3 ml).
Fragen Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin oder das medizinische Fachpersonal, wenn Sie nicht genau wissen, wieviele ml der Injektionslösung Sie aufziehen müssen.
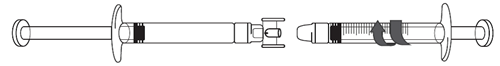
1.Entfernen Sie die Schutzkappen an beiden Enden des Anschlussstücks.
Achten Sie darauf, die Enden des Anschlussstücks und die Spritzenspitzen nicht zu berühren. So vermeiden Sie Verunreinigungen.
2.Schrauben Sie das Anschlussstück auf die Icatibant Spirig HC Fertigspritze.
3.Befestigen Sie die 3 ml-Spritze mit Graduierung am anderen Ende des Anschlussstücks. Beide Verbindungen müssen festsitzen.
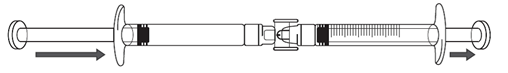
Die Icatibant-Lösung in die Spritze mit Graduierung umfüllen:
1.Um mit dem Umfüllen der Icatibant-Lösung zu beginnen, drücken Sie den Kolben der Fertigspritze (ganz links in der Abbildung unten).
2.Falls die Icatibant-Lösung noch nicht in die Spritze mit Graduierung übergeht, ziehen Sie den Kolben der Spritze mit Graduierung leicht, bis die Icatibant-Lösung in die Spritze mit Graduierung zu fliessen beginnt (siehe Abbildung unten).
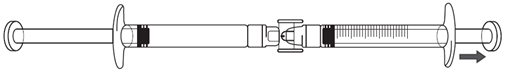
3.Drücken Sie den Kolben der Fertigspritze solange, bis das benötigte Injektionsvolumen in ml (Dosis) in die Spritze mit Graduierung umgefüllt worden ist. Hinweise zur Dosis und Injektionsvolumen siehe Tabelle 1 oben.
Falls sich in der Spritze mit Graduierung Luft befindet:
·Drehen Sie die miteinander verbundenen Spritzen um, sodass die Fertigspritze oben ist (siehe Abbildung unten).
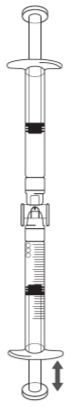
·Drücken Sie den Kolben der Spritze mit Graduierung, sodass eventuell vorhandene Luft in die Fertigspritze zurückgeleitet wird (diesen Schritt bei Bedarf mehrmals wiederholen).
·Entnehmen Sie das benötigte Volumen an Icatibant-Lösung.
4.Nehmen Sie die Fertigspritze und das Anschlussstück von der Spritze mit Graduierung ab.
5.Entsorgen Sie die Fertigspritze und das Anschlussstück in ein Behältnis für spitzige Gegenstände.
2b) Vorbereiten der Spritze (Fertigspritze für Erwachsene, Spritze mit Graduierung für Jugendliche und Kinder) und der Injektionsnadel
Alle Patienten (Erwachsene, Jugendliche und Kinder)
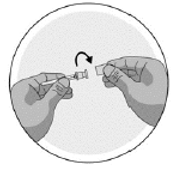
·Nehmen Sie die Nadelkappe aus der Blisterpackung heraus. Nehmen Sie die Nadel nicht aus der Nadelkappe.
·Drehen Sie den Deckel der Nadelkappe, um das Siegel zu brechen (die Nadel sollte sich noch in der Nadelkappe befinden).
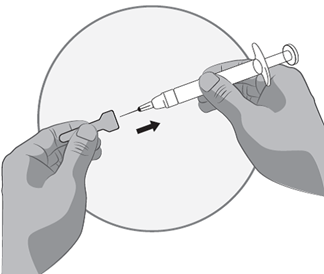
·Nehmen Sie die Spritze (Fertigspritze für Erwachsene, Spritze mit Graduierung für Jugendliche und Kinder bis 65 kg) fest in die Hand. Befestigen Sie dann die Nadel, die immer noch in der Nadelkappe ist, sorgfältig an der Spritze mit der farblosen Lösung.
