Eigenschaften/WirkungenATC-Code
L04AC21
Wirkungsmechanismus
Bimekizumab ist ein humanisierter monoklonaler IgG1/κ-Antikörper der selektiv und mit hoher Affinität an die IL-17A-, IL-17F- und IL-17AF-Zytokine bindet und so deren Interaktion mit dem IL-17RA/IL-17RC-Rezeptorkomplex blockiert. Erhöhte Konzentrationen von IL-17A und IL-17F wurden mit der Pathogenese von verschiedenen immunvermittelten entzündlichen Erkrankungen, einschliesslich Plaque Psoriasis, Psoriasis-Arthritis und axialer Spondyloarthritis, in Verbindung verbracht. IL-17A und IL-17F kooperieren und/oder synergieren mit anderen entzündlichen Zytokinen bei der Auslösung einer Entzündung. IL-17F wird in erheblichen Mengen von Zellen des angeborenen Immunsystemszellen produziert. Diese Produktion kann unabhängig von IL-23 erfolgen. Bimekizumab hemmt die proinflammatorischen Zytokine, was zu einer Normalisierung der entzündeten Haut und einer deutlichen Abnahme der lokalen und systemischen Entzündung sowie in der Folge zu einer Besserung der klinischen Anzeichen und Symptome der Psoriasis, Psoriasis-Arthritis und axialen Spondyloarthritis führt. In In-vitro-Modellen zeigte Bimekizumab eine stärkere hemmende Wirkung auf die mit Psoriasis verbundene Genexpression, Zytokinproduktion, Migration der Entzündungszellen und pathologische Osteogenese im Vergleich zu einer alleinigen IL-17A vermittelten Hemmung.
Pharmakodynamik
Es wurden keine formalen Studien zur Pharmakodynamik von Bimekizumab durchgeführt.
Klinische Wirksamkeit
Plaque Psoriasis
Die Sicherheit und Wirksamkeit von Bimekizumab wurde bei 1'480 Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis in drei multizentrischen, randomisierten, Placebo- und/oder aktiv kontrollierten Phase-III-Studien beurteilt. Die Patienten waren mindestens 18 Jahre alt, hatten einen PASI (Psoriasis Area and Severity Index)-Score ≥12 und einen IGA (Investigators Global Assessment)-Score ≥3 auf einer 5-Punkte-Skala, mindestens 10 % ihrer Körperoberfläche war von Psoriasis betroffen (BSA [Body Surface Area]) ≥10 %) und sie waren Kandidaten für eine systemische Psoriasis-Therapie und/oder Phototherapie. Die Wirksamkeit und Sicherheit von Bimekizumab wurden gegenüber Placebo und Ustekinumab (BE VIVID – PS0009), gegenüber Placebo (BE READY – PS0013) und gegenüber Adalimumab (BE SURE – PS0008) beurteilt.
In der Studie BE VIVID wurden 567 Patienten über 52 Wochen untersucht. Die Patienten wurden auf Bimekizumab (320 mg alle 4 Wochen), Ustekinumab (je nach Gewicht des Patienten 45 mg oder 90 mg zu Baseline, in Woche 4 und dann alle 12 Wochen) oder Placebo für die ersten 16 Wochen, gefolgt von Bimekizumab (320 mg alle 4 Wochen) randomisiert.
In der Studie BE READY wurden 435 Patienten über 56 Wochen untersucht. Die Patienten wurden auf Bimekizumab 320 mg alle 4 Wochen oder Placebo randomisiert. In Woche 16 traten Patienten, die ein PASI-90-Ansprechen erreicht hatten, in die 40-wöchige randomisierte Absetz-Phase ein. Patienten, die anfänglich auf Bimekizumab 320 mg alle 4 Wochen randomisiert worden waren, wurden erneut randomisiert und erhielten dann Bimekizumab 320 mg alle 4 Wochen, Bimekizumab 320 mg alle 8 Wochen oder Placebo (d.h. Absetzen von Bimekizumab). Patienten, die anfänglich auf Placebo randomisiert worden waren, erhielten weiterhin Placebo, wenn sie PASI-90-Responder waren. Patienten, die in Woche 16 kein PASI-90-Ansprechen erreicht hatten, gingen in einen unverblindeten Escape-Arm über und erhielten Bimekizumab 320 mg alle 4 Wochen für 12 Wochen. Patienten mit Rezidiv (kein PASI-75-Ansprechen) während der randomisierten Absetz-Phase gingen ebenfalls in den 12-wöchigen Escape-Arm über.
Die Studie BE SURE untersuchte 478 Patienten über 56 Wochen. Die Patienten wurden auf Bimekizumab 320 mg alle 4 Wochen bis Woche 56, Bimekizumab 320 mg alle 4 Wochen bis Woche 16, gefolgt von Bimekizumab 320 mg alle 8 Wochen bis Woche 56, oder Adalimumab gemäss Zulassungsempfehlung bis Woche 24, gefolgt von Bimekizumab 320 mg alle 4 Wochen bis Woche 56, randomisiert.
Die Baseline-Charakteristika waren in allen 3 Studien ähnlich. Im Median waren 20 % der Körperoberfläche der Patienten von Psoriasis betroffen (BSA 20 %), der mediane PASI-Score lag bei 18 und anhand des IGA-Scores war die Erkrankung bei 33 % der Patienten als schwerwiegend einzustufen. Die medianen Baseline-Scores für Schmerzen, Juckreiz und Schuppung lagen gemäss Patienten-Symptom-Tagebuch (Patient Symptoms Diary, PSD) zwischen 6 und 7 auf einer Punkteskala von 0–10, und der mediane Gesamtscore des Dermatology Life Quality Index (DLQI) betrug bei Baseline 9.
In allen 3 Studien zusammen hatten 38 % der Patienten eine vorherige biologische Therapie erhalten, 23 % mindestens einen Anti-IL17-Wirkstoff und 13 % mindestens einen TNF-Antagonisten. 22 % hatten keinerlei systemische Therapie (nicht-biologische und biologische Wirkstoffe eingeschlossen) erhalten und 39 % der Patienten waren zuvor mit Phototherapie oder Photochemotherapie behandelt worden.
Die Wirksamkeit von Bimekizumab wurde bezüglich der Auswirkung auf die Hauterkrankung insgesamt, auf spezifische Körperstellen (Kopfhaut, Nägel, Handflächen und Fusssohlen), auf die von den Patienten berichteten Symptome und bezüglich der Auswirkungen auf die Lebensqualität bewertet. Die beiden co-primären Endpunkte in allen 3 Studien waren der Anteil der Patienten, die 1) ein PASI-90-Ansprechen und 2) ein IGA-Ansprechen «erscheinungsfrei oder fast erscheinungsfrei» (IGA 0/1 mit Verbesserung um mindestens zwei Skalenpunkte im Vergleich zu Baseline) in Woche 16 erreichten. Das PASI-100- und IGA-0-Ansprechen zu Woche 16 und das PASI-75-Ansprechen zu Woche 4 waren die wichtigsten sekundären Endpunkte in allen 3 Studien.
Hauterkrankung insgesamt
Die Behandlung mit Bimekizumab führte zu einer signifikanten Verbesserung aller Krankheitsaktivitätsparameter im Vergleich zu Placebo, Ustekinumab oder Adalimumab in Woche 16. Die wichtigsten Ergebnisse zur Wirksamkeit sind in Tabelle 2 zusammengefasst.
