ZusammensetzungWirkstoffe
Setmelanotid
Hilfsstoffe
Benzylalkohol (10 mg/ml), mPEG-2000-DSPE, Carmellose-Natrium, Mannitol (E421), Phenol, Natriumedetat, Natriumhydroxid (E524) und / oder Salzsäure (E507) zur pH-Wert-Einstellung, Wasser für Injektionszwecke
1ml Injektionslösung enthält 1.64 mg Natrium.
Indikationen/AnwendungsmöglichkeitenIMCIVREE wird angewendet bei Erwachsenen und Kindern ab 2 Jahren zur Behandlung von Adipositas und zur Kontrolle des Hungergefühls im Zusammenhang mit genetisch bestätigtem Bardet-Biedl-Syndrom (BBS), durch Funktionsverlustmutationen bedingtem biallelischem Proopiomelanocortin (POMC)-Mangel (einschliesslich PCSK1) oder biallelischem Leptinrezeptor (LEPR)-Mangel.
Dosierung/AnwendungDie Behandlung mit IMCIVREE sollte von einem Arzt mit Erfahrung im Bereich Adipositas mit zugrunde liegender genetischer Ätiologie verordnet und überwacht werden.
Dosierung
POMC-Mangel, einschliesslich PCSK1, und LEPR-Mangel
Erwachsene und Kinder ab 12 Jahren
Für Erwachsene und Kinder von 12 bis 17 Jahren ist die Anfangsdosis eine subkutane Injektion von 1 mg einmal täglich über einen Zeitraum von 2 Wochen. Wenn Setmelanotid gut vertragen wird (siehe «Warnhinweise und Vorsichtsmassnahmen»), kann die Dosis nach 2 Wochen auf eine subkutane Injektion von 2 mg einmal täglich erhöht werden (Tabelle 1). Wenn die Dosiseskalation nicht vertragen wird, kann weiterhin die Dosis von 1 mg einmal täglich angewendet werden.
Wenn bei erwachsenen Patienten eine zusätzliche Gewichtsabnahme gewünscht ist, kann die Dosis auf eine subkutane Injektion von 2.5 mg einmal täglich erhöht werden. Wenn die Dosis von 2.5 mg einmal täglich gut vertragen wird, kann die Dosis auf 3 mg einmal täglich erhöht werden (Tabelle 1).
Wenn bei Patienten von 12 bis 17 Jahren mit der subkutanen Injektion von 2 mg einmal täglich das Gewicht über dem 90. Perzentil bleibt und eine zusätzliche Gewichtsabnahme gewünscht wird, kann die Dosis auf 2.5 mg erhöht werden, mit einer Höchstdosis von 3 mg einmal täglich (Tabelle 1).
Tabelle 1 Dosistitration bei erwachsenen und pädiatrischen Patienten ab 12 Jahren
|
Woche
|
Tagesdosis
|
Zu injizierendes
Volumen
| |
Wochen 1–2
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
Ab Woche 3
|
2 mg einmal täglich
|
0.2 ml einmal täglich
| |
Wenn das klinische Ansprechen unzureichend ist und die Dosis von 2 mg einmal täglich gut vertragen wird
|
2.5 mg einmal täglich
|
0.25 ml einmal täglich
| |
Wenn das klinische Ansprechen unzureichend ist und die Dosis von 2.5 mg
einmal täglich gut vertragen wird
|
3 mg einmal täglich
|
0.3 ml einmal täglich
|
Kinder und Jugendliche (Kinder von 6 bis < 12 Jahren)
Für Patienten im Alter von 6 bis < 12 Jahren ist die Anfangsdosis eine subkutane Injektion von 0.5 mg einmal täglich über einen Zeitraum von 2 Wochen. Wenn die Dosis gut vertragen wird, kann die Dosis nach 2 Wochen auf 1 mg einmal täglich erhöht werden. Wenn die Dosiseskalation nicht vertragen wird, kann bei Kindern und Jugendlichen weiterhin die Dosis von 0.5 mg einmal täglich angewendet werden. Wenn die Dosis von 1 mg einmal täglich gut vertragen wird, kann nach 2 Wochen die Dosis auf 2 mg einmal täglich erhöht werden. Wenn mit der subkutanen Injektion von 2 mg einmal täglich das Gewicht über dem 90. Perzentil bleibt und eine zusätzliche Gewichtsabnahme gewünscht wird, kann die Dosis auf 2.5 mg einmal täglich erhöht werden (Tabelle 2).
Tabelle 2 Dosistitration für Kinder und Jugendliche von 6 bis < 12 Jahren
|
Woche
|
Tagesdosis
|
Zu injizierendes Volumen
| |
Patienten im Alter von 6 bis < 12 Jahren
| |
Wochen 1–2
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Wochen 3–4
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
Ab Woche 5
|
2 mg einmal täglich
|
0.2 ml einmal täglich
| |
Wenn das klinische Ansprechen unzureichend ist und die Dosis von 2 mg einmal täglich gut vertragen wird
|
2.5 mg einmal täglich
|
0.25 ml einmal täglich
|
Kinder und Jugendliche (Kinder von 2 bis < 6 Jahren)
Bei Patienten im Alter von 2 bis < 6 Jahren sollte die Dosistitration in Tabelle 3 befolgt werden.
Bei Patienten im Alter von 2 bis < 6 Jahren ist die Anfangsdosis eine subkutane Injektion von 0.5 mg einmal täglich über einen Zeitraum von 2 Wochen. Wenn die Anfangsdosis von 0.5 mg nicht vertragen wird, auf 0.25 mg (0.025 ml) einmal täglich reduzieren. Wenn die einmal tägliche Dosis von 0.25 mg vertragen wird, die Dosistitration fortsetzen.
Tabelle 3 Dosistitration für pädiatrische Patienten von 2 bis < 6 Jahren
|
Körpergewicht/Behandlungswoche
|
Tagesdosis
|
Zu injizierendes Volumen
| |
< 20 kg
| |
Ab Woche 1
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
20 bis < 30 kg
| |
Woche 1–2
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Ab Woche 3 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.5 mg gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
30 bis < 40 kg
| |
Woche 1–2
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Woche 3–4 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0..5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
Ab Woche 5 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 1 mg einmal täglich gut vertragen wird)
|
1.5 mg einmal täglich
|
0.15 ml einmal täglich
| |
≥ 40 kg
| |
Woche 1–2
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Woche 3–4 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
Woche 5–6 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 1 mg einmal täglich gut vertragen wird)
|
1.5 mg einmal täglich
|
0.15 ml einmal täglich
| |
Woche 7–8 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 1.5 mg einmal täglich gut vertragen wird)
|
2 mg einmal täglich
|
0.2 ml einmal täglich
| |
Ab Woche 9 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 2 mg einmal täglich gut vertragen wird)
|
2.5 mg einmal täglich
|
0.25 ml einmal täglich
|
Der verordnende Arzt sollte das Ansprechen auf die Setmelanotid-Therapie regelmässig bewerten. Bei im Wachstum befindlichen Kindern sind die Auswirkungen der Gewichtsabnahme auf Wachstum und Reifung zu bewerten (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Die mit Setmelanotid verbundene Gewichtsabnahme und Kontrolle des Hungergefühls können so lange aufrechterhalten werden, wie die Therapie ohne Unterbrechung fortgesetzt wird. Wenn die Behandlung abgesetzt oder das Dosierungsschema nicht eingehalten wird, kehren die Symptome der durch POMC- und LEPR-Mangel bedingten Adipositas zurück.
Bardet-Biedl-Syndrom
Erwachsene und Kinder im Alter von über 16 Jahren
Bei Erwachsenen und Kindern im Alter von 16 bis 17 Jahren ist der Dosistitration in Tabelle 4 zu folgen.
Tabelle 4 Dosistitration bei Erwachsenen und pädiatrischen Patienten ab 16 Jahren
|
Woche
|
Tagesdosis
|
Zu injizierendes Volumen
| |
Wochen 1-2
|
2 mg einmal täglich
|
0.2 ml einmal täglich
| |
Ab Woche 3 (wenn die Dosis von 2 mg einmal täglich gut vertragen wird)
|
3 mg einmal täglich
|
0.3 ml einmal täglich
|
Wenn die Anfangsdosis von 2 mg nicht vertragen wird, auf 1 mg (0.1 ml) einmal täglich reduzieren. Die Dosistitration fortsetzen, wenn die Dosis von 1 mg einmal täglich vertragen wird.
Wenn nach der Anfangsdosis eine nachfolgende Dosis nicht vertragen wird, auf die vorherige Dosisstufe reduzieren. Dosistitration fortsetzen, wenn reduzierte Dosis vertragen wird.
Kinder und Jugendliche (Kinder im Alter von 6 bis < 16 Jahren)
Bei Patienten im Alter von 6 bis < 16 Jahren ist der Dosistitration in Tabelle 5 zu folgen.
Tabelle 5 Dosistitration für pädiatrische Patienten von 6 bis < 16 Jahren
|
Woche
|
Tagesdosis
|
Zu injizierendes Volumen
| |
Woche 1
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
Woche 2 (wenn die Dosis von 1 mg einmal täglich gut vertragen wird)
|
2 mg einmal täglich
|
0.2 ml einmal täglich
| |
Ab Woche 3 (wenn die Dosis von 2 mg einmal täglich gut vertragen wird)
|
3 mg einmal täglich
|
0.3 ml einmal täglich
|
Wenn die Anfangsdosis von 1 mg nicht vertragen wird, auf 0.5 mg (0.05 ml) einmal täglich reduzieren. Wenn die Dosis von 0.5 mg einmal täglich vertragen wird, die Dosis auf 1 mg einmal täglich erhöhen und die Dosistitration fortsetzen.
Wenn nach der Anfangsdosis eine nachfolgende Dosis nicht vertragen wird, auf die vorherige Dosisstufe reduzieren. Die Dosistitration fortsetzen, wenn die reduzierte Dosis vertragen wird.
Kinder und Jugendliche (Kinder von 2 bis < 6 Jahren)
Bei Patienten im Alter von 2 bis < 6 Jahren sollte die Dosistitration in Tabelle 6 befolgt werden.
Bei Patienten im Alter von 2 bis < 6 Jahren ist die Anfangsdosis eine subkutane Injektion von 0.5 mg einmal täglich über einen Zeitraum von 2 Wochen. Wenn die Anfangsdosis von 0.5 mg nicht vertragen wird, auf 0.25 mg (0.025 ml) einmal täglich reduzieren. Wenn die einmal tägliche Dosis von 0.25 mg vertragen wird, die Dosistitration fortsetzen.
