ZusammensetzungWirkstoffe
Atogepant
Hilfsstoffe
Copovidon K 28, Tocofersolan, Mannitol (E 421), mikrokristalline Cellulose, Natriumchlorid, Croscarmellose-Natrium, hochdisperses Siliciumdioxid, Natriumstearylfumarat.
1 Tablette à 10 mg bzw. 60 mg enthält 5,26 mg Natrium bzw. 31,48 mg Natrium.
Indikationen/AnwendungsmöglichkeitenProphylaktische Behandlung der Migräne bei Erwachsenen, sofern diese indiziert ist.
Dosierung/AnwendungDie Indikation für die Therapie soll durch einen Arzt oder eine Ärztin mit Erfahrung auf dem Gebiet der Migränebehandlung gestellt und durch diese in der weiteren Behandlung begleitet werden.
Übliche Dosierung
Die empfohlene orale Dosis für AQUIPTA beträgt 60 mg einmal täglich.
Dosisanpassung aufgrund Interaktionen
Dosisanpassungen bei gleichzeitiger Anwendung bestimmter Arzneimittel sind in Tabelle 1 aufgeführt (siehe «Interaktionen»).
Tabelle 1: Dosisanpassungen bei Interaktionen und für spezielle Patientengruppen
|
Dosisanpassungen
|
Empfohlene Dosis (einmal täglich)
| |
Starke CYP3A4-Inhibitoren
|
10 mg
| |
OATP-Inhibitoren
|
10 mg
| |
Schwere Nierenfunktionsstörung und terminales Nierenversagen (CLcr <30 ml/min)
|
10 mg
|
Spezielle Dosierungsanweisungen
Ältere Patienten
Die populationspharmakokinetische Modellierung deutet darauf hin, dass es keine klinisch signifikanten pharmakokinetischen Unterschiede zwischen älteren (≥65 Jahre) und jüngeren Studienteilnehmern gibt. Bei älteren Patienten ist keine Dosisanpassung von AQUIPTA erforderlich.
Patienten mit Leberfunktionsstörungen
Die Anwendung von AQUIPTA ist bei Patienten mit schwerer Leberinsuffizienz zu vermeiden. Bei Patienten mit leichter oder mittelschwerer Leberinsuffizienz wird keine Dosisanpassung empfohlen (siehe «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit schwerer Niereninsuffizienz (CLcr 15–29 ml/min) und bei Patienten mit terminalem Nierenversagen (end-stage renal disease, ESRD) (CLcr < 15 ml/min) beträgt die empfohlene Dosis von AQUIPTA 10 mg einmal täglich. Bei Patienten mit ESRD, die eine intermittierende Dialyse erhalten, ist AQUIPTA vorzugsweise nach der Dialyse einzunehmen. Bei Patienten mit leichter oder mittelschwerer Niereninsuffizienz wird keine Dosisanpassung empfohlen (siehe «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von AQUIPTA bei Kindern und Jugendlichen unter 18 Jahren ist bisher noch nicht erwiesen. Es liegen keine Daten vor. AQUIPTA ist für die Anwendung in der pädiatrischen Population nicht zugelassen.
Verspätete Dosisgabe
Wenn eine Einnahme versäumt wurde, ist diese so schnell wie möglich nachzuholen. Wenn jedoch die Einnahme einen ganzen Tag lang versäumt wurde, ist die verpasste Dosis auszulassen und die nächste Dosis wie geplant einzunehmen.
Art der Anwendung
AQUIPTA wird einmal täglich oral mit oder ohne Mahlzeiten eingenommen.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe (siehe «Zusammensetzung») (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Warnhinweise und VorsichtsmassnahmenÜberempfindlichkeitsreaktionen
Überempfindlichkeitsreaktionen, einschliesslich Anaphylaxie, Dyspnoe, Hautausschlag, Pruritus, Urtikaria und Gesichtsödem, wurden bei der Anwendung von AQUIPTA gemeldet (siehe «Unerwünschte Wirkungen»). Einige Überempfindlichkeitsreaktionen können Tage nach der Verabreichung auftreten. Beim Auftreten einer Überempfindlichkeitsreaktion ist AQUIPTA abzusetzen und eine geeignete Therapie muss eingeleitet werden.
Leberfunktionsstörungen
AQUIPTA wird nicht empfohlen bei Patienten mit stark eingeschränkter Leberfunktion (siehe «Dosierung/Anwendung»).
Patienten, die in den klinischen Phase-3-Studien nicht untersucht wurden
Patienten mit klinisch relevanten kardiovaskulären oder zerebrovaskulären Erkrankungen wie ischämische Herzerkrankung, Herzrhythmus- oder Reizleitungsstörungen, Myokardinfarkt, transitorische ischämische Attacke, Herzinsuffizienz, oder unkontrolliertem Bluthochdruck waren von den pivotalen Studien ausgeschlossen worden. Zu diesen Patienten liegen keine Sicherheitsdaten vor.