Tabelle 2: Zusammenfassung des klinischen Ansprechens in BE VIVID, BE READY und BE SURE
|
|
BE VIVID
|
BE READY
|
BE SURE
| |
|
Placebo
(n = 83)
n (%)
|
BKZ 320 mg Q4W
(n = 321)
n (%)
|
Ustekinumab
(n = 163)
n (%)
|
Placebo
(n = 86)
n (%)
|
BKZ 320 mg Q4W
(n = 349)
n (%)
|
BKZ 320 mg Q4W
(n = 319)
n (%)
|
Adalimumab
(n = 159)
n (%)
| |
PASI 100
Woche 16
|
0 (0,0)
|
188 (58,6)a
|
34 (20,9)
|
1 (1,2)
|
238 (68,2)a
|
194 (60,8)a
|
38 (23,9)
| |
PASI 90
Woche 16
|
4 (4,8)
|
273 (85,0)a, b
|
81 (49,7)
|
1 (1,2)
|
317 (90,8)a
|
275 (86,2)a
|
75 (47,2)
| |
PASI 75
Woche 4
Woche 16
|
2 (2,4)
6 (7,2)
|
247 (76,9)a, b
296 (92,2)
|
25 (15,3)
119 (73,0)
|
1 (1,2)
2 (2,3)
|
265 (75,9)a
333 (95,4)
|
244 (76,5)a
295 (92,5)
|
50 (31,4)
110 (69,2)
| |
IGA 0
Woche 16
|
0 (0,0)
|
188 (58,6)a
|
36 (22,1)
|
1 (1,2)
|
243 (69,6)a
|
-
|
-
| |
IGA 0/1
Woche 16
|
4 (4,8)
|
270 (84,1)a, b
|
87 (53,4)
|
1 (1,2)
|
323 (92,6)a
|
272 (85,3)a
|
91 (57,2)
| |
Absoluter PASI ≤2
Woche 16
|
3 (3,6)
|
273 (85,0)
|
84 (51,5)
|
1 (1,2)
|
315 (90,3)
|
280 (87,8)
|
86 (54,1)
| |
Schmerzen PSD (n)
Woche 16
|
(n = 54)
9 (16,7)
|
(n = 229)
177 (77,3)a
|
(n = 107)
73 (68,2)
|
(n = 67)
6 (9,0)
|
(n = 255)
201 (78,8)a
|
-
|
-
| |
Juckreiz PSD (n)
Woche 16
|
(n = 61)
8 (13,1)
|
(n = 244)
187 (76,6)a
|
(n = 117)
77 (65,8)
|
(n = 72)
4 (5,6)
|
(n = 278)
210 (75,5)a
|
-
|
-
| |
Schuppung PSD (n)
Woche 16
|
(n = 63)
8 (12,7)
|
(n = 246)
193 (78,5)a
|
(n = 116)
69 (59,5)
|
(n = 70)
4 (5,7)
|
(n = 286)
223 (78,0)a
|
-
|
-
|
BKZ 320 mg Q4W = Bimekizumab alle 4 Wochen. Non-Responder-Imputation (NRI) wurde verwendet.
Ansprechen IGA 0/1 war definiert als erscheinungsfrei (0) oder fast erscheinungsfrei (1) mit einer Besserung um mindestens 2 Kategorien in Woche 16 gegenüber Baseline. Ansprechen IGA 0 war definiert als erscheinungsfrei (0) mit einer Besserung um mindestens 2 Kategorien in Woche 16 gegenüber Baseline.
PSD ist das Patienten-Symptom-Tagebuch (Patient Symptoms Diary). PSD-Ansprechen ist definiert als Veränderung von Baseline bis Woche 16 ≥ einen definierten Schwellenwert (1,98, 2,39 bzw. 2,86 für Schmerzen, Juckreiz und Schuppung).
a) p < 0,001 gegenüber Placebo (BE VIVID und BE READY), gegenüber Adalimumab (BE SURE), auf Multiplizität adjustiert.
b) p < 0,001 gegenüber Ustekinumab (BE VIVID), auf Multiplizität adjustiert.
Die Behandlung mit Bimekizumab war mit einem schnellen Wirkeintritt assoziiert. In der Studie BE VIVID waren die PASI-90-Ansprechraten in Woche 2 bzw. Woche 4 unter Bimekizumab (12,1 % bzw. 43,6 %) höher als unter Placebo (1,2 % bzw. 2,4 %) und Ustekinumab (1,2 % bzw. 3,1 %).
Abbildung 1: PASI-90-Ansprechraten im Zeitverlauf der Studie BE VIVID
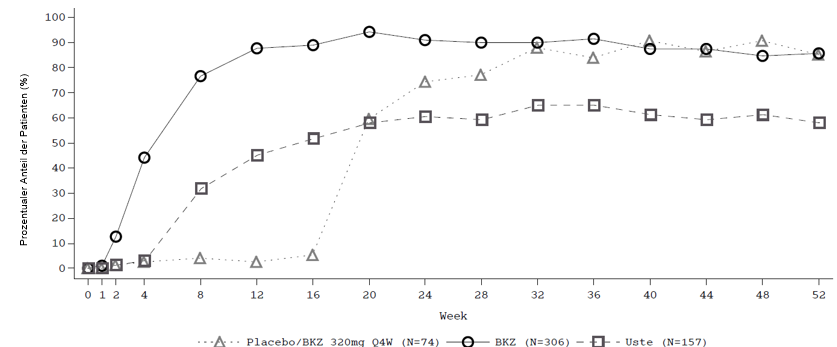
BKZ = Bimekizumab, Uste = Ustekinumab; NRI wird verwendet.
Hinweis: Die Patienten aus der Gruppe Placebo/BKZ wechselten in der Erhaltungsbehandlungsphase ab Woche 16 von Placebo zu BKZ.
In der BE VIVID-Studie erreichten zu Woche 52, im Vergleich zu Ustekinumab, Patienten unter Bimekizumab höhere PASI-90- (81,6 % Bimekizumab gegenüber 55,8 % Ustekinumab, p < 0,001), IGA 0/1- (77,9 % Bimekizumab gegenüber 60,7 % Ustekinumab,p < 0,001) und PASI-100-Ansprechraten (64,2 % Bimekizumab gegenüber 38,0 % Ustekinumab).
In der BE SURE-Studie erreichte zu Woche 24 ein höherer Prozentsatz der mit Bimekizumab behandelten Patienten ein PASI-90-Ansprechen und IGA0/1-Ansprechen im Vergleich zu Adalimumab (85,6 % bzw. 86,5 % gegenüber 51,6 % bzw. 57,9 %,p < 0,001). Unter den 65 Adalimumab-Non-Respondern in Woche 24 (< PASI 90) erreichten 78,5 % nach 16 Behandlungswochen mit Bimekizumab ein PASI-90-Ansprechen. Bei Patienten, die von Adalimumab auf Bimekizumab umgestellt wurden, ergaben sich keine neuen Befunde bezüglich der Sicherheit. In Woche 56 erreichten 70,2 % der mit Bimekizumab behandelten Patienten ein PASI-100-Ansprechen.
Abbildung 2: PASI-90-Ansprechraten im Zeitverlauf der Studie BE SURE
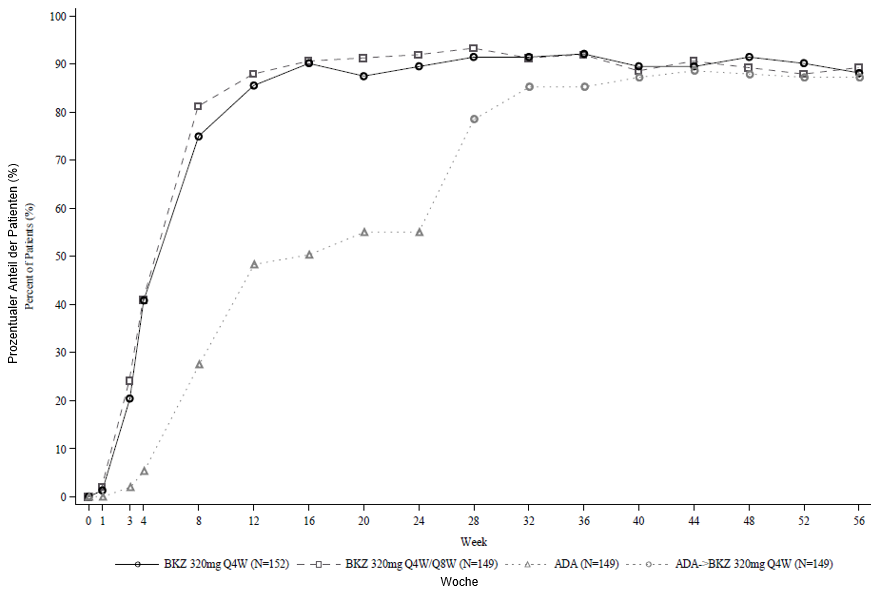
BKZ 320 mg Q4W = Bimekizumab alle 4 Wochen; BKZ 320 mg Q8W = Bimekizumab alle 8 Wochen; ADA = Adalimumab.
Hinweis: Enthalten sind nur Patienten, die in Woche 24 oder später Bimekizumab erhielten. Patienten aus der Gruppe BKZ Q4W/Q8W wechselten in Woche 16 von einer Q4W- zu einer Q8W-Dosierung. Patienten aus der Gruppe ADA/BKZ 320 mg Q4W wechselten in Woche 24 von ADA zu BKZ Q4W. NRI wird verwendet.
Die Wirksamkeit von Bimekizumab wurde unabhängig von Alter, Geschlecht, ethnischer Zugehörigkeit, Krankheitsdauer, Körpergewicht, PASI-Schweregrad bei Baseline und vorheriger Behandlung mit einem Biologikum nachgewiesen. Bimekizumab war wirksam bei Patienten mit vorheriger Biologikum-Exposition, einschliesslich Anti-TNF/Anti-IL-17, und bei Patienten ohne vorherige systemische Therapie. Basierend auf populationspharmakokinetischen/-pharmakodynamischen (PK/PD-)Analysen und gestützt durch klinische Daten, profitierten Patienten mit höherem Körpergewicht (≥120 kg), die in Woche 16 keine vollständige Symptomfreiheit der Haut erreicht hatten, von einer fortgesetzten Behandlung mit Bimekizumab 320 mg alle vier Wochen (Q4W) nach den ersten 16 Behandlungswochen.