Tabelle 6 Dosistitration für pädiatrische Patienten von 2 bis < 6 Jahren
|
Körpergewicht/Behandlungswoche
|
Tagesdosis
|
Zu injizierendes Volumen
| |
< 20 kg
| |
Ab Woche 1
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
20 bis < 30 kg
| |
Woche 1–2
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Ab Woche 3 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.5 mg gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
30 bis < 40 kg
| |
Woche 1–2
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Woche 3–4 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
Ab Woche 5 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 1 mg einmal täglich gut vertragen wird)
|
1.5 mg einmal täglich
|
0.15 ml einmal täglich
| |
≥ 40 kg
| |
Woche 1–2
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Woche 3–4 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
Woche 5–6 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 1 mg einmal täglich gut vertragen wird)
|
1.5 mg einmal täglich
|
0.15 ml einmal täglich
| |
Woche 7–8 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 1.5 mg einmal täglich gut vertragen wird)
|
2 mg einmal täglich
|
0.2 ml einmal täglich
| |
Ab Woche 9 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 2 mg einmal täglich gut vertragen wird)
|
2.5 mg einmal täglich
|
0.25 ml einmal täglich
|
Der verordnende Arzt sollte das Ansprechen auf die Setmelanotid-Therapie regelmässig beurteilen. Bei im Wachstum befindlichen Kindern sind die Auswirkungen der Gewichtsabnahme auf Wachstum und Reifung zu bewerten (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Die mit Setmelanotid verbundene Gewichtsabnahme und Kontrolle des Hungergefühls können so lange aufrechterhalten werden, wie die Therapie ohne Unterbrechung fortgesetzt wird. Wenn die Behandlung abgesetzt oder das Dosierungsschema nicht eingehalten wird, kehren die Symptome der Adipositas und/oder des Hungergefühls bei BBS zurück.
Versäumte Dosis
Wenn eine Dosis versäumt wird, ist das Behandlungsschema mit einmal täglicher Anwendung mit der nächsten geplanten Anwendung in der verordneten Dosis wiederaufzunehmen.
Besondere Patientengruppen
Nierenfunktionsstörung
POMC-Mangel, einschliesslich PCSK1, und LEPR-Mangel
Bei Erwachsenen und pädiatrischen Patienten von 2 bis 17 Jahren mit leichter oder mittelschwerer Nierenfunktionsstörung (siehe «Pharmakokinetik») sind keine Dosisanpassungen erforderlich.
Bei Erwachsenen und Kindern im Alter von 12 bis 17 Jahren mit schwerer Nierenfunktionsstörung (siehe «Pharmakokinetik») ist der Dosistitration in Tabelle 7 zu folgen.
Tabelle 7 Dosistitration bei Erwachsenen und pädiatrischen Patienten ab 12 Jahren mit schwerer Nierenfunktionsstörung
|
Woche
|
Tagesdosis
|
Zu injizierendes Volumen
| |
Wochen 1-2
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Ab Woche 3 (wenn die Dosis von 0.5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich.
|
0.1 ml einmal täglich
| |
Wenn das klinische Ansprechen unzureichend ist und die Dosis von 1 mg
einmal täglich gut vertragen wird
|
2 mg einmal täglich.
|
0.2 ml einmal täglich
| |
Wenn das klinische Ansprechen unzureichend ist und die Dosis von 2 mg
einmal täglich gut vertragen wird
|
2.5 mg einmal täglich.
|
0.25 ml einmal täglich
| |
Wenn das klinische Ansprechen unzureichend ist und die Dosis von 2.5 mg einmal täglich gut vertragen wird
|
3 mg einmal täglich
|
0.3 ml einmal täglich
|
Wenn die Anfangsdosis von 0.5 mg nicht vertragen wird, auf 0.25 mg (0.025 ml) einmal täglich reduzieren. Die Dosistitration fortsetzen, wenn die Dosis von 0.25 mg einmal täglich vertragen wird.
Wenn nach der Anfangsdosis eine nachfolgende Dosis nicht vertragen wird, auf die vorherige Dosisstufe reduzieren. Die Dosistitration fortsetzen, wenn die reduzierte Dosis vertragen wird.
Bei Patienten im Alter von 6 bis < 12 Jahren mit schwerer Nierenfunktionsstörung ist der Dosistitration in Tabelle 8 zu folgen.
Tabelle 8 Dosistitration für pädiatrische Patienten von 6 bis < 12 Jahren mit schwerer Nierenfunktionsstörung
|
Woche
|
Tagesdosis
|
Zu injizierendes Volumen
| |
Wochen 1-2
|
0.25 mg einmal täglich.
|
0.025 ml einmal täglich
| |
Wochen 3-4 (wenn die Dosis von 0.25 mg einmal täglich gut vertragen wird)
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Ab Woche 5 (wenn 0.5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich.
|
0.1 ml einmal täglich
| |
Wenn das klinische Ansprechen unzureichend ist und die Dosis von 1 mg einmal täglich gut vertragen wird
|
2 mg einmal täglich.
|
0.2 ml einmal täglich
|
Wenn die Anfangsdosis von 0.25 mg nicht vertragen wird, muss die Behandlung abgebrochen werden.
Wenn nach der Anfangsdosis eine nachfolgende Dosis nicht vertragen wird, auf die vorherige Dosisstufe reduzieren. Die Dosistitration fortsetzen, wenn die reduzierte Dosis vertragen wird.
Setmelanotid wurde bei Patienten im Alter von 2 bis < 6 Jahren mit schwerer Nierenfunktionsstörung nicht untersucht. Die Dosistitration sollte sich nach der Verträglichkeit (Tabelle 9) richten, und die Patienten sind engmaschig zu überwachen.
Tabelle 9 Dosistitration für pädiatrische Patienten von 2 bis < 6 Jahren mit schwerer Nierenfunktionsstörung
|
Körpergewicht/Behandlungswoche
|
Tagesdosis
|
Zu injizierendes Volumen
| |
< 20 kg
| |
Ab Woche 1
|
0.25 mg einmal täglich
|
0.025 ml einmal täglich
| |
20 bis < 30 kg
| |
Woche 1–2
|
0.25 mg einmal täglich
|
0.025 ml einmal täglich
| |
Ab Woche 3 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.25 mg gut vertragen wird)
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
30 bis < 40 kg
| |
Woche 1–2
|
0.25 mg einmal täglich
|
0.025 ml einmal täglich
| |
Woche 3–4 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.25 mg einmal täglich gut vertragen wird)
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Ab Woche 5 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
≥ 40 kg
| |
Woche 1–2
|
0.25 mg einmal täglich
|
0.025 ml einmal täglich
| |
Woche 3–4 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.25 mg einmal täglich gut vertragen wird)
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Woche 5–6 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
Ab Woche 7 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 1 mg einmal täglich gut vertragen wird)
|
1.5 mg einmal täglich
|
0.15 ml einmal täglich
|
Wenn die Anfangsdosis von 0.25 mg nicht vertragen wird, sollte die Behandlung abgebrochen werden.
Wenn nach der Anfangsdosis eine nachfolgende Dosis nicht vertragen wird, auf die vorherige Dosisstufe reduzieren. Die Dosistitration fortsetzen, wenn die reduzierte Dosis vertragen wird.
Die Anwendung von Setmelanotid wurde bei Patienten mit terminaler Niereninsuffizienz nicht untersucht. Setmelanotid sollte bei Patienten mit terminaler Niereninsuffizienz nicht angewendet werden (siehe «Pharmakokinetik»).
Bardet-Biedl-Syndrom
Bei Erwachsenen und pädiatrischen Patienten von 2 bis 17 Jahren mit leichter oder mittelschwerer Nierenfunktionsstörung (siehe «Pharmakokinetik») sind keine Dosisanpassungen erforderlich.
Bei Erwachsenen und Kindern im Alter von 16 bis 17 Jahren mit schwerer Nierenfunktionsstörung (siehe «Pharmakokinetik») ist der Dosistitration in Tabelle 10 zu folgen.
Tabelle 10 Dosistitration bei Erwachsenen und pädiatrischen Patienten ab 16 Jahren mit schwerer Nierenfunktionsstörung
|
Woche
|
Tagesdosis
|
Zu injizierendes Volumen
| |
Wochen 1-2
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Ab Woche 3 (wenn die Dosis von 0.5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
Wenn das klinische Ansprechen unzureichend ist und die Dosis von 1 mg einmal täglich gut vertragen wird
|
2 mg einmal täglich
|
0.2 ml einmal täglich
| |
Wenn das klinische Ansprechen unzureichend ist und die Dosis von 2 mg einmal täglich gut vertragen wird
|
2.5 mg einmal täglich
|
0.25 ml einmal täglich
| |
Wenn das klinische Ansprechen unzureichend ist und die Dosis von 2.5 mg einmal täglich gut vertragen wird
|
3 mg einmal täglich
|
0.3 ml einmal täglich
|
Wenn die Anfangsdosis von 0.5 mg nicht vertragen wird, auf 0.25 mg (0.025 ml) einmal täglich reduzieren. Die Dosistitration fortsetzen, wenn die Dosis von 0.25 mg einmal täglich vertragen wird.
Wenn nach der Anfangsdosis eine nachfolgende Dosis nicht vertragen wird, auf die vorherige Dosisstufe reduzieren. Die Dosistitration fortsetzen, wenn die reduzierte Dosis vertragen wird.
Bei Patienten im Alter von 6 bis < 16 Jahren mit schwerer Nierenfunktionsstörung ist der Dosistitration in Tabelle 11 zu folgen.
Tabelle 11 Dosistitration für pädiatrische Patienten von 6 bis < 16 Jahren mit schwerer Nierenfunktionsstörung
|
Woche
|
Tagesdosis
|
Zu injizierendes Volumen
| |
Wochen 1-2
|
0.25 mg einmal täglich
|
0.025 ml einmal täglich
| |
Wochen 3-4 (wenn die Dosis von 0.25 mg einmal täglich gut vertragen wird)
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Ab Woche 5 (wenn 0.5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
Wenn das klinische Ansprechen unzureichend ist und die Dosis von 1 mg einmal täglich gut vertragen wird
|
2 mg einmal täglich
|
0.2 ml einmal täglich
|
Wenn die Anfangsdosis von 0.25 mg nicht vertragen wird, sollte die Behandlung abgebrochen werden.
Wenn nach der Anfangsdosis eine nachfolgende Dosis nicht vertragen wird, auf die vorherige Dosisstufe reduzieren. Die Dosistitration fortsetzen, wenn die reduzierte Dosis vertragen wird.
Setmelanotid wurde bei Patienten im Alter von 2 bis < 6 Jahren mit schwerer Nierenfunktionsstörung nicht untersucht. Die Dosistitration sollte sich nach der Verträglichkeit (Tabelle 12) richten, und die Patienten sind engmaschig zu überwachen.
