ZusammensetzungWirkstoffe
Ritlecitinibum (ut Ritlecitinibi Tosilas).
Hilfsstoffe
Inhalt der Hartkapseln: Cellulosum microcristallinum, lactosum monohydricum (21.27 mg), crospovidonum, glyceroli dibehenas.
Hülle der Hartkapseln: Hypromellosum (E 464), titanii dioxidum (E 171), ferrum oxydatum flavum (E 172), Brilliantblau (E 133).
Drucktinte: Lacca, propylenglycolum, ammoniae solutio concentrata, ferrum oxydatum nigrum (E 172), kalii hydroxidum.
Indikationen/AnwendungsmöglichkeitenLitfulo wird angewendet für die Behandlung von schwerer Alopecia areata (≥50% der Kopfhaut betroffen) bei Erwachsenen bis maximal 65 Jahre und Jugendlichen ab einem Alter von 12 Jahren, die für eine systemische Therapie in Frage kommen (siehe «Eigenschaften/Wirkungen, Klinische Wirksamkeit»).
Dosierung/AnwendungDie Behandlung sollte von in der Diagnose und Behandlung von Alopecia areata erfahrenen Dermatologen nach individueller Nutzen-Risiko-Abwägung eingeleitet und überwacht werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Dosierung
Die empfohlene Dosis beträgt 50 mg einmal täglich.
Das Nutzen-Risiko-Verhältnis der Behandlung ist in regelmässigen Abständen auf individueller Basis neu zu bewerten.
Bei Patienten, bei denen nach 36 Behandlungswochen kein therapeutischer Nutzen (mindestens 30% Verbesserung im SALT Score) nachgewiesen werden kann, ist eine Beendigung der Behandlung in Betracht zu ziehen.
Die Kombination mit biologischen Immunmodulatoren, anderen JAK-Inhibitoren, Ciclosporin oder anderen starken Immunsuppressiva wurde nicht untersucht und wird nicht empfohlen.
Überwachung der Laborwerte
Tabelle 1: Laborkontrollen und Leitfaden zur Überwachung
|
Laborkontrollen
|
Routine-Überwachung
|
Massnahme
| |
Thrombozytenzahl
|
Vor Behandlungsbeginn, 4 Wochen nach Behandlungsbeginn und danach im Rahmen der Routineüberwachung des Patienten.
|
Bei Thrombozytenzahlen <50 × 103/mm3 sollte die Behandlung beendet werden.
| |
Lymphozyten
|
Bei einer ALC <0.5 × 103/mm3 sollte die Behandlung unterbrochen werden. Bei einer ALC über diesem Wert kann die Behandlung wieder aufgenommen werden.
|
Abkürzung: ALC = absolute Lymphozytenzahl (absolute lymphocyte count)
Behandlungsbeginn
Bei Patienten mit einer absoluten Lymphozytenzahl (ALC) <0.5 × 103/mm3 oder einer Thrombozytenzahl <100 × 103/mm3 sollte keine Behandlung mit Ritlecitinib eingeleitet werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Behandlungsunterbrechung oder Absetzen der Behandlung
Wenn es bei einem Patienten zu einer schwerwiegenden oder opportunistischen Infektion kommt, sollte die Behandlung mit Ritlecitinib unterbrochen werden, bis die Infektion unter Kontrolle ist (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Eine Unterbrechung oder ein Absetzen der Behandlung kann aufgrund von hämatologischen Laborwertveränderungen erforderlich werden, wie in Tabelle 1 beschrieben.
Wenn eine Unterbrechung der Behandlung erforderlich ist, ist das Risiko eines signifikanten Verlusts von nachgewachsenem Kopfhaar nach einer vorübergehenden Behandlungsunterbrechung von weniger als 6 Wochen gering.
Auslassen einer Dosis
Wenn eine Dosis ausgelassen wurde, sollten Patienten angewiesen werden, die Dosis so bald wie möglich einzunehmen, es sei denn, es bleiben weniger als 8 Stunden bis zur nächsten vorgesehenen Dosis. In diesem Fall sollte die ausgelassene Dosis nicht mehr eingenommen werden. Anschliessend sollte die Einnahme zu den regulär geplanten Zeitpunkten fortgesetzt werden.
Besondere Patientengruppen
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter, mittelschwerer oder schwerer Nierenfunktionsstörung ist keine Dosisanpassung erforderlich (siehe «Pharmakokinetik»).
Ritlecitinib wurde bei Patienten mit terminaler Niereninsuffizienz (endstage renal disease, ESRD) oder Patienten nach Nierentransplantation nicht untersucht und wird daher für die Anwendung bei solchen Patienten nicht empfohlen.
Patienten mit Leberfunktionsstörungen
Bei Patienten mit leichter (Child-Pugh-Klasse A) oder mittelschwerer Leberfunktionsstörung (Child-Pugh-Klasse B) ist keine Dosisanpassung erforderlich (siehe «Pharmakokinetik»). Bei Patienten mit schwerer Leberfunktionseinschränkung (Child-Pugh-Klasse C) ist Ritlecitinib kontraindiziert (siehe «Kontraindikationen»).
Ältere Patienten
Die klinische Erfahrung bei Patienten über 65 Jahren ist sehr begrenzt, so dass diese Patienten nicht mit Ritlecitinib behandelt werden sollten.
Kinder und Jugendliche
Bei Jugendlichen im Alter von 12 bis <18 Jahren ist keine Dosisanpassung erforderlich.
Die Sicherheit und Wirksamkeit von Litfulo bei Kindern unter einem Alter von 12 Jahren ist bisher nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Litfulo wird einmal täglich unabhängig von den Mahlzeiten eingenommen.
Die Kapseln sollten im Ganzen eingenommen werden. Sie sollten nicht zerdrückt, geteilt oder gekaut werden, da solche Arten der Anwendung nicht in klinischen Studien untersucht wurden.
Kontraindikationen·Überempfindlichkeit gegen den Wirkstoff oder einen der in «Zusammensetzung» genannten Hilfsstoffe.
·aktive, schwerwiegende Infektionen, einschliesslich Tuberkulose (TB) (siehe «Warnhinweise und Vorsichtsmassnahmen»).
·schwere Leberfunktionsstörung (siehe «Dosierung/Anwendung»).
·Schwangerschaft und Stillzeit (siehe «Schwangerschaft, Stillzeit»).
Warnhinweise und VorsichtsmassnahmenSchwerwiegende Infektionen
Bei Patienten, die mit Ritlecitinib behandelt wurden, wurden schwerwiegende Infektionen berichtet. Die häufigsten schwerwiegenden Infektionen waren Appendizitis, COVID-19-Infektion (einschliesslich Pneumonie) und Sepsis. Bei Patienten mit einer aktiven schwerwiegenden Infektion darf die Behandlung mit Ritlecitinib nicht eingeleitet werden (siehe «Kontraindikationen»).
Die Risiken und Nutzen der Behandlung sollten abgewogen werden bei Patienten:
·mit chronischer oder rezidivierender Infektion.
·die Tuberkulose (TB) ausgesetzt waren.
·mit schwerwiegender oder opportunistischer Infektion in der Vorgeschichte.
·die sich in Gebieten mit endemischer TB oder endemischen Mykosen aufgehalten oder diese bereist haben.
·mit Grunderkrankungen, die sie für eine Infektion prädisponieren können.