Kopfschmerzen durch Medikamentenübergebrauch (MOH, medication overuse headache)
Ein übermässiger Gebrauch von Arzneimitteln zur Akutbehandlung von Kopfschmerzen kann diese verschlimmern. Es liegen zwar keine Hinweise vor, dass die einmal tägliche Einnahme von Atogepant zur vorbeugenden Behandlung zu MOH führen kann, dennoch sollte eine MOH-Diagnose bei Patienten vermutet werden, die regelmässig oder täglich Kopfschmerzen haben trotz (oder wegen) der regelmässigen Einnahme von Arzneimitteln zur Akutbehandlung. Wenn dies der Fall ist oder ein solcher Verdacht besteht, sollte das Arzneimittel zur Akutbehandlung abgesetzt werden.
Natrium
AQUIPTA 10 mg Tabletten enthalten weniger als 1 mmol Natrium (23 mg) pro Tablette, d.h. sie sind nahezu «natriumfrei».
AQUIPTA 60 mg Tabletten enthalten 31,48 mg Natrium pro Tablette, entsprechend 1,6% der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
InteraktionenWirkung anderer Arzneimittel auf AQUIPTA
Atogepant wird hauptsächlich über den Stoffwechsel eliminiert, vor allem durch CYP3A4.
In vitro ist Atogepant ein Substrat von Pgp, BCRP, OATP1B1, OATP1B3 und OAT1. Atogepant ist kein Substrat von OAT3, OCT2 oder MATE1.
Wechselwirkungsstudien wurden mit den in der folgenden Tabelle aufgelisteten gleichzeitig angewendeten Arzneimitteln durchgeführt.
Tabelle 2. Klinische Auswirkungen anderer Arzneimittel auf Atogepant
|
Begleitmedikation (Enzym oder Transporter)
|
Dosierung der Begleitmedikation
|
Dosierung von Atogepant
|
GMRa (90% KIb)
|
Dosisempfehlung für Atogepant
| |
Cmax
|
AUC
| |
Itraconazol
(starker CYP3A4 Inhibitor)
|
200 mg 1x täglich für 7 Tage
|
60 mg Einzeldosis
|
2,15 (1,95, 2,37
|
5,51 (5,09, 5,96)
|
10 mg einmal täglich
| |
Rifampicin
(OATP Inhibitor)
|
600 mg Einzeldosis
|
60 mg Einzeldosis
|
2,23 (1,99, 2,50)
|
2,85 (2,60, 3,12)
|
10 mg einmal täglich
| |
Rifampicin
(starker CYP3A4 Induktor)
|
600 mg 1x täglich für 7 Tage
|
60 mg Einzeldosis
|
0,70 (0,60, 0,81)
|
0,39 (0,35, 0,44)
|
Keine Dosisanpassung von Atogepant ist empfohlen
| |
Topiramat
(schwacher CYP3A4 Induktor)
|
100 mg 2x täglich für 5 Tage
|
60 mg 1x täglich für 17 Tage
|
0,76 (0,68, 0,85)
|
0,75 (0,69, 0,81)
| |
Chinidin
(Pgp Inhibitor)
|
648 mg 2x täglich für 4 Tage
|
60 mg Einzeldosis
|
1,04 (0,89, 1,22)
|
1,26 (1,11, 1,43)
| |
Esomeprazol
(Protonenpumpen-hemmer)
|
40 mg 1x täglich für 7 Tage
|
60 mg Einzeldosis
|
0,77 (0,68, 0,86)
|
0,92 (0,84, 1,01)
| |
Famotidin
(H2-Rezeptor-Antagonist)
|
2x 20 mg
|
60 mg Einzeldosis
|
0,51
(0,41, 0,63)
|
0,79
(0,67, 0,93)
| |
Sumatriptan
(5-HT1B/1D-Rezeptor-Agonist)
|
100 mg Einzeldosis
|
60 mg Einzeldosis
|
0,78 (0,69, 0,89)
|
0,95 (0,86, 1,05)
| |
Ubrogepant
(CGRP-Rezeptor-Antagonist)
|
100 mg an Tag 1 und an jedem 3.Tag von Tag 7-28
|
60 mg 1x täglich von Tag 2-28
|
1,04 (0,94, 1,15)
|
1,04 (0,98, 1,12)
| |
Naproxen
(NSAR)
|
500 mg Einzeldosis
|
60 mg Einzeldosis
|
1,00 (0,91, 1,11)
|
0,99 (0,92, 1,06)
| |
Paracetamol
(Analgetikum, Anti-pyretikum)
|
1000 mg Einzeldosis
|
60 mg Einzeldosis
|
1,00 (0,90, 1,11)
|
1,13 (1,04, 1,22)
|
aGMR – Verhältnis der geometrischen Mittelwerte definiert als die Exposition (maximale Konzentration oder Fläche unter der Kurve AUC) gegenüber Atogepant bei Anwendung mit Begleitmedikation dividiert durch die Exposition gegenüber Atogepant ohne Begleitmedikation.
