Eigenschaften/WirkungenATC-Code
L04AJ03
Pegcetacoplan ist ein symmetrisches Molekül, das aus zwei identischen Pentadecapeptiden besteht, die kovalent an die Enden eines linearen Polyethylenglykol (PEG)-Moleküls gebunden sind. Das Molekulargewicht von Pegcetacoplan beträgt etwa 43,5 Kilodalton (kDa). Die Peptideinheiten binden an Komplement C3 und C3b und üben eine breite Hemmung auf die Komplementkaskade aus. Der 40-kDa-PEG-Anteil sorgt für eine bessere Löslichkeit und eine längere Verweildauer im Körper nach Verabreichung des Arzneimittels.
Wirkungsmechanismus
Pegcetacoplan bindet mit hoher Affinität an das Komplementprotein C3 und sein Aktivierungsfragment C3b und reguliert dadurch die Spaltung von C3 und die Bildung von nachgeschalteten Effektoren der Komplementaktivierung. Bei PNH wird die extravaskuläre Hämolyse (EVH) durch die C3b-Opsonisierung gefördert, während die intravaskuläre Hämolyse (IVH) durch den nachgeschalteten Membranangriffskomplex (MAC) vermittelt wird. Pegcetacoplan übt eine umfassende Regulierung der Komplementkaskade aus, indem es proximal zur C3b- und MAC-Bildung wirkt und so die Mechanismen kontrolliert, die zu EVH und IVH führen.
Bei C3G und primärer IC-MPGN kommt es zu einer übermässigen Aktivierung von C3, die über alle (alternativen, klassischen und Lektin-) Komplementwege ausgelöst werden kann, mit exzessiver Ablagerung von C3-Abbauprodukten in den Glomeruli der Niere. Dies führt zu einer Schädigung des Nierenparenchyms und einer Beeinträchtigung der Nierenfunktion. Pegcetacoplan zielt auf die vorgelagerten Effektoren der Komplementaktivierung (C3 und C3b) ab und hemmt dadurch die Aktivierung, die über alle (alternativen, klassischen und Lektin-) Komplementwege ausgelöst wird. Durch die Hemmung von C3 greift Pegcetacoplan direkt in die fehlgesteuerte C3-Aktivierung ein und beeinflusst die zugrunde liegende Erkrankung, indem es die übermässige Ablagerung von C3-Abbauprodukten in den Glomeruli reduziert. Durch die Blockade von C3b hemmt Pegcetacoplan zudem über einen zusätzlichen Wirkmechanismus in der Komplementkaskade die Aktivität der C3-Konvertase des alternativen Komplementwegs (AP). Dadurch wird die Ablagerung von C3-Abbauprodukten in den Glomeruli weiter vermindert.
Pharmakodynamik
PNH
In der Studie APL2-302 stieg die mittlere C3-Konzentration im Serum in der Pegcetacoplan-Gruppe von 0,94 g/l bei Studienbeginn auf 3,83 g/l in Woche 16 und verblieb auf diesem Niveau bis Woche 48. In der Studie APL2-308 stieg die mittlere C3-Konzentration im Serum von einem Ausgangswert von 0,95 g/l auf 3,56 g/l in Woche 26.
In der Studie APL2-302 stieg der mittlere prozentuale Anteil an PNH-Typ II + PNH-Typ III Erythrozyten von einem Ausgangswert von 66,80 % auf 93,85 % in Woche 16 und verblieb auf diesem Niveau bis Woche 48. In der Studie APL2 308 stieg der mittlere prozentuale Anteil an PNH-Typ II + PNH-Typ III Erythrozyten von einem Ausgangswert von 42,4 % auf 90,0 % in Woche 26. In der Studie APL2302 sank der mittlere prozentuale Anteil an PNH-TypII + PNH-Typ III Erythrozyten mit C3-Ablagerung von einem Ausgangswert von 17,73 % auf 0,20 % in Woche 16 und verblieb auf diesem Niveau bis Woche 48. In der Studie APL2308 sank der mittlere prozentuale Anteil an PNH-TypII+PNH-Typ III Erythrozyten mit C3 Ablagerung von einem Ausgangswert von 2,85 % auf 0,09 % in Woche 26.
C3G und primäre IC-MPGN
In der Studie APL2-C3G-310 stieg die mittlere C3-Konzentration im Serum in der Pegcetacoplan-Gruppe von 0,62 g/l bei Studienbeginn auf 3,71 g/l in Woche 26 und der Effekt wurde bis Woche 52 aufrechterhalten. In der Placebo-Gruppe blieben die C3-Konzentrationen bis Woche 26 stabil (0,57 g/l bei Studienbeginn; 0,58 g/l in Woche 26) und nach dem Wechsel zu Pegcetacoplan stiegen diese auf 3,59 g/l in Woche 52 an.