Die Patienten sollten während und nach der Behandlung mit Ritlecitinib engmaschig auf die Entwicklung von Anzeichen und Symptomen einer Infektion überwacht werden. Die Behandlung sollte unterbrochen werden, wenn ein Patient eine schwerwiegende oder opportunistische Infektion entwickelt. Ein Patient, der während der Behandlung mit Ritlecitinib eine neue Infektion entwickelt, sollte umgehend einer für einen immungeschwächten Patienten geeigneten, umfassenden diagnostischen Untersuchung unterzogen werden. Eine geeignete antimikrobielle Therapie sollte eingeleitet und der Patient engmaschig überwacht werden. Wird die Behandlung mit Ritlecitinib unterbrochen, kann sie wieder aufgenommen werden, sobald die Infektion unter Kontrolle ist.
Da bei älteren Menschen und Diabetes-Patienten generell eine höhere Inzidenz für Infektionen vorliegt, ist bei der Behandlung von älteren Menschen und Diabetikern Vorsicht geboten und besondere Aufmerksamkeit auf das Auftreten von Infektionen zu richten.
Tuberkulose
Patienten sollten vor Beginn der Therapie mit Ritlecitinib auf TB untersucht werden. Ritlecitinib darf Patienten mit aktiver TB nicht gegeben werden (siehe «Kontraindikationen»). Bei Patienten mit neu diagnostizierter latenter TB oder zuvor unbehandelter latenter TB sollte vor der Einleitung der Behandlung mit Ritlecitinib eine Anti-TB-Therapie begonnen werden. Bei Patienten mit negativem Test auf latente TB aber hohem Risiko sollte vor Beginn der Behandlung mit Ritlecitinib dennoch eine Anti-TB-Therapie in Betracht gezogen werden. Bei Patienten mit hohem TB-Risiko sollte während der Behandlung mit Ritlecitinib ein TB-Screening erwogen werden.
Virusreaktivierung
Virusreaktivierungen, einschliesslich Fälle der Reaktivierung von Herpes-Viren (z.B. Herpes zoster), wurden berichtet (siehe «Unerwünschte Wirkungen»). Falls ein Patient eine Herpes zoster-Infektion entwickelt, kann eine vorübergehende Unterbrechung der Behandlung in Betracht gezogen werden, bis die Infektion abgeklungen ist.
Vor Beginn der Behandlung mit Ritlecitinib sollte ein Screening auf eine Virushepatitis entsprechend den klinischen Leitlinien durchgeführt werden. Patienten mit Nachweis einer Hepatitis-B- oder -C-Infektion waren aus Studien mit Ritlecitinib ausgeschlossen. Während der Behandlung mit Ritlecitinib wird eine Überwachung auf eine Reaktivierung einer Virushepatitis gemäss den klinischen Leitlinien empfohlen. Bei Anzeichen einer Reaktivierung sollte ein Hepatologe hinzugezogen werden.
Gesamtmortalität
In einer grossen, randomisierten Sicherheitsstudie nach Markteinführung eines anderen JAK-Inhibitors bei Patienten mit rheumatoider Arthritis (RA) 50 Jahre und älter mit mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurde bei Patienten, die mit dem JAK-Inhibitor behandelt wurden, eine höhere Gesamtmortalitätsrate einschliesslich plötzlichem kardiovaskulärem Tod, im Vergleich zu mit TNF-Inhibitoren behandelten Patienten, beobachtet. Ritlecitinib ist für RA nicht indiziert. Wägen Sie Nutzen und Risiken für den einzelnen Patienten ab, bevor Sie eine Therapie mit Ritlecitinib beginnen oder fortsetzen.
Maligne Erkrankungen (einschliesslich nichtmelanozytärer Hautkrebs)
Maligne Erkrankungen, einschliesslich nichtmelanozytärer Hautkrebs (non-melanoma skin cancer, NMSC), wurden bei Patienten berichtet, die mit Ritlecitinib behandelt wurden.
In einer grossen randomisierten, aktiv kontrollierten Studie zu Tofacitinib (einem weiteren JAK-Inhibitor) bei Patienten mit RA ab einem Alter von 50 Jahren mit mindestens einem zusätzlichen kardiovaskulären Risikofaktor, wurde unter Tofacitinib im Vergleich zu Tumornekrosefaktor-Inhibitoren (TNF-Inhibitoren) ein Anstieg der Rate maligner Erkrankungen beobachtet, insbesondere von Lungenkrebs, Lymphom und NMSC. Bei aktuellen oder ehemaligen Rauchern, die mit dem JAK-Inhibitor behandelt wurden, wurde eine höhere Rate an Lungenkrebsfällen beobachtet als bei denjenigen, die mit TNF-Inhibitoren behandelt wurden. In dieser Studie hatten aktuelle oder ehemalige Raucher ein zusätzlich erhöhtes Risiko für maligne Erkrankungen insgesamt.
Vor Beginn oder Fortsetzung der Behandlung von Patienten mit bekannter maligner Erkrankung, ausser erfolgreich behandeltem NMSC oder Zervixkarzinom, sollten Risiken und Nutzen einer Behandlung mit Ritlecitinib abgewogen werden.
Regelmässige Hautuntersuchungen werden bei Patienten mit erhöhtem Risiko für Hautkrebs empfohlen.
Schwerwiegende unerwünschte kardiovaskuläre Ereignisse (Major adverse cardiovascular events, MACE), tiefe Venenthrombose (TVT) und Lungenembolie (LE)
Venöse und arterielle Thromboembolien, einschliesslich MACE, wurden bei Patienten berichtet, die mit Ritlecitinib behandelt wurden.
In einer grossen randomisierten, aktiv kontrollierten Studie zu Tofacitinib (einem weiteren JAK-Inhibitor) bei Patienten mit RA ab einem Alter von 50 Jahren mit mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurde unter Tofacitinib im Vergleich zu TNF-Inhibitoren eine höhere Rate von MACE beobachtet, definiert als kardiovaskuläre Mortalität, nicht tödlicher Myokardinfarkt und nicht tödlicher Schlaganfall, sowie eine dosisabhängige höhere Rate venöser Thromboembolien, einschliesslich TVT und LE. Patienten, die Raucher sind oder waren, haben ein zusätzlich erhöhtes Risiko.
Ritlecitinib sollte bei Patienten mit bekannten Risikofaktoren für Thromboembolien mit Vorsicht angewendet werden. Bei Patienten mit Verdacht auf ein thromboembolisches Ereignis wird empfohlen, die Behandlung mit Ritlecitinib abzusetzen und eine sofortige erneute Untersuchung einzuleiten. Risiken und Nutzen der Behandlung mit Ritlecitinib sollten vor der Einleitung einer Therapie bei Patienten gegeneinander abgewogen werden, insbesondere bei Patienten, die Raucher sind oder waren, und bei Patienten mit anderen kardiovaskulären Risikofaktoren.
Neurologische Ereignisse
Eine durch Ritlecitinib bedingte axonale Dystrophie wurde in Studien zur chronischen Toxizität bei Beagle-Hunden beobachtet (siehe «Präklinische Daten»). Die Behandlung mit Ritlecitinib sollte abgebrochen werden, wenn ungeklärte neurologische Symptome auftreten.