In der BE SURE-Studie erhielten die Patienten Bimekizumab 320 mg Q4W bis Woche 16, gefolgt von einer Dosierung Q4W oder alle acht Wochen (Q8W) bis Woche 56, unabhängig vom Responder-Status in Woche 16. Bei Patienten in der Gruppe ≥120 kg (N=37) verbesserte sich im Arm mit Q4W-Erhaltungstherapie das PASI-100-Ansprechen zwischen Woche 16 (23,5 %) und Woche 56 (70,6 %) stärker als im Arm mit Q8W-Erhaltungstherapie (Woche 16: 45,0 % vs. Woche 56: 60,0 %).
Aufrechterhaltung des Ansprechens
Tabelle 3: Fortdauer des Ansprechens in Woche 52 bei Respondern in Woche 16*
|
PASI 100
|
PASI 90
|
IGA 0/1
|
Absoluter PASI ≤2
| |
BKZ 320 mg Q4W/Q4W
(N = 355)
n (%)
|
BKZ 320 mg Q4W/Q8W
(N = 182)
n (%)
|
BKZ 320 mg Q4W/Q4W
(N = 516)
n (%)
|
BKZ 320 mg Q4W/Q8W
(N = 237)
n (%)
|
BKZ 320 mg Q4W/Q4W
(N = 511)
n (%)
|
BKZ 320 mg Q4W/Q8W
(N = 234)
n (%)
|
BKZ 320 mg Q4W/Q4W
(N = 511)
n (%)
|
BKZ 320 mg Q4W/Q8W
(N = 238)
n (%)
| |
295 (83,1)
|
161 (88,5)
|
464 (89,9)
|
214 (90,3)
|
447 (87,5)
|
214 (91,5)
|
460 (90,0)
|
215 (90,3)
|
* Integrierte Analyse von BE VIVID, BE READY und BE SURE. NRI wird verwendet.
BKZ 320 mg Q4W: Bimekizumab 320 mg alle 4 Wochen, gefolgt von Bimekizumab 320 mg alle 4 Wochen ab Woche 16. BKZ 320 mg Q8W: Bimekizumab 320 mg alle 4 Wochen, gefolgt von Bimekizumab 320 mg alle 8 Wochen ab Woche 16.
Aufrechterhaltung des PASI-90-Ansprechens (nach Absetzen von Bimekizumab)
Abbildung 3: PASI-90-Responder-Raten im Zeitverlauf – randomisierte Auswasch-Phase in BE READY
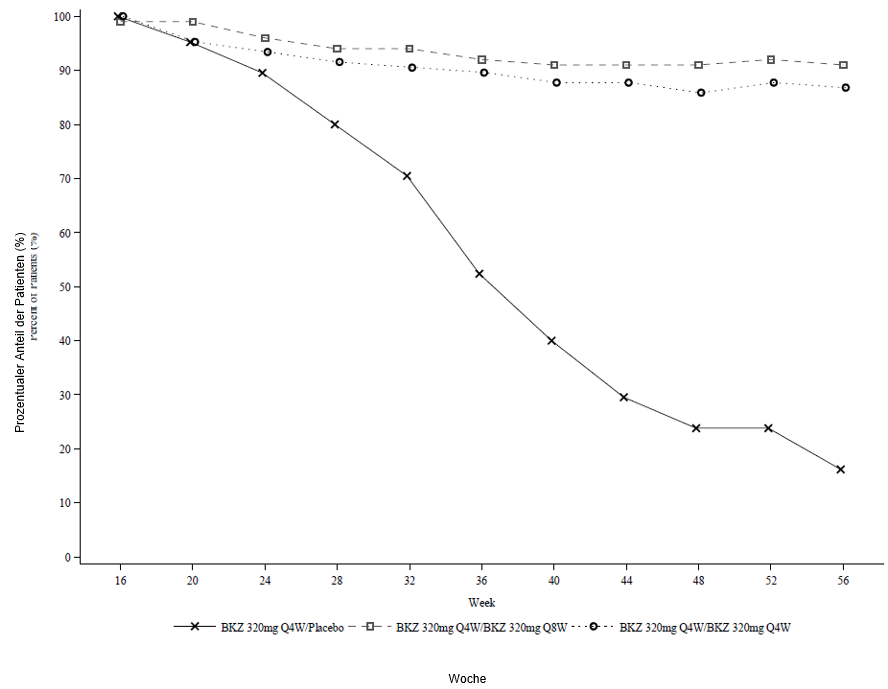
NRI wird verwendet.
In BE READY betrug für PASI-90-Responder aus Woche 16, die dann auf Placebo randomisiert wurden und nicht länger Bimekizumab erhielten, die mediane Zeit bis zum Rezidiv, definiert als Verlust des PASI-75-Ansprechens, ungefähr 28 Wochen (32 Wochen nach der letzten Bimekizumab-Dosis). Von diesen Patienten erreichten 88,1 % innerhalb von 12 Wochen nach Wiederaufnahme der Behandlung mit Bimekizumab 320 mg alle 4 Wochen wieder ein PASI-90-Ansprechen.
Einzelne Körperregionen
Bei Patienten, die mit Bimekizumab behandelt wurden, zeigte sich in Woche 16 in den Studien BE VIVID und BE READY gegenüber Placebo eine signifikante Verbesserung der Psoriasis an Kopfhaut, Nägeln, Handflächen und Fusssohlen (siehe Tabelle 4).
Tabelle 4: Ansprechen in einzelnen Körperregionen in BE VIVID und BE READY in Woche 16
|
|
BE VIVID
|
BE READY
| |
|
Placebo
|
BKZ 320 mg Q4W
|
Ustekinumab
|
Placebo
|
BKZ 320 mg
Q4W
| |
Kopfhaut IGA (n)a
Kopfhaut IGA 0/1, n (%)
|
(72)
11 (15,3)
|
(285)
240 (84,2)b
|
(146)
103 (70,5)
|
(74)
5 (6,8)
|
(310)
286 (92,3)b
| |
pp-IGA (n)a
pp-IGA 0/1, n (%)
|
(29)
7 (24,1)
|
(105)
85 (81,0)
|
(47)
39 (83,0)
|
(31)
10 (32,3)
|
(97)
91 (93,8)
| |
mNAPSI 100 (n)a
mNAPSI 100, n (%)
|
(51)
4 (7,8)
|
(194)
57 (29,4)
|
(109)
15 (13,8)
|
(50)
3 (6,0)
|
(210)
73 (34,8)
|
NRI wird verwendet.
a) Nur Patienten mit einem Investigator Global Assessment (IGA)-Score der Kopfhaut von 2 oder höher, einem palmoplantaren IGA von 2 oder höher und einem mNAPSI (modifizierten Nail Psoriasis and Severity Index)-Score > 0 bei Baseline. Kopfhaut-Ansprechen IGA 0/1 und pp-Ansprechen IGA 0/1 waren definiert als erscheinungsfrei (0) oder fast erscheinungsfrei (1) mit einer Besserung um ≥2 Kategorien gegenüber Baseline.
b) p < 0,001 gegenüber Placebo, für Multiplizität korrigiert.
Das IGA-Ansprechen an der Kopfhaut und der palmoplantaren Region blieb bis Woche 52/56 erhalten. Die Nagel-Psoriasis besserte sich über Woche 16 hinaus. In BE VIVID erreichte in Woche 52 ein höherer Anteil der mit Bimekizumab behandelten Patienten eine völlige Symptomfreiheit der Nägel (mNAPSI 100) im Vergleich zu mit Ustekinumab behandelten Patienten (60,3 % gegenüber 40,4 %). In BE READY erreichten in Woche 56 unter Bimekizumab 320 mg alle 8 Wochen bzw. Bimekizumab 320 mg alle 4 Wochen 67,7 % bzw. 69,8 % der PASI-90-Responder aus Woche 16 eine völlige Symptomfreiheit der Nägel.
Gesundheitsbezogene Lebensqualität / von Patienten berichtete Ergebnisse
In allen 3 Studien erreichte in Woche 16, im Vergleich zu Placebo oder einem aktiven Vergleichsmedikament, ein grösserer Anteil der mit Bimekizumab behandelten Patienten eine nicht mehr durch die Psoriasis eingeschränkte Lebensqualität (gemessen anhand des Dermatology Life Quality Index, DLQI).