Die mittlere sC5b-9-Konzentration im Serum sank in der Pegcetacoplan-Gruppe von 902,5 ng/ml bei Studienbeginn auf 290,2 ng/ml in Woche 26 und der Effekt wurde bis Woche 52 aufrechterhalten. In der Placebo-Gruppe blieben die sC5b-9-Konzentrationen bis Woche 26 stabil (768,3 ng/ml bei Studienbeginn; 759,9 ng/ml in Woche 26) und nach dem Wechsel zu Pegcetacoplan sanken diese auf 272,9 ng/ml in Woche 52.
In Woche 26 betrug der Anteil der Patienten mit einer Verringerung der C3c-Färbeintensität bei der Nierenbiopsie um mindestens zwei Grössenordnungen gegenüber dem Ausgangswert 74,3 % in der Pegcetacoplan-Gruppe, wobei 71,4 % einen Färbungswert von Null erreichten, verglichen mit 11,8 % der Patienten mit einem Rückgang um zwei Grössenordnungen und 8,8 %, die einen Färbungswert von Null in der Placebo-Gruppe erreichten.
In der Studie APL2-C3G-204 stieg bei Patienten mit Krankheitsrezidiv nach der Transplantation die mittlere C3-Konzentration im Serum von 0,70 g/l bei Studienbeginn auf 2,80 g/l in Woche 52 an, und die mittlere sC5b-9-Konzentration im Serum sank von 525,4 ng/ml bei Studienbeginn auf 151,0 ng/ml in Woche 52.
Herz-Elektrophysiologie
Es wurden keine spezifischen Studien durchgeführt, um das Potenzial von Pegcetacoplan zur Verzögerung der kardialen Repolarisation zu bestimmen. Pegcetacoplan ist eine PEGylierte Peptidstruktur und zeigte keine Hemmung im humanen Ether-agogo-Gen (hERG)-Ionenkanaltest. Die Analyse der Konzentration-QTc bestätigte keine Auswirkungen auf die kardiale Repolarisation (QT-Intervall um die Herzfrequenz korrigiert).
Klinische Wirksamkeit
PNH
Die Wirksamkeit und Sicherheit von Pegcetacoplan bei Patienten mit PNH wurde in zwei offenen, randomisierten, kontrollierten Phase-3-Studien untersucht: in Studie APL2-302 an Patienten, die bereits mit einem Komplementinhibitor vorbehandelt waren, und in Studie APL2-308 an Patienten, die zuvor mit keinem Komplementinhibitor behandelt wurden. In beiden Studien lag die Dosis von Pegcetacoplan bei zweimal wöchentlich 1080 mg. Bei Bedarf konnte die Dosis von Pegcetacoplan auf 1080 mg alle 3 Tage angepasst werden.
Studie an erwachsenen Patienten, die bereits mit einem Komplementinhibitor vorbehandelt waren (APL2-302)
Bei der Studie APL2-302 handelte es sich um eine offene, randomisierte Studie mit einem aktiven, Vergleichspräparat-kontrollierten Zeitraum von 16 Wochen, gefolgt von einem 32-wöchigen offenen Zeitraum (OLP). An dieser Studie nahmen Patienten mit PNH teil, die mindestens in den vorangegangenen drei Monaten mit einer stabilen Eculizumab-Dosis behandelt worden waren und einen Hb-Wert <10,5 g/dl aufwiesen.
Die in Frage kommenden Patienten nahmen an einer vierwöchigen Vorlaufphase teil, während der sie zusätzlich zu ihrer bestehenden Eculizumab-Dosis zweimal wöchentlich 1080 mg Pegcetacoplan subkutan erhielten. Anschliessend wurden die Patienten im Verhältnis 1:1 randomisiert und erhielten entweder zweimal wöchentlich 1080 mg Pegcetacoplan oder ihre bestehende Eculizumab-Dosis für die Dauer des 16-wöchigen randomisierten Kontrollzeitraums (RCP). Die Randomisierung wurde anhand der Anzahl der Transfusionen von Erythrozytenkonzentraten innerhalb der letzten 12 Monate vor Tag -28 (<4; ≥4) und der Thrombozytenzahl beim Screening (<100’000/μl; ≥100’000/μl) stratifiziert. Patienten, die die RCP abschlossen, kamen anschliessend in die OLP, in der alle Patienten Pegcetacoplan für bis zu 32 Wochen erhielten (Patienten, die während der RCP Eculizumab erhalten hatten, kamen zunächst in eine 4-wöchige Vorlaufphase, bevor sie zur Pegcetacoplan-Monotherapie wechselten).