Hämatologische Anomalien
Die Behandlung mit Ritlecitinib wurde mit einer Verringerung der Lymphozyten und Blutplättchen in Verbindung gebracht (siehe «Unerwünschte Wirkungen»). Vor Beginn der Behandlung mit Ritlecitinib sollten die absolute Lymphozytenzahl (absolute lymphocyte count, ALC) und die Thrombozytenzahl bestimmt werden. Bei Patienten mit einer ALC <0.5 × 103/mm3 oder einer Thrombozytenzahl <100 × 103/mm3 sollte die Behandlung mit Ritlecitinib nicht eingeleitet werden. Nach Beginn der Behandlung mit Ritlecitinib wird bei Anomalien der ALC und der Thrombozytenzahl eine Unterbrechung oder Beendigung der Behandlung empfohlen (siehe «Dosierung/Anwendung»). Es wird empfohlen, die ALC und die Thrombozytenzahl 4 Wochen nach Beginn der Therapie mit Ritlecitinib und danach entsprechend der Routineversorgung des Patienten zu bestimmen.
Impfungen
Es liegen keine Daten zum Ansprechen auf Impfungen bei Patienten unter Behandlung mit Ritlecitinib vor. Die Anwendung von attenuierten Lebendimpfstoffen sollte während oder unmittelbar vor der Behandlung mit Ritlecitinib vermieden werden. Vor Beginn der Behandlung mit Ritlecitinib wird empfohlen, bei den Patienten alle Immunisierungen in Übereinstimmung mit den geltenden Impfempfehlungen auf den aktuellen Stand zu bringen. Dies gilt auch für prophylaktische Herpes-zoster-Impfungen.
Ältere Patienten
Die klinische Erfahrung bei Patienten über 65 Jahren ist sehr begrenzt, so dass diese Patienten nicht mit Ritlecitinib behandelt werden sollten.
Hilfsstoffe von besonderem Interesse
Lactose
Patienten mit der seltenen hereditären Galactose-Intoleranz, völligem Lactasemangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht anwenden.
InteraktionenEinfluss anderer Substanzen auf die Pharmakokinetik von Ritlecitinib
Andere Interaktionen
Die Metabolisierung von Ritlecitinib erfolgt über mehrere Stoffwechselwege, wobei kein einzelner Clearance-Weg mehr als 25% beiträgt (siehe «Pharmakokinetik»). Daher ist es unwahrscheinlich, dass Arzneimittel, die einen selektiven Stoffwechselweg hemmen, die systemische Exposition von Ritlecitinib beeinflussen. Es ist auch unwahrscheinlich, dass spezifische Transporter-Inhibitoren zu klinisch relevanten Veränderungen der Bioverfügbarkeit von Ritlecitinib führen.
CYP3A-Inhibitoren: Die gleichzeitige Anwendung mehrerer 200 mg-Dosen von Itraconazol, einem starken CYP3A-Inhibitor, erhöhte die Fläche unter der Kurve (area under the curve, AUC)inf von Ritlecitinib um etwa 15%. Dies wird als klinisch nicht signifikant angesehen, sodass eine Dosisanpassung bei gleichzeitiger Anwendung von Ritlecitinib und CYP3A-Inhibitoren nicht erforderlich ist.
CYP-Induktoren: Die gleichzeitige Anwendung mehrerer 600 mg-Dosen von Rifampicin, einem starken Induktor von CYP-Enzymen, verringerte die AUCinf von Ritlecitinib um etwa 44%. Dies wird als klinisch nicht signifikant angesehen, sodass eine Dosisanpassung bei gleichzeitiger Anwendung von Ritlecitinib und Induktoren von CYP-Enzymen nicht erforderlich ist.
In vitro ist Ritlecitinib ein Substrat des P-Glykoproteins (Pgp) und von BCRP. Da Ritlecitinib jedoch eine hohe absorbierte Fraktion (fa) aufweist mit einem Anstieg von sowohl Cmax als auch AUC in dosisproportionaler Weise (20-200 mg Einzeldosisbereich), dürften Pgp und BCRP keinen bedeutenden Einfluss auf die Resorption von Ritlecitinib haben.
Einfluss von Ritlecitinib auf die Pharmakokinetik anderer Substanzen
Andere Interaktionen
Ritlecitinib ist ein kovalenter Inhibitor, der nachweislich an Off-Target-Proteine wie MAP2K7, DOCK10, Albumin, CYP1A2, CYP3A, UGT1A1 und UGT1A4 bindet, von denen einige für Arzneimittelwechselwirkungen klinisch relevant sein könnten.
CYP3A-Substrate: Die mehrfache Anwendung von 200 mg Ritlecitinib einmal täglich erhöhten die AUCinf bzw. Cmax von Midazolam, einem CYP3A4-Substrat, etwa um das 2.7- bzw. 1.8fache. Ritlecitinib ist ein mässig starker Inhibitor von CYP3A. Vorsicht ist geboten bei gleichzeitiger Anwendung von Ritlecitinib mit CYP3A-Substraten (z.B. Chinidin, Ciclosporin, Dihydroergotamin, Ergotamin, Pimozid), bei denen mässig starke Konzentrationsveränderungen zu schwerwiegenden Nebenwirkungen führen können. Empfehlungen zur Dosisanpassung für das CYP3A-Substrat (z.B. Colchicin, Everolimus, Tacrolimus, Sirolimus) sollten beachtet werden.
CYP1A2-Substrate: Die mehrfache Anwendung von 200 mg Ritlecitinib einmal täglich erhöhten die AUCinf bzw. Cmax von Koffein, einem CYP1A2-Substrat, etwa um das 2.7- bzw. 1.1fache. Ritlecitinib ist ein mässig starker Inhibitor von CYP1A2. Vorsicht ist geboten bei gleichzeitiger Anwendung von Ritlecitinib mit anderen CYP1A2-Substraten (z.B. Tizanidin), bei denen mässig starke Konzentrationsveränderungen zu schwerwiegenden Nebenwirkungen führen können. Empfehlungen zur Dosisanpassung für das CYP1A2-Substrat (z.B. Theophyllin, Pirfenidon) sollten beachtet werden.
OCT1-Substrate: Die gleichzeitige Anwendung einer Einzeldosis von 400 mg Ritlecitinib erhöhte die AUCinf von Sumatriptan (einem organischen Kationentransporter organic cation transporter, [OCT]1-Substrat) etwa um das 1.3- bis 1.5fache im Vergleich zu einer allein angewendeten Sumatriptan-Dosis. Der Anstieg der Sumatriptan-Exposition wird als klinisch nicht relevant angesehen. Vorsicht ist geboten bei gleichzeitiger Anwendung von Ritlecitinib mit OCT1-Substraten, bei denen geringe Konzentrationsveränderungen zu schwerwiegenden Nebenwirkungen führen können.
Ritlecitinib führte nicht zu klinisch signifikanten Veränderungen der Exposition gegenüber oralen Kontrazeptiva (z.B. Ethinylestradiol oder Levonorgestrel), CYP2B6-Substraten (z.B. Efavirenz), CYP2C-Substraten (z.B. Tolbutamid) oder Substraten des organischen Anionentransporters (OAT) P1B1, des Breast Cancer Resistance-Proteins (BCRP) oder von OAT3 (z.B. Rosuvastatin).
Kinder und Jugendliche
Studien zur Erfassung von Wechselwirkungen wurden nur bei Erwachsenen durchgeführt.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Ritlecitinib wird bei Frauen im gebärfähigen Alter, die keine Verhütungsmethode anwenden, nicht empfohlen. Frauen im gebärfähigen Alter müssen während der Behandlung und für 1 Monat nach der letzten Dosis Litfulo eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Es liegen keine oder nur begrenzte Daten zur Anwendung von Ritlecitinib bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe «Präklinische Daten»). Litfulo ist während der Schwangerschaft kontraindiziert (siehe «Kontraindikationen»).
