Eigenschaften/WirkungenATC-Code
D11AH12
Wirkungsmechanismus
Nemolizumab ist ein humanisierter monoklonaler IgG2-Antikörper, der die Interleukin-31-(IL-31-)-Signaltransduktion hemmt, indem er selektiv an den Interleukin-31-Rezeptor alpha (IL-31 RA) bindet. IL-31 ist ein natürlich vorkommendes Zytokin, das an Pruritus, Entzündung, epidermaler Dysregulation und Fibrose beteiligt ist. Nemolizumab hemmt IL-31-induzierte Reaktionen, einschließlich der Freisetzung von proinflammatorischen Zytokinen und Chemokinen.
Pharmakodynamik
Klinische Wirksamkeit
1) ) Klinische Wirksamkeit und Sicherheit bei Erwachsenen und Jugendlichen mit atopischer Dermatitis
Die Wirksamkeit und Sicherheit von Nemluvio mit begleitender topischer Hintergrundtherapie wurde in zwei randomisierten, doppelblinden, placebokontrollierten Pivotstudien (ARCADIA 1 und ARCADIA 2) beurteilt, in die total 1’728 Studienteilnehmer ab 12 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis aufgenommen wurden, die durch topische Behandlungen nicht ausreichend kontrolliert werden konnte. Der Schweregrad der Erkrankung wurde durch einen IGA (Investigator's Global Assessment)-Score von 3 (mittelschwer) und 4 (schwer) in der Gesamtbeurteilung der atopischen Dermatitis, einen EASI (Eczema Area and Severity Index)-Score von ≥ 16, eine minimale betroffene Körperoberfläche (BSA) von ≥ 10 % und einen PP-NRS (Peak Pruritus Numeric Rating Scale)-Score von ≥ 4 definiert.
Die Patienten in den Studien erhielten entweder eine erste subkutane Injektion von Nemolizumab 60 mg gefolgt von 30 mg Injektionen alle 4 Wochen (Q4W) oder entsprechendes Placebo. Begleitende TCS mit niedriger und/oder mittlerer Wirkstärke (gemäss United States classification) und/oder TCI wurden sowohl in den Nemolizumab- als auch in den Placebo-Gruppen für mindestens 14 Tage vor Baseline verabreicht und während der Studie fortgesetzt. Je nach Krankheitsaktivität konnten diese Begleittherapien nach Ermessen des Prüfarztes ausgeschlichen und/oder abgesetzt werden.
Nach 16 Wochen setzten die Patienten, die einen Erfolg nach EASI-75 oder IGA erzielten, für weitere 32 Wochen die Erhaltungsphase der Studie fort, um die Aufrechterhaltung des in Woche 16 erreichten Ansprechens zu beurteilen. Nemluvio-Responder wurden erneut randomisiert und erhielten entweder Nemluvio 30 mg alle 8 Wochen oder Placebo alle 4 Wochen (alle Gruppen setzten die Behandlung mit TCS/TCI fort). Patienten, die im anfänglichen Behandlungszeitraum zu Placebo randomisiert wurden und in Woche 16 das gleiche klinische Ansprechen erreichten, erhielten weiterhin alle 4 Wochen Placebo. Non-Responder in Woche 16, Patienten, die im Erhaltungszeitraum das klinische Ansprechen verloren, und Patienten, die den Erhaltungszeitraum absolviert hatten, hatten die Möglichkeit, in die offene Studie (ARCADIA LTE) aufgenommen zu werden und bis zu 200 Wochen lang alle 4 Wochen eine Behandlung mit Nemluvio 30 mg zu erhalten.
Endpunkte
Sowohl ARCADIA 1 als auch ARCADIA 2 bewerteten die primären Endpunkte bezüglich:
§Anteil der Patienten mit einem IGA-Erfolg (definiert als ein IGA von 0 [befallsfrei] oder 1 [nahezu befallsfrei] und einer Reduktion um ≥2 Punkte gegenüber Studienbeginn) in Woche 16
§Anteil der Patienten mit EASI-75 (≥75 % Verbesserung des EASI gegenüber Studienbeginn) in Woche 16
Sekundäre Hauptendpunkte waren PP-NRS-Verbesserung ≥4 gegenüber Baseline in den Wochen 1, 2, 4 und 16, PP-NRS <2 in Woche 4 und Woche 16, Verbesserung auf der SD-NRS (Sleep Disturbance Numeric Rating Scale) ≥4 gegenüber Baseline in Woche 16, Patienten mit einer Verbesserung sowohl des EASI-75 als auch des PP-NRS ≥4 gegenüber Baseline in Woche 16 und Patienten mit sowohl IGA-Erfolg als auch Verbesserung des PP-NRS ≥4 gegenüber Baseline in Woche 16.