Die Patienten wurden gegen Streptococcus pneumoniae, Neisseria meningitidis Typ A, C, W, Y und B sowie Haemophilus influenzae Typ B (Hib) geimpft, und zwar entweder innerhalb von zwei Jahren vor dem ersten Tag der Behandlung oder innerhalb von zwei Wochen nach Beginn der Behandlung mit Pegcetacoplan. Patienten, die nach Tag 1 geimpft wurden, erhielten bis 2 Wochen nach der Impfung eine prophylaktische Behandlung mit geeigneten Antibiotika. Darüber hinaus wurde eine prophylaktische Antibiotikatherapie nach Ermessen des Prüfarztes in Übereinstimmung mit den lokalen Behandlungsrichtlinien für Patienten mit PNH, die mit einem Komplementärinhibitor behandelt werden, verabreicht. Pegcetacoplan wurde als subkutane Infusion verabreicht; die Infusionszeit betrug etwa 20 bis 40 Minuten.
Der primäre und die sekundären Wirksamkeitsendpunkte wurden in Woche 16 bewertet. Der primäre Wirksamkeitsendpunkt war die Veränderung des Hb-Spiegels vom Ausgangswert bis zur Woche 16 (während der RCP). Der Ausgangswert wurde als der Durchschnitt der Messwerte definiert, die vor der Verabreichung der ersten Dosis von Pegcetacoplan aufgezeichnet wurden. Wichtige sekundäre Wirksamkeitsendpunkte waren die Vermeidung von Transfusionen, definiert als der Anteil der Patienten, die während der RCP keine Transfusion benötigten, sowie die Veränderung der absoluten Retikulozytenzahl (ARC), des LDH-Spiegels und des Scores der FACIT-Fatigue-Skala (Functional Assessment of Chronic Illness Therapy – Fatigue) vom Ausgangswert bis zur Woche 16. Insgesamt wurden 80 Patienten in die Vorlaufphase aufgenommen. Am Ende der Vorlaufphase wurden alle 80 Patienten randomisiert, 41 für Pegcetacoplan und 39 für Eculizumab. Die demografischen Daten und die Ausgangskrankheitscharakteristika waren im Allgemeinen zwischen den Behandlungsgruppen ausgewogen (siehe Tabelle 3). Insgesamt 38 Patienten in der mit Pegcetacoplan behandelten Gruppe und 39 Patienten in der Eculizumab-Gruppe schlossen die 16-wöchige RCP ab und setzten die 32-wöchige offene Behandlungsphase fort. Insgesamt brachen 12 von 80 (15 %) Patienten, die Pegcetacoplan erhielten, die Studie wegen unerwünschter Ereignisse ab. Gemäss Prüfplan wurde die Dosis bei 15 Patienten auf 1 080 mg alle 3 Tage angepasst. Bei 12 Patienten wurde der Nutzen beurteilt und bei 8 von 12 Patienten erwies sich die Dosisanpassung als wirksam.
Tabelle 3: Demografische Grunddaten und Merkmale der Patienten in der Studie APL2-302
|
Parameter
|
Statistik
|
Pegcetacoplan (n=41)
|
Eculizumab (n=39)
| |
Alter (Jahre)
|
Mittelwert (SD)
|
50,2 (16,3)
|
47,3 (15,8)
| |
Baseline-Dosis von Eculizumab
Jede 2. Woche i.v. 900 mg
Jeden 11. Tag i.v. 900 mg
Jede 2. Woche i.v. 1200 mg
Jede 2. Woche i.v. 1500 mg
|
n (%)
n (%)
n (%)
n (%)
|
26 (63,4)
1 (2,4)
12 (29,3)
2 (4,9)
|
29 (74,4)
1 (2,6)
9 (23,1)
0
| |
Frauen
|
n (%)
|
27 (65,9)
|
22 (56,4)
| |
Zeit seit PNH Diagnose (Jahre) bis Tag -28
|
Mittelwert (SD)
|
8,7 (7,4)
|
11,4 (9,7)
| |
Hb-Spiegel (g/dl)
|
Mittelwert (SD)
|
8,7 (1,1)
|
8,7 (0,9)
| |
ARC (109 /l)
|
Mittelwert (SD)
|
218 (75,0)
|
216 (69,1)
| |
LDH-Spiegel (U/l)
|
Mittelwert (SD)
|
257,5 (97,6)
|
308,6 (284,8)
| |
FACIT-Fatigue Gesamt-Score*
|
Mittelwert (SD)
|
32,2 (11,4)
|
31,6 (12,5)
| |
Anzahl Transfusionen in den letzten 12 Monaten vor Tag -28
|
Mittelwert (SD)
|
6,1 (7,3)
|
6,9 (7,7)
| |
<4
|
n (%)
|
20 (48,8)
|
16 (41,0)
| |
≥4
|
n (%)
|
21 (51,2)
|
23 (59,0)
| |
Thrombozytenzahl beim Screening (109 /l)
|
Mittelwert (SD)
|
167 (98,3)
|
147 (68,8)
| |
Thrombozytenzahl beim Screening
<100,000/μl
|
n (%)
|
12 (29,3)
|
9 (23,1)
| |
Thrombozytenzahl beim Screening
≥100,000/μl
|
n (%)
|
29 (70,7)
|
30 (76,9)
| |
Aplastische Anämie in der Anamnese
|
n (%)
|
11 (26,8)
|
9 (23,1)
| |
Myelodysplastisches Syndrom in der Anamnese
|
n (%)
|
1 (2,4)
|
2 (5,1)
|
* Der FACIT-Fatigue-Score wird auf einer Skala von 0 bis 52 gemessen, wobei höhere Werte eine geringere Müdigkeit (Fatigue) anzeigen.