In BE READY lag der Prozentsatz der Patienten mit einem DLQI von 0/1 (keine Auswirkungen der Psoriasis auf die gesundheitsbezogene Lebensqualität) in Woche 16 bei 75,6 % in der Bimekizumab-Gruppe bzw. bei 5,8 % in der Placebo-Gruppe.
In BE VIVID betrug die DLQI 0/1 Ansprechrate in Woche 16 in der Bimekizumab-, Ustekinumab- und Placebogruppe 67,3 %, 42,3 % bzw. 12,0 %. Die DLQI 0/1-Ansprechraten stiegen über Woche 16 hinaus an und blieben bis Woche 52 erhalten (74,8 % bei mit Bimekizumab 320 mg Q4W behandelten Patienten).
In BE SURE betrug die DLQI 0/1-Ansprechrate in Woche 16 in der Bimekizumab- und Adalimumab-Gruppe 63,0 % bzw. 46,5 %. Die DLQI 0/1-Ansprechrate in Woche 56 betrug 78,9 % bei mit Bimekizumab 320 mg Q8W bzw. 74,1 % bei mit Bimekizumab 320 mg Q4W behandelten Patienten.
Offene Verlängerungsstudie der Phase III
Patienten, die eine der Zulassungsstudien der Phase III («Feeder-Studien») abgeschlossen haben, konnten in eine 144-wöchige offene Verlängerungsstudie (PS0014) zur Beurteilung der langfristigen Sicherheit und Wirksamkeit von Bimekizumab eintreten.
344 Patienten, die während der Feeder-Studien mit Bimekizumab 320 mg alle 8 Wochen (BKZ 320 mg Q8W) oder alle 4 Wochen (BKZ 320 mg Q4W) behandelt wurden und am Ende der Feeder-Studien ein PASI-90-Ansprechen erreicht hatten, erhielten für die gesamte Dauer der Studie PS0014 weiterhin Bimekizumab 320 mg Q8W. Von diesen schlossen 293 (85,2 %) Patienten die 144-wöchige Behandlung mit Bimekizumab 320 mg Q8W ab. 48 Patienten (14,0 %) brachen die Studie während des Behandlungszeitraums ab, davon 21 (6,1 %) wegen eines unerwünschten Ereignisses und 4 (1,2 %) wegen mangelnder Wirksamkeit.
Bei den in der Studie verbliebenen Patienten wurden die Verbesserungen, die mit Bimekizumab in den Feeder-Studien bei den Wirksamkeitsendpunkten PASI-90-Ansprechen und IGA 0/1 erreicht wurden, im Verlauf der zusätzlichen 144 Wochen der offenen Behandlung erhalten.
Offener Verlängerungszeitraum der Phase IIIb
In Woche 48 durften die Patienten in den 96-wöchigen offenen Verlängerungszeitraum (open label extension period = OLE) eintreten und abhängig von ihrem PASI-90-Ansprechen in Woche 48 die Behandlung mit Bimekizumab in einer Dosis von 320 mg Q4W oder 320 mg Q8W fortsetzen oder beginnen. Studienteilnehmer, die im Rahmen der OLE Bimekizumab ursprünglich in einer Dosis von 320 mg Q4W erhalten hatten, wurden in der Woche 72 oder danach auf Bimekizumab in einer Dosis von 320 mg Q8W umgestellt.
231 Patienten, die mit Bimekizumab in einer Dosis von 320 mg Q8W oder Bimekizumab in einer Dosis von 320 mg Q4W behandelt wurden und in der Woche 48 PASI 90 erreichten, erhielten während des gesamten OLE Bimekizumab in einer Dosis von 320 mg Q8W. Von diesen Patienten brachen 31 (13,4 %) die Studie während des OLE ab, 10 (4,3 %) aufgrund einer Nebenwirkung und 1 (0,4 %) wegen mangelnder Wirksamkeit.
116 Patienten, die mit Secukinumab behandelt wurden und in der Woche 48 PASI 90 erreichten, erhielten während des gesamten OLE Bimekizumab in einer Dosis von 320 mg Q8W. Von diesen Patienten brachen 16 (13,8 %) die Studie während des OLE ab, 6 (5,2 %) aufgrund einer Nebenwirkung und 1 (0,9 %) wegen mangelnder Wirksamkeit.
Bei den in der Studie verbliebenen Patienten blieben die in der Woche 48 mit Bimekizumab oder Secukinumab erzielten Verbesserungen in Bezug auf die Wirksamkeitsendpunkte PASI-100-, PASI-90-, PASI-75-Ansprechen und PASI ≤2 unter Behandlung mit Bimekizumab in einer Dosis von 320 mg Q8W im Rahmen der offenen Behandlung über weitere 96 Wochen erhalten.
Das Sicherheitsprofil von Bimekizumab bis Woche 144 entsprach dem bis Woche 48 beobachteten Sicherheitsprofil.
Psoriasis-Arthritis (PsA)
Die Sicherheit und Wirksamkeit von Bimekizumab wurden bei 1112 erwachsenen Patienten (mindestens 18 Jahre alt) mit aktiver Psoriasis-Arthritis (PsA) in zwei multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studien (PA0010 – BE OPTIMAL und PA0011 – BE COMPLETE) untersucht. Die BE OPTIMAL-Studie umfasste einen Arm mit aktiver Referenzbehandlung (Adalimumab) (N = 140).
In beiden Studien war bei den Patienten vor mindestens 6 Monaten eine aktive Psoriasis-Arthritis gemäss den CASPAR-Kriterien (Classification Criteria for Psoriatic Arthritis) diagnostiziert worden, und die Krankheit war aktiv mit einer Anzahl druckschmerzempfindlicher Gelenke (tender joint count, TJC) ≥3 und einer Anzahl geschwollener Gelenke (swollen joint count, SJC) ≥3. Die Patienten hatten die Diagnose PsA in BE OPTIMAL im Median seit 3,6 Jahren und in BE COMPLETE seit 6,8 Jahren. In diese Studien wurden Patienten mit allen Subtypen der PsA aufgenommen, darunter polyartikuläre symmetrische Arthritis, oligoartikuläre asymmetrische Arthritis, Arthritis der distalen Interphalangealgelenke, prädominante Spondylitis und Arthritis mutilans. Bei Baseline hatten 55,9 % der Patienten ≥3 % Körperoberfläche (Body Surface Area, BSA) mit aktiver Plaque-Psoriasis. 10,4 % der Patienten hatten eine mittelschwere bis schwere Plaque-Psoriasis, und 31,9 % bzw. 12,3 % hatten bei Baseline eine Enthesitis bzw. eine Daktylitis. Der primäre Wirksamkeitsendpunkt in beiden Studien war das ACR-(American College of Rheumatology-)-50-Ansprechen in Woche 16.
Die wichtigsten sekundären Endpunkte in Woche 16 waren in beiden Studien wie folgt: Veränderung gegenüber Baseline des Health Assessment Questionnaire - Disability Index (cfB HAQ-DI), Verringe-rung des Psoriasis Area and Severity Index um 90 % gegenüber Baseline (PASI90), Veränderung gegenüber Baseline des Scores im Short Form-36-Gesundheitsfragebogen (Short Form 36-item Health Survey (SF-36) Physical Component Summary (PCS)), Ansprechen auf die minimale Krankheitsaktivität (Minimal Disease Activity (MDA)), sowie Enthesitis- und Daktylitis-freier Zustand, welche auf gepoolten Daten aus beiden Studien beruhen. In der BE OPTIMAL-Studie war die Veränderung gegenüber Base-line des Van der Heijde modified Total Sharp Score (vdHmTSS) ebenfalls ein wichtiger sekundärer Endpunkt.
In der BE OPTIMAL-Studie wurden 852 Patienten untersucht, die zuvor kein biologisches krankheitsmodifizierendes Antirheumatikum (bDMARD) zur Behandlung von Psoriasis-Arthritis oder Psoriasis erhalten hatten. Die Patienten wurden randomisiert (3:2:1) und erhielten entweder Bimekizumab 160 mg alle 4 Wochen bis Woche 52 oder Placebo bis Woche 16, gefolgt von Bimekizumab 160 mg alle 4 Wochen bis Woche 52 oder einem aktiven Referenzbehandlungarm (Adalimumab 40 mg alle 2 Wochen) bis Woche 52. In dieser Studie hatten 78,3 % der Patienten eine Vorbehandlung mit ≥1 cDMARD erhalten, 21,7 % der Patienten hatten keine Vorbehandlung mit cDMARDs erhalten. Bei Baseline erhielten 58,2 % der Patienten gleichzeitig Methotrexat (MTX), 11,3 % erhielten gleichzeitig cDMARDs ausser MTX, und 30,5 % erhielten keine cDMARDs.