bKI = Konfidenzintervall
Wirkung von AQUIPTA auf andere Arzneimittel
In vitro ist Atogepant in klinisch relevanten Konzentrationen kein Inhibitor der CYP3A4, 1A2, 2B6, 2C8, 2C9, 2C19 oder 2D6. Atogepant hemmt in klinisch relevanten Konzentrationen weder MAO-A noch UGT1A1. Es ist nicht zu erwarten, dass Atogepant durch die Hemmung von CYP450, MAO-A oder UGT1A1 klinisch signifikante Arzneimittelwechselwirkungen verursacht. Atogepant ist in klinisch relevanten Konzentrationen kein Induktor von CYP1A2, CYP2B6 oder CYP3A4.
In vitro ist Atogepant in klinisch relevanten Konzentrationen kein Inhibitor von Pgp, BCRP, OAT1, OAT3, NTCP, BSEP, MRP3 oder MRP4. Atogepant ist ein schwacher Inhibitor von OATP1B1, OATP1B3, OCT1 und MATE1. Es ist nicht zu erwarten, dass Atogepant klinische Arzneimittelwechselwirkungen mit diesen Transportern verursacht.
Wechselwirkungsstudien wurden mit den in der folgenden Tabelle aufgelisteten gleichzeitig angewendeten Arzneimitteln durchgeführt.
Tabelle 3. Klinische Auswirkungen von Atogepant auf andere Arzneimittel
|
Begleitmedikation (Enzym oder Transporter)
|
Dosierung der Begleitmedikation
|
Dosierung von Atogepant
|
GMRa (90% KIb)
|
Dosisempfehlung für die Begleitmedikation
| |
Cmax
|
AUC
| |
Topiramat
(schwacher CYP3A4 Induktor)
|
100 mg 2x täglich für 11 Tage
|
60 mg 1x täglich für 7 Tage
|
0,94 (0,87, 1,01)
|
0,94 (0,88, 1,01)
|
Keine Dosisanpassung der Begleitmedikation ist empfohlen
| |
Sumatriptan
(5-HT1B/1D-Rezeptor-Agonist)
|
100 mg Einzeldosis
|
60 mg Einzeldosis
|
0,95 (0,85, 1,07)
|
1,02 (0,97, 1,08)
| |
Ubrogepant
(CGRP-Rezeptor-Antagonist)
|
100 mg an Tag 1 und an jedem 3.Tag von Tag 7-28
|
60 mg 1x täglich von Tag 2-28
|
1,26 (1,06, 1,49)
|
1,19 (1,09, 1,30)
| |
Ethinylestradiol
(Östrogen)
|
0,03 mg Einzeldosis
|
60 mg 1x täglich für 17 Tage
|
0,90 (0,84, 0,96)
|
1,00 (0,96, 1,05)
| |
Levonorgestrel
(Gestagen)
|
0,15 mg Einzeldosis
|
60 mg 1x täglich für 17 Tage
|
1,09 (1,03, 1,17)
|
1,19 (1,13, 1,26)
| |
Naproxen
(NSAR)
|
500 mg Einzeldosis
|
60 mg Einzeldosis
|
0,94 (0,90, 0,97)
|
0,98 (0,96, 1,00)
| |
Paracetamol
(Analgetikum, Anti-pyretikum)
|
1000 mg Einzeldosis
|
60 mg Einzeldosis
|
0,89 (0,81, 0,97)
|
0,94 (0,89, 0,99)
|
aGMR – Verhältnis der geometrischen Mittelwerte definiert als die Exposition (maximale Konzentration oder Fläche unter der Kurve AUC) gegenüber der Begleitmedikation bei Anwendung mit Atogepant dividiert durch die Exposition gegenüber der Begleitmedikation ohne Atogepant.
bKI = Konfidenzintervall
Schwangerschaft, StillzeitSchwangerschaft
Es liegen nur begrenzte Daten zur Anwendung von Atogepant bei schwangeren Frauen vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe «Präklinische Daten»). Die Anwendung von AQUIPTA während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen.
Stillzeit
In einer Studie mit 12 stillenden Frauen, die eine einmalige orale Dosis von 60 mg Atogepant erhielten, wurde ein minimaler Übergang von Atogepant in die Muttermilch festgestellt. Die geschätzte relative Dosis für den Säugling betrug etwa 0,19% bezogen auf die gewichtsadaptierte Dosis der Mutter, mit einem Milch-zu-Plasma-Verhältnis von 0,08. Die über einen Zeitraum von 24 Stunden in die Muttermilch abgegebene kumulative Menge von Atogepant betrug weniger als 0,01 mg.
Es liegen keine Daten über die Auswirkungen von Atogepant auf den gestillten Säugling oder auf die Milchproduktion vor. Die entwicklungs- und gesundheitsfördernden Vorteile des Stillens sollten zusammen mit dem klinischen Bedarf der Mutter an Atogepant und möglichen negativen Auswirkungen auf den gestillten Säugling aufgrund von Atogepant oder der mütterlichen Erkrankung berücksichtigt werden.