Pegcetacoplan war Eculizumab in Bezug auf den primären Endpunkt der Hämoglobinveränderung gegenüber dem Ausgangswert überlegen (p < 0,0001). Die korrigierte mediane Veränderung des Hb-Wertes gegenüber dem Ausgangswert betrug 2,4 g/dl in der mit Pegcetacoplan behandelten Gruppe gegenüber -1,5 g/dl in der Eculizumab-Gruppe, was einen korrigierten medianen Anstieg von 3,8 g/dl unter Pegcetacoplan im Vergleich zu Eculizumab in Woche 16 entspricht (Abbildung 1).
Abbildung 1: Korrigierter Mittelwert (± SE) der Veränderung des Hämoglobins (g/dl) von Baseline bis Woche 16 in APL2-302

Die Nicht-Unterlegenheit (noninferiority) für die wichtigen sekundären Endpunkte Transfusionsvermeidung und ARC im Vergleich zu den Ausgangswerten wurde nachgewiesen. Bei 85 % der Patienten in der mit Pegcetacoplan behandelten Gruppe konnten Transfusionen vermieden werden, gegenüber 15 % in der Eculizumab-Gruppe.
Die Nicht-Unterlegenheit wurde bei der Veränderung der LDH gegenüber dem Ausgangswert nicht nachgewiesen.
Aufgrund des hierarchischen Testablaufs wurde die Veränderung des FACIT-Fatigue-Scores gegenüber dem Ausgangswert nicht formal getestet.
Die korrigierten Mittelwerte, der Behandlungsunterschied, die Konfidenzintervalle und die statistischen Analysen, die für die wichtigsten sekundären Endpunkte durchgeführt wurden, sind in Abbildung 2 dargestellt.
Abbildung 2: Analyse wichtiger sekundärer Endpunkte in APL2-302

Die Ergebnisse waren in allen bestätigenden Analysen der primären und wichtigen sekundären Endpunkte konsistent, einschliesslich aller beobachteten Daten, die auch Daten nach der Transfusion enthielten.
Bei den mit Pegcetacoplan behandelten Patienten zeigten die primären und wichtigen sekundären Wirksamkeitsanalysen keine nennenswerten Unterschiede in Bezug auf Geschlecht, Rasse oder Alter.
Eine Hb-Normalisierung wurde in Woche 16 bei 34 % der Patienten in der Pegcetacoplan -Gruppe gegenüber 0 % in der Eculizumab-Gruppe erreicht. Eine Normalisierung des ARC wurde bei 78 % der Patienten in der mit Pegcetacoplan behandelten Gruppe gegenüber 3 % in der Eculizumab-Gruppe erreicht. Eine Normalisierung der LDH wurde bei 71 % der Patienten in der mit Pegcetacoplan behandelten Gruppe erreicht, gegenüber 15 % in der Eculizumab-Gruppe.
Insgesamt 77 Patienten nahmen an der 32-wöchigen OLP teil, während der alle Patienten Pegcetacoplan erhielten; die Gesamtexposition betrug bis zu 48 Wochen. Die Ergebnisse in Woche 48 stimmten im Allgemeinen mit denen in Woche 16 überein und belegen eine anhaltende Wirksamkeit.
Studie an erwachsenen Patienten, die mit keinem Komplementinhibitor vorbehandelt wurden (APL2-308)
Bei der Studie APL2-308 handelte es sich um eine offene, randomisierte, kontrollierte Studie, an der Patienten mit PNH teilnahmen, die in den letzten 3 Monaten vor Studienbeginn nicht mit einem Komplementinhibitor behandelt worden waren und deren Hb-Wert unter der unteren Grenze des Normwerts (LLN) lag. Geeignete Patienten wurden im Verhältnis 2:1 randomisiert und erhielten entweder Pegcetacoplan oder eine supportive Behandlung (z. B. Transfusionen, Kortikosteroide, Supplementierung mit z. B. Eisen, Folsäure und Vitamin B12), die im Folgenden als Kontrollgruppe bezeichnet wird, während des gesamten 26wöchigen Behandlungszeitraums.
Die Randomisierung wurde anhand der Anzahl der Transfusionen von PRBC innerhalb der letzten 12 Monate vor Tag -28 (< 4; ≥ 4) stratifiziert. Zu jedem Zeitpunkt während der Studie konnte ein Patient, der der Kontrollgruppe zugewiesen wurde und dessen Hb-Wert ≥ 2 g/dl unter dem Ausgangswert lag oder der ein PNH-assoziiertes thromboembolisches Ereignis hatte, gemäss Prüfplan für den Rest der Studie auf Pegcetacoplan umgestellt werden.