In der BE COMPLETE-Studie wurden 400 Patienten eingeschlossen, die auf eine Behandlung mit einem oder zwei Tumornekrosefaktor-alpha-Inhibitoren (Anti-TNFα-IR) zur Behandlung von Psoriasis-Arthritis oder Psoriasis unzureichend angesprochen (mangelnde Wirksamkeit) oder diese nicht vertragen hatten. Die Patienten wurden auf Bimekizumab 160 mg alle 4 Wochen oder Placebo bis Woche 16 randomisiert (2:1). Bei Baseline erhielten 42,5 % der Patienten gleichzeitig MTX, 8,0 % erhielten gleichzeitig cDMARDs ausser MTX, und 49,5 % erhielten keine cDMARDs. In dieser Studie sprachen 76,5 % der Teilnehmer unzureichend auf einen TNFα-Hemmer an, 11,3 % sprachen unzureichend auf zwei TNFα-Hemmer an und 12,3 % vertrugen TNFα-Hemmer nicht.
Anzeichen und Symptome
Bei bDMARDs-naiven Patienten (BE OPTIMAL) und anti-TNFα-IR-Patienten (BE COMPLETE) führte die Behandlung mit Bimekizumab im Vergleich zu Placebo in Woche 16 zu einer signifikanten Verbesserung der Symptome und der Krankheitsaktivität, wobei in beiden Patientenpopulationen ähnliche Ansprechraten beobachtet wurden (siehe Tabelle 5). In der Beurteilung nach ACR 50, MDA, PASI 90 blieb das klinische Ansprechen in BE OPTIMAL bis zur Woche 52 erhalten.
Tabelle 5: Klinisches Ansprechen in den Studien BE OPTIMAL und BE COMPLETE
|
|
BE OPTIMAL (bDMARD-naiv)
|
BE COMPLETE (anti-TNFα-IR)
| |
|
Placebo
(N = 281)
n (%)
|
BKZ 160 mg Q4W
(N = 431)
n (%)
|
Unterschied gegenüber Placebo (95%-KI)(d)
|
Referenz-behandlung(e) (Adalimumab)
(N = 140)
n (%)
|
Placebo
(N = 133)
n (%)
|
BKZ 160 mg Q4W
(N = 267)
n (%)
|
Unterschied gegenüber Placebo (95%-KI)(d)
| |
ACR 50
|
|
|
|
|
|
|
| |
Woche 16
|
28 (10,0)
|
189 (43,9)*
|
33,9 (27,4; 40,4)
|
64 (45,7)
|
9 (6,8)
|
116 (43,4)*
|
36,7 (27,7; 45,7)
| |
Woche 24
|
-
|
196 (45,5)
|
|
66 (47,1)
|
|
|
| |
Woche 52
|
|
235 (54,5)
|
|
70 (50,0)
|
|
|
| |
MDA(a)
|
|
|
|
|
|
|
| |
Woche 16
|
37 (13,2)
|
194 (45,0)*
|
31,8 (25,2; 38,5)
|
63 (45,0)
|
8 (6,0)
|
118 (44,2)*
|
38,2 (29,2; 47,2)
| |
Woche 24
|
-
|
209 (48,5)
|
|
67 (47,9)
|
|
|
| |
Woche 52
|
|
237 (55,0)
|
|
74 (52,9)
|
|
|
| |
Patienten mit ≥3 % BSA
|
(N = 140)
|
(N = 217)
|
|
(N = 68)
|
(N = 88)
|
(N = 176)
|
| |
PASI 90
|
|
|
|
|
|
|
| |
Woche 16
|
4 (2,9)
|
133 (61,3)*
|
58,4 (49,9; 66,9)
|
28 (41,2)
|
6 (6,8)
|
121 (68,8)*
|
61,9 (51,5; 72,4)
| |
Woche 24
|
-
|
158 (72,8)
|
|
32 (47,1)
|
|
|
| |
Woche 52
|
|
155 (71,4)
|
|
41 (60,3)
|
|
|
| |
Patienten mit LDI > 0 (b)
|
(N = 47)
|
(N = 90)
|
|
|
| |
Frei von Daktylitis (b)
Woche 16
|
24 (51,1)
|
68 (75,6)***
|
24,5 (8,4; 40,6)
|
| |
Patienten mit LEI > 0 (c)
|
(N = 106)
|
(N = 249)
|
|
| |
Frei von Enthesitis (c)
Woche 16
|
37 (34,9)
|
124 (49,8)**
|
14,9 (3,7; 26,1)
|
|
|
BKZ 160 mg Q4W = Bimekizumab 160 mg alle 4 Wochen. KI = Konfidenzintervall, NC = nicht berechenbar (not calculable).
(a) Ein Patient wurde mit minimaler Krankheitsaktivität (Minimal Disease Activity, MDA) eingestuft, wenn er 5 der 7 folgenden Kriterien erfüllte: Anzahl druckschmerzempfindlicher Gelenke < 1; Anzahl geschwollener Gelenke < 1; Psoriasis Activity and Severity Index < 1 oder Körperoberfläche < 3; vom Patienten angegebene Schmerzen auf Visueller Analogskala (VAS) < 15; globale Krankheitsaktivität des Patienten auf VAS < 20; Health Assessment Questionnaire Disability Index < 0,5; schmerzende entheseale Punkte < 1
(b) Auf der Grundlage gepoolter Daten aus den Studien BE OPTIMAL und BE COMPLETE für Patienten mit einem Leeds Dactylitis Index (LDI) > 0 zu Baseline. Frei von Daktylitis bedeutet LDI = 0
(c) Auf der Grundlage gepoolter Daten aus den Studien BE OPTIMAL und BE COMPLETE für Patienten mit einem Leeds Enthesitis Index (LEI) > 0 zu Baseline. Frei von Enthesitis bedeutet LEI = 0
(d) Unbereinigte Unterschiede werden angezeigt
(e) Kein statistischer Vergleich mit Bimekizumab oder Placebo durchgeführt
* p < 0,001 gegenüber Placebo, multiplizitätsbereinigt. ** p = 0,008 im Vergleich zu Placebo, multiplizitätsbereinigt. *** p = 0,002 im Vergleich zu Placebo, multiplizitätsbereinigt. NRI wird verwendet. Andere Endpunkte in Woche 16 und alle Endpunkte in Woche 24 und Woche 52 waren nicht Teil der sequenziellen Testhierarchie und alle Vergleiche sind nominal.
In BE OPTIMAL waren die Ergebnisse für mit cDMARD vorbehandelte Patienten ähnlich (ACR50 Woche 16: Bimekizumab 160mg Q4W: 43.5%, Placebo: 9.5%).
In BE OPTIMAL wurden unter Bimekizumab in Woche 16 bei jeder der einzelnen ACR-Komponenten Verbesserungen gegenüber Baseline festgestellt, die bis zu Woche 52 anhielten.
Das Ansprechen auf die Behandlung mit Bimekizumab war in Woche 4 bei ACR 50 (BE OPTIMAL 17,6 % gegenüber 3,2 %, nominal p < 0,001 und BE COMPLETE 16,1 % gegenüber 1,5 %, nominal p < 0,001) signifikant höher als unter Placebo.
Bei 87,2 % der mit Bimekizumab behandelten Patienten, die in BE OPTIMAL in Woche 16 ein ACR-50-Ansprechen erreichten, blieb dieses Ansprechen bis Woche 52 erhalten.
Die Wirksamkeit und Sicherheit von Bimekizumab wurde unabhängig von Alter, Geschlecht, ethnischer Zugehörigkeit, Baseline-Körpergewicht, Psoriasis-Beteiligung zu Baseline, CRP zu Baseline, Krankheitsdauer und vorheriger Anwendung von cDMARDs nachgewiesen. In beiden Studien wurde ein vergleichbares Ansprechen auf Bimekizumab beobachtet, unabhängig davon, ob die Patienten gleichzeitig cDMARDs, einschliesslich MTX, erhielten oder nicht.
Radiologisches Ansprechen
In BE OPTIMAL wurde die Hemmung des Fortschreitens der strukturellen Schädigung radiologisch beurteilt und als Veränderung des Van der Heijde Modified Total Sharp Scores (vdHmTSS) und seiner Komponenten, Erosion Score (ES) und Joint Space Narrowing Score (JSN) in Woche 16 gegenüber Baseline ausgedrückt (siehe Tabelle 6).