Fertilität
Es liegen keine Daten zur Wirkung von Atogepant auf die Fertilität beim Menschen vor. Tierstudien zeigten keine Auswirkungen der Behandlung mit Atogepant auf die weibliche oder männliche Fertilität.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine entsprechenden Studien durchgeführt. AQUIPTA kann jedoch bei einigen Patienten Fatigue/Somnolenz verursachen. Beim Führen von Fahrzeugen oder dem Bedienen von Maschinen ist Vorsicht geboten, bis die Patienten hinreichend sicher sind, dass AQUIPTA ihre Leistungsfähigkeit nicht beeinträchtigt.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die Sicherheit von AQUIPTA wurde bei 2'657 Migränepatienten untersucht, die mindestens eine Dosis AQUIPTA erhielten. Davon wurden 1'225 Patienten mindestens 6 Monate lang und 826 Patienten mindestens 12 Monate lang mit AQUIPTA behandelt.
In 12wöchigen placebokontrollierten klinischen Studien erhielten 678 Patienten mindestens eine Dosis AQUIPTA 60 mg einmal täglich, und 663 Patienten erhielten Placebo.
Die am häufigsten berichteten unerwünschten Wirkungen waren Übelkeit (9%), Verstopfung (8%) und Fatigue/Somnolenz (5%). Die meisten unerwünschten Wirkungen waren leichter oder mittelschwerer Natur. Die am häufigsten zu einem Behandlungsabbruch führende unerwünschte Wirkung war Übelkeit (0,4%).
Liste der unerwünschten Wirkungen
Die unerwünschten Wirkungen sind nachstehend nach Systemorganklasse und Häufigkeit aufgeführt.
Die Häufigkeitskategorien sind wie folgt definiert: sehr häufig (≥1/10), häufig (≥1/100, < 1/10), gelegentlich (≥1/1'000, < 1/100), selten (≥1/10'000, < 1/1'000) oder sehr selten (< 1/10'000), nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
|
Systemorganklasse
|
Häufigkeit
|
Unerwünschte Wirkung
| |
Erkrankungen des Immunsystems
|
Häufig
|
Überempfindlichkeitsreaktionen*** (z.B. Dyspnoe, Hautausschlag, Pruritus, Urtikaria, Gesichtsödem)
| |
Selten
|
Anaphylaxie***
| |
Stoffwechsel- und Ernährungsstörungen
|
Häufig
|
Verminderter Appetit,
Gewichtsabnahme*
| |
Erkrankungen des Gastrointestinaltrakts
|
Häufig
|
Übelkeit, Verstopfung
| |
Leber- und Gallenerkrankungen
|
Gelegentlich
|
ALT/AST erhöht**
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Häufig
|
Fatigue/Somnolenz
|
* Definiert in klinischen Studien als Gewichtsabnahme von mindestens 7 % zu einem beliebigen Zeitpunkt.
** In klinischen Studien wurden Fälle von erhöhten ALT-/AST-Werten (in klinischen Studien definiert als ≥3 x des oberen normalen Grenzwerts [upper limit of normal, ULN]) im zeitlichen Zusammenhang mit Atogepant beobachtet, darunter Fälle mit positiver Dechallenge-Geschichte, die innerhalb von 8 Wochen nach Absetzen zurückgingen. Die Gesamthäufigkeit war jedoch in den Behandlungsarmen mit Atogepant und Placebo vergleichbar.
*** Siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen», Abschnitt «Überempfindlichkeitsreaktionen».
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIn klinischen Studien wurde Atogepant als Einzeldosis bis zu 300 mg und als Mehrfachgabe bis zu 170 mg einmal täglich verabreicht. Die Nebenwirkungen waren vergleichbar mit solchen bei niedrigerer Dosierung und es wurden keine spezifischen Toxizitäten erkannt. Es ist kein Gegenmittel gegen AQUIPTA bekannt. Die Behandlung einer Überdosierung mit AQUIPTA sollte aus allgemein unterstützenden Massnahmen bestehen, u.a. der Überwachung der Vitalparameter und der Beobachtung des klinischen Status des Patienten.
Eigenschaften/WirkungenATC-Code
N02CD07
Wirkungsmechanismus/Pharmakodynamik
Atogepant ist ein Calcitonin Gene-Related Peptide (CGRP)-Rezeptorantagonist, der die Bindung von CGRP an den Rezeptor blockiert und der CGRP-Rezeptorfunktion entgegenwirkt. CGRP ist ein Neuropeptid, das mit der Pathophysiologie der Migräne in Zusammenhang gebracht wurde. Im trigeminovaskulären System moduliert CGRP die nozizeptive Signalgebung und Entzündung und fungiert zusätzlich als Vasodilatator.