Insgesamt wurden 53 Patienten randomisiert: 35 Patienten in die Pegcetacoplan-Gruppe und 18 Patienten in die Kontrollgruppe. Die demografischen Daten und die Krankheitsmerkmale beim Ausgangswertwaren im Allgemeinen zwischen den Behandlungsgruppen ausgewogen. Das Durchschnittsalter betrug 42,2 Jahre in der Pegcetacoplan-Gruppe und 49,1 Jahre in der Kontrollgruppe. Die durchschnittliche Anzahl von PRBC-Transfusionen in den 12 Monaten vor der Voruntersuchung betrug 3,9 in der Pegcetacoplan-Gruppe und 5,1 in der Kontrollgruppe. Fünf Patienten in jeder Gruppe (14,3 % in der Pegcetacoplan-Gruppe und 27,8 % in der Kontrollgruppe) wiesen eine aplastische Anämie in der Anamnese auf. Weitere Ausgangswerte waren: mittlerer Hb-Ausgangswert (Pegcetacoplan-Gruppe: 9,4 g/dl vs. Kontrollgruppe: 8,7 g/dl), ARC (Pegcetacoplan-Gruppe: 230,2 × 109/l vs. Kontrollgruppe: 180,3 × 109/l), LDH-Wert (Pegcetacoplan-Gruppe: 2 151,0 E/l vs. Kontrollgruppe: 1 945,9 E/l) und Thrombozytenzahl (Pegcetacoplan-Gruppe: 191,4 × 109/l vs. Kontrollgruppe: 125,5 × 109/l). 11 der 18 Patienten, die der Kontrollgruppe zugewiesen wurden, wechselten zu Pegcetacoplan, weil ihr Hb-Wert um ≥ 2 g/dl unter den Ausgangswert sank. Von den 53 randomisierten Patienten erhielten 52 (97,8 %) eine prophylaktische Antibiotikatherapie gemäss den örtlichen Verschreibungsleitlinien.
Der primäre und die sekundären Wirksamkeitsendpunkte wurden in Woche 26 bewertet. Die beiden ko-primären Wirksamkeitsendpunkte waren die Stabilisierung des Hb-Werts, definiert als Vermeidung eines Abfalls der Hb-Konzentration > 1 g/dl gegenüber dem Ausgangswert ohne Transfusion, und die Veränderung der LDH-Konzentration gegenüber dem Ausgangswert.
In der mit Pegcetacoplan behandelten Gruppe erreichten 30 von 35 Patienten (85,7 %) eine Stabilisierung des Hb-Werts gegenüber 0 Patienten in der Kontrollgruppe. Die bereinigte Differenz zwischen Pegcetacoplan und der Kontrollgruppe betrug 73,1 % (95 %-KI: 57,2 % bis 89,0 %; p < 0,0001).
Die kleinste quadratische (LS) mittlere (SE) Veränderung der LDH-Konzentration gegenüber dem Ausgangswert in Woche 26 betrug -1 870 E/l in der mit Pegcetacoplan behandelten Gruppe gegenüber -400 E/l in der Kontrollgruppe (p < 0,0001). Die Differenz zwischen Pegcetacoplan und der Kontrollgruppe betrug -1 470 (95 %-KI: -2 113 bis -827). Die Behandlungsunterschiede zwischen der Pegcetacoplan-Gruppe und der Kontrollgruppe zeigten sich in Woche 2 und blieben bis Woche 26 bestehen (Abbildung 3). Die LDH-Konzentrationen in der Kontrollgruppe verblieben erhöht.
Abbildung 3: Mittlere (±SE) LDH-Konzentration (E/l) im Zeitverlauf nach Behandlungsgruppe in der Studie APL2-308

Bei den ausgewählten wichtigen sekundären Wirksamkeitsendpunkten Hb-Ansprechen ohne Transfusion, Veränderung des Hb-Spiegels und Veränderung der ARC zeigte sich in der mit Pegcetacoplan behandelten Gruppe ein signifikanter Behandlungsunterschied gegenüber der Kontrollgruppe (Tabelle 4).