Tabelle 12 Dosistitration für pädiatrische Patienten von 2 bis < 6 Jahren mit schwerer Nierenfunktionsstörung
|
Körpergewicht/Behandlungswoche
|
Tagesdosis
|
Zu injizierendes Volumen
| |
< 20 kg
| |
Ab Woche 1
|
0.25 mg einmal täglich
|
0.025 ml einmal täglich
| |
20 bis < 30 kg
| |
Woche 1–2
|
0.25 mg einmal täglich
|
0.025 ml einmal täglich
| |
Ab Woche 3 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.25 mg gut vertragen wird)
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
30 bis < 40 kg
| |
Woche 1–2
|
0.25 mg einmal täglich
|
0.025 ml einmal täglich
| |
Woche 3–4 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.25 mg einmal täglich gut vertragen wird)
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Ab Woche 5 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
≥ 40 kg
| |
Woche 1–2
|
0.25 mg einmal täglich
|
0.025 ml einmal täglich
| |
Woche 3–4 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.25 mg einmal täglich gut vertragen wird)
|
0.5 mg einmal täglich
|
0.05 ml einmal täglich
| |
Woche 5–6 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 0.5 mg einmal täglich gut vertragen wird)
|
1 mg einmal täglich
|
0.1 ml einmal täglich
| |
Ab Woche 7 (wenn das klinische Ansprechen unzureichend ist und die Dosis von 1 mg einmal täglich gut vertragen wird)
|
1.5 mg einmal täglich
|
0.15 ml einmal täglich
|
Die Behandlung abbrechen, wenn die Anfangsdosis von 0.25 mg nicht vertragen wird.
Wenn nach der Anfangsdosis eine nachfolgende Dosis nicht vertragen wird, auf die vorherige Dosisstufe reduzieren. Die Dosistitration fortsetzen, wenn die reduzierte Dosis vertragen wird.
Setmelanotid wurde bei Patienten mit terminaler Niereninsuffizienz nicht untersucht. Setmelanotid sollte bei Patienten mit terminaler Niereninsuffizienz nicht angewendet werden (siehe «Pharmakokinetik»).
Patienten mit Leberfunktionsstörungen
Setmelanotid wurde bei Patienten mit Leberfunktionsstörung nicht untersucht. Setmelanotid sollte bei Patienten mit Leberfunktionsstörung nicht angewendet werden.
Kinder und Jugendliche (< 2 Jahre)
Die Sicherheit und Wirksamkeit von Setmelanotid bei Kindern unter 2 Jahren ist bisher noch nicht erwiesen. Es liegen keine Daten vor.
Ältere Patienten
Auch wenn keine offensichtlichen altersbedingten Unterschiede beobachtet wurden, reichen die von älteren Patienten gewonnenen Daten nicht aus, um festzustellen, ob ihr Ansprechen sich von dem jüngerer Patienten unterscheidet. Es gibt keine Hinweise darauf, dass bei der Behandlung älterer Patienten besondere Vorsichtsmassnahmen erforderlich sind (siehe «Pharmakokinetik»).
Art der Anwendung
Zur subkutanen Anwendung.
Setmelanotid ist einmal täglich zu Tagesbeginn (für eine maximale Reduzierung des Hungergefühls während des Wachzeitraums) ohne Berücksichtigung der Essenszeiten zu injizieren.
Setmelanotid ist subkutan in das Abdomen zu injizieren, wobei die abdominelle Injektionsstelle täglich zu wechseln ist.
Vor Einleitung der Therapie sind Patienten von dem medizinischen Fachpersonal in der richtigen Injektionstechnik zu unterweisen, um das Risiko für Anwendungsfehler wie Nadelstichverletzungen und unvollständige Dosisgabe zu reduzieren. Vollständige Anweisungen zur Anwendung mit Illustrationen sind der Packungsbeilage zu entnehmen.
Für die Anwendung von Setmelanotid sollten die in Tabelle 13 dargestellten Spritzenvolumina und Nadelgrössen verwendet werden.
Tabelle 13 Zur Anwendung verwendete Spritze und Nadelgrösse nach Setmelanotid-Dosis
|
Setmelanotid-Dosis
|
Spritze
|
Nadelgrösse (Gauge) und länge
| |
Für Dosen von:
0.25 mg (0.025 ml oder 2.5 Einheiten) einmal täglich
|
0.3-ml-Spritze, mit Skalierung von 0.5 (halben) Einheiten
|
29 bis 31 Gauge
6- bis 13-mm-Nadel
| |
Für Dosen von:
0.5 mg bis 3 mg (0.05 ml bis 0.3 ml) einmal täglich
|
1-ml-Spritze mit 0.01-ml-Skalierung für Dosierung
|
28 bis 29 Gauge
6- bis 13-mm-Nadel
|
Anweisungen zur Handhabung von IMCIVREE, siehe «Sonstige Hinweise».
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der in der Rubrik «Zusammensetzung» genannten Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenKontrolle der Haut
Setmelanotid kann aufgrund seiner pharmakologischen Wirkung zu einer generalisierten erhöhten Hautpigmentierung und Verdunkelung bereits vorhandener Nävi führen (siehe «Unerwünschte Wirkungen» und «Eigenschaften/Wirkungen»). Vor und jährlich während der Behandlung mit Setmelanotid sollten den gesamten Körper umfassende Hautuntersuchungen erfolgen, um bereits vorhandene und neue Pigmentläsionen der Haut zu überwachen.
Kontrolle von Herzfrequenz und Blutdruck
Herzfrequenz und Blutdruck sollten bei mit Setmelanotid behandelten Patienten im Rahmen der standardmässigen klinischen Praxis bei jedem Arzttermin (mindestens alle 6 Monate) kontrolliert werden.
Verlängerte Peniserektion
In klinischen Prüfungen zu Setmelanotid wurden spontane Peniserektionen berichtet (siehe «Unerwünschte Wirkungen»). Patienten, deren Peniserektion länger als 4 Stunden andauert, sind anzuweisen, sich für eine eventuelle Behandlung eines Priapismus in ärztliche Notfallbehandlung zu begeben.
Depression
In klinischen Prüfungen wurden bei mit Setmelanotid behandelten Patienten Depressionen berichtet (siehe «Unerwünschte Wirkungen»).
Patienten mit Depressionen sind während der Behandlung mit IMCIVREE bei jedem Arzttermin zu überwachen. Wenn bei Patienten Suizidgedanken oder suizidale Verhaltensweisen auftreten, sollte in Erwägung gezogen werden, die Behandlung mit IMCIVREE abzusetzen.
Kinder und Jugendliche
Der verordnende Arzt sollte das Ansprechen auf die Setmelanotid-Therapie regelmässig bewerten. Bei im Wachstum befindlichen Kindern sind die Auswirkungen der Gewichtsabnahme auf Wachstum und Reifung zu bewerten. Der verordnende Arzt sollte das Wachstum (Körpergrösse und -gewicht) anhand alters- und geschlechtsspezifischer Wachstumskurven überwachen.
Sonstige Bestandteile
Dieses Arzneimittel enthält 10 mg Benzylalkohol pro ml. Benzylalkohol kann allergische Reaktionen hervorrufen.
Die intravenöse Anwendung von Benzylalkohol war mit schwerwiegenden Nebenwirkungen und Todesfällen bei Neugeborenen ("Gasping- Syndrom") verbunden. Die minimale Menge Benzylalkohol, bei der Toxizität auftritt, ist nicht bekannt. Bei Kleinkindern besteht aufgrund von Akkumulation ein erhöhtes Risiko. Grosse Mengen sollten wegen des Risikos der Akkumulation und Toxizität ("metabolische Azidose") nur mit Vorsicht und wenn absolut nötig angewendet werden, insbesondere bei Personen mit eingeschränkter Leber- oder Nierenfunktion.
Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro Dosis, d. h. es ist nahezu „natriumfrei“.
InteraktionenEs wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
In-vitro-Studien haben gezeigt, dass Setmelanotid ein geringes Potenzial für pharmakokinetische Wechselwirkungen im Zusammenhang mit Cytochrom-P450(CYP)-Transportern und Plasmaproteinbindung aufweist.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine Erfahrungen mit der Anwendung von Setmelanotid bei Schwangeren vor.
Tierexperimentelle Studien ergaben keine Hinweise auf direkte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität. Die Verabreichung von Setmelanotid an trächtige Kaninchen führte jedoch zu einer verminderten Nahrungsaufnahme durch das Muttertier, was wiederum embryofetale Wirkungen nach sich zog (siehe «Präklinische Daten»).
Als Vorsichtsmassnahme sollte die Behandlung mit IMCIVREE nicht während der Schwangerschaft oder während des Versuchs, schwanger zu werden, begonnen werden, da eine Gewichtsabnahme während der Schwangerschaft den Fötus schädigen könnte.
Wenn eine Patientin, die mit Setmelanotid behandelt wird, ein stabiles Gewicht erreicht hat und schwanger wird, sollte in Erwägung gezogen werden, die Behandlung mit Setmelanotid aufrechtzuerhalten, da die nichtklinischen Daten keine Hinweise auf eine Teratogenität ergaben. Wenn eine Patientin, die mit Setmelanotid behandelt wird und aktuell noch an Gewicht verliert, schwanger wird, ist Setmelanotid entweder abzusetzen oder die Dosis zu reduzieren und gleichzeitig eine Überwachung bezüglich der während der Schwangerschaft empfohlenen Gewichtszunahme durchzuführen. Der behandelnde Arzt sollte bei einer Patientin, die mit Setmelanotid behandelt wird, das Gewicht während der Schwangerschaft sorgfältig überwachen.
Schwangere Patientinnen sollten auf das potenzielle Risiko durch den sonstigen Bestandteil Benzylalkohol hingewiesen werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Stillzeit
Es ist nicht bekannt, ob Setmelanotid in die Muttermilch übergeht. In einer nichtklinischen Studie wurde gezeigt, dass Setmelanotid in die Milch säugender Ratten übergeht. Im Plasma gesäugter Jungtiere wurden keine quantifizierbaren Setmelanotid-Konzentrationen nachgewiesen (siehe «Präklinische Daten»).
Ein Risiko für das neugeborene Kind / den Säugling kann nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit IMCIVREE verzichtet werden soll / die Behandlung mit IMCIVREE zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Mutter zu berücksichtigen.
Stillende Patientinnen sollten auf das potenzielle Risiko durch den sonstigen Bestandteil Benzylalkohol hingewiesen werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Fertilität
Humandaten über die Auswirkungen von Setmelanotid auf die Fertilität liegen nicht vor. Tierexperimentelle Studien liessen nicht auf schädliche Auswirkungen in Bezug auf die Fertilität schliessen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenIMCIVREE hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die häufigsten unerwünschten Wirkungen sind Hyperpigmentierungsstörungen (56%), Reaktionen an der Injektionsstelle 45%), Übelkeit (31%) und Kopfschmerzen (20%).