Kardiale Elektrophysiologie
Bei einer Dosis, die 5mal höher ist als die empfohlene maximale Tagesdosis, verlängert AQUIPTA das QT-Intervall nicht.
Klinische Wirksamkeit
AQUIPTA wurde in zwei Zulassungsstudien zur Prophylaxe von Migräne über das gesamte Spektrum der chronischen und episodischen Migräne hinweg untersucht. In die Studie zur episodischen Migräne (ADVANCE) wurden Patienten aufgenommen, welche die Kriterien der International Classification of Headache Disorders (ICHD) für die Diagnose einer Migräne mit oder ohne Aura erfüllten. In die Studie zur chronischen Migräne (PROGRESS) wurden Patienten aufgenommen, welche auch die ICHD-Kriterien für chronische Migräne erfüllten. Beide Studien schlossen Patienten mit Myokardinfarkt, Schlaganfall oder transitorischen ischämischen Attacken innerhalb von sechs Monaten vor dem Screening aus.
Episodische Migräne
AQUIPTA wurde in einer randomisierten, multizentrischen, doppelblinden, placebokontrollierten Studie zur Prophylaxe episodischer Migräne (4 bis 14 Migränetage pro Monat) untersucht (ADVANCE). Die Patienten wurden randomisiert zu AQUIPTA 60 mg (N = 235) oder Placebo (N = 223) einmal täglich während 12 Wochen. Die Akutbehandlung von Kopfschmerzen (d.h. Triptane, Ergotaminderivate, NSAR, Paracetamol und Opioide) war den Patienten erlaubt.
Insgesamt schlossen 88% der Patienten die 12wöchige doppelblinde Studienphase ab. Das mittlere Alter der Patienten betrug 42 Jahre (zwischen 18 und 73); 89% waren weiblich und 83% waren weiss. Die mittlere Migränehäufigkeit bei Baseline betrug etwa 8 Migränetage pro Monat und war in beiden Behandlungsarmen vergleichbar.
Der primäre Endpunkt zur Wirksamkeit war die Veränderung der mittleren Anzahl monatlicher Migränetage (MMD) gegenüber Baseline über die 12wöchige Behandlungsphase.
Die Behandlung mit AQUIPTA zeigte in ADVANCE im Vergleich zu Placebo statistisch signifikante Verbesserungen der primären und bei wichtigen multiplizitätskontrollierten sekundären Ergebnissen zur Wirksamkeit, wie in Tabelle 4 zusammengefasst.
Tabelle 4: Endpunkte zur Wirksamkeit in ADVANCE
|
|
AQUIPTA 60 mg
N = 226
|
Placebo
N = 216
| |
Anzahl monatlicher Migränetage (MMD) über 12 Wochen
| |
Baseline
|
7,8
|
7,5
| |
Mittlere Veränderung gegenüber Baseline
|
-4,1
|
-2,5
| |
Unterschied gegenüber Placebo
|
-1,7
|
| |
p-Wert
|
< 0,001
|
| |
Monatliche Kopfschmerztage über 12 Wochen
| |
Baseline
|
9,0
|
8,5
| |
Mittlere Veränderung gegenüber Baseline
|
-4,2
|
-2,5
| |
Unterschied gegenüber Placebo
|
-1,7
|
| |
p-Wert
|
< 0,001
|
| |
Monatliche Behandlungstage mit Akutmedikation über 12 Wochen
| |
Baseline
|
6,9
|
6,5
| |
Mittlere Veränderung gegenüber Baseline
|
-3,8
|
-2,3
| |
Unterschied gegenüber Placebo
|
-1,4
|
| |
p-Wert
|
< 0,001
|
| |
≥50% MMD-Responder über 12 Wochen
| |
Responder (%)
|
59
|
29
| |
Odds-Ratio (95%-CI)
|
3,55 (2,39; 5,28)
|
| |
p-Wert
|
< 0,001
|
| |
MSQ v.2.1 RFRa in Woche 12
| |
Baseline
|
46,6
|
46,6
| |
Mittlere Veränderung gegenüber Baseline
|
31,0
|
20,0
| |
Unterschied gegenüber Placebo
|
11,0
|
| |
p-Wert
|
< 0,001
|
|
aSocre auf dem Migraine Specific Quality of Life Questionnaire Version 2.1 im Bereich Role Function Restrictive
Abbildung 1 zeigt die mittlere Veränderung der MMD gegenüber Baseline in ADVANCE. Patienten, die mit AQUIPTA 60 mg einmal täglich behandelt wurden, wiesen im Vergleich zu Patienten, die Placebo erhielten, während der 12wöchigen Behandlungsphase eine grössere mittlere Verringerung der MMD gegenüber Baseline auf. Während des ersten Behandlungsmonats ergab sich unter AQUIPTA 60 mg einmal täglich im Vergleich zu Placebo eine grössere mittlere Verringerung der Anzahl wöchentlicher Migränetage gegenüber Baseline.