Tabelle 4: Analyse wichtiger sekundärer Endpunkte in Studie APL2-308
|
Parameter
|
Pegcetacoplan
(N = 35)
|
Kontrollgruppe
(N = 18)
|
Differenz
(95%-KI)
p-Wert
| |
Hb-Ansprechen ohne Transfusiona
(n, %)
|
25 (71 %)
|
1 (6 %)
|
54 % (34 %; 74 %)
p < 0,0001
| |
Veränderung des Hb-Spiegels (g/dl) vom Ausgangswert bis Woche 26
LS-Mittelwert (SE)
|
2,9 (0,38)
|
0,3 (0,76)
|
2,7 (1,0; 4,4)
| |
Veränderung der ARC (109/l) vom Ausgangswert bis Woche 26
LS-Mittelwert (SE)
|
-123 (9,2)
|
-19 (25,2)
|
-104 (-159; -49)
|
a Das Hb-Ansprechen war definiert als Anstieg des Hämoglobins um ≥ 1 g/dl vom Ausgangswert bis Woche 26. ARC = Absolute Retikulozytenzahl, KI = Konfidenzintervall, LS = Kleinste Quadrate (least square), SE = Standardfehler (standard error)
C3G und primäre IC-MPGN
Die Wirksamkeit und Sicherheit von Pegcetacoplan bei Patienten mit C3G oder primärer IC-MPGN wurde in der randomisierten, placebokontrollierten, doppelblinden Phase-III-Studie APL2-C3G-310 untersucht; eingeschlossen waren Erwachsene und Jugendliche mit einer Erkrankung der nativen Niere oder einem Rezidiv von C3G oder primärer IC-MPGN nach Nierentransplantation.
Die Dosis von Pegcetacoplan betrug 1 080 mg zweimal wöchentlich für Erwachsene oder Jugendliche mit einem Körpergewicht ≥ 50 kg oder gewichtsabhängig für Jugendliche mit einem Körpergewicht unter 50 kg.
Studie bei erwachsenen und jugendlichen Patienten mit C3G oder primärer IC-MPGN (APL2-C3G-310)
Die Studie APL2-C3G-310 war eine randomisierte, doppelblinde Studie mit einer placebokontrollierten Behandlungsphase von 26 Wochen, gefolgt von einer 26-wöchigen offenen Behandlungsphase (OLP). An dieser Studie nahmen Jugendliche im Alter von 12 bis 17 Jahren und Erwachsene mit C3G oder primärer IC-MPGN teil. Dabei wurden sowohl Patienten mit einer Erkrankung der nativen Niere als auch solche mit einem Krankheitsrezidiv nach Transplantation aufgenommen, die eine Proteinurie von ≥ 1 g/Tag und eine eGFR von ≥ 30 ml/min/1,73 m2 aufwiesen. Die Patienten erhielten mindestens 12 Wochen vor der Randomisierung eine stabile und optimierte Dosierung für die Behandlung von C3G/primärer IC-MPGN (z. B. RAS-Hemmer, Natrium-Glukose-Cotransporter-2-Hemmer [SGLT-2-Hemmer], Immunsuppressiva, systemische Kortikosteroide in einer Dosierung von maximal 20 mg/Tag Prednison-Äquivalent).
Die in Frage kommenden Patienten wurden im Verhältnis 1:1 randomisiert und erhielten während der 26-wöchigen placebokontrollierten Behandlungsphase (RCP) zweimal wöchentlich Pegcetacoplan oder Placebo subkutan. Bei der Randomisierung wurden zwei Stratifizierungsfaktoren angewandt: Patienten mit einem Krankheitsrezidiv nach einer Transplantation im Vergleich zu Patienten mit einer Erkrankung der nativen Niere, und Patienten mit Nierenbiopsien zu Beginn der Studie (entweder während des Screenings oder innerhalb von 28 Wochen vor der Randomisierung) im Vergleich zu Patienten ohne Nierenbiopsien zu Beginn der Studie. Während des RCP wurden Änderungen an den Ausgangsbehandlungsschemata für C3G/primäre IC-MPGN auf ein Minimum beschränkt und nur dann vorgenommen, wenn dies für das Wohlbefinden des Patienten erforderlich war. Die Patienten, die die RCP abgeschlossen hatten, wechselten in die 26-wöchige offene Behandlungsphase (OLP), in der alle Teilnehmenden zweimal wöchentlich mit Pegcetacoplan behandelt wurden.
Insgesamt wurden 124 Patienten randomisiert, davon erhielten 63 Pegcetacoplan und 61 Placebo. Die demografischen Daten und die Ausgangsmerkmale der Erkrankung waren zwischen den beiden Gruppen im Allgemeinen ausgewogen verteilt (siehe Tabelle 5). Insgesamt schlossen 118 Patienten die 26-wöchige RCP ab, und von diesen schlossen 114 Patienten die OLP-Behandlungsphase mit Pegcetacoplan ab (N = 59 Pegcetacoplan-zu-Pegcetacoplan; N = 55 Placebozu-Pegcetacoplan).