Stillzeit
Verfügbare Daten zur Pharmakodynamik/Toxikologie aus tierexperimentellen Studien zeigen einen Übergang von Ritlecitinib in die Muttermilch (siehe «Präklinische Daten»). Ein Risiko für Neugeborene/Säuglinge kann nicht ausgeschlossen werden. Litfulo ist während der Stillzeit kontraindiziert (siehe «Kontraindikationen»).
Fertilität
Die Wirkung von Ritlecitinib auf die Fertilität beim Menschen wurde nicht untersucht. In tierexperimentellen Studien wurden bei klinisch relevanten Expositionen keine Auswirkungen auf die Fertilität festgestellt (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine entsprechenden Studien durchgeführt. Litfulo kann Schwindelgefühle hervorrufen. Der klinische Zustand des Patienten und das Nebenwirkungsprofil von Litfulo sollten bei der Beurteilung der Fähigkeit des Patienten, ein Fahrzeug zu führen oder Maschinen zu bedienen, in Betracht gezogen werden.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Insgesamt wurden 1'630 Patienten mit Ritlecitinib behandelt, was einer Exposition von 2'303 Patientenjahren entspricht. Drei placebokontrollierte Studien (130 Teilnehmende mit 50 mg täglich und 213 Teilnehmende mit Placebo) wurden zur Bewertung der Sicherheit von Ritlecitinib im Vergleich zu Placebo für bis zu 24 Wochen ab Behandlungsbeginn herangezogen.
Die am häufigsten gemeldeten unerwünschten Wirkungen sind Diarrhoe (9.2%), Akne (6.2%), Infektionen der oberen Atemwege (6.2%), Urtikaria (4.6%), Ausschlag (3.8%), Follikulitis (3.1%) und Schwindelgefühl (2.3%).
Liste der unerwünschten Wirkungen
Alle in Studien zu Alopecia areata beobachteten unerwünschten Wirkungen sind hier nach Systemorganklasse und Häufigkeit aufgeführt. Die Häufigkeitskategorien sind nach folgender Konvention definiert: sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1'000, <1/100), selten (≥1/10'000, <1/1'000) und sehr selten (<1/10'000). Innerhalb jeder Häufigkeitskategorie sind die unerwünschten Wirkungen nach abnehmendem Schweregrad angegeben.
Infektionen und parasitäre Erkrankungen
Häufig: Herpes zoster, Follikulitis, Infektionen der oberen Atemwege.
Erkrankungen des Nervensystems
Häufig: Schwindelgefühl.
Erkrankungen des Gastrointestinaltrakts
Häufig: Diarrhoe.
Erkrankungen der Haut und des Unterhautgewebes
Häufig: Akne, Urtikaria, Ausschlag.
Untersuchungen
Häufig: Kreatinphosphokinase im Blut erhöht.
Gelegentlich: Thrombozytenzahl erniedrigt, Lymphozytenzahl erniedrigt, Alaninaminotransferase erhöht auf >3 × ULNa, Aspartataminotransferase erhöht auf >3 × ULNa.
a Beinhaltet bei routinemässigen Laborkontrollen gemessene Veränderungen
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Infektionen
In den placebokontrollierten Studien wurden über einen Zeitraum bei insgesamt von bis zu 24 Wochen bei 31% der mit Placebo behandelten Patienten (80.35 pro 100 Patientenjahre) und bei 33% der mit Ritlecitinib 50 mg behandelten Patienten (74.53 pro 100 Patientenjahre) Infektionen gemeldet. In der Studie AA-I wurden über einen Zeitraum von bis zu 48 Wochen bei 51% der Patienten (89.32 pro 100 Patientenjahre), die mit Ritlecitinib in einer Dosis von 50 mg oder höher behandelt wurden, Infektionen gemeldet.
Von allen Patienten, die in der integrierten Sicherheitsanalyse mit Ritlecitinib behandelt wurden, einschliesslich einer Langzeitstudie und einer Studie zu Vitiligo, wurden bei 45.4% der Patienten (50.02 pro 100 Patientenjahre), die mit Ritlecitinib in einer Dosis von 50 mg oder höher behandelt wurden, Infektionen gemeldet. Die meisten Infektionen hatten einen leichten oder mittleren Schweregrad.
In den placebokontrollierten Studien lag der Prozentsatz der Patienten, die über infektionsbedingte Nebenwirkungen von Herpes zoster berichteten, bei 1.5% in der Gruppe mit 50 mg Ritlecitinib im Vergleich zu 0 in der Placebogruppe. Keines der Herpes-zoster-Ereignisse war schwerwiegend. Bei einem Patienten, der Ritlecitinib 200/50 mg erhielt (200 mg einmal täglich für 4 Wochen, gefolgt von 50 mg einmal täglich), trat eine Varizella-zoster-Virusinfektion auf, welche die Kriterien für eine opportunistische Infektion erfüllte (Herpes zoster multipler Dermatome). In der Studie AA-I berichteten 2.3% der Patienten (2.61 pro 100 Patientenjahre), die mit Ritlecitinib in einer Dosis von 50 mg oder höher über einen Zeitraum von bis zu 48 Wochen behandelt wurden, über Herpes zoster. Bei allen Patienten, die in der integrierten Sicherheitsanalyse mit Ritlecitinib behandelt wurden, einschliesslich einer Langzeitstudie und einer Studie zu Vitiligo, lag die Rate von Herpes zoster bei 1.10 pro 100 Patientenjahre bei Patienten, die mit Ritlecitinib in einer Dosis von 50 mg oder höher behandelt wurden.
In den placebokontrollierten Studien wurden über einen Zeitraum von bis zu 24 Wochen keine schwerwiegenden Infektionen bei Patienten gemeldet, die mit Placebo oder Ritlecitinib 50 mg behandelt wurden. Der Anteil und die Rate schwerwiegender Infektionen bei Patienten, die 200/50 mg Ritlecitinib erhielten, lag bei 0.9% (2.66 pro 100 Patientenjahre). In der Studie AA-I wurden über einen Zeitraum von bis zu 48 Wochen bei 0.8% der Patienten (0.86 pro 100 Patientenjahre), die mit Ritlecitinib in einer Dosis von 50 mg oder höher behandelt wurden, schwerwiegende Infektionen gemeldet. Bei allen Patienten der integrierten Sicherheitsanalyse, einschliesslich einer Langzeitstudie und einer Studie zu Vitiligo, die mit Ritlecitinib behandelt wurden, lagen der Anteil und die Rate schwerwiegender Infektionen unter Ritlecitinib in einer Dosis von 50 mg oder höher bei 0.8% (0.59 pro 100 Patientenjahre).
Opportunistische Infektionen
Opportunistische Infektionen eines multidermatomalen Herpes zoster wurden bei 1 Patienten (0.50 pro 100 Patientenjahre), der in den placebokontrollierten Studien mit Ritlecitinib 200/50 mg behandelt wurde, bei keinem Patienten in der Studie AA-I über einen Zeitraum von bis zu 48 Wochen und bei 2 Patienten (0.09 pro 100 Patientenjahre) der integrierten Sicherheitsanalyse, einschliesslich der Langzeitstudie und einer Studie bei Vitiligo, die mit Ritlecitinib in einer Dosis von 50 mg oder höher behandelt wurden, gemeldet. Die Fälle von opportunistischem Herpes zoster waren von einem leichten oder mittelschweren Schweregrad.