Merkmale bei Studienbeginn
In diesen Studien waren bei Studienbeginn 51,0 % der Patienten männlich, 79,9 % waren weiss und 15,4 % waren 12 bis 17 Jahre alt. 70 % der Patienten hatten bei Studienbeginn einen IGA-Score von 3 (mittelschwere AD) und 30 % der Patienten einen IGA-Score von 4 (schwere AD). Der mittlere EASI-Wert bei Studienbeginn lag bei 27,5, der wöchentliche PP-NRS-Wert bei Studienbeginn betrug 7,1 (schwerer Juckreiz) und der wöchentliche SD-NRS-Wert lag bei Studienbeginn bei 5,8. Gesamthaft hatten 63,3 % der Patientinnen und Patienten andere vorgängige systemische Behandlungen für atopische Dermatitis erhalten.
Klinische Wirksamkeit - ARCADIA 1 und ARCADIA 2 – Erwachsene und Jugendliche – Induktionsphase, Woche 0 bis Woche 16
Nemluvio war im Vergleich zu Placebo hinsichtlich der hautbezogenen koprimären Endpunkte IGA-Erfolg und EASI-75 über 16 Wochen statistisch signifikant überlegen (Tabelle 2). Die Ergebnisse für beide koprimären Endpunkte waren bei der Population mit starkem Juckreiz (Baseline PP NRS ≥7) konsistent.
Tabelle 2 – Wirksamkeitsergebnisse für Nemluvio (30 mg Q4W) mit begleitenden TCS/TCI in ARCADIA 1 und ARCADIA 2 in Woche 16
|
|
ARCADIA 1
|
ARCADIA 2
| |
Nemluvio + TCS/TCI
|
Placebo +
TCS/TCI
|
Nemluvio + TCS/TCI
|
Placebo +
TCS/TCI
| |
Anzahl der randomisierten und behandelten Patienten (PP NRS bei Studienbeginn ≥4)
|
620
|
321
|
522
|
265
| |
% der Patienten mit IGA 0 oder 1a
|
35,6#
|
24,6
|
37,7#
|
26,0
| |
% der Patienten mit EASI-75a
|
43,5*
|
29,0
|
42,1#
|
30,2
|
a Patienten, die eine Bedarfsbehandlung erhielten, oder mit fehlenden Daten wurden als Non-Responder eingeordnet
* p-Wert <0,0001, #p-Wert <0,001Der schichtbereinigte p-Wert basiert auf dem CMH-Test, stratifiziert nach PP NRS und IGA-Score bei Studienbeginn
Abbildung 1 – Anteil der Patienten mit IGA-Erfolg und EASI-75 von Studienbeginn bis Woche 16 in ARCADIA 1 und ARCADIA 2
Eine signifikante Verbesserung des Pruritus bei Patienten, die in ARCADIA 1 und ARCADIA 2 mit Nemluvio behandelt wurden, im Vergleich zu Placebo, basierend auf Verbesserung der PP NRS von ≥4 gegenüber Studienbeginn wurde ab Woche 1 beobachtet und bis Woche 16 aufrechterhalten (Tabelle 3). Die Ergebnisse waren bei der Population mit schwerem Juckreiz (Ausgangswert PP-NRS ≥7) konsistent.
Tabelle 3 – Wirksamkeitsergebnisse zu Juckreiz bei Nemluvio mit begleitenden TCS/TCI in ARCADIA 1 und ARCADIA 2 bis Woche 16
|
|
ARCADIA 1
|
ARCADIA 2
| |
Nemluvio + TCS/TCI
|
Placebo + TCS/TCI
|
Nemluvio + TCS/TCI
|
Placebo + TCS/ TCI
| |
Anzahl der randomisierten und behandelten Patienten (PP NRS bei Studienbeginn ≥4)a
|
620
|
321
|
522
|
265
| |
% der Patienten mit PP-NRS-Verbesserung ≥4a
| |
In Woche 1
|
4,7§
|
1,2
|
6,7*
|
0,4
| |
In Woche 2
|
17,7*
|
3,1
|
16,9*
|
1,9
| |
In Woche 4
|
27,4*
|
6,5
|
26,1*
|
5,3
| |
In Woche 16
|
42,7*
|
17,8
|
41,0*
|
18,1
| |
% der Patienten mit PP NRS <2a
| |
In Woche 4
|
16,0*
|
3,7
|
15,9*
|
2,6
| |
In Woche 16
|
30,6*
|
11,2
|
28,4*
|
11,3
|
a Patienten, die eine Bedarfsbehandlung erhielten, oder mit fehlenden Daten wurden als Non-Responder eingeordnet
* p-Wert <0,0001, # p-Wert <0,001, § p-Wert <0,05 Der schichtbereinigte p-Wert basiert auf dem CMH-Test, stratifiziert nach PP NRS und IGA-Score bei Studienbeginn.