Tabelle 5: Demographische Ausgangsdaten und Krankheitsmerkmale der Patienten in der Studie APL2-C3G-310
|
Parameter
|
Statistik
|
Pegcetacoplan
(N = 63)
|
Placebo
(N = 61)
| |
Alter (Jahre)
|
Mittelwert (SA)
|
28,2 (17,1)
|
23,6 (14,3)
| |
Jugendliche (12–17 Jahre)
|
n (%)
|
28 (44,4)
|
27 (44,3)
| |
Erwachsene ≥18 Jahre
|
n (%)
|
35 (55,6)
|
34 (55,7)
| |
Geschlecht
Männlich
Weiblich
|
n (%)
n (%)
|
26 (41,3)
37 (58,7)
|
28 (45,9)
33 (54,1)
| |
Erkrankung beim Screening
|
|
|
| |
C3G
|
n (%)
|
51 (81,0)
|
45 (73,8)
| |
C3GN
|
n (%)
|
45 (71,4)
|
41 (67,2)
| |
DDD
|
n (%)
|
4 (6,3)
|
4 (6,6)
| |
Unbestimmt
|
n (%)
|
2 (3,2)
|
0
| |
IC-MPGN
|
n (%)
|
12 (19,0)
|
16 (26,2)
| |
Zeit seit der Diagnose von C3G/IC-MPGN (Jahre)
|
Mittelwert (SA)
|
3,64 (3,47)
|
3,76 (3,62)
| |
Vorherige Nierentransplantation
|
n (%)
|
5 (7,9)
|
4 (6,6)
| |
Zeit seit der letzten Nierentransplantation (Jahre)
|
Mittelwert (SA)
|
11,4 (6,7)
|
5,8 (6,4)
| |
Zeit seit dem letzten Krankheitsrezidiv nach Transplantation (Jahre)
|
Mittelwert (SA)
|
1,47 (1,49)
|
1,38 (1,64)
| |
uPCR im dreifach gemessenen Morgenurin bei Baseline (mg/g)
|
Mittelwert (SA)
|
3124 (2408)
|
2541 (2015)
| |
eGFR bei Baseline (ml/min/1,73 m2)
|
Mittelwert (SA)
|
78,5 (34,1)
|
87,2 (37,2)
| |
C3c-Färbung in der Baseline-Biopsie
|
|
|
| |
3+
|
n (%)
|
51 (81,0)
|
51 (83,6)
| |
2+
|
n (%)
|
12 (19,0)
|
10 (16,4)
| |
Serumalbumin bei Baseline (g/dl)
|
Mittelwert (SA)
|
3,31 (0,61)
|
3,39 (0,70)
| |
Serum-C3 bei Baseline (mg/dl)
|
Mittelwert (SA)
|
60,6 (45,7)
|
56,3 (35,6)
| |
Manifestationen der Krankheit
|
|
|
| |
Ödeme
|
n (%)
|
45 (71,4)
|
32 (52,5)
| |
Müdigkeit (Fatigue)
|
n (%)
|
16 (25,4)
|
8 (13,1)
| |
Hämaturie
|
n (%)
|
37 (58,7)
|
39 (63,9)
| |
Hoher Blutdruck
|
n (%)
|
35 (55,6)
|
29 (47,5)
| |
Nephrotisches Syndrom
|
n (%)
|
32 (50,8)
|
27 (44,3)
| |
Begleitbehandlungen bei Studienbeginn*
|
|
|
| |
Wirkstoffe, die auf das Renin-Angiotensin-System wirken
|
n (%)
|
59 (93,7)
|
54 (88,5)
| |
Immunsuppressiva
|
n (%)
|
49 (77,8)
|
45 (73,8)
| |
Glukokortikoide
|
n (%)
|
29 (46,0)
|
27 (44,3)
|
* innerhalb von 12 Wochen vor Studienbeginn.
C3G = C3-Glomerulopathie, C3GN = C3-Glomerulonephritis, DDD = Dense Deposit Desease, IC MPGN = Immunkomplex-vermittelte membranoproliferative Glomerulonephritis, FMU = Morgenurin, uPCR = Urin-Protein-Kreatinin-Verhältnis, eGFR = Geschätzte glomeruläre Filtrationsrate, SA = Standardabweichung
Der primäre Wirksamkeitsendpunkt war das log-transformierte Verhältnis des uPCR im Morgenurin (FMU) in Woche 26 im Vergleich zum Ausgangswert.
Pegcetacoplan erwies sich als dem Placebo überlegen, mit einer statistisch signifikanten Reduktion des uPCR von 68,1 % (95 %-KI: 57,3 % bis 76,2 %, p < 0,0001) im Vergleich zum Ausgangswert gegenüber Placebo nach 26-wöchiger Behandlung (-67,2 % [95 %-KI: -74,9 % bis -57,2 %] bzw. + 2,9 % [95 %-KI: -8,6 % bis 15,9 %] für Pegcetacoplan bzw. Placebo. Eine ähnliche Wirksamkeit wurde in Untergruppen unabhängig von Alter (Jugendliche vs. Erwachsene), Krankheitstyp (C3G vs. primäre IC-MPGN), Krankheitsstatus (native vs. rezidivierende Erkrankung nach Transplantation) und gleichzeitiger Anwendung von Immunsuppressiva/Glukokortikoiden (ja vs. nein) beobachtet. Die Wirkung von Pegcetacoplan auf das uPCR hielt bis Woche 52 an (-67,2 % gegenüber dem Ausgangswert). Patienten, die in Woche 26 von Placebo auf Pegcetacoplan umgestellt wurden (Abbil-dung 4), zeigten in Woche 52 eine ähnliche Verringerung (-51,3 %).