Liste der unerwünschten Wirkungen
Die in klinischen Prüfungen beobachteten unerwünschten Wirkungen sind nachstehend nach Systemorganklasse und Häufigkeit gemäss der MedDRA-Konvention der Häufigkeiten aufgeführt, die wie folgt definiert ist: sehr häufig (≥ 1/10), häufig (≥1/100 bis <1/10), gelegentlich (≥1/1000 bis <1/100).
Tabelle 14 unerwünschten Wirkungen
|
Systemorganklasse
gemäss MedDRA
|
Häufigkeit
| |
Sehr häufig
|
Häufig
|
Gelegentlich
| |
Erkrankungen der Haut und des Unterhautzellgewebes
|
Hauthyperpigmentierung
|
Pruritus,
trockene Haut,
Hyperhidrose,
Hautverfärbung,
Hautläsion,
Alopezie
|
Ephelides,
Erythem,
Ausschlag,
Hautstriae,
Änderungen der
Haarfarbe,
Lentigo,
Macula,
Hautzyste,
Dermatitis,
Nagelerkrankung,
Nagelverfärbung,
Ausschlag papulös
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Reaktionen an der Injektionsstelle
|
Ermüdung,
Asthenie,
Schmerz
|
Brustkorbschmerz,
Temperaturintoleranz,
Pruritus an der Applikationsstelle,
Schüttelfrost,
Kältegefühl,
Wärmegefühl
| |
Erkrankungen des Gastrointestinaltrakts
|
Übelkeit,
Erbrechen
|
Diarrhö,
Abdominalschmerz,
Mundtrockenheit,
Dyspepsie,
Obstipation,
abdominale Beschwerden
|
Zahnfleischverfärbung,
Bauch aufgetrieben,
Hypersalivation,
Flatulenz,
Gastroösophageale
Refluxerkrankung
| |
Erkrankungen des Nervensystems
|
Kopfschmerzen
|
Schwindelgefühl
|
Somnolenz,
Hyperästhesie,
Migräne,
Parosmie,
Geschmacksstörung,
Angst,
Stimmungsänderung
| |
Erkrankungen der Geschlechtsorgane und der Brustdrüse
|
Spontane Peniserektion
|
Erektion erhöht,
Störung der sexuellen Erregung,
Libido gesteigert
|
Sexuelle Erregungsstörung der Frau,
Beschwerden im Genitalbereich,
Erkrankung der weiblichen Geschlechtsorgane,
genitale Hyperästhesie,
Ejakulationsstörung,
Libido vermindert
| |
Psychiatrische Erkrankungen
|
|
Depression,
Schlaflosigkeit
|
Depressive Verstimmung,
Schlafstörungen,
Alptraum
| |
Gutartige, bösartige und unspezifische Neubildungen (einschl. Zysten und Polypen)
|
|
Melanozytischer Nävus
|
Dysplastischer Nävus,
Augennävus
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
|
|
Rückenschmerzen,
Myalgie,
Muskelspasmen, Schmerz in einer Extremität
|
Arthralgie,
muskuloskelettale Brustschmerzen
| |
Erkrankungen der Atemwege, des Brustraums und Mediastinums
|
|
|
Gähnen,
Husten,
Rhinorrhoe
| |
Augenerkrankungen
|
|
|
Skleraverfärbung,
okulärer Ikterus
| |
Gefässerkrankungen
|
|
Hitzewallung
|
| |
Erkrankungen des Ohrs und des Labyrinths
|
|
Vertigo
|
| |
Stoffwechsel- und Ernährungsstörungen
|
Polydipsie
|
|
|
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Reaktionen an der Injektionsstelle
Reaktionen an der Injektionsstelle traten bei 45% der mit Setmelanotid behandelten Patienten auf. Die häufigsten Reaktionen an der Injektionsstelle waren Erythem an der Injektionsstelle (27%), Jucken an der Injektionsstelle (21%), Verhärtung an der Injektionsstelle (13%) und Schmerzen an der Injektionsstelle (13%). Diese Reaktionen waren in der Regel mild, von kurzer Dauer und schritten nicht fort oder führten nicht zum Abbruch der Therapie. Reaktionen an der Injektionsstelle umfassen folgende die Injektionsstelle betreffende Ereignisse: Erythem, Pruritus, Ödem, Schmerzen, Verhärtung, blaue Flecken, Reaktion, Schwellung, Blutung, Überempfindlichkeit, Hämatom, Knötchen, Verfärbung, Erosion, Entzündung, Reizung, Wärme, Atrophie, Unbehagen, Trockenheit, Raumforderungen, Hypertrophie, Ausschlag, Narbe, Abszess und Urtikaria.
Hyperpigmentierung
Eine Verdunkelung der Haut wurde bei 56% der mit Setmelanotid behandelten Patienten beobachtet. Dies trat im Allgemeinen innerhalb von 2 bis 3 Wochen nach Therapiebeginn auf, setzte sich für die Dauer der Behandlung fort und klang nach Absetzen der Behandlung ab. Diese Verdunkelung der Haut ist auf den Wirkmechanismus zurückzuführen und ist die Folge einer Stimulation des MC1-Rezeptors. Hyperpigmentierungserkrankungen umfassen Hauthyperpigmentierung, Hautverfärbung, Ephelides, Änderungen der Haarfarbe, Lentigo, Macula, Nagelverfärbung, Melanodermie, Pigmentierungsstörungen, Hauthypopigmentierung, Lentigo solaris, Acanthosis nigricans, Caféaulait Flecken, melanozytische Hyperplasie, melanozytischer Nävus, Nagelpigmentation, Zahnfleischverfärbung, Pigmentation der Lippe, Zungenverfärbung, Hyperpigmentierung des Zahnfleischs, Mundschleimhautverfärbung und Augennävus.
Gastrointestinale Störungen
Übelkeit und Erbrechen wurden bei 31 % bzw. 12 % der mit Setmelanotid behandelten Patienten berichtet. Übelkeit und Erbrechen traten im Allgemeinen zu Therapiebeginn (innerhalb des ersten Monats) auf, waren leicht und führten nicht zum Absetzen der Therapie. Diese Wirkungen waren vorübergehend und hatten keine Auswirkungen auf die Therapietreue im Hinblick auf die empfohlenen täglichen Injektionen.
Peniserektionen
Spontane Peniserektion und Erektion erhöht wurden bei 20% bzw. < 8% der mit Setmelanotid behandelten männlichen Patienten berichtet; keiner dieser Patienten meldete eine verlängerte Erektionsdauer (länger als 4 Stunden), die eine dringende medizinische Untersuchung erforderlich gemacht hätte (siehe «Warnhinweise und Vorsichtsmassnahmen»). Diese Wirkung kann auf die Nervenstimulation des Melanocortin 4(MC4)-Rezeptors zurückzuführen sein.
Immunogenität
Aufgrund der potenziell immunogenen Eigenschaften von Arzneimitteln, die Proteine oder Peptide enthalten, können Patienten nach der Behandlung mit Setmelanotid Antikörper entwickeln. Es wurden keine Fälle von einem schnellen Abfallen der Setmelanotid-Konzentrationen beobachtet, das auf das Vorhandensein von Anti-Drug-Antikörper hinweisen würde. In klinischen Prüfungen (RM-493-012 und RM-493-015) wurden im Screening auf Antikörper gegen Setmelanotid 68% (19 von 28) der erwachsenen und pädiatrischen Patienten mit POMC- oder LEPR-Mangel positiv und 32% negativ getestet. Bei den 68% der Patienten, die im Screening ein positives Ergebnis für Antikörper gegen Setmelanotid aufwiesen, war das Ergebnis für Antikörper gegen Setmelanotid im Bestätigungstest unschlüssig.
Bei ca. 13% der erwachsenen und pädiatrischen Patienten von 6 bis < 18 Jahren mit LEPR-Mangel (3 Patienten) wurde ein positives Ergebnis für Antikörper gegen Alpha-MSH bestätigt, die jedoch als niedrigtitrig und nicht persistent eingestuft wurden. Von diesen 3 Patienten (13%) wurden 2 nach der IMCIVREE-Behandlung positiv getestet und 1 wurde vor der Behandlung positiv getestet. Bei keinem der Patienten mit POMC-Mangel wurden Antikörper gegen Alpha-MSH bestätigt.
Bei einem pädiatrischen Patienten mit BBS im Alter von ≥12 Jahren wurde ein positiver Test auf Setmelanotid-Anti-Drug-Antikörper mit einem sehr niedrigen Titer bestätigt.
Kinder und Jugendliche
Insgesamt 124 pädiatrische Patienten (n = 12 im Alter von 2 bis < 6 Jahren; n = 26 im Alter von 6 bis < 12 Jahren, n = 86 im Alter von 12 bis < 18 Jahren) waren gegenüber Setmelanotid exponiert, einschließlich 21 pädiatrischer Patienten mit POMC- oder LEPR-Mangel-bedingter Adipositas, die an den pivotalen klinischen Prüfungen teilnahmen (n = 7 im Alter von 2 bis < 6 Jahren; n = 6 im Alter von 6 bis < 12 Jahren, n = 8 im Alter von 12 bis < 18 Jahren) und 33 pädiatrische Patienten mit BBS (n = 5 im Alter von 2 bis < 6 Jahren; n = 8 im Alter von 6 bis < 12 Jahren, n = 20 im Alter von 12 bis < 18 Jahren). Die Häufigkeit, Art und Schwere der Nebenwirkungen waren in den erwachsenen und pädiatrischen Populationen ähnlich.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungDie Symptome einer Setmelanotid-Überdosierung können unter anderem Übelkeit und Peniserektion sein. Im Falle einer Überdosierung ist eine angemessene unterstützende Behandlung entsprechend den klinischen Anzeichen und Symptomen des Patienten einzuleiten. Im Falle einer Überdosierung sind Blutdruck und Herzfrequenz über einen Zeitraum von 48 Stunden oder so lange wie klinisch relevant regelmässig zu überwachen.
Eigenschaften/WirkungenATC-Code
A08AA12, Antiadiposita, exkl. Diätetika, zentral wirkende Antiadiposita
Wirkungsmechanismus
Setmelanotid ist ein selektiver MC4-Rezeptor-Agonist. MC4-Rezeptoren im Gehirn sind an der Regulierung von Hunger- und Sättigungsgefühl sowie Energieumsatz beteiligt. Es wird davon ausgegangen, dass Setmelanotid bei genetischen Formen der Adipositas, die mit einer unzureichenden Aktivierung des MC4-Rezeptors zusammenhängen, die Aktivität des MC4-Rezeptor-Signalwegs wiederherstellt, um das Hungergefühl zu reduzieren und durch eine reduzierte Kalorienzufuhr und einen erhöhten Energieumsatz eine Gewichtsabnahme herbeizuführen.