Abbildung 1: Veränderung der Anzahl monatlicher Migränetage gegenüber Baseline in ADVANCE
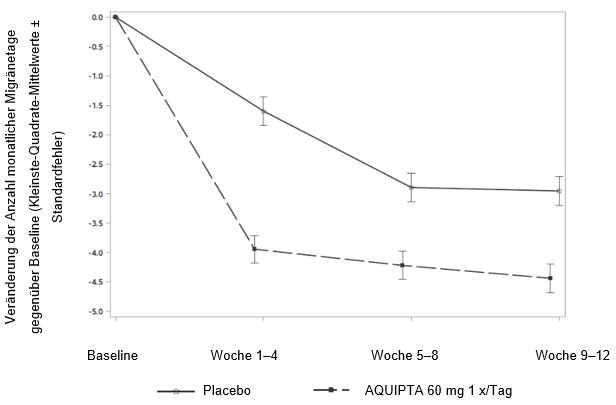
Langfristige Wirksamkeit
In einer offenen Studie, in der Patienten mit episodischer Migräne AQUIPTA 60 mg einmal täglich erhielten, wurde die Wirksamkeit bis zu 1 Jahr aufrechterhalten. 68,4% der Patienten schlossen die Behandlungsphase ab. Die Verringerung der mittleren Anzahl monatlicher Migränetage (Methode der kleinsten Quadrate) betrug im ersten Monat (Woche 1–4) -3,8 Tage und verbesserte sich im letzten Monat (Woche 49–52) auf eine mittlere Verringerung (Methode der kleinsten Quadrate) von -5,2 Tagen. Etwa 84%, 70% bzw. 48% der Patienten gaben in Woche 49–52 eine Verringerung der Anzahl monatlicher Migränetage um ≥50%, ≥75% bzw. 100% an.
Patienten mit vorherigem Versagen von zwei bis vier Klassen oraler prophylaktischer Behandlungen
In der ELEVATE-Studie wurden 315 erwachsene Patienten mit episodischer Migräne, bei denen zuvor zwei bis vier Klassen oraler prophylaktischer Behandlungen (z.B. Topiramat, trizyklische Antidepressiva, Betablocker) im Hinblick auf Wirksamkeit und/oder Verträglichkeit versagt hatten, im Verhältnis 1:1 randomisiert und erhielten zwölf Wochen lang entweder Atogepant 60 mg (n = 157) oder Placebo (n = 158). Die Ergebnisse dieser Studie stimmten mit den wichtigsten Ergebnissen früherer Studien zur Wirksamkeit bei episodischer Migräne überein und waren hinsichtlich der primären und sekundären Wirksamkeitsendpunkte statistisch signifikant, darunter mehrere Patientenfragebögen zur Bewertung der Funktionsfähigkeit. Die Behandlung mit Atogepant führte zu einer Verringerung der Anzahl MMD um 4,2 Tage im Vergleich zu 1,9 Tagen unter Placebo (p < 0,001) 50,6 % (78/154) der Patienten im Behandlungsarm mit Atogepant erreichten eine mindestens 50 %ige Reduktion der MMD gegenüber Baseline, verglichen mit 18,1 % (28/155) unter Placebo (Odds-Ratio [95%-CI]: 4,82 [2,85; 8,14]; p < 0,001).
Chronische Migräne
AQUIPTA wurde in einer randomisierten, multizentrischen, doppelblinden, placebokontrollierten Studie (PROGRESS) zur Prophylaxe chronischer Migräne (15 oder mehr Kopfschmerztage pro Monat mit mindestens 8 Migränetagen) untersucht. Die Patienten wurden randomisiert zu AQUIPTA 60 mg (N = 262) oder Placebo (N = 259) einmal täglich während 12 Wochen. Einer Untergruppe von Patienten (11%) wurde die Anwendung begleitender Migräneprophylaxe Medikamente (z.B. Amitriptylin, Propranolol, Topiramat) erlaubt. Die Akutbehandlung von Kopfschmerzen (d.h. Triptane, Ergotaminderivate, NSAR, Paracetamol und Opioide) war den Patienten erlaubt. Es wurden auch Patienten mit übermässiger Medikamentenanwendung der Akutmedikation und Kopfschmerzen infolge der übermässigen Medikamentenanwendung eingeschlossen.
Insgesamt schlossen 463 (89%) Patienten die 12wöchige doppelblinde Studie ab. Das mittlere Alter der Patienten betrug 42 Jahre (zwischen 18 und 74); 87% waren weiblich und 59% waren weiss. Die mittlere Migränehäufigkeit bei Baseline betrug etwa 19 Migränetage pro Monat und war in beiden Behandlungsarmen vergleichbar.
Der primäre Endpunkt zur Wirksamkeit war die Veränderung der mittleren MMD gegenüber Baseline über die 12wöchige Behandlungsphase.
Die Behandlung mit AQUIPTA zeigte in PROGRESS im Vergleich zu Placebo statistisch signifikante Verbesserung der primären und wichtigen multiplizitätskontrollierten sekundären Ergebnissen zur Wirksamkeit, wie in Tabelle 5 zusammengefasst.