Die numerische Bewertungsskala für Schlafsörungen (SD NRS) ist eine Tagesskala, die von den Probanden verwendet wird, um den Grad ihres Schlafverlustes im Zusammenhang mit atopischer Dermatitis zu melden. In der Woche 16 wurde im Vergleich zu Placebo eine signifikante Verbesserung der Schlafstörung beobachtet (Tabelle 4). Die Ergebnisse waren bei der Population mit starken Juckreiz (Ausgangswert PP NRS ≥7) konsistent.
Tabelle 4 – Wirksamkeit bei Schlafstörungen mit Nemluvio mit begleitenden TCS/TCI in ARCADIA 1 und ARCADIA 2 in Woche 16
|
|
ARCADIA 1
|
ARCADIA 2
| |
Nemluvio + TCS/TCI
|
Placebo + TCS/TCI
|
Nemluvio + TCS/TCI
|
Placebo + TCS/ TCI
| |
Anzahl der randomisierten und behandelten Patienten (PP NRS bei Studienbeginn ≥4)a
|
620
|
321
|
522
|
265
| |
% der Patienten mit SD-NRS-Verbesserung ≥4a
Mittlere Veränderung gegenüber Studienbeginn (%)
|
37,9*
-64,6
|
19,9
-38,1
|
33,5*
-59,7
|
16,2
-35,4
|
a Teilnehmende, die eine Bedarfsbehandlung erhielten, oder mit fehlenden Daten wurden als Non-Responder eingeordnet
* p-Wert <0,0001Der schichtbereinigte p-Wert basiert auf dem CMH-Test, stratifiziert nach PP NRS und IGA-Score bei Studienbeginn
Jugendliche mit atopischer Dermatitis (12 bis 17 Jahre)
Die Ergebnisse zur Wirksamkeit der Studien ARCADIA 1 und ARCADIA 2 in Woche 16 für pädiatrische Probanden im Alter von 12 bis 17 Jahren sind in Tabelle 5 dargestellt. Die Ergebnisse in der pädiatrischen Probandengruppe stimmten im Allgemeinen mit den Ergebnissen in der erwachsenen Probandengruppe überein. Die Ergebnisse in den koprimären und wichtigsten sekundären Endpunkten waren in der Gruppe mit starkem Juckreiz (Baseline PP NRS ≥7) konsistent.
Tabelle 5 – Ergebnisse zur Wirksamkeit von Nemolizumab (30 mg Q4W) mit gleichzeitiger Gabe von TCS/TCI in ARCADIA 1 und ARCADIA 2 in Woche 16 bei pädiatrischen Probanden im Alter von 12 bis 17 Jahren
|
|
ARCADIA 1 AND ARCADIA 2
| |
Nemluvio+ TCS/TCI
|
Nemoluvio + TCS/TCI
| |
Anzahl der behandelten und randomisierten Patienten (Baseline PP NRS ≥4)
|
179
|
90
| |
% der Patienten mit IGA 0 or 1 a
|
48.9*
|
34.4
| |
% der Patienten mit EASI-75 a
|
53.4§
|
43.3
|
a Probanden, die eine Notfallbehandlung erhielten oder bei denen Daten fehlten, wurden als Non-Responder betrachtet
≠p-Wert <0,0001, #p-Wert <0,001, *p-Wert <0,05, ∞p-Wert =0,0591, §p-Wert =0,1824
Der stratifizierte p-Wert basiert auf dem CMH-Test, der nach PP-NRS- und IGA-Score zu Studienbeginn stratifiziert wurde.