·Schrauben Sie dazu die Spritze auf die Nadel, die immer noch fest in der Nadelkappe sitzt. Die Nadel mit der Nadelkappe ist nun an der Spritze befestigt.
·Halten Sie die Spritze am Spritzengehäuse und ziehen Sie die Nadel von der Nadelkappe ab, indem Sie am Gehäuse der Spritze ziehen. Nicht am Kolben ziehen.
·Die Spritze ist nun fertig für die Injektion.
3) Vorbereiten der Injektionsstelle
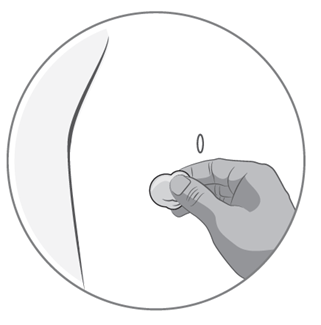
·Wählen Sie die Injektionsstelle. Dies sollte eine Hautfalte an einer Seite des Bauches sein, etwa 5-10 cm unterhalb Ihres Nabels. Der Bereich sollte mindestens 5 cm von etwaigen Narben entfernt sein. Wählen Sie keine Stelle, die schmerzt, oder an der sich Blutergüsse oder Schwellungen befinden.
·Reinigen Sie die Injektionsstelle mit einem mit Desinfektionsalkohol getränkten Wattebausch. Warten Sie, bis die Stelle getrocknet ist.
4) Injizieren der Lösung
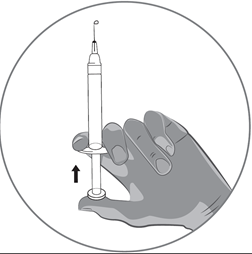
·Halten Sie die Spritze vertikal zwischen zwei Fingern einer Hand, wobei sich der Daumen unter dem Kolben befindet.
·Sorgen Sie dafür, dass sich keine Luftblase in der Spritze befindet, indem Sie den Kolben drücken, bis der erste Tropfen der Lösung an der Nadelspitze sichtbar wird.
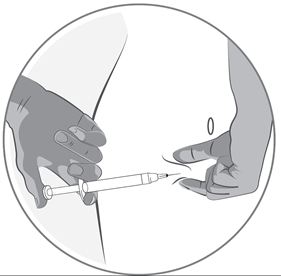
·Halten Sie die Spritze in einem Winkel von 45-90 Grad zu Ihrer Haut, wobei die Nadel in Richtung Haut zeigt.
·Während Sie mit einer Hand die Spritze halten, fassen Sie mit der anderen Hand an der zuvor desinfizierten Injektionsstelle vorsichtig eine Hautfalte zwischen Daumen und Fingern.
·Während Sie die Hautfalte festhalten, führen Sie die Spritze in Richtung Haut und stechen Sie die Nadel schnell in die Hautfalte ein.
·Drücken Sie langsam den Kolben der Spritze. Halten Sie dabei Ihre Hand ruhig, bis die gesamte Flüssigkeit in die Haut injiziert wurde und sich keine Flüssigkeit mehr in der Spritze befindet.
·Drücken Sie den Kolben so langsam, dass der Vorgang etwa 30 Sekunden dauert.
·Lassen Sie die Hautfalte los und ziehen Sie vorsichtig die Nadel heraus.
5) Entsorgung des Injektionsmaterials
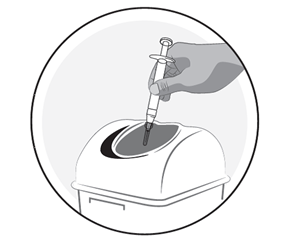
·Entsorgen Sie die Spritze, die Nadel und die Nadelkappe in ein Behältnis für spitzige Gegenstände, um zu vermeiden, dass sich jemand damit verletzt.
|