Pharmakodynamik
Hautpigmentierung
Setmelanotid ist ein selektiver MC4-Rezeptor-Agonist mit geringerer Aktivität am Melanocortin-1(MC1)-Rezeptor. Der MC1-Rezeptor wird auf Melanozyten exprimiert, und die Aktivierung dieses Rezeptors führt unabhängig von ultraviolettem Licht zur Akkumulation von Melanin und einer erhöhten Hautpigmentierung (siehe «Warnhinweise und Vorsichtsmassnahmen» und «Unerwünschte Wirkungen»).
Klinische Wirksamkeit
POMC-Mangel, einschliesslich PCSK1, und LEPR-Mangel
Die Sicherheit und Wirksamkeit von Setmelanotid bei der Behandlung von POMC- und LEPR-Mangel-bedingter Adipositas wurde bei zwei identisch aufgebauten, 1jährigen offenen pivotalen Studien ermittelt, die jeweils eine doppelblinde, placebokontrollierte Absetzphase umfassten:
·In Studie 1 (RM-493-012) wurden Patienten ab einem Alter von 6 Jahren mit genetisch bestätigter POMC-Mangel-bedingter (einschliesslich PCSK1) Adipositas aufgenommen.
·In Studie 2 (RM-493-015) wurden Patienten ab einem Alter von 6 Jahren mit genetisch bestätigter LEPR-Mangel-bedingter Adipositas aufgenommen.
In beiden Studien hatten die erwachsenen Patienten einen Body-Mass-Index (BMI) von ≥ 30 kg/m2. Das Gewicht bei Kindern lag gemäss Beurteilung anhand der Wachstumstabelle im 95. Perzentil oder darüber.
Über einen Zeitraum von 2 bis 12 Wochen wurde eine Dosistitration vorgenommen, gefolgt von einer 10-wöchigen offenen Behandlungsphase. Patienten, die am Ende der offenen Behandlungsphase eine Gewichtsabnahme von mindestens 5 kg (oder eine Gewichtsabnahme von mindestens 5 %, wenn das Körpergewicht zum Studienbeginn < 100 kg betrug) erreichten, setzen die Studie in einer doppelblinden, placebokontrollierten Absetzphase von 8 Wochen (4wöchige Behandlung mit Placebo und 4wöchige Behandlung mit Setmelanotid) fort. Nach der Absetzphase nahmen die Patienten die aktive Behandlung mit Setmelanotid bei der therapeutischen Dosis wieder auf und setzten diese bis zu 32 Wochen lang fort. 21 Patienten (10 in Studie 1 und 11 in Studie 2) wurden seit mindestens 1 Jahr behandelt und sind in den Wirksamkeitsanalysen enthalten.
In einer Studie unter Leitung des Prüfarztes und in einer noch laufenden Verlängerungsstudie wurden zusätzliche unterstützende Daten gesammelt.
Studie 1 (RM-493-012)
In Studie 1 erreichten 80 % der Patienten mit POMC-Mangel-bedingter Adipositas den primären Endpunkt mit einer Gewichtsabnahme von ≥ 10 % nach 1-jähriger Behandlung mit Setmelanotid, und 50 % der Patienten mit POMC-Mangel-bedingter Adipositas erreichten nach 1 Jahr eine vorher festgelegte, klinisch bedeutsame Verbesserung des Hungergefühl-Scores von ≥ 25 % gegenüber Studienbeginn (Tabelle 15).
Für Studie 1 wurden statistisch signifikante und klinisch bedeutsame mittlere prozentuale Abnahmen des Körpergewichts gegenüber Studienbeginn von 25.6 % gemeldet. Bei Patienten im Alter von ≥ 12 Jahren wurden nach 1 Jahr die Veränderungen des Hungergefühls anhand eines täglich auszufüllenden Fragebogens für Patienten und Betreuungspersonen beurteilt, der den Aspekt „stärkstes Hungergefühl im Laufe der letzten 24 Stunden“ behandelte. Für Studie 1 wurden statistisch signifikante und klinisch bedeutsame mittlere prozentuale Abnahmen des Hungergefühls in den letzten 24 Stunden als wöchentlicher Durchschnittswert gegenüber Studienbeginn von 27.1 % gemeldet (Tabelle 16).
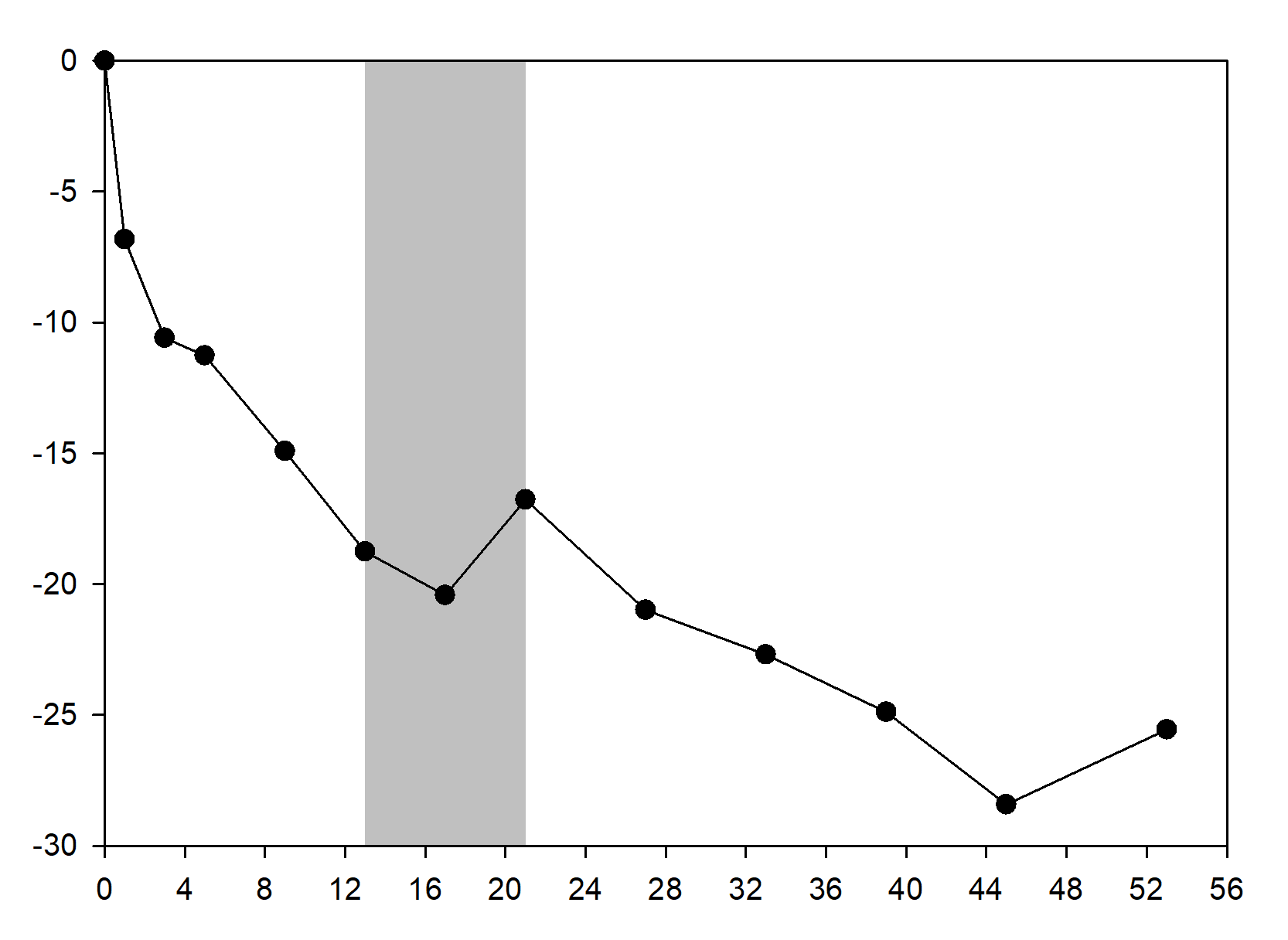
Als die Behandlung mit Setmelanotid bei Patienten mit Gewichtsabnahme während der 10wöchigen offenen Phase abgesetzt wurde, nahmen diese Patienten wieder an Gewicht zu (Abbildung 1), und die mittleren Hungergefühl-Scores stiegen über die 4-wöchige Behandlung mit Placebo wieder an.
Tabelle 15 Anteil der Patienten, die eine Gewichtsabnahme von mindestens 10 % erreichten, und Anteil von Patienten, die nach 1 Jahr eine Verbesserung des täglichen Hungergefühls von mindestens 25 % gegenüber Studienbeginn erreichten, in Studie 1
|
Parameter
|
Statistik
| |
Patienten, die nach 1 Jahr eine Gewichtsabnahme von mindestens 10 % erreichten
(n = 10)
|
n (%)
|
8 (80.0)
| |
90-%-KI1
|
(49.31, 96.32)
| |
p-Wert2
|
<0.0001
| |
Patienten, die nach 1 Jahr eine Verbesserung des Hungergefühls von mindestens 25 % gegenüber Studienbeginn erreichten (n = 8)
|
n (%)
|
4 (50.0)
| |
90-%-KI1
|
(19.29, 80.71)
| |
p-Wert1
|
0.0004
| |
Hinweis: Das Analysekollektiv umfasst Patienten, die mindestens 1 Dosis des Prüfpräparats erhielten und mindestens 1
Untersuchung zum Studienbeginn unterzogen wurden.
1 Mittels der exakten Methode nach Clopper-Pearson
2 Nullhypothesentest: Anteil = 5%
|
Tabelle 16 Prozentuale Veränderung von Gewicht und Hungergefühl nach 1 Jahr gegenüber Studienbeginn in Studie 1
|
Parameter
|
Statistik
|
Körpergewicht (kg)
(n = 9)
|
Hungergefühl-Score1
(n = 7)
| |
Studienbeginn
|
Mittelwert (SA)
|
115.0 (37.77)
|
8.1 (0.78)
| |
|
Median
|
114.7
|
8.0
| |
|
Min, Max
|
55.9, 186.7
|
7, 9
| |
1 Jahr
|
Mittelwert (SA)
|
83.1 (21.43)
|
5.8 (2.02)
| |
|
Median
|
82.7
|
6.0
| |
|
Min, Max
|
54.5, 121.8
|
3, 8
| |
Prozentuale Veränderung von Studienbeginn bis 1 Jahr (%)
|
Mittelwert (SA)
|
-25.6 (9.88)
|
-27.06 (28.11)
| |
|
Median
|
-27.3
|
-14.29
| |
|
Min, Max
|
-35.6, -2.4
|
-72.2, -1.4
| |
|
KQ-Mittelwert
|
-25.39
|
-27.77
| |
|
90-%-KI
|
(-28.80, -21.98)
|
(-40.58, -14.96)
| |
|
p-Wert
|
<0.0001
|
0.0005
| |
Hinweis: Diese Analyse umfasst Patienten, die mindestens eine Dosis des Prüfpräparats erhalten haben und mindestens
einer Untersuchung zu Studienbeginn unterzogen wurden sowie über die 12-wöchige offene Behandlungsphase eine
Gewichtsabnahme von ≥ 5 kg (oder 5% des Körpergewichts, wenn das Gewicht zu Studienbeginn < 100 kg betrug)
erreichten und mit der doppelblinden, placebokontrollierten Absetzphase fortfuhren.