Klinische Wirksamkeit - ARCADIA 1 und ARCADIA 2 – Erwachsene und Jugendliche – Erhaltungsphase, Woche 16 bis Woche 48
Das klinische Ansprechen bei Nemolizumab-Respondern (IGA 0/1 oder EASI-75 in Woche 16) wurde zwischen Woche 16 und Woche 48 in den Studien ARCADIA 1 und ARCADIA 2 ausgewertet. Für die Erhaltungsbehandlung wurden 338 Nemolizumab-Responder erneut randomisiert und erhielten entweder Nemolizumab 30 mg Q8W oder Placebo Q4W (Nemolizumab-Absetzen) mit gleichzeitiger TCS/TCI. In Woche 48 erreichten in der Nemolizumab 30 mg + TCS/TCI Q8W Gruppe 60.4% der Patienten ein IGA 0/1 und 75.7% ein EASI-75, verglichen mit 49.7% ein IGA 0/1 und 63.9% ein EASI-75 in der Placebo + TCS/TCI Q4W Gruppe.
2) Klinische Wirksamkeit und Sicherheit bei Erwachsenen mit Prurigo nodularis
Die Wirksamkeit und Sicherheit von Nemluvio als Monotherapie wurde in zwei randomisierten, doppelblinden, placebokontrollierten Pivotstudien (OLYMPIA 1 und OLYMPIA 2) untersucht, in die total 560 Teilnehmende ab 18 Jahren mit Prurigo nodularis aufgenommen wurden. Der Schweregrad der Erkrankung wurde anhand einer allgemeinen Beurteilung durch die Prüfärztin/den Prüfarzt (IGA) bei der Gesamtbeurteilung der Prurigo nodularis-Knoten auf einer Schweregradskala von 0 bis 4 definiert. Die in diese beiden Studien aufgenommenen Patienten hatten einen IGA-Score von ≥3, schweren Pruritus, definiert durch einen wöchentlichen Durchschnitt des Wertes auf der numerischen Bewertungsskala für den Pruritus-Spitzenwert (PP NRS) von ≥7 auf einer Skala von 0 bis 10 sowie mindestens 20 noduläre Läsionen. OLYMPIA 1 und OLYMPIA 2 beurteilten die Wirkung der Nemluvio-Monotherapie auf die Anzeichen und Symptome von Prurigo nodularis und zielten auf die Verbesserung der Hautläsionen und des Pruritus über 16 Wochen ab. OLYMPIA 1 hatte einen 24-wöchigen und OLYMPIA 2 einen 16-wöchigen Behandlungszeitraum.
Teilnehmende, die OLYMPIA 1 und OLYMPIA 2 abschlossen, hatten die Möglichkeit, in die offene Studie (OLYMPIA LTE) aufgenommen zu werden und bis zu 184 Wochen lang alle 4 Wochen mit Nemluvio behandelt zu werden.
Teilnehmende mit einem Körpergewicht von weniger als 90 kg in der Nemluvio-Monotherapiegruppe erhielten in Woche 0 subkutane Injektionen von Nemluvio 60 mg (2 Injektionen mit 30 mg), gefolgt von Injektionen mit 30 mg alle 4 Wochen. Teilnehmende mit einem Körpergewicht von 90 kg oder mehr in der Nemluvio-Monotherapiegruppe erhielten in Woche 0 subkutane Injektionen von Nemluvio 60 mg (2 Injektionen mit 30 mg) sowie alle 4 Wochen.
Endpunkte
Sowohl OLYMPIA 1 als auch OLYMPIA 2 bewerteten die gleichen beiden primären Endpunkte:
§Anteil der Patienten mit einer Verbesserung von ≥4 gegenüber Studienbeginn auf der numerischen Bewertungsskala für den Pruritus-Spitzenwert (NRS) in Woche 16
§Anteil der Patienten mit einem IGA-Erfolg (definiert als ein IGA von 0 [befallsfrei] oder 1 [nahezu befallsfrei] und einer Verbesserung um ≥2 Punkte gegenüber Studienbeginn) in Woche 16
Die wichtigsten sekundären Endpunkte umfassten eine PP-NRS-Verbesserung ≥4 gegenüber Studienbeginn in Woche 4, PP-NRS <2 in Woche 4 und Woche 16, eine SD-NRS-Verbesserung ≥4 gegenüber Studienbeginn in Woche 4 und 16.
Merkmale bei Studienbeginn
In diesen Studien waren zu Studienbeginn 59,6 % der Patienten weiblich, 81,4 % waren weiss und 25,4 % waren älter als 65 Jahre. Der wöchentliche durchschnittliche PP-NRS-Score bei Studienbeginn betrug im Mittel (SD) 8,4 (0,9). 58 % der Patienten hatten bei Studienbeginn einen IGA-Score von 3 (mässige PN) und 42 % der Patienten hatten bei Studienbeginn einen IGA von 4 (schwere PN).