Verringerung der Lymphozytenzahl
In den placebokontrollierten Studien über einen Zeitraum von bis zu 24 Wochen und in der Studie AA-I über einen Zeitraum von bis zu 48 Wochen ging die Behandlung mit Ritlecitinib mit einer Verringerung der Lymphozytenzahl einher. Die stärkste Wirkung auf die Lymphozyten wurde innerhalb von 4 Wochen beobachtet, danach blieb die Lymphozytenzahl bei fortgesetzter Therapie auf einem niedrigeren Niveau stabil. Unter allen Patienten der integrierten Sicherheitsanalyse, die mit Ritlecitinib behandelt wurden, einschliesslich der Langzeitstudie und einer Studie bei Vitiligo, trat bei 2 Teilnehmenden (<0.1%), die mit Ritlecitinib 50 mg behandelt wurden, eine bestätigte ALC <0.5 × 103/mm3 auf.
Verringerung der Thrombozytenzahl
In den placebokontrollierten Studien über einen Zeitraum von bis zu 24 Wochen und in der Studie AA-I über einen Zeitraum von bis zu 48 Wochen ging die Behandlung mit Ritlecitinib mit einer Verringerung der Thrombozytenzahl einher. Die stärkste Wirkung auf die Thrombozyten wurde innerhalb von 4 Wochen beobachtet, danach blieb die Thrombozytenzahl bei fortgesetzter Therapie auf einem niedrigeren Niveau stabil. Unter allen Patienten der integrierten Sicherheitsanalyse, die mit Ritlecitinib behandelt wurden, einschliesslich der Langzeitstudie und einer Studie bei Vitiligo, trat bei 1 Patienten (<0.1%), der mit Ritlecitinib in einer Dosis von 50 mg oder höher behandelt wurde, eine bestätigte Thrombozytenzahl von <100 × 103/mm3 auf.
Erhöhung der Kreatinphosphokinase (creatine phosphokinase, CPK)
In den placebokontrollierten Studien mit bis zu 24 Wochen Dauer wurden bei 2 Patienten (1.5%), die mit Ritlecitinib 50 mg behandelt wurden, Ereignisse mit erhöhter CPK im Blut gemeldet. In der Studie AA-I mit einer Dauer von bis zu 48 Wochen wurde bei 3.8% der Patienten, die mit Ritlecitinib in einer Dosis von 50 mg oder höher behandelt wurden, über Ereignisse mit erhöhter CPK im Blut berichtet. Bei 2 mit Placebo behandelten Patienten (0.9%) und bei 5 Patienten (3.9%), die 50 mg Ritlecitinib erhielten, kam es zu CPK-Erhöhungen über das 5fache der oberen Normgrenze (>5 x upper limit of normal, ULN). In Studie AA-I mit einer Dauer von bis zu 48 Wochen wurde bei 6.6% der Patienten, die mit Ritlecitinib in einer Dosis von 50 mg oder höher behandelt wurden, über CPK-Erhöhungen >5 x ULN berichtet. Die Erhöhungen waren in den meisten Fällen vorübergehend, und keine der Erhöhungen erforderte ein Absetzen der Behandlung.
Erhöhung der Transaminasen
In den placebokontrollierten Studien über einen Zeitraum von bis zu 24 Wochen wurden bei 3 Patienten (0.9%) bzw. 2 Patienten (0.6%), die mit Ritlecitinib in einer Dosis von 50 mg oder höher behandelt wurden, Erhöhungen der ALT- bzw. AST-Werte (>3 × ULN) gemeldet. Die meisten Erhöhungen waren vorübergehend, und keine führte zum Absetzen der Behandlung.
Pädiatrische Population
Insgesamt 181 Jugendliche (im Alter von 12 bis unter 18 Jahren) wurden in Studien zu Ritlecitinib bei Alopecia areata aufgenommen.
Das bei Jugendlichen beobachtete Sicherheitsprofil war dem der erwachsenen Population ähnlich.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungRitlecitinib wurde in placebokontrollierten Studien bis zu einer peroralen Einzeldosis von 800 mg und peroralen Mehrfachdosen von 400 mg täglich über einen Zeitraum von 14 Tagen angewendet. Es wurden keine spezifischen Toxizitäten erkannt.
Im Falle einer Überdosierung wird empfohlen, den Patienten auf Anzeichen und Symptome von Nebenwirkungen zu überwachen (siehe «Unerwünschte Wirkungen»). Es gibt kein spezifisches Antidot für eine Überdosierung mit Ritlecitinib. Die Behandlung sollte symptomatisch und unterstützend erfolgen.
Pharmakokinetische Daten zu einer peroralen Einzeldosis bis einschliesslich 800 mg bei gesunden erwachsenen Probanden deuten darauf hin, dass mehr als 90% der angewendeten Dosis voraussichtlich innerhalb von 48 Stunden ausgeschieden werden.
Eigenschaften/WirkungenATC-Code
L04AF08.
Wirkungsmechanismus
Ritlecitinib hemmt irreversibel und selektiv die Januskinase (JAK) 3 und die Tyrosinkinase, die auch in der Familie der hepatozellulären Karzinome (TEC) exprimiert wird, indem es die Adenosintriphosphat (ATP)-Bindungsstelle blockiert. In zellulären Versuchsansätzen hemmt Ritlecitinib spezifisch die Signaltransduktion der Zytokine mit γc-Kette (common γ, γc, IL-2, IL-4, IL-7, IL-15 und IL-21) über JAK3-abhängige Rezeptoren für γc-Kette. Darüber hinaus hemmt Ritlecitinib die Kinasen der TEC-Familie, was zu einer verringerten zytolytischen Aktivität von NK-Zellen und CD8+ T-Zellen führt.
Sowohl die JAK3- als auch die über die TEC-Familie vermittelten Signalwege sind an der Pathogenese der Alopecia areata beteiligt, wenn auch die vollständige Pathophysiologie noch nicht verstanden ist.
Pharmakodynamik
Lymphozyten-Untergruppen
Bei Patienten mit Alopecia areata war die Behandlung mit Ritlecitinib mit einem dosisabhängigen frühen Rückgang der absoluten Lymphozytenzahlen sowie der T-Lymphozyten (CD3) und der Untergruppen von T-Lymphozyten (CD4 und CD8) verbunden. Nach dem anfänglichen Rückgang erholten sich die Werte teilweise und blieben bis zu 48 Wochen lang stabil. Eine Veränderung der B-Lymphozyten (CD19) wurde in keiner Behandlungsgruppe beobachtet. Bei den NK-Zellen (CD16/56) kam es zu einer dosisabhängigen frühen Verringerung, die bis zur 48. Woche auf dem niedrigeren Niveau stabil blieb.
Immunglobuline
Bei Patienten mit Alopecia areata ging die Behandlung mit Ritlecitinib bis zur 48. Woche nicht mit klinisch bedeutsamen Veränderungen der Immunglobuline (Ig)G, IgM oder IgA einher, was auf eine fehlende systemische humorale Immunsuppression hinweist.