1 Das Hungergefühl wird auf einer Skala vom Likert-Typ von 0 bis 10 eingestuft, wobei 0 = überhaupt kein Hungergefühl und 10 = stärkstes mögliches Hungergefühl. Der Hungergefühl-Score wurde in einem täglich auszufüllenden Tagebuch erfasst und wurde gemittelt, um für die Analyse einen wöchentlichen Score zu berechnen.
|
Abbildung 1 Prozentuale Veränderung des Körpergewichts gegenüber Studienbeginn nach Termin (Studie 1 [n = 9])
Studie 2 (RM-493-015)
In Studie 2 erreichten 46 % der Patienten mit LEPR-Mangel-bedingter Adipositas den primären Endpunkt mit einer Gewichtsabnahme von ≥ 10 % nach 1-jähriger Behandlung mit Setmelanotid, und 73 % der Patienten mit LEPR-Mangel-bedingter Adipositas erreichten nach 1 Jahr eine vorher festgelegte, klinisch bedeutsame Verbesserung des Hungergefühl-Scores von ≥ 25 % gegenüber Studienbeginn (Tabelle 17).
Für Studie 2 wurden statistisch signifikante und klinisch bedeutsame mittlere prozentuale Abnahmen des Körpergewichts gegenüber Studienbeginn von 12.5 % gemeldet. Bei Patienten im Alter von ≥ 12 Jahren wurden nach 1 Jahr die Veränderungen des Hungergefühls anhand eines täglich auszufüllenden Fragebogens für Patienten und Betreuungspersonen beurteilt, der den Aspekt „stärkstes Hungergefühl im Laufe der letzten 24 Stunden“ behandelte. Für Studie 2 wurden statistisch signifikante und klinisch bedeutsame mittlere prozentuale Abnahmen des Hungergefühls in den letzten 24 Stunden als wöchentlicher Durchschnittswert gegenüber Studienbeginn von 43.7 % gemeldet (Tabelle 18).
Als die Behandlung mit Setmelanotid bei Patienten mit Gewichtsabnahme während der 10-wöchigen offenen Phase abgesetzt wurde, nahmen diese Patienten wieder an Gewicht zu (Abbildung 2), und die mittleren Hungergefühl-Scores stiegen über die 4-wöchige Behandlung mit Placebo wieder an.
Tabelle 17 Anteil der Patienten, die eine Gewichtsabnahme von mindestens 10 % erreichten, und Anteil von Patienten, die nach 1 Jahr eine Verbesserung des täglichen Hungergefühls von mindestens 25 % gegenüber Studienbeginn erreichten, in Studie 2
|
Parameter
|
Statistik
| |
Patienten, die nach 1 Jahr eine Gewichtsabnahme von mindestens 10 % erreichten
(n = 11)
|
n (%)
|
5 (45.5)
| |
90-%-KI1
|
(19.96, 72.88)
| |
p-Wert2
|
0.0002
| |
Patienten, die nach 1 Jahr eine Verbesserung des Hungergefühls von mindestens 25 % gegenüber Studienbeginn erreichten (n = 11)
|
n (%)
|
8 (72.7)
| |
90-%-KI1
|
(43.56, 92.12)
| |
p-Wert1
|
< 0.0001
| |
Hinweis: Das Analysekollektiv umfasst Patienten, die mindestens 1 Dosis des Prüfpräparats erhielten und mindestens 1
Untersuchung zum Studienbeginn unterzogen wurden.
1 Mittels der exakten Methode nach Clopper-Pearson
2 Nullhypothesentest: Anteil = 5%
|
Tabelle 18 Prozentuale Veränderung von Gewicht und Hungergefühl nach 1 Jahr gegenüber Studienbeginn in Studie 2
|
Parameter
|
Statistik
|
Körpergewicht (kg)
(n = 7)
|
Hungergefühl-Score1
(n = 7)
| |
Studienbeginn
|
Mittelwert (SA)
|
131.7 (32.6)
|
7.0 (0.77)
| |
|
Median
|
120.5
|
7.0
| |
|
Min, Max
|
89.4, 170.4
|
6, 8
| |
1 Jahr
|
Mittelwert (SA)
|
115.0 (29.6)
|
4.1 (2.09)
| |
|
Median
|
104.1
|
3.0
| |
|
Min, Max
|
81.7, 149.9
|
2, 8
| |
Prozentuale Veränderung von Studienbeginn bis 1 Jahr (%)
|
Mittelwert (SA)
|
-12.5 (8.9)
|
-43.7 (23.69)
| |
|
Median
|
-15.3
|
-52.7
| |
|
Min, Max
|
-23.3, 0.1
|
-67, 0
| |
|
KQ-Mittelwert
|
-12.47
|
-41.93
| |
|
90-%-KI
|
(-16.10, -8.83)
|
(-54.76, -29.09)
| |
|
p-Wert
|
<0.0001
|
<0.0001
| |
Hinweis: Diese Analyse umfasst Patienten, die mindestens eine Dosis des Prüfpräparats erhalten haben und mindestens
einer Untersuchung zu Studienbeginn unterzogen wurden sowie über die 12-wöchige offene Behandlungsphase eine Gewichtsabnahme von ≥ 5 kg (oder 5 % des Körpergewichts, wenn das Gewicht zu Studienbeginn <100 kg betrug) erreichten
und mit der doppelblinden, placebokontrollierten Absetzphase fortfuhren.
1 Das Hungergefühl wird auf einer Skala vom Likert-Typ von 0 bis 10 eingestuft, wobei 0 = überhaupt kein Hungergefühl und 10 = stärkstes mögliches Hungergefühl. Der Hungergefühl-Score wurde in einem täglich auszufüllenden Tagebuch erfasst und wurde gemittelt, um für die Analyse einen wöchentlichen Score zu berechnen.
|
Abbildung 2 Prozentuale Veränderung des Körpergewichts gegenüber Studienbeginn nach Termin (Studie 2 [n = 7])

Bardet-Biedl-Syndrom
Studie 3 (RM-493-023)
Sicherheit und Wirksamkeit von IMCIVREE bei der Behandlung von Patienten ab 6 Jahren mit Adipositas aufgrund von BBS wurden in einer einjährigen klinischen Studie mit einer 14wöchigen placebokontrollierten Phase (Studie 3 [RM-493-023]) beurteilt. An der Studie nahmen Patienten ab 6 Jahren mit Adipositas und BBS teil. Die erwachsenen Patienten hatten einen BMI von ≥ 30 kg/m2. Die pädiatrischen Patienten hatten gemäss Beurteilung anhand der Wachstumstabelle für Alter und Geschlecht einen BMI im 97. Perzentil oder darüber.
Geeignete Patienten nahmen an einer 14wöchigen, randomisierten, doppelblinden, placebokontrollierten Behandlungsphase (Phase 1) teil, an die sich eine 38-wöchige offene Behandlungsphase (Phase 2) anschloss, in der alle Patienten Setmelanotid erhielten. Um die Verblindung während der Phase 2 aufrechtzuerhalten, wurde die Dosis in den ersten 2 Wochen sowohl der Phase 1 als auch der Phase 2 auf eine feste Dosis von 3 mg titriert. Zweiunddreissig Patienten wurden mindestens 1 Jahr lang behandelt und sind in die Wirksamkeitsanalysen eingeschlossen.
In Studie 3 erreichten 35.7 % der Patienten mit BBS im Alter von ≥ 12 Jahren und 46.7 % der Patienten mit BBS im Alter von ≥ 18 Jahren den primären Endpunkt, das heisst einen Gewichtsverlust von ≥ 10 % nach 1 Jahr Behandlung mit Setmelanotid (Tabelle 19). Die Wirkung von IMCIVREE auf das Körpergewicht war bei Patienten, die vom Prüfer als kognitiv beeinträchtigt eingestuft wurden, ähnlich wie bei Patienten ohne kognitive Beeinträchtigung.
In Studie 3 führte eine ~52wöchige Behandlung mit Setmelanotid bei 100 % der BBS-Patienten im Alter von < 12 Jahren zu einer klinisch bedeutsamen Verringerung des BMI-Z-Scores, wobei bei Patienten im Alter von ≥ 12 und < 18 Jahren konsistente Ergebnisse beobachtet wurden. Bei den Patienten im Alter von < 18 Jahren betrug die mittlere Verringerung des BMI-Z-Scores gegenüber Studienbeginn 0.75, und die mittlere Verringerung des prozentualen Anteils im 95. Perzentil des BMI nach Alter und Geschlecht betrug 17.3 % gegenüber Studienbeginn.
Patienten ab 12 Jahren, die in der Lage waren, ihr Hungergefühl selbst einzuschätzen, protokollierten ihr tägliches maximales Hungergefühl in einem Tagebuch, das dann anhand des Daily Hunger Questionnaire (Fragebogen zum täglichen Hungergefühl), Item 2, beurteilt wurde. Das Hungergefühl wurde auf einer 11-Punkte-Skala von 0 („überhaupt kein Hungergefühl“) bis 10 („stärkstes mögliches Hungergefühl“) bewertet. In Studie 3 wurden für das grösste/schlimmste Hungergefühl nach 1 Jahr statistisch signifikante und klinisch bedeutsame mittlere prozentuale Verringerungen gegenüber Studienbeginn von 30.5 % berichtet (Tabelle 20).