Klinische Wirksamkeit
Monotherapiestudien (OLYMPIA 1 und OLYMPIA 2) – Woche 0 bis Woche 16
Die Ergebnisse der Pivotstudien zur Beurteilung der Behandlung von Nemluvio in OLYMPIA 1 und OLYMPIA 2 werden in Tabelle 6 dargestellt und zeigen eine signifikante Verbesserung bei mit Nemluvio behandelten Patienten im Vergleich zu Placebo sowohl für die primären Endpunkte (Abbildung 2 und Abbildung 3) als auch für die wichtigsten sekundären Endpunkte.
Tabelle 6 – Wirksamkeitsergebnisse für Nemluvio-Monotherapie (Q4W) in OLYMPIA 1 und OLYMPIA 2
|
|
OLYMPIA 1
|
OLYMPIA 2
| |
Nemluvio
|
Placebo
|
Nemluvio
|
Placebo
| |
Anzahl der randomisierten Patienten
|
190
|
96
|
183
|
91
| |
% der Patienten mit einer PP-NRS-Verbesserung ≥4 gegenüber Studienbeginna
| |
Woche 4
|
41,1*
|
6,3
|
41,0*
|
7,7
| |
Woche 16
|
58,4*
|
16,7
|
56,3*
|
20,9
| |
% der Patienten mit IGA 0 oder 1 in Woche 16a
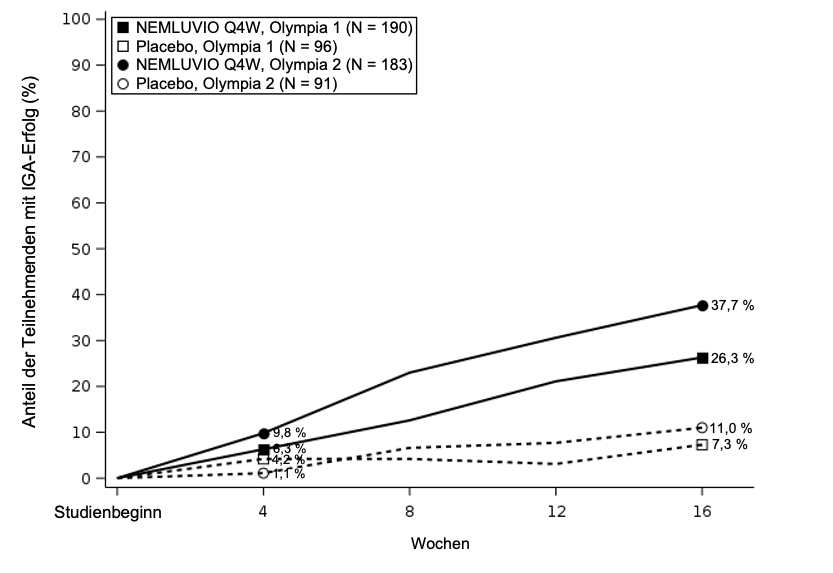
|
26,3#
|
7,3
|
37,7*
|
11
| |
% der Patienten mit PP NRS <2a
| |
Woche 4
|
21,6*
|
1,0
|
19,7*
|
2,2
| |
Woche 16
|
34,2*
|
4,2
|
35,0*
|
7,7
| |
% der Patienten mit einer SD-NRS-Verbesserung ≥4 gegenüber Studienbeginna
| |
Woche 4
|
31,1*
|
5,2
|
37,2*
|
9,9
| |
Woche 16
|
50,0*
|
11,5
|
51,9*
|
20,9
|
a Wenn Teilnehmende eine Bedarfstherapie erhalten haben, wird eine Strategie mit zusammengesetzten Variablen angewandt: Die zugrunde liegenden Daten bei/nach Erhalt der Bedarfstherapie werden als schlechtest möglicher Wert festgelegt, und das Ansprechen wird aus dem zugrunde liegenden Datenwert abgeleitet. Teilnehmende mit fehlenden Ergebnissen gelten als Non-Responder.
b Nicht angepasst für Multiplizität
* p-Wert <0,0001, # p-Wert = 0,0025 Strata angepasst anhand der randomisierten Stratifizierungsvariablen (Analysenzentrum und Körpergewicht bei Studienbeginn (<90 kg, ≥90 kg)
§ p-Wert <0,0001 Strata-bereinigt vs. Placebo (ANCOVA MI-MAR)
|
Abbildung 2 – Anteil der Patienten mit PP-NRS-Verbesserung von ≥4 gegenüber Studienbeginn bis Woche 16
| |
| |
Abbildung 3 – Anteil der IGA-Responder von Studienbeginn bis Woche 16
| |
|
|
|