Tabelle 5: Endpunkte zur Wirksamkeit in PROGRESS
|
|
AQUIPTA 60 mg
N = 257
|
Placebo
N = 249
| |
Anzahl monatlicher Migränetage (MMD) über 12 Wochen
| |
Baseline
|
19,2
|
19,0
| |
Mittlere Veränderung gegenüber Baseline
|
-6,8
|
-5,1
| |
Unterschied gegenüber Placebo
|
-1,7
|
| |
p-Wert
|
0,002
|
| |
Monatliche Kopfschmerztage über 12 Wochen
| |
Baseline
|
21,5
|
21,4
| |
Mittlere Veränderung gegenüber Baseline
|
-6,9
|
-5,2
| |
Unterschied gegenüber Placebo
|
-1,7
|
| |
p-Wert
|
0,002
|
| |
Monatliche Behandlungstage mit Akutmedikation über 12 Wochen
| |
Baseline
|
15,5
|
15,3
| |
Mittlere Veränderung gegenüber Baseline
|
-6,2
|
-4,1
| |
Unterschied gegenüber Placebo
|
-2,1
|
| |
p-Wert
|
0,002
|
| |
≥50% MMD-Responder über 12 Wochen
| |
Responder (%)
|
40
|
27
| |
Unterschied gegenüber Placebo (%)
|
14
|
| |
p-Wert
|
0,002
|
| |
MSQ v.2.1 RFRa in Woche 12
| |
Baseline
|
43,3
|
44,1
| |
Mittlere Veränderung gegenüber Baseline
|
23,1
|
17,3
| |
Unterschied gegenüber Placebo
|
5,8
|
| |
p-Wert
|
0,002
|
|
aScore auf dem Migraine Specific Quality of Life Questionnaire Version 2.1 im Bereich Role Function Restrictive
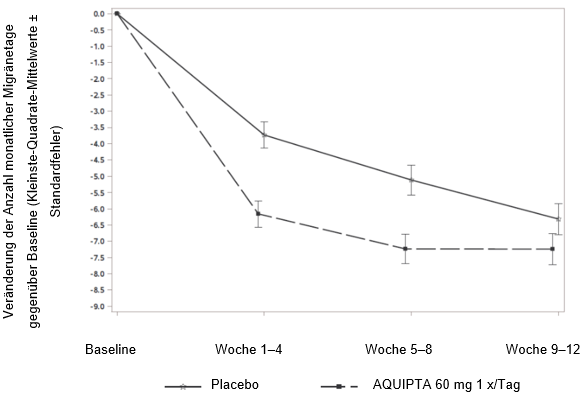
Abbildung 2 zeigt die mittlere Veränderung der MMD gegenüber Baseline in PROGRESS. Patienten, die mit AQUIPTA 60 mg einmal täglich behandelt wurden, wiesen im Vergleich zu Patienten, die Placebo erhielten, während der 12wöchigen Behandlungsphase eine grössere mittlere Verringerung der MMD gegenüber Baseline auf.
Abbildung 2: Veränderung der Anzahl monatlicher Migränetage gegenüber Baseline in PROGRESS
Pädiatrie
Es liegen keine Daten zur Anwendung bei Kindern und Jugendlichen unter 18 Jahren vor.
PharmakokinetikAbsorption
Nach oraler Verabreichung von AQUIPTA wird Atogepant schnell resorbiert und die Tmax-Werte liegen im Median zwischen 1 und 2 Stunden. Nach einmal täglicher Dosierung zeigt Atogepant bis zu 170 mg (etwa 3mal höher als die höchste empfohlene Dosis) eine Pharmakokinetik proportional zur Dosis ohne Akkumulation.
Einfluss von Nahrungsmitteln
Bei Anwendung von AQUIPTA zusammen mit einer fettreichen Mahlzeit war der Einfluss von Nahrungsmitteln nicht signifikant (AUC und Cmax wurden um ca. 18% bzw. 22% verringert, ohne Einfluss auf die mediane Dauer bis zur maximalen Atogepant-Konzentration im Plasma). AQUIPTA wurde in klinischen Studien zur Wirksamkeit unabhängig von einer Mahlzeit verabreicht.
Distribution
Die Plasmaproteinbindung von Atogepant war im Bereich von 0,1 bis 10 μM nicht konzentrationsabhängig; der ungebundene Anteil von Atogepant im menschlichen Plasma betrug etwa 4,7%. Das mittlere apparente Verteilungsvolumen von Atogepant (Vz/F) nach oraler Verabreichung beträgt etwa 292 l.
Metabolismus
Atogepant wird hauptsächlich über den Stoffwechsel eliminiert, vor allem durch CYP3A4. Die Muttersubstanz (Atogepant) und ein Glucuronidkonjugatmetabolit (M23) waren die häufigsten zirkulierenden Bestandteile im menschlichen Plasma.