Tabelle 19 Körpergewicht (kg) – Anteil aller Patienten, der Patienten mit BBS im Alter von ≥ 12 Jahren und der Patienten mit BBS im Alter von ≥ 18 Jahren, die nach 1 Jahr einen Gewichtsverlust von mindestens 10 % gegenüber Studienbeginn erreichten (Studie 3 [vollständiges Analysekollektiv])
|
Parameter
|
Statistik1
|
Patienten
≥ 12 Jahre
|
BBS
≥ 18 Jahre
| |
Patienten, die nach 1 Jahr eine Gewichtsabnahme von mindestens 10 % erreichten
|
N
|
28
|
15
| |
%
|
35.7
|
46.7
| |
95 % KI1
|
(18.6, 55.9)
|
(21.3, 73.4)
| |
p-Wert
|
0.0002
|
0.0003
| |
1
Geschätzter Prozentsatz, 95%iges Konfidenzintervall und p-Wert basieren auf Rubins Regel. Der p-Wert ist einseitig und wird mit alpha = 0.025 verglichen.
|
Tabelle 20 Tägliche Hungergefühl-Scores – Veränderung nach 1 Jahr gegenüber Studienbeginn bei allen Patienten und Patienten mit BBS im Alter von ≥ 12 Jahren (Studie 3 [Vollständiges Analysekollektiv])
|
Zeitpunkt
|
Statistik
|
Patienten
≥ 12 Jahre
| |
Studienbeginn
|
N
|
14
| |
Mittel (SA)
|
6.99 (1.893)
| |
Median
|
7.29
| |
Min, Max
|
4.0, 10.0
| |
Woche 52
|
N
|
14
| |
Mittel (SA)
|
4.87 (2.499)
| |
Median
|
4.43
| |
Min, Max
|
2.0, 10.0
| |
Veränderung in Woche 52
|
N
|
14
| |
Mittel (SA)
|
-2.12 (2.051)
| |
Median
|
-1.69
| |
Min, Max
|
-6.7, 0.0
| |
95 % KI1
|
-3.31, -0.94
| |
p-Wert 1
|
0.0010
| |
Prozentuale Veränderung in Woche 52
|
N
|
14
| |
Mittel (SA)
|
-30.45 (26.485)
| |
Median
|
-25.00
| |
Min, Max
|
-77.0, 0.0
| |
95 % KI1
|
-45.74, -15.16
| |
p-Wert 1
|
0.0004
| |
Abkürzungen: KI = Konfidenzintervall, Max = Maximum, Min = Minimum, SA = Standardabweichung.
1 95%iges KI und p-Wert basieren auf Rubins Regel. Der p-Wert ist einseitig.
Hinweis: Studienbeginn bezeichnet in beiden Studien die letzte Beurteilung vor Einleitung der Setmelanotid-Gabe.
Hinweis: Das Daily Hunger Questionnaire wird nicht an Patienten <12 Jahren oder an Patienten durchgeführt, die gemäss dem Urteil des Prüfers kognitiv beeinträchtigt sind.
|
Die Wirkung von IMCIVREE auf die Gewichtsabnahme wurde durch allgemeine numerische Verbesserungen bei kardiometabolischen Parametern wie Blutdruck, Lipide, glykämische Parameter und Taillenumfang unterstützt.
Kinder und Jugendliche
Studie 4 (RM-493-033)
Die Sicherheit und Wirksamkeit von Setmelanotid bei der Behandlung von Patienten im Alter von 2 bis < 6 Jahren mit durch einen POMC- und LEPR-Mangel-bedingter Adipositas oder BBS wurden in einer 1jährigen offenen nicht-kontrollierten Studie (Studie 4 [RM-493-033]) untersucht. An der Studie nahmen Patienten im Alter von 2 bis < 6 Jahren mit einem BMI ≥ 97. Perzentile anhand von Wachstumstabellen für Alter und Geschlecht und mit einem Körpergewicht von mindestens 15 kg bei Studienbeginn teil.
Geeignete Patienten nahmen an der Studie teil und erhielten Setmelanotid. Zwölf Patienten wurden in die Studie aufgenommen und in die Analysen der Wirksamkeit einbezogen. In Anbetracht des Studiendesigns und der kleinen Fallzahl sind die Wirksamkeitsergebnisse vorsichtig zu interpretieren.
In Studie 4 erreichten 85.7 % der Patienten mit durch POMC- und LEPR-Mangel bedingter Adipositas und 80.0 % der Patienten mit BBS den primären Endpunkt und erzielten nach 1-jähriger Behandlung mit Setmelanotid eine Verringerung des BMI-Z-Scores um ≥ 0,2 (Tabelle 21). Die mittlere prozentuale Veränderung des BMI zwischen Studienbeginn und Woche 52 betrug -25.597 % bei Patienten mit durch POMC- und LEPR-Mangel bedingter Adipositas und -9.719 % bei Patienten mit BBS (Tabelle 22).
Table 21 BMI-Z-Score – Anteil aller Patienten, Patienten mit durch POMC- und LEPR-Mangel bedingter Adipositas sowie Patienten mit BBS im Alter von 2 bis < 6 Jahren, die nach 1 Jahr eine Verringerung des BMI-Z-Scores gegenüber dem Studienbeginn um mindestens 0,2 erreichten (Studie 4 [Sicherheitskollektiv])
|
Parameter
|
Statistik1
|
Patienten mit POMC oder LEPR
(n = 7)
|
Patienten mit BBS
(n = 5)
|
Gesamt
(N = 12)
| |
Patienten, die nach 1 Jahr eine Reduzierung des BMI-Z-Scores um mindestens 0,2 erreichten
|
N
|
6
|
4
|
10
| |
%
|
85.7
|
80.0
|
83.3
| |
95 %-KI1
|
(54.1; 100)
|
(28.4; 99.5)
|
(58.7; 99.8)
| |
1 Das zweiseitige 95%ige KI wurde mit der Methode nach Clopper-Pearson berechnet.
|
Tabelle 22 Prozentuale Veränderung des BMI von Studienbeginn bis 1 Jahr (Studie 4 [Sicherheitskollektiv])
|
|
|
Patienten mit POMC oder LEPR
(n = 7)
|
Patienten mit BBS
(n = 5)
|
Gesamt
(N = 12)
| |
Parameter
|
Statistik
|
%
|
%
|
%
| |
Studienbeginn
|
N
|
7
|
5
|
12
| |
|
Mittelwert (SA)
|
34.347 (7.0673)
|
23.716 (3.5184)
|
29.918 (7.8559)
| |
|
Median
|
32.196
|
22.986
|
28.670
| |
|
Min, Max
|
25.99; 42.54
|
19.31; 29.04
|
19.31; 42.54
| |
Tatsächliche Veränderung von Studienbeginn bis 1 Jahr
|
N
|
6
|
5
|
11
| |
|
Mittelwert (SA)
|
-8.250 (3.2392)
|
-2.363 (2.1579)
|
-5.574 (4.0697)
| |
|
Median
|
-9.237
|
-2.191
|
-4.940
| |
|
Min, Max
|
-11.16; -2.65
|
-4.94; 0.58
|
-11.16; 0.58
| |
Prozentuale Veränderung von Studienbeginn bis 1 Jahr (%)
|
N
|
6
|
5
|
11
| |
|
Mittelwert (SA)
|
-25.597 (11.4911)
|
-9.719 (8.8383)
|
-18.380 (12.8851)
| |
|
95 %-KI1
|
(-37.66; -13.54)
|
(-20.69; 1.26)
|
(-27.04; -9.72)
| |
|
Median
|
-23.237
|
-8.978
|
-21.624
| |
|
Min, Max
|
-39.28; -8.24
|
-21.62; 2.54
|
-39.28; 2.54
| |
1
Das zweiseitige 95%ige KI wurde mithilfe einer Student-t-Verteilung berechnet.
|
|
In Studie 4 führte eine ca. 52-wöchige Behandlung mit Setmelanotid zu einer klinisch bedeutsamen Verringerung des BMI-Z-Scores um -5.185 bei Patienten mit durch POMC- oder LEPR-Mangel bedingter Adipositas und um -1.331 bei Patienten mit BBS. Die mittlere Verringerung gegenüber dem Studienbeginn in Prozent des 95. Perzentils für den BMI nach Alter und Geschlecht betrug -47.595 % bei Patienten mit durch POMC- oder LEPR-Mangel bedingter Adipositas und -14.462 % bei Patienten mit BBS.
In klinischen Studien waren 44 der mit Setmelanotid behandelten Patienten zum Studienbeginn 2 bis 17 Jahre alt (21 Patienten mit POMC-, PCSK1- oder LEPR-Mangel und 33 Patienten mit BBS). Insgesamt waren im Hinblick auf Wirksamkeit und Sicherheit bei diesen jüngeren Patienten ähnliche Trends wie bei den untersuchten älteren Patientenfestzustellen, wobei ein scheinbar deutlicher Rückgang des BMInachgewiesen wurde. Bei Patienten, deren Wachstum noch nicht abgeschlossen war, wurde während der Studienphase ein Trend zu einem angemessenen Fortschreiten der pubertären Entwicklung und eine Zunahme der Körpergröße beobachtet.
PharmakokinetikDie mittlere Cmax,ss, AUCtau, und Talkonzentration im Steady-State bei einmal täglicher subkutaner Anwendung einer 3-mg-Dosis bei ansonsten gesunden Probanden mit Adipositas (n=6) über einen Zeitraum von 12 Wochen betrugen 37.9 ng/ml, 495 h*ng/ml bzw. 6.77 ng/ml. Steady-State-Plasmakonzentrationen von Setmelanotid wurden bei täglicher Gabe von 1 mg bis 3 mg Setmelanotid innerhalb von 2 Tagen erreicht. Die Akkumulation von Setmelanotid im systemischen Kreislauf bei einmal täglicher Gabe über 12 Wochen betrug ca. 30%. Die AUC und Cmax von Setmelanotid stiegen nach subkutaner Anwendung mehrerer Dosen im vorgeschlagenen Dosisbereich (1-3 mg) proportional an.
Es wurde ein populationspharmakokinetisches Modell untersucht, das 410 Teilnehmer in 11 Studien umfasste. Diese Teilnehmer ergaben 7 087 Beobachtungen, von denen 6 847 Proben quantifizierbare Setmelanotid-Konzentrationen aufwiesen. Die PK-Daten stammten überwiegend von 271 Erwachsenen und 87 Jugendlichen (im Alter von 12 bis < 18 Jahren). Es gab auch 41 Kinder im Alter von 6 bis < 12 Jahren und 11 Kinder im Alter von 2 bis < 6 Jahren. Die Studienpopulation umfasste 166 männliche und 244 weibliche Teilnehmer im Alter von 2 bis 78 Jahren (Mittelwert = 29.7 Jahre) und mit einem Körpergewicht von 17.8 kg bis 246 kg (Mittelwert = 113 kg). Das gepoolte Kollektiv umfasste 329 Teilnehmer mit POMC-, PCSK1- oder LEPR-Mangel, BBS oder anderen seltenen genetischen Adipositas verursachenden Erkrankungen (80.2 %) und 81 Teilnehmer ohne POMC-, PCSK1- oder LEPR-Mangel, BBS oder andere seltene genetische Adipositas verursachende Erkrankungen (19.8 %); alle Teilnehmer ohne POMC-, PCSK1- oder LEPR-Mangel, BBS oder andere seltene genetische Adipositas verursachende Erkrankungen waren Erwachsene.