Tabelle 6: Veränderung des vdHmTSS in BE OPTIMAL in Woche 16
|
|
Placebo
|
BKZ 160 mg Q4W
|
Unterschied gegenüber Placebo (95%-KI)a)
| |
Population mit erhöhtem hs-CRP und/oder mindestens 1 Knochenerosion zu Baseline
|
(N = 227)
|
(N = 361)
|
| |
Mittlere Veränderung gegenüber Baseline (SE)
|
0,36 (0,10)
|
0,04 (0,05)*
|
-0,32 ( -0,35; -0,30)
| |
Gesamtpopulation
|
(N = 269)
|
(N = 420)
|
| |
Mittlere Veränderung gegenüber Baseline (SE)
|
0,32 (0,09)
|
0,04 (0,04)*
|
-0,26 ( -0,29; -0,23)
|
*p = 0,001 im Vergleich zu Placebo. p-Werte basieren auf einer referenzbasierten Imputation unter Verwendung des Unterschieds im Kleinste-Quadrate-Mittelwert anhand eines ANCOVA-Modells mit der Behandlung, der Knochenerosion zu Baseline und der Region als feste Effekte und dem Baseline-Score als Kovariate.
Die zusammenfassenden Daten für Woche 16 basieren auf dem ersten Wertesatz für die Primäranalyse.
a) Unbereinigte Unterschiede werden angezeigt
Bimekizumab hemmte das Fortschreiten der Gelenkschädigung bis Woche 16 sowohl in der Population mit erhöhtem hs-CRP und/oder mindestens einer Knochenerosion zu Baseline als auch in der Gesamtpopulation im Vergleich zu Placebo signifikant. Während die referenzbasierte Imputation als Methode für den Umgang mit fehlenden Daten im statistischen Testverfahren zum Vergleich von Bimekizumab und Placebo festgelegt wurde, wurden die Veränderungen gegenüber Baseline sowohl in der Population mit erhöhtem hs-CRP und/oder mindestens einer Knochenerosion zu Baseline als auch in der Gesamtpopulation in Woche 16 im Bimekizumab-Arm (mittlere Veränderung gegenüber Baseline 0,01 bzw. 0,01) und im Adalimumab-Arm (mittlere Veränderung gegenüber Baseline -0,05 bzw. -0,03) ebenfalls mittels standardmässiger multipler Imputation berechnet. Die Hemmung des Fortschreitens der Gelenkschädigung wurde sowohl in der Population mit erhöhtem hs-CRP und/oder mindestens einer Knochenerosion zu Baseline als auch in der Gesamtpopulation bis Woche 52 sowohl im Bimekizumab-Arm (mittlere Veränderung gegenüber Baseline 0,10 bzw. 0,10) als auch im Adalimumab-Arm (mittlere Veränderung gegenüber Baseline -0,17 bzw. -0,12) aufrechterhalten.
Der beobachtete Anteil der Patienten, die von der Randomisierung bis Woche 52 kein radiologisch nachweisbares Fortschreiten der Gelenkschädigung (definiert als eine Veränderung des mTSS von ≤0,5 gegenüber Baseline) aufwiesen, betrug in der Population mit erhöhtem hs-CRP und/oder mindestens einer Knochenerosion 87,9 % (N = 276/314) für Bimekizumab und 84,8 % (N = 168/198) für Studienteilnehmer unter Placebo, die auf Bimekizumab umgestellt wurden, sowie 94,1 % (N = 96/102) für Adalimumab. Vergleichbare Raten wurden in der Gesamtpopulation beobachtet (89,3 % (N = 326/365) für Bimekizumab und 87,3 % (N = 207/237) für Studienteilnehmer unter Placebo, die auf Bimekizumab umgestellt wurden, sowie 94,1 % (N = 111/118) für Adalimumab).
Körperliche Funktionsfähigkeit und weitere gesundheitsbezogene Ergebnisse
Sowohl bDMARD-naive (BE OPTIMAL) als auch Anti-TNFα-IR-Patienten (BE COMPLETE), die Bimekizumab erhielten, zeigten in Woche 16 eine signifikante Verbesserung der körperlichen Funktionsfähigkeit gegenüber Baseline im Vergleich zu Placebo-Patienten (p < 0,001), bewertet anhand des HAQ-DI (Veränderung des Kleinste-Quadrate-Mittelwerts gegenüber Baseline: -0,3 versus -0,1 in BE OPTIMAL bzw. -0,3 versus 0 in BE COMPLETE). In beiden Studien erreichte ein grösserer Anteil der Patienten in der Bimekizumab-Gruppe in Woche 16 eine klinisch bedeutsame Verringerung des HAQ-DI-Wertes um mindestens 0,35 gegenüber Baseline als in der Placebo-Gruppe.
Mit Bimekizumab behandelte Patienten zeigten in Woche 16 im Vergleich zu Placebo eine signifikante Verbesserung des Scores im Short Form-36-Gesundheitsfragebogen (Short Form-36 item Health Survey Physical Component Summary, SF-36 PCS) (Veränderung des Kleinste-Quadrate-Mittelwerts gegenüber Baseline: 6,3 versus 1,9, p < 0,001 in BE OPTIMAL und 6,2 versus 0,1, p < 0,001 in BE COMPLETE).
In beiden Studien berichteten die mit Bimekizumab behandelten Patienten im Vergleich zur Placebogruppe in Woche 16 über eine deutliche Verringerung der Müdigkeit gegenüber Baseline, gemessen anhand des FACIT-(Functional Assessment of Chronic Illness Therapy-)Fatigue-Scores. In der mit Bimekizumab behandelten Gruppe wurde im Vergleich zur Placebogruppe in Woche 16 auch eine deutliche Verbesserung des Psoriasis-Arthritis-Impact-of-Disease-12-(PsAID-12-)Scores gegenüber Baseline beobachtet.
Bei Patienten mit axialer Beteiligung zu Baseline, etwa 74 % der Patienten (definiert als ein BASDAI-Wert [Bath Ankylosing Spondylitis Disease Activity Index] ≥4), hatte sich der BASDAI-Wert in Woche 16 gegenüber Baseline im Vergleich zu Placebo stärker verbessert.
Die bis Woche 16 erzielten Verbesserungen bei allen Messgrössen der körperlichen Funktionsfähigkeit und anderen oben erwähnten gesundheitsbezogenen Ergebnisse (HAQ-DI-, SF-36 PCS-, FACIT-Fatigue-, PsAID-12-Werte und BASDAI) blieben in BE OPTIMAL bis Woche 52 erhalten.
In der BE OPTIMAL-Studie erreichten in Woche 52 65,5 % der mit Bimekizumab behandelten Patienten eine völlige Symptomfreiheit der Nägel (mNAPSI-Abheilung bei Patienten mit einem mNAPSI-Wert von über 0 zu Baseline).
Axiale Spondyloarthritis (nr-axSpA und AS)
Die Sicherheit und Wirksamkeit von Bimekizumab wurde bei 586 erwachsenen Patienten (mindestens 18 Jahre alt) mit aktiver axialer Spondyloarthritis (axSpA) in zwei multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studien untersucht, eine bei nicht-röntgenologischer axialer Spondyloarthritis (nr-axSpA) und eine bei ankylosierender Spondylitis (AS), auch als röntgenologische axiale Spondyloarthritis (axSpA) bezeichnet. Der primäre Endpunkt war in beiden Studien der Anteil der Patienten, die in Woche 16 ein ASAS (Assessment of SpondyloArthritis International Society)-40-Ansprechen erreichten. Beide Patientengruppen hatten übereinstimmende Ergebnisse.
In der Studie BE MOBILE 1 (AS0010) wurden 254 Patienten mit aktiver nr-axSpA untersucht. Die Patienten hatten eine axSpA (Alter bei Beginn der Symptome < 45 Jahre), die den ASAS-Klassifizierungskriterien entsprach, und eine aktive Erkrankung, definiert als Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 und Wirbelsäulenschmerzen ≥4 auf einer numerischen Bewertungsskala von 0 bis 10 (nach BASDAI Frage 2), sowie keine Anzeichen für radiologisch nachweisbare Veränderungen in den Iliosakralgelenken, die den modifizierten New-York-Klassifikationskriterien der AS entsprechen würden. Die Patienten zeigten auch objektive Anzeichen einer Entzündung, festgestellt durch erhöhtes C-reaktives Protein (CRP) und/oder mittels Nachweis einer Sakroiliitis in der Magnetresonanztomographie (MRT) sowie ein unzureichendes Ansprechen auf zwei verschiedene nicht-steroidale Antirheumatika (NSARs oder non-steroidal anti-inflammatory drugs, NSAIDs) beziehungsweise eine Unverträglichkeit oder Gegenanzeige gegen NSARs in der Vorgeschichte. Die Patienten wurden randomisiert (1:1) und erhielten entweder Bimekizumab 160 mg alle 4 Wochen bis Woche 52 oder Placebo bis Woche 16, gefolgt von Bimekizumab 160 mg alle 4 Wochen bis Woche 52. Bei Studienbeginn betrug die mittlere Dauer der nr-axSpA-Symptome 9 Jahre (Median 5,5 Jahre). 10,6 % der Patienten wurden zuvor mit einem Anti-TNFα-Wirkstoff behandelt.