Elektrophysiologie des Herzens
Bei der 12-fachen mittleren maximalen Exposition der 50-mg-Dosis einmal täglich bei Patienten mit Alopecia areata gab es keine klinisch relevante Wirkung auf das QTc-Intervall.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von Ritlecitinib wurden in einer zulassungsrelevanten, randomisierten, doppelblinden, placebokontrollierten Studie (Studie AA-I) bei Patienten mit Alopecia areata ab einem Alter von 12 Jahren mit ≥50% Kopfhaarverlust, einschliesslich Alopecia totalis und Alopecia universalis, untersucht. Das Dosisansprechen auf Ritlecitinib wurde in dieser Studie ebenfalls untersucht. Der Behandlungszeitraum der Studie bestand aus einer placebokontrollierten Phase von 24 Wochen und einer Verlängerungsphase von weiteren 24 Wochen. In Studie AA-I wurden insgesamt 718 Patienten untersucht, die für 48 Wochen auf eines der folgenden Behandlungsschemata randomisiert wurden: 1) 200 mg einmal täglich für 4 Wochen, gefolgt von 50 mg einmal täglich für 44 Wochen; 2) 200 mg einmal täglich für 4 Wochen, gefolgt von 30 mg einmal täglich für 44 Wochen; 3) 50 mg einmal täglich für 48 Wochen; 4) 30 mg einmal täglich für 48 Wochen; 5) 10 mg einmal täglich für 48 Wochen; 6) Placebo für 24 Wochen, gefolgt von 200 mg einmal täglich für 4 Wochen und 50 mg einmal täglich für 20 Wochen; 7) Placebo für 24 Wochen, gefolgt von 50 mg für 24 Wochen.
In dieser Studie wurde als primärer Endpunkt der Anteil der Patienten bewertet, die in Woche 24 einen SALT-Score (Severity of Alopecia Tool) von ≤10 erreichten (d.h. mindestens 90%ige Bedeckung der Kopfhaut mit Haaren). Darüber hinaus wurden in dieser Studie als wichtiger sekundärer Endpunkt das Ansprechen in Bezug auf die «Patient's Global Impression of Change» (PGI-C) in Woche 24 und ausserdem als sekundärer Endpunkt ein SALT-Score von ≤20 (d.h. mindestens 80%ige Bedeckung der Kopfhaut mit Haaren) in Woche 24 und Verbesserungen des Nachwachsens von Augenbrauen und/oder Wimpern in Woche 24 ermittelt.
Patientencharakteristika bei Baseline
In Studie AA-I wurden Patienten und Patientinnen ab einem Alter von 12 Jahren untersucht. Alle Patienten hatten eine Alopecia areata mit ≥50% Kopfhaarverlust (SALT-Score ≥50) ohne Anzeichen eines Nachwachsens von Terminalhaar in den vorhergehenden 6 Monaten. Die aktuelle Episode des Kopfhaarverlusts bestand seit ≤10 Jahren und es war keine andere Ursache für den Haarausfall bekannt (z.B. androgenetische Alopezie).
Über alle Behandlungsgruppen hinweg waren 62.1% der Teilnehmenden weiblich, 68.0% waren Kaukasier, 25.9% Asiaten und 3.8% waren schwarz oder afroamerikanisch. Das mittlere Alter der Patienten lag bei 33.7 Jahren und die meisten Patienten (85.4%) waren Erwachsene (im Alter von ≥18 Jahren). Insgesamt 105 Patienten (14.6%) im Alter von 12 bis <18 Jahren und 20 Patienten (2.8%) im Alter ab 65 Jahren wurden eingeschlossen. Der mittlere (SD) absolute SALT-Score bei Baseline lag zwischen 88.3 (16.87) und 93.0 (11.50) über alle Behandlungsgruppen hinweg. Der mittlere SALT-Score bei Patienten ohne Alopecia totalis/Alopecia universalis bei Baseline lag zwischen 78.3 und 87.0. Über alle Behandlungsgruppen hinweg hatten die meisten Patienten bei Baseline anormale Augenbrauen (83.0%) und Wimpern (74.7%). Die mediane Dauer seit Diagnose der Alopecia areata betrug 6.9 Jahre und die mediane Dauer der aktuellen Alopecia-areata-Episode 2.5 Jahre. Die Randomisierung wurde nach dem Status der Alopecia totalis/Alopecia universalis stratifiziert, wobei 46% der Patienten mit einem Baseline-SALT-Score von 100 als Patienten mit Alopecia totalis/Alopecia universalis eingestuft wurden.
Klinisches Ansprechen
Ein signifikant grösserer Anteil an Patienten erreichte unter Ritlecitinib 50 mg im Vergleich zu Placebo in Woche 24 ein Ansprechen im Sinne eines SALT-Scores von ≤10 (siehe Tabelle 2). Die Ansprechrate im Sinne eines SALT-Scores ≤10 unter Ritlecitinib 50 mg stieg in Woche 48 weiter an (siehe Abbildung 1).
Ein signifikant grösserer Anteil an Patienten erreichte unter Ritlecitinib 50 mg im Vergleich zu Placebo in Woche 24 ein PGI-C-Ansprechen (Patient's Global Impression of Change, siehe Tabelle 2), und die Ansprechrate nahm bis Woche 48 weiter zu (siehe Abbildung 1).
Ein signifikant grösserer Anteil an Patienten erreichte unter Ritlecitinib 50 mg im Vergleich zu Placebo in Woche 24 ein Ansprechen im Sinne eines SALT-Scores von ≤20 (siehe Tabelle 2). Die Ansprechrate im Sinne eines SALT-Scores ≤20 stieg in Woche 48 weiter an.
Eine Verbesserung des Nachwachsens von Augenbrauen und/oder Wimpern wurde in Woche 24 (siehe Tabelle 2) mit Ritlecitinib 50 mg bei Patienten mit anormalen Augenbrauen und/oder Wimpern bei Baseline beobachtet. Ein weiterer Anstieg zeigte sich in Woche 48.
Die Behandlungseffekte in Woche 24 in den Subgruppen (Alter, Geschlecht, ethnische Herkunft, Region, Gewicht, Dauer der Erkrankung seit der Diagnose, Dauer der aktuellen Episode, vorherige pharmakologische Behandlung) waren mit den Ergebnissen in der Gesamtpopulation der Studie konsistent. Die Behandlungseffekte in Woche 24 in der Subgruppe mit Alopecia totalis/Alopecia universalis waren geringer als in der Subgruppe mit Nicht-Alopecia totalis/Nicht-Alopecia universalis. Die Behandlungseffekte in Woche 24 bei Jugendlichen im Alter von 12 bis unter 18 Jahren waren mit den Ergebnissen in der Gesamtpopulation der Studie konsistent.
Tabelle 2: Wirksamkeitsergebnisse von Ritlecitinib in Woche 24
|
Endpunkt
|
Ritlecitinib 50 mg einmal täglich
(n = 130)
% Responder
|
Placebo
(n = 131)
% Responder
|
Differenz zu
Placebo
(95%-KI)
| |
SALT-Ansprechen ≤10a,b
|
13.4
|
1.5
|
11.9
(5.4; 18.3)
| |
PGI-C-Ansprechenb,c
|
49.2
|
9.2
|
40.0
(28.9; 51.1)
| |
SALT-Ansprechen ≤20d,e
|
23.0
|
1.6
|
21.4
(13.4; 29.5)
| |
EBA-Ansprechenf
|
29.0
|
4.7
|
24.3
(14.8; 34.5)
| |
ELA-Ansprecheng
|
28.9
|
5.2
|
23.7
(13.6; 34.5)
|
Abkürzungen: EBA = Bewertung der Augenbrauen (eyebrow assessment), ELA = Bewertung der Wimpern (eyelash assessment), KI = Konfidenzintervall, n = Gesamtanzahl Patienten, PGI-C = Patient's Global Impression of Change (allgemeine Einschätzung der Veränderung durch den Patienten), SALT = Severity of Alopecia Tool (Skala zur Bewertung des Schweregrads der Alopezie)
a.Als Responder mit SALT ≤10 galten Patienten mit einem Kopfhaarverlust von ≤10%. Die Skala der SALT-Scores reicht von 0 bis 100, wobei 0 = kein Kopfhaarverlust und 100 = vollständiger Kopfhaarverlust bedeutet.