Absorption
Nach subkutaner Injektion von Setmelanotid stiegen die Steady-State-Plasmakonzentrationen von Setmelanotid langsam an und erreichten nach einer medianen tmax von 8.0 Stunden die Höchstkonzentrationen. Die absolute Bioverfügbarkeit nach subkutaner Anwendung von Setmelanotid wurde beim Menschen nicht untersucht.
Die Schätzung der interindividuellen Variabilität (VK%) aus dem finalen populationspharmakokinetischen Modell betrug 39.9 % (CL/F).
Die PK von Setmelanotid bei Patienten mit BBS war ähnlich wie bei Patienten mit POMC-, PCSK1- und LEPR-Mangel, was darauf hindeutet, dass der Krankheitszustand allein keinen Einfluss auf die PK von Setmelanotid hat.
Distribution
Das mittlere apparente Verteilungsvolumen von Setmelanotid nach subkutaner Anwendung von Setmelanotid 3 mg einmal täglich wurde anhand des populationspharmakokinetischen Modells auf 75.2 l geschätzt. Setmelanotid bindet zu 79.1% an menschliches Plasmaprotein.
In-vitro-Versuche lassen darauf schliessen, dass Setmelanotid kein Substrat von OATP1B1, OATP1B3, OAT1, OAT3 oder OCT2 ist.
In-vitro-Daten legen die Vermutung nahe, dass es sehr unwahrscheinlich ist, dass Setmelanotid ein Pgp- oder BCRP-Substrat ist.
Metabolismus
Setmelanotid schien von Lebermikrosomen oder -zellen oder von Nierenmikrosomen von Ratten, Affen oder Menschen nicht metabolisiert zu werden.
Elimination
Die effektive Eliminationshalbwertszeit (t½) von Setmelanotid betrug ca. 11 Stunden. Die apparente Gesamtclearance von Setmelanotid im Steady-State nach subkutaner Anwendung von Setmelanotid 3 mg einmal täglich wurde anhand des populationspharmakokinetischen Modells auf 7.15 l/h geschätzt.
Ca. 39% der angewendeten Setmelanotid-Dosis wurden nach subkutaner Anwendung von 3 mg einmal täglich während des 24stündigen Dosierungsintervalls unverändert mit dem Urin ausgeschieden.
Linearität/Nicht Linearität
Die AUC und Cmax von Setmelanotid stiegen nach subkutaner Anwendung mehrerer Dosen im Bereich von 0.5 mg bis 5 mg annähernd linear an.
Kinetik spezieller Patientengruppen
Kinder und Jugendliche
Setmelanotid wurde bei pädiatrischen Patienten (im Alter von 2 bis 17 Jahren) untersucht. Simulationen auf Grundlage der populationspharmakokinetischen Analysen lassen auf eine leicht höhere Exposition bei jüngeren Patienten (die auch ein geringeres Körpergewicht haben) schliessen und stützen das Dosierungsschema bei Patienten ab einem Alter von 2 Jahren.
Ältere Patienten
Die verfügbaren Daten aus einer kleinen Stichprobe älterer Patienten deuten nicht darauf hin, dass sich die Setmelanotid-Exposition mit zunehmendem Alter wesentlich verändert. Diese Daten sind jedoch zu begrenzt, um klare Schlussfolgerungen zu ziehen.
Nierenfunktionsstörungen
Die pharmakokinetische Analyse ergab im Vergleich zu Patienten mit normaler Nierenfunktion eine um 12%, 26% bzw. 49% geringere Clearance (CL/F) von Setmelanotid bei Patienten mit leichter, mittelschwerer bzw. schwerer Nierenfunktionsstörung.
POMC-Mangel, einschliesslich PCSK1, und LEPR-Mangel
Für Patienten mit leichter (geschätzte glomeruläre Filtrationsrate [eGFR] von 60-89 ml/min/1.73 m2) oder mittelschwerer Nierenfunktionsstörung (eGFR von 30-59 ml/min/1.73 m2) sind keine Dosisanpassungen erforderlich. Für Patienten mit schwerer Nierenfunktionsstörung (eGFR 15-29 ml/min/1.73 m2) wird eine Dosisanpassung empfohlen (siehe «Dosierung/Anwendung»). Setmelanotid sollte bei Patienten mit terminaler Niereninsuffizienz (eGFR < 15 ml/min/1.73 m2) nicht angewendet werden (siehe «Dosierung/Anwendung»).
Bardet-Biedl-Syndrom
Für Patienten mit leichter (geschätzte glomeruläre Filtrationsrate [eGFR] von 60-89 ml/min/1.73 m2) oder mittelschwerer Nierenfunktionsstörung (eGFR von 30-59 ml/min/1.73 m2) sind keine Dosisanpassungen erforderlich. Für Patienten mit schwerer Nierenfunktionsstörung (eGFR 15-29 ml/min/1.73 m2) wird eine Dosisanpassung empfohlen (siehe «Dosierung/Anwendung»). Setmelanotid sollte bei Patienten mit terminaler Niereninsuffizienz (eGFR < 15 ml/min/1.73 m2) nicht angewendet werden (siehe «Dosierung/Anwendung »).
Leberfunktionsstörungen
Setmelanotid ist in Leberzellen von Menschen, Ratten und Affen stabil; daher wurden keine Studien unter Beteiligung von Patienten mit Leberfunktionsstörung durchgeführt. Setmelanotid darf nicht bei Patienten mit Leberfunktionsstörung angewendet werden.
Körpergewicht
Die CL/F von Setmelanotid variierte je nach Körpergewicht und gemäss einer festen allometrischen Beziehung.
Geschlecht
Basierend auf dem Geschlecht wurden keine klinisch signifikanten Unterschiede hinsichtlich der Pharmakokinetik von Setmelanotid beobachtet.
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Genotoxizität, Karzinogenität, Fertilität, Teratogenität und postnatalen Entwicklung lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Eine Studie zur Entwicklungs- und Reproduktionstoxizität an Kaninchen zeigte eine erhöhte embryofetale Resorption und eine höhere Anzahl von Abgängen nach der Einnistung bei mit Setmelanotid behandelten trächtigen Kaninchen. Diese Wirkungen wurden auf eine extreme Reduzierung der Nahrungsaufnahme durch das Muttertier aufgrund der primären pharmakodynamischen Wirkung von Setmelanotid zurückgeführt. In einer Studie zur Entwicklungs- und Reproduktionstoxizität an Ratten wurden keine vergleichbaren Reduzierungen der Nahrungsaufnahme und keine damit zusammenhängenden embryofetalen Abgänge beobachtet. In keiner der Tierarten wurden teratogene Wirkungen beobachtet.
In der Phase vor der Entwöhnung in einer Studie zur prä- und postnatalen Entwicklung bei Ratten wurden 2 Stunden nach der subkutanen Injektion dosisabhängige Setmelanotid-Konzentrationen in der Milch beobachtet. Im Plasma gesäugter Jungtiere wurden bei keiner Dosis quantifizierbare Setmelanotid-Konzentrationen nachgewiesen.
Anders als bei Primaten wurden bei Ratten und Minischweinen variable kardiovaskuläre Wirkungen, wie z. B. eine erhöhte Herzfrequenz und ein erhöhter Blutdruck, beobachtet. Die Gründe für diese Unterschiede zwischen diesen Tierarten sind bisher nicht geklärt. Bei Ratten waren die dosisabhängigen Wirkungen von Setmelanotid auf Herzfrequenz und Blutdruck mit einem erhöhten Tonus des Sympathikus verbunden, und man stellte fest, dass diese Wirkungen bei wiederholter täglicher Gabe immer weiter abnahmen.
Nach langfristiger Anwendung bei erwachsenen Ratten und Affen wurde eine minimale zytoplasmatische Vakuolisierung im Zusammenhang mit dem Hilfsstoff mPEG-DSPE im Plexus choroideus beobachtet. Bei jugendlichen Ratten, die an den Tagen 7 bis 55 nach der Geburt auf einer mg/m2/Tag-Basis mit Setmelanotid/mPEG-DSPE behandelt wurden, dessen Dosis 9.5fach höher war als die humantherapeutische Dosis von mPEG-DSPE aus 3 mg Setmelanotid, wurde keine Vakuolisierung des Plexus choroideus beobachtet.
Die verfügbaren Daten zur Karzinogenität bei Tg.rasH2-Mäusen legen die Vermutung nahe, dass Setmelanotid/mPEG-DSPE bei der klinischen Dosis von 3 mg/Tag kein karzinogenes Risiko für Patienten darstellt, mit einer Sicherheitsspanne von 17 für Setmelanotid basierend auf der AUC und einer Dosisspanne von 16 für mPEG-DSPE auf einer mg/m²/Tag-Basis. Mangels besorgniserregender, auf ein karzinogenes Potenzial hinweisender Anzeichen aus den nichtklinischen und klinischen Daten zu Setmelanotid wurde keine 2-Jahres-Karzinogenitätsstudie bei Ratten durchgeführt.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit „EXP“ bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
28 Tage oder bis zum Verfalldatum (je nachdem, was früher eintritt).
Nicht über 30°C lagern.
Die chemische und physikalische Stabilität der gebrauchsfertigen Zubereitung (siehe «Hinweise für die Handhabung») wurde für 28 Tage bei 2°C bis 30°C nachgewiesen.
Aus mikrobiologischer Sicht kann das Arzneimittel nach Anbruch maximal 28 Tage bei 2°C bis 30°C aufbewahrt werden. Andere Aufbewahrungszeiten und -bedingungen liegen in der Verantwortung des Anwenders.
Besondere Lagerungshinweise
Ungeöffnete Durchstechflasche
Im Kühlschrank (2-8°C) lagern.
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Ungeöffnete Durchstechflaschen können bis zu 30 Tage lang bei Raumtemperatur (maximal 30°C) gelagert werden.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Vorbereitung
IMCIVREE sollte ca. 15 Minuten vor der Anwendung aus dem Kühlschrank genommen werden. Alternativ können Patienten das Arzneimittel vor der Anwendung erwärmen, indem sie die Durchstechflasche 60 Sekunden lang vorsichtig zwischen den Handflächen rollen.
IMCIVREE ist vor jeder Injektion zu überprüfen, und die Lösung darf nicht verwendet werden, wenn sie trüb ist oder Partikel enthält.
Wenn IMCIVREE Temperaturen von >30°C ausgesetzt wurde, ist es zu verwerfen und darf nicht verwendet werden.
Zur Vermeidung von Kontaminationen ist für jede Injektion stets eine neue Spritze zu verwenden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den lokalen Anforderungen zu beseitigen.
PackungenMehrdosen-Durchstechflasche aus Klarglas Typ I mit einem Stopfen aus Brombutyl-Gummi und einer Kappe aus Aluminium.
Packungsgrössen: 1 Mehrdosen-Durchstechflasche (B)
Zulassungsinhaberinpharma services Oehler GmbH, Wollerau
Stand der InformationMärz 2025
|