In der BE MOBILE 2-Studie (AS0011) wurden 332 Patienten mit aktiver AS mit dokumentiertem radiologischem Nachweis (Röntgen) untersucht, die die modifizierten New-York-Klassifikationskriterien der AS erfüllten. Die Patienten hatten eine aktive Erkrankung, definiert als BASDAI ≥4 und Wirbelsäulenschmerzen ≥4 auf einer numerischen Bewertungsskala von 0 bis 10 (nach BASDAI Frage 2). Die Patienten mussten in der Vergangenheit unzureichend auf 2 verschiedene NSARs angesprochen haben oder eine Unverträglichkeit oder eine Kontraindikation für NSARs aufweisen. Die Patienten wurden randomisiert (2:1) und erhielten entweder Bimekizumab 160 mg alle 4 Wochen bis Woche 52 oder Placebo bis Woche 16, gefolgt von Bimekizumab 160 mg alle 4 Wochen bis Woche 52. Bei Studienbeginn betrug die mittlere Dauer der AS-Symptome 13,5 Jahre (Median 11 Jahre). 16,3 % der Patienten wurden zuvor mit einem Anti-TNFα-Wirkstoff behandelt.
Klinisches Ansprechen
Die Behandlung mit Bimekizumab führte sowohl in der Patientengruppe mit nr-axSpA als auch in jener mit AS im Vergleich zu Placebo in Woche 16 zu einer signifikanten Besserung der Symptome und der Krankheitsaktivität (siehe Tabelle 7). Das klinische Ansprechen blieb in der Beurteilung gemäss allen in Tabelle 7 dargestellten Endpunkten in beiden Patientengruppen bis Woche 52 erhalten.
Tabelle 7: Klinisches Ansprechen in BE MOBILE 1 und BE MOBILE 2
|
|
BE MOBILE 1 (nr-axSpA)
|
BE MOBILE 2 (AS)
| |
|
Placebo
(N = 126)
n (%)
|
BKZ 160 mg Q4W
(N = 128)
n (%)
|
Unterschied gegenüber Placebo (95%-KI)a)
|
Placebo
(N = 111)
n (%)
|
BKZ 160 mg Q4W
(N = 221)
n (%)
|
Unterschied gegenüber Placebo (95%-KI)a)
| |
ASAS 40
|
|
|
|
|
|
| |
Woche 16
Woche 52
|
27 (21,4)
|
61 (47,7)*
78 (60,9)
|
26,2 (14,9; 37,5)
|
25 (22,5)
|
99 (44,8)*
129 (58,4)
|
22,3 (11,5; 33,0)
| |
ASAS-40 ohne anti-TNFα-Vorbehandlung
|
|
|
|
|
|
| |
Woche 16
Woche 52
|
(N = 109)
25 (22,9)
|
(N = 118)
55 (46,6)
73 (61,9)
|
24,8 (12,4; 37,1)
|
(N = 94)
22 (23,4)
|
(N = 184)
84 (45,7)*
108 (58,7)
|
22,3 (10,5; 34,0)
| |
ASAS 20
|
|
|
|
|
|
| |
Woche 16
Woche 52
|
48 (38,1)
|
88 (68,8)*
94 (73,4)
|
30,7 (19,0; 42,3)
|
48 (43,2)
|
146 (66,1)*
158 (71,5)
|
22,8 (11,8; 33,8)
| |
ASDAS – erhebliche Verbesserung
|
|
|
|
|
|
| |
Woche 16
Woche 52
|
9 (7,1)
|
35 (27,3)*
47 (36,7)
|
20,2 (11,2; 29,3)
|
6 (5,4)
|
57 (25,8)*
71 (32,1)
|
20,4 (11,7; 29,1)
| |
BASDAI-50
|
|
|
|
|
|
| |
Woche 16
Woche 52
|
27 (21,4)
|
60 (46,9)
69 (53,9)
|
25,3 (14,0; 36,6)
|
29 (26,1)
|
103 (46,6)
119 (53,8)
|
20,5 (9,6; 31,4)
|
BKZ 160 mg Q4W = Bimekizumab 160 mg alle 4 Wochen. ASDAS = Ankylosing Spondylitis Disease Activity Score.
NRI wird verwendet.
a) Unbereinigte Unterschiede werden dargestellt.
* p < 0,001 gegenüber Placebo, für Multiplizität korrigiert.
Der Anteil der Patienten in BE MOBILE 1, die in Woche 16 einen ASDAS < 2,1 (eine Kombination aus ASDAS-ID [inactive disease] und ASDAS-LD [low disease]) erreichten, betrug in der Bimekizumab-Gruppe 46,1 % und 21,1 % in der Placebogruppe (multiple Imputation). In Woche 52 erreichten 61,6 % der Patienten in der Bimekizumab-Gruppe einen ASDAS < 2,1, davon 25,2 % mit inaktiver Krankheit (ASDAS < 1,3).
Der Anteil der Patienten in BE MOBILE 2, die in Woche 16 einen ASDAS < 2,1 (eine Kombination aus ASDAS-ID und ASDAS-LD) erreichten, betrug in der Bimekizumab-Gruppe 44,8 % und 17,4 % in der Placebogruppe (multiple Imputation). In Woche 52 erreichten 57,1 % der Patienten in der Bimekizumab-Gruppe einen ASDAS < 2,1, davon 23,4 % mit inaktiver Krankheit (ASDAS < 1,3).
Alle vier ASAS-40-Komponenten (Gesamtwirbelsäulenschmerzen, Morgensteifigkeit, Bath Ankylosing Spondylitis Functional Index [BASFI] und Patient's Global Assessment of Disease Activity [PGADA]) verbesserten sich unter der Bimekizumab-Behandlung und trugen zum gesamten ASAS-40-Ansprechen in Woche 16 bei. Diese Verbesserungen blieben in beiden Patientengruppen bis Woche 52 erhalten.
Die Verbesserungen bei anderen Wirksamkeitsparametern sind in Tabelle 8 dargestellt.
Tabelle 8: Weitere Wirksamkeitsparameter in BE MOBILE 1 und BE MOBILE 2
|
|
BE MOBILE 1 (nr-axSpA)
|
BE MOBILE 2 (AS)
| |
|
Placebo
(N = 126)
|
BKZ 160 mg Q4W
(N = 128)
|
Placebo
(N = 111)
|
BKZ 160 mg Q4W
(N = 221)
| |
Nächtliche Wirbelsäulenschmerzen
|
|
|
|
| |
Baseline
Mittlere Veränderung in Woche 16 gegenüber Baseline
Mittlere Veränderung in Woche 52 gegenüber Baseline
|
6,7
-1,7
|
6,9
-3,6*
-4,3
|
6,8
-1,9
|
6,6
-3,3*
-4,1
| |
BASDAI
|
|
|
|
| |
Baseline
Mittlere Veränderung in Woche 16 gegenüber Baseline
Mittlere Veränderung in Woche 52 gegenüber Baseline
|
6,7
-1,5
|
6,9
-3,1*
-3,9
|
6,5
-1,9
|
6,5
-2,9*
-3,6
| |
BASMI
|
|
|
|
| |
Baseline
Mittlere Veränderung in Woche 16 gegenüber Baseline
Mittlere Veränderung in Woche 52 gegenüber Baseline
|
3,0
-0,1
|
2,9
-0,4
-0,6
|
3,8
-0,2
|
3,9
-0,5**
-0,7
| |
hs-CRP (mg/l)
|
|
|
|
| |
Baseline (geometrisches Mittel)
Verhältnis zu Baseline in Woche 16
Verhältnis zu Baseline in Woche 52
|
5,0
0,8
|
4,6
0,4
0,4
|
6,7
0,9
|
6,5
0,4
0,3
|
BASMI = Bath Ankylosing Spondylitis Metrology Index. Hs-CRP = hochsensitives C-reaktives Protein
MI wird verwendet.
*p < 0,001 referenzbasierte Imputation, gegenüber Placebo, für Multiplizität korrigiert. **p < 0,01 referenzbasierte Imputation, gegenüber Placebo, für Multiplizität korrigiert.
Die Behandlung mit Bimekizumab war sowohl in der nr-axSpA- als auch in der AS-Patientengruppe mit einem schnellen Wirkeintritt assoziiert.