b.Statistisch signifikant mit Adjustierung für Mehrfachtestung
c.Als PGI-C-Responder galten Patienten mit einer Bewertung von «mässig verbessert» oder «stark verbessert» auf einer 7-Punkte-Skala von «stark verbessert» bis «stark verschlechtert».
d.Als Responder mit SALT ≤20 galten Patienten mit einem Kopfhaarverlust von ≤20%. Die Skala der SALT-Scores reicht von 0 bis 100, wobei 0 = kein Kopfhaarverlust und 100 = vollständiger Kopfhaarverlust bedeutet.
e.Statistisch signifikant
f.Ein EBA-Ansprechen wurde definiert als eine Verbesserung um mindestens 2 Stufen gegenüber Baseline oder ein normaler EBA-Score auf einer 4-Punkte-Skala bei Patienten mit anormalen Augenbrauen bei Baseline.
g.Ein ELA-Ansprechen wurde definiert als eine Verbesserung um mindestens 2 Stufen gegenüber Baseline oder ein normaler ELA-Score auf einer 4-Punkte-Skala bei Patienten mit anormalen Wimpern bei Baseline.
Abbildung 1. SALT ≤10 und PGI-C-Ansprechen bis Woche 48
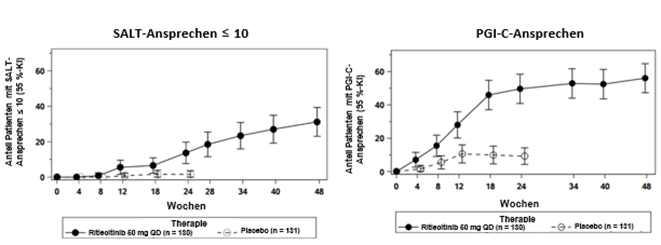
Abkürzungen: KI = Konfidenzintervall, n = Gesamtanzahl Patienten, PGI-C = Patient's Global Impression of Change (allgemeine Einschätzung der Veränderung durch den Patienten), QD = einmal täglich, SALT = Severity of Alopecia Tool (Skala zur Bewertung des Schweregrads der Alopezie)
PharmakokinetikAbsorption
Die absolute orale Bioverfügbarkeit von Ritlecitinib liegt bei etwa 64%. Bei oraler und intravenöser Verabreichung des markierten Wirkstoffs betrug die relative Wiederfindungsrate von markierten Bestandteilen im Urin (oral/intravenös) etwa 89%, was auf eine hohe absorbierte Fraktion (fa) hinweist. Nach oraler Verabreichung von Mehrfachdosen wird die Spitzenkonzentration im Plasma innerhalb von 1 Stunde erreicht. Nahrungsmittel haben keinen klinisch signifikanten Einfluss auf das Ausmass der Resorption von Ritlecitinib, da eine fettreiche Mahlzeit die Cmax von Ritlecitinib um etwa 32% verringerte und die AUCinf um etwa 11% erhöhte. In placebokontrollierten Studien wurde Ritlecitinib unabhängig von den Mahlzeiten angewendet (siehe «Dosierung/Anwendung»).
Distribution
Nach intravenöser Anwendung beträgt das Verteilungsvolumen von Ritlecitinib etwa 74 l. Ungefähr 14% des zirkulierenden Ritlecitinibs werden an Plasmaproteine gebunden, und zwar primär an Albumin. Die Blut-/Plasma-Verteilungsrate von Ritlecitinib beträgt 1.62.
Metabolismus
Der Metabolismus von Ritlecitinib wird durch mehrere Isoformen der Glutathion-S-Transferasen (GST: zytosolische GST A1/3, M1/3/5, P1, S1, T2, Z1 und mikrosomale Membranproteine, die am Eicosanoid- und Glutathion-Stoffwechsel beteiligt sind [membrane-associated proteins in eicosanoid and glutathione metabolism, MAPEG]1/2/3) und über CYP-Enzyme (CYP3A, CYP2C8, CYP1A2 und CYP2C9) vermittelt, wobei kein einzelner Clearance-Weg zu mehr als 25% beiträgt.
In einer Humanstudie mit radioaktiver Markierung war Ritlecitinib nach oraler Gabe die vorherrschende zirkulierende Substanz (30.4% der zirkulierenden Radioaktivität), mit einem Cystein-Konjugat als pharmakologisch inaktivem Hauptmetaboliten M2 (16.5%).
Elimination
Ritlecitinib wird hauptsächlich durch metabolische Clearance-Mechanismen eliminiert, wobei etwa 4% der Dosis als unveränderter Wirkstoff im Urin ausgeschieden werden. Etwa 66% der radioaktiv markierten Ritlecitinib-Dosis werden im Urin und 20% in den Fäzes ausgeschieden. Nach peroraler Mehrfachdosierung wurde der Steady-State aufgrund der nichtstationären PK ungefähr an Tag 4 erreicht. Die Steady-State-PK-Parameter AUCtau und Cmax schienen bis zu einer Dosis von 200 mg ungefähr dosisproportional anzusteigen. Die mittlere terminale Halbwertszeit lag zwischen 1.3 und 2.3 Stunden.
Kinetik spezieller Patientengruppen
Körpergewicht, Geschlecht, Genotyp, ethnische Herkunft und Alter
Körpergewicht, Geschlecht, GST-P1-, -M1- und -T1-Genotyp, ethnische Herkunft und Alter hatten keine klinisch bedeutsamen Auswirkungen auf die Ritlecitinib-Exposition.
Jugendliche (≥12 bis <18 Jahre)
Populationspharmakokinetische Analysen zeigten keinen klinisch relevanten Unterschied der Ritlecitinib-Exposition bei jugendlichen Patienten im Vergleich zu Erwachsenen.
Kinder (<12 Jahre)
Die PK von Ritlecitinib bei Kindern unter einem Alter von 12 Jahren ist bisher noch nicht erwiesen.
Nierenfunktionsstörungen
Bei Patienten mit schwerer Nierenfunktionsstörung (geschätzte glomeruläre Filtrationsrate [estimated glomerular filtration rate, eGFR] <30 ml/min) lagen AUC24 und Cmax um etwa 55% bzw. 44% höher als bei entsprechenden Teilnehmenden mit normaler Nierenfunktion. Dies wurde in einer populationspharmakokinetischen (popPK) Analyse bestätigt. Diese Unterschiede werden als nicht klinisch relevant eingestuft. Ritlecitinib wurde bei Patienten mit leichter (eGFR 60 bis <90 ml/min) oder mittelschwerer (eGFR 30 bis <60 ml/min) Nierenfunktionsstörung nicht untersucht. Die eGFR und die Klassifikation des Nierenfunktionsstatus der Teilnehmenden wurden anhand der MDRD-Formel (Modification of Diet in Renal Disease) berechnet.