Elimination
Die Eliminationshalbwertszeit von Atogepant beträgt etwa 11 Stunden. Die mittlere apparente orale Clearance (CL/F) von Atogepant beträgt etwa 19 l/h. Nach Anwendung einer einzelnen oralen Dosis von 50 mg 14C-Atogepant wurden bei gesunden männlichen Probanden 42% bzw. 5% der Dosis unverändert über den Stuhl bzw. Urin ausgeschieden.
Kinetik spezieller Patientengruppen
Basierend auf einer populationsbezogenen pharmakokinetischen Analyse hatten Alter (18 bis 78 Jahre), Geschlecht, ethnische Zugehörigkeit (kaukasisch vs. japanisch oder chinesisch) und Körpergewicht (40,7 bis 196 kg) keinen signifikanten Einfluss auf die Pharmakokinetik (Cmax und AUC) von Atogepant. Daher sind aufgrund dieser Faktoren keine Dosisanpassungen erforderlich.
Nierenfunktionsstörungen
Der Eliminationsweg über die Nieren spielt bei der Clearance von Atogepant eine untergeordnete Rolle. Basierend auf populationsbezogenen pharmakokinetischen Analysen gibt es keinen signifikanten Unterschied in der Pharmakokinetik von Atogepant bei Patienten mit leichter oder mittelschwerer Niereninsuffizienz (CLcr 30–89 ml/min) im Vergleich zu solchen mit normaler Nierenfunktion (CLcr > 90 ml/min). Da Patienten mit schwerer Niereninsuffizienz oder terminalem Nierenversagen (end-stage renal disease, ESRD; CLcr < 30 ml/min) nicht untersucht wurden, wird für diese Patienten Atogepant 10 mg empfohlen.
Leberfunktionsstörungen
Bei Patienten mit vorbestehender leichter (Child-Pugh-Stadium A), mittelschwerer (Child-Pugh-Stadium B) oder schwerer Leberinsuffizienz (Child-Pugh-Stadium C) stieg die Gesamtexposition gegenüber Atogepant um 24%, 15% bzw. 38% an. Die Exposition gegenüber ungebundenem Atogepant war jedoch bei Patienten mit schwerer Leberinsuffizienz etwa 3mal höher. Die Anwendung von AQUIPTA ist bei Patienten mit schwerer Leberinsuffizienz zu vermeiden.
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Gentoxizität, Kanzerogenität und Fertilität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Reproduktionstoxizität
Die orale Verabreichung von Atogepant (0, 5, 15, 125 oder 750 mg/kg/Tag) an trächtige Ratten während der Organogenese führte bei den beiden höchsten untersuchten Dosen (125 und 750 mg/kg) zu einem verminderten Körpergewicht der Föten und einer erhöhten Inzidenz von skelettalen Veränderungen bei den Föten, was nicht mit einer maternalen Toxizität in Zusammenhang stand. Bei der Dosis ohne unerwünschte Wirkungen auf die embryofetale Entwicklung (15 mg/kg/Tag) war die Plasmaexposition (AUC) etwa 4mal höher als bei Menschen unter 60 mg/Tag-Dosis.
Die orale Verabreichung von Atogepant (0, 30, 90 oder 130 mg/kg/Tag) an trächtige Kaninchen während der Organogenese führte bei der hohen Dosis (130 mg/kg/Tag) zu einer erhöhten Inzidenz von viszeralen und skelettalen Veränderungen. Bei der Dosis ohne unerwünschte Wirkungen auf die Entwicklung bei Kaninchen (90 mg/kg/Tag) war die Plasmaexposition (AUC) etwa 3mal höher als bei Menschen unter einer Dosis von 60 mg/Tag.
Die orale Verabreichung von Atogepant (0, 15, 45 oder 125 mg/kg/Tag) an Ratten während der Trächtigkeit und Säugezeit führte in der höchsten Dosis (125 mg/kg/Tag) zu einem geringeren Körpergewicht der Nachkommen. Die Plasmaexposition (AUC) bei der höchsten untersuchten Dosis ohne Effekte auf die Entwicklung der Nachkommen (45 mg/kg/Tag) war etwa 5mal höher als die bei Menschen unter der 60 mg/Tag-Dosis.
Toxizitätsprüfungen mit juvenilen Tieren
Die Anwendung von Atogepant in Form einer einmal täglichen orale Gabe von 10, 30 oder 200 mg/kg/Tag an junge Ratten von Tag 28 bis Tag 70 nach Geburt war nicht mit unerwünschten Wirkungen auf die Entwicklung assoziiert.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Nicht über 30°C lagern.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer69128 (Swissmedic)
PackungenAQUIPTA 10 mg: Blisterpackungen mit 28 Tabletten (B)
AQUIPTA 60 mg: Blisterpackungen mit 28 Tabletten (B)
ZulassungsinhaberinAbbVie AG, 6330 Cham
Stand der InformationApril 2025
|