Abbildung 4: ASAS-40-Ansprechen im Zeitverlauf bis Woche 52 in BE MOBILE 1 (NRI)
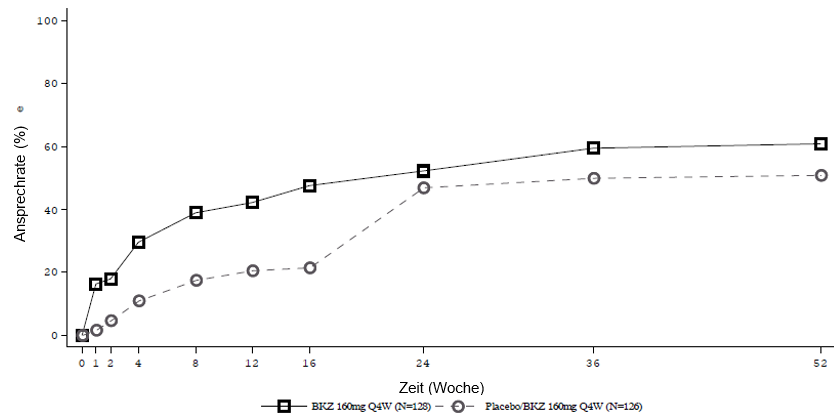
Patienten, die Placebo erhielten, wurden in Woche 16 auf Bimekizumab 160 mg Q4W umgestellt.
Abbildung 5: ASAS-40-Ansprechen im Zeitverlauf bis Woche 52 in BE MOBILE 2 (NRI)
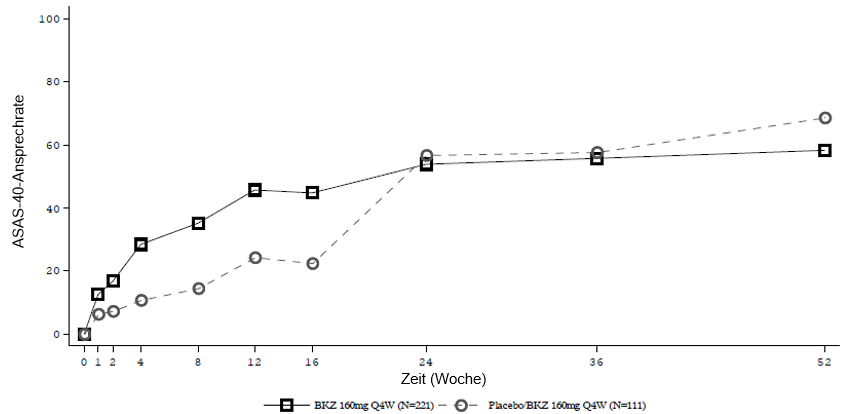
Patienten, die Placebo erhielten, wurden in Woche 16 auf Bimekizumab 160 mg Q4W umgestellt.
In einer integrierten Analyse zu BE MOBILE 1 und BE MOBILE 2 blieb bei den mit Bimekizumab behandelten Patienten, die in Woche 16 ein ASAS-40-Ansprechen erreichten, dieses Ansprechen bei 82,1 % bis Woche 52 erhalten.
Die Wirksamkeit von Bimekizumab wurde unabhängig von Alter, Geschlecht, ethnischer Zugehörigkeit, Krankheitsdauer, Entzündungsstatus zu Baseline, Baseline-ASDAS und begleitenden cDMARDs nachgewiesen.
In Woche 16 war bei Patienten mit Enthesitis bei Baseline der Anteil der Patienten (NRI) mit abklingender Enthesitis gemäss Maastricht Ankylosing Spondylitis Enthesitis Index (MASES) unter Bimekizumab grösser als unter Placebo (BE MOBILE 1: 51,1 % gegenüber 23,9 % und BE MOBILE 2: 51,5 % gegenüber 32,8 %). Das Abklingen der Enthesitis blieb unter Bimekizumab in beiden Studien bis zur Woche 52 erhalten (BE MOBILE 1: 54,3 % und BE MOBILE 2: 50,8 %).
Verringerung der Entzündung
Bimekizumab verringerte die Entzündungswerte gemäss hs-CRP (siehe Tabelle 9) und gemäss MRT in einer Bildgebungs-Substudie. Die Entzündungszeichen wurden zu Baseline und in Woche 16 mittels MRT beurteilt und als Veränderung im SPARCC (Spondyloarthritis Research Consortium of Canada)-Score für die Iliosakralgelenke und im ASspiMRI-a(Ankylosing Spondylitis spine Magnetic Resonance Imagine-activity)-Score in der Berlin-Modifikation für die Wirbelsäule gegenüber Baseline ausgedrückt. Eine Verringerung der Entzündungszeichen sowohl in den Iliosakralgelenken als auch in der Wirbelsäule wurde bei den mit Bimekizumab behandelten Patienten im Vergleich zu Placebo beobachtet (siehe Tabelle 9). Die Verringerung der Entzündung gemäss hs-CRP und gemäss MRT blieb bis Woche 52 erhalten.
Tabelle 9: Verringerung der Entzündung gemäss MRT in BE MOBILE 1 und BE MOBILE 2
|
|
BE MOBILE 1 (nr-axSpA)
|
BE MOBILE 2 (AS)
| |
|
Placebo
|
BKZ 160 mg Q4W
|
Placebo
|
BKZ 160 mg Q4W
| |
SPARCC-Score
|
|
|
|
| |
Mittlere Veränderung in Woche 16 gegenüber Baseline a)
|
-1,56
(N = 62)
|
-6,15
(N = 78)
|
0,59
(N = 46)
|
-4,51
(N = 81)
| |
Mittlere Veränderung in Woche 52 gegenüber Baseline a)
|
|
-7,57
(N = 67)
|
|
-4,67
(N = 78)
| |
ASspiMRI-a-Score (in der Berlin Modifikation)
|
|
|
|
| |
Mittlere Veränderung in Woche 16 gegenüber Baseline a)
|
0,03
(N = 60)
|
-0,36
(N = 74)
|
-0,34
(N = 46)
|
-2,23
(N = 81)
| |
Mittlere Veränderung in Woche 52 gegenüber Baseline a)
|
|
-0,70
(N = 65)
|
|
-2,38
(N = 77)
|
a)Die Werte für die Veränderung gegenüber Baseline basieren auf den beobachteten Fällen und wurden durch die zentrale Auswertung des Datensatzes für Woche 52 ermittelt.
Körperliche Funktionsfähigkeit und weitere gesundheitsbezogene Ergebnisse
Die mit Bimekizumab behandelten Patienten zeigten eine signifikante Verbesserung der körperlichen Funktion gemäss BASFI-Beurteilung gegenüber Baseline im Vergleich zu Placebo (Veränderung des Kleinste-Quadrate-Mittelwerts in Woche 16 gegenüber Baseline in BE MOBILE 1: -2,4 gegenüber - 0,9, p < 0,001 und in BE MOBILE 2: -2,0 gegenüber -1,0, p < 0,001). Die mit Bimekizumab behandelten Patienten zeigten eine signifikante Verbesserung im SF-36 PCS-Score gegenüber Baseline im Vergleich zu Placebo (Veränderung des Kleinste-Quadrate-Mittelwerts in Woche 16 gegenüber Baseline in BE MOBILE 1: 9,3 gegenüber 5,4, p < 0,001 und in BE MOBILE 2: 8,5 gegenüber 5,2, p < 0,001).
Die mit Bimekizumab behandelten Patienten zeigten eine signifikante Verbesserung der gesundheitsbezogenen Lebensqualität gemäss ASQoL (AS Quality of Life Questionnaire) gegenüber Baseline im Vergleich zu Placebo (Veränderung des Kleinste-Quadrate-Mittelwerts in Woche 16 gegenüber Baseline in BE MOBILE 1: -4,9 gegenüber -2,3, p < 0,001 und in BE MOBILE 2: -4,6 gegenüber -3,0, p < 0,001) sowie eine bedeutsame Verringerung der Ermüdung gemäss FACIT-Fatigue-Score (mittlere Veränderung in Woche 16 gegenüber Baseline in BE MOBILE 1: 8,5 für Bimekizumab gegenüber 3,9 für Placebo und in BE MOBILE 2: 8,4 für Bimekizumab gegenüber 5,0 für Placebo).
Die bis Woche 16 erzielten Verbesserungen bei allen Messgrössen der körperlichen Funktion und den anderen zuvor erwähnten gesundheitsbezogenen Ergebnissen (BASFI-, SF-36 PCS-, ASQoL- und FACIT-Fatigue-Score) blieben in beiden Studien bis Woche 52 erhalten.
Extraartikuläre Manifestation
In gepoolten Daten aus BE MOBILE 1 (nr-axSpA) und BE MOBILE 2 (AS) war in Woche 16 der Anteil der Patienten, die eine Uveitis entwickelten, unter Bimekizumab (0,6 %) geringer als unter Placebo (4,6 %). Die Uveitis-Inzidenz blieb unter Langzeitbehandlung mit Bimekizumab gering (1,2/100 Patientenjahre in den gepoolten Phase-II/III-Studien).
|