Leberfunktionsstörungen
Bei Patienten mit mittelschwerer Leberfunktionsstörung (Child-Pugh-Klasse B) wurde ein Anstieg der AUC24 von Ritlecitinib um 18.5% im Vergleich zu Patienten mit normaler Leberfunktion gemessen. Ritlecitinib wurde bei Patienten mit leichter Leberfunktionsstörung (Child-Pugh-Klasse A) nicht untersucht. Aufgrund der Ergebnisse bei Patienten mit mittelschwerer Leberfunktionsstörung ist ein klinisch signifikanter Anstieg der Ritlecitinib-Exposition bei diesen Patienten jedoch nicht zu erwarten. Ritlecitinib wurde bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh-Klasse C) nicht untersucht (siehe «Kontraindikationen»).
Präklinische DatenAllgemeine Toxizität
Verminderte Lymphozytenzahlen sowie eine verringerte lymphoide Zellularität von Organen und Geweben des Immunsystems und des hämatolymphopoetischen Systems wurden in präklinischen Toxizitätsstudien beobachtet und auf die pharmakologischen Eigenschaften (JAK3-/TEC-Hemmung) von Ritlecitinib zurückgeführt.
Die chronische Verabreichung von Ritlecitinib an Beagle-Hunde führte zum Auftreten einer axonalen Dystrophie bei systemischen Expositionen von mindestens dem 7.4fachen der erwarteten Exposition bei Patienten, die mit 50 mg pro Tag behandelt werden (basierend auf der ungebundenen AUC24). Die axonale Dystrophie steht vermutlich im Zusammenhang mit der Bindung an neuronale Off-Target-Proteine. Es ist nicht bekannt, ob bei niedrigeren systemischen Expositionen bei Hunden eine axonale Dystrophie auftrat. Bei einer systemischen Exposition, die 33-mal höher war als die erwartete Exposition bei Patienten, die mit 50 mg pro Tag behandelt werden (basierend auf der ungebundenen AUC24), war die axonale Dystrophie mit einem neurologischen Hörverlust verbunden. Zwar bildeten sich diese Befunde nach Beendigung der Ritlecitinib-Dosierung bei Hunden nachweislich zurück, aber bei chronischer Dosierung kann ein Risiko für Patienten nicht vollständig ausgeschlossen werden.
Genotoxizität
Ritlecitinib war im bakteriellen Mutagenitätstest (Ames-Test) nicht mutagen. Gemäss den Ergebnissen des In-vivo-Mikronukleustests an Rattenknochenmark ist Ritlecitinib bei Expositionen, die dem 130fachen der MRHD basierend auf der ungebundenen AUC entsprachen, nicht aneugen oder klastogen.
Kanzerogenität
In einer 6-monatigen Studie an Tg.ras H2-Mäusen, denen Ritlecitinib bei Expositionen verabreicht wurde, die dem 11fachen der MRHD basierend auf der ungebundenen AUC entsprachen, wurden keine Hinweise auf eine Tumorigenität festgestellt. In einer 2-Jahres-Studie zur Karzinogenität bei Ratten wurde nach Verabreichung von Ritlecitinib bei Expositionen, die dem 29fachen der MRHD auf Basis der ungebundenen AUC entsprachen, bei weiblichen Ratten eine höhere Inzidenz von gutartigen Thymomen und bei männlichen Ratten von gutartigen follikulären Schilddrüsenadenomen festgestellt. Bei dieser Ritlecitinib-Exposition kann eine höhere Inzidenz maligner Thymome bei weiblichen Ratten nicht ausgeschlossen werden. Bei Expositionen, die dem 6.3fachen der MRHD auf Basis der ungebundenen AUC entsprachen, wurden keine Ritlecitinib-bedingten Thymome oder follikulären Schilddrüsenadenome beobachtet.
Reproduktions- und Entwicklungstoxizität
Bei Expositionen, die dem 55fachen der MRHD auf Basis der ungebundenen AUC entsprachen, wurden keine Auswirkungen von Ritlecitinib auf die Fertilität weiblicher Ratten festgestellt. Auswirkungen auf die Fertilität männlicher Ratten (höhere Präimplantationsverluste, die zu einer Verringerung der Implantationsstellen und einer entsprechend geringeren Wurfgrösse bei unbehandelten Weibchen führten, die mit Ritlecitinib behandelten Männchen gepaart wurden) wurden bei einer Exposition in Höhe des 55fachen der MRHD auf Basis der ungebundenen AUC beobachtet. Bei Expositionen, die dem 14fachen der MRHD auf Basis der ungebundenen AUC entsprachen, wurden keine Auswirkungen auf die männliche Fertilität festgestellt. In der Fertilitätsstudie an Ratten wurden bei keiner Dosis Auswirkungen auf die Spermatogenese (Spermienzahl, Spermienproduktionsrate, Motilität und Morphologie) festgestellt.
In Studien zur embryofetalen Entwicklung an trächtigen Ratten führte die orale Verabreichung von Ritlecitinib zwischen dem 6. und 17. Trächtigkeitstag bei Expositionen, die grösser oder gleich dem 49fachen der ungebundenen AUC bei der MRHD waren, zu fetalen Skelettfehlbildungen und -variationen sowie zu einem niedrigeren fetalen Körpergewicht. Expositionen, die dem 16fachen der ungebundenen AUC bei der MRHD entsprachen, hatten keine Auswirkungen auf die embryofetale Entwicklung.
In einer Studie zur embryofetalen Entwicklung an trächtigen Kaninchen führte die orale Verabreichung von Ritlecitinib zwischen dem 7. und 19. Trächtigkeitstag bei Expositionen, die dem 55fachen der ungebundenen AUC bei der MRHD entsprachen, zu einem geringeren mittleren Körpergewicht der Feten und höheren Inzidenzen von viszeralen und Skelettfehlbildungen sowie Skelettvariationen. Expositionen, die dem 12fachen der ungebundenen AUC bei der MRHD entsprachen, hatten keine Auswirkungen auf die embryofetale Entwicklung.
In einer prä- und postnatalen Entwicklungsstudie an Ratten führte die orale Verabreichung von Ritlecitinib bei einer Exposition in Höhe des 41fachen der ungebundenen AUC bei der MRHD ab dem 6. Trächtigkeitstag bis zum 20. Laktationstag zu einer Entwicklungstoxizität mit geringerer postnataler Überlebensrate, geringerem Körpergewicht der Nachkommen und sekundären Entwicklungsverzögerungen. Weibliche Nachkommen der F1-Generation wiesen bei Expositionen, die dem 41fachen der ungebundenen AUC bei der MRHD entsprachen, eine geringere mittlere Anzahl von Corpora lutea auf. Expositionen, die dem 14fachen der ungebundenen AUC bei der MRHD entsprachen, hatten keine Auswirkungen auf die prä- und postnatale Entwicklung.
In einer Studie zur Toxizität bei juvenilen Ratten führte die orale Verabreichung von Ritlecitinib ab dem 10. bis zum 60. postnatalen Tag (vergleichbar mit dem Alter eines Säuglings bis zum Jugendalter beim Menschen) nicht zu Auswirkungen auf das Nerven- oder Skelettsystem.
Laktation
Nach Verabreichung von Ritlecitinib an laktierende Ratten waren die Ritlecitinib-Konzentrationen in der Muttermilch im Zeitverlauf höher als diejenigen im Plasma. Das mittlere AUC-Verhältnis zwischen Muttermilch und Plasma betrug 2.2.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Nicht über 30 °C lagern.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer69695 (Swissmedic).
PackungenLitfulo 50 mg Hartkapseln: 30 (Blisterpackung). [B]
ZulassungsinhaberinPfizer AG, Zürich.
Stand der InformationOktober 2024
LLD V003
|