Abbildung 4: Geometrisches Mittelwertverhältnis (95%-KI) des FMU uPCR im Vergleich zum Ausgangswert im Zeitverlauf nach Behandlungsgruppe, basierend auf dem MMRM-Modell in der Studie APL2-C3G-310
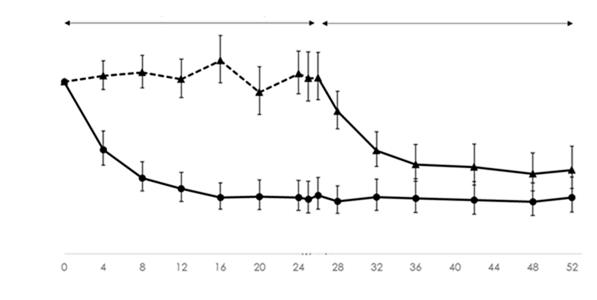
Hinweis: Geometrisches Mittelwertverhältnis berechnet aus reexponierten LS-Mittelwerten
KI = Konfidenz-Intervall, LS = Methode der kleinsten Quadrate (Least-Squares), FMU = Morgenurin, uPCR = Urin-Protein-Kreatinin-Verhältnis, MMRM = Gemischtes Modell der wiederholten Messung
Die 26-wöchige Behandlung mit Pegcetacoplan zeigte eine statistisch signifikante Verbesserung des wichtigen sekundären Endpunkts: 60,3 % der mit Pegcetacoplan behandelten Patienten erreichten eine ≥ 50%ige Reduktion des uPCR, verglichen mit 4,9 % in der Placebo-Gruppe (p < 0,0001).
Die 26-wöchige Behandlung mit Pegcetacoplan führte zu einem höheren Anteil an Patienten, die eine Reduktion um zwei Grössenordnungen oder mehr auf einer Skala von 0 bis 3 in der C3-Färbungsintensität der Nieren erreichten, mit 26 (74,3 %) Patienten unter Pegcetacoplan gegenüber 4 (11,8 %) unter Placebo (nominal p < 0,0001), was auf eine Krankheitsmodifikation bei den mit Pegcetacoplan behandelten Patienten hindeutet.
Die 26-wöchige Behandlung mit Pegcetacoplan zeigte eine Stabilisierung der eGFR mit einer Ver-änderung gegenüber dem Ausgangswert von -1,497 (2,242) unter Pegcetacoplan gegenüber -7,808 (1,919) unter Placebo (nominal p = 0,0333). Die Wirkung von Pegcetacoplan auf die eGFR hielt bis Woche 52 an.
Eine ähnliche Wirksamkeit wurde hinsichtlich einer Proteinurie-Reduktion von ≥ 50 %, einer C3-Färbungsclearance und einer eGFR-Stabilisierung in allen relevanten Untergruppen in Woche 26 beobachtet.
Studie bei Erwachsenen mit Rezidiv von C3G oder primärer IC-MPGN nach Transplantation (APL2 C3G-204)
Die Studie APL2-C3G-204 war eine offene, randomisierte Phase-II-Studie bei 13 erwachsenen Patienten mit Krankheitsrezidiv von C3G (N = 10) oder primärer IC-MPGN (N = 3) nach Transplantation über einen Zeitraum von 52 Wochen.
Während der ersten 12 Wochen der Studie erhielten 10 Patienten Pegcetacoplan zusätzlich zur Standardtherapie (SOC), und 3 Patienten nur Standardtherapie. Alle Patienten erhielten Pegcetacoplan von Woche 13 bis Woche 52.
Der primäre Endpunkt, eine Verringerung der C3-Färbungsintensität bei der Nierenbiopsie in Woche 12, wurde bei 50 % der mit Pegcetacoplan behandelten Patienten (5 von 10 Patienten, von denen 4 einen Färbungsscore von Null erreichten) und bei 33,3 % der Patienten in der Kontrollgruppe (1 von 3 Patienten, wobei dieser Patient einen Färbungsscore von 1 erreichte) beobachtet.
Im Allgemeinen waren die Veränderungen und prozentualen Veränderungen der eGFR gegenüber dem Ausgangswert (sekundärer Endpunkt) gering. Die mittlere (SA) eGFR veränderte sich von 52,3 (12,11) ml/min/1,73 m2 bei Studienbeginn auf 57,3 (25,12) ml/min/1,73 m2 in Woche 52, und die mediane eGFR veränderte sich von 50,5 ml/min/1,73 m2 bei Studienbeginn auf 58,5 ml/min/1,73 m2 in Woche 52. Die meisten Patienten (9 von 13 Patienten [69,2 %]) in allen Gruppen erreichten bis Woche 52 eine Stabilisierung oder Verbesserung der eGFR.
|